

Abstract
Exposure to house dust mite (HDM) is highly associated with the development of allergic asthma. The adaptive immune response to HDM is largely T helper cell type 2 and type 17 (Th2 and Th17) dominant and a number of innate immune receptors have been identified which recognize HDM to initiate these responses. Nucleotide-binding Oligomerization Domain-containing Protein 2 (NOD2) is a cytosolic sensor of peptidoglycan which is important for Th2 and Th17 polarization. NOD2 mediates its signaling through its downstream effector kinase, Receptor-interacting Serine/Threonine Protein Kinase 2 (RIP2). We have previously shown that RIP2 promotes HDM-associated allergic airway inflammation and Th2 and Th17 immunity, acting early in the HDM response and likely, within airway epithelial cells. However, the consequences of inhibiting RIP2 during this critical period has not yet been examined. In this study, we pharmacologically inhibited RIP2 activity during the initial exposure to allergen in an acute HDM model of asthma and determined the effect on the subsequent development of allergic airway disease. We show that early inhibition of RIP2 was sufficient to reduce lung histopathology and local airway inflammation while reducing the Th2 immune response. Using a chronic HDM asthma model, we demonstrate that inhibition of RIP2, despite attenuating airway inflammation and airway remodeling, was insufficient to reduce airway hyperresponsiveness. These data demonstrate the potential of pharmacological targeting of this kinase in asthma and support further development and optimization of RIP2 targeted therapies.
Keywords: RIP2, NOD2, allergic asthma, house dust mite, Th2
Introduction
Asthma is a chronic inflammatory disease of the airways which affects up to 300 million people worldwide (1). It is characterized by narrowing of the airways, mucus overproduction, airway hyperresponsiveness (AHR) and pathological airway remodeling. Two endotypes (subgroups with similar pathophysiology) are generally recognized for asthma: Type 2 (or type 2 high) and non-type 2 (or type 2 low) (2). Around half of all asthmatics have type 2 inflammation and, of these, the majority suffer from allergic asthma (3). Exposure and sensitization to indoor allergens such as pet dander, cockroaches and dust mites are consistently associated with the development of allergic asthma (4–6). Of these aeroallergens, house dust mites (HDM) are the most frequent sensitizer, with up to 70% of asthmatics showing reactivity to this allergen (7–11).
Type 2 or type-2 helper T cell (Th2) immunity is distinguished by elevated production of the canonical type 2 cytokines IL-4, IL-5 and IL-13 (12). In the pathologic setting of allergic asthma, these cytokines are critical mediators which collectively result in the observed asthmatic symptoms. The large number of therapeutics developed to target these type 2 cytokines and their receptors, or type 2-induced IgE and IgE receptors, as well as clinical translation of such biologics, reflect their importance in the progression of type 2 disease (13). However, the limited indication for such biologics (type 2 asthmatics who have severe disease or high eosinophil counts) as well as the high cost and need for chronic administration, point to the need for novel therapeutic targets which lie upstream of type 2 cytokine production and which have the potential to modulate the type 2 response.
Initiation of type 2 immunity in response to HDM is a process orchestrated by multiple cell types, including epithelial cells, dendritic cells (DCs), group 2 innate lymphoid cells (ILC2s), mast cells, and basophils (12). Likewise, numerous receptors and pathways in innate and structural cells have been implicated in the initial recognition and response to HDM (14). Using knock-out models, our own laboratory has previously demonstrated that Receptor-interacting Serine/Threonine Protein Kinase 2 (RIPK2 or RIP2) is involved in promoting allergic airway inflammation in response to HDM (15). Importantly, multiple lines of evidence, including HDM-induced activation of RIP2 within airway epithelial cells, suggested that the actions of RIP2 were crucial at a very early timepoint. RIP2 is a kinase which mediates signaling downstream of Nucleotide-binding and Oligomerization Domain-Protein 2 (NOD2), a cytosolic receptor for bacterial peptidoglycan (16, 17). Apart from allergic airway inflammation, RIP2 has also been associated with the pathogenesis of various inflammatory diseases including inflammatory bowel disease (IBD), arthritis, and experimental autoimmune encephalomyelitis (EAE) (18–20). These findings have spurred the development and preclinical testing of numerous RIP2 inhbitors as well as initiation of a Phase I clinical trial (21). In the current work, we examine the efficacy of early and acute pharmacological inhibition of RIP2 in the setting of a HDM-induced allergic asthma model. The resulting findings may lend further support for RIP2 as being a viable druggable target in the setting of allergic asthma.
Materials and Methods
Mice.
C57BL/6J mice (000664) were obtained from Jackson Laboratories (Bar Harbor, Maine) and were housed in specific pathogen free (SPF) and AALAC-accredited animal facility at the UCF Health Science Campus at Lake Nona. Eight week old male and female mice were used for the experiments. All animal procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Central Florida and using a reviewed and approved animal protocol.
House Dust Mite Asthma Models and GSK Intervention.
Lyophilized Dermatophagoides pteronyssinus (house dust mite, HDM) was obtained from Greer Laboratories (XPB82D3A2.5, Lot no. 346230) (Lenoir, NC), resuspended in sterile PBS at necessary concentrations, and frozen in aliquots at −20C until use. For all models, irradiated GSK583 chow (0.25g GSK583/kg chow, C19062701i, Research Diets, New Brunswick, NJ) or irradiated vehicle chow (C13513i, Research Diets, New Brunswick, NJ) was provided ad libitum on days −2 to day 2. This amount will deliver approximately 30 mg/kg/day based on a 3g/day consumption per mouse. On day 3, all mice were switched to vehicle chow.
The acute house dust mite model was conducted as previously published (15). On day 0, 40 μL of a 1.5mg/ml HDM mixture was delivered intratracheally (i.t.) while 25 μL of a 0.5mg/ml HDM mixture was delivered i.t. from days 7–11. For the chronic house dust mite model, mice were additionally administered 25 μL of a 0.5mg/ml HDM mixture on days 14, 16, 18, 21, 23, and 25. All intratracheal instillations of HDM were performed using an endotracheal instillation kit with animals under isoflurane anesthesia.
Mice were euthanized on day 14 or day 28 (depending on the model) for collection of tissues. Depending on the experiment and model duration, the following tissues were collected: bronchoalveolar lavage and lung for analysis of infiltrating immune subsets, blood for serum analysis, lung for measurement of local cytokines, lung for analysis of Th subsets and lung for histological assessment.
Histopathological scoring.
Lung tissue was harvested from euthanized mice for histology. Tissue was fixed in 10% buffered formalin and sent to AML labs (St. Augustine, FL) for paraffin embedding, sectioning, and Hematoxylin & Eosin (H&E), Periodic Acid-Schiff (PAS), and Masson’s Trichrome staining to assess total inflammation, mucus production and collagen deposition/fibrosis, respectively. All sections were scored blindly using a modified histopathological scoring system as previously published (15, 22). Bronchoarterial inflammation, pulmonary vein inflammation, amuscular blood vessel inflammation, interalveolar space inflammation and pleural inflammation were combined into one Inflammatory Index with a maximum score of 16. The maximum score for mucus production was 4, and the maximum score for trichrome staining was 6 (the maximum score being the most severe). For the chronic model, an alternative scoring system to assess chronic airway remodeling was used, for a maximum combined score of 22. The following parameters were assessed for incidence throughout the section: epithelial folding/distortion (0–2), goblet cell metaplasia (0–2), smooth muscle cell hypertrophy (0–2), subepithelial fibrosis (0–2), peribronchial trichrome blue staining (0–3), and parenchymal collagen deposition (0–3). In addition, the following parameters were assessed for severity: epithelial folding/distortion (0–2), goblet cell metaplasia (0–2), smooth muscle cell hypertrophy (0–2), and subepithelial fibrosis (0–2).
Flow cytometry.
On day 14, bronchoalveolar lavage (BAL) and a portion of the right lung lobe were collected and processed as previously described. To assess cellular infiltrate, one million cells of lung or BAL were stained using an antibody cocktail against the following mouse antigens: Siglec-F (clone E50–2440), CD11b (clone M1/70), Ly-6G (clone 1A8), CD11c (clone N418), CD45 (clone 30-F11), CD3 (clone 145–2C11) and B220 (clone RA3–6B2). All antibodies were from Biolegend, eBioscience/ThermoFisher and BD Biosciences. The gating strategies were performed as follows. Lung or BAL cells were gated for singlets. Of these, an FSC vs CD45 plot was used to further gate CD45+ cells. A plot of CD11c versus Siglec-F gated on CD45+ cells was used to discriminate alveolar macrophages (CD11c+Siglec-F+) from eosinophils (CD11c−Siglec-F+). A plot of CD11b versus Ly-6G gated on CD45+ cells was used to identify neutrophils (CD11b+Ly-6G+). A plot of SSC versus CD3 and B220 gated on CD45+ cells was used to identify lymphocytes (CD3+ and B220+, SSClow).
For intracellular cytokine staining, cell suspensions were obtained from lung tissue, stimulated with PMA (5ng/mL) and ionomycin (500ng/mL) in the presence of Brefeldin A and monensin and stained for cell surface antigens and intracellular cytokines. For cell surface staining, antibodies against mouse CD45 (clone 30-F11) and mouse CD4 (clone GK1.5) were used (Biolegend). The cells were then fixed and permeabilized using a Fixation/Permeabilization kit (BD Biosciences) and stained intracellularly using antibodies against mouse IL-4 (clone 11B11), mouse IL-5 (clone TRFK5), mouse IL-17A (clone TC11–18H10.1) and mouse IFN-γ (clone XMG 1.2) (all from Biolegend). All samples were acquired using a Novocyte flow cytometer (ACEA Biosciences, San Diego, CA) and analyzed using the NovoExpress Software.
Lung homogenate cytokine analysis.
A portion of the right lobe of the lung was homogenized in T-PER buffer with protease inhibitors (ThermoFisher, Waltham, MA). Samples were normalized to 0.5 mg/mL protein in Legendplex assay buffer (Biolegend, San Diego, CA) using a Bradford Assay (BioRad, Hercules, CA). A 13-plex bead-based immunoassay was performed using the manufacturer’s instructions (Legendplex Th Panel, Biolegend, San Diego, CA). All samples were acquired using Novocyte flow cytometer (ACEA Biosciences, San Diego, CA) and analyzed using the Legendplex Data Analysis software (v8, Vigenetech, Carlisle, MA). Using the provided standards, the analysis software automatically performs curve fitting and returns values for experimental samples only if these fall within the bounds of the standard curve generated (i.e. invalid samples do not return a value or are expressed by inequalities).
Serum antibody analysis.
Mice were subjected to cardiac puncture for collection of blood, which was clotted for 30 minutes in serum separator tubes prior to centrifugation for 10 mins at 4C for collection of serum. Serum was diluted 1:10 in assay diluent. Murine anti-HDM IgG1 and IgG ELISA kits were obtained from Chrondrex, Inc. (Redmont, WA) and used as directed by the manufacturer. Assays were developed and analyzed as previously described (15).
Flexivent analysis for airway hyperresponsiveness.
On day 28, mice were anesthetized using a cocktail of Ketamine/Xylazine/Acepromazine (65 mg/kg, 13 mg/kg, 2mg/kg, respectively). Once appropriate plane of anesthesia was achieved, mice were cannulated intratracheally and connected to a FlexiVent mechanical ventilator (Scireq). Basal lung mechanics and airway hyperresponsiveness to increasing doses of inhaled aerosolized methacholine (0, 3.125, 6.25, 12.5, 25, 50, and 100mg/mL) was measured using pre-set scripts. Deep Inflation, SnapShot-150, Quick Prime-3, and PVs-P perturbations were collected at baseline three times. For each dose, SnapShot-150 and Quick Prime-3 perturbations were collected 12 times. Analysis was conducted using the FlexiWare 7 Software (Scireq).
Statistical analysis.
Statistical analysis was conducted using GraphPad Prism. Significance levels were fixed at 5% for each measured response. Figure legends indicate specific tests used for analysis of each dataset, number of animals per group, and number of times the experiment was repeated. Bar heights indicate means and error bars indicate SEM.
Results
Early inhibition of RIP2 reduces lung pathology in mice subjected to an acute house dust mite asthma model.
We have previously shown that genetic loss of RIP2 attenuates lung pathology in response to HDM exposure (15). In the prior study, we additionally had evidence to indicate that RIP2 was important in the HDM-induced pro-inflammatory chemokine release in the lung very early during exposure (within 24hrs). Follow-up bone marrow chimera studies suggested that RIP2 in the non-hematopoetic compartment was important for mediating this effect (Supplemental Fig 1.). However, in the absence of conditional, inducible, or floxed RIP2 strains, we chose instead to investigate the role of RIP2 specifically during the early reponse to HDM through pharmacological inhibition. We subjected mice to an acute HDM model of asthma and provided a selective RIP2 inhibitor (GSK583) via chow at a dose of 30 mg/kg/day on the 5 days during and surrounding the initial exposure to HDM (days −2 to 2, Figure 1A). This would allow us to distinguish early involvement of RIP2 within structural cells and APCs versus possible direct effects on adaptive immunity. The specificity, mechanism and in vivo inhibitory activity of GSK583 have been previously reported elsewhere (23). On day 14 of this model, mice were euthanized and lungs were harvested and fixed for histological examination. Paraffin-embedded sections were stained using Hematoxylin & Eosin (H&E), Periodic Acid Schiff (PAS), or Trichrome stains, then scored blindly. We used a combination of parameters to create an inflammatory index (maximum score of 16) comprised of individual scores for bronchoarterial inflammation, amuscular blood vessel inflammation, inter-alveolar space inflammation, pleural inflammation, and pulmonary vein inflammation. Early inhibition of RIP2 in GSK583-treated mice was sufficient to reduce overall inflammation compared to their vehicle-treated counterparts (Figure 1B, with corresponding graph in Figure 1C). Similarly, this early inhibition of RIP2 using GSK583 was sufficient to reduce mucus production as seen with PAS staining (magenta in PAS stain in Figure 1B, with corresponding graph in Figure 1D) and collagen deposition as seen with trichrome staining (blue in trichrome stain in Figure 1B, with corresponding graph in Figure 1G) compared to vehicle-treated mice. In addition, GSK583-treated mice demonstrated reduced lumen narrowing (increased lumen area) (Figure 1E), and decreased epithelial thickness (Figure 1F) compared to vehicle-treated mice subjected to an acute HDM asthma model. Collectively, these data show that RIP2 inhibition using GSK583 during the initial exposure to HDM is sufficient to improve lung pathology in an acute house dust mite asthma model.
Figure 1. Early prophylactic inhibition of RIP2 reduces lung pathology in an acute house dust mite (HDM) model of asthma.
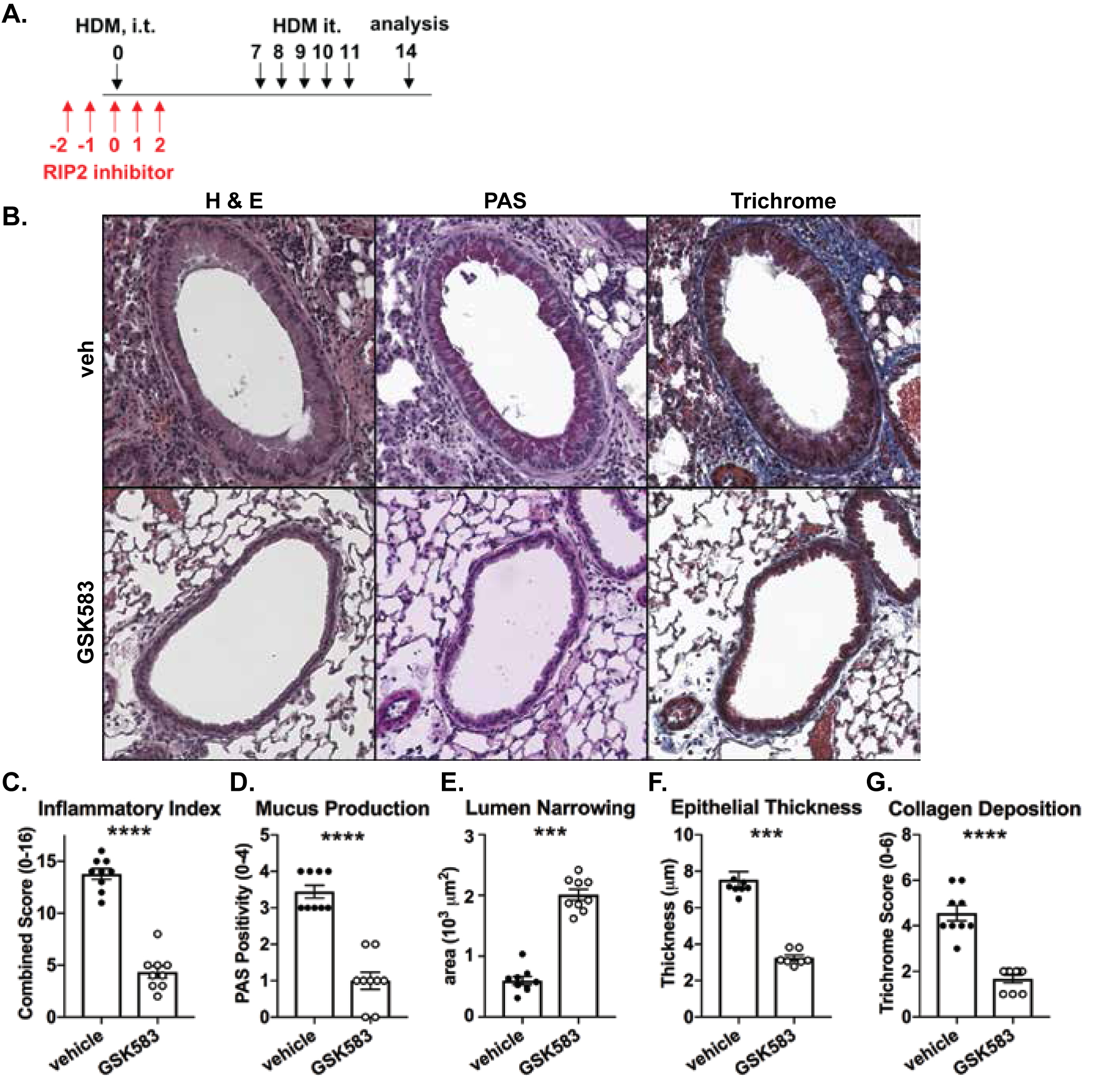
A.) WT C57BL/6 mice were subjected to an acute HDM asthma model and were administered either regular chow or chow containing RIP2 inhibitor (GSK583 at 30mg/kg/day) for the 5 days during and surrounding the initial exposure to HDM (red arrows). Mice were euthanized on day 14 and lungs were harvested, fixed in 10% formalin, and embedded in paraffin. B.) Consecutive sections were stained with either H&E, PAS, or Trichrome. Severity of lung pathology was scored based on C.) inflammatory index, D.) mucus production, E.) lumen narrowing, F.) epithelial thickness, and G.) collagen deposition and was performed by a blinded observer. Data are presented as scatterplots with bars where bar heights represent means ± SEM. Filled in circles represent individual datapoints for vehicle/control chow-treated mice and open circles represent individual datapoints for GSK583-treated mice. Data are pooled from 3 independent experiments for a total of n=9 mice per group. Statistical analysis was performed using an unpaired, two-tailed Student’s t-test. *** = p<0.001, **** = p<0.0001.
Early prophylactic inhibition of RIP2 reduces eosinophilia and lung neutrophilia in a HDM model of asthma.
To assess the effect of early pharmacological inhibition of RIP2 in an HDM asthma model on the recruitment of inflammatory cells to the airway, we harvested bronchoalveolar lavage (BAL) and lung cells and stained these using a panel of antibodies which would allow discrimination of hematopoietic cells, lymphocytes, neutrophils, alveolar macrophages, and eosinophils when subjected to flow cytometric analysis. Figure 2A shows that mice undergoing pharmacological inhibition of RIP2 during initial exposure to HDM allergen (GSK583-treated mice) have significantly reduced numbers of CD45+ cells and eosinophils recovered from the BAL (corresponding representative gating strategies shown in Figure 2B). There was a trend for decreased numbers of lymphocytes and neutrophils recovered in the GSK583-treated BAL compared to vehicle-treated mice; although this difference was not significant. Similar to the BAL, early inhibition of RIP2 led to significantly reduced numbers of CD45+ cells and eosinophils recruited to the lung compared to vehicle treated mice (Figure 2C, with corresponding representative gating strategies shown in Figure 2D). Additionally, recruitment of neutrophils were also significantly decreased in the lung (Fig 2C and 2D). Overall, these results indicate that early inhibition of RIP2 can reduce eosinophilia and lung neutrophilia during an acute house dust mite asthma model.
Figure 2. Early prophylactic inhibition of RIP2 reduces eosinophilia and lung neutrophilia in an acute HDM model of asthma..
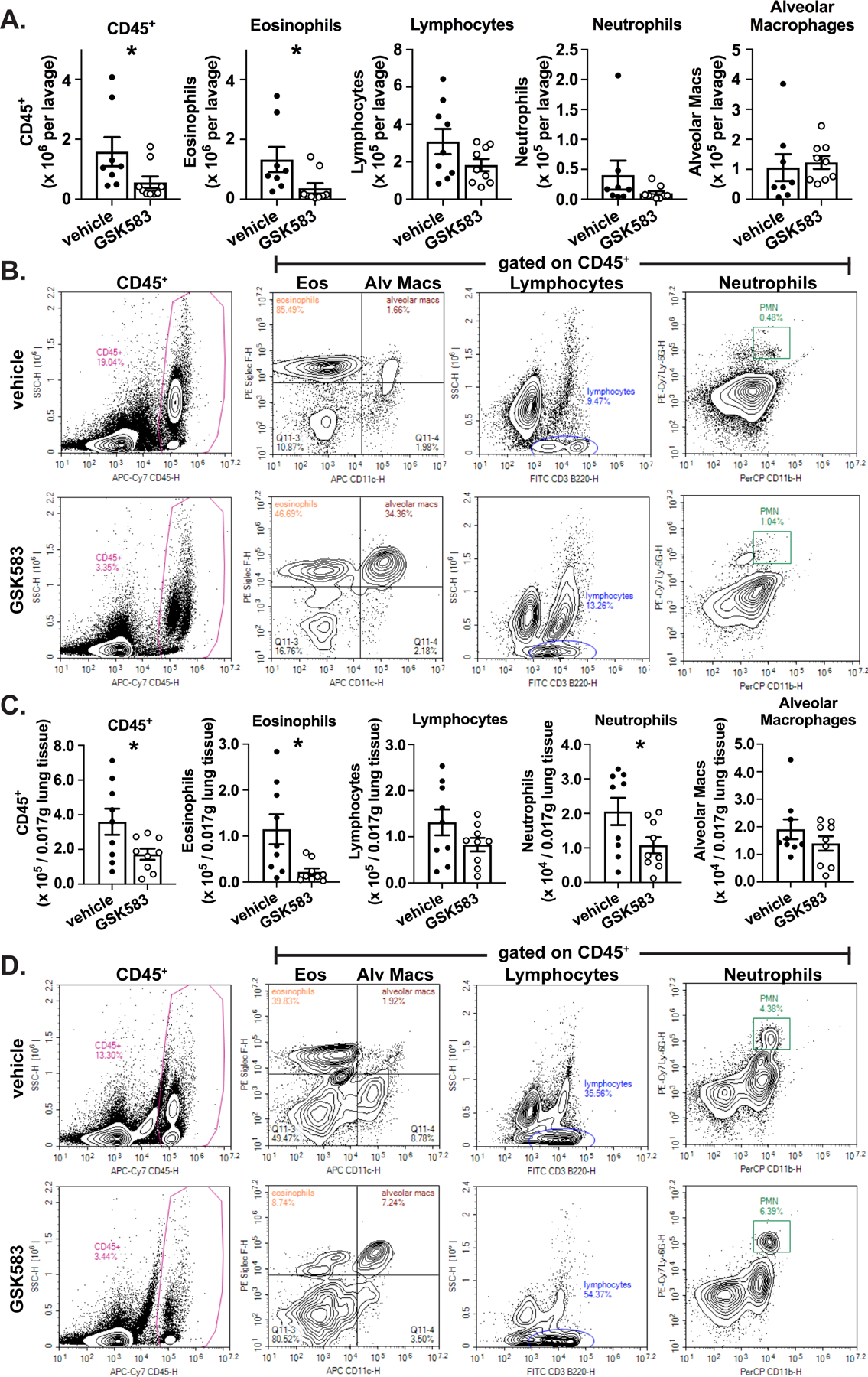
WT C57BL/6 mice were subjected to an acute HDM asthma model and were administered either regular chow or chow containing RIP2 inhibitor (GSK583) as indicated in Fig 1A. On day 14, mice were euthanized and bronchoalveolar lavage (BAL) cells or dissociated lung cells were isolated, stained for immune cell markers, and subjected to flow cytometric analysis. A). The total number of each cellular population within the BAL for vehicle compared to GSK583-treated mice. B.) Flow cytometry gating strategy for obtaining BAL cell counts in A.). C.) The numbers of each cellular population within a standardized amount of lung tissue is shown for vehicle compared to GSK583-treated mice. D.) Flow cytometry gating strategy for obtaining lung cell numbers in C.). Data are presented as scatterplots with bars where bar heights represent means ± SEM. Filled in circles represent individual datapoints for vehicle/control chow-treated mice and open circles represent individual datapoints for GSK583-treated mice. Data are pooled from 3 independent experiments for a total of n=8–9 mice per group (bloody lavage were excluded). Statistical analysis was performed using an unpaired, two-tailed Student’s t-test. *= p<0.05.
Early pharmacological inhibition of RIP2 downregulates Th2 and Th17 immunity in an acute HDM model of asthma.
The adaptive immune response during an HDM asthma model is largely Th2 and Th17 dominated. To assess the effects of early pharmacological inhibition of RIP2 on the resulting adaptive immune response, we collected lung cells at day 14 of the acute HDM asthma model, stimulated these with PMA+ionomycin and performed intracellular cytokine staining to assess production of Th1, Th2, or Th17-associated cytokines. We observed a reduction in Th2 (CD4+IL-4+IL-5+) as well as Th17 (CD4+IL-17+) cell numbers within the lung in GSK583-treated mice, which is consistent with the previously observed decrease in eosinophilia and neutrophilia, while Th1 (CD4+IFN-γ+) cell numbers were not significantly altered (Figure 3A). Corresponding representative gating strategies are shown in Figure 3B.
Figure 3. Pharmacological inhibition of RIP2 downregulates Th2 and Th17 immunity in a HDM model of asthma.
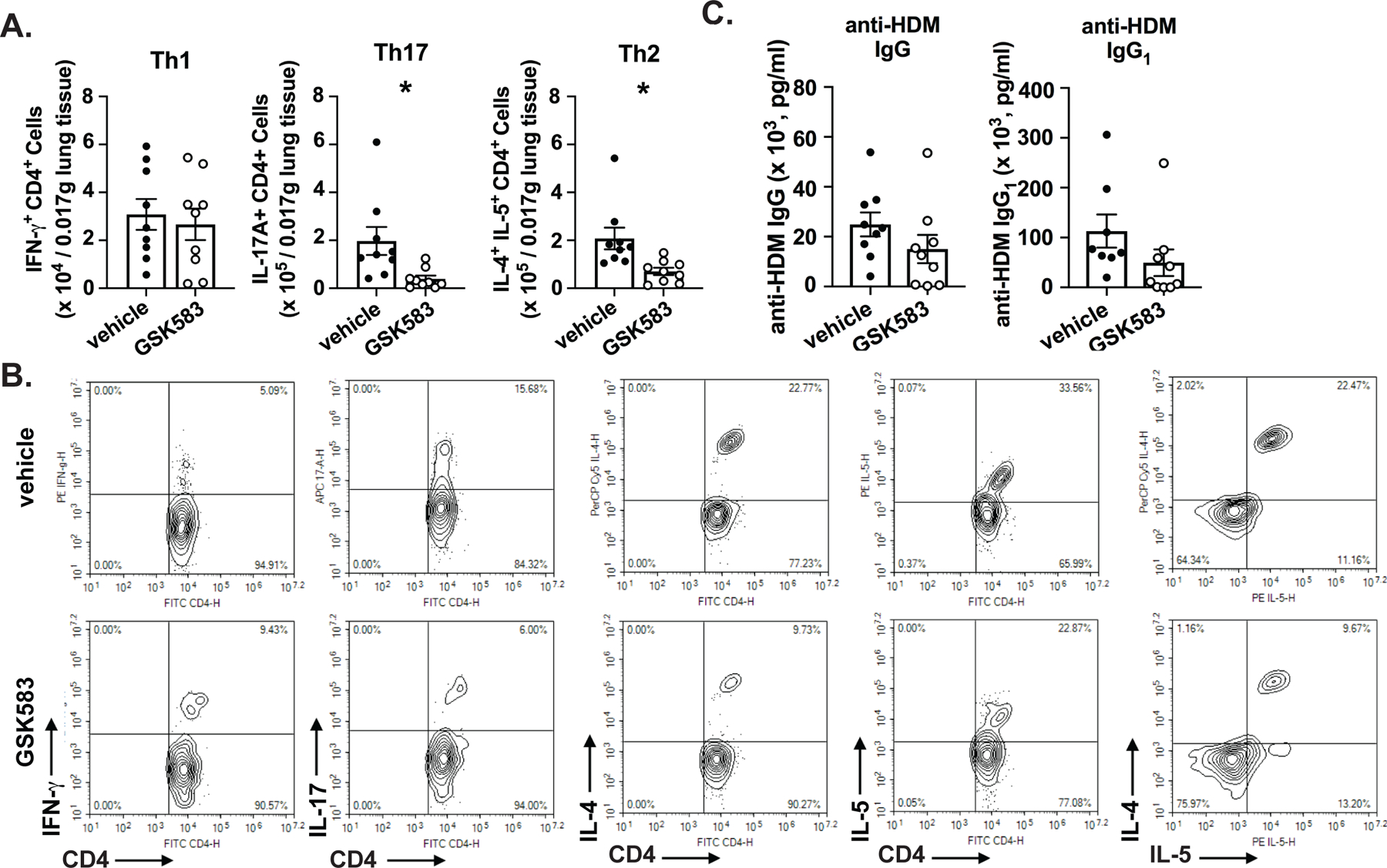
WT C57BL/6 mice were subjected to an acute HDM asthma model and were administered either regular chow or chow containing RIP2 inhibitor (GSK583) as indicated in Fig 1A. On day 14, mice were euthanized and dissociated lung cells were stimulated with PMA and ionomycin and subjected to intracellular cytokine staining (ICS). A.) The numbers of CD4+IFN-γ+(Th1), CD4+IL-17+(Th17), or CD4+IL-4+IL-5+(Th2) within a standardized amount of lung tissue are shown for vehicle compared to GSK583-treated mice. B.) Flow cytometry gating strategy for ICS cell numbers shown in A.). C.) Levels of HDM-specific total IgG and IgG1 antibodies in the serum of vehicle chow or RIP2 inhibitor (GSK583) chow-treated mice subjected to an acute HDM asthma model as measured by ELISA. Data are presented as scatterplots with bars where bar heights represent means ± SEM. Filled in circles represent individual datapoints for vehicle/control chow-treated mice and open circles represent individual datapoints for GSK583-treated mice. Data are pooled from 3 independent experiments for a total of n=9 mice per group. Statistical analysis was performed using an unpaired, two-tailed Student’s t-test. *= p<0.05.
Furthermore, we also assessed the effect of early and acute RIP2 inhibition on the production of HDM-specific antibodies. We have previously shown that on the C57BL/6J background (without the use of an additional adjuvant such as alum), the HDM asthma model leads primarily to an increase in the production of HDM-specific total IgG and the Th2-associated IgG1 subclass of antibodies (15). Changes in levels of these antibodies in the serum of GSK583 or vehicle-treated animals suggested a trend towards decreased levels of HDM-specific total IgG and IgG1; however, this was not statistically significant (Figure 3C).
To additionally examine the local adaptive immune response within the lung, we performed a bead-based multiplex assay with lung homogenates from GSK583 and vehicle-treated mice (Figure 4). This assay measures cytokines collectively secreted by the major Th lineages (Th1, Th2, Th9, Th17, Th22 and T follicular cells). These data indicate that there was a significant reduction in the levels of IL-4 and IL-5 in the lungs of GSK583-treated mice compared to vehicle-treated mice on an HDM asthma model without appreciable alteration of any of the other 11 Th cytokines tested (Figure 4). Collectively, these data indicate that pharmacological inhibition of RIP2 during the initial exposure to HDM is enough to decrease the numbers of Th2 and Th17 cells in the lung and to reduce the production of local Th2-derived cytokines.
Figure 4. Early in vivo inhibition of RIP2 reduces Th2 associated cytokines in the lung.
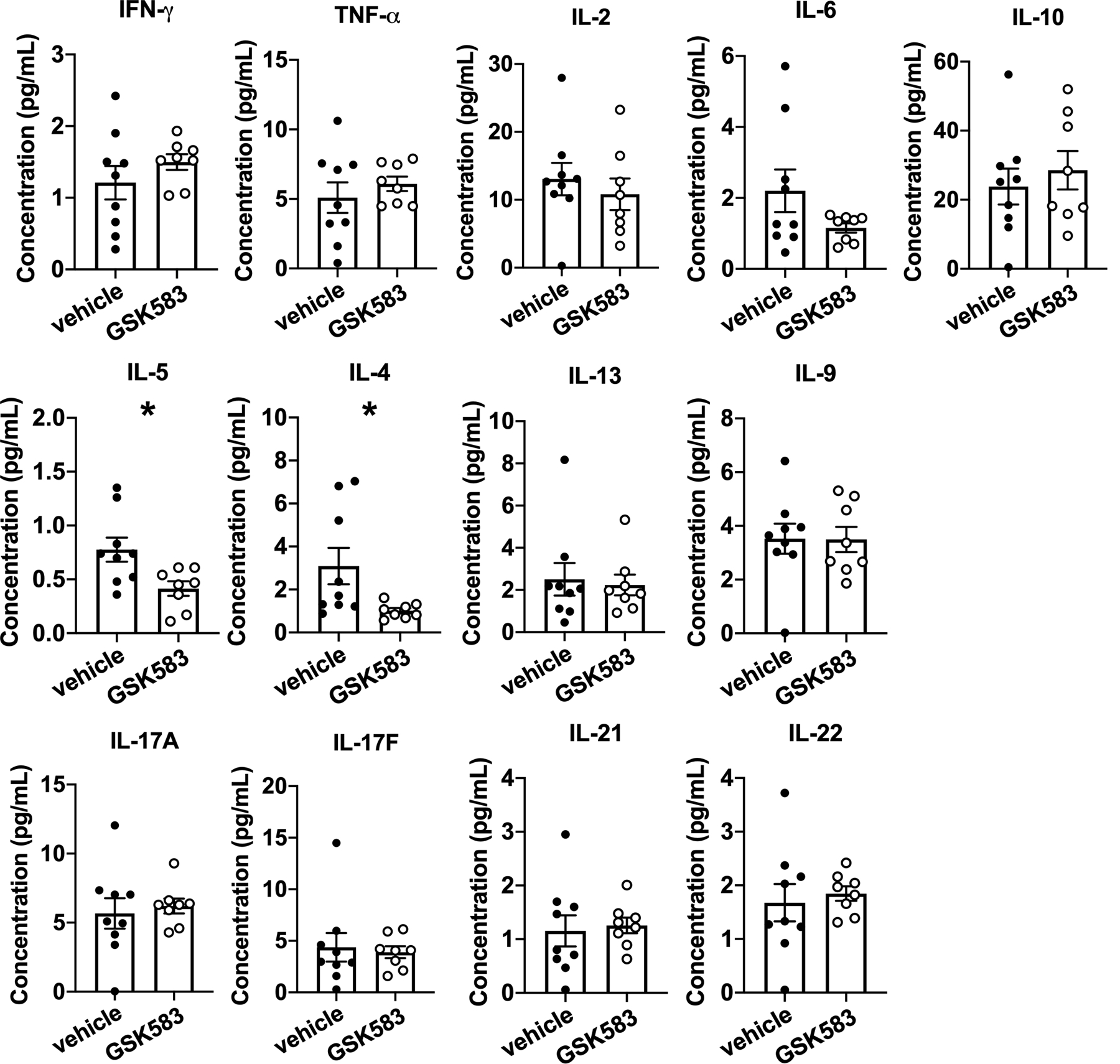
WT C57BL/6 mice were subjected to an acute HDM asthma model and were administered either regular chow or chow containing RIP2 inhibitor (GSK583) as indicated in Fig 1A. On day 14, mice were euthanized and lung tissue was collected and lysed in protein extraction buffer containing inhibitors. Supernatants were subjected to multiplexed bead-based immunoassay for measurement of various Th-associated cytokines. Data are presented as scatterplots with bars where bar heights represent means ± SEM. Filled in circles represent individual datapoints for vehicle/control chow-treated mice and open circles represent individual datapoints for GSK583-treated mice. Data are pooled from 3 independent experiments for a total of n=8–9 mice per group (outlier by Grubb’s test was excluded). Statistical analysis was performed using an unpaired, two-tailed Student’s t-test. *= p<0.05.
Early inhibition of RIP2 in mice undergoing a chronic HDM asthma model exhibits disparate effects on lung pathology and airway hyperresponsiveness (AHR).
Given that acute models of asthma do not recapitulate many important features of this chronic disease such as airway remodeling and airway hyperresponsiveness (AHR), we also wanted to utilize a chronic HDM asthma model to determine whether early pharmacological inhibition of RIP2 had beneficial effects which would still be evident even at the later stages of this disease. As such, we treated mice with GSK583 or vehicle chow for 5 days around the time of initial exposure to HDM allergen and continued to expose them to HDM for a period of 4 weeks as depicted in Figure 5A. On day 28 of the model, mice were euthanized and lungs were harvested, fixed, paraffin-embedded and stained for histological examination. H&E, PAS, and trichrome staining was performed and sections were scored by a blinded observer. Similar to the acute model, early inhibition of RIP2 using GSK583 was sufficient to reduce the total inflammatory index (Figure 5B, with corresponding graph in Figure 5C) and mucus production as shown by PAS positivity (magenta in PAS staining in Figure 5B, with corresponding graph in Figure 5E) compared to vehicle treated mice when mice were subjected to a chronic HDM model. In addition to evaluating inflammation, we also assessed airway remodeling. Using a combination of parameters encompassing severity and incidence of epithelial folding/distortion, goblet cell metaplasia, smooth muscle cell hypertrophy, subepithelial fibrosis, peribronchial trichrome blue staining, and parenchymal trichrome blue staining, we report an airway remodeling index (maximum score of 22). Early inhibition of RIP2 using GSK583 exhibited significantly reduced airway remodeling compared to vehicle-treated mice (11.22 ± 0.88 compared to 21 ± 0.29) when mice were subjected to a chronic model of asthma (Figure 5B, with corresponding graph in Figure 5D). These results indicate that by several histological parameters, early inhibition of RIP2 is enough to improve lung pathology and airway remodeling even during a chronic model of asthmatic disease.
Figure 5. Mice undergoing a chronic HDM asthma model exhibit improved lung pathology with early RIP2 inhibition but minimal effects on HDM-specific antibody responses.
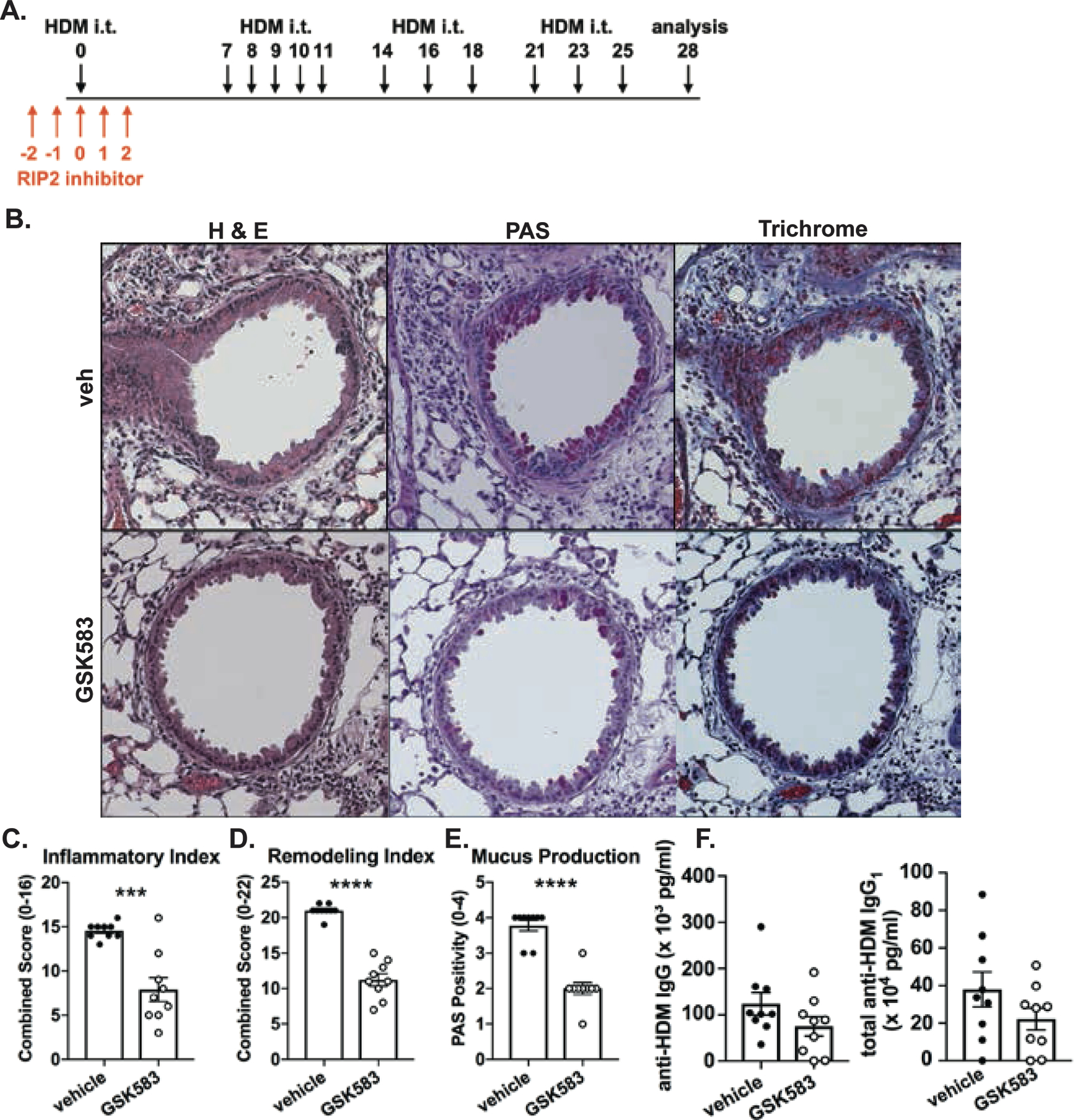
A.) WT C57BL/6 mice were subjected to a chronic HDM asthma model and were administered either regular chow or chow containing RIP2 inhibitor (GSK583 at 30mg/kg/day) for the 5 days during and surrounding the initial exposure to HDM (red arrows). On day 28, mice were euthanized and lungs were harvested, fixed in 10% formalin, and embedded in paraffin. B.) Consecutive sections were stained with either H&E, PAS, or Trichrome. Severity of lung pathology was blindly scored based on C.) inflammatory index, D.) remodeling index and E.) mucus production. F.) Levels of HDM-specific total IgG and IgG1 antibodies in the serum of vehicle chow or RIP2 inhibitor (GSK583) chow-treated mice subjected to a chronic HDM asthma model as measured by ELISA. Data are presented as scatterplots with bars where bar heights represent means ± SEM. Filled in circles represent individual datapoints for vehicle/control chow-treated mice and open circles represent individual datapoints for GSK583-treated mice. Data are pooled from 3 independent experiments for a total of n=9 mice per group. Statistical analysis was performed using an unpaired, two-tailed Student’s t-test. ***= p<0.001, ****= p<0.0001
In addition, we assessed whether treatment with GSK583 would lead to deficient HDM-specific antibody responses in the chronic HDM asthma model. Similar to the acute model, there was a trend for reduced HDM-specific total IgG and IgG1 antibody levels in GSK583-treated mice compared to vehicle-treated mice; however, this was not significant (Figure 5F).
Lastly, to determine whether pharmacological inhibition of RIP2 during initial exposure to allergen affected AHR, mice treated with either GSK583 or vehicle chow underwent a chronic HDM asthma model. On day 28, mice were anesthetized, cannulated, connected to the FlexiVent ventilator and subjected to various mechanical perturbations in a customized asthma challenge script which included administration of nebulized methacholine at increasing doses in between measruments. Resistance of the total respiratory system (Rrs), central airway Newtonian resistance (RN), Elastance of the respiratory system (Ers), tissue damping (Max G), and tissue elastance (Max H) were calculated for each mouse at each dose of methacholine. No significant changes in Rrs (Figure 6A) or RN (Figure 6B) were observed with GSK583 treatment compared to vehicle treatment. Ers (Figure 6C), Max G (Figure 6D), and Max H (Figure 6E) were slightly decreased in GSK583-treated mice compared to vehicle-treated mice at a the highest dose of MCh (100mg/mL); however, these changes were not significant. Overall, these results indicate that early inhibition of RIP2, while sufficient to reduce lung pathology and airway remodeling, is by itself not sufficient to decrease AHR in a chronic house dust mite asthma model.
Figure 6. GSK583-treated mice did not exhibit changes in HDM-induced airway hyperresponsiveness.
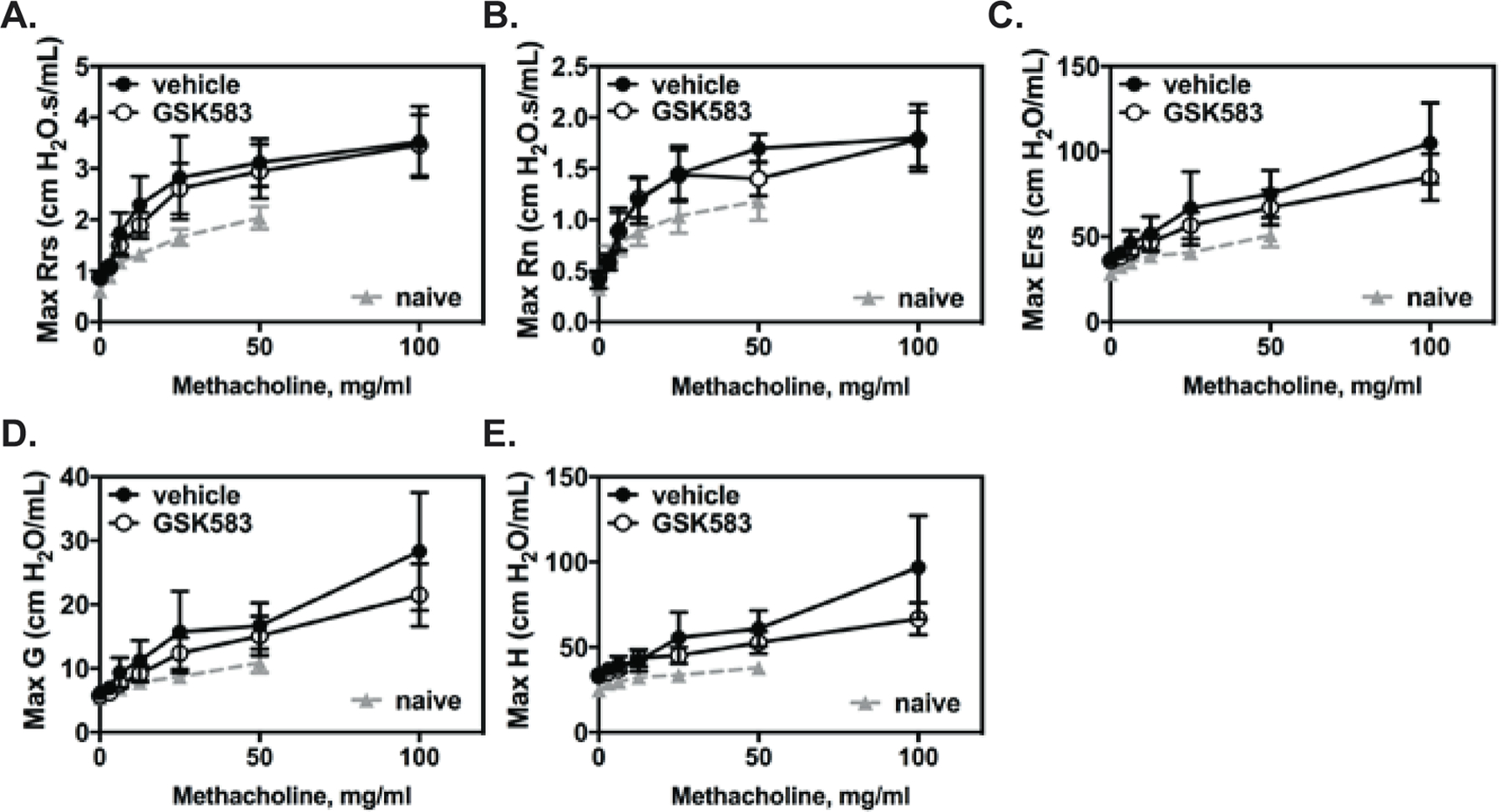
WT C57BL/6 mice were subjected to a chronic HDM asthma model and were administered either regular chow or chow containing RIP2 inhibitor (GSK583) as indicated in Fig 5A. On day 28, mice were anesthetized, intratracheally cannulated, and connected to a FlexiVent mechanical ventilator. Baseline lung function was obtained, after which, mice were subjected to increasing doses of methacholine to measure airway hyperresponsiveness. Dose dependent changes in A.) Resistance of the respiratory system (Rrs), B.) Resistance of the central airways (Rn), C.) Elastance of the respiratory system (Ers), D.) Tissue damping and E.) Tissue elastance (H) are shown. Lines connected by filled in circles represent vehicle/control chow-treated mice and lines connected by open circles represent individual datapoints for GSK583-treated mice. Data are pooled from 2 independent experiments for a total of n=5–6 mice per group. The area under the curve (AUC) was calculated for each treatment and an unpaired, two-tailed Student’s t-test was performed comparing the two.
Discussion
Currently, the only disease modifying treatment available for asthmatics is allergen immunotherapy (AIT) (24). AIT has a long history of efficacy primarily in the form of subcutaneous immunotherapy (SCIT or “allergy shots”) and, more recently, as sublingual formulations (SLIT). The proposed mechanisms for the effectiveness of AIT include induction of T cell anergy, induction of regulatory T cells (Tregs) or immune deviation (25). The result is the production of antibodies of the IgG4 or IgG isotype with the ability to block allergen specific IgE. Although this treatment has been available for some time, it is generally underused due to lack of standardization (allergen preparation, potency, delivery system, route of delivery) and is contraindicated for patients with uncontrolled asthma (25). In contrast, current pharmacotherapeutic approaches, though safe and effective, are only capable of managing the allergic asthmatic symptoms. This would mean that maintaining the effect would require continuous administration. Ideally, an emerging therapy for allergic asthma would have the lasting, disease-modifying efficacy of AIT with the ease and safety of pharmacotherapeutic approaches. The capacity for AIT to induce long-lived effects relies on its ability to modulate or reprogram adaptive immunity by affecting upstream cues such as the generation of a regulatory or immature DC phenotype. In the present study, we sought to determine whether interfering with early upstream inflammatory cues, such as those emanating from activation of the kinase RIP2, would be capable of modulating the immune response with lasting effect.
Our previous study using RIP2 KO mice in the context of a HDM model of allergic asthma identified RIP2 as a kinase important in promoting not only the Th2 and Th17 response to HDM, but also the accompanying airway eosinophilia and lung pathology (15). RIP2 is widely expressed in various innate immune populations such as neutrophils, macrophages and dendritic cells where it serves important functions in mediating host defense. Some reports also indicate a role for RIP2 within T cells (26, 27). Additionally, expression and function of RIP2 as well as its upstream receptor NOD2 has been demonstrated within non-hematopoietic cells such as airway epithelial cells (28). We suspected that involvement of RIP2 was crucial during the very early stages of allergic sensitization and likely to be primarily acting within airway epithelial cells since loss of RIP2 inhibited the early HDM-induced pro-inflammatory chemokine response in the lung and in primary airway epithelial cells without influencing ILC2 expansion (15). Subsequent bone marrow chimera experiments confirmed the importance of RIP2 within the non-hematopoietic compartment in mediating these early responses. In our previous studies, RIP2 was absent allthroughout the asthma model (global RIP2 KO) and early effects of RIP2 within innate or structural cells could not be distinguished from any potential late effects on adaptive immunity. In the absence of any inducible and conditional RIP2 mouse strains which would allow us to tease apart these roles, in the current study, we decided to intervene using pharmacological means. Using GSK583, a specific RIP2 inhibitor, we were able to verify and isolate the importance of RIP2 during the initial period leading to the establishment of T cell immunity by inactivating RIP2 during a crucial window (initial allergen exposure). We demonstrate that early acute inhibition of RIP2 is beneficial and immune modulating and that the effects on cellular inflammation and lung pathology (airway remodeling) persist long after treatment is discontinued. Although the lack of efficacy of RIP2 inhibition on airway hyperresponsiveness is somewhat disappointing (no change in central airway resistance, RN), the trend in the reduction of tissue damping and elastance are suggestive for a potential role of RIP2 in influencing localized peripheral effects in the lung tissue, changes which may be more apparent if using a different genetic background, sensitization procedure (alum adjuvanted) or timing of inhibitor administration.
The duration of RIP2 inhibition (during and up to 2 days after initial allergen exposure) will have most likely impacted airway structural cells, antigen presenting cells and ILC2 cells within the lung. Although ILC2 numbers increase in the setting of HDM asthma, our previous studies did not find any differences in ILC2 frequencies in the absence of RIP2 and, therefore, we suspect that involvement of this cell type in the observed reduction in Th2 and Th17 responses is unlikely. We have previously reported a role for RIP2 in AEC-derived production of CCL2/MCP-1, a chemokine which has been shown by others to be important in DC recruitment into the lung (29–31) and in establishing a Th2 promoting melieu (32). In DCs, activation of NOD2:RIP2 in synergy with activation of other TLRs, has been demonstrated to promote production of IL23p19, IL-1α, IL-1β and Th17 immunity (33). Thus, a possible model is that during the early period of allergic sensitization to HDM, RIP2 acts within AECs to promote recruitment of DCs and support polarization of Th2 immunity and within DCs to promote Th17 immunity. Consequently, interfering with these early signals leads to a reduction in Th2 and Th17 numbers, a decrease in eosinophilia, lung neutrophilia and an improvement in the histological parameters of inflammation.
One of the limitations of these studies is that we did not assess the therapeutic efficacy of RIP2 inhibition. This was intentional as we wanted to confirm the importance of RIP2 activity during the early response to HDM which we demonstrate influences the later establishment of adaptive immunity and allows for a sustained decrease in airway inflammation and a protective effect on airway remodeling. How early RIP2 inhibition promotes sustained immune modulation is currently unknown. We did not assess the generation of tolerogenic DCs or Tregs in this study which could be responsible for the protective effects in the face of repeated allergen exposure. However, data arguing against this mechanism include no significant differences observed in local IL-10 production within the lung and that RIP2 (at least downstream of NOD2 engagement) has been shown to be important for IL-10 production (in direct opposition of this hypothesis) (34, 35). We cannot rule out that RIP2 inhibition may have directly or indirectly influenced other phenotypes of DCs by affecting the production of secreted regulatory molecules such as retinoic acid (RA), TGF-β and indoleamine 2,3-dioxygenase (IDO) or expression of cell surface markers important for polarization to Th2 immunity (OX40L, Jagged 2 etc.) or influencing DC migration. These would be interesting avenues to pursue in the future to identify important cellular mechanisms for the observed sustained efficacy of early RIP2 inhibition.
In practicality, it would be difficult to capture the period at which sensitization to allergens occurs. Given our model above, how would therapeutic targeting of RIP2 potentially work in a sensitized individual (an established model of allergic asthma)? Subsequent allergen exposure also results in re-activation of the airway epithelium and production of various chemokines which result in an influx of DCs important in re-stimulation of memory T cell responses. Thus, RIP2 inhibition may still be beneficial in preventing DC recruitment, memory T cell reactivation and potentially promote immune modulation or deviation. However, this direction should also be approached cautiously as RIP2 has also been described to play a T cell intrinsic role (regulating pathogenic Th17 immunity in certain models of inflammation). We suspect that although a similar benefit in attenuation of allergic airway inflammation and Th2 immunity may be observed when RIP2 inhibitors are administered therapeutically, whether such therapies also increase pathogenic Th17 responses remains to be be seen. Given the widespread expression and importance of RIP2 not only within epithelial cells and DCs but also in various immune cell subsets present during the effector phase, we consider this beyond the scope of the current study.
Use of an immunomodulatory small molecule inhibitor (SMI) needing only intermittent or brief adminstration for treatment of asthma would be very appealing. A short therapeutic regimen would additionally reduce any potential concerns about immunosuppression given the fact that RIP2 does carry out many host protective functions. As a potential therapeutic target, RIP2 has garnered a great deal of interest as beneficial effects of genetic loss or pharmacological inhibition of this kinase has been demonstrated in models of inflammatory bowel disease (19, 36), ex vivo sarcoidosis tissues (37), a model of multiple sclerosis (18, 38) and various cancers (39, 40). This work is the first to demonstrate the efficacy and modulatory activity of a RIP2 targeting compound using a clinically relevant model of allergic asthma. Numerous inhibitors have both been discovered and developed for modulation of RIP2 activity (23, 41–49). The RIP2 inhibitor utilized in this study, GSK583, has been reported to be highly selective and exhibits strong potency even in vivo (23, 45). Numerous reports have emerged recently regarding the mechanism of action of such inhibitors in attenuating the downstream inflammatory signaling emanating from RIP2. Although many RIP2-targeted therapies strongly inhibit the kinase function of RIP2, the ability of RIP2 to bind XIAP and undergo ubiquitination rather than its kinase activity per se appears to be crucial for propagation of NOD2 inflammatory signaling (45, 46). GSK583 has been demonstrated to disrupt XIAP:RIP2 binding and prevent RIP2 ubiquitination and optimized derivatives of this compound (GSK2983559) briefly entered into human clinical trials in 2017 (21). However, this trial was terminated in 2019 due to “non clinical toxicology findings and reduced safety margins” (21). The data presented in this current study, demonstrating a clear benefit of acute and early RIP2 inhibition for attenuation of allergic asthma and modulation of Th2 immunity, will hopefully provide the needed rationale and support for continued development of what will likely be very useful compounds for asthma and other inflammatory diseases.
Supplementary Material
Acknowledgments
We would like to thank Pamela Haile, Bart Votta, Allison Beal and John Lich for their assistance in selection of the GSK583 RIP2 inhibitor, determination of the appropriate dose in chow and for providing the compound for use in this study. We additionally would like to thank them for helpful comments during revision of the manuscipt.
We would like to thank UCF Core facilities for use of the GentleMacs tissue dissociator and Madelyn’s thesis committee for discussions regarding her work.
Support:
This study was supported by an NIH 5R00HL122365 grant and start-up funds from Univ. of Central Florida to J.T.T-A and by a gift of GSK583 in chow from GSK to J.T.T-A.
Footnotes
Conflict of Interest Statement.
M.H.M, M.G.S and J.T.T-A have no conflicts of interest to disclose.
References
- 1.Organization, W. H. 2018. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2016. WHO Press, Geneva. [Google Scholar]
- 2.Kuruvilla ME, Lee FE, and Lee GB 2019. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin Rev Allergy Immunol 56: 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fahy JV 2015. Type 2 inflammation in asthma--present in most, absent in many. Nat Rev Immunol 15: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaffin JM, and Phipatanakul W 2009. The role of indoor allergens in the development of asthma. Current opinion in allergy and clinical immunology 9: 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Illi S, von Mutius E, Lau S, Niggemann B, Gruber C, Wahn U, and g. Multicentre Allergy Study. 2006. Perennial allergen sensitisation early in life and chronic asthma in children: a birth cohort study. Lancet 368: 763–770. [DOI] [PubMed] [Google Scholar]
- 6.Kusel MM, de Klerk NH, Kebadze T, Vohma V, Holt PG, Johnston SL, and Sly PD 2007. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol 119: 1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sporik R, Chapman MD, and Platts-Mills TA 1992. House dust mite exposure as a cause of asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology 22: 897–906. [DOI] [PubMed] [Google Scholar]
- 8.Sears MR, Herbison GP, Holdaway MD, Hewitt CJ, Flannery EM, and Silva PA 1989. The relative risks of sensitivity to grass pollen, house dust mite and cat dander in the development of childhood asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology 19: 419–424. [DOI] [PubMed] [Google Scholar]
- 9.Sporik R, Holgate ST, Platts-Mills TA, and Cogswell JJ 1990. Exposure to house-dust mite allergen (Der p I) and the development of asthma in childhood. A prospective study. N Engl J Med 323: 502–507. [DOI] [PubMed] [Google Scholar]
- 10.Celedon JC, Milton DK, Ramsey CD, Litonjua AA, Ryan L, Platts-Mills TA, and Gold DR 2007. Exposure to dust mite allergen and endotoxin in early life and asthma and atopy in childhood. J Allergy Clin Immunol 120: 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tovey ER, Almqvist C, Li Q, Crisafulli D, and Marks GB 2008. Nonlinear relationship of mite allergen exposure to mite sensitization and asthma in a birth cohort. J Allergy Clin Immunol 122: 114–118, 118 e111–115. [DOI] [PubMed] [Google Scholar]
- 12.Lambrecht BN, Hammad H, and Fahy JV 2019. The Cytokines of Asthma. Immunity 50: 975–991. [DOI] [PubMed] [Google Scholar]
- 13.Doroudchi A, Pathria M, and Modena BD 2020. Asthma biologics: Comparing trial designs, patient cohorts and study results. Ann Allergy Asthma Immunol 124: 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang FL, Liao EC, and Yu SJ 2018. House dust mite allergy: Its innate immune response and immunotherapy. Immunobiology 223: 300–302. [DOI] [PubMed] [Google Scholar]
- 15.Miller MH, Shehat MG, Alcedo KP, Spinel LP, Soulakova J, and Tigno-Aranjuez JT 2018. Frontline Science: RIP2 promotes house dust mite-induced allergic airway inflammation. Journal of leukocyte biology 104: 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, and Sansonetti PJ 2003. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. The Journal of biological chemistry 278: 8869–8872. [DOI] [PubMed] [Google Scholar]
- 17.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, and Nuñez G 2003. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. The Journal of biological chemistry 278: 5509–5512. [DOI] [PubMed] [Google Scholar]
- 18.Nachbur U, Stafford CA, Bankovacki A, Zhan Y, Lindqvist LM, Fiil BK, Khakham Y, Ko HJ, Sandow JJ, Falk H, Holien JK, Chau D, Hildebrand J, Vince JE, Sharp PP, Webb AI, Jackman KA, Mühlen S, Kennedy CL, Lowes KN, Murphy JM, Gyrd-Hansen M, Parker MW, Hartland EL, Lew AM, Huang DC, Lessene G, and Silke J 2015. A RIPK2 inhibitor delays NOD signalling events yet prevents inflammatory cytokine production. Nature communications 6: 6442. [DOI] [PubMed] [Google Scholar]
- 19.Tigno-Aranjuez JT, Benderitter P, Rombouts F, Deroose F, Bai X, Mattioli B, Cominelli F, Pizarro TT, Hoflack J, and Abbott DW 2014. In vivo inhibition of RIPK2 kinase alleviates inflammatory disease. The Journal of biological chemistry 289: 29651–29664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vieira SM, Cunha TM, França RF, Pinto LG, Talbot J, Turato WM, Lemos HP, Lima JB, Verri WA Jr., Almeida SC, Ferreira SH, Louzada-Junior P, Zamboni DS, and Cunha FQ 2012. Joint NOD2/RIPK2 signaling regulates IL-17 axis and contributes to the development of experimental arthritis. Journal of immunology (Baltimore, Md. : 1950) 188: 5116–5122. [DOI] [PubMed] [Google Scholar]
- 21.GlaxoSmithKline. 2017. GSK2983559 First Time in Human Study. ClinicalTrials.gov, Identifier: NCT03358407.
- 22.Wachtel MS, Shome G, Sutherland M, and McGlone JJ 2009. Derivation and validation of murine histologic alterations resembling asthma, with two proposed histologic grade parameters. BMC Immunol 10: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haile PA, Votta BJ, Marquis RW, Bury MJ, Mehlmann JF, Singhaus R Jr., Charnley AK, Lakdawala AS, Convery MA, Lipshutz DB, Desai BM, Swift B, Capriotti CA, Berger SB, Mahajan MK, Reilly MA, Rivera EJ, Sun HH, Nagilla R, Beal AM, Finger JN, Cook MN, King BW, Ouellette MT, Totoritis RD, Pierdomenico M, Negroni A, Stronati L, Cucchiara S, Ziolkowski B, Vossenkamper A, MacDonald TT, Gough PJ, Bertin J, and Casillas LN 2016. The Identification and Pharmacological Characterization of 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine (GSK583), a Highly Potent and Selective Inhibitor of RIP2 Kinase. J Med Chem 59: 4867–4880. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Lin C, Sampath V, and Nadeau K 2018. Impact of allergen immunotherapy in allergic asthma. Immunotherapy 10: 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shamji MH, and Durham SR 2017. Mechanisms of allergen immunotherapy for inhaled allergens and predictive biomarkers. J Allergy Clin Immunol 140: 1485–1498. [DOI] [PubMed] [Google Scholar]
- 26.Shimada K, Porritt RA, Markman JL, O’Rourke JG, Wakita D, Noval Rivas M, Ogawa C, Kozhaya L, Martins GA, Unutmaz D, Baloh RH, Crother TR, Chen S, and Arditi M 2018. T-Cell-Intrinsic Receptor Interacting Protein 2 Regulates Pathogenic T Helper 17 Cell Differentiation. Immunity 49: 873–885 e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Napier RJ, Lee EJ, Davey MP, Vance EE, Furtado JM, Snow PE, Samson KA, Lashley SJ, Brown BR, Horai R, Mattapallil MJ, Xu B, Callegan MC, Uebelhoer LS, Lancioni CL, Vehe RK, Binstadt BA, Smith JR, Caspi RR, and Rosenzweig HL 2020. T cell-intrinsic role for Nod2 in protection against Th17-mediated uveitis. Nature communications 11: 5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bist P, Dikshit N, Koh TH, Mortellaro A, Tan TT, and Sukumaran B 2014. The Nod1, Nod2, and Rip2 axis contributes to host immune defense against intracellular Acinetobacter baumannii infection. Infect Immun 82: 1112–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao L, Yang W, Yang X, Lin Y, Lv J, Dou X, Luo Q, Dong J, Chen Z, Chu Y, and He R 2014. Chemerin suppresses murine allergic asthma by inhibiting CCL2 production and subsequent airway recruitment of inflammatory dendritic cells. Allergy 69: 763–774. [DOI] [PubMed] [Google Scholar]
- 30.Osterholzer JJ, Curtis JL, Polak T, Ames T, Chen GH, McDonald R, Huffnagle GB, and Toews GB 2008. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. Journal of immunology (Baltimore, Md. : 1950) 181: 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robays LJ, Maes T, Lebecque S, Lira SA, Kuziel WA, Brusselle GG, Joos GF, and Vermaelen KV 2007. Chemokine receptor CCR2 but not CCR5 or CCR6 mediates the increase in pulmonary dendritic cells during allergic airway inflammation. Journal of immunology (Baltimore, Md. : 1950) 178: 5305–5311. [DOI] [PubMed] [Google Scholar]
- 32.Gu L, Tseng S, Horner RM, Tam C, Loda M, and Rollins BJ 2000. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 404: 407–411. [DOI] [PubMed] [Google Scholar]
- 33.van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat SA, Kapsenberg ML, and de Jong EC 2007. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 27: 660–669. [DOI] [PubMed] [Google Scholar]
- 34.Noguchi E, Homma Y, Kang X, Netea MG, and Ma X 2009. A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat Immunol 10: 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagener J, Malireddi RK, Lenardon MD, Koberle M, Vautier S, MacCallum DM, Biedermann T, Schaller M, Netea MG, Kanneganti TD, Brown GD, Brown AJ, and Gow NA 2014. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog 10: e1004050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe T, Minaga K, Kamata K, Sakurai T, Komeda Y, Nagai T, Kitani A, Tajima M, Fuss IJ, Kudo M, and Strober W 2019. RICK/RIP2 is a NOD2-independent nodal point of gut inflammation. Int Immunol 31: 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talreja J, Talwar H, Ahmad N, Rastogi R, and Samavati L 2016. Dual Inhibition of Rip2 and IRAK1/4 Regulates IL-1beta and IL-6 in Sarcoidosis Alveolar Macrophages and Peripheral Blood Mononuclear Cells. Journal of immunology (Baltimore, Md. : 1950) 197: 1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi H, Mak TW, and Kanneganti TD 2011. Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity 34: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaafar R, Mnich K, Dolan S, Hillis J, Almanza A, Logue SE, Samali A, and Gorman AM 2018. RIP2 enhances cell survival by activation of NF-kB in triple negative breast cancer cells. Biochem Biophys Res Commun 497: 115–121. [DOI] [PubMed] [Google Scholar]
- 40.Cai X, Yang Y, Xia W, Kong H, Wang M, Fu W, Long M, Hu Y, and Xu D 2018. RIP2 promotes glioma cell growth by regulating TRAF3 and activating the NFkappaB and p38 signaling pathways. Oncol Rep 39: 2915–2923. [DOI] [PubMed] [Google Scholar]
- 41.Haffner CD, Charnley AK, Aquino CJ, Casillas L, Convery MA, Cox JA, Elban MA, Goodwin NC, Gough PJ, Haile PA, Hughes TV, Knapp-Reed B, Kreatsoulas C, Lakdawala AS, Li H, Lian Y, Lipshutz D, Mehlmann JF, Ouellette M, Romano J, Shewchuk L, Shu A, Votta BJ, Zhou H, Bertin J, and Marquis RW 2019. Discovery of Pyrazolocarboxamides as Potent and Selective Receptor Interacting Protein 2 (RIP2) Kinase Inhibitors. ACS Med Chem Lett 10: 1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haile PA, Casillas LN, Votta BJ, Wang GZ, Charnley AK, Dong X, Bury MJ, Romano JJ, Mehlmann JF, King BW, Erhard KF, Hanning CR, Lipshutz DB, Desai BM, Capriotti CA, Schaeffer MC, Berger SB, Mahajan MK, Reilly MA, Nagilla R, Rivera EJ, Sun HH, Kenna JK, Beal AM, Ouellette MT, Kelly M, Stemp G, Convery MA, Vossenkamper A, MacDonald TT, Gough PJ, Bertin J, and Marquis RW 2019. Discovery of a First-in-Class Receptor Interacting Protein 2 (RIP2) Kinase Specific Clinical Candidate, 2-((4-(Benzo[d]thiazol-5-ylamino)-6-(tert-butylsulfonyl)quinazolin-7-yl)oxy)ethyl Dihydrogen Phosphate, for the Treatment of Inflammatory Diseases. Journal of medicinal chemistry 62: 6482–6494. [DOI] [PubMed] [Google Scholar]
- 43.Haile PA, Casillas LN, Bury MJ, Mehlmann JF, Singhaus R Jr., Charnley AK, Hughes TV, DeMartino MP, Wang GZ, Romano JJ, Dong X, Plotnikov NV, Lakdawala AS, Convery MA, Votta BJ, Lipshutz DB, Desai BM, Swift B, Capriotti CA, Berger SB, Mahajan MK, Reilly MA, Rivera EJ, Sun HH, Nagilla R, LePage C, Ouellette MT, Totoritis RD, Donovan BT, Brown BS, Chaudhary KW, Gough PJ, Bertin J, and Marquis RW 2018. Identification of Quinoline-Based RIP2 Kinase Inhibitors with an Improved Therapeutic Index to the hERG Ion Channel. ACS Med Chem Lett 9: 1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salla M, Aguayo-Ortiz R, Danmaliki GI, Zare A, Said A, Moore J, Pandya V, Manaloor R, Fong S, Blankstein AR, Gibson SB, Garcia LR, Meier P, Bhullar KS, Hubbard BP, Fiteh Y, Vliagoftis H, Goping IS, Brocks D, Hwang P, Velazquez-Martinez CA, and Baksh S 2018. Identification and Characterization of Novel Receptor-Interacting Serine/Threonine-Protein Kinase 2 Inhibitors Using Structural Similarity Analysis. J Pharmacol Exp Ther 365: 354–367. [DOI] [PubMed] [Google Scholar]
- 45.Goncharov T, Hedayati S, Mulvihill MM, Izrael-Tomasevic A, Zobel K, Jeet S, Fedorova AV, Eidenschenk C, deVoss J, Yu K, Shaw AS, Kirkpatrick DS, Fairbrother WJ, Deshayes K, and Vucic D 2018. Disruption of XIAP-RIP2 Association Blocks NOD2-Mediated Inflammatory Signaling. Molecular cell 69: 551–565 e557. [DOI] [PubMed] [Google Scholar]
- 46.Hrdinka M, Schlicher L, Dai B, Pinkas DM, Bufton JC, Picaud S, Ward JA, Rogers C, Suebsuwong C, Nikhar S, Cuny GD, Huber KV, Filippakopoulos P, Bullock AN, Degterev A, and Gyrd-Hansen M 2018. Small molecule inhibitors reveal an indispensable scaffolding role of RIPK2 in NOD2 signaling. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suebsuwong C, Pinkas DM, Ray SS, Bufton JC, Dai B, Bullock AN, Degterev A, and Cuny GD 2018. Activation loop targeting strategy for design of receptor-interacting protein kinase 2 (RIPK2) inhibitors. Bioorg Med Chem Lett 28: 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canning P, Ruan Q, Schwerd T, Hrdinka M, Maki JL, Saleh D, Suebsuwong C, Ray S, Brennan PE, Cuny GD, Uhlig HH, Gyrd-Hansen M, Degterev A, and Bullock AN 2015. Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem Biol 22: 1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tigno-Aranjuez JT, Asara JM, and Abbott DW 2010. Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes & development 24: 2666–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.