

Kinetic environment controls cell fate.
Abstract
Exposure of cells to diverse types of stressful environments differentially regulates cell fate. Although many types of stresses causing this differential regulation are known, it is unknown how changes over time of the same stressor regulate cell fate. Changes in extracellular osmolarity are critically involved in physiological and pathophysiological processes in several tissues. We observe that human cells survive gradual but not acute hyperosmotic stress. We find that stress, caspase, and apoptosis signaling do not activate during gradual stress in contrast to acute treatments. Contrary to the current paradigm, we see a substantial accumulation of proline in cells treated with gradual but not acute stresses. We show that proline can protect cells from hyperosmotic stress similar to the osmoprotection in plants and bacteria. Our studies found a cell fate switch that enables cells to survive gradually changing stress environments by preventing caspase activation and protect cells through proline accumulation.
INTRODUCTION
All cells use signal transduction pathways to respond to physiologically relevant changes in extracellular stressors, nutrient levels, hormones, and morphogens. These environments vary as functions of both concentration and time in healthy and diseased states (1). Cell signaling and cell fate responses to the environment are commonly studied using acute concentration changes (1). Only a few pioneering studies have explored the effects of the concentration and time, which is a gradual change of stimuli as a function of time on cell signaling in microbes (2) and in mammalian cells (3). Thus, the impact of the rate of environmental change on cell signaling, cell fate, and phenotype is a fundamental and poorly understood cell biological property (Fig. 1A). We address this lack in knowledge by thoroughly measuring molecular changes in cells exposed to gradual environmental changes.
Fig. 1. Human cell fate decisions are regulated differently upon step or ramp treatment conditions.
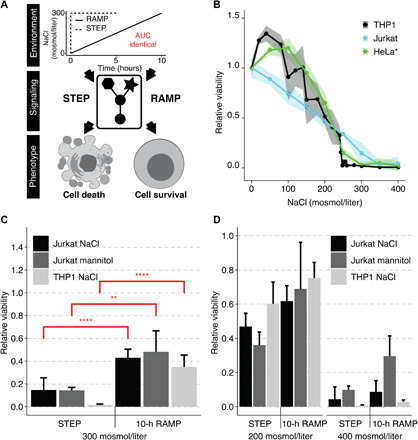
(A) Environments such as concentration ramps, as observed in different physiological relevant conditions, may differentially modulate cell signaling, cell fate, and phenotype even if the final concentration and total amount of stress are identical. Step experiments finish after 5 hours and ramp experiments finish after 10 hours to account for the same total exposure or AUC. (B) We measured relative cell viability after exposure to instant hyperosmotic stress (NaCl for 5 hours for Jurkat and THP1) or 24 hours (HeLa cells). Cell viability was determined by measuring intracellular adenosine triphosphate (Jurkat and THP1) or cell counts (HeLa). The shaded area represents the SD (Jurkat and THP1) or SE (HeLa) (11). (C and D) Relative cell viability was determined for step and 10-hour ramp treatment after addition of (C) 300 mosmol/liter osmolyte or (D) 200 and 400 mosmol/liter osmolyte. We determined viability at the end of the experiment after reaching the same cumulative exposure of additional NaCl. Bars represent data from at least three independent experiments for each condition. Error bars represent SD. Two-sided unpaired Student’s t test: **P < 0.01 and ****P < 0.001.
To begin to understand how the rate of environmental change regulates human cell fate decisions, we systematically expose cells to varying temporal profiles of increasing NaCl concentrations. NaCl is a ubiquitous osmolyte in the human body and causes cells to experience hypertonic stress at concentrations that change over time (4). While all tissues can experience increased NaCl concentrations in their microenvironment, measurements of osmolytes in the kidney have revealed very high physiological NaCl concentrations (5). In the kidney, spatial gradients of different osmolytes exist that change over time under normal and pathophysiological conditions (6). Hypertonicity changes over time are also known to occur in the intestinal system (7), the cerebrovascular discs (8), and the skin (9). In many of these high osmolarity tissues, resident immune cells provide basal protection or require migration upon an immune response of additional immune cells (10). Therefore, immune cells need to have the ability to survive such harsh high osmolarity environments that change over time. We choose immune cells as a model to systematically investigate how both rapidly and slowly increasing hypertonic yet physiological environments affect cell survival, signaling, and metabolism.
RESULTS
The rate of environmental change regulates cellular phenotype
We compared cell viability, cell signaling, and metabolism in cells exposed to either linear (ramp) or acute (step) concentration changes in the environments in which the final concentration and the total amount of osmotic stress [area under the curve (AUC)] is identical (Fig. 1A and fig. S1A). We identified the dynamic range of cell viability by determining the tolerance of monocytes (THP1 cell line, male, and acute monocytic leukemia), T cells (Jurkat, male, and acute T cell leukemia), and cervical cells [HeLa, female, and cervical adenocarcinoma (11)] to step increases in NaCl concentrations (Fig. 1B). In the nonstress control condition, cells were grown in culture under physiological NaCl concentrations of about 280 mosmol/liter NaCl to which we added the hypertonic osmolytes NaCl and mannitol. To stress the cells and mimic in vivo osmolyte changes, we added up to 400 mosmol/liter NaCl to the cells (Fig. 1 and fig. S1). We observed that cell viability decreases with an increased NaCl concentration of up to 300 mosmol/liter. At and above of 300 mosmol/liter NaCl, cell viability is below 15% for all cell lines. Our results with the abovementioned cell lines are consistent with previous studies in HeLa cells (Fig. 1B) (11), indicating that different cell types respond similarly to hypertonic stress.
We then quantified the response of different cell lines (Jurkat and THP1) to different rates and final NaCl concentrations (Fig. 1, C and D, and fig. S1). To compare the different conditions for the same final NaCl concentration, we exposed cells to the same cumulative exposure by integrating the total amount of NaCl over the entire profile (AUC). We performed experiments for each NaCl concentration for ramp durations of up to 10 hours. For experiments with ramp durations of less than 10 hours, cells stayed at the final NaCl concentration until the AUC is identical to the 10-hour ramp experiment (fig. S1A). When we exposed Jurkat cells to 300 mosmol/liter hypertonic osmolyte, viability improves from 15 to 40% for a ramp duration of 10 hours [Fig. 1C (black) and fig. S1B (cyan)]. In comparison, a step increase of 200 mosmol/liter NaCl to the media for 5 hours reduced viability to around 50% and showed only minor improvement with increases in ramp duration [Fig. 1D (black) and fig. S1B (magenta)]. For the step condition of added 400 mosmol/liter NaCl for 5 hours, cell viability was below 5% and showed only minor improvement with increasing ramp durations [Fig. 1D (black) and fig. S1B (green and yellow)]. These observations are consistent in THP1 cells, indicating that this effect is reproducible in a different cell line and cell type [Fig. 1, C and D (light gray), and fig. S1C]. To distinguish the effect on cell viability between NaCl toxicity and changes in external osmolarity, we repeated the experiments with mannitol in the Jurkat cell line at the same osmolar concentrations [Fig. 1, C and D (dark gray), and fig. S1D]. Mannitol is not able to easily pass through the cell membrane and is known to have low cell toxicity. When we added 300 mosmol/liter mannitol to the medium, Jurkat cells survive better during the ramp compared to the step treatment. This comparison shows no difference between cells treated with NaCl or mannitol, indicating that extracellular hypertonicity and not NaCl-specific toxicity drives these effects. These results strongly suggest that cell viability improvements while slowly increasing NaCl concentration are a robust cell type– and cell line–independent hypertonic stress response.
A functional temporal screen identifies regulators of cell viability in step and ramp conditions
Authors of previous studies argued that up-regulation of genes encoding proteins responsible for the accumulation of cell internal osmolytes such as taurine (TauT), betaine (BGT1), sorbitol (AR), and inositol (SMIT) is the cause for improved viability in kidney cells exposed to a linear increase in osmolarity (12). To address whether indeed these osmolytes are increased in our experiments, we determined the change in osmolyte levels in the cell by mass spectrometry measured in 5-hour step and 10-hour ramp conditions both to a final osmolarity of additional 300 mosmol/liter NaCl. We found that sorbitol, inositol, betaine, taurine, and urea do not change compared to unstimulated cells (Fig. 2A).
Fig. 2. Temporal functional quantitative flow cytometry screen identifies differential regulation of stress and caspase signaling during step and ramp hyperosmotic stress conditions.
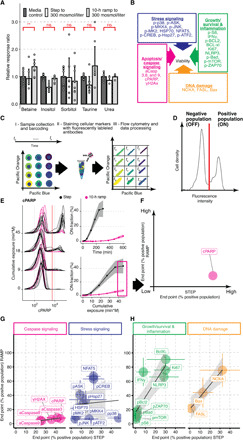
(A) Mean response ratio of cellular osmolytes relative to media measured in Jurkat cells exposed to an additional 300 mosmol/liter NaCl and determined by mass spectrometry. Two-sided unpaired Student’s t test: **P < 0.01; ns, not significant. (B) Overview of protein markers in this study. (C) Multiplex flow cytometry workflow to quantify dynamic changes in protein activity over time. (D) Flow cytometry distribution is threshold-gated (red line). (E) Representative flow cytometry distributions for cleaved PARP (cPARP) at selected time points for step (black) and 10-hour ramp (magenta) experiments (left), positive cells (ON-fraction) over time (right, top), or cumulative NaCl exposure (right, bottom). Mean (solid line) and SD (shaded area) of >3 biological replicas. (F) End point measurement (magenta box in E) to determine ON-fractions to compare changes for step and ramp conditions. (G and H) Comparison of end point measurement for individual markers of groups as indicated in (B). Circles represent mean of ON-fractions after exposing cells for 5 hours (step) or 10 hours (ramp) to 300 mosmol/liter NaCl. N ≥ 3, colored lines represent SD. Black lines indicate linear regression fits. Shaded area represents 95% confidence interval.
To understand which cellular mechanisms contribute to improved viability during the slow ramp, we performed a temporal functional screen using a selected set of 27 well-established and validated markers of cell state and signaling that contribute to cell viability (Fig. 2B). We grouped these into four cellular processes known to have an impact on cellular viability: stress signaling (blue), caspase signaling (magenta), DNA damage (orange), and growth/survival and inflammation (green) (Fig. 2B). Each of these processes is known to be affected by increased NaCl concentrations (4). The process “stress signaling” (blue) consists of markers belonging to stress/mitogen-activated protein kinases (SAPK/MAPK) pathways (13) such as phosphorylated proteins p38, JNK, MK2, ASK1, MKK4, HSP27, CREB, ATF2, as well as protein levels of HSP70 and NFAT5/TonEBP (14). MAPK pathways are known to convey stress signals to alter gene expression and cell phenotype (15). Proteins in the “caspase signaling” group are initiator caspases (16), such as activated caspase 8 (extrinsic pathway) and caspase 9 (intrinsic pathway), effector caspase 3 (17), cleaved poly (ADP-ribose) polymerase (cPARP) (18) as a substrate of caspase 3, and histone H2AX (γH2AX) (19) as a marker for the excessive DNA damage caused by DNA degradation during apoptosis. The “growth/survival and inflammation” group contains proteins that counteract apoptotic responses or indicate growth, proliferation, and inflammatory stimulation. The group contains phosphorylated forms of Bad and Bcl2, two antiapoptotic proteins (20); mechanistic target of rapamycin (mTOR) (21), a key node in the cell growth pathway; ribosomal protein S6 (22), a marker for active translation; and p-ZAP70, a marker for activated inflammatory signaling. The group also contains proteins Bcl-XL (23), an antiapoptotic protein; Ki67, a general marker of proliferative activity; NLRP3 (24), a marker for the inflammasome; and intracellular interferon-γ (IFNγ), a marker for inflammatory cytokine production. In response to DNA damage (25, 26), proteins such as Noxa, Fas-L, and BAX are expressed and fall into the group DNA damage.
We used fluorescent cell barcoding for multiplex quantitative flow cytometry to identify differentially regulated markers over time in step or ramp conditions (27). This functional temporal screen allows us to uniquely encode each time point sample with a combination of two dye concentrations (Fig. 2C). We pooled barcoded samples and then split them again into different tubes to stain each split sample with specific antibodies. The advantages of first barcoding and then sample splitting are as follows: reduced variability between samples, increased throughput, and reduced cost for different markers. Using this approach, we screened protein markers in Jurkat cells for their change over time in step versus 10 hours of ramp experiments to an additional concentration of 300 mosmol/liter NaCl. After data collection, we demultiplexed each sample with one or two protein markers to extract the individual time points (Fig. 2, C and D). To quantify each marker’s response, we next computed the fraction of positive cells for this marker and called this population “ON-fraction” (Fig. 2, D and E). We then plotted the ON-fraction of each marker at the end of the time course experiment between the ramp and the step treatment to understand the correlation between the markers in each group (Fig. 2F). This analysis revealed several distinct response patterns: (i) We observed strong activation in step but not ramp condition of proteins of the caspase signaling group and p38 of the stress signaling group (Fig. 2G and fig. S2). (ii) We observed minimal activation in step but strong activation in ramp conditions for some markers of stress response (pASK, NFAT5, and HSP70) [Fig. 2G (blue) and fig. S3], growth (Ki67), antiapoptotic (Bcl-XL), inflammation (IFNγ and NLRP3) [Fig. 2H (green) and fig. S4], and markers of DNA damage [Fig. 2H (orange) and fig. S5]. (iii) A screen for other markers of cell survival, growth, and DNA damage reveals no substantial differential changes over time. On the basis of this temporal functional screen, we focused on protein markers of the caspase signaling group.
Caspases differentially regulate step and ramp conditions
Activated caspases 3, 8, 9, cPARP, and γH2AX all showed strong activation (ON-fraction) during the 300 mosmol/liter NaCl step treatment [Fig. 3, A to E (black) and fig. S2E). Notably, caspase and γH2AX activation, as well as PARP cleavage, are negligible during the 10-hour ramp treatment condition to the same final concentration [Fig. 3, A to E (magenta), and fig. S2E). To test whether the time of exposure to the final concentration is critical for caspase-dependent apoptosis, we measured caspase activation for 5 hours after the 10-hour ramp. These results demonstrate that caspases and PARP cleavage are not activated after the ramp supporting our hypothesis that ramp treatment prevents caspase activation (fig. S2, G to I). To better understand the induction of caspase-mediated apoptosis during step increase of NaCl, we added 200 and 400 mosmol/liter NaCl as a 5-hour step and measured caspase 3 and cPARP activation (fig. S2, J to L). After adding 200 mosmol/liter NaCl, we found a modest induction in caspase 3 activity and PARP cleavage [fig. S2, K and L (black lines)], suggesting low apoptosis in agreement with the relatively small decrease in cell viability under this condition (Fig. 1D). After adding 400 mosmol/liter NaCl, we see a stronger induction of caspase 3 activation and PARP cleavage [fig. S2, K and L (gray lines)], explaining the strongly reduced viability observed for this condition (Fig. 1D). The temporal dynamics are very similar between these different step experiments while the fraction of cells undergoing caspase-mediated apoptosis is dependent on the strength of the hypertonic stress, suggesting that the underlying cellular mechanisms of apoptosis induction are similar across these experiments. Phosphorylation of γH2AX is also entirely prevented when caspase activity is inhibited during step NaCl treatment by a pan-caspase inhibitor (fig. S6A), which suggests prevention of apoptosis-associated destruction of DNA. Next, we investigated the contributions of caspases 3, 8, and 9 to the cell viability phenotype by quantifying the time course of activation for each member of the caspase signaling group relative to cleavage of PARP (Fig. 3F). We found that caspase 3 (gray) is activated slightly before its target cPARP (purple), as expected (Fig. 3F). Unexpectedly, we found activation of the initiator caspases 8 (magenta) and 9 (cyan) after caspase 3 and cPARP. These results suggest that caspase 3 contributes to the induction of apoptosis but not cleaved caspases 8 and 9. To understand whether these population-level effects are indeed observable in the same cell, we costained cells with antibodies for activated caspase 9 and cPARP (Fig. 4A). We found that single cells that are negative for cPARP are never positive for activated caspase 9 at any point during the treatment (Fig. 4B). Cells positive for activated caspase 9 already have a high level of cPARP, suggesting that caspase 9 cleavage is not causative for apoptosis induction in single cells. As caspase 9 can also be activated by dimerization (28, 29), it is still possible that apoptosis activation is mediated by caspase 9. Similarly, single cells costained for cPARP and activated caspase 8 are never negative for cPARP and positive for activated caspase 8, at the same time throughout the time course (Fig. 4C). These results indicate no activation of caspase 8 before apoptosis induction (Fig. 4D). In summary, these results suggest that activated caspase 3 but not cleaved caspase 8 and 9 contribute to the induction of apoptosis (Fig. 4D).
Fig. 3. Differential caspase signaling regulates cell viability.
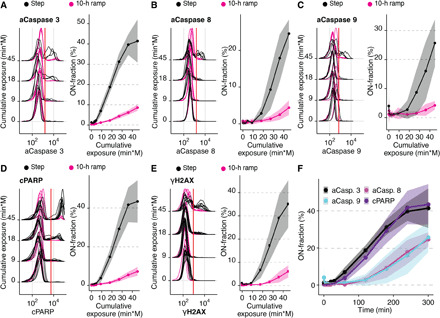
(A to E) Differential regulation of (A) cleaved caspase 3, (B) cleaved caspase 8, (C) cleaved caspase 9, (D) cPARP, and (E) γH2AX in Jurkat cells exposed to 300 mosmol/liter NaCl by a step (black) or a 10-hour ramp (magenta). The left panel shows selected single-cell distributions over the cumulative exposure with individual lines representing independent experiments. Red line indicates the threshold for determining the ON-fraction. Right panels represent ON-fraction mean and SD of 3 to 10 independent experiments as a function of cumulative exposure of NaCl. (F) ON-fraction kinetics of caspase signaling markers over time indicate early (caspase 3 and cPARP) and late (caspase 8 and 9) activation. Lines indicate mean and SD of 3 to 10 independent experiments.
Fig. 4. Activated caspase 8 or 9 are not initiating apoptosis in hyperosmotic stress.
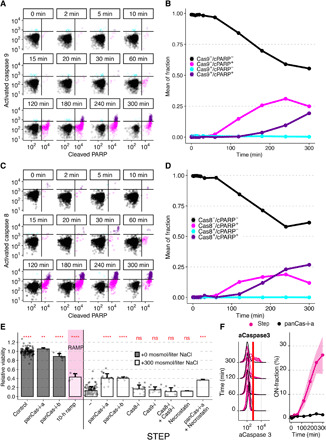
(A to D) Jurkat cells costained for cPARP and activated caspase 9 (A) or 8 (C) measured by flow cytometry after step exposure to 300 mosmol/liter NaCl for 5 hours. Black lines indicate thresholds to determine individual fractions of activated caspase 9 or 8 and cPARP. Circles represent single cells. Quantification of fraction of cells stained for caspase 9 (B) or 8 (D) activation and PARP cleavage over the time course using thresholds indicated in (A) or (C). (E) Relative viability after 10-hour ramp (magenta) or 5-hour step treatment to 300 mosmol/liter NaCl (white) or control (gray) in the presence of the following: “panCas-i-a” (Z-VAD-FMK, 100 μM), “panCas-i-b” (Q-VD-OPH, 100 μM), “Cas8-i” (Z-IETD-FMK, 100 μM), “Cas9-i” (Z-LEHD-FMK, 100 μM), and necrostatin (10 μM). Bars indicate mean and SD, N ≥ 3. Two-sided unpaired Student’s t test: **P < 0.01, ***P < 0.001, and ****P < 0.00001. (F) Activated caspase 3 (aCaspase 3) in Jurkat cells step exposure to 300 mosmol/liter NaCl in the presence (black) or absence (magenta) of pan-caspase inhibitor (Z-VAD-FMK, 20 μM). Left panel shows single-cell distributions over cumulative exposure with individual lines representing independent experiments. Red line indicates ON-fraction threshold. Right panels represent mean and SD of one to four independent experiments over cumulative exposure.
Caspase signaling is the main contributor to cell death in step conditions
We next tested whether these different caspases contributed to cell viability and addressed their mechanism in an attempt to link dynamics in caspase activation to apoptosis and cell phenotype (Fig. 4E). In our ramp treatment condition to additional 300 mosmol/liter NaCl in 10 hours, we found that cell viability increases about two- to threefold to 40% in comparison to 15% in step treatment of the same final concentration and the total amount of NaCl relative to cells grown in control conditions (100% viability) [Fig. 4E (magenta area)]. We asked whether this increase in viability is entirely related to the lack of caspase activation and PARP cleavage, as observed in Figs. 3 and 4. To test this idea, we treated cells with a step of 300 mosmol/liter NaCl in the presence of different, potent pan-caspase inhibitors (panCas-i-a = Z-VAD-FMK, panCas-i-b = Q-VD-OPH) (Fig. 4E). We observed an increase in cell viability to 40%, which is the same as for the ramp treatment. This result suggests that caspase activation and caspase-mediated apoptosis are necessary to explain the reduction in viability during the step treatment relative to the ramp treatment. We then asked whether other pathways of cell death (30) could explain these results. We first focused on necroptosis, by preincubation of cells with necrostatin, an inhibitor for this process (31). In the presence of necrostatin, we did not see an improvement in viability relative to step-exposed cells neither in the presence nor in the absence of caspase inhibition (Fig. 4E and fig. S6B). In addition, we analyzed the temporal dynamics of predisposition to cell death during the 5-hour step by quantifying nonapoptotic, cPARP-negative cells along the time course (comparable to analysis in Fig. 3F) and independently by quantifying the decrease in cell viability [intracellular ATP (adenosine triphosphate) content, CellTiter-Glo]. We find that cell viability sharply drops within the first hour to about 50%. The remaining decrease from 50 to 15% is correlated with the activation dynamics of caspase-dependent apoptosis and the decrease of nonapoptotic cells (fig. S6C). This suggests that caspase-dependent apoptosis is mainly responsible for the viability decrease after the first hour of exposure. Therefore, we hypothesize that caspase-dependent apoptosis is the main contributor to the difference in viability between the step and the long ramp treatment conditions.
We predicted that early caspase 3 activation triggers PARP cleavage and apoptosis compared to late caspase 8 and 9 activation (Figs. 3 and 4, A to D). To test this prediction, we exposed cells to inhibitors of caspase 8, caspase 9 alone, or in combination. We found that inhibitors for caspase 8 and 9 do not substantially improve viability after step exposure to 300 mosmol/liter NaCl (Fig. 4E). As expected, we found that pan-caspase inhibition prevents the cleavage of caspase 3 during the step treatment (Fig. 4F).
Through our functional temporal screen, we also observed that p38 is strongly activated in NaCl step treatment condition, as previously reported to occur in other mammalian cells (Fig. 5A) (32, 33). However, during a 10-hour ramp treatment, we found that p38 is only slightly activated, perhaps playing a role in the decreased cell viability phenotype following step stimulation relative to the ramp stimulation. MAPKs, such as p38, have been reported to mediate paraptosis (34). We found that the inhibition of all p38 protein isoforms by using a pan-p38 inhibitor (BIRB 796) had a statistically significant but biologically small effect on cell viability following step treatment condition (Fig. 5B and fig. S6B). Inhibition of c-Jun N-terminal kinase (JNK) had no effect on cell viability (Fig. 5B). When combining MAPK and pan-caspase inhibition, we found an additive effect on improving cell viability (fig. S6B). From these results, we conclude that the rate of hypertonic stress addition differentially regulates p38 but that p38 activity is not essential for the apoptosis-dependent decrease of cell viability and may mediate paraptosis-associated decreases in cell viability following step treatment.
Fig. 5. Contribution of p38 to apoptosis in hypertonic stress is minimal.
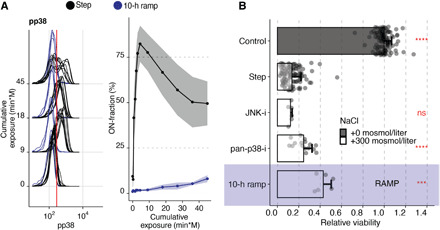
(A) Phosphorylation of p38 in Jurkat cells exposed to 300 mosmol/liter NaCl by a step (black) or a 10-hour ramp (blue). The left panel shows selected single-cell distributions over the cumulative exposure with individual lines representing independent experiments. The red line indicates the threshold for determining a cell that is p38 phosphorylation positive (ON-fraction). The right panel represents the ON-fraction mean and SD of 3 to 10 independent experiments as a function of cumulative exposure. (B) Viability of Jurkat cells relative to untreated cells (control) exposed to an additional 0 (gray) or 300 mosmol/liter (white) NaCl for 5 hours (step) or 10 hours (ramp, purple), respectively. Pan p38 inhibitor (pan-p38-i, BIRB796, 10 μM) and JNK inhibitor (SP600125, 10 μM) were added 30 min before NaCl. Circles represent single experiments. Bars indicate the mean and SD of at least three replicates. Two-sided unpaired Student’s t test: ***P < 0.01 and ****P < 0.0001.
Compared to p38, pASK, NFAT5, and HSP70, signals are reduced in step but not in ramp conditions shortly after osmotic stress (fig. S3, C to E). Followed by this initial drop are similar temporal profiles for step and ramp conditions. These results demonstrate that the dynamics of NFAT5, pASK, and HSP70 are not differentially regulated. We also observed similar dynamics for markers of the growth (Ki67), antiapoptosis (Bcl-xL), inflammation (IFNγ and NLRP3), and the DNA damage (BAX, NOXA, and Fas-L) signaling groups (fig. S4, B and D to F). Markers that did change over time but not strongly between step and ramp conditions are the proliferation markers p-S6 and p-mTOR and anti-apoptotic protein p-BAD (fig. S4, A, G, and H). We observed no change given the error in the measurements between step and ramp conditions for selected markers of stress signaling (p-MK2, p-JINK, p-MKK4, p-HSP27, p-ATF2, and p-CREB) and an antiapoptotic protein p-Bcl2 (figs. S3, A, B, F to H, and P, and S4C).
Intracellular proline levels improve viability in ramp stress conditions
To better understand the protective mechanisms contributing to improved viability during the ramp condition, we analyzed the abundance and fold changes of metabolites that may function as cell internal osmolytes (Fig. 6A and fig. S7). We found that among the most abundant metabolites are the amino acid proline, glutamic acid, and arginine. In comparison, traditional osmolytes such as betaine, inositol, sorbitol, taurine, or urea are significantly less abundant in the cell (fig. S7). Of these amino acids, only proline is differentially regulated in step and ramp conditions, rejecting the possibility that these amino acids are the only by-products of protein degradation (Fig. 6A). This result suggests that proline may act as an osmoprotective molecule in human cells in ramp treatment conditions. The increase in abundance of cell internal proline levels relative to other amino acids and organic molecules suggests that cells import proline from the growth media. Elevated protein degradation in the cell would presumably result in an equal distribution of increased amino acid abundance. We then tested whether intracellular proline levels are independent of the activation of the caspase pathway or whether preventing caspase-mediated cell death results in higher levels of proline in the cells. In these experiments, we exposed cells to a step treatment of NaCl with or without pan-caspase inhibitor Z-VAD-FMK (Fig. 6B). As in previous experiments, we exposed cells to the same cumulative exposure of NaCl for the same final NaCl concentration and compared the results. We found that regardless of pan-caspase inhibition, cells accumulated significantly less proline during the step treatment than cells exposed to the ramp treatment (Fig. 6B). We conclude that caspase inhibition during hypertonic stress does not result in additional proline accumulation during the step treatment. This result indicates that caspase activation and proline accumulation are independent. To test whether extracellular levels of proline can improve cell viability in the step treatment to 300 mosmol/liter NaCl, we added free l-proline to the media of the cells before applying hypertonic stress (Fig. 6C). We found a more than twofold increase (13 to 31%) in viability due to added proline (16 mM), in comparison to cells where no additional proline was added (Fig. 6C). This result suggests that proline is transported into the cells and can protect mammalian cells from hypertonic stress. It is well established that hyperosmotic stress up-regulates transporters for glutamine and proline (35–37). Because free glutamine is generally more abundant than free proline in cell culture medium and in the human body (38, 39), we tested whether additional external l-glutamine, a precursor of proline (40), can also improve viability. When we added additional l-glutamine to the media before adding NaCl, we observed a significant improvement in cell viability, similar to adding proline. To understand the longer-term osmoprotective effects of proline, we determined the cell viability after a 10-hour step to 300 mosmol/liter NaCl in the presence and absence of either l-glutamine or l-proline. We find that viability is significantly decreased after a 10-hour step when compared to a 5-hour step in cells not preincubated with proline (fig. S8B). However, when proline or glutamine is present, viability improves to about the same level we observed after a 5-hour step in the presence of proline or glutamine (fig. S8B). These experiments suggest that extracellular proline and glutamine provide longer-term protection in hypertonic stress. Because proline is a yet unidentified compound in the mammalian response to hyperosmotic stress, we tested the effect of typical mammalian osmolytes on cell viability (41). When we added compounds identified as physiological osmoprotectants to the media, such as taurine, sorbitol, or betaine, we observe that these compounds seem to provide less or the same protection as proline or glutamine during hypertonic stress. These results demonstrate that proline and glutamine are as effective as traditional osmolytes in protecting the cell from osmotic stress (fig. S8).
Fig. 6. Intracellular proline protects human cells during ramp stress conditions.
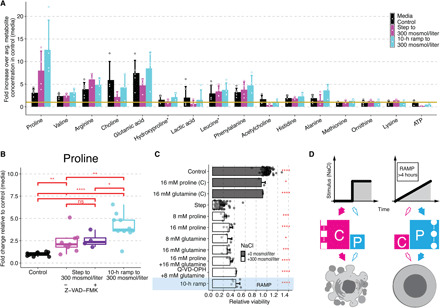
(A) Fifteen most abundant metabolites detected in Jurkat cells without stimulation (black), after treatment with step (magenta) or 10-hour ramp (cyan) to 300 mosmol/liter NaCl. Bars represent mean and SD of the fold change of each metabolite to the average metabolite concentration in the control condition (yellow line) with circles representing replicates. (B) Change of proline levels in Jurkat cells relative to control (no additional NaCl) in 0 (black) or step without (magenta) or with pan-caspase inhibitor “a” (Z-VAD-FMK, 100 μM) (purple) or 10-hour ramp to 300 mosmol/liter NaCl (cyan). Circles represent replicates, N ≥ 4. (C) Viability in Jurkat cells exposed to step or 10-hour ramp (blue) of 300 mosmol/liter NaCl relative to control (gray). Pan-caspase inhibitor (Q-VD-OPH, 100 μM) was added 30 min and amino acids 60 min before NaCl. Bars indicate mean and SD, N ≥ 3. Two-sided unpaired Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (D) Model: Instant stress conditions cause activation of caspase signaling (C) and cell death (magenta), whereas the gradual increased stress to the same final concentration does not activate caspase signaling but instead increases intracellular proline (P) as an osmolyte to protect cells against increasing stress (cyan).
DISCUSSION
Previous studies have established that acute changes in environmental stimulus concentrations can control cell fate. However, cells in physiological environments may not necessarily experience such acute concentration changes. It is conceivable that typical solute concentration changes are gradual over time with different kinetics (42). However, there is a limited understanding of how a gradual change of stimulus concentrations affect cellular responses. We investigated stress responses of human immune cells to ramp increases in the concentrations of different osmolytes to address the key question of how varying the kinetics of stimulation affect cellular responses. We found that in comparison to instantly changing osmolyte concentrations, slow changes protect human immune cells from otherwise lethal insults (Fig. 1, B to D, and fig. S1). These results indicated that sensitivity to the rate of change of external osmolyte concentrations is a fundamental feature of human cells. These results are important because they demonstrate that immune cells that migrate into and through hypertonic tissues such as renal, intestinal, or epidermal tissue can survive hypertonic conditions better if these changes occur at a low rate over time. These results are consistent with pioneering studies indicating partial protection of renal medullary cells from slowly increasing external osmolytes (5, 12). The authors of these pioneering studies postulate that an increase in cell internal organic osmolytes is responsible for protecting cells exposed to gradually increasing osmolyte gradients (12). Perhaps unexpectedly, we found that well-established osmolytes such as betaine, inositol, sorbitol, taurine, or urea did not increase at the end of our experiments (Fig. 2A). One reason for this observation is likely that kidney cells respond differently to hypertonic stress than immune cells. Another reason is that we quantify cell internal osmolytes at the end of the 10-hour ramp experiment, whereas the previous study analyzed the response of cells 24 hours after the ramp treatment. We hypothesize that increases in traditional cell internal osmolytes after 24 hours may indeed function as a secondary and long-term protection against osmotic stress but are not significant for short-term protection. Because the step and ramp conditions do not differentially regulate the concentrations of these osmolytes (Fig. 2), we studied the cellular pathways that are important in the regulation of cell viability during hyperosmotic stress. We found differential regulation between ramp and step conditions of caspases 3, 8, and 9 (Fig. 3, A to C). In step conditions, a large fraction of cleaved caspases is observed, whereas in ramp conditions, only a small fraction of cells show cleaved caspases (Fig. 3, A to C). This mechanism enables a population of cells to respond gradually to stresses that change over time without changing the ability of individual cells to undergo apoptosis. It is conceivable that in the kidney or the intestine, immune cells need to adjust not only to the absolute change but also to the rate of change in hypertonicity to avoid apoptosis. A property of an adapting system is to distinguish between a rapid and a slow increase of a stimulus. Adaptation has been studied in several important model systems, such as in yeast osmotic stress response signaling (43), chemotaxis signaling in bacteria (2), mitogen (44), and developmental (3) signaling. These studies demonstrate that differential regulation of cell signaling between step and ramp stimulation might be a universal feature of signal transduction pathways by determining the presence or absence of a response to changes in the environment over time.
To better understand the mechanism underlying this observation, we analyzed the timing of caspase activation in single cells. Caspases are the main transducer of regulated cell death in mammalian cells. They are divided into initiator caspases (caspase 2, 8, 9, and 10) and effector caspases (caspase 3, 6, and 7). Caspases exist as zymogens in the cell and are cleaved at specific sites by processing enzymes into an activated form. A special case is caspase 9, which does not need to be cleaved for activation. Caspase 9, which mediates the intrinsic apoptotic pathway, dimerizes and binds Apaf-1 to form the apoptosome (29, 45). These early binding events have been reported to result in a thousand-fold activation (28), whereas caspase 9 cleavage only results in a 10-fold increase in activity (28). We found that activated caspase 3 and cPARP increase before activated caspase 8 and 9 (Fig. 3F). These findings support previous studies demonstrating that activated caspase 3 cleaves PARP (18). This observation is consistent with published studies of apoptosis induction through caspase 9 protein recruitment but not its cleavage (28, 29). Recruited caspase 9 then cleaves caspase 3, which subsequently cleaves PARP (16, 17, 46). However, these cell population experiments cannot determine whether indeed in a single cell, PARP is cleaved before caspase 8 or 9 (Fig. 4, A to D). To test whether indeed PARP is cleaved before caspase 8 and 9 in single cells, we quantified costained cells for cleaved caspase 8, 9, and cPARP. Our single-cell analysis demonstrates that PARP gets cleaved before caspase 8 or 9, supporting our results and are consistent with previous cell population studies (16, 17). From these single-cell time course experiments, we predicted that inhibition of caspase signaling in step conditions increases cell viability similar to ramp conditions in single cells (Fig. 4E). Because PARP is cleaved before caspases 8 and 9, we predicted that these caspases do not contribute significantly to cell death. We indeed found that inhibiting caspase 8 or 9 individually or together does not improve viability (Fig. 4E).
To better understand which proteins contribute to differential caspase activation and cell survival, we analyzed changes in protein levels and/or phosphorylation states of upstream markers for proteins contributing to and indicating stress, growth, proapoptosis, antiapoptosis, inflammation, and DNA damage. We separated these proteins into three groups. In the first group of protein markers of stress (NFAT5, pASK, and HSP70), growth (Ki67), antiapoptosis (Bcl-xL), inflammation (IFNγ and NLRP3), and DNA damage (BAX, NOXA, and Fas-L) drop rapidly in step but not in ramp conditions. These results could indicate that these markers can sense the difference in the type of stress gradient in a switch-like manner, although the dynamics of their distributions do not change overall. The second group of markers, such as proliferation markers p-S6 and p-mTOR and anti-apoptotic protein p-BAD, decrease over time but showed no differences between step and ramp conditions relative to the cumulative osmolyte exposure. These results indicate that a strong reduction of these markers is independent of the stress kinetics. The third group of proteins, such as stress signaling (p-MK2, p-JNK, p-MKK4, p-HSP27, p-ATF2, and p-CREB), and the antiapoptotic protein p-Bcl2 did not show a clear difference between step and ramp treatments given the experimental constraints.
We also investigated the well-established link between osmotic stress and p38 signaling. We observed that p38 phosphorylation and phosphorylation of its target histone γH2AX are also differentially regulated in ramp and step conditions (Figs. 3E and 5). However, inhibition of p38 does not contribute to cell viability improvement as much as caspase inhibition (Fig. 5B). These results are consistent with previous studies in macrophages where inhibition of stress response pathways such as p38 or JNK did not contribute to caspase signaling (33). This large temporal functional screen establishes caspase signaling as the main contributor to differential regulation in step versus ramp stress condition compared to alternative signaling pathways of stress, proliferation, antiapoptosis, proapoptosis, inflammation, and DNA damage.
Together these results indicate that human immune cells can survive shallow gradients to high osmolarity. This protective capability might be important because monocytes need to migrate inside the kidney from the low osmolarity cortex to the very high osmolarity medulla to prevent bacterial infection (47). These results then beg the question of how do cells survive gradients of osmotic stresses that would otherwise be deadly?
We extended our initial analysis of cell internal organic osmolytes to a wide range of metabolites measured in step and ramp conditions. Although we detected many well-established osmolytes, their concentration is significantly lower than many other metabolites that we detected (Fig. 2A and fig. S7). In addition, none of these osmolytes change significantly in step and ramp conditions (Fig. 6). Instead, from this analysis, we found disproportional proline increases compared to the other amino acids. This disproportional increase for one amino acid excludes differential global protein degradation as a mechanism to increase proline levels (Fig. 6, A and B). Supplementing external proline or one of its precursors, glutamine, protected cells from acute hypertonic stress, similar to stress protection in ramp conditions (Fig. 6C). Proline and glutamine are transported into the cell by sodium-dependent amino acid transporters (48). Some of these transporters, such as SNAT2/SLC38A2 (specific for neutral amino acids) and SLC6A7 (proline specific), have been found to be up-regulated during hypertonic stress in mouse (36, 49, 50) and human cells (fig. S8C) (35, 37). Elevated levels of amino acids or sodium, as present in our experiments, favor amino acid import into the cell following mass-action kinetics and chemical gradients. Although not well established in mammalian cells, in plants, proline acts as an osmoprotective molecule, and its accumulation is a well-described mechanism applied by plants to endure droughts and other stresses (51). Our results strongly suggest that the accumulation of intracellular proline plays a role in the protection of human immune cells from slowly increasing hypertonicity and the prevention of apoptosis (Fig. 6C and fig. S8).
In summary, we propose a model (Fig. 6D) in which step increases in hypertonicity activate caspase signaling, PARP cleavage, and cause cell death. In contrast, slowly increasing hypertonicity did not activate caspase signaling but instead caused accumulation of intracellular proline. Proline is known to be up-regulated during hypertonic stress in plants and bacteria and to have an osmoprotective function. Proline functions as an organic osmolyte, molecular chaperone, metal chelator, and reactive oxygen species scavenger independent of caspase activation (52). These properties make proline an efficient stress response molecule. We argue that proline has an underestimated and critical role in protecting human cells from cell death in hypertonic conditions and could explain how immune cells can survive in microenvironments within the body that have extreme osmolarities that change over time such as the renal papilla or the intestine.
MATERIALS AND METHODS
Human cell culture
THP1 [American Type Culture Collection (ATCC) TIB-202] cells were cultured at 0.5 to 1 × 106 cells/ml in RPMI 1640 media (Corning, catalog no. 15-040-CV) containing 10% heat-inactivated fetal bovine serum (FBS) (Gibco, catalog no. 16140-071), penicillin-streptomycin (100 U/ml) (Gibco, catalog no. 15140-122), 2 mM l-alanyl-l-glutamine dipeptide (GlutaMAX, Gibco, catalog no. 35050-061), and 0.05 mM 2-mercaptoethanol (Sigma-Aldrich, catalog no. M3148) at 37°C in a 5% CO2 humidity-controlled environment. Jurkat cells (clone E6-1, ATCC TIB-152) were cultured at 0.5 to 1.5 × 106 cells/ml in RPMI 1640 media (Corning, catalog no. 15-040-CV) containing 10% heat-inactivated FBS (Gibco, catalog no. 16140-071), penicillin-streptomycin (100 U/ml) (Gibco, catalog no. 15140-122), and 2 mM l-alanyl-l-glutamine dipeptide (GlutaMAX, Gibco, catalog no. 35050-061) at 37°C in a 5% CO2 humidity-controlled environment.
Experimental procedure for step and ramp treatment
A programmable pump (New Era Syringe Pump Systems, NE-1200) was used to apply gradually increasing (ramp) profiles. Briefly, the pumping rate and dispensed volume per interval were calculated as described (42) and uploaded to the pump via a computer. A syringe pump driving a syringe (BD, catalog no. 309628) filled with 5 M NaCl (Corning, catalog no. 46-032-CV) solution connected to a needle (Jensen Global, catalog no. JG21-1.0x) with tubing (Scientific Commodities, catalog no. BB31695-PE/4). The tubing was inserted into a foam stopper on an autoclaved glass flask (Pyrex, catalog no. 4980-500) holding the suspension cells. Cells were shaken at 100 rpm during the entire experiment using a CO2 resistant shaker, ensuring proper mixing (Thermo Fisher Scientific, catalog no. 88881101). For step stimulation, appropriate amount of 5 M NaCl (Corning; 500 ml of 5 M sodium chloride; no. 46-032-CV) solution was added by a syringe within 5 s to reach the desired final concentration. Five milliliters of cells was removed with a syringe (BD, catalog no. 309628) through autoclaved silicone tubing (Thermo Fisher Scientific, catalog no. 8600-0020) to collect time point samples.
Cell viability assay
Cell viability was measured with CellTiter-Glo (Promega, catalog no. G7571). Cells were transferred to a white 96-well plate according to the manufacturer’s instructions and equilibrated to room temperature for 10 min. CellTiter-Glo reagent was added in a ratio 1:8 to cell suspension. Luminescence was measured using a plate reader (Promega, GloMax Discover plate reader, GM3000). Relative viability was calculated by dividing luminescence values for each replicate by mean luminescence of media control for each experiment.
Quantitative flow cytometry
Cells are fixed with 1.6% formaldehyde (Fisher, catalog no. F79-4) in a 15-ml Falcon tube. Fixation was quenched by adding 200 mM glycine after 8 min. Cells were washed with phosphate-buffered saline (PBS) (Corning, catalog no. 46-013-CM) and permeabilized with methanol (Thermo Fisher Scientific, catalog no. A454-4) for 15 min on ice. Cells were washed with PBS and stained with Pacific Blue NHS ester (Pacific Blue Succinimidyl Ester, Thermo Fisher Scientific, no. P10163) and Pacific Orange NHS ester (Pacific Orange Succinimidyl Ester, triethylammonium salt, Thermo Fisher Scientific, no. P30253) for 30 min. Cells are blocked with 1% bovine serum albumin (RPI, catalog no. A30075-100.0) in PBS. Cells are washed and stained with a primary monoclonal antibody for 60 min at room temperature. Quantitative flow cytometry was performed on BD LSR II (five lasers). All antibodies used in this study are listed in table S1.
Quantitative flow cytometry analysis
Flow cytometry data were analyzed with custom R software. The primary cell population was gated on FSC-A versus SSC-A by using the “flowCore” package (53). The cell populations are automatically debarcoded, and the resulting data were analyzed using custom software in R applying the following packages: “ggplot2,” “data.table,” “plyr,” “dplyr,” “flowViz,” “flowCore,” “flowStats,” “ggcyto,” “RcppEigen,” “fields,” “ggridges,” “viridis,” “scales,” and “xml2”. The distributions between independent experiments with similar shapes are aligned for their 0-min time point so that their means are identical. This offset was applied to all the distributions in each experiment. Experiments are performed so that the total exposure to NaCl is identical between step and ramp experiments. The distributions and ON-fraction are plotted as a function of the cumulative exposure. Plotting data as a function of the cumulative NaCl expose helps to distinguish between changes related to the total NaCl exposure compared to the temporal change in the NaCl concentration.
Inhibitor studies
All inhibitors used in this study are listed in table S2. Inhibitors were dissolved in dimethyl sulfoxide and added 30 min before the start of the experiment to the cell culture media at indicated concentrations.
Targeted metabolomics methodology
Five milliliters of cell suspension was pelleted; the supernatant was removed and resuspended in 90% methanol. Analysis of metabolites was performed at the Vanderbilt University Mass Spectrometry Research Center using an Acquity ultra performance liquid chromatography (UPLC) system (Waters, Milford, MA) interfaced with a TSQ Quantum triple-stage quadrupole mass spectrometer (Thermo Fisher Scientific, San Jose, CA), using heated electrospray ionization operating in multiple reaction monitoring mode. Five hundred microliters of each cell lysate sample was blown to dryness with N2 and reconstituted with 150 μl of acetonitrile/H2O (2:1) solution containing stable isotope-labeled internal standards: tyrosine-d2 and lactate-13C3 (Cambridge Isotope Lab, Tewksbury, MA). We centrifuged the cell lysate at 10,000g for 20 min and injected 90 μl of the supernatant into UPLC. The supernatant was chromatographically separated with a Zic-cHILIC column (3 μm; 150 × 2.1 mm; Merck SeQuant, Darmstadt, Germany) at a flow rate of 300 μl/min. The mobile phases were (A) 15 mM ammonium acetate with 0.2% acetic acid in water/acetonitrile (90:10, v/v) and (B) 15 mM ammonium acetate with 0.2% acetic acid in acetonitrile/water/methanol (90:5:5, v/v). The gradient was as follows: 0 min, 85% B; 2 min, 85% B; 5 min, 30% B; 9 min, 30% B; 11 min, 85% B; and 20 min, 85% B. We set the spray voltage to 5 kV and the capillary and vaporizer temperatures to 300° and 185°C, with sheath gas and auxiliary gas set to 414 and 310 kPa, respectively. The skimmer offset was −10 V, and the collision energy varied for each transition. Metabolites were identified on the basis of predetermined peaks and elution times. The response ratio was calculated for each detected metabolite relative to the internal standard.
Acknowledgments
We thank R. H. Markowitz, Y. Perevalova, and M. H. Tran for technical assistance. We thank the Bachmann lab for technical assistance in setting up fluorescent cell barcoding for multiplex quantitative flow cytometry and the Vanderbilt Flow Cytometry and Mass Spectrometry Core. We thank D. G. Harrison, J. M. Titze, J. A. Capra, A. Kirabo, M. S. Madhur, V. Gama, and J. A. Gomez for useful discussions and B. K. Kesler, J. J. Hughes, H. Jashnsaz, A. N. Johnson, R. Ali, R. Colbran, D. Cortez, S. S. Zinkel, P. A. Weil, A. Page-McCaw, and K. S. Lau for feedback on the manuscript. Funding: This work was supported by an NIH Director’s New Innovator DP2 award (GM11484901) to G.N., an American Heart Association (AHA) Pre-doctoral Fellowship award (18PRE34050016) to A.T., Vanderbilt Institute for Clinical and Translational Research (VICTR) awards VR53716 to A.T., and Vanderbilt Startup Funds. Author contributions: G.N. and A.T. conceived the study and designed the experiments. A.T. performed the experiments and the data analysis. G.N. and A.T. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The flow cytometry data will be made publicly available on https://flowrepository.org/ upon publication. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabe1122/DC1
REFERENCES AND NOTES
- 1.W. Lim, B. Meyer, T. Pawson, Cell Signaling: Principles and Mechanisms (Garland Science, 2014). [Google Scholar]
- 2.Young J. W., Locke J. C. W., Elowitz M. B., Rate of environmental change determines stress response specificity. Proc. Natl. Acad. Sci. U.S.A. 110, 4140–4145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorre B., Warmflash A., Brivanlou A. H., Siggia E. D., Encoding of temporal signals by the TGF-β pathway and implications for embryonic patterning. Dev. Cell 30, 334–342 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burg M. B., Ferraris J. D., Dmitrieva N. I., Cellular response to hyperosmotic stresses. Physiol. Rev. 87, 1441–1474 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Cai Q., Michea L., Andrews P., Zhang Z., Rocha G., Dmitrieva N., Burg M. B., Rate of increase of osmolality determines osmotic tolerance of mouse inner medullary epithelial cells. Am. J. Physiol. Renal Physiol. 283, F792–F798 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Firsov D., Bonny O., Circadian rhythms and the kidney. Nat. Rev. Nephrol. 14, 626–635 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Overduin J., Tylee T. S., Frayo R. S., Cummings D. E., Hyperosmolarity in the small intestine contributes to postprandial ghrelin suppression. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G1108–G1116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang L.-B., Cao L., Yin X.-F., Yasen M., Yishake M., Dong J., Li X.-L., Activation of autophagy via Ca2+-dependent AMPK/mTOR pathway in rat notochordal cells is a cellular adaptation under hyperosmotic stress. Cell Cycle 14, 867–879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jantsch J., Schatz V., Friedrich D., Schröder A., Kopp C., Siegert I., Maronna A., Wendelborn D., Linz P., Binger K. J., Gebhardt M., Heinig M., Neubert P., Fischer F., Teufel S., David J.-P., Neufert C., Cavallaro A., Rakova N., Küper C., Beck F.-X., Neuhofer W., Muller D. N., Schuler G., Uder M., Bogdan C., Luft F. C., Titze J., Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 21, 493–501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller S., Quast T., Schröder A., Hucke S., Klotz L., Jantsch J., Gerzer R., Hemmersbach R., Kolanus W., Salt-dependent chemotaxis of macrophages. PLOS ONE 8, e73439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.N. I. Dmitrieva, M. B. Burg, Analysis of DNA breaks, DNA damage response, and apoptosis produced by high NaCl. 295, F1678–F1688 (2008). [DOI] [PMC free article] [PubMed]
- 12.Cai Q., Ferraris J. D., Burg M. B., Greater tolerance of renal medullary cells for a slow increase in osmolality is associated with enhanced expression of HSP70 and other osmoprotective genes. Am. J. Physiol. Renal Physiol. 286, F58–F67 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R. III, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J., ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Cheung C. Y., Ko B. C., NFAT5 in cellular adaptation to hypertonic stress - regulations and functional significance. J. Mol. Signal. 8, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kyriakis J. M., Avruch J., Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 92, 689–737 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Slee E. A., Harte M. T., Kluck R. M., Wolf B. B., Casiano C. A., Newmeyer D. D., Wang H.-G., Reed J. C., Nicholson D. W., Alnemri E. S., Green D. R., Martin S. J., Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9–dependent manner. J. Cell Biol. 144, 281–292 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McComb S., Chan P. K., Guinot A., Hartmannsdottir H., Jenni S., Dobay M. P., Bourquin J.-P., Bornhauser B. C., Efficient apoptosis requires feedback amplification of upstream apoptotic signals by effector caspase-3 or -7. Sci. Adv. 5, eaau9433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazebnik Y. A., Kaufmann S. H., Desnoyers S., Poirier G. G., Earnshaw W. C., Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371, 346–347 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Kuo L. J., Yang L.-X., γ-H2AX – a novel biomarker for DNA double-strand breaks. In Vivo 22, 305–309 (2008). [PubMed] [Google Scholar]
- 20.Pihán P., Carreras-Sureda A., Hetz C., BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 24, 1478–1487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peña-Oyarzun D., Troncoso R., Kretschmar C., Hernando C., Budini M., Morselli E., Lavandero S., Criollo A., Hyperosmotic stress stimulates autophagy via polycystin-2. Oncotarget 8, 55984–55997 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magnuson B., Ekim B., Fingar D. C., Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441, 1–21 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Clem R. J., Cheng E. H.-Y., Karp C. L., Kirsch D. G., Ueno K., Takahashi A., Kastan M. B., Griffin D. E., Earnshaw W. C., Veliuona M. A., Hardwick J. M., Modulation of cell death by BCl-xL through caspase interaction. Proc. Natl. Acad. Sci. U.S.A. 95, 554–559 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma P., Zha S., Shen X., Zhao Y., Li L., Yang L., Lei M., Liu W., NFAT5 mediates hypertonic stress-induced atherosclerosis via activating NLRP3 inflammasome in endothelium. Cell Commun. Signal 17, 102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dmitrieva N. I., Cai Q., Burg M. B., Cells adapted to high NaCl have many DNA breaks and impaired DNA repair both in cell culture and in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 2317–2322 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roos W. P., Kaina B., DNA damage-induced cell death by apoptosis. Trends Mol. Med. 12, 440–450 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Krutzik P. O., Nolan G. P., Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat. Methods 3, 361–368 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Stennicke H. R., Deveraux Q. L., Humke E. W., Reed J. C., Dixit V. M., Salvesen G. S., Caspase-9 can be activated without proteolytic processing. J. Biol. Chem. 274, 8359–8362 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S., Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. U.S.A. 98, 14250–14255 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galluzzi L., Vitale I., Aaronson S. A., Abrams J. M., Adam D., Agostinis P., Alnemri E. S., Altucci L., Amelio I., Andrews D. W., Annicchiarico-Petruzzelli M., Antonov A. V., Arama E., Baehrecke E. H., Barlev N. A., Bazan N. G., Bernassola F., Bertrand M. J. M., Bianchi K., Blagosklonny M. V., Blomgren K., Borner C., Boya P., Brenner C., Campanella M., Candi E., Carmona-Gutierrez D., Cecconi F., Chan F. K.-M., Chandel N. S., Cheng E. H., Chipuk J. E., Cidlowski J. A., Ciechanover A., Cohen G. M., Conrad M., Cubillos-Ruiz J. R., Czabotar P. E., D’Angiolella V., Dawson T. M., Dawson V. L., De Laurenzi V., De Maria R., Debatin K.-M., Deberardinis R. J., Deshmukh M., Di Daniele N., Di Virgilio F., Dixit V. M., Dixon S. J., Duckett C. S., Dynlacht B. D., El-Deiry W. S., Elrod J. W., Fimia G. M., Fulda S., García-Sáez A. J., Garg A. D., Garrido C., Gavathiotis E., Golstein P., Gottlieb E., Green D. R., Greene L. A., Gronemeyer H., Gross A., Hajnoczky G., Hardwick J. M., Harris I. S., Hengartner M. O., Hetz C., Ichijo H., Jäättelä M., Joseph B., Jost P. J., Juin P. P., Kaiser W. J., Karin M., Kaufmann T., Kepp O., Kimchi A., Kitsis R. N., Klionsky D. J., Knight R. A., Kumar S., Lee S. W., Lemasters J. J., Levine B., Linkermann A., Lipton S. A., Lockshin R. A., López-Otín C., Lowe S. W., Luedde T., Lugli E., MacFarlane M., Madeo F., Malewicz M., Malorni W., Manic G., Marine J.-C., Martin S. J., Martinou J.-C., Medema J. P., Mehlen P., Meier P., Melino S., Miao E. A., Molkentin J. D., Moll U. M., Muñoz-Pinedo C., Nagata S., Nuñez G., Oberst A., Oren M., Overholtzer M., Pagano M., Panaretakis T., Pasparakis M., Penninger J. M., Pereira D. M., Pervaiz S., Peter M. E., Piacentini M., Pinton P., Prehn J. H. M., Puthalakath H., Rabinovich G. A., Rehm M., Rizzuto R., Rodrigues C. M. P., Rubinsztein D. C., Rudel T., Ryan K. M., Sayan E., Scorrano L., Shao F., Shi Y., Silke J., Simon H.-U., Sistigu A., Stockwell B. R., Strasser A., Szabadkai G., Tait S. W. G., Tang D., Tavernarakis N., Thorburn A., Tsujimoto Y., Turk B., Vanden Berghe T., Vandenabeele P., Vander Heiden M. G., Villunger A., Virgin H. W., Vousden K. H., Vucic D., Wagner E. F., Walczak H., Wallach D., Wang Y., Wells J. A., Wood W., Yuan J., Zakeri Z., Zhivotovsky B., Zitvogel L., Melino G., Kroemer G., Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Degterev A., Hitomi J., Germscheid M., Ch’en I. L., Korkina O., Teng X., Abbott D., Cuny G. D., Yuan C., Wagner G., Hedrick S. M., Gerber S. A., Lugovskoy A., Yuan J., Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han J., Lee J. D., Bibbs L., Ulevitch R. J., A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265, 808–811 (1994). [DOI] [PubMed] [Google Scholar]
- 33.Ip W. K. E., Medzhitov R., Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 6, 6931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sperandio S., Poksay K., de Belle I., Lafuente M. J., Liu B., Nasir J., Bredesen D. E., Paraptosis: Mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 11, 1066–1075 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Franchi-Gazzola R., Gaccioli F., Bevilacqua E., Visigalli R., Dall’Asta V., Sala R., Varoqui H., Erickson J. D., Gazzola G. C., Bussolati O., The synthesis of SNAT2 transporters is required for the hypertonic stimulation of system A transport activity. Biochim. Biophys. Acta 1667, 157–166 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Izumi Y., Yang W., Zhu J., Burg M. B., Ferraris J. D., RNA-Seq analysis of high NaCl-induced gene expression. Physiol. Genomics 47, 500–513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krokowski D., Jobava R., Guan B.-J., Farabaugh K., Wu J., Majumder M., Bianchi M. G., Snider M. D., Bussolati O., Hatzoglou M., Coordinated regulation of the neutral amino acid transporter SNAT2 and the protein phosphatase subunit GADD34 promotes adaptation to increased extracellular osmolarity. J. Biol. Chem. 290, 17822–17837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curi R., Lagranha C. J., Doi S. Q., Sellitti D. F., Procopio J., Pithon-Curi T. C., Corless M., Newsholme P., Molecular mechanisms of glutamine action. J. Cell. Physiol. 204, 392–401 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Canepa A., Divino Filho J. C., Gutierrez A., Carrea A., Forsberg A.-M., Nilsson E., Verrina E., Perfumo F., Bergström J., Free amino acids in plasma, red blood cells, polymorphonuclear leukocytes, and muscle in normal and uraemic children. Nephrol. Dial. Transplant. 17, 413–421 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Li H., Jiang W., Liu Y., Jiang J., Zhang Y., Wu P., Zhao J., Duan X., Zhou X., Feng L., The metabolites of glutamine prevent hydroxyl radical-induced apoptosis through inhibiting mitochondria and calcium ion involved pathways in fish erythrocytes. Free Radic. Biol. Med. 92, 126–140 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Phang J. M., Liu W., Hancock C., Christian K. J., The proline regulatory axis and cancer. Front. Oncol. 2, 60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiemicke A., Jashnsaz H., Li G., Neuert G., Generating kinetic environments to study dynamic cellular processes in single cells. Sci. Rep. 9, 10129 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muzzey D., Gómez-Uribe C. A., Mettetal J. T., van Oudenaarden A., A systems-level analysis of perfect adaptation in yeast osmoregulation. Cell 138, 160–171 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albeck J. G., Mills G. B., Brugge J. S., Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 49, 249–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Twiddy D., Cain K., Caspase-9 cleavage, do you need it? Biochem. J. 405, e1 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi S. Y., Lee-Kwon W., Lee H. H., Lee J. H., Sanada S., Kwon H. M., Multiple cell death pathways are independently activated by lethal hypertonicity in renal epithelial cells. Am. J. Physiol. Cell Physiol. 305, C1011–C1020 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Berry M. R., Mathews R. J., Ferdinand J. R., Jing C., Loudon K. W., Wlodek E., Dennison T. W., Kuper C., Neuhofer W., Clatworthy M. R., Renal sodium gradient orchestrates a dynamic antibacterial defense zone. Cell 170, 860–874.e19 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Bröer S., The SLC38 family of sodium-amino acid co-transporters. Pflugers Arch. Eur. J. Physiol. 466, 155–172 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Farabaugh K. T., Krokowski D., Guan B.-J., Gao Z., Gao X.-H., Wu J., Jobava R., Ray G., de Jesus T. J., Bianchi M. G., Chukwurah E., Bussolati O., Kilberg M., Buchner D. A., Sen G. C., Cotton C., McDonald C., Longworth M., Ramakrishnan P., Hatzoglou M., PACT-mediated PKR activation acts as a hyperosmotic stress intensity sensor weakening osmoadaptation and enhancing inflammation. eLife 9, e52241 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferreiro I., Joaquin M., Islam A., Gomez-Lopez G., Barragan M., Lombardía L., Domínguez O., Pisano D. G., Lopez-Bigas N., Nebreda A. R., Posas F., Whole genome analysis of p38 SAPK-mediated gene expression upon stress. BMC Genomics 11, 144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang X., Zhang L., Natarajan S. K., Becker D. F., Proline mechanisms of stress survival. Antioxid. Redox Signal. 19, 998–1011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szabados L., Savouré A., Proline: A multifunctional amino acid. Trends Plant Sci. 15, 89–97 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Le Meur N., Hahne F., Ellis B., Haaland P., flowCore: Data structures package for flow cytometry data. Bioconductor Proj., 1–34 (2007). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabe1122/DC1