

Machine learning and single-cell data inform personalized drug combinations that selectively co-inhibit cancer cell populations.
Abstract
The extensive drug resistance requires rational approaches to design personalized combinatorial treatments that exploit patient-specific therapeutic vulnerabilities to selectively target disease-driving cell subpopulations. To solve the combinatorial explosion challenge, we implemented an effective machine learning approach that prioritizes patient-customized drug combinations with a desired synergy-efficacy-toxicity balance by combining single-cell RNA sequencing with ex vivo single-agent testing in scarce patient-derived primary cells. When applied to two diagnostic and two refractory acute myeloid leukemia (AML) patient cases, each with a different genetic background, we accurately predicted patient-specific combinations that not only resulted in synergistic cancer cell co-inhibition but also were capable of targeting specific AML cell subpopulations that emerge in differing stages of disease pathogenesis or treatment regimens. Our functional precision oncology approach provides an unbiased means for systematic identification of personalized combinatorial regimens that selectively co-inhibit leukemic cells while avoiding inhibition of nonmalignant cells, thereby increasing their likelihood for clinical translation.
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous disease, characterized by a broad spectrum of molecular alterations that influence the patient’s clinical outcomes (1–3). Despite the recent increase in molecularly targeted treatment options, primary and acquired drug resistance poses a substantial challenge for most patients with AML (4). Monotherapy resistance has remained a major clinical challenge even in patients with a confirmed disease target (2, 5). For example, nearly 60% of patients with relapsed/refractory AML with FLT3 mutation show resistance to gilteritinib therapy (6). This is partly due to the activation of RAS/MAPK (mitogen-activated protein kinase) pathway genes through acquisition of new mutations (7). Multitargeted therapies provide an opportunity for synergistic inhibition of multiple resistance mechanisms, including patient-specific cancer rescue pathways and phenotypic redundancy across heterogeneous cancer subclones (8–13). However, the emergence of resistance is a dynamic process influenced by both genetic and molecular factors, together with selective pressure from the administered therapeutics; this can be seen, for example, in the emergence of NRAS-mutated subclones in the acquired resistance to FLT3 inhibition (7) or intrinsic molecular and metabolic properties of monocytic subclones of patient cells resistant to venetoclax therapy (14).
Identification of patient-specific drug combinations that target specific cell subpopulations poses a combinatorial challenge. Although genomic analyses have led to improvements in understanding of the molecular landscape of AML, patient-specific therapeutic responses are often associated with unique clusters of nonrecurrent and co-occurring mutations, making even the large-scale genomic profiling resources underpowered for identifying patient-customized combinatorial mechanisms. Furthermore, many of the most frequent AML mutations generate broad changes in the epigenome, RNA splicing, and translation (15, 16), suggesting that precision medicine approaches based solely on mutation signature may fail to predict clinically useful combinations (2). An additional clinical challenge comes from intolerable, drug-induced toxicities, especially in older patients with AML (11, 17, 18). Identification of both effective and safe combinations requires capturing the molecular heterogeneity of the disease progression and differential responses of combinations between cell subpopulations at various stages of pathogenesis, using assays that are practical for translational applications in scarce primary patient cells.
In this work, we implemented a functional precision medicine approach to prioritize AML patient–specific and cell subpopulation–targeting drug combinations that uses only limited numbers of patient cells. Our mechanism-agnostic approach exploits the power of single-cell RNA sequencing (scRNA-seq) technology to identify various cell subpopulations in the complex patient samples at baseline and in various disease stages. Using an efficient machine learning approach, together with compound-target interaction networks, we combined the single-cell transcriptomic profiles of the patient cells with their ex vivo single-agent treatment viability responses to predict synergistic combinatorial inhibition of AML cells with a low likelihood for toxic effects. Using this approach, we identified both common and patient-specific synergies for four treatment-naïve or treatment-refractory AML patient samples, each presenting with different molecular backgrounds and synergy mechanisms. Subsequent flow cytometry experiments in the same patient cells confirmed that many of the predicted combinations led to minimal inhibition of nonmalignant cells, hence reducing the likelihood of broadly toxic combination effects. To our knowledge, this is the first translational approach for systematic tailoring of personalized drug combinations that takes into account both the molecular heterogeneity of AML and toxic effects of combinations.
RESULTS
Prediction of subpopulation-targeting drug combinations using limited patient cells
We applied our computational-experimental approach (Fig. 1) to four bone marrow aspirates from three patients with AML (Table 1). Similar to our previous work (19), the computational search approach makes use of both on- and off-targets of the compounds to narrow down the combinatorial search space of more than 100,000 pairwise combinations, far beyond what could be tested systematically in limited patient cells. To prioritize the top patient-specific combinations, we first compressed the genome-wide scRNA-seq transcriptomic profiles of each cell in individual patient samples into compound-target expression enrichment scores using gene set variation analysis (GSVA) (20). The GSVA results in compound-specific enrichment scores among the cell types and protein targets of the 456 compounds tested ex vivo with whole-well viability assay (see Materials and Methods; fig. S1). In the prediction step (Fig. 1A), we trained an XGBoost machine learning model with conformal prediction (CP) (21) to predict the most synergistic drug combinations for each patient sample with confidence. The model makes use of the target expression levels of the compounds both in malignant and nonmalignant cells (here, nonlymphocytes and lymphocytes, respectively). This enables us to control not only the predicted overall synergistic effects but also the predicted differential co-inhibition effects between AML and nonmalignant cells.
Fig. 1. Schematic representation of the drug combination prediction approach.
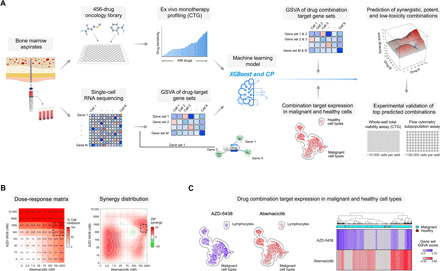
(A) Prediction of combinations with high synergy and potency in malignant cells and low toxicity in nonmalignant cells based on high-throughput ex vivo single-agent profiling of viability responses of individual patient sample to 456 compounds, combined with the 3′ end whole-transcriptome scRNA-seq of enriched mononuclear cells, compressed to compound-target enrichment score matrix (GSVA based on the single-agent targets). These input data were used for training of an integrated machine learning model (XGBoost combined with CP) for the prediction of compound-induced overall cell viability inhibition at the concentration nearest to the relative half-maximal inhibitory concentration (IC50) of each single agent and each patient sample separately (see fig. S2). CTG, CellTiter-Glo. (B) An example of a predicted drug combination (AZD-5438 and abemaciclib) for AML2 patient sample. Combination dose-response matrix and the corresponding synergy distribution confirmed the predicted synergistic effect in the region around IC50 of the two compounds (dashed rectangle). (C) Uniform manifold approximation and projection (UMAP) projection of the scRNA-seq profiles informs about the compound target enrichment scores across cell types of the patient sample before the ex vivo treatments. In this example, GSVA revealed complementary low-overlapping expression scores for the targets of the two compounds, explaining their synergistic co-inhibition effect in various populations of malignant cells. The prediction algorithm was trained on all the leukemic cells (blast and nonblasts), and lymphocytes were considered as nonmalignant “healthy” cells in the current combination predictions.
Table 1. Clinical and molecular characteristics of the patients with AML and samples.
Flow cytometry–based clinical immunophenotyping was performed in fresh red blood cell–depleted bone marrow samples at Helsinki University Hospital, whereas the scRNA-seq profiling was done on the basis of thawed Ficoll-treated and cryopreserved AML cells at the FIMM (Institute for Molecular Medicine Finland) Single-Cell Unit. The blast percentages differ between the two readouts because of different cell isolation protocols and different blast identification approaches. ICD-O, International Classification of Diseases for Oncology.
|
Patient sample |
Disease stage |
Age | Sex |
scRNA-based blast (%) |
Clinical morphological blast (%) |
Diagnosis (ICD-O) |
FAB type* |
Previous malignancies or predisposing conditions |
Risk class at the time of diagnosis |
Potential driver mutations |
Treatment history |
| AML1_D | Diagnosis | 35 | M | 68 | 65 | AML, C92, 9874 | M2 | No | Low |
WT1, CCND2, and CEBPA |
Cytarabine- idarubicin, lenalidomide |
| AML2_R | Refractory | 68 | M | 26 | 40 | AML C92, 9920 | NA | Non-Hodgkin’s lymphoma |
Intermediate |
DNMT3A, ERG, U2AF1, and BCOR |
Cytarabine- idarubicin, azacytidine |
| AML3_D | Diagnosis | 70 | F | 35 | 70 | AML C92 | M1 | Non-Hodgkin’s lymphoma |
Intermediate | NPM1 and TET2 | Azacitidine |
| AML3_R | Refractory | 71 | F | 56 | 42 | AML, C92 | M1 | Non-Hodgkin’s lymphoma |
Intermediate |
NPM1, TET2, and HDAC 1,2,7 |
Azacitidine- venetoclax |
*FAB, the French-American-British classification.
In the experimental step (Fig. 1B), we tested the most promising predicted combinations for each patient sample in 8 × 8 dose-response combination matrices using ex vivo viability assays to confirm whether the predicted combinations truly act synergistically, i.e., jointly inhibit patient cells more than what is expected from their single-agent effects, as quantified using the zero interaction potency (ZIP) synergy score (22). After confirming the overall synergistic effects in the whole-well viability assays, we further tested a subset of validated synergistic combinations using high-throughput flow cytometry assays in the same patient cells to differentiate between combinatorial responses observed in the malignant and nonmalignant primary cell subpopulations. This experimental step was important to prioritize those combinations that selectively target AML cells and to avoid toxic co-inhibition of lymphocytes that were considered as nonmalignant cells in the prediction model (23). The number of combinations selected for both whole-well and flow cytometry assays was based on the number of cells available in each patient primary sample. The combination target expression patterns from the whole-transcriptome scRNA profiles provide hypotheses for the molecular determinants of drug combination efficacy and toxicity in each patient sample (Fig. 1C).
Patient-specific combinations show high overall synergy and wide interpatient variability
We first investigated a subset of the combinations that were shared among the top 5% of predicted combinations in at least two patient samples. The predicted combinations resulted in co-inhibition of various targets and biological pathways (Fig. 2A). Two such common combinations that showed high synergy (ZIP score > 5) (22) in the whole-well viability assay across all the samples included camptothecin (topoisomerase I inhibitor) combined with etoposide (topoisomerase II inhibitor) and venetoclax (Bcl-2 inhibitor) combined with vistusertib [mechanistic target of rapamycin (mTOR) inhibitor]. While simultaneous blocking of both topoisomerase I and II may lead to overlapping toxicities (24), it has been shown that cotargeting of both Bcl-2 and mTOR pathway triggers synergistic apoptosis and enhances drug-induced cytotoxicity by suppressing MCL-1 in leukemic cells (25). The predictive approach also identified the ruboxistaurin-ipatasertib combination as synergistic in all but one sample (in the refractory AML3 sample, the combination was additive with ZIP = 2.3). This combination is likely to target multiple monotherapy-resistant AML subclones, as co-inhibition of protein kinase C (PKC) and AKT promotes blast cell death by down-regulation of antiapoptotic Bcl-2 family proteins in leukemic cells (26, 27).
Fig. 2. Experimental validation of the combination predictions using combinatorial CTG viability assay.
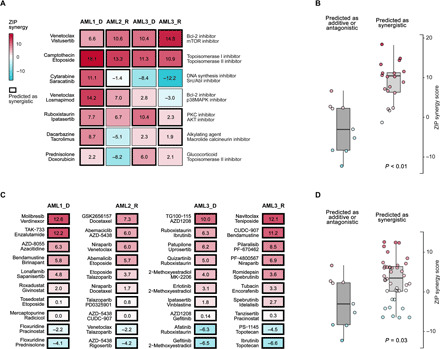
(A) Top seven shared combinations predicted to have synergy and AML cell selectivity in at least two samples. The numbers correspond to the ZIP synergy score (22), calculated for the dose region around IC50 values of each drug in combination, separately for each patient sample and combination. (B) Comparison of the synergy score distributions of the combinations predicted to be either synergistic (ZIP > 5), antagonistic (ZIP < −5), or additive (−5 < ZIP < 5) in the patient samples (P < 0.01; Wilcoxon rank sum test). (C) Top 10 patient-specific combinations predicted uniquely for each patient sample. (D) The measured synergies of the patient-specific combination predictions were higher compared with those that were predicted to be only additive or antagonistic (P = 0.03; Wilcoxon rank sum test). Overall, 53% of the 59 predicted synergistic combinations were experimentally confirmed to show synergy, and 83% were nonantagonistic (ZIP > −5).
In general, across 28 common combinations (7 combinations tested in each of the four samples), those combinations that were predicted to have synergy and AML selectivity showed significantly higher synergy in the combinatorial viability assay, compared with those that were predicted to be only additive or antagonistic (P < 0.01, Wilcoxon rank sum test; Fig. 2B). This demonstrates the importance of patient specificity of the predictions, even for those combinations resulting in shared synergy among multiple patient cases. For instance, we identified a strong overlapping synergy between venetoclax and the p38 MAPK inhibitor losmapimod in the two samples where the combination was predicted to be synergistic and AML selective (AML1 and AML2), while exhibiting an additive effect in the two other patient samples. It has been shown that co-inhibition of Bcl-2 and p38 MAPK leads to synergistic decrease of phosphorylated Bcl-2 because inhibition of p38 MAPK activity alone cannot stop phosphorylation of Bcl-2 (28), causing resistance to venetoclax in leukemic cells (29). Patients with AML with blast cells lacking Bcl-2 phosphorylation survive longer than those patients with blast cells with phosphorylated Bcl-2 (26).
The measured synergies of the patient-specific unique combination predictions were higher than those of shared combinations predicted as antagonistic or additive (P = 0.03; Wilcoxon rank sum test; Fig. 2D). Notably, we observed that the shared combinations that were predicted to act synergistically across multiple patient samples showed higher synergies than the patient-specific combinations (P = 0.0002; Fig. 2B), but this difference was mainly due to the two broadly synergistic combinations, venetoclax-vistusertib and camptothecin-etoposide (Fig. 2A). The patient-specific combinations revealed a wide spectrum of co-inhibitors of multiple biological pathways active in the AML patient cells (Fig. 2C). Although these one-off combinations are increasingly difficult to predict for single patient cases, 40% (16 of 40) of the predicted unique combinations were experimentally confirmed to show synergy in the whole-well viability assays (ZIP > 5). Among the 28 shared combinations (Fig. 2A), the true-positive rate of the experimental validations was much higher, namely, 79% (15 of 19). Among the 68 the tested combinations, there was only one synergistic combination that was not predicted by the model, leading to <2% false-negative rate, indicating high precision of the predictive approach in this challenging personalized prediction task.
Flow cytometry assay confirms cell subpopulation–specific combinatorial inhibition effects
Although patient-specific combination designs arguably exclude broadly toxic combinations, the whole-well viability assay cannot effectively discriminate between AML cell killing and potential toxic effects of combinations. We therefore next investigated the degree of which the predicted combinations that showed high overall cell inhibition synergy led to the co-inhibition of specific cell populations using combinatorial flow cytometry assays (Fig. 3). To quantify the AML-selective effects, we compared the relative co-inhibition of lymphocytes [specifically T and natural killer (NK) cells] against the other cell populations in each of the AML patient samples separately (marked as AML cells in Fig. 3B). The average co-inhibition of the nonmalignant cell subpopulations across the combinations predicted for the three patient cases was 40 ± 22%, significantly lower compared to the combinatorial inhibition of the AML cells (average, 60 ± 22%; P = 3.9 × 10−6, Wilcoxon signed-rank test). When using a cutoff of 50% for the relative inhibition of T and NK cells, 67% (12 of 18) of the predicted combinations showed low toxicity, indicating that the patient-specific combinations resulted in relatively selective killing of AML cells.
Fig. 3. Co-inhibition effects of the predicted combinations selected for flow cytometry experiments in each patient sample.
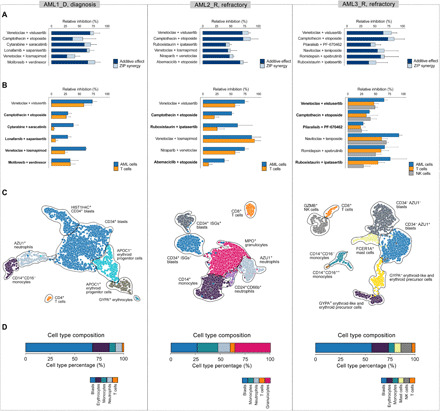
(A) The combined dark and light blue bars together indicate the relative combinatorial inhibition, compared to nontreated cells, based on the whole-well viability assays, which was used in the prediction model for single-agent total viability responses. The dark blue parts of each bar indicate the expected additive inhibition from the combinations, and the light blue parts mark the unselective co-inhibition synergy based on the whole-well viability assays (excess inhibition % based on the ZIP synergy model). (B) Relative inhibition of malignant AML cell subpopulations compared to inhibition of nonmalignant cells (T and NK cells) in the patients based on the flow cytometry assay. Boldfacing indicates those 12 combinations (67%) with low toxicity (less than 50% of relative inhibition of T and NK cells). The co-inhibition of the nonmalignant cell subpopulations was significantly lower compared to that of the AML cells (P = 3.9 × 10−6; Wilcoxon signed-rank test). Note: No cells were available from the AML3 patient diagnostic sample for the flow cytometry experiments. (C) UMAP visualizations of the cell subpopulations based on the scRNA-seq transcriptomic profiles of the patient cells extracted before ex vivo compound testing. Cell clusters were identified using our ScType scRNA-seq processing pipeline (31), with the Louvain clustering implemented in Seurat v3.1.0 (32). The identified clusters were annotated on the basis of cell-specific marker information from our ScType marker database, and unassigned cell types were manually identified (detailed in Materials and Methods). (D) Cell type composition of the patient samples based on their scRNA-seq transcriptomic profiles. Erythrocyte cell type corresponds to erythroid-like and erythroid precursor cells; see Materials and Methods. See fig. S3 for an extended version, where also the cell subpopulations identified in the patient sample AML3_D are shown.
Notably, we revealed marked differences in the AML cell–selective co-inhibition effects both between the predicted combinations and patient samples. In particular, combinations involving venetoclax had relatively high co-inhibition effects on T cells, where their potential toxic effects depended on the particular patient sample and the combination partner (Fig. 3B). This suggests that the immune composition of the bone marrow of these patients with AML may become reshaped during the venetoclax treatment, depending on the specific combination (30). We identified also unique combinations in single patients, such as the combination between saracatinib (SRC and ABL inhibitor) and cytarabine (DNA synthesis inhibitor), which was confirmed to be highly synergistic, low toxic, and selective in AML1 patient only. Many of these subpopulation-specific co-inhibition effects cannot be captured by the whole-well assay and therefore required additional validation with flow cytometry assay. However, we note that the whole-well viability assay was informative for confirming the overall co-inhibition synergy, which remained high across all the patient-specific combinations (average ZIP = 9.50 ± 3.58%; Fig. 3A).
Cellular heterogeneity of the patient samples explains variable treatment responses
We next charted the cell type composition of each patient sample using the whole-transcriptome scRNA-seq profiling of the primary patient cells, extracted before the ex vivo compound testing (Fig. 3, C and D). Using a highly specific scRNA-seq data processing pipeline and our comprehensive cell marker database (31, 32), we observed a wide spectrum of cellular heterogeneity between and within the AML patient samples that provided further insights into the compound-induced cell subpopulation co-inhibition effects ex vivo and in vivo. For instance, the scRNA-seq transcriptomes identified NK cells both in the diagnostic and refractory samples of the AML3 patient case, therefore enabling the evaluation of co-inhibition effects of the patient-specific combinations on the NK cell population as well (fig. S3b). We also identified two cell clusters of rare CD34− blast cells both in the diagnostic and refractory AML3 samples based on the scRNA-seq profiles (figs. S4 and S5).
When further comparing the two longitudinal samples from the AML3 patient, we revealed a decrease in the numbers of erythroid-like and NK cells, as well as the emergence of mast cell population in the refractory stage of the AML3 patient, as compared to the diagnosis stage of the same patient (Fig. 3D). By averaging the importance of individual cells within distinct cell types for the prediction model construction, we observed that the GYPA− erythroid-like cell type had high similarity both in the diagnostic and refractory AML3 patient samples (Fig. 4), hence making them less important for the model predictions. In contrast, GYPA+ erythroid-like cells were required for more accurate predictions in the AML3 diagnostic sample, while having a considerably lower relative importance in the refractory sample of AML3. This suggests that GYPA+ erythroid-like cells were more diverse in the diagnosis patient sample, possibly denoting cells at different stages of erythropoiesis.
Fig. 4. Contribution of single-cell features across various cell types to the predictive modeling.
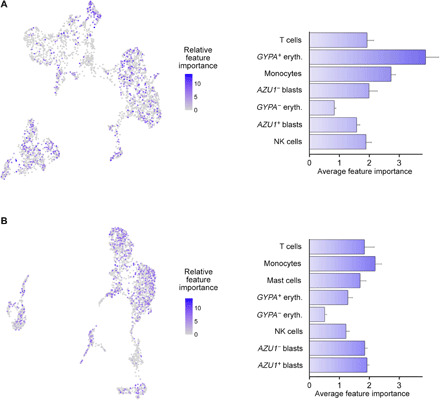
(Left) Relative importance of each single cell for the predictive contribution of the model, define by the features’ predictive contribution for each regression tree of the XGBoost algorithm. (Right) Average feature importance (AFI) of each cell type for generating the model predictions, calculated by averaging the feature importance within the cell types. (A) AML3_D and (B) AML_R. Higher AFI values indicate that higher percentage of cells within the cluster are distinct from each other and are required for more accurate predictions, while low AFI values imply that cells within a cell type are highly similar from the model perspective, and only few cells are enough for model construction. The error bars denote SEM of the scaled AFI.
We identified a number of potential molecular predictors of the treatment outcomes. For instance, the CD34+ blast cell clusters of AML1 and AML2 patient samples showed a high expression of two prognostic markers, ankyrin repeat domain 28 (ANKRD28) and guanine nucleotide binding protein 15 (GNA15) (fig. S6A), which are associated with a poor overall survival in patients with AML (33). Specifically in the AML2 refractory sample, we identified a population of CD34+ blast cells that were further classified into two subpopulations (fig. S6B): those expressing interferon-stimulated genes (ISGs), such as ISG15, IFIT2, and IFIT3, or not. Because the patient was not earlier treated with IFN-α/β (34) and he did not have a known history of virus infection during the time of sampling (35), it is likely that the ISGs+ CD34+ blast cells are induction therapy–induced drug-resistant quiescent (G0) leukemic cells (36), causing the therapy failure, as indicated by change to azacitidine therapy after two cycles of induction therapy (Table 1).
Furthermore, we revealed a number of molecular determinants of ex vivo synergies. For example, venetoclax and losmapimod combination was validated to be synergistic both in AML1 and AML2 patients but neither in the diagnosis nor the refractory samples of AML3 (Figs. 2 and 3). The observed synergy of this combination, as well as its AML selectivity, may be explained by the higher expression level of multiple genes in the apoptotic and p38 MAPK signaling pathways both in the AML1 and AML2 patients’ AML cells in comparison to the two other samples (table S2). In particular, we observed a significantly higher expression of the primary target of losmapimod, MAPK14 (table S3), when comparing malignant and nonmalignant cells of the patient samples (P < 2.2 × 10−16; Fig. 5A). To illustrate the complexity of the molecular interactions behind the combination response, we connected the drug targets with differentially expressed genes in the target pathways (Fig. 6A), highlighting potential determinants of the combination effect. The below subsections describe translationally the most interesting single-drug regimens and combinations identified in each patient case separately, together with their potential molecular determinants.
Fig. 5. Target expression of compounds identified as effective and less toxic combinations.
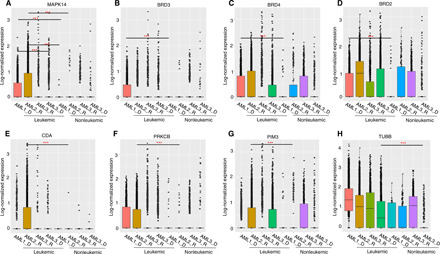
(A) Comparison of the expression of losmapimod primary target MAPK14 in the leukemic cells of AML1 and AML2 patients, against leukemic cells of AML3 patient, and nonleukemic cells of AML1 and AML2. (B to D) Comparison of expression levels of molibresib targets BRD2, BRD3, and BRD4 between leukemic and nonleukemic cells of AML1. (E) Comparison of the cytarabine-deactivating enzyme cytidine deaminase (CDA) expression in the leukemic cells of AML2 patient with leukemic cells of other samples and nonleukemic cells of AML2. (F and G) Comparison of the expression levels of ruboxistaurin targets PRKCB and PIM3 in the leukemic cells of AML2 patient with leukemic cells of other samples and nonleukemic cells of AML2. (H) Comparison of expression levels of patupilone target tubulin β (TUBB) in the leukemic cells of AML3 patient with leukemic cells of other samples and nonleukemic cells of AML3. In each panel, y axis shows log-normalized count values after sctransform (32), where P values were calculated with Wilcoxon rank sum test. ***P < 0.001.
Fig. 6. Interaction networks for the predicted and validated patient-specific combinations.
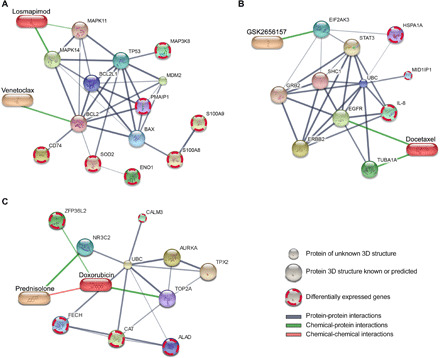
(A) Venetoclax and losmapimod, (B) GSK2656157 and docetaxel, and (C) doxorubicin and prednisolone. The protein nodes in the networks include the primary and secondary targets of the compounds in the combinations, along with differentially expressed genes in the target pathways that could potentially explain the observed combination effects in the particular patient samples [(A) AML1 and AML2; (B) AML2; (C) AML3_D]. The visualization was performed using the STITCH web tool (67). The edge width and color darkness indicate the degree of data support for the connection. The chemical-protein interactions include both direct targets of the compounds and their downstream targets and other molecular modifiers of the compounds’ responses. 3D, three-dimensional.
AML1 patient (diagnosis stage)
We first applied the integrated platform to predict patient-specific combinations for a 35-year-old male patient with AML (M2 subtype), with mutated CEBPA, WT1, and CCND2, and who was treated with induction chemotherapy (cytarabine-idarubicin; Table 1). scRNA-seq analysis showed that the patient sample was characterized by a high proportion of blasts (68%; Table 1). The ex vivo single-agent responses revealed high selective activity [selective drug sensitivity score (sDSS) > 20] of crenolanib (FLT3 and platelet-derived growth factor receptor inhibitor) and ruxolitinib (Janus kinase 1 and 2 inhibitor) (table S1). In general, the patient sample showed highly selective sensitivity to multiple kinase inhibitors in addition to cytotoxic chemotherapy (fig. S7).
For combinatorial targeting of AML1 patient leukemic cells, we identified and confirmed selective synergy between molibresib (BET family inhibitor) and verdinexor (XPO1/CRM1 inhibitor) (Fig. 3B). This combination was unique to the AML1 patient sample (Fig. 2C), possibly because the leukemic cells in this sample expressed a higher level of the molibresib targets (BRD2, BRD3, and BRD4; table S3), when compared to the nonleukemic cells of AML1 sample (P < 0.0001, Wilcoxon rank sum test; Fig. 5, B to D). This unanticipated combinatorial effect would have remained unpredictable on the basis of the single-agent testing results only.
AML2 patient (refractory stage)
The second patient case was a 68-year-old male AML patient with a history of non-Hodgkin’s lymphoma and mutated DNMT3A, U2AF1, and BCOR (Table 1). Before sampling, the patient was treated with induction therapy (cytarabine-daunorubicin), followed by azacitidine, as the patient did not respond to the induction therapy (fig. S8). The monocytic blast cell population of the AML2 patient had the highest expression of cytidine deaminase, a known cytarabine-inactivating enzyme that converts cytarabine to cytarabine-uracil (37), possibly explaining the lack of the patient’s response to induction therapy (Fig. 5E). The single-agent screening revealed sensitivity to epigenetic modifiers and apoptotic and heat shock protein inhibitors (fig. S7). Furthermore, the sample showed sensitivity to BAY87-2243 (mitochondrial complex I inhibitor), pevonedistat (NAE inhibitor), and plicamycin (RNA synthesis inhibitor), as the top monotherapies (table S1).
We identified a highly synergistic combination between GSK2656157 [protein kinase R-like endoplasmic reticulum kinase (PERK) inhibitor] and docetaxel (microtubule disassembly inhibitor), which was unique to the AML2 sample (Fig. 2C). This selective combination synergy could be due to the up-regulation of several genes in the PERK-mediated signaling and microtubule polymerization or depolymerization pathways in the AML2 patient AML cells when comparing to the three other samples (table S2). A network visualization reveals a complex interplay between the drug targets and genes differentially expressed in the target pathways such as interleukin-8 [IL-8 or chemokine (C-X-C motif) ligand 8 (CXCL8); Fig. 6B]. We identified also a highly synergistic and low-toxic combination between ruboxistaurin (PKCβ inhibitor) and ipatasertib (AKT inhibitor) only in the AML2 sample (Fig. 3B). This selective combination may appear because the nonleukemic cells of the AML2 sample uniquely expressed the ruboxistaurin targets PRKCB and PIM3 (table S3), as compared to the other samples that expressed neither of the targets in their AML cells (Fig. 5, F and G).
Furthermore, we revealed a strong patient-specific synergy among cyclin-dependent kinase (CDK) inhibitors and other single agents for this patient case. One such combination with a strong synergy was between AZD-5438 (a small-molecule CDK1/2/9 inhibitor) and abemaciclib (CDK4/6 inhibitor). This combination is interesting especially in the mutated DNMT3A AML2 patient sample because simultaneous inhibition of CDK4 and CDK2 has shown to induce strong senescence response and growth arrest in leukemic cells with low DNMT3A expression (38, 39).
AML3 patient (diagnosis stage)
As the final application case, we predicted potential drug combinations for the diagnosis stage of a 70-year-old male AML3 patient (M1 subtype), with mutated NPM1, and previous history of non-Hodgkin’s lymphoma (Table 1). In support of earlier observations that blast cells in AMLs with NPM1 mutation subtype are often CD34 negative (40), the presence of CD34− leukemic cells was confirmed with our scRNA-seq analysis. Further analysis revealed a high proportion of erythroid lineage cells in the bone marrow mononuclear cells (35.8%), characterized by up-regulation of hemoglobin-related genes (e.g., HBA2 and HBB). The diagnostic stage ex vivo responses to the 456 single agents revealed navitoclax (Bcl-2/Bcl-xL inhibitor), dexamethasone, and methylprednisolone (glucocorticoids) as the top three drugs with the highest activity in the patient sample (table S1). The potency of navitoclax is in line with our earlier observation of higher activity of Bcl-2 inhibitors in the M1 subtype AML (41). In general, the patient sample showed a high selective sensitivity (sDSS range, 13 to 21) to a number of targeted compounds, including kinase inhibitors, heat shock protein inhibitors, and apoptotic modulators (fig. S7).
Using the predictive model, we identified a strong and unique synergy between patupilone (microtubule disassembly inhibitor) and uprosertib (AKT kinase inhibitor) in the AML3 patient diagnostic sample (Fig. 2C). It has previously been shown that a dual inhibition of microtubules and AKT/mTOR pathways can synergistically enhance apoptosis of cancerous cells, particularly in patients with cancer resistant to mitotic inhibitors because phosphatidylinositol 3-kinase/Akt/mTOR pathway is associated with resistance in multiple cancers including leukemia (42, 43). In line with this observation, patupilone primary target, tubulin β (TUBB; table S3), showed higher expression in the leukemic cells of AML3 patient diagnosis sample, compared to nonleukemic cells (P < 0.0001, Wilcoxon rank sum test; Fig. 5H), suggesting that the synergistic co-inhibition might be achieved through selective inhibition of the tubulins involved in cancer chemoresistance and metastasis (44, 45).
We also identified that the combination between doxorubicin (topoisomerase II inhibitor) and prednisolone (glucocorticoid) showed a selective synergy in AML3 patient diagnostic sample. As potential explanations for this patient-specific combination effect, we found several overexpressed genes in the pathways related to the doxorubicin pharmacokinetics and response to corticosteroid in the AML3 patient diagnostic sample (table S2). The complex interactions underlying this combination may provide potential explanations for the patient-specific observed synergy (Fig. 6C). However, we note that these interactions should be considered as potential hypotheses to be tested in larger patient populations.
AML3 patient (refractory stage)
The leukemia of patient AML3 was refractory to azacitidine, as observed after a single 28-day cycle of azacitidine treatment (7 days on drug), and the patient was therefore transferred to an azacitidine-venetoclax regimen after 1 month (fig. S8). Consistent with the clinical treatment responses, our predictive model with conformal learning did not prioritize either azacitidine monotherapy or any combinations involving azacytidine as partner because of its low ex vivo response in the diagnostic sample (fig. S9A), together with a relatively low predicted response and a relatively high prediction error (fig. S9b). In particular, we note the azacitidine-venetoclax combination was not among the combinations predicted by the model, and leukemia was, in fact, clinically refractory to this combinatorial therapy (fig. S8). However, the ex vivo drug response after 72 hours of drug treatment may not completely represent the clinical response for azacitidine that is expected to come primarily from the hypomethylating effects observed only after multiday daily treatments.
The ex vivo single-agent responses for the other inhibitors revealed that the AML3 refractory-stage sample was still sensitive to dexamethasone (glucocorticoid), BMS-754807, and GSK-1904529A (insulin-like growth factor 1 receptor inhibitor) (table S1), similar to the diagnostic sample (fig. S7). However, the refractory sample became more sensitive both to epigenetic modifiers (P = 1.5 × 10−3) and chemotherapies (P = 1.2 × 10−3, Wilcoxon rank sum test). The synergy between ruboxistaurin (PKCβ inhibitor) and ipatasertib (AKT inhibitor), which was observed only in the diagnostic samples, was lost as the disease progressed from the diagnosis to refractory stage, possibly because of the lower expression ruboxistaurin target PIM3 in the leukemic cells of the refractory sample (Fig. 5G). This case example demonstrates the need for longitudinal patient sampling and subpopulation-level prediction machinery to dynamically tailor AML-selective combinations at different disease stages.
DISCUSSION
AML cells harbor a number of genetically and epigenetically heterogeneous subpopulations, each of which may be associated with a distinct cellular function or phenotype such as self-renewal, drug sensitivity, or resistance. To address this heterogeneity in a personalized treatment design, we implemented a computational-experimental platform and applied it to AML patient primary cells to rationally combine drugs that preemptively inhibit multiple AML-related dysfunctions or resistance mechanisms in individual patients. The selective approach avoids a severe co-inhibition of nonmalignant cells, with the aim to improve both combination efficacy and tolerability. To the best of our knowledge, this is the first systematic approach to personalized drug combinations selection that takes into account both the molecular heterogeneity of AML cells and the possible toxic effects of combinations, thereby increasing their likelihood for clinical translation. The approach requires only a limited number of patient cells and is widely applicable to hematological cancers that are amenable to scRNA-seq profiling and systematic ex vivo compound testing. In the present work, we used a total of 30 to 40 million cells per patient for both the primary single-agent screening and the scRNA-seq profiling, as well as for the combinatorial validation experiments with whole-well and flow cytometry assays (a vial of 10 million cells for each assay). The implementation of the experimental-computational approach takes less than 3 weeks per sample, where scRNA-seq is the current bottleneck, demonstrating that the approach can be applied within a clinically actionable time frame, especially once the sequencing analysis becomes faster.
In cancer treatment, there is an urgent clinical need to develop rational and systematic strategies for designing anticancer combinatorial treatment that can target cancer heterogeneity and drug-resistant cell populations (46). Earlier approaches have used experimental testing of single-drug efficacies in genetically variant cell subpopulations, together with RNA interference to model the functional diversity and perturbation effects of heterogeneous tumors, followed by computational optimization algorithm that predicts how drug combinations will affect heterogeneous tumors (47). While these optimized design principles can offer useful insights into intratumoral heterogeneity, profiling each component subpopulation with single drugs together with knockdown experiments in patient cells is arguably not amenable to routine clinical application. Recently, scRNA-seq has been widely used to chart the cellular composition of complex samples and to design combinatorial regimens in solid tumors that show increased treatment efficacy over monotherapy (48). Although scRNA-seq enables efficient mapping of primary AML cell populations at baseline (23), when used alone, it lacks the possibility to profile functional responses of drug treatments. Another recent study used nested-effects modeling combined with mass cytometry time of flight (CyTOF) to simultaneously profile single-agent responses and to catalog signaling heterogeneity at the single-cell level, with the aim to suggest drug combinations that lead to maximal desired intracellular effects (49). However, this method requires CyTOF profiling before and after the drug treatment, hence limiting its predictive clinical value.
Next-generation functional precision medicine approaches combine molecular and genomic characterization of individual patient samples at various disease stages with the whole-well ex vivo phenotypic profiling of the patient samples using large compound libraries. We and others have demonstrated that these integrated approaches based on bulk profiling of both drug sensitivity and genomic changes provide a relatively fast and practical platform for translational discoveries in hematological malignancies, informing both monotherapy regimes and combination discoveries (50–53). Recently, high-throughput flow cytometry–based drug response profiling approaches have been established for higher-resolution evaluation of ex vivo sensitivity of various cell populations in primary AML samples (41), where especially refractory patient samples often have lower blast cell percentages, making the whole-well bulk drug assays less reliable for drug response profiling. However, high-throughput flow cytometry profiling of a larger number of drug combinations in multiple doses is not feasible in scarce primary patient cells; instead, the current translational strategies rely on testing only selected drugs or combinations in single concentrations (54). Because the predefined combinations and fixed concentrations may easily miss novel synergies, there is a need for efficient data-driven approaches to systematically prioritize the most potent and selective combinations for each patient to be tested in subpopulation-level drug combination assays using dose-combination matrix designs, before entering into further preclinical validation.
Our experimental results demonstrated that the bulk viability single-agent screening assays have an unexpectedly high predictive power for the AML cell subpopulation co-inhibition effects, when combined with the scRNA-seq transcriptomic data and machine learning modeling. However, because of its cell population unspecificity, we used the bulk viability assay in the prediction approach only as the first filtering phase, following the model predictions, which enrich a smaller number of the most synergistic and selective combinations to be validated in the flow cytometry–based subpopulation assays. This is because we could not perform testing of all the predicted combinations with the more high-resolution flow cytometry assay due to the scarcity of the primary patient cells. However, in applications where one could perform flow cytometry–based drug viability assessment for all the drugs in the first experimental phase as the model input, one could potentially further improve the model performance and selectivity against blast cells. We also demonstrated that combinations having high synergy in the whole-well viability assay may sometimes lead to high likelihood of toxic effects, especially in the patient samples with limited amounts of nonleukemic cells, making the subpopulation-specific flow cytometry response assays highly informative for translational applications. Notably, the flow cytometry–based experiments revealed a wide range of interpatient variability in the combinations involving venetoclax. For instance, the combination of venetoclax and losmapimod showed a relatively variable ex vivo synergy-efficacy-toxicity profiles between diagnostic AML1 and refractory AML2 samples (Fig. 3).
The benefits of the integrated approach were demonstrated in multiple case examples, including the combination between saracatinib (SRC and ABL inhibitor) and cytarabine (DNA synthesis inhibitor), which was confirmed to be highly synergistic, low toxic, and selective for AML1 patient (Figs. 2 and 3). The combination is likely to remain effective also in more complex disease systems because the SRC family kinases are activated in AML stem/progenitor cells, known to contribute to their survival and proliferation (55). Furthermore, an in vivo study has shown that SRC kinase inhibitor treatment enhances chemotherapy-induced targeting of primary murine AML stem cells, capable of regenerating leukemia in secondary recipients by activating p53 (56). Therefore, this combination may be best suited for cancers that contain wild-type p53, as was the case with the AML1 patient (Table 1). On the other hand, a cytarabine-saracatinib combination showed synergy in the whole-well viability assay only in one of the two predicted samples (Fig. 2A), indicating that part of the heterogeneity in the combinatorial response either is not captured by the whole-well viability readouts or remains unpredictable by the current integrated approach. This could be due to the patient and sample specificity of the current model, designed for personalized oncology applications. The predictive modeling could also be done in a batch mode by making use of the different disease stages (e.g., diagnostic and refractory) of the same patient, when these are available, or even combining measurements from multiple patients into a single prediction model.
Given that drug combination synergy is a rare phenomenon, with estimated likelihood of synergy being around 5% in large-scale combination screens (19, 57, 58), our predictive platform led to highly accurate combination prioritization, achieving a higher than 50% success rate in the validation experiments. For specific patient cases, the success rate was even higher. For instance, in the AML1 case, we tested 17 of the predicted combinations using the whole-well viability assay, and the validations revealed that 10 (59%) of the combinations showed strong synergistic effect (ZIP > 5) and 15 (88%) of the predicted combination were either synergistic or additive (ZIP > 0). We acknowledge that some of the ex vivo predictions may not lead to clinically actionable therapies. For instance, simultaneous blocking of both topoisomerase I and II showed synergies in all the patient samples. While this combination might provide an opportunity to effectively cotarget AML rescue pathways, as inhibition of either of the enzymes alone may lead to increase in the expression of the other, hence resulting in treatment of resistance (24), the sequential or simultaneous co-inhibition of topoisomerase I and II by compounds forming DNA lesions has shown severe side effects (59). Therefore, dual inhibitors with a longer half-life and improved binding efficacy (e.g., tafluposide and batracylin) are being designed to tackle treatment resistance and toxicity (24). Similarly, the combination of AZD-5438 (CDK1/2 inhibitor) and abemaciclib (CDK4/6 inhibitor) showed patient AML2–specific synergy; however, this combination is unlikely to be translated to clinics, as the AZD-5438 monotherapy trial was discontinued because of intolerable toxicities when administered continuously in patients with advanced solid tumors (38).
These AML case studies also demonstrated that in addition to the overall co-inhibition synergy, it is important to also consider the relative differences between the combination efficacy and toxicity because these are critical determinants for the clinical success of a therapy (11, 60). As a unique component of our machine learning model, it makes use of the information from scRNA-seq on the target expression levels of the compounds both in malignant and nonmalignant cells, when predicting not only synergistic effects but also the differential co-inhibition effects between malignant and nonmalignant cells. In the present work, the prediction algorithm was trained on all the leukemic cells, which makes the predictions more robust, compared to using only the blast cells. For instance, even with a small scRNA-seq blast percentage in some cases (e.g., in the refractory AML2_R sample), we showed that the prediction algorithm led to highly accurate combination predictions, in terms of both efficacy and toxicity, as validated by the cell subpopulation–level flow cytometry experiments (Fig. 3). In general, the flow cytometry assay validations showed that 67% of the predicted and synergistic combinations had a highly selective co-inhibition effect (<50% toxicity to T and NK cells), indicating that the predictive approach effectively avoids suggesting only broadly toxic and potentially synergistic combinations that unselectively kill various cell types. To our knowledge, there are no large-scale studies of such selective co-inhibition effects in cancer cells, but this is arguably even a rarer phenomenon than an overall combination synergy, hence further demonstrating the high precision of the current prediction model.
In the present work, we used the T and NK cells as nonmalignant cell types in the combination predictions because these lymphocytes were robustly identified in the AML patient samples using their scRNA-seq profiles (Fig. 3D). Lymphocytes can be identified using distinct surface markers, and they are considered nonmalignant cells across all the subtypes of myeloid leukemia. Furthermore, a recent study demonstrated the absence of AML-related mutations in lymphocytes using single-cell genotyping of 16 patients with AML (23). However, we note that drug toxicities are not derived from the activity on lymphocytes alone, or even solely on cells of the hematopoietic compartment, and therefore, inhibition of T and NK cells is not an absolute measure of toxicity. We recommend that the choice of healthy cell types should be made on the basis of the application and experimental setup for the assessment of predicted combinations in terms of efficacy and toxicity. Further, we note that our method provides merely an initial hypothesis for effective, synergistic, and low-toxicity combinations based on the patient-derived primary cells; however, these predictions should be further tested in more complex preclinical model systems to assess their potential toxicity in other organs (e.g., hepatotoxicity or cardiotoxicity). In future applications, one can freely choose also other cell populations in the prediction pipeline and in the ScType platform to define both the therapeutic and toxic effects in other applications, especially when going beyond AML to other hematological cancers.
In conclusion, combination synergy, efficacy, and toxicity profiling each provide complementary information on the combinations’ mode of action, and therefore, integrating these data should become informative for translational applications. Our machine learning model enabled also possibilities to identify potential molecular markers for the low-toxic and synergistic combinations in each patient case based on their scRNA-seq profiles, including several apoptotic and p38 MAPK signaling pathway genes for the venetoclax and losmapimod combination, which warrant further testing as predictive biomarkers in larger AML patient cohorts.
MATERIALS AND METHODS
Experimental design
The computational and experimental steps of the study design are illustrated in Fig. 1. The study material and methodological details are described in the below sections.
Patient samples
Four bone marrow aspirates from three patients with AML (indexed as AML1, AML2, and AML3, the latter with both diagnostic and refractory samples) were collected at Helsinki University Hospital and The Finnish Hematology Registry and Clinical Biobank (FHRB) after informed consent, using protocols approved by a local institutional review board in accordance with the Declaration of Helsinki. Patient clinical characteristics and mutation profiles are shown in Table 1.
Single-cell RNA sequencing
Enriched (Ficoll-Paque PREMIUM, GE Healthcare) mononuclear cells were used for Chromium single-cell 3′ RNA-seq. The gel beads in emulsion generation, cDNA amplification, and library preparation were performed according to the manufacturer’s instructions using Chromium Single Cell 3′ v2 Reagent Kit (10X Genomics) aiming for a 4000– to 6000–target cell capture per sample. The sample libraries were sequenced in Illumina HiSeq 2500 using Rapid mode aiming for sequencing depth of 50,000 reads per cell. The Cell Ranger v1.3 mkfastq and count analysis pipelines (10X Genomics) were used to demultiplex and convert Chromium single-cell 3′ RNA-seq barcode and read data to FASTQ files and to generate align reads and gene-cell matrices. The gene-barcode matrices were analyzed using the ScType scRNA-seq processing pipeline (31) with the Louvain cell clustering as implemented in Seurat v3.1.0 (32). Cells with either a low (below 100 to 200) or unexpectedly high number (3000 to 4000, depending on the sample) of detected genes, or cells with more than 8 to 10% of mitochondrial UMI (unique molecular modifier) counts, were classified as low-quality or uninteresting cells and were excluded from the analysis.
Cell type identification and manual annotation
We used our ScType cell type annotation web tool (31) for the initial cell cluster annotation. ScType platform uses the currently two largest resources of human cell population markers (61, 62) to classify cell types by considering all the gene expression changes among the cell clusters. More specifically, the ScType method calculates a cumulative sum of marker specificity scores of all the genes supporting a cell type to make an automated decision of the cell type annotation. Next, we manually validated the ScType cell type classifications by visualizing a number of established marker genes that were used to define the major cell types in the present AML cases (Table 2).
Table 2. Immune cell markers used for the cell type annotations in the present application.
The slash (/) indicates that either one or another marker set was used, depending on the cell cluster.
| Cell types | Markers |
| Monocytes | CD14+ |
| T cells | CD3+, CD4+/CD8+ |
| Erythroid-like and erythroid precursor cells |
GYPA+ HBB+, CD71+ |
| Granulocytes | MPO+ |
| Blasts | CD34+/CD34− CD33− CD38_low CD105+ CD99_high |
| NK cells | CD56+ GZMB+/CD335+ GZMB+ |
| Neutrophils | MPO+ AZU1+ |
| Mast cells | FCER1A+ CD203c+ |
Compound sensitivity testing
We used a library consisting of 456 single compounds from the Institute for Molecular Medicine Finland (FIMM) oncology set, which is a comprehensive and evolving compound collection of both approved drugs and investigational compounds (fig. S1). For the single-agent response testing, 20 μl of fresh AML cell (approximately 10,000) suspension in mononuclear cell medium was added per well to predrugged plates with 10-fold dilution series of five concentrations, and the whole-well cell viability was measured with CellTiter-Glo (CTG; Promega) in duplicate, as previously described (19, 50). The concentration ranges were selected for each compound separately to investigate their full dynamic range of the dose-response relationships. After 72 hours of incubation at 37°C and 5% CO2, cell viability of each well was measured using the CTG luminescent assay and a PHERAstar FS (BMG Labtech) plate reader. The percentage inhibition was calculated by normalizing the cell viability to negative control wells containing only 0.1% dimethyl sulfoxide (DMSO) and positive control wells containing 100 μM cell killing benzethonium chloride (BzCl).
The dose-response curve of each drug was fitted using the four-parameter nonlinear logistic regression, also called the Hill equation
where g is a response of a single agent at dose d, a is the minimum asymptote, b is the maximum asymptote, c is the half-maximal inhibitory concentration (relative IC50), and s is the slope of the curve. The fitting was done using the drc package (version, 3.0-1) in R. The maximal relative inhibition level closest to relative IC50 concentration was used as a single-agent response outcome measure when training the machine learning model (fig. S2A). We quantified the summary response of each compound in the patient samples using the DSS, as described previously (63). sDSS was calculated by subtracting from the patient-specific DSS the average of six healthy bone marrow samples’ DSS values (table S1).
Compound-target information
We retrieved comprehensive compound-target information from two publicly available databases: Drug Target Commons v2.0 (drugtargetcommons.fimm.fi) (64) and DGIdb version 3.0 (www.dgidb.org) (65). We included in the compound-target networks only those protein targets with a relatively potent binding affinity (Kd, Ki, IC50 < 1000 nM) from the Drug Target Commons database and protein targets with a reference score >2 in the DGIdb database. Using this rather liberal potency cutoff, we retrieved 898 unique protein targets for the 456 compounds, including both their nominal on-targets and potent off-targets. The predictive modeling made use of the comprehensive compound-target interaction networks, ranging from primary targets to downstream targets and other modifiers of the compounds’ responses to make systematic predictions of their coactivities based on a wide spectrum of binding affinities and inhibitory levels of the compounds against target pathways.
We used the Molecular Signatures Database (MSigDB) v7.2 (66) to map the target pathways for the differential expression analyses (table S2). The visualization of the chemical-compound and protein-protein interactions was performed using the STITCH v5 platform (67), which extracts a wide range of molecular interactions in addition to direct compound-target binding interactions (Fig. 6). However, when making more stringent analysis of the identified co-inhibition effects of the combinations in specific patient samples (Fig. 5), we only focused on the primary targets and those off-targets with similar affinity or inhibition than those of the intended targets, using the curated target binding information from DrugBank (www.drugbank.ca/) (68), IUPHAR (www.guidetopharmacology.org/) (69), and MedChemExpress (www.medchemexpress.com/) (table S3).
Predictive modeling
Because systematic testing of all the potential combinations among the 456 compounds is not experimentally feasible (>100,000 pairwise combinations), we adopted an efficient computational strategy that combines the multiple compound-target expression levels from scRNA-seq data into a single compound-specific target score for each cell using the GSVA approach (20). More specifically, we reduced the dimensionality of the genome-wide scRNA-seq transcriptomic profiles and calculated the target enrichment score for individual cells using GSVA, which calculates the normalized difference in empirical cumulative distribution functions of gene expression ranks inside and outside the drug proteins targets (here, gene set), as an enrichment statistic per cell, which is further normalized by the range of the statistic values across all the gene sets. These compound-target scores were used as input for training a regularized boosted regression trees (XGBoost) algorithm to predict the single-agent viability inhibition (measured as response nearest to the relative IC50 concentration; see fig. S2A) for each compound and each patient separately. The predictive model was optimized using Bayesian optimization with repeated fivefold cross-validation (CV) approach within the 456 compounds in each patient sample separately, and the optimized model was then used to predict the compound combination responses in the particular patient sample. The union of targets of individual compounds was used to construct a combination target score, similar to our previous work (19), and this combination score was used as input for the trained patient-specific XGBoost model for the drug combination response prediction. The ZIP synergy model (22) was used to identify the most synergistic predicted combinations for each patient. To eliminate low-confidence predictions, we used a CP approach (21). In short, CP uses absolute values of the repeated CV residuals as a dependent variable for the error model to predict how unlikely (conformal) each prediction is, which subclusters the prediction space into various confidence level regions. Specifically, we removed all the XGBoost predictions with a nonconformity score lower than 0.8. In doing so, we used in combination predictions only those single agents whose response could be predicted confidently using their target expression levels.
The prediction algorithm was trained on all the leukemic cells (blast and nonblasts), and lymphocytes were considered as nonmalignant “healthy” cells in the current combination predictions. To individually prioritize the most promising and clinically actionable combinations for each patient, the predicted combinations were further ranked on the basis of their predicted toxic effect on nonmalignant cells, along with their predicted capacity to target multiple cancer cell subpopulations. First, we selected the top 10% combination based on the predicted ZIP synergy score. Out of the selected synergistic combinations, we further selected the top 10% combinations with the highest predicted efficacy on the malignant cells. Last, we prioritized the top 10% of the remaining combinations with the lowest target enrichment scores in the nonmalignant cells (i.e., lowest number of cells with positive GSVA values), thus excluding those synergistic and effective combinations that could potentially result in toxic effects. The underlying principle is that the model attempts to select those compounds and combinations, which follow our assumption of their mode of action. Namely, whenever a compound’s target gene set is highly enriched across all cell types, it is expected to have cell type–independent inhibition effects, which should lead to high-CTG ex vivo responses. In contrast, the compounds with low target gene set enrichment in all cell types are assumed to show lower-CTG ex vivo responses. We note that not all the compounds follow this assumption, and those compounds as well as the others that the model is not able to learn properly during Bayesian optimization are removed with the conformal learning. Ultimately, the model is used to predict which combinations will produce the highest cell killing effect, and then we select among the top predictions those with a lower likelihood of toxic effects (e.g., deprioritize combinations whose target gene set is highly enriched in healthy cell types).
Validation of the predicted combination using CTG viability assay
The predicted combinations involving 64 single agents were subsequently tested on the bone marrow mononuclear cells of each patient in an 8 × 8 dose-response matrix using CTG viability assay, similarly as described before (19, 50) and above (see the “Compound sensitivity testing” section). The combination synergy in the experimental validations was quantified using the ZIP model (22), calculated on the basis of the dose region around IC50 values of each compound in combination, separately for each patient and combination, because the machine learning model predicted synergistic inhibition at doses closest to IC50 concentration.
Validation of the combination using high-throughput flow cytometry assay
We performed high-throughput flow cytometry assays to assess the effects of drugs on multiple cell populations in primary AML patient samples, as described previously (41). Briefly, the compounds were dissolved in 100% DMSO or an aqueous solution and dispensed on tissue culture–grade Corning V-bottom 96-well plates using an Echo 550 liquid handler (Labcyte) in four concentrations of 10-fold dilution series around the IC50 (fig. S2B). The conditioned medium suspended cells were seeded at 100,000 cells per well on the drug plates and incubated for 3 days at 37°C and 5% CO2. The cells were washed with a cell staining buffer (phosphate-buffered saline with 2% fetal bovine serum), centrifuged at 600g for 5 min. To profile cell subpopulation responses, the cells were stained with BV605 Mouse Anti-Human CD56, BV421 Mouse Anti-Human CD19, BV421 Mouse Anti-Human CD3, FITC Mouse Anti-Human CD45, APC Mouse Anti-Human CD34, and BV786 Mouse Anti-Human CD38 antibodies (all antibodies from BD Biosciences) for 30 min at room temperature in the dark. After antibody staining, the cells were washed in cell staining buffer and resuspended in 25 μl of annexin V binding buffer containing 0.5 μl of phycoerythrin annexin V and 7-aminoactinomycin D (7AAD) and incubated for 10 min at room temperature in the dark. The cells were analyzed on an iQue Plus instrument (IntelliCyt). The data were analyzed by using FlowJo software v10.6.2 (TreeStar) and collected 50 to 80,000 events. Briefly, cell singlets were identified on the basis of FSC-A (forward-scattered area) versus FSC-H ratio, and live cells were identified using annexin V and 7AAD markers followed by identification of leukocytes on the basis of CD45 and FSC-A. We further characterized NK cells (CD56 positive and CD3/CD19 negative), leukemic stem cells (CD34 and CD38 positive), and T/B cells [based on SSCA (side-scattered area] and CD3/19) from the leukocytes. Details of the gating strategy are illustrated in figs. S4B and S10.
Statistical analysis and data availability
The specific statistical test performed for each experiment and the significance values are included in the figure legends or results text. The R-package (sc-comb) and its source code are freely available at GitHub (https://github.com/IanevskiAleksandr/scComb), with user instructions for installation and usage of the package. The R-package makes it possible to reproduce the current results and modify the codes in applications of the predictive model in other similar studies. The single-compound testing data from the four AML patient samples are available in table S1. The scRNA-seq raw data generated in this study for the AML2 patient and AML3 refractory sample are available at European Genome-phenome Archive (EGA) (EGAS00001004614). scRNA-seq data from AML1 patient and AML3 diagnostic sample are available at EGA (EGAS00001004444) (70).
Acknowledgments
We are grateful to the patients who have donated samples to the study. We thank the FIMM High Throughput Biomedicine and Single-Cell Analytics units supported by HiLIFE and Biocenter Finland for technical support, D. Malani (FIMM) for information about the patient samples, O. Kallioniemi (Science for Life Laboratory, Sweden) for financial support for the molecular and compound profiling, and S. Potluri (Senior Principal Scientist at Pfizer Rinat Laboratories, SF, USA) for the scRNA-seq data generation. Funding: The work was partially funded by the Academy of Finland (grants 310507, 313267, 326238, and 344698 to T.A.); iCAN Digital Precision Cancer Medicine Flagship (grant 1320185 to T.A. and C.A.H.); Cancer Society of Finland (T.A., K.W., and C.A.H.); Sigrid Jusélius Foundation (K.P., T.A., and K.W.); Helse Sør-Øst (grant 2020026 to T.A.); Helsinki University Hospital and Business Finland joint project eCare4Me/Block 2 (BF grant 6767/31/2018 to K.P.); Novo Nordisk Foundation (grant NNF17CC0027852 to K.W.); and Finnish Medical Foundation, Finnish Cancer Institute (M.K.). Author contributions: Study conception: A.I., A.K.G., and T.A. Data generation: J.L., K.K.J., P.S., P.G., L.T., and N.L. Data analysis: A.I., B.R.G., H.K., and A.K.G. Clinical information: M.K. and K.P. Study supervision: C.A.H., P.M., K.W., and T.A. Manuscript writing: A.I., A.K.G., and T.A. Critical review: M.V.-K. and K.W. Competing interests: The senior authors have received collaborative research grants for other projects as listed: K.P., honoraria and research funding from Bristol-Myers Squibb, Celgene, Novartis, and Pfizer. C.A.H., research funding from Celgene, Kronos Bio, Novartis, Oncopeptides, Orion Pharma, Pfizer, and the IMI2 projects HARMONY and HARMONY Plus. K.W., research funding from Novartis and Pfizer. M.K., research funding from AbbVie. The other authors declare that they have no competing interests. Data and materials availability: The scRNA-seq raw data generated in this study for the AML2 patient and AML3 refractory sample are available at EGA (EGAS00001004614). scRNA-seq data from AML1 patient and AML3 diagnostic sample are available at EGA (EGAS00001004444). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabe4038/DC1
REFERENCES AND NOTES
- 1.Cancer Genome Atlas Research Network, Ley T. J., Miller C., Ding L., Raphael B. J., Mungall A. J., Robertson A. G., Hoadley K., Triche T. J. Jr., Laird P. W., Baty J. D., Fulton L. L., Fulton R., Heath S. E., Kalicki-Veizer J., Kandoth C., Klco J. M., Koboldt D. C., Kanchi K.-L., Kulkarni S., Lamprecht T. L., Larson D. E., Lin L., Lu C., McLellan M. D., McMichael J. F., Payton J., Schmidt H., Spencer D. H., Tomasson M. H., Wallis J. W., Wartman L. D., Watson M. A., Welch J., Wendl M. C., Ally A., Balasundaram M., Birol I., Butterfield Y., Chiu R., Chu A., Chuah E., Chun H.-J., Corbett R., Dhalla N., Guin R., He A., Hirst C., Hirst M., Holt R. A., Jones S., Karsan A., Lee D., Li H. I., Marra M. A., Mayo M., Moore R. A., Mungall K., Parker J., Pleasance E., Plettner P., Schein J., Stoll D., Swanson L., Tam A., Thiessen N., Varhol R., Wye N., Zhao Y., Gabriel S., Getz G., Sougnez C., Zou L., Leiserson M. D. M., Vandin F., Wu H.-T., Applebaum F., Baylin S. B., Akbani R., Broom B. M., Chen K., Motter T. C., Nguyen K., Weinstein J. N., Zhang N., Ferguson M. L., Adams C., Black A., Bowen J., Gastier-Foster J., Grossman T., Lichtenberg T., Wise L., Davidsen T., Demchok J. A., Shaw K. R. M., Sheth M., Sofia H. J., Yang L., Downing J. R., Eley G., Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quek L., David M. D., Kennedy A., Metzner M., Amatangelo M., Shih A., Stoilova B., Quivoron C., Heiblig M., Willekens C., Saada V., Alsafadi S., Vijayabaskar M. S., Peniket A., Bernard O. A., Agresta S., Yen K., Beth K. M., Stein E., Vassiliou G. S., Levine R., De Botton S., Thakurta A., Penard-Lacronique V., Vyas P., Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 24, 1167–1177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen A., Hu S., Wang Q.-F., Tumor heterogeneity of acute myeloid leukemia: Insights from single-cell sequencing. Blood Sci. 1, 73–76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Short N. J., Konopleva M., Kadia T. M., Borthakur G., Ravandi F., DiNardo C. D., Daver N., Advances in the treatment of acute myeloid leukemia: New drugs and new challenges. Cancer Discov. 10, 506–525 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Intlekofer A. M., Shih A. H., Wang B., Nazir A., Rustenburg A. S., Albanese S. K., Patel M., Famulare C., Correa F. M., Takemoto N., Durani V., Liu H., Taylor J., Farnoud N., Papaemmanuil E., Cross J. R., Tallman M. S., Arcila M. E., Roshal M., Petsko G. A., Wu B., Choe S., Konteatis Z. D., Biller S. A., Chodera J. D., Thompson C. B., Levine R. L., Stein E. M., Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559, 125–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perl A. E., Martinelli G., Cortes J. E., Neubauer A., Berman E., Paolini S., Montesinos P., Baer M. R., Larson R. A., Ustun C., Fabbiano F., Erba H. P., Stasi A. D., Stuart R., Olin R., Kasner M., Ciceri F., Chou W.-C., Podoltsev N., Recher C., Yokoyama H., Hosono N., Yoon S.-S., Lee J.-H., Pardee T., Fathi A. T., Liu C., Hasabou N., Liu X., Bahceci E., Levis M. J., Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 381, 1728–1740 (2019). [DOI] [PubMed] [Google Scholar]
- 7.McMahon C. M., Ferng T., Canaani J., Wang E. S., Morrissette J. J. D., Eastburn D. J., Pellegrino M., Durruthy-Durruthy R., Watt C. D., Asthana S., Lasater E. A., De Filippis R. A., Peretz C. A. C., McGary L. H. F., Deihimi S., Logan A. C., Luger S. M., Shah N. P., Carroll M., Smith C. C., Perl A. E., Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 9, 1050–1063 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karjalainen R., Pemovska T., Popa M., Liu M., Javarappa K. K., Majumder M. M., Yadav B., Tamborero D., Tang J., Bychkov D., Kontro M., Parsons A., Suvela M., Safont M. M., Porkka K., Aittokallio T., Kallioniemi O., Cormack E. M., Gjertsen B. T., Wennerberg K., Knowles J., Heckman C. A., JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood 130, 789–802 (2017). [DOI] [PubMed] [Google Scholar]
- 9.DiNardo C. D., Pratz K., Pullarkat V., Jonas B. A., Arellano M., Becker P. S., Frankfurt O., Konopleva M., Wei A. H., Kantarjian H. M., Xu T., Hong W.-J., Chyla B., Potluri J., Pollyea D. A., Letai A., Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133, 7–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Witte T., Suciu S., Meert L., Halkes C., Selleslag D., Bron D., Amadori S., Willemze R., Muus P., Baron F., Idarubicin and cytarabine in combination with gemtuzumab ozogamicin (IAGO) for untreated patients with high-risk MDS or AML evolved from MDS: A phase II study from the EORTC and GIMEMA Leukemia Groups (protocol 06013). Ann. Hematol. 94, 1981–1989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giri A. K., Ianevski A., Aittokallio T., Genome-wide off-targets of drugs: Risks and opportunities. Cell Biol. Toxicol. 35, 485–487 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Pützer S., Varghese L., von Jan J., Braun T., Giri A. K., Mayer P., Riet N., Timonen S., Oberbeck S., Kuusanmäki H., Mustjoki S., Stern M.-H., Aittokallio T., Newrzela S., Schrader A., Herling M., Reinstated p53 response and high anti-T-cell leukemia activity by the novel alkylating deacetylase inhibitor tinostamustine. Leukemia 34, 2513–2518 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Cortes J. E., Smith B. D., Wang E. S., Merchant A., Oehler V. G., Arellano M., De Angelo D. J., Pollyea D. A., Sekeres M. A., Robak T., Ma W. W., Zeremski M., Shaik M. N., Laird A. D., O’Connell A., Chan G., Schroeder M. A., Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am. J. Hematol. 93, 1301–1310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei S., Pollyea D. A., Gustafson A., Stevens B. M., Minhajuddin M., Fu R., Riemondy K. A., Gillen A. E., Sheridan R. M., Kim J., Costello J. C., Amaya M. L., Inguva A., Winters A., Ye H., Krug A., Jones C. L., Adane B., Khan N., Ponder J., Schowinsky J., Abbott D., Hammes A., Myers J. R., Ashton J. M., Nemkov T., D’Alessandro A., Gutman J. A., Ramsey H. E., Savona M. R., Smith C. A., Jordan C. T., Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 10, 536–551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li S., Mason C. E., Melnick A., Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr. Opin. Genet. Dev. 36, 100–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassan C., Afshinnekoo E., Li S., Wu S., Mason C. E., Genetic and epigenetic heterogeneity and the impact on cancer relapse. Exp. Hematol. 54, 26–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erba H. P., Finding the optimal combination therapy for the treatment of newly diagnosed AML in older patients unfit for intensive therapy. Leuk. Res. 39, 183–191 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Ianevski A., Timonen S., Kononov A., Aittokallio T., Giri A. K., SynToxProfiler: An interactive analysis of drug combination synergy, toxicity and efficacy. PLOS Comput. Biol. 16, e1007604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L., Tang J., Andersson E. I., Timonen S., Koschmieder S., Wennerberg K., Mustjoki S., Aittokallio T., Patient-customized drug combination prediction and testing for t-cell prolymphocytic leukemia patients. Cancer Res. 78, 2407–2418 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Hänzelmann S., Castelo R., Guinney J., GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shafer G., Vovk V., A tutorial on conformal prediction. J. Mach. Learn. Res. 9, 371–421 (2008). [Google Scholar]
- 22.Yadav B., Wennerberg K., Aittokallio T., Tang J., Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 13, 504–513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Galen P., Hovestadt V., Ii M. H. W., Hughes T. K., Griffin G. K., Battaglia S., Verga J. A., Stephansky J., Pastika T. J., Story J. L., Pinkus G. S., Pozdnyakova O., Galinsky I., Stone R. M., Graubert T. A., Shalek A. K., Aster J. C., Lane A. A., Bernstein B. E., Single-cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell 176, 1265–1281.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hevener K., Verstak T. A., Lutat K. E., Riggsbee D. L., Mooney J. W., Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 8, 844–861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirabilii S., Ricciardi M. R., Allegretti M., Mirabilii S., Licchetta R., Bergamo P., Rinaldo C., Zeuner A., Foà R., Milella M., McCubrey J. A., Martelli A. M., Tafuri A., Co-targeting of Bcl-2 and mTOR pathway triggers synergistic apoptosis in BH3 mimetics resistant acute lymphoblastic leukemia. Oncotarget 6, 32089–32103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurinna S., Konopleva M., Palla S. L., Chen W., Kornblau S., Contractor R., Deng X., May W. S., Andreeff M., Ruvolo P. P., Bcl2 phosphorylation and active PKC α are associated with poor survival in AML. Leukemia 20, 1316–1319 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Ruvolo P. P., Zhou L., Watt J. C., Ruvolo V. R., Burks J. K., Jiffar T., Kornblau S., Konopleva M., Andreeff M., Targeting PKC-mediated signal transduction pathways using enzastaurin to promote apoptosis in acute myeloid leukemia-derived cell lines and blast cells. J. Cell. Biochem. 112, 1696–1707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Chiara G., Marcocci M. E., Torcia M., Lucibello M., Rosini P., Bonini P., Higashimoto Y., Damonte G., Armirotti A., Amodei S., Palamara A. T., Russo T., Garaci E., Cozzolino F., Bcl-2 Phosphorylation by p38 MAPK: Identification of target sites and biologic consequences. J. Biol. Chem. 281, 21353–21361 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Song T., Chai G., Liu Y., Yu X., Wang Z., Zhang Z., Bcl-2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT-737, ABT-263 and ABT-199 by impeding direct binding. Br. J. Pharmacol. 173, 471–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Weerdt I., Hofland T., de Boer R., Dobber J. A., Dubois J., van Nieuwenhuize D., Mobasher M., de Boer F., Hoogendoorn M., Velders G. A., van der Klift M., Remmerswaal E. B. M., Bemelman F. J., Niemann C. U., Kersting S., Levin M.-D., Eldering E., Tonino S. H., Kater A. P., Distinct immune composition in lymph node and peripheral blood of CLL patients is reshaped during venetoclax treatment. Blood Adv. 17, 2642–2652 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ianevski A., Giri A. K., Aittokallio T., Fully-automated cell-type identification with specific markers extracted from single-cell transcriptomic data. bioRxiv, 812131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Jonge H. J. M., Woolthuis C. M., Vos A. Z., Mulder A., van den Berg E., Kluin P. M., van der Weide K., de Bont E. S. J. M., Huls G., Vellenga E., Schuringa J. J., Gene expression profiling in the leukemic stem cell-enriched CD34+ fraction identifies target genes that predict prognosis in normal karyotype AML. Leukemia 25, 1825–1833 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Schoggins J. W., Rice C. M., Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1, 519–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X., Thi V. L. D., Huang Y., Billerbeck E., Saha D., Hoffmann H.-H., Wang Y., Silva L. A. V., Sarbanes S., Sun T., Andrus L., Yu Y., Quirk C., Li M., MacDonald M. R., Schneider W. M., An X., Rosenberg B. R., Rice C. M., Intrinsic immunity shapes viral resistance of stem cells. Cell 172, 423–438.e25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S., Vasudevan S., JNK MAPK regulates IFN-stimulated genes and cell adhesion in chemoresistant, quiescent leukemic cells. bioRxiv, 689570 (2019). [Google Scholar]
- 37.Lamba J. K., Genetic factors influencing cytarabine therapy. Pharmacogenomics 10, 1657–1674 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boss D. S., Schwartz G. K., Middleton M. R., Amakye D. D., Swaisland H., Midgley R. S., Ranson M., Danson S., Calvert H., Plummer R., Morris C., Carvajal R. D., Chirieac L. R., Schellens J. H. M., Shapiro G. I., Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclin-dependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann. Oncol. 21, 884–894 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Acevedo M., Vernier M., Mignacca L., Lessard F., Huot G., Moiseeva O., Bourdeau V., Ferbeyre G., A CDK4/6-dependent epigenetic mechanism protects cancer cells from PML-induced senescence. Cancer Res. 76, 3252–3264 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Taussig D. C., Vargaftig J., Miraki-Moud F., Griessinger E., Sharrock K., Luke T., Lillington D., Oakervee H., Cavenagh J., Agrawal S. G., Lister T. A., Gribben J. G., Bonnet D., Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34− fraction. Blood 115, 1976–1984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuusanmäki H., Leppä A.-M., Pölönen P., Kontro M., Dufva O., Deb D., Yadav B., Brück O., Kumar A., Everaus H., Gjertsen B. T., Heinäniemi M., Porkka K., Mustjoki S., Heckman C. A., Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 105, 708–720 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.VanderWeele D. J., Zhou R., Rudin C. M., Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol. Cancer Ther. 3, 1605–1613 (2004). [PubMed] [Google Scholar]
- 43.Morishita N., Tsukahara H., Chayama K., Ishida T., Washio K., Miyamura T., Yamashita N., Oda M., Morishima T., Activation of Akt is associated with poor prognosis and chemotherapeutic resistance in pediatric B-precursor acute lymphoblastic leukemia. Pediatr. Blood Cancer 59, 83–89 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Chen X., Yang C., Xu Y., Zhou H., Liu H., Qian W., The microtubule depolymerizing agent CYT997 effectively kills acute myeloid leukemia cells via activation of caspases and inhibition of PI3K/Akt/mTOR pathway proteins. Exp. Ther. Med. 6, 299–304 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Decker T., Marschner N., Muendlein A., Welt A., Hagen V., Rauh J., Schröder H., Jaehnig P., Potthoff K., Lerchenmüller C., VicTORia: A randomised phase II study to compare vinorelbine in combination with the mTOR inhibitor everolimus versus vinorelbine monotherapy for second-line chemotherapy in advanced HER2-negative breast cancer. Breast Cancer Res. Treat. 176, 637–647 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Boshuizen J., Peeper D. S., Rational cancer treatment combinations: An urgent clinical need. Mol. Cell 78, 1002–1018 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Zhao B., Pritchard J. R., Lauffenburger D. A., Hemann M. T., Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov. 4, 166–174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim K.-T., Lee H. W., Lee H.-O., Song H. J., Jeong D. E., Shin S., Kim H., Shin Y., Nam D.-H., Jeong B. C., Kirsch D. G., Joo K. M., Park W.-Y., Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 17, 80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anchang B., Davis K. L., Fienberg H. G., Williamson B. D., Bendall S. C., Karacosta L. G., Tibshirani R., Nolan G. P., Plevritis S. K., DRUG-NEM: Optimizing drug combinations using single-cell perturbation response to account for intratumoral heterogeneity. Proc. Natl. Acad. Sci. U.S.A. 115, E4294–E4303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pemovska T., Kontro M., Yadav B., Edgren H., Eldfors S., Szwajda A., Almusa H., Bespalov M. M., Ellonen P., Elonen E., Gjertsen B. T., Karjalainen R., Kulesskiy E., Lagström S., Lehto A., Lepistö M., Lundán T., Majumder M. M., Lopez Marti J. M., Mattila P., Murumägi A., Mustjoki S., Palva A., Parsons A., Pirttinen T., Rämet M. E., Suvela M., Turunen L., Västrik I., Wolf M., Knowles J., Aittokallio T., Heckman C. A., Porkka K., Kallioniemi O., Wennerberg K., Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 3, 1416–1429 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Snijder B., Vladimer G. I., Krall N., Miura K., Schmolke A.-S., Kornauth C., de la Fuente O. L., Choi H.-S., van der Kouwe E., Gültekin S., Kazianka L., Bigenzahn J. W., Hoermann G., Prutsch N., Merkel O., Ringler A., Sabler M., Jeryczynski G., Mayerhoefer M. E., Simonitsch-Klupp I., Ocko K., Felberbauer F., Müllauer L., Prager G. W., Korkmaz B., Kenner L., Sperr W. R., Kralovics R., Gisslinger H., Valent P., Kubicek S., Jäger U., Staber P. B., Superti-Furga G., Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: Interim results from a singlearm, open-label, pilot study. Lancet Haematol. 4, e595–e606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurtz S. E., Eide C. A., Kaempf A., Khanna V., Savage S. L., Rofelty A., English I., Ho H., Pandya R., Bolosky W. J., Poon H., Deininger M. W., Collins R., Swords R. T., Watts J., Pollyea D. A., Medeiros B. C., Traer E., Tognon C. E., Mori M., Druker B. J., Tyner J. W., Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid-derived hematologic malignancies. Proc. Natl. Acad. Sci. U.S.A. 114, E7554–E7563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyner J. W., Tognon C. E., Bottomly D., Wilmot B., Kurtz S. E., Savage S. L., Long N., Schultz A. R., Traer E., Abel M., Agarwal A., Blucher A., Borate U., Bryant J., Burke R., Carlos A., Carpenter R., Carroll J., Chang B. H., Coblentz C., d’Almeida A., Cook R., Danilov A., Dao K.-H. T., Degnin M., Devine D., Dibb J., Edwards D. K. V, Eide C. A., English I., Glover J., Henson R., Ho H., Jemal A., Johnson K., Johnson R., Junio B., Kaempf A., Leonard J., Lin C., Liu S. Q., Lo P., Loriaux M. M., Luty S., Macey T., Maniman J. M., Martinez J., Mori M., Nelson D., Nichols C., Peters J., Ramsdill J., Rofelty A., Schuff R., Searles R., Segerdell E., Smith R. L., Spurgeon S. E., Sweeney T., Thapa A., Visser C., Wagner J., Watanabe-Smith K., Werth K., Wolf J., White L., Yates A., Zhang H., Cogle C. R., Collins R. H., Connolly D. C., Deininger M. W., Drusbosky L., Hourigan C. S., Jordan C. T., Kropf P., Lin T. L., Martinez M. E., Medeiros B. C., Pallapati R. R., Pollyea D. A., Swords R. T., Watts J. M., Weir S. J., Wiest D. L., Winters R. M., McWeeney S. K., Druker B. J., Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spinner M. A., Aleshin A., Santaguida M. T., Schaffert S. A., Zehnder J. L., Patterson A. S., Gekas C., Heiser D., Greenberg P. L., Ex vivo drug screening defines novel drug sensitivity patterns for informing personalized therapy in myeloid neoplasms. Blood Adv. 4, 2768–2778 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Grandis M., Bardin F., Fauriat C., Zemmour C., El-Kaoutari A., Sergé A., Granjeaud S., Pouyet L., Montersino C., Chretien A.-S., Mozziconacci M.-J., Castellano R., Bidaut G., Boher J.-M., Collette Y., Mancini S. J. C., Vey N., Aurrand-Lions M., JAM-C identifies Src family kinase-activated leukemia-initiating cells and predicts poor prognosis in acute myeloid leukemia. Cancer Res. 77, 6627–6640 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Dos Santos C., McDonald T., Ho Y. W., Liu H., Lin A., Forman S. J., Kuo Y. H., Bhatia R., The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood 122, 1900–1913 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yilancioglu K., Cokol M., Design of high-order antibiotic combinations against M. tuberculosis by ranking and exclusion. Sci. Rep. 9, 11876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griner L. A. M., Guha R., Shinn P., Young R. M., Keller J. M., Liu D., Goldlust I. S., Yasgar A., Knight C. M., Boxer M. B., Duveau D. Y., Jiang J.-K., Michael S., Mierzwa T., Huang W., Walsh M. J., Mott B. T., Patel P., Leister W., Maloney D. J., Leclair C. A., Rai G., Jadhav A., Peyser B. D., Austin C. P., Martin S. E., Simeonov A., Ferrer M., Staudt L. M., Thomas C. J., High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 111, 2349–2354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Skok Ž., Zidar N., Kikelj D., Ilaš J., Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J. Med. Chem. 63, 884–904 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Arrowsmith J., Trial watch: Phase II failures: 2008-2010. Nat. Rev. Drug Discov. 10, 328–329 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., Ping Y., Li F., Shi A., Bai J., Zhao T., Li X., Xiao Y., CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franzén O., Gan L.-M., Björkegren J. L. M., PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019, baz046 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yadav B., Pemovska T., Szwajda A., Kulesskiy E., Kontro M., Karjalainen R., Majumder M. M., Malani D., Murumägi A., Knowles J., Porkka K., Heckman C., Kallioniemi O., Wennerberg K., Aittokallio T., Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep. 4, 5193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanoli Z., Alam Z., Vaha-Koskela M., Ravikumar B., Malyutina A., Jaiswal A., Tang J., Wennerberg K., Aittokallio T., Drug Target Commons 2.0: A community platform for systematic analysis of drug-target interaction profiles. Database (Oxford) 2018, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cotto K. C., Wagner A. H., Feng Y.-Y., Kiwala S., Coffman A. C., Spies G., Wollam A., Spies N. C., Griffith O. L., Griffith M., DGIdb 3.0: A redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 46, D1068–D1073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J. P., Tamayo P., The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szklarczyk D., Santos A., von Mering C., Jensen L. J., Bork P., Kuhn M., STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 44, D380–D384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wishart D. S., Feunang Y. D., Guo A. C., Lo E. J., Marcu A., Grant J. R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Knox C., Wilson M., DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Armstrong J. F., Faccenda E., Harding S. D., Pawson A. J., Southan C., Sharman J. L., Campo B., Cavanagh D. R., Alexander S. P. H., Davenport A. P., Spedding M., Davies J. A.; NC-IUPHAR , The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: Extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 48, D1006–D1021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dufva O., Pölönen P., Brück O., Keränen M. A. I., Klievink J., Mehtonen J., Huuhtanen J., Kumar A., Malani D., Siitonen S., Kankainen M., Ghimire B., Lahtela J., Mattila P., Vähä-Koskela M., Wennerberg K., Granberg K., Leivonen S.-K., Meriranta L., Heckman C., Leppä S., Nykter M., Lohi O., Heinäniemi M., Mustjoki S., Immunogenomic landscape of hematological malignancies. Cancer Cell 38, 380–399.e13 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabe4038/DC1