

Naked mole rat TRF1 enhances telomeric and metabolic functions in hypoxia through an evolutionary gain of function.
Abstract
The naked mole rat (NMR), a long-lived and cancer-resistant rodent, is highly resistant to hypoxia. Here, using robust cellular models wherein the mouse telomeric protein TRF1 is substituted by NMR TRF1 or its mutant forms, we show that TRF1 supports maximal glycolytic capacity under low oxygen, shows increased nuclear localization and association with telomeres, and protects telomeres from replicative stress. We pinpoint this evolutionary gain of metabolic function to specific amino acid changes in the homodimerization domain of this protein. We further find that NMR TRF1 accelerates telomere shortening. These findings reveal an evolutionary strategy to adapt telomere biology for metabolic control under an extreme environment.
INTRODUCTION
Aging and cancer are intricately related; however, notable differences exist among species, e.g., humans live much longer and are less prone to cancer than mice. During aging, a gradual loss of telomeric DNA in dividing somatic cells contributes to replicative senescence (1, 2). These dynamics play an important role in determining cell fate and influence cancer incidence. In human germ, embryonic, and cancer cells, this loss is counteracted by telomerase that replenishes telomeric DNA at each round of replication, whereas in the mouse telomerase is active in all tissues. However, differences in life span and disease susceptibility among species, as related to telomere biology, are not fully understood. In this regard, an interesting model is the naked mole rat (NMR; Heterocephalus glaber), whose maximum life span exceeds 30 years, making it the longest-lived rodent. Age-associated physiological decline in physiological function is delayed in the NMR (3), and its mortality rate does not increase with age (4). Furthermore, the NMR has a low cancer incidence (5), and induction of tumorigenesis by methods that efficiently work in murine models has also been unsuccessful (6, 7). Moreover, living underground, the NMR can survive up to 15 min in complete anoxia, suggesting a remarkable adaptation to hypoxia (8).
Sequencing of the NMR genome revealed several positively selected mutations, one of which (Ala75Thr) is present in telomeric repeat binding factor 1 (TRF1), within its TRF homology (TRFH)–like domain (9). TRF1 is a component of the shelterin complex that caps telomeres (10). This protein and another shelterin component TRF2 bind duplex TTAGGG DNA repeats. Other shelterin subunits include POT1, which binds single-strand telomeric repeats, TIN2 and TPP1, which bridge TRF1 and TRF2 to POT1, and Rap1, the latter directly interacting with TRF2 (10). Protected telomeres preserve genome integrity, whereas their dysfunction sets off DNA damage response (DDR), leading to senescence and other aging phenotypes (11, 12). DDR is known to limit tumor formation (13); however, when checkpoints are compromised, DDR supports tumorigenesis (14). Within the shelterin complex, TRF1 facilitates a proper progression of the replication fork through the telomeric chromatin (15, 16) and represses telomerase (17). However, without homodimerization, TRF1 cannot efficiently bind telomeres (18).
The shelterin component TIN2 plays an important role in TRF1 nuclear localization and stability (19–22). A subset of dyskeratosis congenita (a progeroid syndrome) patients features mutations in this gene (Tinf2) (23, 24). TIN2 induces telomere shortening (TRF1-TIN2) (25) or telomerase recruitment (TIN2-TPP1) (26). In addition, TIN2 contains a signal peptide at its N terminus and may translocate to mitochondria under hypoxic conditions, followed by changes in glycolysis (27). It was also reported that helicases BLM and WRN resolve topological constraints and can unfold G-quadruplexes, the secondary structures formed in G-rich strands of telomeres during progression of the replicative fork (1). TRF1 recruits BLM to support telomeric replication and POT1 to blunt the ATR-mediated DDR to replicative stress (1, 16). Thus, in the absence of TRF1, telomeres become fragile. TRF1 expression decreases with age (28), and its overexpression was reported to delay aging (29). On the basis of these observations, we sought to examine the function of NMR TRF1. Our analysis revealed that this protein enhances telomeric and metabolic functions specifically under conditions of hypoxia and does it through an evolutionary gain of function involving its homodimerization domain.
RESULTS
Unique sequences in TRF1
Sequence alignment of TRF1 revealed unique amino acid changes within two highly conserved sequence blocks (Fig. 1A), including Ala75Thr (position based on Protein Data Bank no. P54274), which was found to be positively selected in the NMR (9) and located in the homodimerization domain, and Ala105Thr, which was implicated in TIN2 binding. Ala75Thr is located in helix 1 within a hydrophobic region involved in the interaction with helix 9, which shows no compensatory mutations. Replacement of alanine with threonine may slightly bend the helix containing this site and affect hydrophobicity of this area, influencing TRFH homodimerization. Ala105Thr is located in the center of a deep hydrophobic pocket of the TRFH domain in the TIN2 binding area, suggesting that it may affect TRFH homodimerization and TIN2 binding (Fig. 1B).
Fig. 1. NMR TRF1 improved glycolytic capacity at low oxygen.
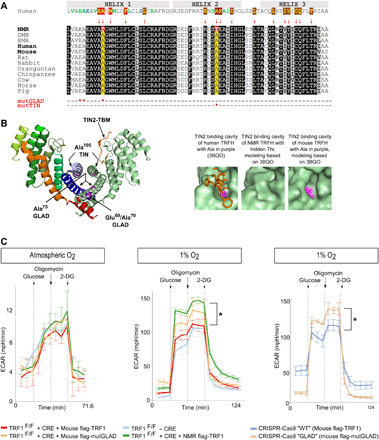
(A) Unique amino acid changes of NMR TRF1 (in red) in conserved sequences. The first row shows amino acids involved in TRFH dimerization (in red) and TIN2 binding cavity (in brown). The last two rows show mutGLAD and mutTIN mutations in the mouse sequence. NMR, naked mole rat; DMR, Damaraland mole rat; BMR, blind mole rat. (B) Dimerization of human TRFH, with the TRF binding motif (TBM) of TIN2 shown in orange. Enlarged details of TRFH of TRF1 show the surface of TIN2 binding cavity, with Ala105 in purple and TIN2 TBD in orange sticks. Models were prepared with I-TASSER (iterative threading assembly refinement) based on 3BQO. (C) Analysis of glycolysis: Kinetics of ECAR in MEFs with TRF1F/F + recombinase (CRE) + ectopic TRF1 variants and of MEFs with p53−/− CRISPR-Cas9 “WT” and “GLAD” in response to glucose and oligomycin and 2-DG at atmospheric oxygen and 1% oxygen. The ECAR value was not normalized, and each data point represents the mean ± SD (n = 3 to 5). *P ≤ 0.05 (Wilcoxon-Mann-Whitney test), calculated glycolytic capacity based on the difference between the maximum ECAR following oligomycin injection and the last rate measurement before glucose injection in the case of NMR TRF1 and mouse TRF1 and CRISPR-Cas9 GLAD and WT.
We investigated the functional consequences of NMR TRF1 sequence changes by studying properties of respective mutant proteins in a heterologous cell system. Specifically, we compared, in transformed mouse embryonic fibroblasts (tMEFs), the effects of NMR TRF1, mouse TRF1, and mouse TRF1 forms containing NMR sequence changes designated as mutGLAD (with Glu69Lys, Val70Ala, and Ala75Thr in the mouse TRF1 context), mutTIN (with Ala105Thr), and mutGLAD/TIN (see the Materials and Methods and fig. S1). Inasmuch as NMRs and mice live in different environmental niches, the oxygen environment and its fluctuations are different for these two organisms. To account for these differences, at the cellular level, our experiments were carried out at both the atmospheric level and low oxygen levels (1 to 3%).
NMR TRF1 improves glycolytic capacity in low oxygen
We first examined whether NMR TRF1 can contribute to the glycolytic adaptation to low oxygen. We found that glycolysis, as measured by the extracellular acidification rate (ECAR), is increased in tMEFs expressing NMR TRF1, compared to isogenic cells expressing mouse TRF1, when cells were grown under low oxygen, but not at atmospheric oxygen (Fig. 1C). This glycolytic enhancement was due to the mutGLAD mutation because the introduction of this sequence into the mouse TRF1 gene also improved glycolysis at low oxygen (Fig. 1C). Because NMR TRF1 regulates glycolysis, we examined the expression level of molecules engaged in this pathway and found that NMR TRF1 improves the glycolytic flux through a higher expression of Glu-1, a glucose and fructose receptor (>2-fold log2 higher than mouse TRF1). mutGLAD also tends to improve Glut-1 expression (fig. S2A).
As NMRs can switch from glucose to fructose as the hexose source in response to hypoxia and anoxia (8), we examined ECAR following fructose addition. tMEFs did not respond to fructose stimuli (fig. S2B), and we failed to detect any expression of Glut-5 mRNA, a specific fructose transporter. Nevertheless, after exposure to glucose and under 1% oxygen versus atmospheric oxygen, tMEFs increased the expression of ketohexokinase (KHK-C), which supports fructose metabolism (30). NMR TRF1 as well as mouse TRF1 carrying mutGLAD or the double mutGLAD/TIN sequences further enhanced the expression of KHK-C (>4-fold log2 higher than mouse TRF1) (fig. S2A). We further investigated gene expression changes in tMEFs expressing various TRF1 constructs (with NMR mutations) by RNA sequencing (RNA-seq). Genes involved in glycolysis and hypoxia-inducible factor 1α (HIF1α) pathways, which were found to be significantly different between cells exposed at atmospheric oxygen and 1% oxygen, are shown in fig. S2C. We found these pathways to be up-regulated upon exposure to hypoxia. Compared to mutTIN, mutGLAD increased the expression of genes involved in glycolysis. No clear HIF1a transcriptomic signature could be deduced from the RNA-seq results. Noteworthy, mutGLAD decreased the expression of Cdkn1a, which encodes p21, while this gene was markedly up-regulated in mutTIN cells, suggesting cell-cycle dysregulation due to mutTIN mutation. Hence, the presence of NMR TRF1 in mouse cells improves glycolysis at low oxygen, revealing a metabolic gain associated with the unique mutGLAD sequence.
Improved binding of TRF1 to telomeres
Next, we examined the capacity of TRF1 to protect telomeres. First, we investigated the presence of NMR TRF1 at telomeres in comparison to mouse TRF1 at both atmospheric and low oxygen. On the basis of the flag-tag signal, almost 95% of telomeres colocalized with NMR TRF1 versus 80% with mouse TRF1 (Fig. 2, A and B, and fig. S3, A, D, and F) in normoxia. These results demonstrate that NMR TRF1 localizes to telomeres more efficiently than mouse TRF1. We found that mutGLAD was responsible for the improved telomere binding (Fig. 2, B and C). However, the opposite was found in the case of mutTIN cells, with fewer than 50% of telomeres associated to TRF1 (Fig. 2, A and B). The mutGLAD/mutTIN version behaved similar to mutGLAD, indicating that mutGLAD compensates for the loss of telomere function caused by mutTIN (Fig. 2B). To confirm TRF1 localization at telomeres, chromatin immunoprecipitation (ChIP) through the flag tag was performed. Unequivocally, more NMR TRF1 was bound to telomeres than mouse TRF1 (Fig. 2, D to F). mutGLAD also appeared to favor telomere binding.
Fig. 2. NMR TRF1 displays enhanced localization at and binding to telomeres compared to mouse TRF1.
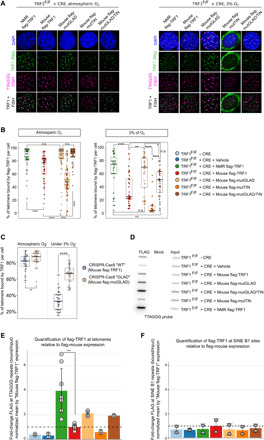
(A and B) Representative images (A) and quantification (B and C) of colocalization between flag-TRF1 foci (green) and telomeric repeats foci (purple) under atmospheric O2 and 3% O2 for MEFs with TRF1F/F + CRE + ectopic TRF1 variants (B) and for MEFs with p53−/− CRISPR-Cas9 WT and “GLAD” (C) . (D) Representative image and (E and F) quantification of ChIP analyses for TRF1 binding to telomeric repeats (E) or to SINE B elements (F) in MEFs with SV40-LT TRF1F/F + CRE + TRF1 rescue based on NMR or mouse TRF1 orthologs or TRF1 variants. Immunoprecipitation with flag antibody; bead controls were spotted onto slot blots and hybridized with a telomeric probe. Quantification of four independent experiments (in duplicate for each) that were performed for NMR and mouse TRF1 samples. Two independent experiments (in duplicate) were performed for mutGLAD and for telomeric repeats, and one experiment (in duplicate) was performed for other samples. Statistical analyses: Student’s t test, n ≥ 30 samples, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001; n.s. not significant.
If the overall rate of TRF1-telomere colocalization decreased when the cells were grown under 3% O2, differences in the levels of telomere association between NMR TRF1, mouse TRF1, and the mutGLAD/mutTIN mutants presented the same trend (Fig. 2, A and B). Noteworthy, mutTIN cells showed even greater deleterious effects under 3% O2, with the TRF1-Flag signals being almost totally excluded from telomeres and the nucleus forming a crown around the 4′,6-diamidino-2-phenylindole (DAPI) staining (Fig. 2, A and B). Although the mutGLAD/TIN form was not significantly different from the mouse TRF1, mutGLAD could compensate for the mutTIN effect. Moreover, mutGLAD was not significantly different from NMR TRF1 in both normoxia and hypoxia.
NMR TRF1 exhibits improved telomere protection during replication under low oxygen
Because NMR TRF1 shows improved telomere binding, we hypothesized that NMR TRF1 may also better protect telomeres during replication. Without DDR induction in normoxia, tMEFs expressing NMR or mouse TRF1 showed similar growth characteristics (fig. S1, D and E). There was also no difference in DDR activation, based on 53BP1 and γH2A.X analyses (fig. S4B). However, the telomeres of dividing cells treated with drugs that induce replicative stress (camptothecin, aphidicolin, and hydroxyurea) were better protected from DDR by NMR TRF1 than the mouse protein, as measured by the telomere dysfunction-induced focus (TIF) assay (fig. S4, A, C, and E to G). This effect was observed in different cell systems and with two different DDR markers (γH2A.X and 53BP1). In all analyses, NMR TRF1 showed 2- to 10-fold fewer TIFs than mouse TRF1 (fig. S4, C, F, and G). Thus, the NMR protein confers a better telomere protection during cell replication than mouse TRF1.
Notably, mouse telomeres are almost unprotected under hypoxic condition. Without the drug, the level of DNA damage at telomeric sites in the case of mouse TRF1 was higher than with NMR TRF1 (Fig. 3D). In contrast, NMR TRF1 and mutGLAD TRF1 generated by CRISPR-Cas9 presented very low TIFs under hypoxic conditions even under replicative stress (Fig. 3, A and E). In addition, the level of TIFs in cells expressing mouse TRF1 subjected to replicative stress was closer to that of TRF1 knockout (KO) tMEFs than tMEFs expressing NMR TRF1 (Fig. 3A). This suggested that NMR TRF1 exhibits a gain of function with regard to telomere protection (capping and replicative stress) when cells are exposed to low oxygen. Clearly, mutGLAD is responsible, at least in part, for this adaptation to protect telomeres in physioxia (3% O2). These effects of NMR TRF1 do not only concern the detection of telomere damage in interphase cells but also during mitosis. We observed that in metaphase spreads, chromosomes under aphidicolin show statistically fewer multitelomeric signals (MTSs) when NMR TRF1 is expressed compared to mouse TRF1 (Fig. 3, B and C). MTS is a typical phenotype of telomere replication damage and fragility (1, 16). mutGLAD is sufficient to reinforce telomeres during replication when introduced into mouse TRF1, suggesting that the improved telomere replication conferred by NMR TRF1 results from its improved telomere association.
Fig. 3. NMR TRF1 enhances telomere protection under low oxygen and during replication.
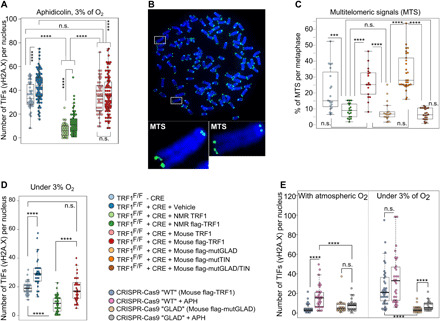
(A and D) Quantification of TIF of cells under 3% O2 and after aphidicolin treatment revealed by γH2A.X staining (A) or without drug treatment (D). (B and C) Representative images (B) and quantification (C) of the MTS rate per chromosome spreads. Cells for (A) to (D) experiences: MEFs with SV40-LT TRF1F/F + CRE + TRF1 rescue based on NMR or mouse TRF1 orthologs or TRF1 variants. (E) Quantification of TIFs, without the drug and with aphidicolin (+ APH) treatment revealed by γH2A.X staining at atmospheric oxygen and under 3% oxygen in MEFs with p53−/− CRISPR-Cas9 WT and GLAD. Statistical analyses: Student, Wilcoxon-Mann-Whitney, and ANOVA tests, n ≥ 30 samples, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
NMR TRF1 improves TIN2 nuclear localization
We discovered that the Tinf2 gene was duplicated in the NMR genome after the divergence of Hystricognathi rodents, which include guinea pigs and mole rats (fig. S5) (31). This duplication results in a higher Tinf2 gene expression in NMR brain and kidney than in the corresponding organs of mice and rats (fig. S5B). The Tinf2 expression level in the NMR is roughly similar to that in longer-lived mammals, such as primates. Therefore, we examined the possibility that the higher level of TIN2 in NMR synergizes with the unique sequences of NMR TRF1 to enhance the binding of TRF1 to telomeres.
First, we examined the association of NMR TRF1 to TIN2. For this, we counted the number of myc-TIN2 foci colocalized with flag-TRF1. In tMEFs, this rate was higher for NMR TRF1 than for mouse, mutGLAD, and mutTIN versions of TRF1 (Fig. 4, A and B). The higher rate of TIN2 association observed for NMR TRF1 correlated with a higher level of TRF1 and TIN2 nuclear localization (Fig. 4, C and D). Noteworthy, an ectopic myc-TIN2 expression increased the nuclear localization of mutTIN and of mouse TRF1 to a level similar to the one of mutGLAD without reaching the one of NMR TRF1 (Fig. 4C). The fact that TIN2 overexpression rescues the nuclear localization of the mouse TRF1 to a level similar to that of its mutant form carrying the mutGLAD sequence suggests that an improved interaction of TIN2 to TRF1 plays a role in the TRF1 nuclear localization efficiency depending on mutGLAD.
Fig. 4. NMR TRF1 supports improved TRF1 and TIN2 nuclear localization and limits telomere elongation.
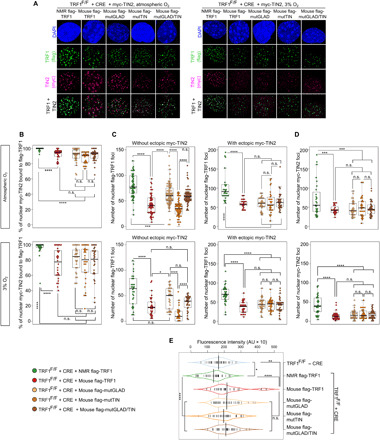
(A to D) Representative images (A) and quantification (B and D) of colocalization between flag-TRF1 foci (green) and myc-TIN2 (purple) (B); quantification of total number of flag-TRF1 foci in nucleus without and with ectopic myc-TIN2 expression (C) and of total number of myc-TIN2 foci in nucleus (D) in MEFs with SV40-LT TRF1F/F following KO of endogenous TRF1 by CRE and rescue by ectopic NMR or mouse TRF1 orthologs under atmospheric oxygen or at 3% oxygen. (E) Telomere length distribution averages are represented by the shape of each beanplot, with small vertical lines that represent the mean of telomere length for each metaphase spread. Statistics: Student and ANOVA tests, n ≥ 30 samples, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001. AU, arbitrary units.
Overall, these results suggest a model wherein NMR TRF1 exhibits an increased association with TIN2, leading to a tighter telomere binding and nuclear localization. This is in agreement with the fact that the Ala75Thr change, being located at the TRF1 dimer interface, could alter the intersubunit interaction to facilitate the association with TIN2. The above findings raise the possibility that a functionally important feature of NMR TRF1 is to increase its association with TIN2. Because Chen et al. (27) revealed that, in hypoxic conditions, TIN2 is sent to mitochondria where it regulates glycolysis, we hypothesize that the metabolic gain offered by NMR TRF1 relies on its complex association with TIN2. In agreement with this view, when compared to atmospheric oxygen conditions (Fig. 4, A to D), nuclear localization of TRF1 and TIN2 decreases under 3% of oxygen, suggesting that TIN2 may be sent to mitochondria in NMR cells.
Because TPP1 not only can modulate mitochondrial TIN2 (27) but also is essential for shelterin stability (32), we investigated the proportion of nuclear TPP1 tagged with V5 (fig. S6, D and E). Without ectopic myc-TIN2, mutGLAD, but not mutTIN, increased the number of TPP1-V5 foci, suggesting that mutTIN mutation may affect the formation of TRF1-TIN2-TPP1 subcomplexes. However, because TIN2 is expressed at a higher level in NMR than in mouse cells, the extra TIN2 may help TPP1 association. We propose that the elevated level of TIN2 expression in the NMR together with its enhanced association with TRF1 maintained a balanced distribution of TIN2 between the nucleus and mitochondria under hypoxic conditions, consequently preserving telomere protection.
NMR TRF1 promotes telomere shortening
Because TRF1 and TIN2 play a role in telomeric DNA length (TL), we determined whether NMR TRF1, which affects nuclear TRF1 and TIN2, may compete with telomerase. By measuring TL with quantitative fluorescence in situ hybridization (Q-FISH), we found that NMR TRF1 induced accelerated telomere shortening compared to mouse TRF1 (Fig. 4E and fig. S6A). TL was assessed in tMEFs after maintaining these cells long term in culture (fig. S1A). This effect likely results from the stronger binding of NMR TRF1 to telomeres, which is expected to down-regulate telomerase activity. The difference in the association with TIN2 between NMR TRF1 and mutGLAD may explain why we did not observe accelerated telomere shortening with mutGLAD, because TIN2 is also a regulator of telomerase activity (25).
DISCUSSION
We found that NMR TRF1 binds telomeres better than its mouse ortholog and that this effect was even more pronounced under conditions of low oxygen, a situation mimicking the subterranean life of NMR. NMR TRF1 offers a gain of function in protecting telomeres during replication under hypoxia, the conditions where mouse TRF1 fails, and a gain a function with regard to glycolytic capacity, supporting a basal energy fuel under extreme low oxygen conditions. This, together with the positive selection of the Ala75Thr site in mutGLAD, strongly suggests that NMR TRF1 has been selected during evolution as an adaptation to hypoxic conditions.
The relevance of our findings is twofold. First, we carried out a comparative analysis of NMR and mouse TRF1 forms using highly controlled cell models. This revealed the intrinsic properties of NMR TRF1 that support improved telomere shuttering, which, in turn, protects against replication stress and favors telomere shortening. Second, this is the first evidence for an effect of hypoxia on TRF1 function. Our work demonstrates that, in hypoxia, nuclear TRF1 in mouse cells offers imperfect telomere protection. This finding highlights the need for TRF1 to protect NMR telomeres from hypoxia while limiting telomerase activity.
Our results also point to a complex role of the TRF1-TIN2 axis in TRF1 nuclear localization and metabolic adaptation to hypoxia. In addition to its telomeric functions, TIN2 was reported to be located in mitochondria, and the reduction of TIN2 levels inhibits glycolysis (27). This is also related to the observed duplication of Tinf2 in the NMR genome, leading to a higher mRNA Tinf2 expression in the NMR. Hence, NMR TRF1 and TIN2 can synergize to regulate glucose metabolism, allowing NMR cells to survive in low oxygen conditions. Together, the TRF1-TIN2 axis appears as an important mechanism to support NMR longevity.
Critically short telomeres and replicative senescence are akin to tumor suppressor functions, and the capacity to limit telomere elongation appears as an evolutionary strategy in humans to decrease cancer susceptibility (33). If long telomeres can increase the risk of some types of cancers, telomere replication stress can also be a driver of malignant transformation (34). Therefore, the ability of NMR TRF1 to limit telomere elongation and prevent the replication stress–induced damage may contribute to the exceptional resistance of the NMR to cancer. Because NMR TRF1 offers strong protection to telomeres during replication, it limits DNA damage that otherwise contributes to aging and tumor development (35–37) and also restricts telomere elongation. We propose that the combination of low telomerase activity of NMR cells (fig. S6B) and strong association of NMR TRF1 with telomeres resulting in shorter telomeres contributes to low cancer incidence in the NMR, because long telomeres and high telomerase activity are tumor promoting. The ability of TRF1 to be highly localized at telomeres seems unique to NMR cells (fig. S6C). To summarize, functional and/or protected telomere is a determinant for cell fate, and TRF1 acts as a major modulator for aging and cancer.
Our discovery that enhanced TRF1 functions were selected during evolution in the NMR and likely contribute to its extreme longevity is in line with the wealth of data indicating that TRF1 exhibits antiaging properties. On the one hand, a decrease in TRF1 expression leads to bone marrow failure (38, 39) and lymphoma formation in aging mice (40, 41). On the other hand, an increase in TRF1 expression prevents an age-related decline in neuromuscular function, glucose tolerance, cognitive function, and chronic anemia, all without increases in cancer incidence (29). In summary, our work has revealed that a positively selected site in NMR TRF1 results in gain of function in physioxia by facilitating association of this protein with telomeres, protecting telomeres from replicative damage, inhibiting telomerase, and supporting adaptation to glucose metabolism.
MATERIALS AND METHODS
Cell construction strategy and artifacts
To prepare constructs for cell culture, we first used p53−/− MEFs. We knocked down the endogenous mouse TRF1 and rescued its function by the ectopic expression of the entire coding sequence (CDS) of NMR or mouse TRF1 genes. Because of a high homology between mouse and NMR TRF1s, we designed a short hairpin RNA (shRNA) to target the 3′ untranslated region (3′UTR) of mouse TRF1 gene and another shRNA with nine mismatches between mouse and NMR sequences to target the coding region. We also expressed the ectopic forms in the presence of scramble shRNA and identified conditions that support expression of mouse and NMR proteins and at the level equivalent to the expression of endogenous TRF1. This was accomplished by using green fluorescent protein (GFP)–containing lentiviral expression vectors at a multiplicity of infection (MOI) of 0.5 and by sorting the cells with a low GFP level. TRF1 expression was assessed by immunoblotting and quantitative polymerase chain reaction (qPCR) (fig. S1, A to H). Noteworthy, NMR TRF1 expressed from an infection at MOI = 2 showed an aberrant phenotype of the TRF1/telomeric signal (fig. S1F), further justifying the adjustment of the transgene expression to a physiological level.
Upon targeting by shRNAs, p53−/− MEFs expressing NMR TRF1 showed almost 80% of the telomere signal colocalized with NMR protein, whereas only 40% in the case of mouse TRF1 (fig. S3F). The ratio of telomeres colocalized with the ectopic NMR or mouse TRF1 was similar in the presence of scramble shRNA and following knockdown of endogenous TRF1 with shRNA #1 or #2. These observations suggest that the antibody (developed against human TRF1) preferentially binds NMR TRF1 over mouse TRF1 independent of cell context. To exclude the possibility of a preferential binding of anti-TRF1 antibodies (developed against human TRF1; referred to as “endo” in the figures) to the NMR protein, we carried out experiments with tMEFs wherein the endogenous TRF1 is replaced by N-terminal flag-tagged NMR TRF1, mouse TRF1, or mutated mouse TRF1. Therefore, we also used MEFs with simian virus 40 (SV40) large T TRF1F/F, applied the Cre-Lox system to knock out the endogenous TRF1, and integrated Flag tag at the N-terminal sequence of ectopic TRF1. The colocalization rate between TRF1 loci and telomere signal was not influenced by telomere length because we observed no deviation in telomere signal between the samples (fig. S3, E and G).
We were unable to obtain mutTIN and mutGLAD/TIN mutation within mouse Terf1 gene using CRISPR-Cas9 constructions despite many attempts. We also attempted to obtain a reverse mutation, named mutDALG mutNIT and mutDALG/NIT, in NMR cells but could only obtain a wild-type (WT) flag-TRF1 (fig. S1, J and K).
Plasmid constructs
Design of shRNA sequences to target mouse Terf1 mRNA was based on web service from Massachusetts Institute of Technology (MIT) (http://jura.wi.mit.edu/bioc/siRNAext/). Sequences were further analyzed by BLAST to ensure that they did not have matches in the NMR genome. Only sequences with a high number of mismatches between mouse and NMR Terf1 sequences were chosen. After annealing oligos flanked with sticky ends of Eco RI and Age I, oligos were inserted inside pLKO.1-TRC containing hygromycin resistance (Addgene, #24150) upon digestion/ligation with Eco RI and Age I restriction enzymes and T4 ligase. These plasmids were tested by reverse transcription qPCR (RT-qPCR), and two shRNAs showing high efficiency were used and designated as TRF1#1 shRNA (5′-CCGGGAACGCCTTATCGCAGTTAAACTCGAGTTTAACTGCGATAAGGCGTTCTTTTTTG-3′) and TRF1#2 shRNA (5′-CCGGAAATACTTGGATCACTACACTCTCGAGAGTGTAGTGATCCAAGTATTTTTTTTTG-3′). For rescue experiments with ectopic TRF1, CDS of mouse and NMR Terf1s were extracted by PCR from MEFs and NMR embryonic fibroblast (NEF) mRNA with mouse primers [5′-AGGCACGGCGAGCGCTTT-3′ (forward) and 5′-CCACCACCACCATAATGCTT-3′ (reverse)] and NMR primers [5′-GCGTGAGAGTTTCAACATGG-3′ (forward) and 5′-TCAAAGACCAGCAAGTTTCAA-3′ (reverse)]. Primers were designed using the National Center for Biotechnology Information (NCBI) primer and/or Primer3 websites. PCR products were included in pWPI lentivirus vector (Addgene, #12254) using Swa I and Pac I restriction enzyme sites with Gibson Assembly NEB kit using the following primers: forward NMR without flag-tag, 5′-TGAGGAATTTCGACATTTAAATGGCGGAGGATACCTCC-3′; forward NMR with flag-tag, 5′-TGAGGAATTTCGACATTTAAATGGACTACAAGGATGACGATGACAAGGCGGAGGATACCTCCGTG-3′; reverse NMR, 5′-GATACCGTCGAGATTAATTAATCAGTCTTCATTGTCTGAGCAAAT-3′; forward mouse without flag-tag, 5′-TGAGGAATTTCGACATTTAAATGGCGGAGACGGTCTCC-3′; forward mouse with flag-tag, 5′-TGAGGAATTTCGACATTTAAATGGACTACAAGGATGACGATGACAAGGCGGAGACGGTCTCCTCA-3′; reverse mouse, 5′-GATACCGTCGAGATTAATTAATCAGCTAATCAGTTTCAGTCTCTTCATTG-3′.
Mutagenesis was performed by PCR targeting the flag-mouse TRF1 vector with the following primers: mutGLAD, 5′-CGTGGCTACGGGCTGGATGCTCGACTTCCTC-3′ (forward) and 5′-GCCTCCGCCTTAGCCACAAGCTCCGCGTTCTC-3′ (reverse); mutTIN, 5′-TCGTGACAGCACCGAGGCTATTATTCATGGAC-3′ (forward) and 5′-GTACGACGAAAGCTGTCACGAGTACGACG-3′ (reverse).
For CRISPR-Cas9 coupled with homologous recombination (HR) experiment, several plasmids were generated: for guide RNAs (gRNAs) to cut the genomic DNA (gDNA) close to ATG-methionine GLAD and TIN positions and plasmids for HR to generate mutations. gRNAs were designed based on service from MIT (https://zlab.bio/guide-design-resources) for mouse genome and with https://crispr.dbcls.jp/ for NMR genome. gRNAs were inserted in PX458 pCas9 GFP (Addgene, #48138) according to the protocol from Ran et al. (42). To generate HR sequence including the CDS of the first exon of Terf1 with 300 to 425 nucleotides of left and right intronic arms, gDNA extractions from MEFs or NEFs were assessed by PCR in two steps with first outer primers and then with inner primers that contain gRNA sequence of ATG-methionine and Xho I sequence. PCR products were inserted in plasmid lacZ DsRed (Addgene, #99914). Then, mutagenesis was processed directly on plasmid containing gDNA of Terf1.
Mouse cutting sequences [gRNA + protospacer adjacent motif (PAM)]: ATG, 5′-GCGCTTTCGGTTTAACATGGCGG-3′; mutGLAG, 5′-GAGGTGGAGGCCGTGGCTGCGGG-3′; mutTIN, 5′-TCGTACTCGTGACAGCGCCGAGG-3′.
Mouse primers for pCas9 with gRNA: ATG, 5′-CACCgGCGCTTTCGGTTTAACATGG-3′ (forward) and 5′-AAACCCATGTTAAACCGAAAGCGCc-3′ (reverse); mutGLAD, 5′-CACCgGAGGTGGAGGCCGTGGCTGC-3′ (forward) and 5′-AAACGCAGCCACGGCCTCCACCTCc-3′ (reverse); mutTIN, 5′-CACCgTCGTACTCGTGACAGCGCCG-3′ (forward) and 5′-AAACCGGCGCTGTCACGAGTACGAc-3′ (reverse).
Mouse primers for HR PCR: Outer primers, 5′-GAGCCTGGGGGTAATATGCC-3′ (forward) and 5′–TCTCTCACCAACACACAGGC-3′ (reverse); inner primers for plasmidic integration with sgRNA-ATG, 5′-ATAACTCGAGGCGCTTTCGGTTTAACATGGCGGCATAACGCTCAAGTGCTCCA-3′ (forward) and 5′–ATAACTCGAGGCGCTTTCGGTTTAACATGGCGGCATAGGCTCTGGGGATCAAA-3′ (reverse).
Mouse primers for HR mutagenesis: ATG, 5′-GATGATGATAAAGCGGAGACGGTCTCCTCA-3′ (forward) and 5′-ATCTTTATAATCCATGTTAAACCGAAAGCGCTC-3′ (reverse); mutGLAD, 5′-CGTGGCTACGGGCTGGATGCTCGACTTC-3′ (forward) and 5′-GCCTCCGCCTTAGCCACAAGCTCCGCGTT-3′ (reverse); mutTIN, 5′-GAGTGCGGGCTCCCGGGCCGGAGGCTG-3′ (forward) and 5′-ACCCTCGGTGCTGTCACGAGTACGACGAAAGTCCTC-3′ (reverse).
NMR cutting sequences (gRNA + PAM): ATG, 5′-GCGTGAGAGTTTCAACATGGCGG-3′; mutDALG, 5′-AAGGCCGAGGCGGTGGCTACCGG-3′; mutNIT, 5′-CCGCATCCGCGACAGCACCGAGG-3′.
NMR primers for pCas9 with gRNA: ATG, 5′-CACCgGCGTGAGAGTTTCAACATGG-3′ (forward) and 5′-AAACCCATGTTGAAACTCTCACGCc-3′ (reverse); mutDALG, 5′-CACCgAAGGCCGAGGCGGTGGCTAC-3′ (forward) and 5′-AAACGTAGCCACCGCCTCGGCCTTc-3′ (reverse); mutNIT, 5′-CACCgCCGCATCCGCGACAGCACCG-3′ (forward) and 5′-AAACCGGTGCTGTCGCGGATGCGGc-3′ (reverse).
NMR primers for HR PCR: Outer primers, 5′-CATTCCCCTGCTCTCCCTTC-3′ (forward) and 5′-TCTGAAAGCCCGTAGGAGGA-3′ (reverse); inner primers for plasmidic integration with sgRNA-ATG, 5′- ATAACTCGAGGCGTGAGAGTTTCAACATGGCGGTTTCCTATGCCAATGCCTTC-3′ (forward) and 5′- ATAACTCGAGGCGTGAGAGTTTCAACATGGCGGAAGCTGGACCCTGTCTCAAA-3′ (reverse).
NMR primers for HR mutagenesis: ATG, 5′-gatgatgataaaGCGGAGGATACCTCCGTG-3′ (forward) and 5′-atctttataatcCATGTTGAAACTCTCACGCC-3′ (reverse); DALG, 5′- ggtggctgccGGGTGGATGCTGGATTTCCTCTGC-3′ (forward) and 5′-gcctccacctcCGCCACCAGGTCCGCGTC-3′ (reverse); NIT, 5′-tgagtgcGGCAGATGCTTGCAGGATCC-3′ (forward) and 5′-ccctcggcGCTGTCGCGGATGCGGTG-3′ (reverse).
All constructs were verified by sequencing.
Cell culture, virus production, transduction/transfection, CRISPR-Cas9/HR, and reagents
Immortalized MEFs with p53 KO (p53−/− MEF) were obtained from J. Jacobs laboratory. Immortalized SV40-LT MEF TRF1F/F were from the American Type Culture Collection (ATCC; #CRL-3316). Immortalized NEFs were described previously (43). Human MRC5 fibroblasts (ATCC; #CCL-171) were immortalized by pLenti CMV/TO SV40 small + large T (Addgene, #22298) (SV40-tT MRC5). Human fibroblasts human embryonic kidney (HEK) 293T/17 were used for virus production (ATCC; #CRL-11268). Cells were grown in DEM-GlutaMAX (Gibco, #10569-010) with 10% fetal calf serum (Gibco, #10437-028) and antibiotic-antimycotic 1× (Gibco, 15240-062) or antimicrobial agent (100 μg/ml) (InvivoGen, #ant-pm-2).
To create lentiviruses, we transfected HEK 293T/17 cells on 10-cm dishes with 8.6 μg of pCMVΔR8.91, 2.8 μg of phCMV-G, and 8.6 μg of lentiviral transfer plasmid encoding insert of interest. The list of plasmids is as follows: pLenti plasmid (SV40), pHAGE2 (CRE-IRES-puroR), pWPI-IRES-GFP plasmid containing different forms of the TRF1 CDS, and pLKO.1 containing mouse TRF1 and scrambled shRNA. Plasmids were transfected by calcium phosphate precipitation. Viral supernatants were collected 24 hours after transfection. The MOI was investigated by determining the percentage of GFP-positive cells using fluorescence-activated cell sorting (FACS) analysis 3 days after transfection or by the number of clones after 1 week of selection with an appropriated antibiotic.
For selection of p53−/− MEFs after mouse TRF1 shRNA transduction (MOI = 5), cells were selected on hygromycin (125 μg/ml) (InvivoGen, #ant-hg-1) for 5 days. SV40-LT MEF TRF1 F/F cells were transduced (MOI = 5) with CRE-IRES-puroR (Addgene, #30205) with puromycin selection (3 μg/ml) (InvivoGen, #ant-pr-1) for 3 days. Rescue of p53−/− MEFs and SV40-LT MEF TRF1 F/F cells was done with ectopic TRF1 (MOI = 0.5). SV40-LT MEF TRF1 F/F cells (1 × 106 cells) were transfected with myc-TIN2 or TPP1-V5 plasmid (5 μg) using Neon Transfection System (Invitrogen) or Nucleofector 2b (Lonza). Cell sorting was done based on GFP fluorescence using BD FACSAria I with 488-nm laser.
For CRISPR-Cas9/HR experiences, cells were transfected by electroporation (Neon Transfection System, Invitrogen) with a total of 4 μg for plasmids with gRNA and 8 μg for HR plasmid for a total of 1 × 5 106 MEFs or 4 × 106 NEFs. Directly after electroporation, cells were seeded in medium containing the nonhomologous end joining inhibitor Scr7 pyrazine (30 μM) (Sigma-Aldrich, #SML1546-5MG). Two days after transfection, cells were isolated (one cell per well onto 96-well plates) and sorted based on GFP and RFP (red fluorescent protein) fluorescence using BD FACSAria I. The gDNA of each clone was analyzed based on TRF1 PCR amplification and denaturation/annealing/digestion of PCR products with T7 endonuclease, which cleaves DNA mismatches (NEB, #E3321). Then, homoduplexes were sequenced to discriminate WT sequences from mutated sequences.
To induce replicative stress, cells were treated with camptothecin (5 nM) for 2 hours (Sigma-Aldrich, #C9911), aphidicolin (0.2 μM) for 16 hours (Calbiochem, #504744), or hydroxyurea (5 μM) for 48 hours (Sigma-Aldrich, #H8627). For further analyses, cells were fixed after a 2-hour release.
For normoxic conditions, p53−/− MEFs, SV40-LT MEF TRF1F/F cells, and SV40-tT MRC5 cells were cultured at 37°C, 5% CO2 at the atmospheric oxygen. To induce hypoxia, these cells were grown at 37°C, 5% CO2 and 3% O2 and then placed in a hypoxia chamber at 37°C, 5% CO2. SV40-LT NEF cells were cultured at 32°C, 5% CO2 and 3% O2.
Chromatin immunoprecipitation
A total of 107 cells/immunoprecipitation were collected and suspended in 15 ml of Dulbecco’s modified Eagle’s medium (DMEM). After wash with 1× phosphate-buffered solution (PBS), cells were cross-linked in PBS with 1% paraformaldehyde (Sigma-Aldrich), incubated 12 min at room temperature while rocking, and quenched with 0.125 M glycine for 5 min. Cells were then washed with cold PBS and lysed on ice for 10 min with 1 ml of extraction buffer [0.25 M sucrose, 10 mM tris-Cl (pH 8.0), 10 mM MgCl2, and 1% Triton X-100] plus protease (cOmplete Mini EDTA-free, Roche) and phosphatase (PhosSTOP, Roche) PPIC (phosphatase protease inhibitor cocktail) inhibitors. After spinning down at 3000 rpm at 4°C, the nuclear pellet was incubated with 1.5 ml of nuclear lysis buffer [50 mM tris-Cl (pH 8.0), 10 mM EDTA, and 1% SDS] plus PPIC and sonicated using Diagenode Bioruptor with 12 cycles of 30-s pulses and 60-s intervals to shear DNA to ~500–base pair fragments. Lysates were then cleared by centrifugation at 11,500 rpm for 10 min at 4°C and diluted 10-fold with ChIP dilution buffer [1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM tris-Cl (pH 8.0), and 167 mM NaCl] plus PPIC. Protein A/G Sepharose beads (30 μl; Invitrogen) were added, and the lysate was precleared for 1 hour at 4°C. Immunoprecipitation was carried out by adding 5 μg of anti-FLAG_M2 antibody (F3165-SIGMA) and rotating overnight at 4°C. Protein G beads (25 μl) blocked with bovine serum albumin (BSA) (1 mg/ml) were mixed with the immunoprecipitated chromatin 4 hours at 4°C while rotating. Beads were successively washed two times with (i) low-salt buffer [150 mM NaCl, 0.1% SDS, 1% Triton X-100, 20 mM EDTA, and 20 mM tris-Cl (pH 8.0)], (ii) high-salt buffer (500 mM NaCl, 1% Triton X-100, and 0.1% SDS), (iii) LiCl buffer [0.25 M LiCl, 1% IGEPAL CA-630, 1% sodium deoxycholate, 1 mM EDTA, and 10 mM tris-Cl (pH 8.0)], and (iv) tris-EDTA (TE) buffer. Beads were resuspended in 50 μl of elution buffer and incubated with 2 μl of ribonuclease (Sigma-Aldrich, R-4642), 1 μl of glycogen (20 mg/ml) for 30 min at 37°C, and 2.5 μl of proteinase K (Invitrogen) for 2 hours at 37°C. Reverse cross-linking was performed by incubating overnight at 65°C, and DNA was isolated using 2.2× AMPure XP beads (Beckman Coulter) according to the manufacturer’s instruction. ChIP-DNA was quantified using the Qubit dsDNA High Sensitivity Assay Kit (Thermo Fisher Scientific).
Slot blot
A total of 100 ng of telo-probe DNA fragment was denatured and radioactively labeled with 2 μl of NEB2 buffer, 4 μl of 10 mM 2′-deoxyadenosine 5′-triphosphate (dATP), 3′-deoxythymidine 5′-triphosphate (dTTP), and 2′-deoxyguanosine 5′-triphosphate (dGTP) mix (Promega, U1518), 2 μl of hexanucleotide 10× (Roche), 4 μl of 32P-labeled 2′-deoxycytidine 5′-triphosphate (dCTP) (40 μCi), and 1 μl of Klenow large fragment enzyme (M0212L BioLabs) for 1 hour at 37°C. The unincorporated radioactivity was removed using ProbeQuant G50 microcolumns (GE Healthcare). ChIP-DNA samples were incubated for 10 min at room temperature with 3× denaturation solution (1.5 M NaOH and 1 M NaCl), diluted 10-fold in dilution buffer (0.1 SSC and 0.125 M NaOH), and blotted on Amersham Hybond N+ membrane. Cross-linked membrane was incubated with SSC 6×, 0.5% SDS, and 1% milk at 50°C for 1 hour and hybridized with the radioactive probe at 50°C overnight. The membrane was washed once in 2× SSC at 50°C for 10 min, once in 2× SSC, 1% SDS at 50°C for 30 min, and twice in 2× SSC, 0.1% SDS at 50°C for 30 min. The membranes were exposed onto Phosphorimager screens, and the signal intensity was quantified with ImageQuant software.
Metaphase spread preparation, FISH, and Q-FISH analysis
For FISH, cells were incubated with KaryoMax (0.2 μg/ml) (Life Technologies, #15212) for 2 hours, trypsinized, washed with medium containing serum and PBS, swollen in 0.075 M KCl for 15 min at 37°C, fixed in ice-cold 3:1 methanol:acetic acid, and stored overnight (or up to 1 week) at 4°C. Metaphase spreads were dropped onto slides, aged overnight, rehydrated in PBS, fixed in 4% formaldehyde, treated with 1 mg of pepsin per 1 ml of 20 mM glycine (pH 2.0) for 10 min at 37°C, and again fixed with 4% formaldehyde. Then, slides were washed, dehydrated in an ethanol series, air-dried, and hybridized for 2 hours or overnight at room temperature with the FAM-TelC PNA (peptide nucleic acid) probe (PNA Bio, #F1001) in formamide, blocking reagent (1 mg/ml; Roche, #11096176001), and 10 mM tris-HCl (pH 7.2) after a 3-min denaturation at 80°C. Slides were washed with 70% formamide in 10 mM tris-HCl (pH 7.2) (twice for 15 min) and in 0.1 M tris-HCl (pH 7.0), 0.15 M NaCl, and 0.08% Tween 20 (three times for 5 min). Slides were dehydrated in ethanol and then mounted in Vectashield mounting medium with DAPI (Vector, #H-1200).
Images were produced using Zeiss Axio Vert.A1 microscope with X-Cite Series 120Q (Lumen Dynamics) light source and with Plan-Apochromat ×63 [numerical aperture (NA) 1.4, oil-immersion] lens. For Q-FISH, setting of fluorescence intensity was assessed with FluoSpheres carboxylate, 0.2 μm, yellow-green 505/515 (Life Technologies, #F8811). Analyses were performed using TFL-TeloV2 software developed by P. Lansdorp (https://wflintbox.com/public/offering/1895/).
Immunostaining and immunofluorescence detection
Cells were incubated with 5-ethylnyl-2’deoxyuridine (EdU) in the culture medium at a final concentration of 10 mM for 30 min before fixation in 4% formaldehyde. EdU staining was performed according to the manufacturer’s instructions (Life Technologies, #C10640) before immunofluorescence (IF)–FISH. For IF, slides were fixed with 4% formaldehyde at room temperature for 15 min and then incubated for 1 hour with the blocking buffer (0.8× PBS, 50 mM NaCl, 0.5% Triton X-100, and 3% milk), followed by incubation overnight at 4°C with primary antibody. Primary antibody included mouse monoclonal anti-human TRF1 (Abcam, #ab10579; dilution 1:300), mouse monoclonal anti-flag M2 (Sigma-Aldrich, #F3165; dilution 1:300), rabbit monoclonal anti-myc (Cell Signaling Technology, #71D10; dilution 1:200), mouse monoclonal anti–phospho-γH2A.X (Ser139) (EMD Millipore, #05-636; dilution 1:300), and rabbit polyclonal anti-53BP1 (Novus Biologicals, #NB100-305; dilution 1:300).
The cells were then washed with 0.8× PBS, 50 mM NaCl, and 0.1% Triton X-100 and incubated with anti-mouse Alexa488 (Life Technologies, #A11029), anti-rabbit Alexa488 (Life Technologies, #A11034), or anti-rabbit Alexa568 (Life Technologies, #A11036) antibodies (1:500) for 1 hour. Then, slides were washed with 0.8× PBS, 50 mM NaCl, and 0.1% Triton X-100. After IF, slides were mounted in Vectashield mounting medium with DAPI or additional staining by FISH was performed. After washing, slides were rinsed with PBS and dehydrated with successive ethanol solutions (3 × 3 min, 50, 75, and 100%). PNA probe was hybridized for 3 min at 80°C and for 2 hours or overnight at room temperature with the TelC Cy3 PNA probe (PNA Bio, #F1002) in a moist chamber in the dark as mentioned above.
Confocal images were produced using a Zeiss LSM 700 Axio Observer confocal microscope. Optical sections were recorded at an interval of 5 μm, with a Plan-Apochromat ×63 (NA 1.4, oil-immersion) lens. The excitation wavelengths were 405, 488, 555, and 639 nm for detection of DAPI, secondary antibodies Alexa488, secondary antibody Alexa568 and/or PNA TelC-Cy3, and Edu-Alexa647, respectively. Colocalizations were scored in each optical section by scrolling through the z-stack. Cropped images were done by ImageJ and Inkscape software.
Reverse transcription quantitative polymerase chain reaction
RNA was extracted from harvested cells using an RNA extraction kit according to the manufacturer’s instructions (Macherey Nagel, #740955.50) and reverse-transcribed using a complementary DNA (cDNA) reverse transcription kit (Bio-Rad, #1708840). qPCR amplification was done with the SYBR Green Supermix System (Bio-Rad, #1708880). The geometrical mean of two reference genes was used for gene expression normalization (reference genes: mouse Rpl19 and Ppia). Quantification of mTerf1 was done based on a small sequence with 100% homology between mouse and NMR sequences. The list of primer sequences is as follows: mRpl19, 5′-ATGAGTATGCTCAGGCTACAGA-3′ (forward) and 5′-GCATTGGCGATTTCATTGGTC-3′ (reverse); mPpia, 5′-GAGCTGTTTGCAGACAAAGTTC-3′ (forward) and 5′-CCCTGGCACATGAATCCTGG-3′ (reverse); mTerf1, 5′-CATGGCTTTGGGAAGAAGACA-3′ (forward) and 5′-GACACTTGTTCGGTTGTTGAA-3′ (reverse); mKHK-C, 5′-GCTGACTTCAGGCAGAGG-3′ (forward) and 5′-CCTTCTCAAAGTCCTTAGCAG-3′ (reverse); mGlut1, 5′-CAGTTCGGCTATAACACTGGTG-3′ (forward) and 5′-GCCCCCGACAGAGAAGAT-3′ (reverse).
Telomerase activity assay
Telomerase activity was measured in vitro by a qPCR telomerase repeat amplification protocol (TRAP) assay according to (44).
Immunoblotting
Cell extracts were made using radioimmunoprecipitation assay buffer [150 mM NaCl, 1% NP-40, 0.1% Triton X-100, 50 mM tris-HCl (pH 8.0)] and protease inhibitors (Roche, #05056489001). Lysates were digested for 30 min on ice and homogenized with a 21-gauge needle and then centrifuged for 20 min at maximum speed. Protein concentrations from supernatants were determined with “the DC Protein Assay” (Bio-Rad, #5000112). Samples were suspended in 3× Laemmli loading buffer (375 mM tris-HCl, 9% SDS, 50% glycerol, 9% β-mercaptoethanol, and 0.03% bromophenol blue) and heated at 95°C for 10 min. Electrophoresis was carried out with NuPAGE 10% bis-tris gel (Invitrogen, #NP0310BOX) and Mops or MES-SDS Running Buffer (Boston BioProducts, #BP-170; Invitrogen, #NP0002). Semidry transfers to polyvinylidene difluoride membrane (Life Technologies, #L2005) were done with a transfer buffer (Boston BioProducts, #BP-190) supplemented with 20% ethanol and 0.1% SDS. After blocking with 5% nonfat dry milk or BSA in PBS–Tween 20 (PBST) for 1 hour at room temperature, membranes were incubated in PBST at 4°C overnight with a primary antibody. Membranes were washed three times with PBS or TBST (tris-buffered saline–Tween 20) and then incubated with secondary antibody for 1 hour at room temperature. After washes, membrane revelations were performed by chemiluminescence (Bio-Rad, #170-5061) and developed with ChemiDoc XRS+ Imaging System (Bio-Rad). Images were analyzed with ImageJ software. The following antibodies were used: mouse monoclonal anti-human TRF1 (Abcam, #ab10579; dilution 1:500), mouse monoclonal anti-flag M2 (Sigma-Aldrich, #F3165; dilution 1:1000), rabbit monoclonal anti-myc (Cell Signaling Technology, #71D10; dilution 1:500), rabbit polyclonal anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Abcam, #ab9485; dilution 1:1000), enhanced chemiluminescence (ECL) horseradish peroxidase (HRP)–conjugated anti-mouse (GE Healthcare, #NXA931; dilution 1:5000), and ECL HRP-conjugated anti-rabbit (GE Healthcare, #NA934; dilution 1:2000).
ECAR by Seahorse
ECAR measurements were performed using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience). MEFs were seeded at 30,000 per well before the day of analysis. The cells were incubated in a humidified 37°C incubator with 5% CO2 with regular medium (DMEM). For hypoxic condition, cells were incubated in 37°C, 5% CO2, 1% O2 humidified chamber. One hour before performing an assay, DMEM was removed and replaced by appropriated medium without glucose in XF cell plates in a 37°C/non-CO2 incubator. For glycolysis analyses, after three baseline measurements of ECAR, successive injections of glucose (final concentration, 100 mM), oligomycin (final concentration, 2 μM), and 2-deoxyglucose (2-DG) (final concentration, 500 mM) were performed. Three measurements were assessed after injections.
Tinf2 analyses in mammals
Publicly available genome assemblies of mammals were downloaded from NCBI. These were then searched using Selenoprofiles, a family-based gene finder (45), using a profile alignment built ad hoc from mammalian Tinf2 sequences. Gene predictions were completed, when possible, by open reading frame extension and then manually inspected. While most mammals had a single Tinf2 gene, Hystricognathi had two copies. We further analyzed Tinf2 genes in this group, including mouse as outgroup. We built a gene tree based on Tinf2 protein sequences, using the “phylomedb4” workflow in ETE3 (46) (including the meta-alignment of protein sequences by M-coffee (47), multiple neighbor-joining routines for evolutionary model testing, and finally maximum likelihood phylogenetic reconstruction with PhyML (48)). The resulting protein tree suggested independent duplications in the various Hystricognathi species: The two copies in the NMR are more similar to each other than to any other homolog. However, this scenario is not consistent with their pattern of gene occurrence, as well as with their common synteny. We hypothesized that gene conversion has occurred between Tinf2 paralogs, which could result in the observed tree topology. The two Tinf2 paralogs in NMR have the characteristics required for gene conversion: They are highly similar (>98% identity in coding sequences), and they are located on the same chromosome and on the same strand, at ~45-kb distance. To assess the likelihood of gene conversion, we performed phylogenetic reconstruction using an alignment of the estimated 5′UTR sequences of Tinf2 genes, using the “full_fast_modeltest_bootstrap” workflow in ETE3. The 5′UTR tree correctly clustered the two paralogous gene groups, resulting in a tree topology consistent with a single Tinf2 duplication predating the split of Hystricognathi (fig. S5, C to E). Complemented with manual sequence alignment inspection, we concluded that gene conversion occurred between Tinf2 paralogs in the genomic region that comprises their entire coding sequences and introns, but not their full 5′UTRs. Next, we assessed the effect of the Tinf2 duplication on gene expression using an RNA-seq dataset that included diverse mammalian species and spanned multiple tissues (49). We quantified the RNA-seq read mapping to the coding sequences of Tinf2 genes. Because of the high similarity of NMR Tinf2 paralogs, reads coming from either gene cannot be distinguished so that the resulting quantification represents their aggregated expression level. For comparison across species, normalization was performed on the expression levels of all one-to-one orthologs quantified in our dataset (13,784 genes), assuming the same gene expression mean and variance in all samples.
RNA-seq and analysis
RNA quality was checked using Bioanalyzer 2100 (Agilent Technologies). Indexed cDNA libraries were obtained using the Ovation Solo RNA-Seq System (NuGen) following the manufacturer’s recommendations. Multiplexed libraries were loaded on a NovaSeq 6000 system (Illumina) using an S2 flow cell, and sequences were produced using a 200-cycle kit. Paired-end reads were mapped against the mouse reference genome GRCm38 using STAR software (version 2.5.3a) to generate read alignments for each sample. Gene-level counts were obtained using HTSeq (50). Differential gene expression analysis was performed using DESeq2 R package (51). P values were corrected for multiple testing using the Benjamini and Hochberg default method. The adjusted P values were filtered with a cutoff of 0.05 to obtain a list of differentially expressed genes. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed using “gage” and “KEGGREST” R packages (52). The list of differentially expressed genes with their corresponding log2 fold change values was used for this analysis. Gene Expression Omnibus (GEO) accession: GSE163064.
Statistics
Considering size and distribution of samples, we performed parametric Student’s t test and one-way analysis of variance (ANOVA) and nonparametric Wilcoxon-Mann-Whitney test, using R software to test significance of mean differences in each experiment. Boxplots were based on default parameters boxplot in R software. For all experiments, relative TRF1 localization, TIN2-TRF1 association, and replicative stress n = 30 or 50, except for ChIP (n = 8 for NMR-TRF1 and mouse-TRF1 samples, n = 6 for GLAD samples, and n = 2 for others). For metaphase spreads, n = 15 to 30, corresponding to ≥1050 chromosomes. For Seahorse assay, n = 3 to 5 replicates per sample.
Acknowledgments
We thank R. Liao for the use of microscopy and flow cytometry facilities and W. Oldham and J. Loscalzo for the use of a hypoxia chamber and the Seahorse facility. Sequencing was performed at the Brussels Interuniversity Genomics High Throughput core (www.brightcore.be). We also thank F. Miot, X. De Deken, and I. Pirson for some aliquot of antibodies against tag V5 and Flag (M2). Plasmid TPP1-V5 was provided by Z. Songyang and D. Liu. We also thank A. Leon Trapp who coordinated transfer of materials. Funding: The V.N.G. laboratory is supported by NIH grants, and the E.G. laboratory is supported by ANR S-ENCODE, the ARC Fondation (labelized team and the CANC’AIR program Genexposomics) and the Inserm Cross-cutting program AGEMED. The B.B. laboratory is supported by the WELBIO (FRFS-WELBIO-CR-2017S-01) and the Fondation contre le cancer (FCC / ULB 2018-067). V.G. and V.N.G. were supported by the National Institutes of Health grant AG047200. A.A. was supported by postdoctoral fellowships from the French Foundation ARC, EMBO long-term fellowship (EMBO ALTF 896-2013), and Fondation contre le cancer (FCC/ULB 2018_067). Author contributions: A.A. conceived the project; performed plasmid and cell constructions, virus production, all microscopy, RT-qPCR, immunoblotting, and ECAR experiments, modeling of TRFH for NMR TRFH with I-TASSER, telomerase activity, and statistical data; interpreted all data; and coordinated all experiments. M.M. performed Tinf2 gene conversion and duplication analyses. M.P. performed KEGG analysis. D.F. performed ChIP experiments. F.L. performed read alignments of RNA-seq and mapped against mouse genome reference. B.B., V.G., and E.G. contributed to the design and interpretation of experiments. V.N.G. supervised the project. A.A., E.G., and V.N.G. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabe0174/DC1
REFERENCES AND NOTES
- 1.Gilson E., Géli V., How telomeres are replicated. Nat. Rev. Mol. Cell Biol. 8, 825–838 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Harley C. B., Futcher A. B., Greider C. W., Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 (1990). [DOI] [PubMed] [Google Scholar]
- 3.Buffenstein R., The naked mole-rat: A new long-living model for human aging research. J. Gerontol. A Biol. Sci. Med. Sci. 60, 1369–1377 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Ruby J. G., Smith M., Buffenstein R., Naked mole-rat mortality rates defy gompertzian laws by not increasing with age. eLife 7, e31157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor K. R., Milone N. A., Rodriguez C. E., Four cases of spontaneous neoplasia in the naked mole-rat (Heterocephalus glaber), a putative cancer-resistant species. J. Gerontol. A Biol. Sci. Med. Sci. 72, 38–43 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Liang S., Mele J., Wu Y., Buffenstein R., Hornsby P. J., Resistance to experimental tumorigenesis in cells of a long-lived mammal, the naked mole-rat (Heterocephalus glaber). Aging Cell 9, 626–635 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian X., Azpurua J., Hine C., Vaidya A., Myakishev-Rempel M., Ablaeva J., Mao Z., Nevo E., Gorbunova V., Seluanov A., High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park T. J., Reznick J., Peterson B. L., Blass G., Omerbašić D., Bennett N. C., Kuich P. H. J. L., Zasada C., Browe B. M., Hamann W., Applegate D. T., Radke M. H., Kosten T., Lutermann H., Gavaghan V., Eigenbrod O., Bégay V., Amoroso V. G., Govind V., Minshall R. D., Smith E. S. J., Larson J., Gotthardt M., Kempa S., Lewin G. R., Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science 356, 307–311 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Kim E. B., Fang X., Fushan A. A., Huang Z., Lobanov A. V., Han L., Marino S. M., Sun X., Turanov A. A., Yang P., Yim S. H., Zhao X., Kasaikina M. V., Stoletzki N., Peng C., Polak P., Xiong Z., Kiezun A., Zhu Y., Chen Y., Kryukov G. V., Zhang Q., Peshkin L., Yang L., Bronson R. T., Buffenstein R., Wang B., Han C., Li Q., Chen L., Zhao W., Sunyaev S. R., Park T. J., Zhang G., Wang J., Gladyshev V. N., Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palm W., de Lange T., How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Fumagalli M., Rossiello F., Clerici M., Barozzi S., Cittaro D., Kaplunov J. M., Bucci G., Dobreva M., Matti V., Beausejour C. M., Herbig U., Longhese M. P., di Fagagna F. d.’ A., Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hewitt G., Jurk D., Marques F. D. M., Correia-Melo C., Hardy T., Gackowska A., Anderson R., Taschuk M., Mann J., Passos J. F., Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartkova B. J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J. M., Lukas C., Ørntoft T., Lukas J., Bartek J., DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Gorgoulis V. G., Vassiliou L.-V. F., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., DiTullio R. A., Kastrinakis N. G., Levy B., Kletsas D., Yoneta A., Herlyn M., Kittas C., Halazonetis T. D., Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Sfeir A., Kosiyatrakul S. T., Hockemeyer D., MacRae S. L., Karlseder J., Schildkraut C. L., de Lange T., Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmermann M., Kibe T., Kabir S., de Lange T., TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 28, 2477–2491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Steensel B., de Lange T., Control of telomere length by the human telomeric protein TRF1. Nature 385, 740–743 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Fairall L., Chapman L., Moss H., De Lange T., Rhodes D., Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol. Cell 8, 351–361 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Bhanot M., Smith S., TIN2 stability is regulated by the E3 ligase Siah2. Mol. Cell. Biol. 32, 376–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frescas D., de Lange T., TRF2-tethered TIN2 can mediate telomere protection by TPP1/POT1. Mol. Cell. Biol. 34, 1349–1362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu C., Rai R., Huang C., Broton C., Long J., Xu Y., Xue J., Lei M., Chang S., Chen Y., Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Res. 27, 1485–1502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye J. Z. S., De Lange T., TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat. Genet. 36, 618–623 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Frescas D., de Lange T., A TIN2 dyskeratosis congenita mutation causes telomerase-independent telomere shortening in mice. Genes Dev. 28, 153–166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savage S. A., Giri N., Baerlocher G. M., Orr N., Lansdorp P. M., Alter B. P., TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 82, 501–509 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S. H., Kaminker P., Campisi J., TIN2, a new regulator of telomere length in human cells. Nat. Genet. 23, 405–412 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abreu E., Aritonovska E., Reichenbach P., Cristofari G., Culp B., Terns R. M., Lingner J., Terns M. P., TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol. Cell. Biol. 30, 2971–2982 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L. Y., Zhang Y., Zhang Q., Li H., Luo Z., Fang H., Kim S. H., Qin L., Yotnda P., Xu J., Tu B. P., Bai Y., Songyang Z., Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol. Cell 47, 839–850 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hohensinner P. J., Kaun C., Buchberger E., Ebenbauer B., Demyanets S., Huk I., Eppel W., Maurer G., Huber K., Wojta J., Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim. Biophys. Acta Mol. Cell Res. 1863, 360–367 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Derevyanko A., Whittemore K., Schneider R. P., Jiménez V., Bosch F., Blasco M. A., Gene therapy with the TRF1 telomere gene rescues decreased TRF1 levels with aging and prolongs mouse health span. Aging Cell 16, 1353–1368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirtschink P., Krishnan J., Grimm F., Sarre A., Hörl M., Kayikci M., Fankhauser N., Christinat Y., Cortijo C., Feehan O., Vukolic A., Sossalla S., Stehr S. N., Ule J., Zamboni N., Pedrazzini T., Krek W., HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 522, 444–449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macrae S. L., Zhang Q., Lemetre C., Seim I., Calder R. B., Hoeijmakers J., Suh Y., Gladyshev V. N., Seluanov A., Gorbunova V., Vijg J., Zhang Z. D., Comparative analysis of genome maintenance genes in naked mole rat, mouse, and human. Aging Cell 14, 288–291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Connor M. S., Safari A., Xin H., Liu D., Songyang Z., A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc. Natl. Acad. Sci. U.S.A. 103, 11874–11879 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen M. E. B., Hunt S. C., Stone R. C., Horvath K., Herbig U., Ranciaro A., Hirbo J., Beggs W., Reiner A. P., Wilson J. G., Kimura M., De Vivo I., Chen M. M., Kark J. D., Levy D., Nyambo T., Tishkoff S. A., Aviv A., Shorter telomere length in Europeans than in Africans due to polygenetic adaptation. Hum. Mol. Genet. 25, 2324–2330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macheret M., Halazonetis T. D., DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. Mech. Dis. 10, 425–448 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Flach J., Bakker S. T., Mohrin M., Conroy P. C., Pietras E. M., Reynaud D., Alvarez S., Diolaiti M. E., Ugarte F., Forsberg E. C., Le Beau M. M., Stohr B. A., Méndez J., Morrison C. G., Passegué E., Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaillard H., García-Muse T., Aguilera A., Replication stress and cancer. Nat. Rev. Cancer 15, 276–289 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Vermeij W. P., Hoeijmakers J. H. J., Pothof J., Aging: Not all DNA damage is equal. Curr. Opin. Genet. Dev. 26, 124–130 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Beier F., Foronda M., Martinez P., Blasco M. A., Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood 120, 2990–3000 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rufer N., Brümmendorf T. H., Chapuis B., Helg C., Lansdorp P. M., Roosnek E., Accelerated telomere shortening in hematological lineages is limited to the first year following stem cell transplantation. Blood 97, 575–577 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Beccaria M., Martinez P., Mendez-Pertuz M., Martinez S., Blanco-Aparicio C., Canamero M., Mulero F., Ambrogio C., Flores J. M., Megias D., Barbacid M., Pastor J., Blasco M. A., Therapeutic inhibition of TRF1 impairs the growth of p53-deficient K-RasG12V-induced lung cancer by induction of telomeric DNA damage. EMBO Mol. Med. 7, 930–949 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartmann K., Illing A., Leithäuser F., Baisantry A., Quintanilla-Martinez L., Rudolph K. L., Gene dosage reductions of Trf1 and/or Tin2 induce telomere DNA damage and lymphoma formation in aging mice. Leukemia 30, 749–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S. G., Mikhalchenko A. E., Yim S. H., Lobanov A. V., Park J. K., Choi K. H., Bronson R. T., Lee C. K., Park T. J., Gladyshev V. N., Naked mole rat induced pluripotent stem cells and their contribution to interspecific chimera. Stem Cell Rep. 9, 1706–1720 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mender I., Shay J. W., Arabia S., Telomerase repeat amplification protocol (TRAP). Bio Protoc. 5, e1657 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mariotti M., Guigó R., Selenoprofiles: Profile-based scanning of eukaryotic genome sequences for selenoprotein genes. Bioinformatics 26, 2656–2663 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huerta-Cepas J., Serra F., Bork P., ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wallace I. M., O’Sullivan O., Higgins D. G., Notredame C., M-Coffee: Combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34, 1692–1699 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O., New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Fushan A. A., Turanov A. A., Lee S. G., Kim E. B., Lobanov A. V., Yim S. H., Buffenstein R., Lee S. R., Chang K. T., Rhee H., Kim J. S., Yang K. S., Gladyshev V. N., Gene expression defines natural changes in mammalian lifespan. Aging Cell 14, 352–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anders S., Pyl P. T., Huber W., HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanehisa M., Goto S., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabe0174/DC1