

Mitochondrial energetics is responsive to the one-carbon metabolic network.
Abstract
Induction of the one-carbon cycle is an early hallmark of mitochondrial dysfunction and cancer metabolism. Vital intermediary steps are localized to mitochondria, but it remains unclear how one-carbon availability connects to mitochondrial function. Here, we show that the one-carbon metabolite and methyl group donor S-adenosylmethionine (SAM) is pivotal for energy metabolism. A gradual decline in mitochondrial SAM (mitoSAM) causes hierarchical defects in fly and mouse, comprising loss of mitoSAM-dependent metabolites and impaired assembly of the oxidative phosphorylation system. Complex I stability and iron-sulfur cluster biosynthesis are directly controlled by mitoSAM levels, while other protein targets are predominantly methylated outside of the organelle before import. The mitoSAM pool follows its cytosolic production, establishing mitochondria as responsive receivers of one-carbon units. Thus, we demonstrate that cellular methylation potential is required for energy metabolism, with direct relevance for pathophysiology, aging, and cancer.
INTRODUCTION
The activation of one-carbon units through folate-mediated metabolism is required for the biosynthesis of a wide range of metabolites including purines, thymidylates, and S-adenosylmethionine (SAM). SAM is the principal biological methyl group donor and has major implications in development and disease (1). The abundance of methionine and SAM relates to the epigenetic state of the cell through histone methylation (2) and affects stem cell differentiation (3) and cancer progression (4).
SAM is synthesized in the cytosol from adenosine 5′-triphosphate (ATP) and methionine within the methionine cycle. It is linked to one-carbon metabolism by the remethylation of homocysteine using 5-methyl-tetrahydrofolate. Approximately 30% of total cellular SAM has been reported to reside inside mitochondria (5), where it is a cofactor for ubiquinone [coenzyme Q (CoQ)] and lipoic acid synthesis and for modification of mitochondrial RNA (6, 7). There is no described intramitochondrial source of SAM, and the mitochondrial SAM carrier (SAMC), encoded by SLC25A26, imports cytosolic SAM in exchange for mitochondrial S-adenosylhomocysteine (8). Mutations in SLC25A26 led to severe mitochondrial disease in three patients, associated with a range of mitochondrial defects, including CoQ and lipoic acid deficiency, and respiratory chain dysfunction (9).
A unifying metabolic feature upon mitochondrial insult is an induction of the mitochondrial one-carbon route (10–12). However, the underlying molecular pathways are unclear, and it remains uncertain whether one-carbon metabolism by itself can affect mitochondrial function. Here, we describe that cellular SAM levels control mitochondrial energy metabolism through protein methylation in complex I and iron-sulfur cluster (ISC) biosynthesis.
RESULTS
Acute mitoSAM depletion is lethal through loss of mitoSAM-dependent metabolites
To address the function of mitoSAM, we targeted CG4743 (fig. S1A), the putative Drosophila melanogaster (Dm) ortholog of SLC25A26 and the only known route of SAM into mitochondria. Confocal microscopy confirmed the colocalization of tagged CG4743 with ATP5a, a subunit of the mitochondrial ATP synthase (fig. S1A). To titrate mitochondrial SAM (mitoSAM) levels, we replaced the endogenous genomic locus with a control allele (rescue), a translation-deficient (M1fs;M39*) CG4743 null (null) allele, as well as the Dm equivalents of the previously reported human SLC25A26 mutations: p.A123V, p.I172G, or p.P223L (fig. S1, B and C) (9). All mutations are close to or at the substrate binding site of the carrier (fig. S1D), and proline 223 is part of the highly conserved and symmetric PX[DE]XX[RK] motif required for substrate transport (13). Each mutation impaired SAM flux into mitochondria and lowered mitoSAM levels (Fig. 1A and fig. S1E) compared to the wDah genetic background control.
Fig. 1. Acute mitoSAM depletion is lethal through loss of mitoSAM-dependent metabolites.
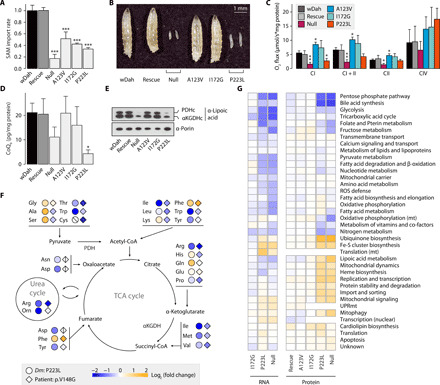
(A) SAM import rates into larval mitochondria (n = 3) relative to the genetic background control wDah. (B) Four-day-old mutant Dm larvae. (C) Mitochondrial oxygen consumption rates in larvae (n = 5) normalized to protein content. (D) CoQ9 levels in larval mitochondria (n = 3) normalized to protein content of input larvae. (E) Immunoblot on mitochondrial lysates from larvae, against lipoylation of pyruvate dehydrogenase (PDHc) and α-ketoglutarate dehydrogenase (αKGDHc) subunit E2, or porin as the loading control. (F) Total amino acid levels in p223l larvae relative to wDah controls (n = 3) or in serum of the previously reported p.V148G patient relative to mean standard values [diamonds; patient 1 in (9)]. Crossed shapes, metabolite not detected. CoA, coenzyme A. TCA, tricarboxylic acid. (G) Mitochondrial subsetting of larval RNA sequencing and proteomics data (n = 5). Expression level relative to wDah controls and vertical hierarchical clustering. UPRmt, mitochondrial unfolded protein response. Bar graphs show means + SD. *P < 0.05, **P < 0.01, and ***P < 0.001 with Dunnett’s test against wDah (A, C, and D). All experiments were performed on 4-day-old Dm larvae.
Only the null and p223l mutations caused larval lethality (Fig. 1B) where we observed respiratory chain dysfunction (Fig. 1C and fig. S1F), markedly reduced CoQ9 steady-state levels (Fig. 1D), and impaired lipoic acid metabolism (Fig. 1E). Targeted metabolomics revealed that amino acids upstream of the lipoic acid–dependent complexes, such as glutamine and glycine, that were accumulated in null and p223l larvae. However, levels of other amino acids were generally depleted, and consistently paralleled levels observed in patient serum (Fig. 1F and data file S1). Differential transcriptomic and proteomic analysis revealed the most prominent compensatory changes in the up-regulation of SAM-dependent pathways CoQ and lipoic acid metabolism, as well as ISC biosynthesis in severely affected mutants, whereas the pentose phosphate pathway and bile acid synthesis were decreased (Fig. 1G). There were no signatures of translational arrest as mitochondrial RNA species and ribosomal proteins were either unchanged or increased (Fig. 1G and fig. S1G), and SAM-dependent modifications on 12S ribosomal RNA (rRNA) were only mildly decreased in null and p223l (fig. S1H). Furthermore, steady-state levels of oxidative phosphorylation (OXPHOS) subunits were unchanged in any of the larval models (Fig. 1G and fig. S1I). Thus, the primary effect of mitoSAM depletion is the loss of metabolites dependent on mitoSAM for synthesis and dysregulation of ISC biosynthesis, resulting in larval lethality.
SAMC is the only mitoSAM carrier and is required for OXPHOS and oxidative tricarboxylic acid (TCA) metabolism
The sensitivity of these pathways to mitoSAM in larvae might be a function of turnover rates and mask further involved processes. To model stable and complete mitoSAM deficiency, we generated a conditional Slc25a26 knockout (KO) mouse model and established Slc25a26−/− (SAMC KO) cell lines from immortalized mouse embryonic fibroblasts (MEFs) (fig. S2, A to C). Hemizygous KO mice (Slc25A26+/−) were viable and fertile, while homozygous disruption (Slc25A26−/−) was embryonically lethal (Fig. 2A). Imaging of mitochondrial ultrastructure revealed swollen organelles with highly irregular cristae and large intramitochondrial vacuoles in SAMC KO cells (fig. S2D). Proteomics showed a profound mitochondrial defect affecting both mitochondrial translation and OXPHOS (fig. S2, E and F) with both nuclear and mitochondrial encoded subunits of complexes I, III, and IV severely decreased (fig. S2, G and H). Complex I, III, and V assembly (Fig. 2B and fig. S2I) and isolated respiratory chain enzyme activities were markedly affected (Fig. 2C), and both mitochondrial rRNA (mt-rRNA) transcripts and protein levels of several mitochondrial ribosome components were reduced, compatible with a mitochondrial translation defect (fig. S2, F and J). Contrary to SAMC null flies, SAM-dependent methylations on 12S rRNA were effectively absent in SAMC KO MEFs (Fig. 2D), demonstrating that SAMC is the only source for SAM in mitochondria.
Fig. 2. SAMC is the only mitoSAM carrier and is required for OXPHOS and oxidative tricarboxylic acid (TCA) metabolism.
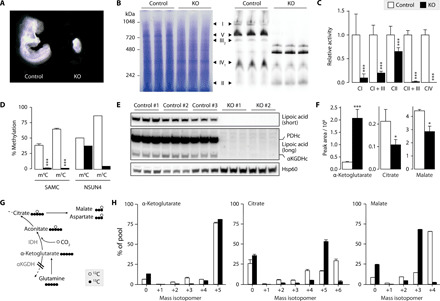
(A) Wild-type (Slc25a26+/+) and Slc25a26 homozygous KO (Slc25A26−/−, KO) embryos at embryonic day 8.5. (B) Blue Native polyacrylamide gel electrophoresis (BN-PAGE) immunoblot on MEF mitochondrial extracts with Coomassie staining (left) and an antibody mix against OXPHOS complex subunits (right) (n = 3). (C) Isolated respiratory chain complex activities (n = 3). (D) Bisulfite pyrosequencing on 12S mt-rRNA from MEFs (n = 3), or control (white) and Nsun4 KO (black) hearts (n = 1), targeting 4′-methylcytosine m4C909 (m4C) or 5′-methylcytosine m5C911 (m5C). (E) Western blot analysis on mitochondrial lysates from MEFs, showing lipoylation of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase E2 subunits. Hsp60 is the loading control. Short and long exposures are shown (n = 3). (F) Intracellular levels of α-ketoglutarate, citrate, and malate in MEFS cultured in 12C medium (n = 3). (G) Schematic of metabolite labeling obtained during reductive carboxylation of glutamine-glutamate–derived α-ketoglutarate. Circles represent 13C (black) and 12C (white) atoms. IDH, isocitrate dehydrogenase. (H) Mass isotopomers of α-ketoglutarate, citrate, and malate in MEFs cultured in 13C medium (n = 3). Bar graphs show means + SD. *P < 0.05 and ***P < 0.001 with two-sided Student’s t test of KO (black) against MEF control (white) cells. (C) and (D) show pooled data from three control and two KO MEF cell lines (n = 3).
Consistent with fly and human data (9), protein lipoylation on pyruvate dehydrogenase and α-ketoglutarate dehydrogenase was not detectable in SAMC KO cells (Fig. 2E), suggesting a disruption of the tricarboxylic acid (TCA) cycle (14). In agreement, α-ketoglutarate levels were increased, while the levels of other TCA cycle related metabolites were decreased (Fig. 2F and fig. S2K). Furthermore, 13C deep labeling (15) of SAMC KO MEFs revealed a metabolite profile that is compatible with disturbed oxidative TCA cycle flux. Although α-ketoglutarate showed nearly full 13C labeling in KO cells, mass isotopomers of citrate and aconitate m+6 and malate and aspartate m+4 were decreased, while citrate and aconitate m+5 and malate and aspartate m+3 were increased (Fig. 2, G and H, and fig. S2L). This is comparable to labeling profiles described during the reductive carboxylation of glutamine-derived α-ketoglutarate (16, 17).
Cytosolic methionine levels coordinate the mitoSAM pool
With SAMC being the only carrier for SAM into mitochondria, we next explored how mitoSAM levels are regulated. MitoSAM levels did not increase upon overexpression of SAMC in larvae, and SAM import was only mildly dependent on mitochondrial membrane potential (Fig. 3, A and B). A p223l intercross, supplemented with different compounds related to mitochondrial function, showed that only methionine and SAM could improve larvae development (Fig. 3, C and D), suggesting that the methionine cycle but not SAMC is rate limiting for physiological mitoSAM levels. This was confirmed by growing larvae on high methionine, which failed to have any beneficial effect on null larvae development despite increased total SAM levels but increased both total and mitoSAM levels in control and p223l larvae (Fig. 3, E and F). Reduced SAM import capacity increased cytosolic SAM levels (Fig. 3G), suggesting that an efficient compensatory down-regulation of the cytosolic methionine cycle does not occur upon acute disruption of SAM import into mitochondria.
Fig. 3. Cytosolic SAM production and not SAMC regulates the mitoSAM pool.
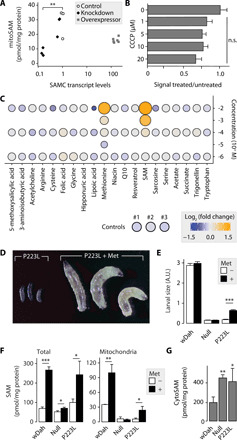
(A) mitoSAM levels upon RNAi knockdown or overexpression of cg4743 with corresponding transcript levels (two knockdown lines, one da-GAL4/+ control line, and two overexpression lines; n = 3). (B) Relative SAM import rates into enriched larval mitochondria upon uncoupling with carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (n = 4). Control is 0 μM CCCP and was treated with dimethyl sulfoxide only. (C) Metabolic rescue of p223l larvae grown on standard yeast food supplemented with metabolites at the given concentration (n ≥ 10). Color indicates log2-transformed fold change of larval size relative to controls, and circle size relates to observed larval size. Controls are larval size means of three independent vials. (D) Six-day-old p223l larvae grown on standard food with or without methionine supplementation. (E) Size quantification of larvae with or without methionine supplementation (n ≥ 10). A.U., arbitrary units. (F) Steady-state levels of SAM in total larval extracts and a mitochondrially enriched fraction with or without methionine supplementation (n = 3). (G) Cytosolic SAM levels (n = 4) in untreated larvae. Bar graphs show means + SD. n.s., not significant. *P < 0.05, **P < 0.01, and ***P < 0.001 with two-sided Student’s t test against wDah controls (E and F) or Dunnett’s test against control (A and B) or wDah (G). (A) to (C) and (E) to (G) performed on 4-day-old larvae. Met, 10 mM methionine.
A validated map of mitochondrial protein methylation
To identify regulatory methylation sites on mitochondrial proteins, we mapped a high-confidence lysine and arginine protein methylome. We developed a methyl stable isotope labeling of amino acids (18) in flies (methyl-SILAF) approach in Dm and combined it with mitochondrial enrichment, tryptic or chymotryptic digestion, and antibody enrichment of methylated peptides (Fig. 4A and fig. S3, A and B). Confidence grading based on mass shift, retention times, and spectral quality resulted in a mitochondrial methyl proteome library with 216 unique modifications on 122 proteins (fig. S3C and data file S2). Most functional categories in mitochondria contained at least one modified protein, with OXPHOS comprising the largest group (Fig. 3B), and no major bias toward any specific category (Fig. 3C). A preferential conservation of methylated over nonmethylated residues (fig. S3D) suggests a structural or functional relevance of mitochondrial protein methylation.
Fig. 4. A methyl-SILAF map of mitochondrial protein methylation.
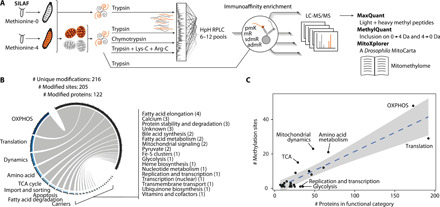
(A) Schematic overview of the workflow to detect the mitochondrial protein methylome. Mitochondrial or total protein extracts of 1:1 mixed unlabeled:labeled larvae were subjected to various protease digestions; in part high-pH reversed-phase liquid chromatography (HpH RPLC); antibody-based peptide enrichments against various l-lysine modifications (pmK) or monomethyl, symmetric, or asymmetric dimethyl l-arginine (mR, sdmR, and admR); liquid chromatography coupled to mass spectrometry; and bioinformatic data processing. (B) Number of modifications, sites, proteins, and their mitochondrial functional categories within the untargeted protein methylation library. (C) Relation of number of methylation events by number of protein members of the respective functional category. Blue dashed line shows the linear regression (R2 = 0.72) with a 99% confidence interval in gray. Functional categories with an absolute standardized residual of more than one are labeled.
Folates have been previously identified as a mitochondrial methyl group donor relevant for mitochondrial transfer RNA maturation (19). This raises the possibility of alternative SAM-independent protein methylation mechanisms. However, using a holidic SILAF food source containing either methionine-[methyl-2H313C] (methionine-4) or serine-[2H213C] (serine-3), labeling SAM- or SAM- and folate-derived groups, respectively, we observed no folate-dependent protein methylation in mitochondria (fig. S3, E to G), highlighting that all detected methyl groups originate from SAM.
We validated a subset of the obtained sites by parallel reaction monitoring (PRM)–based liquid chromatography–tandem mass spectrometry (LC-MS/MS) with synthesized heavy standards (Fig. 5A and data file S3) and confirmed 26 of 27 unique modifications and 26 of 28 unmodified sites (Fig. 5B and fig. S4, A and B) in the fly. Of all identified, modified lysine and arginine residues, 55 and 56%, respectively, are conserved to both human and mouse (Fig. 5C and fig. S4C). Targeting 22 conserved residues in mitochondria from human or mouse fibroblasts revealed extensive conservation of modifications, with 20 and 24 specific modifications confirmed, respectively (Fig. 5D and fig. S5, A to D). We observed spectra for 15 and 21 corresponding unmodified peptides in human and mouse (Fig. 5D), respectively, further suggesting that most proteins are present in both states.
Fig. 5. The mitochondrial methylome is highly conserved from fly to mouse and human.
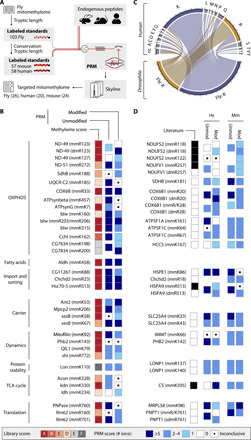
(A) Schematic workflow to confirm the untargeted mitochondrial fly methylome in three species by targeted PRM-based LC-MS/MS. Heavy isotope-labeled synthesized peptide standards were spiked into endogenous peptide samples. (B) Confidence scoring of selected methyl groups targeted with synthesized standards in Dm. (C) Conservation status of 203 methylated Dm residues in human protein homologs. nc, nonconserved amino acid. major ticks are 10, and minor ticks are 2 residues. (D) Scores of conserved residues in human (Hs) and mouse (Mm) fibroblasts as in (C). Listing in PhosphoSitePlus (68) as black squares in the literature column. The best score of at least three experiments is shown in (B) and (D). unmod, unmodified; mod, modified.
ISC assembly is sensitive to mitoSAM loss
We then combined our methyl library with p223l larvae transcriptomics and proteomics and the known interactome (fig. S6A) and observed an enrichment of methylated proteins involved in ISC biosynthesis, namely, mitochondrial cysteine desulfurase Nfs1 (DmNfs1) and ferredoxin 2 (Fdx2) (Fig. 6A). In support for a defect in iron metabolism and ISC biosynthesis in a mitoSAM-depleted state, mitochondrial aconitase activity, a [4Fe–4S] cluster containing enzyme, was decreased to ~50% in null and p223l larvae (Fig. 6B). Moreover, supplementation with CoQ alone did not rescue respiration deficiency in p223l larvae (fig. S6B). We further observed electron-dense inclusions in transmission electron microscopy (TEM) micrographs of null and p223l but not in mitochondria from the milder affected i172g larvae (Fig. 6C), as described in patients with mutations in ISC genes (20). This was corroborated by an increase in total iron content in null and p223l larvae and transcriptional up-regulation of the ISC machinery in SAMC KO MEFs (Fig. 6D and fig. S6C). Nfs1 is a cysteine desulfurase central to the initial stages of ISC biosynthesis (21). The DmNfs1 methylated residue R77 is highly conserved (fig. S6D), and a missense mutation affecting the corresponding human residue HsR72 has been associated with infantile mitochondrial disease (22). HsR72 is a critical residue interacting with the protein ISD11 (23) and in proximity to an integral acyl-chain 4′-phosphopantethiene (4-PPE; Fig. 6E). Hydrophobicity modeling revealed a highly hydrophobic ISD11 pocket around 4-PPE with the prominent exception of R72 of HsNFS1 (human NFS1) (fig. S6E), suggesting that methylation might be required for stable complex formation. Overexpression of the methylated mimetic DmNfs1.R77F, but not the unmethylated mimetic DmNfs1.R77K in fly decreased total iron content (Fig. 6F and fig. S6F). Furthermore, pull-down of flagged mutant DmNfs1 showed a differential enrichment profile, with ISC containing proteins ND-23 and ND-51 coprecipitating with DmNfs1.R77F, while the iron chelatase ferritin 1 heavy chain (Fer1HCH) was enriched with DmNfs1.R77K (Fig. 6G). Likewise, overexpression of the unmethylated mimetic HsNFS1.R72K in cells altered the profile of HsNFS1 containing complexes (Fig. 6H and fig. S6G) and coimmunoprecipitating proteins (fig. S6H). The distinct interactors observed are consistent with a dual function for Nfs1, as proposed in yeast (24).
Fig. 6. ISC assembly is sensitive to mitoSAM loss.
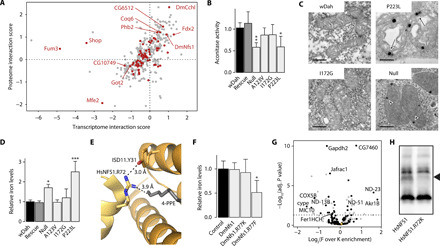
(A) Interaction score of p223l transcriptomics and proteomics. Methylated proteins are shown in dark red. Gene names to methylated targets with an absolute interaction score of >1 in at least one dataset. (B) Mitochondrial aconitase activity (n = 4). (C) Transmission electron micrographs. Arrows indicate suspected iron deposits in null and p223l mitochondria. Scale bar, 0.5 μm. (D) Total cellular iron levels in 4-day-old Dm larvae (n = 4). (E) Interaction site of R72 in HsNFS1 with ISD11 and 4-PPE in gray [Protein Data Bank (PDB): 5USR]. (F) Cellular iron levels in fly models overexpressing DmNfs1 constructs (n = 3). Control is da-GAL4/+. (G) Immunoenrichment of DmNfs1.R77F-FLAG over DmNfs1.R77K-FLAG (n = 4). Black dots, mitochondrial; orange dot, DmNfs1. Significant proteins with known relation to iron metabolism or OXPHOS are labeled. (H) BN-PAGE immunoblot with an antibody against NFS1 in cells overexpressing HsNFS1-FLAG constructs (representative image, n = 3). The missing band in HsNFS1.R72K is marked. Graphs show means + SD. *P < 0.05, **P < 0.01, and ***P < 0.001 with Dunnett’s test against wDah (B and C) or control (F) or false discovery adjusted P value (G). (A) to (D), (F), and (G) performed on 4-day-old larvae.
Reduced complex I methylation is linked to decreased life span
Given that the cytosolic production of SAM directly guides the mitoSAM pool, we asked whether the availability of mitochondrial one-carbon units can control mitochondrial function. Mining codependencies within the Cancer Dependency Map data (25), we found that genes related to mitochondrial translation and OXPHOS correlated highly with SLC25A26 essentiality in 769 cancer cell lines (Fig. 7A). In conjunction with the protein methylome, we identified the complex I subunit gene NDUFS2 and the corresponding methyltransferase NDUFAF7 ranked within the top 10 codepending OXPHOS candidates and highest among complex I subunits.
Fig. 7. Reduced complex I methylation causes premature ageing.
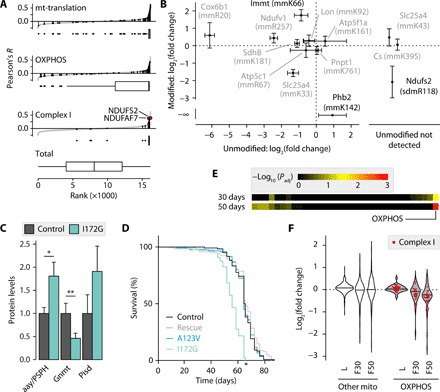
(A) Codependency of SLC25A26 with genes of indicated categories in cancer cell lines as Pearson’s correlation of CERES scores summarized in box plots with median, upper and lower quartile, and 1.5× interquartile range as whiskers. (B) Modification status of the unmodified/modified endogenous peptides in SAMC KO MEFs versus controls relative to standards (n = 3). Significantly changed modified peptide in black. Targets in the right box without quantifiable unmodified peptides. (C) Proteomic levels of enzymes in serine biosynthesis and SAM metabolism in 4-day-old larvae (n = 5). (D) Fly survival curve, one of two experiments shown. (E) Adjusted P values of functional categories in female i172g fly proteomes at 30 or 50 days (n = 3). Significant labels (P < 0.05) are shown. (F) Proteomic changes in i172g insects [n = 5 for larvae (L), n = 3 for flies (F)] as violin plots with median as horizontal line. Graphs show means ±/+ SD. *P < 0.05 and **P < 0.01 with Bonferroni-adjusted P values (C and E) or log-rank test (D).
We then quantified the methylation status of 11 proteins in SAMC null larvae and KO MEFs with targeted probes. While we did not detect any significant changes in null larvae (fig. S7A), the stable MEF KO model revealed a significant decrease in the mitoSAM-dependent dimethylation of R118 of NDUFS2 and monomethylation of K142 on Phb2 (Fig. 7B). The remaining analyzed sites in MEF KO cells were largely unaffected. Evidently, methylation can occur independent of the mitoSAM pool. In support for this notion, we observed an increased density of modifications within the cleavable N-terminal mitochondrial target signal (fig. S7B) across our methyl library, suggesting that the majority of mitochondrial proteins are posttranslationally modified before import.
R118 on NDUFS2 is symmetrically dimethylated by the intra-mitoSAM–dependent methyltransferase NDUFAF7, and knockdown of the transferase has previously been shown to result in loss of adjacent complex I subunits (26). The methylation is essential for complex I assembly and inaccessible to potential demethylation once modified NDUFS2 is integrated into the Q module of the matrix arm. To test whether long-term mild mitoSAM reduction would affect complex I stability, we turned to the mild i172g fly model. Despite normal-sized larvae and unaffected enclosure rates, we observed protein changes related to one-carbon metabolism before pupation (Fig. 7C and fig. S7C). Young a123v and i172g flies showed a mild trend toward reduced climbing efficiency, and comparative life-span analysis revealed that i172g female flies died prematurely (Fig. 7D and fig. S7D). Methylation on 12S rRNA and OXPHOS function were intact in aged i172g flies despite mildly decreased mitoSAM levels (fig. S6, E to H). A proteomic time course at 30 and 50 days of age exposed a progressive loss of OXPHOS subunits (Fig. 7, E and F), predominantly related to complex I. Specifically, we observed a reduction of proteins that physically interact with DmND-49/NDUFS2 (NADH:ubiquinone oxidoreductase core subunit S2), prominently NADH dehydrogenase subunit 1 (ND1) (fig. S7, I and J) that also becomes lost in a human cell model upon NDUFAF7 suppression (26). Thus, we conclude that the one-carbon cycle can control mitochondrial function, via mitoSAM-dependent NDUFS2 methylation and ISC biosynthesis.
DISCUSSION
The SLC25 family is almost exclusively responsible for the transport of metabolites, including amino acids, fatty acids, cofactors, inorganic ions, and nucleotides across the inner mitochondrial membrane. The substrate of around one third of SLC25 carriers has not been identified, and selected metabolites are known to be transported by more than one carrier (27). Here, we identified cg4743 as the ortholog of SLC25A26 in Dm and its deletion in both flies and mice established SAMC as the only route for SAM into mitochondria, with no significant compensatory contribution of mitoSAM via other processes.
Our work demonstrates that mitoSAM levels are determined by the cytosolic methionine cycle and that viable mitoSAM levels have a tight threshold. Severe mitoSAM deficiency first affected the synthesis of ISCs and metabolites, such as ubiquinone and lipoic acid, while mt-rRNA and protein methylations were not affected, suggesting either a hierarchical order for mitoSAM-dependent methyltransferases through different enzyme affinity rates to SAM or alternate turnover rates. Prolonged depletion of mitoSAM, both in vivo and in culture, revealed additional mitochondrial dysfunctions. A moderate reduction in SAM flux into mitochondria in flies led to a shortened life span and progressive depletion of several complex I subunits adjacent to the intramitochondrially methylated NDUFS2. In contrast, SAMC KO MEFs had an almost entirely dissociated OXPHOS system and a remodeled TCA cycle consistent with induction of reductive TCA cycle metabolism, suggesting that the effect of reduced SAM import on mitochondrial processes is dependent on the severity and duration of the depletion.
This can be of pathological relevance upon alterations in folate-mediated one-carbon metabolism, as has been described in cardiovascular and liver disease and several cancer types (1, 28). An up-regulation of the one-carbon cycle was also observed as an early response to mitochondrial dysfunction (10–12). Our results suggest that mitochondrial function is sensitive to one-carbon cycle activity and benefits from adequate SAM levels. Methionine and SAM levels have been connected to the regulation of the epigenome (28, 29). While our data signifies relevance to disease, whether fine-tuning of mitochondrial function occurs through a comparable mechanism at physiological SAM levels needs further investigation.
Few mitochondrial protein methylation sites had been validated by targeted or molecular techniques (26, 30). Our SILAF approach, in combination with targeted proteomics and genetic models, indicates that the mitochondrial protein methylome consists of at least 205 lysine and arginine modifications on 122 proteins in Dm and a large proportion of residues are conserved to the human proteome. All detected methylation events were dependent on SAM and not folate species, and we find that only two of the analyzed protein methyl sites depend on the mitoSAM pool. This strengthens previous reports suggesting a role of cytosolic arginine methyltransferases for mitochondrial protein methylation (31). Our screen identifies a number of methylation events on proteins in the intermembrane space (IMS), such as COX6B1 (cytochrome c oxidase subunit 6B1) or CHCHD2 (coiled-coil-helix-coiled-coil-helix domain containing 2). No IMS-localized protein methyltransferases have been reported in the IMS proteome (32), implying that these too are methylated outside of mitochondria. Conversely, how polypeptide chains can be recognized and methylated before mitochondrial import and whether the abundant methylation within the mitochondrial target sequence serves a localization purpose are unknown. We identified methylation events on residues previously associated with mitochondrial disease, including at least R33 of COX6B1, K33 of ANT1, (adenine nucleotide translocase 1), and R72 of NFS1 (22, 33, 34), and our data suggest that lack of methylation of these residues might be part of disease etiology.
In summary, we demonstrate that import through SAMC is the only source for mitoSAM and that mitochondrial processes show differential sensitivity to mitoSAM depletion and methionine cycle activity. As mitoSAM levels are directly dependent on cytosolic SAM generated in the methionine cycle, secondary mitochondrial dysfunction can be predicted to arise as a consequence of multiple disease states, including nutrient deficiencies, cardiovascular disease, and cancer. Our results add mitochondria to the downstream effectors of one-carbon metabolism and open up new possibilities for mitochondrial protein regulation through posttranslational modifications.
MATERIALS AND METHODS
Materials
Antibodies were purchased from the following sources: NDUFA9 (ab14713) OXPHOS Rodent Wb Cocktail (ab110413), VDAC1 (ab14734), APT5a (ab14748), and DDDDK (ab1257) from Abcam; lipoic acid (437695) from Calbiochem; HSP60 (ADI-SPA-807) from Enzo Life Sciences; and NFS1 (GTX33356) from GeneTex. Dm FLAG-tagged proteins were enriched with the FLAG Immunoprecipitation Kit (Millipore, FLAGIPT1), and FLAG-tagged proteins expressed in human embryonic kidney (HEK) 293T cells were enriched on FLAG M2 magnetic beads (Millipore, M8823). The bacterial artificial chromosome (BAC) to extract the DmSAMC genomic region was obtained from BACPAC Genomics (RP98-9D8). l-Methionine-[methyl-13C,2H2] (299154) and l-serine-13C,15N,2,2,3-2H3 (749877) were purchased from Sigma-Aldrich. The metabolomics standard SAMd3 (D-4093) was purchased from CDN Isotopes, and tritiated SAM-[methyl-2H] at 250 μCi (NET155V250UC) was from PerkinElmer. Proteases were Pierce Trypsin, MS-grade (90059), Pierce Chymotrypsin, MS-grade (90056), and Pierce Lys-C, MS-grade (90051) from Thermo Fisher Scientific and Arg-C, sequencing grade (V1881) from Promega. Methylated peptides were enriched with the PTMScan Mono-Methyl Arginine Motif [mme-RG] (12235), Asymmetric Di-Methyl Arginine Motif [adme-R] (13474), Symmetric Di-Methyl Arginine Motif [sdme-R] (13563), and Pan-Methyl Lysine (14809) kits from Cell Signaling Technology. RNA for sequencing was prepared with a miRNeasy Mini kit from QIAGEN (217004). Pyro-bisulfite sequencing on 12S rRNA involved bisulfite conversion with the EZ RNA Methylation Kit (Zymo Research, R5001), reverse transcription with the QIAGEN PyroMark RT kit (978801), and sequencing on a PyroMark Q24 system (9001514). Plasmids used in this study were pGX-attP (GenBank, FJ791035), pGE-attB-GMR (FH791037), pUAST-attB (EF362409), and pBSSVD2005 (Addgene, 21826). Dm genetic backgrounds were wDah or w1118 in-house stocks. Fly lines obtained from the Vienna Drosophila Resource Center were the CG4743 RNAi lines 9487 and 103950 (35).
Generation of genetically modified flies
KO and knockin fly models were generated as previously described (36, 37). Briefly, two homologous 500–base pair (bp) regions 5′ and 3′ of the CG4743 target region were sequenced and cloned into a pGX-attP vector. The donor template was injected into w118 fly embryos and randomly integrated into the fly genome via P element–mediated germ line transformation using the BestGene Drosophila Embryo Injection Services (Chino Hills, CA, USA). w+ chromosome II transformants were crossed with hs-FLP, hs-I-SceI flies (BL#6934) and heat-shocked at 38°C for 1 hour. Female chimeric offspring was individually crossed to TM3, Sb Ser/TM6B-Tb balancer flies, and a mapping was cross revealed chromosome III insertion. Positive CG4743 KO flies were PCR-screened and sequenced, and a Southern blot was performed. For knockin, the w+ marker in KO flies was first removed by crossing to a Cre expressing fly line (BL#766). A sequenced construct with wild-type or mutant CG4743 including introns and 150-bp 5′ untranslated region was cloned into pGE-GMR-attB and injected with PhiC31 into hemizygous w− CG4743-KO embryos at BestGene. Positive transformants were identified by w+ and backcrossed into a wDah background.
Flies overexpressing CG4743, CG4743-FLAG, NFS1, NFS1-FLAG, NFS1.R77K-FLAG, or NFS.R77F-FLAG were generated by cloning a sequenced cDNA construct into pUAST-attB. The vector was injected together with PhiC31 into y[1] w[67c23]; P{y[+t7.7] = CaryP}attP1 (BL#8621) with a chromosome II attP-landing site. UAS-CG4743 w+ transformants were backcrossed, all others were analyzed relative to w1118 genetic background.
Fly husbandry
All fly lines were maintained, and experiments were performed at 25°C and 60% humidity on a 12-hour:12-hour light:dark cycle and fed on a standard yeast-sugar-agar medium, unless stated differently. All CG4743 fly stocks were backcrossed for at least six generations into the white Dahomey Wolbachia–free background (wDah). The p223l and null parental stocks were kept heterozygous balanced over TM6B-Tb GFP. For experimental crosses, flies were put to lay for 8 hours on grape juice plates with yeast paste. Eggs were rinsed in phosphate-buffered saline (PBS) (pH 7.4) and spread onto standard medium vials. All experiments on larval tissue were performed at 4 days after egg lay, for which larvae were manually picked. Homozygosity was validated by the absence of green fluorescent protein signal under a fluorescent microscope for every p223l and null sample, whereas control, rescue, a123v, and i172g were kept as homozygous stocks. UAS constructs were induced by crossings to a daughterless-GAL4 (;;da-GAL4) driver line.
Mouse husbandry and tissue culture
Slc25a26 conditional KO mice were generated by Taconic Biosciences (Germany). The targeting strategy is based on National Center for Biotechnology Information (NCBI) transcript NM_026255.5. Exon 3 of Slc25a26 was flanked with loxP sites. A puromycin resistance (PuroR) positive selection marker flanked by FRT (flippase recognition target) sites was inserted into intron 2. The targeting vector was generated using BAC clones from the C57BL/6J RPCI-23 BAC library and transfected into the Taconic Biosciences C57BL/6N Tac ES cell line. Homologous recombinant clones were isolated using the positive (PuroR) and negative (thymidine kinase) selections. The conditional KO allele was obtained following in vivo Flp-mediated removal of the positive selection marker. All mice had an inbred C57BL/6N background. Mice were housed in standard individually ventilated cages with a 12-hour:12-hour light:dark cycle under controlled environmental conditions. Mice were fed ad libitum with standard CRM mouse food (Special Diet Services). Animal studies were approved by the local animal welfare ethics committee and performed in compliance with the national and European law. Heterozygous KO (Slc25a26+/−) mice were obtained by crossing Slc25a26loxP/loxP mice with transgenic mice ubiquitously expressing Cre recombinase (+/β-actin cre).
MEFs were derived from embyonic day 13.5 embryos from intercrossed Slc25a26loxp/loxp mice. Embryos were dissected, and dissociated cells were plated in MEF medium comprising Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose, GlutaMAX (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), and 1% penicillin/streptomycin (Thermo Fisher Scientific). MEFs were grown at 37°C and 5% CO2. MEFs were immortalized by transient expression of SV40 large T antigen (Addgene, plasmid #21826). Immortalized Slc25a26−/− cells were obtained by transiently expressing Cre-recombinase (38). Transfected cells were serially diluted to separate single cells, and subsequent clones were screened by PCR analysis. Slc25a26loxP/loxP (n = 3) and Slc25a26−/− (n = 2) clones were isolated from the same process, and medium was supplemented with uridine (50 μg/ml; Sigma-Aldrich).
Generation of stable HEK cell lines
HsNFS1-FLAG and HsNFS1.R72K-FLAG cDNA sequences (GenScript, custom order) were cloned into the retroviral pBABE-puromycin vector using the Gateway cloning system (Thermo Fisher Scientific). Phoenix Amphotropic cells were transfected with the pBABE constructs using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Retrovirus containing medium was passed through a 0.45-μm filter (Sarstedt) and added to HEK293T cells after addition of polybrene (4 μg/ml). Stably transduced cells were cultured with DMEM high glucose, GlutaMAX (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), and 1% penicillin/streptomycin (Thermo Fisher Scientific) and maintained under puromycin selection (1.5 μg/ml).
Hatching rates
Flies were put to lay for 4 hours, and 100 eggs per replica were removed and transferred with a sterile needle onto standard yeast food, with 10 replicates performed per genotype. Fly hatching was monitored at 12-hour intervals.
Life-span analysis
Flies hatching within 12 hours from tubes with controlled larval density were transferred into new tubes for 2 days, before separating into batches of 20 males and 20 females per replica. Vials were kept horizontally in random order. Survival and dropout were counted, and tubes were changed every 2 to 3 days.
Climbing assay
Twenty-five male flies were picked 2 days after hatching and allowed to recover from anesthesia for 24 hours. They were then transferred randomly into 20-cm climbing vials and allowed to acclimatize for 1 hour. Video recording was initiated from the moment of banging the vials mildly onto the table surface. Videos were evaluated blindly, and the number of flies that climbed above 10 cm within 10 s was counted and represented relative to the total number.
Metabolic rescue
Two hundred millimolars of stock solutions of metabolites were diluted and mixed at a 1:20 ratio in 6 ml of liquid standard yeast food. In cases of low solubility, high concentrations were achieved by mixing appropriate quantities of powder into liquid food. Heterozygous p223l eggs were spread onto vials and allowed to grow for 4 days at standard conditions. At least 10 homozygous larvae were photographed under a stereomicroscope at fixed magnification including a size marker. Pictures were blinded, and the area of larvae was assessed with ImageJ 1.52q.
In vivo labeling—Methyl-SILAF and serine-SILAF
For large-scale methyl-SILAF, holidic food was prepared as previously described (39–41) in the FLYAA composition for the light fraction or by substituting l-methionine with equimolar amounts of l-methionine-[methyl-13C,2H3] (Sigma-Aldrich). Serine labeling was achieved by substituting l-serine with equimolar amounts of l-serine–13C3,15N,2,2,3-2H3 (Sigma-Aldrich). Larvae were grown for 7 days with standard conditions before mitochondrial isolation.
Mitochondrial preparations
For enrichment of Dm mitochondria, larvae were picked as described above and homogenized with a Teflon-coated Dounce homogenizer (14 strokes at 700 rpm) on ice in STE-enrichment buffer [250 mM sucrose, 5 mM tris, and 2 mM EGTA (pH 7.4)] with 5% bovine serum albumin (BSA). Detritus and large cellular components were removed by centrifugation at 1000g for 10 min at 4°C, and mitochondria were enriched through differential centrifugation twice at 3000g for 10 min and in STE buffer without BSA at 7000g for 10 min.
For the mitochondrial protein methylome, mitochondria were further purified on a Percoll gradient by layering 3 mg on 8 ml of 20% Percoll in STE buffer and centrifuging for 30 min at 40,000g. The mitochondrial fraction was removed, washed in four volumes of STE, and quantified with a Qubit protein assay.
For enrichment of mitochondria from MEFs, harvested cell pellets were resuspended in 1 ml of ice-cold mitochondria isolation buffer [MIB; 320 mM sucrose, 1 mM EDTA, and 20 mM tris-HCl (pH 7.2)] with 0.1% BSA, transferred to a glass homogenizer, and homogenized with 60 strokes. The homogenate was centrifuged at 1000g for 5 min at 4°C, and the supernatant was collected. The pellet was resuspended in 0.5 ml of MIB with 0.1% BSA, transferred to a glass homogenizer, and homogenized with 20 strokes. The homogenate was centrifuged at 1000g for 5 min, and the supernatants were combined. Mitochondria were enriched by differential centrifugation, twice at 12,000g for 10 min and in MIB without BSA at 12,000g for 10 min.
Mitochondrial respiration
Mitochondrial respiration was assessed in dissected larvae or thoraces from adult flies at 25°C using an Oxygraph-2K respirometer (Oroboros). Samples were resuspended in respiratory buffer [0.5 mM EGTA, 3 mM MgCl2·6H2O, 60 mM lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM Hepes, 110 mM sucrose, and BSA (1 mg/ml) (pH 7.1)] and permeabilized with digitonin. Complex I–dependent respiration was assessed by the addition of pyruvate (5 mM), glutamate (10 mM), and malate (2 mM), followed by the addition of adenosine 5′-diphosphate (2.5 mM). Succinate (10 mM) was added to determine complex I + II–dependent respiration, and then rotenone (0.5 μM) was added to determine complex II–dependent respiration. Nonmitochondrial respiration was determined by the addition of antimycin A (2.5 μM). Last, complex IV activity was assessed by the addition of N,N,N’,N’-tetramethyl-p-phenylenediamine dihydrochloride (TMPD) (0.5 mM) and ascorbate (2 mM), followed by the addition of potassium cyanide (1 mM). Oxygen consumption rate was normalized to protein content measured by the bicinchoninic acid (BCA) assay.
Isolated respiratory chain complex activities
The respiratory chain enzyme activities of complexes I and III NADH:cytochrome c reductase, complex I (NADH:CoQ reductase), complexes II and III (succinate:cytochrome c reductase), complex IV (COX), and citrate synthase (CS) were determined as described (42). The respiratory chain enzyme and CS activities were determined at 35°C using standard methods on an Indiko automated photometer (Thermo Fisher Scientific). All activities were expressed as units per unit of CS activity in the mitochondrial suspension.
Microscopy
Adult flight muscles were rapidly dissected in PBS, fixed for 10 min in 4% formaldehyde, and washed for 5 min. For immunolabeling, tissues were dissected in PBS, permeabilized for 2 hours in 0.5% Triton X-100 in PBS, and saturated for 1 hour in 0.5% BSA and 0.3% Tween 20 in PBS (PBTB). Primary antibody diluted in PBTB was incubated overnight at 4°C and later washed for 1 hour in 0.1% Tween 20 in PBS (PBTW). Secondary antibody was incubated for 2 hours and washed for 1 hour in PBTW. Preparations were mounted in VECTASHIELD/4′,6-diamidino-2-phenylindole. An LSM880 Zeiss confocal microscope was used for imaging.
Transmission electron microscopy
Harvested MEFs were resuspended and fixed in 2.5% glutaraldehyde in phosphate buffer (0.1 M) (pH 7.4) and stored at 4°C. Dissected Dm larvae were fixed in 2.5% glutaraldehyde and 1% paraformaldehyde in phosphate buffer and stored at 4°C. Following the primary fixation, the samples were rinsed with phosphate buffer, followed by postfixation in 2% osmium tetroxide in phosphate buffer at 4°C for 2 hours. The samples were then subjected to stepwise ethanol dehydration followed by stepwise acetone/LX-112 infiltration and lastly embedded in LX-112. Ultrathin sections (approximately 60 to 80 nm) were prepared using an EM UC7 ultramicrotome (Leica Microsystems). The ultrathin sections were contrasted with uranyl acetate followed by Reynolds lead citrate and examined in a Tecnai Spirit BioTWIN TEM (Thermo Fisher Scientific) operated at 100 kV. Digital images were acquired using a 2kx2k Veleta CCD camera (EMSIS GmbH).
DNA isolation
Genomic and mitochondrial DNA was isolated using the DNeasy Blood and Tissue Kit (QIAGEN) according to the manufacturer’s instructions. DNA concentration was determined with the Qubit dsDNA BR assay.
RNA isolation
RNA was isolated using a ToTALLY RNA kit (Ambion) or TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Isolated RNA was treated with a TURBO DNA-free kit (Thermo Fisher Scientific). RNA concentration was determined with the Qubit RNA HS assay.
Qualitative reverse transcription PCR
Reverse transcription using 2 μg of RNA was performed for cDNA synthesis with the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Qualitative reverse transcription PCR was performed on a on a QuantStudio 6 system using TaqMan probes and TaqMan Universal Master Mix II, with uracil-N-glycosylase (UNG) (Thermo Fisher Scientific) or with gene-specific primers and Platinum SYBR Green qPCR SuperMix-UDG. As a loading control, rpl32 was used in flies and 18S in mouse. TaqMan probes and gene-specific primers are listed in data file S3.
RNA sequencing
Dm RNA was extracted, and on-column deoxyribonuclease digested with an miRNeasy Mini kit (QIAGEN). RNA integrity was certified on a Bioanalyzer (Agilent Technologies). Two micrograms of RNA was subjected to 2× 75-bp Ribo-Zero RNA sequencing at a depth of 3 million reads per sample on a HiSeq4000.
Bisulfite pyrosequencing
One microgram of RNA per sample was bisulfite converted with the EZ RNA Methylation Kit (Zymo Research) and transcribed into cDNA and PCR amplified with a PyroMark RT kit (QIAGEN) using an optimized pair of one biotinylated and one unmodified primer. cDNA amplicons were enriched with streptavidin-coupled sepharose beads before following the manufacturer’s recommendations for pyrosequencing.
Aconitase activity
Mitochondrial aconitase activity was measured in larvae using an Aconitase Activity Assay kit (Sigma-Aldrich) according to the manufacturer’s instructions. To normalize, protein content was determined in the crude mitochondrial pellet by the BCA assay, and the assay was performed with equal sample input.
Western blot analysis
Crude mitochondrial or total protein lysates were resuspended in the NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) and loaded onto precast NuPAGE 4 to 12% bis-tris gels (Thermo Fisher Scientific) in an XCell SureLock electrophoresis system (Thermo Fisher Scientific). Following electrophoresis, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and blocked in 5% (w/v) milk in tris buffer saline with 1% (v/v) Tween 20 (TBST; Sigma-Aldrich). Immunoblotting was performed following standard techniques and developed using Clarity Western ECL substrate (Bio-Rad).
Blue Native polyacrylamide gel electrophoresis
Fifty micrograms of fly mitochondria per lane was pelleted, resuspended in lysis buffer [1% digitonin, 0.1 mM EDTA, 50 mM NaCl, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 20 mM tris-HCl (pH 7.4)], and incubated for 15 min on ice. Undissolved material was removed by centrifugation, and the supernatant with 10× sample buffer [5% Brilliant Blue G, 10-mm bis-tris (pH 7.0), and 500 mM aminocaproic acid] was loaded onto a 4 to10% polyacrylamide gel with 4% stacking. In-gel complex activities and loading by Coomassie staining were assessed at room temperature. Complex I was determined after incubation in iodonitrotetrazolium chloride (2.5 mg/ml), 2 mM tris-HCl (pH 7.4), and NADH (0.1 mg/ml). Complex IV activity was determined after incubation in 3.3′-diamidobenzidine tetrahydrochloride (0.5 mg/ml), cytochrome c (1 mg/ml), 50 mM phosphate buffer (pH 7.4), 0.2 M sucrose, and catalase (20 μg/ml).
MEF lysate was run on precast NativePAGE 4 to 16% gels (Thermo Fisher Scientific), transferred onto PVDF membranes, and blocked in 5% milk TBST overnight at 4°C. Immunoblotting was performed following standard techniques and developed using Clarity Western ECL substrate (Bio-Rad). For Coomassie staining, the gel was fixed (40% methanol and 10% acetic acid) for 1 hour at room temperature, followed by incubation with Imperial Protein Stain (Thermo Fisher Scientific) for 1 hour and destaining (8% acetic acid).
HEK293T cells expressing flagged wild-type and mutant HsNFS1 were lysed in lysis buffer. Nonsolubilized material was removed by centrifugation at 12,000g at 4°C. Eluates from Hs.NFS1-FLAG immunoprecipitations or 50 μg of protein from supernatants were loaded on precast NativePAGE 3 to 12% gels (Thermo Fisher Scientific) and ran at 20 mA. Resolved complexes were then transferred to PVDF membranes and blocked in 5% milk TBST. Immunoblotting was performed following standard techniques and developed using Clarity Western ECL substrate (Bio-Rad), or Coomassie staining was performed with Imperial Protein Stain.
Peptide preparation
For whole larvae label-free quantification, larvae were picked as described above, frozen in liquid nitrogen, and homogenized with a Teflon-coated pestle in 6 M guanidine hydrochloride and 50 mM tris (pH 8.0). After sonication and centrifugal removal of larval detritus, proteins were reduced with 5 mM 1,4-dithiothreiotol at 55°C for 30 min, cooled briefly on ice, and alkylated with 15 mM 2-chloroacetamide for 15 min in the dark. One hundred micrograms of protein quantified by BCA was diluted 1:10 in 50 mM tris (pH 8.0) and digested overnight with 2 μg of Pierce MS-grade trypsin (Thermo Fisher Scientific) at 37°C and mild shaking. The digest was stopped by addition of 1.2% formic acid (FA), precipitates were removed by centrifugation, and the supernatant was desalted on prepacked Empore SPE cartridges (3 M) following the manufacturer’s instructions. Peptides were eluted, dried at 30°C in a vacuum concentrator (Eppendorf), and quantified in 0.5% FA using a NanoDrop (Thermo Fisher Scientific).
For the methylome, 30 mg of Percoll-purified mitochondria of both light and heavy fraction were pooled at equal protein mass and lysed in 2 ml of 6 M guanidine hydrochloride and 20 mM tris (pH 8.0). Samples were incubated at room temperature for 10 min, sonicated for 100 s in a 10-s on/off cycle, and then incubated again for 10 min. Without removal of membranes, proteins were reduced and alkylated as above and then digested with either MS-grade trypsin or chymotrypsin overnight and acidified with 1.2% FA. After centrifugal pelleting of precipitated material and membrane detritus, peptides were desalted on Sep-Pak Plus Short 360 mg C18 columns (Waters) according to the manufacturer’s instructions. Eluted peptides were dried at 60°C for up to 6 hours until the precipitation point.
For total cell proteomics of MEF, cell pellets were suspended in 100 μl of 8 M urea and 0.1% ProteaseMAX (Promega) in 250 mM tris (pH 8.5) adding protease and phosphatase inhibitors. The samples were sonicated, lysates were spun down, and protein concentration was determined by BCA assay (Pierce). An aliquot of 50 μg of each sample in 25 μl of lysis buffer was supplemented with 65 μl of 10% acetonitrile in double-distilled H2O (ddH2O) and was reduced by adding 1 μl of 0.5 M dithiothreitol (DTT) at 25°C for 1 hour, alkylated with 3 μl of 0.5 M iodoacetamide for 1 hour at room temperature in the dark, and quenched the excess of iodoacetamide by adding 2 μl of 10 mM DTT before subjected to proteolytic digestion. Lysyl endopeptidase (MS-grade, Waco) at 0.5 μg/μl in water was added in an enzyme to protein ratio of 1:50 (2 μl) and incubated in a thermal shaker at 24°C at 450 rpm overnight. Digestion was continued adding 92 μl of 50 mM tris (pH 8.5) and 1 μg of trypsin (0.5 μg/μl; sequencing grade, Promega) and incubated at 37°C while for 4 hours at 450 rpm. The proteolysis was stopped with 10 μl of FA at a final concentration of 5% (total volume of 212 μl). The samples were cleaned on a C18 Hypersep plate (Thermo Fisher Scientific) and dried using a Vacufuge vacuum concentrator (Eppendorf). Dry samples (20 μg each) were dissolved in 58 μl of 50 mM triethylammonium bicarbonate (pH 8.0), and 100 μg of TMTpro reagents (lot number UK292954, Thermo Fisher Scientific) in 25 μl of dry acetonitrile was added. Samples were incubated at room temperature at 450 rpm for 2 hours. The labeling reaction was stopped by adding hydroxylamine at a final concentration of 0.5% and incubated at room temperature with 550 rpm for 15 min. Individual samples were combined to one analytical sample and dried in vacuum concentrator, followed by cleanup of a 20-μg aliquot on C18 StageTip and dried again.
Fractionation
For high-pH reversed-phase fractionation, peptides within the methylome screen and TMTpro-labeled tryptic peptides from MEFs were dissolved in 100 μl and 90 μl of 20 mM ammonium hydroxide, respectively, loaded onto an XBridge Peptide bridged ethyl hybrid C18 ultraperformance liquid chromatography (UPLC) column (4.6 mm in inner diameter by 250 mm in length, 300 Å, 3.5-μm particle size; Waters), and profiled with a nonlinear gradient of 1 to 23.5% B [20 mM ammonium hydroxide in acetonitrile (pH 10.0)] in 42 min, followed by 23.5 to 42% B in 4 min and 42 to 63% B in 2 min at a flow rate of 200 μl/min. The chromatographic performance was monitored by sampling eluate with an ultraviolet detector (UltiMate 3000 UPLC, Thermo Fisher Scientific) scanning at 214 nm. Fractions were collected at 30-s intervals into a 96-well plate and concatenated into eight pools. Eluates were then dried at 30°C for 10 hours.
For ÄKTA fractionation within the methylome screen, a RESOURCE RPC 3-ml column (GE Healthcare) was washed in buffer B [0.0125% ammonium hydroxide (pH 10.0) and 80% acetonitrile], changed to buffer A [0.0125% ammonium hydroxide (pH 10.0) and 2% acetonitrile], and equilibrated for 30 min. After near-complete drying, peptides were resuspended in 2.0-ml buffer A, and a total of 2.5 ml was loaded in a 5-ml sample loop. After washing, peptides were eluted in a ramp from 5 to 32% B in 62 min, 75% for 5 min, and 87.5% B in 3 min at a constant flow rate of 1 ml/min into 6 (mitochondrial tryptic), 8 (mitochondrial tryptic and chymotryptic), or 12 (total larvae tryptic) concatenated fractions of 1-min length. Eluates were dried at 30°C for 10 hours in a VacuFuge (Eppendorf).
Immunoaffinity enrichment and methyl peptide preparation
Methylated peptides were enriched using commercial kits (Cell Signaling Technology). Briefly, dried peptides were resuspended in 400 μl of immunoaffinity purification (IAP) buffer, and a fraction of two combined antibody-bead slurry vials was added to each tube. After 2 hours on a rotator at 4°C, beads were washed twice in IAP buffer and three times in deionized water, and peptides were eluted twice with 30 μl of 0.15% trifluoroacetic acid. Peptides were desalted on C18-STAGE tips and dried in a Concentrator plus at room temperature (43). Peptides were then either quantified in 0.5% FA before quantification and drying down or digested for 3 hours with either 250 ng of Arg-C in 50 mM tris (pH 7.8), 5 mM CaCl2, and 2 mM EDTA or 250 ng of Lys-C in 100 mM ammonium bicarbonate (pH 8.0) before desalting again on STAGE tips, quantification, and drying down. Peptides were stored at −80°C until further sample processing.
DmNfs1.R77K-FLAG or DmNfs1.R77F-FLAG was pulled down following the manufacturer’s recommendations of the FLAG Immunoprecipitation Kit (Sigma-Aldrich). Proteins were on-bead digested as previously described (44).
HsNFS1-FLAG and HsNFS1.R72K-FLAG containing complexes were pulled down with FLAG M2 magnetic beads (Millipore, M8823) using the standard manufacturer’s protocols. Briefly, 600 μg of protein lysates [0.1% Triton X-100, 0.1 mM EDTA, 50 mM NaCl, 10% glycerol, 1 mM PMSF, and 20 mM tris-HCl (pH7.4)] were incubated overnight with 40 μl of bead suspension at 4°C on a rotator. Nonbound proteins were then washed, and coprecipitated complexes were eluted from beads using a 3xFLAG peptide (Millipore, F4799).
Liquid chromatography–tandem mass spectrometry
For total cell proteomics of MEFs, approximately 2 μg of samples (4 μl of 30 μl) were injected in an UltiMate 3000 nano UPLC online coupled to a Q Exactive HF Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The chromatographic separation of the peptides was achieved using a 50-cm-long C18 EASY-Spray column (Thermo Fisher Scientific) at 55°C, with the following gradient: 4 to 26% of solvent B (98% acetonitrile and 0.1% FA) in 90 min, 26 to 95% of solvent B in 5 min, and 95% of solvent B for 5 min at a flow rate of 300 nl/min. The mass spectrometric acquisition method comprised one survey full mass spectrum ranging from a mass/charge ratio (m/z) of 350 to 1600, acquired with a resolution of R = 120,000 (at m/z 200) targeting 5 × 106 ions for a maximum injection time of 100 ms, followed by data-dependent higher-energy collision dissociation (HCD) fragmentations of maximum 18 most intense precursor ions with a charge state 2+ to 7+, using 45-s dynamic exclusion. The tandem mass spectra were acquired with a resolution of R = 60,000, targeting 2 × 105 ions for a maximum injection time of 54 ms, setting isolation width to m/z 1.4, and normalized collision energy to 33% setting first mass at m/z 100.
For LC-MS/MS method specifications underlying Fig. 4 and fig. S3, refer to data file S3. For PRM validation of methylated targets, stable isotope-labeled peptide standards (SpikeTides L) were obtained from JPT Peptide Technologies GmbH (Berlin, Germany) targeting 10 nmol each (data file S3). Synthesizable modifications were as follows: monomethyl arginine and lysine and asymmetric and symmetric arginine, whereof the symmetric modification was chosen over asymmetric, unless indicated differently. The peptides were dissolved in 100 μl of 20% acetonitrile in 100 mM ammonium bicarbonate and 30-fold diluted with ddH2O (3.3 pmol/μl), and a combined mixture was spiked into digested protein samples. One microgram of sample was injected in an UltiMate 3000 nano UHPLC online coupled to an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). The chromatographic separation of the peptides was achieved using a 25-cm-long C18 EASY-Spray column (Thermo Fisher Scientific) at 45°C, with the following gradient: 4 to 26% of solvent B (98% acetonitrile and 0.1% FA) in 30 min, 26 to 95% of solvent B in 2 min, and 95% of solvent B for 8 min at a flow rate of 300 nl/min. The mass spectrometer was set to acquire tandem mass spectra after isolating precursors defined in an inclusion mass list with m/z 0.7 isolation width in the quadrupole ranging from m/z 350 to 1500 at a resolution of R = 15,000 (at m/z 200) targeting 5 × 104 ions for a maximum injection time of 22 ms, using HCD fragmentations at 28% normalized collision energy.
mitoSAM transport assay
Isolated mitochondria (50 μg) were washed twice at 9000g and resuspended in 50 μl of assay buffer [100 mM mannitol, 10 mM sodium succinate, 80 mM KCl, 5 mM MgCl2, 1 mM KH2PO4, 25 mM Hepes, 5 mM ATP, 200 nM guanosine 5′-triphosphate, 6 mM creatine phosphate, creatine kinase (60 ng/ml), and each amino acids (60 mg/liter): alanine, arginine, apartic acid, asparagine, cysteine, glutamic acid, glutamine, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, and valine (pH 7.4)]. Prewarmed assay buffer (50 μl) supplemented with radiolabeled SAM-[methyl-3H] (PerkinElmer, Waltham, MA, USA) at a final concentration of 1.12 μM was added, and mitochondria were incubated at 30°C for 20 min. Mitochondria were then washed twice in assay buffer containing 1 mM unlabeled SAM. The mitochondrial pellet was resuspended in 50 μl of PBS and added to 3 ml of Ultima Gold scintillation cocktail (PerkinElmer), and mean activity was acquired over 1 min with a liquid scintillation counter (LKB Instruments).
SAM levels
Mitochondria were isolated in 136 mM KCl and 10 mM KH2PO4 (pH 7.25), as described above. Metabolite extraction was performed by ice-cold 80% methanol spiked with labeled internal standard SAM[2H3] and incubated on ice for 15 min, and supernatants were collected after centrifugation (15,000g for 15 min at 4°C) and evaporated to dryness in a Concentrator plus (Eppendorf). Dried extracts were resuspended in mobile phase A. Quantification of SAM was developed in a QQQ instrument (Waters Xevo TQ MS). Metabolite measurements were performed by multiple reaction monitoring in positive mode (ES+). Analyte solutions were directly infused to determine instrument parameters and are detailed in data file S3. Electrospray ionization (ESI) capillary voltage was 3.0 kV, extractor voltage was 3 V, radio frequency (RF) lens voltage was 0.1 V, desolvation gas (hydrogen) flow was 1200 liters/hour at 450°C, and cone gas (argon) flow was 150 liters/hour with source block at 150°C. Separation was achieved by reverse phase in an ACQUITY UPLC BEH C18 of 1.7 μm, 2.1 mm by 100 mm (Waters, 186002352) and equivalent ACQUITY VanGuard columns. Automatic sampler (Waters, 186003975) was at 10°C, and column was at 60°C. Partial loop injection with needle overfill with a volume of 10 μl was performed. A linear gradient of mobile phase A [10 mM ammonium formeate (pH 5.7)] and B (100% acetonitrile) was used. Flow was kept at a steady of 0.6 ml/min (100% A), kept constant for 1 min, followed by an increase in B to 100% in 2.5 min, and kept for 0.6 min. Phase A was set to 100% in 0.1 min and kept for 2 min. Calibration curves (0 to 1000 pmol) were based on determination of analyte/internal standard (IS) peak area ratios versus analyte amount. Analytical procedures were validated for compound and matrixes of interest. Method development, data acquisition, and peak integration were all performed with MassLynx 4.1 software (Waters).
Iron levels
Iron content was determined with a colorimetric ferrozine assay. Larvae were homogenized in 125 μl of PBST (PBS with 0.1% Triton X-100) containing protease inhibitors, and insoluble material was removed by centrifugation at 16,000g for 10 min. Supernatant protein content was determined by the BCA assay. Equal mass protein samples were hydrolyzed with 22% hydrochloric acid at 95°C for 20 min, followed by two centrifugations at 16,000g for 2 min, recovering supernatant. To reduce ferric iron to ferrous iron, 20 μl of ascorbate (75 mM; Sigma-Aldrich) was added to 50 μl of supernatant. Twenty microliters of ferrozine (10 mM; Sigma-Aldrich) and 40 μl of saturated ammonium acetate were added. Absorbance was measured at 562 nm, and iron content was expressed relative to the wDah control.
CoQ9 levels
Isolated mitochondria were resuspended in 200 μl of 1 mM copper sulfate, followed by the addition of 200 μl ethanol. The sample was sonicated at high power for 2 min in a 10-s on/off cycle. Hexane (400 μl) was added, and the sample was vortexed and centrifuged at 2500g for 5 min at 4°C. The hexane soluble upper fraction was collected and dried using a vacuum concentrator (Eppendorf). CoQ9 was measured essentially as described previously (45) and normalized to mitochondrial protein content.
Amino acid plasma levels
Larvae or flies were homogenized in 500 μl ice-cold modified simulated body fluid (46). Three hundred microliters of homogenate was mixed with 300 μl of ice-cold Norleucin (175 μM) and incubated on ice for 1 hour. The sample was centrifuged at 13,600g for 10 min at 4°C to remove insoluble material, and the supernatant was collected. Amino acids were analyzed using ion exchange chromatography (Biochrom 30+ amino acid analyzer) and normalized to protein content (47).
Deep labeling and metabolomics analysis
For isotope tracing, two sets of culture media of the same composition as DMEM high glucose and GlutaMAX (Thermo Fisher Scientific) were synthesized from pure components as previously described (48), with either unlabeled glucose and amino acids (12C medium) or their 13C-labeled counterparts (13C medium) at the same molar concentrations. Glucose and all amino acids except glutamine were U-13C, while glutamine was U-13C,15N. All media were further supplemented with 10% fetal bovine serum and unlabeled uridine. MEFs were cultured in each medium for 48 hours before metabolite extraction with cold methanol, as previously described (48).
Untargeted LC-MS/MS analysis was performed using a Q Exactive Orbitrap mass spectrometer coupled to a Vanquish UPLC system (both Thermo Fisher Scientific). Chromatographic separation of metabolites was achieved using a SeQuant Zic-pHILIC 100 mm by 2.1 mm 5-μm column (Millipore) maintained at 45°C. Compounds were eluted via a 6-min linear gradient starting from 90:10 to 45:55 acetonitrile: 20 mM ammonium bicarbonate (pH 9.6). A Q Exactive Orbitrap (Thermo Fisher Scientific) mass spectrometer was operated in positive and negative ion modes using a heated ESI source at 30,000 resolution, 75-ms ion trap time for MS1, and at 15,000 resolution, 50-ms ion trap time for MS2 collection. Data were collected over a mass range of m/z 65 to 975, using a sheath gas flow rate of 40 U, an auxiliary gas flow rate of 20 U, a sweep gas flow rate of 2 U, a spray voltage of 3.5 kV, a capillary inlet temperature of 275°C, an auxiliary gas heater temperature of 350°C, and a S-lens RF level of 45. For MS2 collection, MS1 ions were isolated using a 1.0 m/z window and fragmented using a normalized collision energy of 30 eV. Fragmented ions were placed on dynamic exclusion for 5 s before being allowed to be fragmented again.
LC-MS peaks for metabolites of interest were identified using m/z and retention time from pure standards. Chromatograms were manually quality checked, and peak areas for all mass isotopomers were integrated directly from instrument data files using the mzAccess data service (49) controlled from Mathematica 11.1 (Wolfram Research Inc.). For aspartate (C4H7NO4), the two 15N mass isotopomers were summed to obtain the 13C mass isotopomer distribution. Correction for naturally occurring 13C was performed using the binomial matrix method (50), assuming a natural 13C abundance of 0.0107%.
Structural representations
The CG4743 structure was modeled with Modeller version 9.21 (51) on the basis of a Bos taurus SLC25A4/ANT1 structure [Protein Data Bank (PDB): 1OKC] (52) at standard settings. Structures were visualized with PyMOL 2.3.4. NFS1-ISD11 interactions are shown on the basis of the human structure (PDB: 5USR) (23). Hydrophobicity overlays were computed with the PyMOL plugin Color_h on the basis of empirically determined amino acid hydrophobicity (53). B. taurus structure (PDB: 5LDW) (54) was used for complex I visualization.
Bioinformatic computations
Unless indicated differently, all data presented were analyzed in R version 3.6.2 (December 2019) running in RStudio 1.2.5001. Computations using Quandenser and MaxQuant were performed on resources provided by Swedish National Infrastructure for Computing (SNIC) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under project SNIC 2019-8-175 and uppstore2018188. MaxQuant was configured in Windows and run with 20 parallel cores on the Uppmax CentOS 7 cluster with the Mono runtime version 5.10.1.27. Critical R code to obtain the untargeted Dm mitochondrial methylome is available in data file S4.
RNA sequencing data processing
Paired-end RNA sequencing reads were quasi-mapped against a cDNA library of BDGP6 (release 94) with salmon v0.11.3 and a --gcbias flag (55). Differential gene expression analysis was performed with DESeq2 version 1.26.0 and Independent Hypothesis Weighting version 1.14.1 (56).
Proteomics data processing
Label-free raw data were processed using Quandenser version 0.061 (57) in batches of wDah, rescue, and a123v; wDah, i172g, p223l, and null; and aged fly samples at 30 and 50 days. Methylation sites were mapped using MaxQuant 1.6.2.1 (58) with enabled post-translational modification (PTM) mode against a Dm FASTA library (UP000000803_7227, downloaded from UniProt in September 2018). Methionine oxidation, N-terminal acetylation, and methionine+0, methionine+4 (+4.02219 Da) and serine-0 or serine-7 (+7.025930 Da, here called serine-3 for donating a +3-Da carbon unit to tetrahydrofuran) were set as fixed modifications, and light or heavy monomethylation (KR, 14.01565, 17.03156 Da from serine-3 or +18.03748 Da from methionine-4), dimethylation (KR, 28.03130, 34.06312 Da from serine-3 or +36.07567 Da from methionine-4), and trimethylation (K, 42.04659, 51.09468 Da from serine-3 or 54.11351 Da from methionine-4) were set as variable modifications. Light and heavy fractions of the same sample were analyzed in two different runs. SAM-derived mass shifts from light to heavy or heavy to light were separately quantified with MethylQuant version 1.0 (59) at standard settings comprising a mass error window at 10 parts per million and a pair overlap search window of ±0.14 min, adjusting for the methionine or serine heavy label. The MaxQuant evidence file and PTM-specific data tables as well as MethylQuant output tables were imported and processed in R. A manually annotated reference data table of Dm mitochondrial proteins and their functions obtained from the mitoXplorer version 1.0 website (60) was used to filter all methylation sites. Quality scoring of methylation sites was performed as described in the results. Large-scale prediction of Dm protein homologs in mouse and human was performed with the Drosophila RNAi Screening Center (DRSC) Integrative Ortholog Prediction Tool version 8.0 (61), and the best fit was defined as the protein with the highest weighted score. Sequence alignment was performed with the msa R package (62). The org.Dm.eg.db or org.Mm.eg.db (version 3.8.2) packages were used for ID conversion in fly or mouse, respectively. Sequence logos were plotted with ggseqlogo version 0.1, and circular visualization plots were generated with the circlize package version 0.4 (63).
MEF spectra from TMTpro-labeled peptides were mapped against UP000000589_10090 (March 2020) with MaxQuant version 1.6.10.43 with manually configured MS2 report ions and adjusting for batch-dependent −2/−1/+1/+2 correction factors. Gene set enrichment analysis was performed with WebGestalt (64) with UniProt Swiss-Prot IDs against indicated nonredundant gene ontology dataset and pathways. Terms for plotting were selected manually by removing highly redundant and strictly nonmitochondrial categories. Mitochondrial proteins were annotated using MitoCarta2.0 (65).
For PRM data analysis, Skyline v19.1.0.193 and 20.1.0.76 was used (66). Raw data files were imported with carbamidomethylation on cysteine and allowing all isotope labeling types (C-terminal lysine-[13C615N2], C-terminal arginine-[13C615N4], proline-[13C515N1], and valine-[13C515N1]). Peptide transitions were filtered for y ions with 1+ and 2+ charge states. The precursor m/z exclusion window was set to 5, allowing automatic selection of all matching transitions. The imported data were manually controlled, and Skyline files are available online. Peak boundaries presented here refer to the standard’s maximum ±1 min.
NFS1-FLAG immunoprecipitation data were mapped with MaxQuant 1.6.10.43. Differential protein expression was assessed with the Differential Expression Proteomics R package release 3.11 (67), using a custom variance stabilization function that normalizes against intensity levels of proteins correlating with NFS1 at an R value of greater than 0.75.
Web-based and computational tools
Information about the annotation status of modified sites was extracted from PhosphoSitePlus v6.5.9.3 (68). Mitochondrial target sequences were computed with MitoProt II (69). Codependency with SLC25A26 was assessed by calculating the Pearson correlation coefficient of all CERES scores per gene with SLC25A26 itself on a local machine. The data were obtained from the Cancer Dependency Map (25, 70).
Data representation and statistical analysis
Details of statistical analysis can be found in the figure legends. All data are represented as means + SD unless indicated differently. A two-sided Student’s t test was used for pairwise comparisons and a one-way analysis of variance (ANOVA) with a Dunnett’s post hoc test when comparing several conditions against one control. *P < 0.05, **P < 0.01, and ***P < 0.001. Only gel images of Western blots are shown without quantitative assessments. Number of replicates was chosen depending on the differences expected in preexperiments. All graphs based on fly material show individual biological replicates that are grown separately and processed in random order. Graphs based on mouse material use three control cell lines and two KO lines obtained from one mother unless indicated differently. n indicates number of replicates. For quantitative proteomics, samples were processed in random order, and experimenters were blinded for experimental conditions.
Acknowledgments
We thank N.-G. Larsson for critical reading of the manuscript, J. Busch for NSUN4 KO samples, R. Palmer and G. Wolfstetter at the University of Gothenburg for advice and fly lines, and the Division of Molecular Metabolism for continuous input and support. Funding: This study was supported by the Swedish Research Council (VR2016-02179 to A.Wr. and VR2016-01082 to A.We.), the European Research Council (715009 to A.Wr.), the Knut and Alice Wallenberg Foundation (KAW 2014.0293 to A.We.), and the Max Planck Society (to I.A.). F.A.S. is part of the mentor program of the Studienstiftung des deutschen Volkes. A.Wr. is a Ragnar Söderberg fellow of medicine. Targeted PRM and MEF expression proteomic analyses were performed by the Proteomics Biomedicum Core Facility at Karolinska Institutet. Fly stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) and Vienna Drosophila Resource Center were used in this study. E.C. and R.N. received funding from the Swedish Foundation for Strategic Research (grant no. ITM17-0245). Author contributions: A.Wr. and A.We. acquired funding. A.Wr., C.F., F.A.S., D.M., I.A., M.F.M., and A.We. conceptualized the study. F.A.S., D.M., I.A., M.F.M., P.C., Á.V., .N.E.F., R.F., A.-L.B., Y.H., M.T., E.H., E.C., A.B., R.W., M.J., R.N., and L.K. developed assays, performed experiments, and analyzed data. A.Wr., C.F., A.We., I.A., Á.V., M.J., R.N., L.K., and Y.H. provided resources. A.Wr., C.F., A.We., and L.K. supervised the study. F.A.S., D.M., C.F., and A.Wr. visualized the data and wrote the manuscript. Competing interest: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (71) with the dataset identifiers PXD019654 (methylome), PXD019550 (Drosophila and mouse total cell expression proteomics), PXD019551 (NFS1 immunoprecipitation), and PXD019494 (targeted proteomics). The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) (72) and are accessible through GEO series accession number GSE151656. The generated CG4743 knockin fly lines are made available through the Bloomington Drosophila Stock Center at the Indiana University.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabf0717/DC1
REFERENCES AND NOTES
- 1.Ducker G. S., Rabinowitz J. D., One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shyh-Chang N., Locasale J. W., Lyssiotis C. A., Zheng Y., Teo R. Y., Ratanasirintrawoot S., Zhang J., Onder T., Unternaehrer J. J., Zhu H., Asara J. M., Daley G. Q., Cantley L. C., Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiraki N., Shiraki Y., Tsuyama T., Obata F., Miura M., Nagae G., Aburatani H., Kume K., Endo F., Kume S., Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Shi X., Tasdogan A., Huang F., Hu Z., Morrison S. J., DeBerardinis R. J., The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci. Adv. 3, eaao5268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farooqui J. Z., Lee H. W., Kim S., Paik W. K., Studies on compartmentation of S-adenosyl-L-methionine in Saccharomyces cerevisiae and isolated rat hepatocytes. Biochim. Biophys. Acta 757, 342–351 (1983). [PubMed] [Google Scholar]
- 6.Suzuki T., Suzuki T., A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 42, 7346–7357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metodiev M. D., Spåhr H., Polosa P. L., Meharg C., Becker C., Altmueller J., Habermann B., Larsson N.-G., Ruzzenente B., NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLOS Genet. 10, e1004110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agrimi G., Noia M. A. D., Marobbio C. M. T., Fiermonte G., Lasorsa F. M., Palmieri F., Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 379, 183–190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kishita Y., Pajak A., Bolar N. A., Marobbio C. M. T., Maffezzini C., Miniero D. V., Monné M., Kohda M., Stranneheim H., Murayama K., Naess K., Lesko N., Bruhn H., Mourier A., Wibom R., Nennesmo I., Jespers A., Govaert P., Ohtake A., Van Laer L., Loeys B. L., Freyer C., Palmieri F., Wredenberg A., Okazaki Y., Wedell A., Intra-mitochondrial methylation deficiency due to mutations in SLC25A26. Am. J. Hum. Genet. 97, 761–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikkanen J., Forsström S., Euro L., Paetau I., Kohnz R. A., Wang L., Chilov D., Viinamäki J., Roivainen A., Marjamäki P., Liljenbäck H., Ahola S., Buzkova J., Terzioglu M., Khan N. A., Pirnes-Karhu S., Paetau A., Lönnqvist T., Sajantila A., Isohanni P., Tyynismaa H., Nomura D. K., Battersby B. J., Velagapudi V., Carroll C. J., Suomalainen A., Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Bao X. R., Ong S.-E., Goldberger O., Peng J., Sharma R., Thompson D. A., Vafai S. B., Cox A. G., Marutani E., Ichinose F., Goessling W., Regev A., Carr S. A., Clish C. B., Mootha V. K., Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, e10575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kühl I., Miranda M., Atanassov I., Kuznetsova I., Hinze Y., Mourier A., Filipovska A., Larsson N.-G., Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 6, e30952 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson A. J., Overy C., Kunji E. R. S., The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc. Natl. Acad. Sci. U.S.A. 105, 17766–17771 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solmonson A., DeBerardinis R. J., Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 293, 7522–7530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grankvist N., Watrous J. D., Lagerborg K. A., Lyutvinskiy Y., Jain M., Nilsson R., Profiling the metabolism of human cells by deep 13C Labeling. Cell Chem. Biol. 25, 1419–1427.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mullen A. R., Wheaton W. W., Jin E. S., Chen P.-H., Sullivan L. B., Cheng T., Yang Y., Linehan W. M., Chandel N. S., De Berardinis R. J., Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaude E., Schmidt C., Gammage P. A., Dugourd A., Blacker T., Chew S. P., Saez-Rodriguez J., O’Neill J. S., Szabadkai G., Minczuk M., Frezza C., NADH shuttling couples cytosolic reductive carboxylation of glutamine with glycolysis in cells with mitochondrial dysfunction. Mol. Cell 69, 581–593.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ong S.-E., Mittler G., Mann M., Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 1, 119–126 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Morscher R. J., Ducker G. S., Li S. H.-J., Mayer J. A., Gitai Z., Sperl W., Rabinowitz J. D., Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554, 128–132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mochel F., Knight M. A., Tong W.-H., Hernandez D., Ayyad K., Taivassalo T., Andersen P. M., Singleton A., Rouault T. A., Fischbeck K. H., Haller R. G., Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 82, 652–660 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lill R., Freibert S.-A., Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu. Rev. Biochem. 89, 471–499 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Farhan S. M. K., Wang J., Robinson J. F., Lahiry P., Siu V. M., Prasad C., Kronick J. B., Ramsay D. A., Rupar C. A., Hegele R. A., Exome sequencing identifies NFS1 deficiency in a novel Fe-S cluster disease, infantile mitochondrial complex II/III deficiency. Mol. Genet. Genom. Med. 2, 73–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cory S. A., Van Vranken J. G., Brignole E. J., Patra S., Winge D. R., Drennan C. L., Rutter J., Barondeau D. P., Structure of human Fe–S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP–ISD11 interactions. Proc. Natl. Acad. Sci. U.S.A. 114, E5325–E5334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakai Y., Umeda N., Suzuki T., Nakai M., Hayashi H., Watanabe K., Kagamiyama H., Yeast Nfs1p Is involved in thio-modification of both mitochondrial and cytoplasmic tRNAs. J. Biol. Chem. 279, 12363–12368 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Ghandi M., Huang F. W., Jané-Valbuena J., Kryukov G. V., Lo C. C., Robert McDonald E. III, Barretina J., Gelfand E. T., Bielski C. M., Li H., Hu K., Andreev-Drakhlin A. Y., Kim J., Hess J. M., Haas B. J., Aguet F., Weir B. A., Rothberg M. V., Paolella B. R., Lawrence M. S., Akbani R., Lu Y., Tiv H. L., Gokhale P. C., de Weck A., Mansour A. A., Oh C., Shih J., Hadi K., Rosen Y., Bistline J., Venkatesan K., Reddy A., Sonkin D., Liu M., Lehar J., Korn J. M., Porter D. A., Jones M. D., Golji J., Caponigro G., Taylor J. E., Dunning C. M., Creech A. L., Warren A. C., McFarland J. M., Zamanighomi M., Kauffmann A., Stransky N., Imielinski M., Maruvka Y. E., Cherniack A. D., Tsherniak A., Vazquez F., Jaffe J. D., Lane A. A., Weinstock D. M., Johannessen C. M., Morrissey M. P., Stegmeier F., Schlegel R., Hahn W. C., Getz G., Mills G. B., Boehm J. S., Golub T. R., Garraway L. A., Sellers W. R., Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhein V. F., Carroll J., Ding S., Fearnley I. M., Walker J. E., NDUFAF7 methylates arginine 85 in the NDUFS2 subunit of human complex I. J. Biol. Chem. 288, 33016–33026 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruprecht J. J., Kunji E. R. S., The SLC25 mitochondrial carrier family: Structure and mechanism. Trends Biochem. Sci. 45, 244–258 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Avila M. A., Berasain C., Torres L., Martín-Duce A., Corrales F. J., Yang H., Prieto J., Lu S. C., Caballería J., Rodés J., Mato J. M., Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J. Hepatol. 33, 907–914 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Mentch S. J., Mehrmohamadi M., Huang L., Liu X., Gupta D., Mattocks D., Padilla P. G., Ables G., Bamman M. M., Thalacker-Mercer A. E., Nichenametla S. N., Locasale J. W., Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 22, 861–873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Małecki J., Ho A. Y. Y., Moen A., Dahl H.-A., Falnes P. Ø., Human METTL20 is a mitochondrial lysine methyltransferase that targets the β subunit of electron transfer flavoprotein (ETFβ) and modulates its activity. J. Biol. Chem. 290, 423–434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shishkova E., Zeng H., Liu F., Kwiecien N. W., Hebert A. S., Coon J. J., Xu W., Global mapping of CARM1 substrates defines enzyme specificity and substrate recognition. Nat. Commun. 8, 15571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hung V., Zou P., Rhee H.-W., Udeshi N. D., Cracan V., Svinkina T., Carr S. A., Mootha V. K., Ting A. Y., Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdulhag U. N., Soiferman D., Schueler-Furman O., Miller C., Shaag A., Elpeleg O., Edvardson S., Saada A., Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 23, 159–164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King M. S., Thompson K., Hopton S., He L., Kunji E. R. S., Taylor R. W., Ortiz-Gonzalez X. R., Expanding the phenotype of de novo SLC25A4-linked mitochondrial disease to include mild myopathy. Neurol. Genet. 4, e256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dietzl G., Chen D., Schnorrer F., Su K.-C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S., Couto A., Marra V., Keleman K., Dickson B. J., A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Huang J., Zhou W., Watson A. M., Jan Y.-N., Hong Y., Efficient ends-out gene targeting in Drosophila. Genetics 180, 703–707 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J., Zhou W., Dong W., Watson A. M., Hong Y., Directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. Proc. Natl. Acad. Sci. U.S.A. 106, 8284–8289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Motori E., Puyal J., Toni N., Ghanem A., Angeloni C., Malaguti M., Cantelli-Forti G., Berninger B., Conzelmann K.-K., Götz M., Winklhofer K. F., Hrelia S., Bergami M., Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 18, 844–859 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Piper M. D. W., Blanc E., Leitão-Gonçalves R., Yang M., He X., Linford N. J., Hoddinott M. P., Hopfen C., Soultoukis G. A., Niemeyer C., Kerr F., Pletcher S. D., Ribeiro C., Partridge L., A holidic medium for Drosophila melanogaster. Nat. Methods 11, 100–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piper M. D. W., Soultoukis G. A., Blanc E., Mesaros A., Herbert S. L., Juricic P., He X., Atanassov I., Salmonowicz H., Yang M., Simpson S. J., Ribeiro C., Partridge L., Matching dietary amino acid balance to the in silico-translated exome optimizes growth and reproduction without cost to lifespan. Cell Metab. 25, 610–621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schober F. A., Atanassov I., Moore D., Wedell A., Freyer C., Wredenberg A., Versatile proteome labelling in fruit flies with SILAF. Biorxiv, 590315 (2019). [Google Scholar]
- 42.Wibom R., Hagenfeldt L., von Döbeln U., Measurement of ATP production and respiratory chain enzyme activities in mitochondria isolated from small muscle biopsy samples. Anal. Biochem. 311, 139–151 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Rappsilber J., Mann M., Ishihama Y., Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Turriziani B., Garcia-Munoz A., Pilkington R., Raso C., Kolch W., von Kriegsheim A., On-beads digestion in conjunction with data-dependent mass spectrometry: A shortcut to quantitative and dynamic interaction proteomics. Biology 3, 320–332 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freyer C., Stranneheim H., Naess K., Mourier A., Felser A., Maffezzini C., Lesko N., Bruhn H., Engvall M., Wibom R., Barbaro M., Hinze Y., Magnusson M., Andeer R., Zetterström R. H., von Döbeln U., Wredenberg A., Wedell A., Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4–dihydroxybensoic acid. J. Med. Genet. 52, 779–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oyane A., Kim H.-M., Furuya T., Kokubo T., Miyazaki T., Nakamura T., Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. A 65, 188–195 (2003). [DOI] [PubMed] [Google Scholar]
- 47.F. A. Hommes, in Techniques in Diagnostic Human Biochemical Genetics (Wiley, 1990). [Google Scholar]
- 48.Grankvist N., Watrous J. D., Jain M., Nilsson R., Metabolic flux analysis in eukaryotic cells, methods and protocols. Methods Mol Biology. 2088, 73–92 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Lyutvinskiy Y., Watrous J. D., Jain M., Nilsson R., A web service framework for interactive analysis of metabolomics data. Anal. Chem. 89, 5713–5718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nilsson R., Validity of natural isotope abundance correction for metabolic flux analysis. Math. Biosci. 330, 108481 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Webb B., Sali A., Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform. 54, 5.6.1–5.6.37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pebay-Peyroula E., Dahout-Gonzalez C., Kahn R., Trézéguet V., Lauquin G. J.-M., Brandolin G., Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Eisenberg D., Schwarz E., Komaromy M., Wall R., Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179, 125–142 (1984). [DOI] [PubMed] [Google Scholar]
- 54.Zhu J., Vinothkumar K. R., Hirst J., Structure of mammalian respiratory complex I. Nature 536, 354–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ignatiadis N., Klaus B., Zaugg J. B., Huber W., Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.The M., Käll L., Focus on the spectra that matter by clustering of quantification data in shotgun proteomics. Nat. Commun. 11, 3234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyanova S., Temu T., Cox J., The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Tay A. P., Geoghegan V., Yagoub D., Wilkins M. R., Hart-Smith G., MethylQuant: A tool for sensitive validation of enzyme-mediated protein methylation sites from heavy-methyl SILAC data. J. Proteome Res. 17, 359–373 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Yim A., Koti P., Bonnard A., Marchiano F., Dürrbaum M., Garcia-Perez C., Villaveces J., Gamal S., Cardone G., Perocchi F., Storchova Z., Habermann B. H., mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Nucleic Acids Res. 48, 605–632 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu Y., Flockhart I., Vinayagam A., Bergwitz C., Berger B., Perrimon N., Mohr S. E., An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 12, 357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bodenhofer U., Bonatesta E., Horejš-Kainrath C., Hochreiter S., msa: An R package for multiple sequence alignment. Bioinformatics 31, 3997–3999 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Gu Z., Gu L., Eils R., Schlesner M., Brors B., circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Wang J., Vasaikar S., Shi Z., Greer M., Zhang B., WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W130–W137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calvo S. E., Clauser K. R., Mootha V. K., MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pino L. K., Searle B. C., Bollinger J. G., Nunn B., MacLean B., MacCoss M. J., The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 39, 229–244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang X., Smits A. H., van Tilburg G. B., Ovaa H., Huber W., Vermeulen M., Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 13, 530–550 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., Skrzypek E., PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Claros M. G., Vincens P., Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 779–786 (1996). [DOI] [PubMed] [Google Scholar]
- 70.Meyers R. M., Bryan J. G., McFarland J. M., Weir B. A., Sizemore A. E., Xu H., Dharia N. V., Montgomery P. G., Cowley G. S., Pantel S., Goodale A., Lee Y., Ali L. D., Jiang G., Lubonja R., Harrington W. F., Strickland M., Wu T., Hawes D. C., Zhivich V. A., Wyatt M. R., Kalani Z., Chang J. J., Okamoto M., Stegmaier K., Golub T. R., Boehm J. S., Vazquez F., Root D. E., Hahn W. C., Tsherniak A., Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., Vizcaíno J. A., The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edgar R., Domrachev M., Lash A. E., Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cornelissen T., Spinazzi M., Martin S., Imberechts D., Vangheluwe P., Bird M., Strooper B. D., Vandenberghe W., CHCHD2 harboring Parkinson’s disease-linked T61I mutation precipitates inside mitochondria and induces precipitation of wild-type CHCHD2. Hum. Mol. Genet. 29, 1096–1106 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabf0717/DC1