

Abstract
The successful identification of promising investigational therapies for the treatment of epilepsy can be credited to the use of numerous animal models of seizure and epilepsy for over 80 years. In this time, the maximal electroshock test in mice and rats, the subcutaneous pentylenetetrazol test in mice and rats, and more recently the 6 Hz assay in mice, have been utilized as primary models of electrically or chemically-evoked seizures in neurologically intact rodents. In addition, rodent kindling models, in which chronic network hyperexcitability has developed, have been used to identify new agents. It is clear that this traditional screening approach has greatly expanded the number of marketed drugs available to manage the symptomatic seizures associated with epilepsy. In spite of the numerous antiseizure drugs (ASDs) on the market today, the fact remains that nearly 30% of patients are resistant to these currently available medications. To address this unmet medical need, the National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP) revised its approach to the early evaluation of investigational agents for the treatment of epilepsy in 2015 to include a focus on preclinical approaches to model pharmacoresistant seizures. This present report highlights the in vivo and in vitro findings associated with the initial pharmacological validation of this testing approach using a number of mechanistically diverse, commercially available antiseizure drugs, as well as several probe compounds that are of potential mechanistic interest to the clinical management of epilepsy.
Keywords: Antiseizure drugs, Animal models, Corneal kindling, N6-cyclopentyladenosine, M-CPP, Valproic acid, 6 Hz test
Introduction
For more than 40 years, the National Institute of Neurological Disorders and Stroke (NINDS) has invested in identifying and developing novel antiseizure medications for the treatment of epilepsy with the Anticonvulsant Screening Program, known since 2016 as the Epilepsy Therapy Screening Program (ETSP). In this effort, numerous therapies have come to market to benefit the person with epilepsy. Although numerous antiseizure drugs (ASDs) are FDA approved, a significant proportion of individuals with epilepsy remain refractory to therapy. In addition, there are currently no FDA-approved drugs to prevent the development of epilepsy in individuals at risk following a brain insult. Given that there are over 65 million people worldwide with epilepsy and 1 in 26 people will develop epilepsy at some point in their lifetime, there is a significant unmet need for improved treatment options for pharmacoresistant epilepsy. Basic science continues to make incredible strides in understanding the pathophysiology of epilepsy. The challenge now is to identify transformative therapies for those patients who remain refractory to available therapies, as well as to identify therapies that may have better safety and tolerability profiles to improve patient adherence [1] and reduce adverse effects liabilities [2], all of which may contribute to uncontrolled or pharmacoresistant epilepsy. To address the recommendations of the 2015 NINDS Working Group report [3], the ETSP revised the testing approach to include a focus on finding effective treatments for pharmacoresistant epilepsy (Fig. 1). As noted in this flow chart in Fig. 1, testing is divided into an initial “Identification” phase followed by a later “Differentiation” phase, the latter being comprised of more resource-intensive tests. As described below, key elements of this new flow chart are (1) overall, a higher threshold for advancing compounds through early Identification screening tests, but also increased flexibility for identifying potential efficacy of novel compounds; and (2) implementation in the Differentiation phase of more resource-intensive, lower-throughput disease models chosen to identify agents that may have improved efficacy relative to existing drugs for treating pharmacoresistant epilepsy.
Fig. 1.
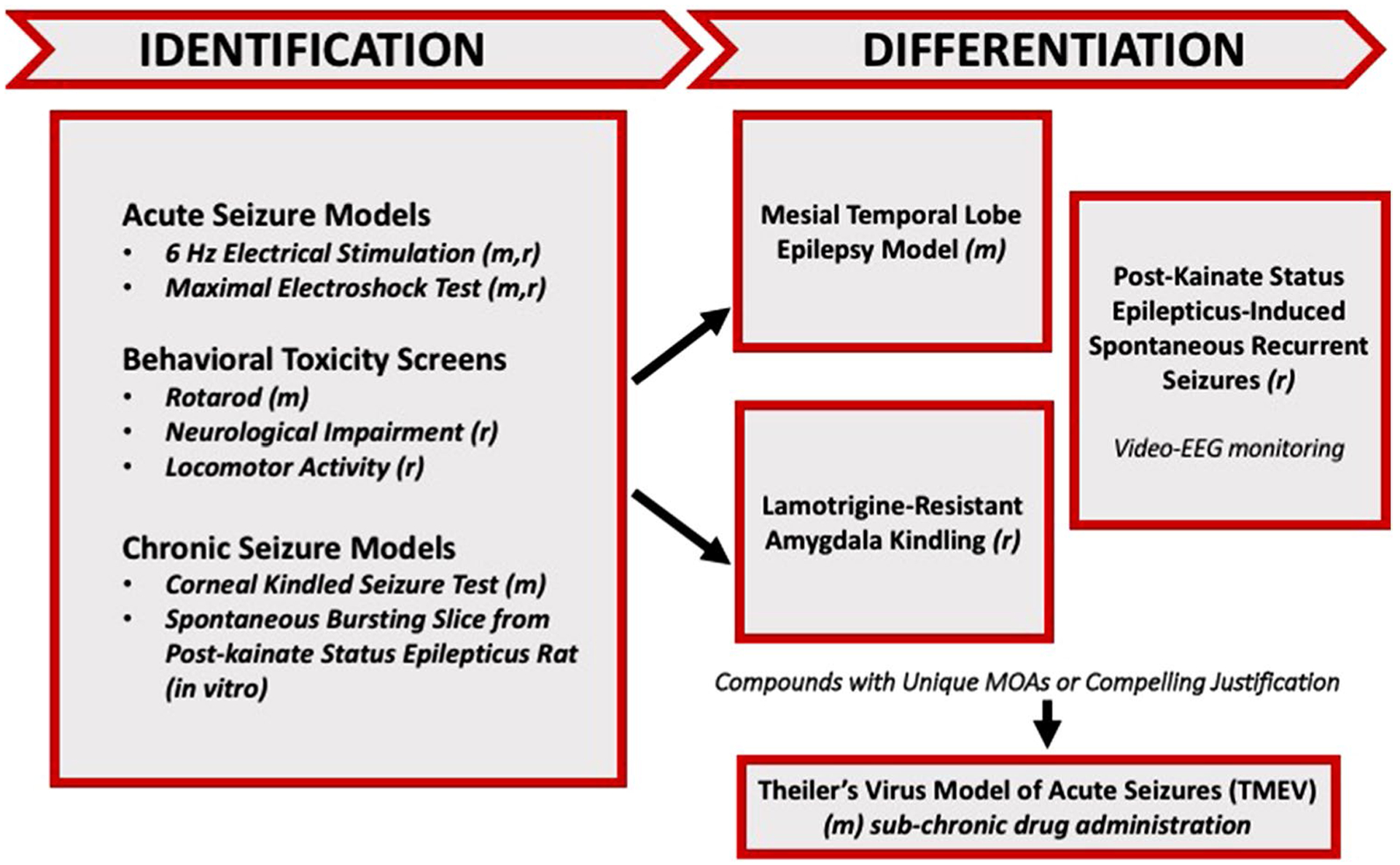
Revised testing approach for the validation of the screening of investigational compounds by the ETSP. *Prototype compounds were presently screened for efficacy only in Identification phase assays. The most promising compounds would be candidates to advance to more advanced, labor-intensive etiologically-relevant models of disease, including the LTG-resistant amygdala-kindled rat or mouse model of mesial temporal lobe epilepsy
While the traditional approaches to drug development for epilepsy have relied primarily on the evaluation of acute anticonvulsant efficacy in models of electrically- or chemically-evoked seizures in neurologically-intact rodents, additional models are now available that may allow for the identification of transformative therapies. This is not to say that the traditional models are no longer useful; there remains utility from a drug screening perspective to employ high-throughput, technically-approachable models that can be employed for the rapid evaluation of numerous compounds of limited quantity [4]. The electrically-evoked seizure models presently employed in this revised testing approach include the maximal electroshock (MES) test in mice and rats, and the 6 Hz model of focal seizures in mice (Fig. 1). MES is a model of generalized tonic-clonic seizures and provides an indication of a compound’s ability to prevent seizure spread. These seizures are highly reproducible and are electrophysiologically consistent with human seizures. Moreover, the MES test was the first clinically-validated animal model of seizure and it was instrumental to Merrit and Putnam’s identification of phenytoin in 1937 [5]; one year later, this ASD was clinically-available. The 6 Hz test, when conducted at a 44 mA stimulus intensity in mice, can differentiate the profile of investigational compounds at the preclinical level [6], regardless of efficacy in the MES test. These seizures are believed to model focal seizures observed in humans [6]. Thus, the initial evaluation of candidate compounds submitted to the ETSP now occurs in two electrically-induced models of seizure in mouse.
In spite of the numerous ASDs that are effective in these models, these electrical seizure tests are conducted in neurologically-intact rodents, wherein network remodeling and behavioral alterations consistent with temporal lobe epilepsy (TLE) are absent. Therefore, the MES and 6 Hz tests do not represent the pathophysiology of epilepsy, and it is possible that novel antiseizure agents (e.g. anti-inflammatory agents [7]) that ameliorate imbalances present in the epileptic brain might not be detected if these tests were the sole gatekeepers for screening. The use of etiologically-relevant disease models of chronic network hyperexcitability and/or spontaneous seizures are thus now incorporated in both the early (Identification) and late (Differentiation) phases of testing for the evaluation of novel therapies at the ETSP. The 60 Hz corneal kindled mouse demonstrates a pharmacological profile consistent with the hippocampal kindled rat model [8] but requires far less compound for the early evaluation of an investigational compound [9]. The corneal kindled mouse exhibits reactive gliosis [10] and behavioral alterations [11] associated with TLE. The pharmacological profile of the corneal kindled mouse is also consistent with human partial epilepsy and effectively identifies the anticonvulsant potential of useful compounds for this condition, such as levetiracetam [8, 12]. Thus, novel compounds that are not active in the MES or 6 Hz assays will nonetheless be tested in the corneal kindled mouse for potential activity. One potential limitation of the 60 Hz corneal kindled mouse is that it does not exhibit a pharmacoresistant profile, as has been demonstrated more recently in a 6 Hz corneal kindling protocol [13]. The 60 Hz corneal kindled mouse exhibits clear and consistent secondarily generalized seizure endpoints after repeated stimulation with an initially-benign current, which is in contrast to the 6 Hz stimulation protocol [13]. Therefore, the 60 Hz corneal kindled mouse is a useful moderate- to high-throughput screening platform to identify compounds that may only work in a hyperexcitable neuronal network. A final option for identifying novel compounds in the Identification phase is an in vitro screen using the medial entorhinal cortex-hippocampal (mEC-HC) slice obtained from kainic acid (KA)-treated rats [14]. The mEC-HC slices collected from KA-treated rats exhibit spontaneous, electrographic “interictal-like” events that are resistant to traditional ASDs [14]. Moreover, the mEC-HC slices obtained from KA-treated rats are hyperexcitable in normal artificial cerebrospinal fluid (ACSF) solution as early as one week following KA-induced SE [14]. This mEC-HC slice model exhibits a profile consistent with in vivo models of pharmacoresistant seizures, thus offering an in vitro surrogate to evaluate compounds for proof-of-concept antiseizure activity in the context that activity is not observed in the above-described in vivo models. The in vitro slice also provides a means of potentially identifying early investigational compounds that may exhibit challenges in brain penetrance, despite the potential for target-based efficacy in the context of an epileptic substrate.
Identification of an active compound in one or more of the Identification phase assays described above, bolstered by information on the compound’s pharmacokinetics, etc., can qualify it to advance to the Differentiation phase, which is presently comprised of three resource-intensive in vivo assays. As an etiologically-relevant model of pharmacoresistant TLE, the intrahippocampal kainic acid (KA) mouse is characterized by an initial focal neurotoxic event: a unilateral intrahippocampal injection of KA into the dorsal hippocampus, which induces non-convulsive SE lasting several hours, and subsequent spontaneous recurrent hippocampal paroxysmal discharges (HPD) that present 2–3 weeks after a latent phase [15–17]. These HPDs are also resistant to several clinical ASDs [18]. Furthermore, mechanistically novel compounds demonstrate efficacy in the intrahippocampal KA model [19], but whether these novel compounds will gain clinical utility remains to be determined [20]. In addition, the lamotrigine (LTG)-resistant amygdala kindled rat is useful to identify compounds effective against secondarily generalized focal seizures [21, 22]; it may also differentiate compounds that may be effective in therapy-resistant patients [23]. The addition of the traditional ASDs, carbamazepine or LTG, during the development of kindled seizures in this model impairs the effectiveness of LTG against a fully expressed kindled seizure [24], whereas valproic acid remains effective in LTG-resistant rats [23]. These findings suggest that the presence of LTG during the epileptogenic process leads to a subsequent resistance to other sodium channel blockers, highlighting the utility of this model for pharmacoresistant seizures. The third model, the post-KA SE rat model of chronic epilepsy [25], uses chronic video-EEG monitoring to assess the effects of administration of the most promising investigational agents on chronic seizure activity [26]. Thus, models of chronic network hyperexcitability, spontaneous electrographic and convulsive seizure activity, and pharmacoresistant seizures have gained a prominent role in the early overall evaluation of investigational therapies submitted to the ETSP.
In an effort to validate the overhaul of the traditional drug evaluation approach of the ETSP, a series of mechanistically diverse compounds were subjected to this screening platform in a blinded fashion over the course of August–December 2015. To mimic investigator-initiated submission protocols of the ETSP, nine compounds were blindly screened through this modified approach: acetaminophen, carbamazepine (CBZ), meta-chlorophenylpiperazine (m-CPP), clobazam (CLB), N6-cyclopentyladenosine (N6-CPA), levetiracetam (LEV), retigabine/ezogabine (RTG), tiagabine (TGB), and valproic acid (VPA). In an effort to model the screening approach, compounds were evaluated in the Identification phases of this modified flow chart at standardized starting doses that would, theoretically, capture as many compounds with potential efficacy as possible. The results of the evaluation of acetaminophen, m-CPP, and N6-CPA from this validation effort are presented herein due to their unique pharmacological profiles in the models in which they were tested. The evaluation of the broadly-acting serotonin agonist, m-CPP, was selected for testing due to the hypothesized efficacy of this mechanistic class in seizures and epilepsy [27, 28]. The adenosine dysfunction hypothesis has also gained a prominent place in the anticonvulsant therapy development pipeline [29, 30], thus N6-CPA was evaluated in this protocol to define activity of this broadly acting agonist in standard models of seizure. Additionally, the in vivo and in vitro activity of acetaminophen is also provided. Where relevant, the activities of CBZ, CLB, LEV and VPA are included for comparison purposes, but the activity of many of these compounds have been disclosed previously and validated on numerous occasions [4, 8, 14, 23, 31]. The activity and profiles of these commercially-available prototypes will also be added to the NINDS-sponsored open access database, PANAChE (https://panache.ninds.nih.gov), which already includes activity information from a number of other ASDs not presently evaluated in this drug screening overhaul (e.g. phenobarbital).
Methods
Animals and Investigational Compound Testing
Male albino CF-1 mice (18–25 g, approximately 4–6 weeks old; Charles River, Kingston, NY) and male albino Sprague–Dawley rats (250–300 g or 9–11 weeks old, kindling tests; 100–150 g, approximately 5–6 weeks old all other tests; Charles River, Raleigh, NC) were used as experimental animals. All animals were allowed free access to both food (Prolab RMH 3000) and water except when they were removed from their cages for the experimental procedure. All rats and mice were housed, fed, and handled in a manner consistent with the recommendations in the National Research Council publication, “Guide for the Care and Use of Laboratory Animals” and animal was approved by the University of Utah Institutional Animal Care and Use Committee (IACUC). No insecticides capable of altering hepatic drug metabolism enzymes were used in the animal facilities. Except for kindling studies, animals were used once.
The investigational compounds were each administered in 0.5% methylcellulose (MC). The test compound was administered either by the intraperitoneal (i.p.) or oral (p.o.) route in a volume of 0.01 ml/g body weight in mice and 0.04 ml/10 g body weight in rats. Testing results were recorded and quantified as number of animals (N) protected/not protected out of the number of mice tested (F). Initial qualitative efficacy screening was conducted in groups of n = 4 mice or rats. Prior to determining the median effective (ED50) or median toxic (TD50) dose of an investigational compound, the time of peak effect (TPE) was determined with n = 4 animals/time point based on the dose used in the qualitative screen that produced the greatest protection at the time points evaluated. Animals (mice or rats) were treated with the test compound (i.p. or p.o.) and evaluated at the following time points (0.25, 0.5, 1.0, and 2.0 h), or as determined empirically necessary. Mice were also checked 72 h after drug administration, to rule out any overt effects of a compound on morbidity or mortality. Quantification of the ED50/TD50 was then conducted at the TPE. Quantification of the ED50/TD50 was conducted in groups of n = 8 animals by administering various doses of the candidate drug until at least two points could be clearly established between the limits of 0 and 100% protection/toxicity. The ED50 and/or TD50, 95% confidence interval, slope of the regression line, and standard error of the mean (S.E.M.) of the slope were calculated by Probit analysis [32].
Maximal Electroshock Test (MES) in Mouse and Rats
For all MES tests, 60 Hz of alternating current was delivered for 0.2 s by corneal electrodes. An electrolyte solution containing an anesthetic agent was applied to the eyes before stimulation (0.5% tetracaine HCl). The current intensity was species-specific, i.e., 50 mA for mice and 150 mA for rats. An animal was considered “protected” from MES-induced seizures in the absence of the hindlimb tonic extension component of the seizure [33–35].
6 Hz Mouse Test
Investigational compounds were screened for their ability to block psychomotor seizures induced by a low-frequency (6 Hz), long-duration (3 s) stimulus delivered through corneal electrodes. Mice were challenged with a 44 mA current (twofold increase from the convulsive current that elicits seizures in 97% of CF-1 mice [6]). The seizure was characterized by an initial momentary stun followed immediately by forelimb clonus, twitching of the vibrissae, and Straub tail [6]; absence of these behaviors were the criteria for a “protected” mouse.
Corneal Kindled Mouse
Male CF-1 mice were kindled electrically with a 3 s, 3 mA, 60 Hz corneal stimulation to a criterion of 5 consecutive Stage 5 seizures (facial clonus and head nodding progressing to forelimb clonus, and finally rearing and falling accompanied by a generalized clonic seizure [36]). Stage 5 was reached after twice daily corneal stimulation for 8–10 days. Twice daily stimulations continued until each mouse had achieved the criterion of 5 consecutive stage 5 seizures, whereby it was considered “fully kindled”. Fully kindled mice were then stimulated every-other day until all other mice within the group reached the criterion of 5 consecutive Stage 5 seizures. Any mouse not achieving the fully kindled state was not included in any evaluation of investigational compounds. Testing of investigational compounds commenced at least 5–7 days after the last corneal stimulation necessary for all mice to be fully kindled. Mice were stimulated the day before testing to ensure the consistency of Stage 5 seizure. On the testing day, mice displaying a seizure score ≤ 3 were considered protected. Unlike acute seizure tests, each corneal kindled mouse was allowed at least 3–4 days between tests to “washout” any investigational compound after testing.
Minimal Motor Impairment
To assess the potential for adverse side effects, animals were visually evaluated for overt impairments of neurological or muscular function. In mice, the rotorod was used to identify minimal motor impairment (MMI [37]). A mouse can maintain its equilibrium for long periods of time on a rod that rotates at a speed of 6 rpm. The animal was considered toxic if it fell off this rotating rod three times during a 1 min period. In rats, MMI was indicated by visual assessment of motor coordination and normal behaviors. All animals are observed for motor coordination and behavior prior to drug administration. Evidence of ataxia, and/or abnormal, uncoordinated gait at the time of peak anticonvulsant efficacy of the compound are sufficient to indicate toxicity. In addition to MMI, animals may exhibit a circular or zigzag gait, abnormal body posture and spread of the legs, tremors, hyperactivity, lack of exploratory behavior, somnolence, stupor, catalepsy, loss of placing response, and changes in muscle tone. A rat was considered impaired if it displayed two or more of these abnormal behaviors, in addition to evidence of ataxia and/or abnormal, uncontrolled gait.
Open Field Activity Monitor of Rats
An automated open field activity assessment was performed by an experimenter blinded to treatment condition following administration of the investigational agent prior to determining the behaviorally-impairing dose (e.g. TD50). In this assay, each rat was administered the test compound and visually evaluated for MMI (approximately 1–2 min of evaluation) at the TPE. Immediately after the subjective MMI determination, the rat was placed into an open field Plexiglas chamber (40L × 40 W × 30 H cm) equipped with infrared sensors to detect animal movement for 10 min. During the 10 min period, the total distance travelled (cm), vertical activity counts, and horizontal activity counts were measured and recorded by the automated computer system [11]. A vehicle-treated control cohort of n = 8 rats was run within 24 h of the candidate compound-treated rodents at the various doses needed to complete a TD50. The effects of an investigational agent on open field activity were compared to MMI scores, as well as vehicle-treated controls, to provide an automated dose–response evaluation of motor performance following administration of an investigational agent.
In Vitro Slice Electrophysiology Studies
Male rats (100–150 g) were treated with systemic low-dose administration of KA until sustained seizure activity (status epilepticus, SE) was achieved [25]. Vehicle (0.9% saline) or KA (5 mg/kg, i.p.) was administered once every hour until animals began to exhibit behaviors consistent with early stage seizures (Stage 1–3). Seizures were scored during the experiment based on the Racine scale [38]. Once an animal began to seize, dosing was ceased or reduced to 2.5 mg/kg (i.p.) to maintain a seizure frequency of least one Stage 4/5 seizure per hour for over 3.5 h. Animals not having at least one Stage 4 or 5 seizure per hour were not included. After 3.5 h of monitoring, rats were given an i.p. injection of lactated Ringer’s solution (1–2 mLs) for hydration and returned to their home cages until sacrifice for in vitro testing 2 weeks later.
A combined medial entorhinal cortex (mEC)/hippocampal (HC) slice preparation was then obtained from surviving rats (150–180 g). On the day of sacrifice, rats were anesthetized with pentobarbital (35 mg/kg), decapitated, and brains quickly removed. The brains were immediately placed, for one minute, in an ice-cold, oxygenated (95% O2/5% CO2) Ringer’s solution containing, (in mM): sucrose (125.0), KCl (3.0), NaPO4 (1.2), MgSO4 (2.0), NaHCO3 (26.0), glucose (10.0), and CaCl2 (2.0) [39]. Horizontal Sect. (400 μm) containing the mEC and HC were taken and placed in a holding chamber for at least 1 h before commencing field potential recording. The oxygenated Ringer’s solution in the holding chamber, and for recording, had NaCl (126 mM) instead of sucrose, pH 7.4 and osmolarity of 300–310 mOsm.
Extracellular field potential recordings were then made in Layer II of mEC with borosilicate glass electrodes (3–6 MΩ) filled with normal Ringer’s solution. A concentric bipolar or twisted nichrome/formvar stimulating electrode placed in the angular bundle was used to elicit field potential responses. Signals were filtered at 1 kHz, sampled at 10 kHz, and acquired for computer storage using a Digidata 1440 A AD Converter (Axon Instruments). Voltage pulses of 1–20 V were administered using a stimulus isolator unit. A PC with pClamp 10 software was used to record all data for analysis. Only slices that generated stable I/O responses throughout the baseline recording period were accepted. The extracellular solution was then switched to one containing 6 mM KCl and 0.1 mM Mg2+ in order to elicit spontaneous, electrographic burst activity (SB). The rate and duration of SB was compared between vehicle- and investigational compound-treated periods of recording. Concentration–response profiles were quantified for median concentration necessary to inhibit bursting activity (IC50) for any compound found to significantly attenuate burst activity in the initial screen.
Investigational Compounds
Investigational compounds were purchased from commercial suppliers and formulated in 0.5% methylcellulose vehicle (Sigma, catalog #M0430). The investigational compounds were: acetaminophen (Spectrum Chemical Company, catalog #A1278); carbamazepine (Sigma, catalog #C4024); levetiracetam (Sigma, catalog #L8668); retigabine (Sigma, catalog #90221); clobazam (Sigma, catalog # C8414); tiagabine (Sigma, catalog #SML0035); N6-cyclopentyladenosine (Sigma, catalog #C8031); meta-chlorophenylpiperazine (Sigma, catalog #125180); valproic acid (Sigma, catalog #P4543). The compounds were formulated as either solutions (valproic acid, levetiracetam) or as suspensions (all others). While all investigational compounds were tested in a blinded fashion and quantified in their entirety within the ETSP, only the results with carbamazepine, clobazam, levetiracetam, and valproic acid will be extensively discussed herein. The results with the remaining compounds, e.g. retigabine and tiagabine, have been discussed previously [6, 8], or will be presented in greater detail elsewhere.
Timeline of Testing
All compounds were evaluated and quantified in their entirety within the ETSP Identification phase of testing during the period of August–December 2015. The entirety of the dataset will be made freely available on the NINDS-sponsored PANAChE database (https://panache.ninds.nih.gov).
Statistics
All median effective/toxic doses were quantified by the Probit method originally described by Finney and colleagues [32]. The frequency and duration of in vitro spontaneous bursts in the presence and absence of each investigational compound was measured for analysis by Student’s t test, with statistical significance defined as p < 0.05. The open field activity assay was quantified by one-way ANOVA, with p < 0.05 considered statistically significant. With the exception of Probit calculations and in vitro bursting activity analysis (pCLAMP), all statistical analysis was conducted in GraphPad Prism version 5.0 or later.
Results
Modified Testing Protocol for Pharmacoresistant Epilepsy
Based on the recommendation from the 2015 NINDS Working Group report to develop novel therapies for drug refractory epilepsy, the ETSP testing approach was revised to identify therapies effective in models of pharmacoresistant seizures. To achieve this goal, the following models were prioritized in the ETSP testing approach (Fig. 1):
Assays in the initial Identification phase (which will be the focus of the present manuscript) include MES (mouse and rats), 6 Hz 44 mA (mouse), corneal kindled mouse, and the spontaneous bursting hippocampal slice (rat).
Assays in the Differentiation phase include the LTG-resistant amygdala-kindled rat, intrahippocampal KA mouse model of MTLE, and the chronically-epileptic rat with video-EEG monitoring.
Other current efforts in the program, including screening in Theiler’s virus-treated mice (a model of viral encephalitis associated epilepsy) and models for identifying antiepileptogenic agents, are also not included in this present manuscript. Prior validation has been previously conducted and reported [40–42].
The revised testing approach (Fig. 1) focuses on early evaluation in mouse models of generalized seizures (MES) or pharmacoresistant seizures (6 Hz 44 mA) with additional opportunities to identify efficacy of novel compounds in more etiologically-relevant rodent models of chronic network hyperexcitability (corneal kindled mouse) and in an in vitro assay (rat spontaneous bursting slice). Successful progression of a compound through the Identification phase would lead to the evaluation for activity against pharmacoresistant seizures in epileptic substrates; e.g. LTG-resistant amygdala-kindled rat, the post-intrahippocampal KA MTLE mouse [18], and rat model of KA status epilepticus-induced spontaneous recurrent seizures.
Identification and Quantification of Activity in Mice
The activity of the investigational compounds was evaluated in a blinded fashion (compounds were assigned the identifiers of A–J only) and provided to technical staff in uniform amber vials. The activity of an investigational compound was first assessed in the MES and 6 Hz 44 mA assay following i.p. and p.o. administration. Oral activity has now also been prioritized to circumvent any potential confounding effects of i.p. formulation (e.g. solution vs. suspension), as well as to provide more clinically-relevant route of administration information early in the drug discovery process. Activity was determined at 0.5 and 2 h post drug administration in these assays using default doses of 30, 100, and 300 mg/kg (i.p. or p.o.; Table 1). It should be noted that NINDS has, at any point, the ability to customize the dose range based on known pharmacology or pharmacokinetic parameters of a candidate compound. Compounds found to exhibit activity in any portion of the initial identification were determined to be “hits” and were then candidates for quantitative evaluation studies in the appropriate assay and route of administration (Table 1).
Table 1.
Effect of prototype compounds in mouse models of seizure and chronic network hyperexcitability. Effective doses are listed where calculated (mg/kg)
| Generic name | ASP ID # | Mouse I.P identification | Mouse P.O. identification | Mouse I.P. quantification | Mouse P.O. quantification | Mouse CKM (I.P.) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MES | 44 mA | TOX | MES | 44 mA | TOX | MES (ED50) | 44 mA (ED50) | TOX (TD50) | MES (ED50) | 44 mA (ED50) | TOX (TD50) | ID | Quant | ||
| Acetaminophen | 490002 | − | − | − | − | − | − | NT | NT | NT | NT | NT | NT | + | >450 |
| Carbamazepine | 490003 | + | + | + | + | + | + | 8.38 | 31.4 | 45.6 | 11.35 | 63.7 | 95.4 | + | 6.42 |
| Levetiracetam | 490004 | − | + | − | − | + | − | NT | >1500 | >1500 | NT | >500 | >500 | + | Inc |
| Retigabine | 490005 | + | + | - | + | + | + | 25.7 | 32.5 | 129 | 57.0 | 75.1 | >500 | + | 13.65 |
| Clobazam* | 490006 | + | + | + | + | + | − | 16.5 | 4.5 | 26.1 | 29.9 | 6.99 | 196.7 | + | 2.53 |
| Tiagabine* | 490007 | − | + | − | − | + | − | NT | 1.03 | 5.84 | NT | 1.87 | 33.2 | NT | NT |
| N6-Cyclopentyladenosine | 490008 | + | + | + | + | + | + | 1.01 | 0.19 | 0.94 | 4.43 | 1.36 | 6.94 | − | NT |
| m-CPP | 490009 | − | − | + | − | − | − | >60 | >55 | 52.6 | NT | NT | NT | − | NC |
| Valproic acid | 490010 | - | + | − | − | − | − | >60 | 239.2 | >300 | >300 | >300 | <300 | + | 94.02 |
NT Not Tested due to inactivity in screen
NC ED50 Could Not Be Calculated
(+) Effect observed at at least one dose and time point tested
(−) No effect observed at any dose and time point tested
Inc. Incomplete, testing started but not completed
Starting doses reduced from default
Acetaminophen exhibited no activity in any mouse screening assay (MES or 6 Hz) following either route of administration (p.o. or i.p.); therefore, this compound was advanced to the corneal kindled mouse, to determine whether protection could be observed in an epileptic substrate (discussed below). The initial screening doses of CLB were reduced to 3, 10 and 30 mg/kg (i.p. and p.o.), as would be standard practice for compounds from known mechanistic classes, such as the benzodiazepines. CLB exhibited activity in the MES and 6 Hz screens following administration by both routes; motor impairment was only detected following i.p. administration. As would be expected, CBZ demonstrated activity, as well as toxicity, in all aspects of the initial screen at the default doses tested (Table 1). Neither LEV nor VPA demonstrated activity in the MES identification screen (i.p. or p.o.), but did demonstrate activity in the 6 Hz assay (i.p.). In contrast to LEV, VPA did not demonstrate activity in the 6 Hz 44 mA assay following administration by the oral route. N6-CPA demonstrated activity in all initial screens, albeit the starting doses were reduced to 3, 10 and 30 mg/kg based on the robust activity of this mechanistic class [43]. Lastly, m-CPP demonstrated adverse effects in the screening assay at the doses tested (reduced to 3, 10, and 30 mg/kg), albeit one out of four mice were protected in the MES test at 10 and 30 mg/kg. Because of this potential activity and the fact that this compound had not yet been evaluated in the ETSP, follow up studies with i.p. administration of m-CPP were conducted to define the potential for activity and toxicity in the initial mouse identification assays (Table 1). Furthermore, m-CPP is an example of a compound of sufficient mechanistic novelty to warrant further evaluation in the revised ETSP testing approach. These data demonstrate that the mouse identification and quantification approach using the MES and 6 Hz 44 mA tests as first-line screens can effectively identify a broad range of mechanistically-rich compounds so as to inform on follow-on evaluations in naïve rats, as well as further evaluations in etiologically-relevant rodent models of epilepsy (e.g. corneal kindled mouse and LTG-resistant rat).
The quantitative assays allow for the determination of protective index (PI; ED50/TD50), which may inform on the extent of an investigational compound’s safety margin. N6-CPA demonstrated robust potency in the mouse assays. In the MES test, the i.p. ED50 for N6-CPA was determined to be 1.01 mg/kg, but the TD50 was 0.94 mg/kg. Therefore, the i.p. PI of N6-CPA was less than 1.0 in the MES test. However, in the 44 mA 6 Hz test, the i.p. ED50 was 0.19 mg/kg, giving an i.p. PI of 4.95 in this assay. The oral route of administration with N6-CPA demonstrated similar activity: MES ED50 of 4.43 mg/kg and TD50 of 6.94 mg/kg (PI of 1.56). In the 6 Hz 44 mA test, the oral route ED50 with N6-CPA was 1.36 mg/kg (PI of 5.1). The ED50 (i.p.) of m-CPP in the MES and 6 Hz 44 mA test was determined to exceed the highest dose tested (60 mg/kg and 55 mg/kg, respectively). The TD50 (i.p.) was determined to be 52.6 mg/kg, therefore an i.p. PI for this compound in this test was less than 1.0.
Any compound found to be safe and effective in this 6 Hz assay would become a candidate for evaluation in subsequent Differentiation phase assays, including the LTG-resistant rat. However, in this validation effort, all compounds that demonstrated efficacy in the initial screening (MES or 6 Hz) of the Identification phase were subjected to evaluation in the corneal kindled mouse. The exception was TGB, which was not evaluated in corneal kindled mouse in this screening assay, but has previously demonstrated activity in this model [8]. Activity with acetaminophen was observed 30 min after drug administration in the corneal kindled mouse, albeit an i.p. ED50 was determined to exceed the highest dose tested of 450 mg/kg (Table 1). Testing was also stopped at 450 mg/kg because 1 out of 4 corneal kindled mice died from this high, potentially hepatotoxic dose [44]. The doses and protection observed with acetaminophen adminstration in the corneal kindled mouse were as follows: 20 mg/kg (1/8 protected); 80 mg/kg (0/8 protected); 120 mg/kg (1/8 protected); 150 mg/kg (2/8 protected); 175 mg/kg (0/12 protected); 450 mg/kg (3/4 protected, 1/4 death 24-hours later). Lastly, N6-CPA was evaluated for activity in this model of chronic network hyperexcitability. However, doses of 0.1 and 2 mg/kg resulted in average seizure scores of 4.75 ± 0.25 (SEM) and 4.88 ± 0.13, respectively. Thus, no further testing was conducted to calculate an ED50 with N6-CPA in this model. Based on the absence of any activity in the mouse screening assays, m-CPP was a candidate for evaluation in the corneal kindled mouse according to the revised ETSP testing flowchart (Table 1). Thus, m-CPP was screened for activity in the corneal kindled mouse model. Upon administration of m-CPP 15 min prior to testing of corneal kindled mice (20 mg/kg, i.p.), no mice (0/8) were protected and 3/8 exhibited significant MMI on the rotarod assay. Administration of 30 mg/kg (n = 2 mice) and 40 mg/kg (n = 1 mouse) m-CPP was also associated with lack of protection, and there was significant MMI in all animals tested. Corneal kindled mice treated with m-CPP exhibited severe adverse side effects, including splaying, tremors, and mortality following seizure at the highest doses tested. Thus, no further quantitative testing was conducted with m-CPP in the corneal kindled mouse nor in any Differentiation phase models. For reference, the prototype ASDs CBZ (6.42 mg/kg, PI of 7.1), CLB (2.53 mg/kg, PI of 10.3) and VPA (94.0 mg/kg, PI of >94) were indeed found effective in this model and ED50s were determined (Table 1). The PI was calculated based on the i.p. TD50 for each compound. Thus, this screening approach in mice can effectively identify prototype ASDs of diverse mechanistic classes, inform on the potential for adverse effects liability, as well as differentiate compounds for subsequent evaluation in models of pharmacoresistant seizures in an epileptic substrate (i.e. Differentiation phase tests).
Identification and Quantification of Activity in Rats
The investigational compounds were screened for activity in the rat MES test following i.p. and p.o. administration (Table 2). In the instance that an investigational compound presently under evaluation was an actual investigator-identified compound submitted to the ETSP, inactivity in the mouse screening assays alone would not be sufficient to stop further testing, given sufficient rationale or scientific justification. As in the mouse assays, acetaminophen did not demonstrate activity in any rat assays, thus further in vivo quantitative testing with this compound in rats was not pursued. N6-CPA demonstrated activity in the rat MES test following both i.p. and p.o. administration. However, only an i.p. ED50 could be quantified (6.7 mg/kg), but the TD50 was determined to exceed 10 mg/kg (i.p.; PI of 1.5). Interestingly, m-CPP did not exhibit activity in the mouse MES test with either route of administration, whereas it was found to have activity in the rat MES test. An i.p. ED50 was calculated for this compound in this model to be 37.1 mg/kg; albeit this dose exceeded the i.p. TD50, which was determined to be less than 10 mg/kg. Interestingly, the standard starting doses used to evaluate VPA (30 and 100 mg/kg) were likely insufficient in the initial screening to identify activity of this compound in rats.
Table 2.
Effect of prototype compounds in rat models of seizure and chronic network hyperexcitability. Effective doses are listed where calculated (mg/kg)
| Generic name | Rat I.P. identification | Rat P.O. identification | Rat I.P. quantification | Rat P.O. quantification | |||||
|---|---|---|---|---|---|---|---|---|---|
| ASP ID # | MES | TOX | MES | TOX | MES (ED50) | TOX (TD50) | MES (ED50) | TOX (TD50) | |
| Acetaminophen | 490002 | − | − | − | − | NT | NT | NT | NT |
| Carbamazepine* | 490003 | + | + | + | + | 6.5 | 22.9 | 10.9 | >500 |
| Levetiracetam | 490004 | − | − | − | − | NT | NT | NT | NT |
| Retigabine* | 490005 | + | + | + | + | 4.01 | 23.4 | 12.1 | 64.4 |
| Clobazam* | 490006 | − | − | − | − | 36.3 | 15.7 | - | NT |
| Tiagabine* | 490007 | − | + | − | − | NT | NT | NT | NT |
| N6-Cyclopentyladenosine* | 490008 | + | + | + | + | 6.7 | <10 | >10 | >10 |
| m-CPP | 490009 | + | − | + | − | 37.1 | <10 | - | NT |
| Valproic acid | 490010 | − | − | − | − | NT | NT | NT | NT |
NC ED50/IC50 Could Not Be Calculated
(+) Effect observed at at least one dose and time point tested
(−) No effect observed at any dose and time point tested
NT Not Tested
Starting Doses Reduced from Default
Quantification of Adverse Effects in Rats
In addition to the quantitative evaluation of MMI in rats for the calculation of a TD50 following i.p. or p.o. administration of an investigational compound, quantitation of locomotor activity in an open field(OF) is now implemented to objectively determine MMI [11]. The doses of each compound in this assay were based on those used for the MMI evaluation in rats following i.p. administration. The activity of m-CPP and CBZ are presented as examples (Fig. 2). Both compounds at the doses tested induced significant reductions in motor activity, thereby providing an additional quantitative evaluation of the extent of motor impairment following administration of an investigational compound. Administration of m-CPP induced a significant, dose-dependent reduction in total distance travelled in the OF (F = 35.97, p < 0.0001; Fig. 2a), with post-hoc Dunnett’s test demonstrating that both 45 and 60 mg/kg significantly reduced exploratory behavior (p < 0.0001, each).
Fig. 2.
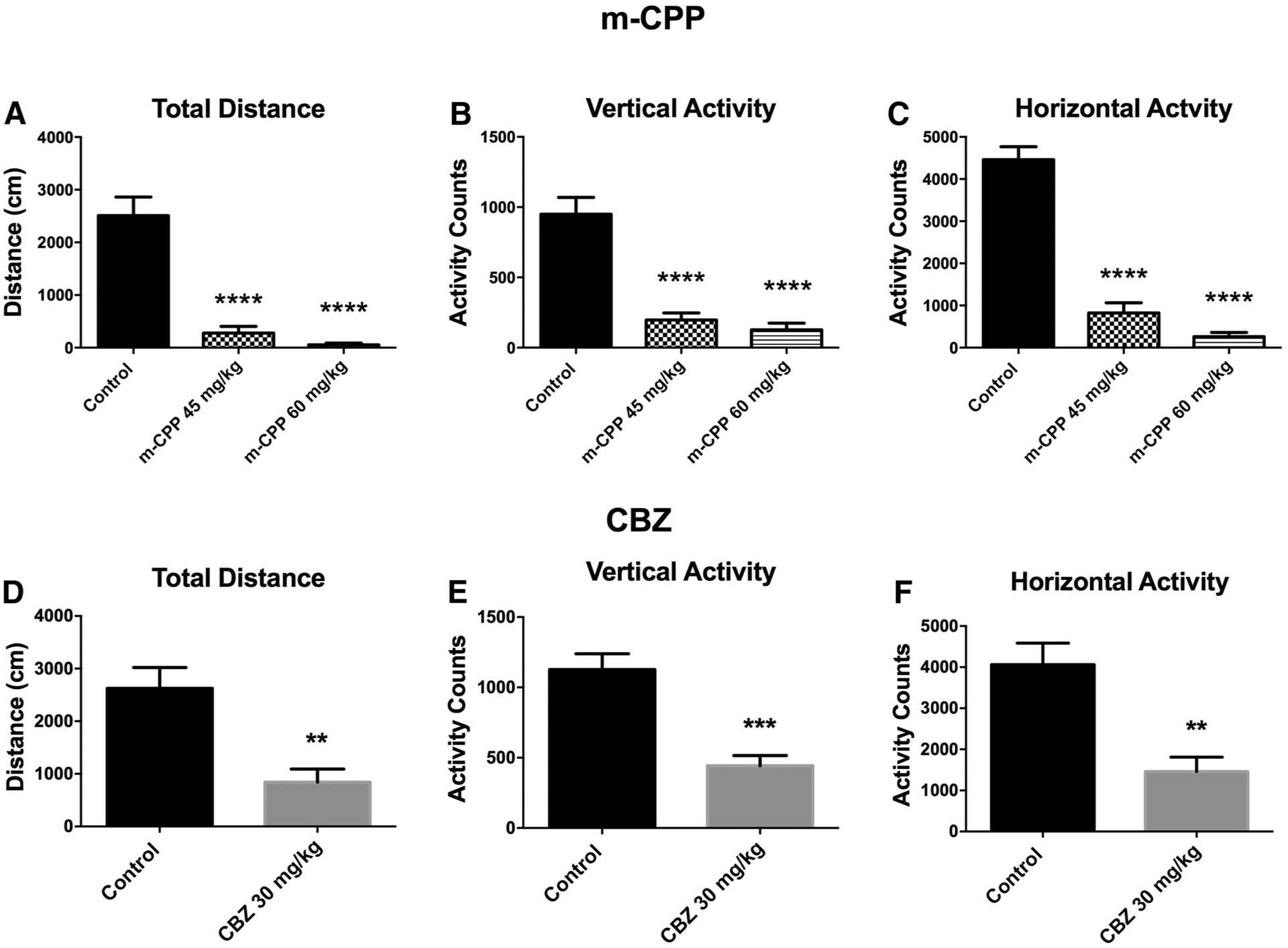
Open field activity of rats was quantified by an automated program following administration of doses of m-CPP (a–c) or CBZ (d–f) that were visually observed to induced minimal motor impairment in the subjective MMI screen. a–c Administration of m-CPP induced dose-dependent suppression of all outcome measures of motor activity. ****Indicates significantly different from control, p < 0.0001. d–f Administration of a single dose of CBZ induced a robust reduction of motor activity in the open field, consistent with previous reports for this compound at a similar (33 mg/kg) dose [11]. **Indicates significantly different from control, p < 0.01; ***p < 0.001
There were similar effects on vertical activity (F = 32.77, p < 0.0001; Fig. 2b) and horizontal (F = 90.29, p < 0.0001; Fig. 2c); post-hoc analysis showed similar effects of dose on both measures (p < 0.0001 all). The effect of CBZ at 30 mg/kg also induced significant reductions in locomotor activity (Fig. 2d–f); effects that are consistent with previously reported observations with CBZ in this assay [11]. Total distance travelled (t = 3.48, p = 0.0045; Fig. 2d), vertical activity (t = 4.73, p = 0.0005; Fig. 2e) and horizontal activity (t = 3.81, p = 0.0025; Fig. 2e) were all significantly reduced in CBZ-treated rats. The effect of other prototype ASDs on rat performance in an open field has been previously presented in greater detail [11]. Thus, the open field activity monitor provides a robust and quantitative means to evaluate effects of investigational compounds on exploratory behavior and motor performance of rats in an unbiased, automated fashion.
Identification and Quantification of Activity Against Spontaneous Bursting in the Hippocampal Slic
The in vitro spontaneous bursting slice from post-KA SE rats is positioned as a “last-chance” approach to identify the activity of an investigational compound against pharmacoresistant seizure-like activity when all other first-line in vivo screening studies have not demonstrated efficacy. Nonetheless, all investigational compounds of this present validation effort were screened for potential activity against spontaneous bursts (100 μM initial concentration; Table 3). Any compound found to be effective was then quantified for activity on spontaneous burst rate and duration (Table 3). Acetaminophen, indeed, demonstrated no effect in the initial 100 μM screen, thus it was not a candidate for further quantification. Of note, VPA also was ineffective in the initial 100 μM screen (Table 3). However, based on the in vivo activity (Tables 1, 2), it would have been a candidate for further quantification in this assay in the situation that VPA was an investigational compound submitted to the ETSP by an outside party. All other investigational compounds were found to be active at the initial screening concentration (100 μM), thus quantification was attempted to define the median inhibitory concentration (IC50) on spontaneous bursts rate and duration (Table 3) if an IC50 for that compound had not previously been reported by our group (e.g. CBZ and RTG [14]). In support of their in vivo efficacy, N6-CPA and m-CPP exhibited inhibitory effects on measures of SB activity. N6-CPA robustly inhibited SB rate, with an IC50 of 0.005 μM; effects on SB duration were also quite potent, with a calculated IC50 of 0.19 μM. The calculated inhibitory concentrations on SB rate and duration for m-CPP were 384 and 405 μM, respectively (Table 3). Thus, the in vitro spontaneous bursting hippocampal slice model exhibits the potential to identify agents that may have antiseizure activity and can effectively differentiate the activity of such investigational compounds to inform on preclinical development efforts.
Table 3.
Effect of prototype compounds on spontaneous bursting of a mEC-HC slice derived from adult rats following KA-induced SE. Effective concentrations are listed where calculated (μM)
| Cmpd ID | Generic name | In vitro Post-KA rat bursting slice | |||
|---|---|---|---|---|---|
| ASP ID # | ID | SB rate: IC50 (uM) | SB duration: IC50 (uM) | ||
| A | Acetaminophen | 490002 | − | NC | NC |
| B | Carbamazepine | 490003 | + | N/A | N/A |
| D | Retigabine | 490,005 | + | N/A | N/A |
| G | N6-Cyclopentyladenosine | 490,008 | + | 0.005 | 0.19 |
| H | m-CPP | 490,009 | + | 384 | 405 |
| J | Valproic acid | 490,010 | − | NC | NC |
NC IC50 not calculated; inactive at 100 uM screen concentration
N/A Activity observed but IC50 not presently reported (see [22])
(+) Effect observed at 100 uM screening concentration
(−) No effect observed at 100 uM screening concentration
Discussion
Based on the 2015 NINDS Working Group recommendations to identify and target novel therapies for pharmacoresistant patient populations, a decision was made to revise the preclinical testing approach in use at the NINDS ETSP (Fig. 1). In an effort to validate the potential suitability of the initial Identification phase of this revised approach, a number of mechanistically-diverse, FDA-approved ASDs were evaluated in a blinded fashion over the course of 4 months in autumn of 2015 (Tables 1, 2, 3). Additional agents were also selected to interrogate specific mechanisms of action that are hypothesized to be of relevance to the management of pharmacoresistant epilepsy [28, 45, 46]. This included the adenosine A1-type receptor selective agonist, N6-cyclopentyladenosine, and the broadly-acting serotonin receptor agonist, meta-chlorophenylpiperazine (m-CPP). Lastly, acetaminophen was originally selected to act as a negative control due to the similar molecular weight (151 g/mol) to valproic acid (144 g/mol), and the fact that it had no known in vivo anticonvulsant activity in established acute models of seizure (i.e. MES) commonly in use for the early identification of promising anticonvulsant agents.
Some of the models presently in use in the revised ETSP testing platform are resistant to sodium channel-blocking agents. However, the barrier to entry into the Differentiation phase is high; the 6 Hz 44 mA test is quite resistant to most available ASDs [6]. Indeed, this present report has confirmed this earlier finding that levetiracetam exhibits limited potency in the 44 mA stimulus intensity 6 Hz test (Table 1). The additional models are included to provide further characterization and comparative differentiation data for any compound that is determined to demonstrate efficacy in this assay, or the MES, corneal kindled mouse, or spontaneous bursting hippocampal slice. With this identification phase data in mind, effective compounds can then be advanced to the Differentiation phase assays, which include the LTG-resistant amygdala-kindled rat, intrahippocampal-KA mouse model, and the post KA-SE rat model of epilepsy. However, the kainate rat is also not without limitations from a drug-discovery and screening perspective: the size of rodents will require large amounts of drug for chronic administration and pharmacological profile is the less well-characterized [26, 47]. Therefore, the Identification and Differentiation phase tests were selected to develop a differentiation profile of a promising investigational antiseizure drug, but this is not to say that selected models available within the ETSP are the only models that should be used for drug discovery and development purposes.
The results of this evaluation of the Identification phase demonstrate a number of opportunities to identify novel pharmacotherapies for the treatment of pharmacoresistant seizures in patients. First, this testing strategy represents a leaner approach to drug screening supported by the ETSP: extraneous in vivo tests are removed, total animal numbers are reduced, compound requirements are minimized, and testing timeframe for investigators is shortened. The Identification phase now places greater emphasis on the early identification of efficacy in the MES test and 6 Hz 44 mA test in mice. However, should a compound fail these initial screens in naïve rodents, “fail safes” in relevant disease models are available (e.g. corneal kindled mouse and/or spontaneous bursting hippocampal slice from rats). Second, this approach may indeed identify mechanistically-diverse pharmacotherapies. This hypothesis is supported by the presently reported results for evaluation of numerous compounds in the Identification phase, which demonstrated differentiation between approved ASDs and mechanistically-relevant agents, including acetaminophen, N6-CPA and m-CPP. By including the in vitro spontaneous bursting hippocampal slice derived from post-SE rats, compounds that may have metabolic or brain penetration challenges may still be evaluated for efficacy with the potential to demonstrate activity in an etiologically-relevant model of pharmacoresistant seizure-like activity [14]. Indeed, should a compound with a unique mechanism of action require proof of concept demonstration of ability to suppress seizure-like activity, the spontaneous bursting slice assay can provide such a suitable platform. Third, emphasis is placed on the earlier identification of oral activity of investigational compounds; this may provide more information to the compound’s sponsor to accelerate the drug development process. Fourth and finally, this screening approach demonstrates greater stringency in compound evaluation. Indeed, LEV and VPA may have been missed in the early Identification phase screens in mice (e.g. MES and 6 Hz assay), but subsequent fail safes such as the corneal kindled mouse and bursting hippocampal slice would provide opportunities to reevaluate potentially efficacious agents in an epileptic substrate. Placing greater reliance on the MES and 6 Hz 44 mA test may miss compounds such as LEV and VPA at the screening doses presently in use [6], but compounds like brivaracetam may not be missed [20, 48]. However, the clinical impact of brivaracetam in pharmacoresistant patient populations awaits further evaluation. Ultimately, this screening approach was designed to attempt to identify compounds efficacious against pharmacoresistant seizures, in both acute models and in models of chronic network hyperexcitability. While acetaminophen, N6-CPA, and m-CPP exhibited interesting activity profiles in the Identification phase of this screening approach, it remains to be determined what their activity profiles will be in the Differentiation phase of this revised testing approach. Given the adverse effects and mortality associated with m-CPP administration to corneal kindled mice, however, further studies with this compound would be approached with strong caution. Moreover, whether other compounds that interrogate the adenosine and serotoninergic system will exhibit improved activity profiles in either portion of this screening approach is undefined. Whether other mechanistic classes, e.g. anti-inflammatory or epigenetic modifiers, may also demonstrate activity in the Identification phase of this revised approach also awaits further study. Nonetheless, the overhaul of the ETSP testing approach now places greater emphasis on the utilization of relevant models of pharmacoresistant seizures to provide a potentially more appropriate platform for the early identification and differentiation of transformative therapies for individuals with epilepsy.
The novel findings of this present report include the anticonvulsant activity profiles of N6-CPA, m-CPP, and acetaminophen in established in vivo and in vitro models of seizures, including pharmacoresistant seizures. We defined the ED50s of N6-CPA in several mouse and rat models of seizures, as well as corresponding activity information in an in vitro model of pharmacoresistant spontaneous bursting. These results align closely with prior reports for activity of adenosine A1-type receptor agonists on seizures [49, 50], as well as clinical evidence that adenosine itself exhibits rapid endogenous anticonvulsant activity [51]. On the contrary, m-CPP demonstrated mixed activity in the in vivo and in vitro assays presently evaluated. In both the mouse and rat assays, motor impairment was detected prior to significant reductions in seizure activity, albeit an ED50 was successfully quantified in male rats in the MES assay. Furthermore, m-CPP was associated with significant adverse side effects in vivo. While m-CPP did demonstrate in vitro efficacy against spontaneous bursts, there is no corresponding output of motor impairment in such a slice preparation. Thus, m-CPP likely possesses antiseizure activity mediated through modulation of the serotonergic system, but this compound also induces significant adverse motor impairment that would likely make it unsuitable for future preclinical studies. Nonetheless, the present in vitro and in vivo results with m-CPP support a growing body of evidence to indicate that stimulation of 5-HT receptors may become a novel means to suppress seizures and seizure-like activity [28, 30]. Indeed, fluoxetine, a selective serotonin reuptake inhibitor, can potentiate the effects of VPA in the MES and subcutaneous pentylenetetrazol tests in mice [52, 53]. Whether augmenting serotoninergic tone in more etiologically-relevant models of seizure, such as the corneal kindled mouse, or Differentiation phase assays, like the LTG-resistant amygdala-kindled rat, will demonstrate an ability to further enhance the activity of traditional ASDs clearly remains to be defined. Future studies with specific serotonin receptor subtype-targeting compounds, whether alone or in combination with traditional ASDs, in these and other models of seizure may indeed further demonstrate the preclinical suitability of this novel mechanistic class for anticonvulsant efficacy.
In contrast to the approved ASDs, m-CPP and N6-CPA, acetaminophen was originally selected to serve as a negative control for activity in these acute seizure models. We presently demonstrate that acetaminophen indeed exhibited no acute effects on MES seizures in rats or mice, as well as no effects against 44 mA 6 Hz seizures in mice. We presently report some acute anticonvulsant activity with acetaminophen in the corneal kindled mouse, albeit an ED50 was determined to exceed the highest dose protected (450 mg/kg; Table 1); a dose which may be associated with hepatotoxicity in mice [44]. Interestingly, acetaminophen has been reported to inhibit status epilepticus (SE)-like activity in vitro at a concentration of 500 μM [54]. The reported mechanism underlying the effects of acetaminophen in that study was postulated to proceed through inhibition of CB1 receptors [54]. However, we did not identify any acute effects of acetaminophen on spontaneous bursting in vitro at concentrations up to 1000 μM, however a concentration of 3000 μM did induce a small, but significant increase in the amplitude of spontaneous bursts (Fig. 2). The physiological relevance of such an amplitude increase at likely hepatotoxic concentrations is ultimately questionable. Furthermore, the study from Deshpande and DeLorenzo utilized cultured hippocampal neurons derived from postnatal day 2 rat brains, whereas the present study utilized spontaneous bursting activity from mEC-HC slices derived from adult rats lesioned in vivo with KA to induce SE prior to slice collection [14]. In light of the in vivo and in vitro inefficacy of the present study, these data suggest that acetaminophen likely does not directly inhibit processes underlying pharmacoresistant seizures at non-toxic doses, either in vivo or in vitro.
These present results further support the diverse screening approach in use by the NINDS ETSP to identify novel therapies for pharmacoresistant epilepsy. Altogether, we now demonstrate the pharmacological validation of the Identification phase of this testing approach using numerous mechanistically-diverse ASDs and compounds of preclinical interest to the treatment of epilepsy. These studies suggest that the Identification phase of this testing platform may identify numerous compounds to be advanced to further differentiation studies in etiologically-relevant models of epilepsy. Importantly, the key modifications that have been implemented by NINDS ETSP include the use of the corneal kindled mouse and bursting hippocampal slice in the Identification phase of testing. As both models exhibit “epilepsy-like” characteristics, they may ultimately prove useful to identify compounds that would otherwise be missed in traditional models of seizure in naïve rodents (e.g. MES and 6 Hz tests). Indeed, we now confirm that the Identification phase itself provides useful differentiation data that would be sufficient to advance the most promising compounds to more labor-intensive, etiologically-relevant models of pharmacoresistant epilepsy. It is anticipated that future screening in this staged approach will likely identify and advance compounds through numerous diverse paths, all of which will carry the potential to be transformative for the patient with pharmacoresistant epilepsy.
Acknowledgements
The authors wish to thank Dr. Cameron Metcalf, University of Utah, and Dr. Shamsi Raessi, NINDS Epilepsy Therapy Screening Program, for helpful discussions and data retrieval. The authors also gratefully acknowledge the assistance of Dr. John Kehne and Dr. Brian Klein, NINDS Epilepsy Therapy Screening Program, for helpful discussions and invaluable review of initial drafts of this manuscript. This work was supported by the National Institute of Neurological Disorder and Stroke’s Epilepsy Therapy Screening Program contract HHSN 271201100029 C to H. Steve White. In December 2015, the contract HHSN 271201100029 C was transferred to KSW. Lastly, the authors acknowledge the intellectual and scientific contributions of Dr. H. Steve White to the conception and implementation of the initial phases of this revised testing flow chart. While not listed as an author, this work would not have been completed without Dr. White’s input and scientific guidance developed through numerous years of experience with the NINDS Epilepsy Therapy Screening Program. This manuscript is submitted as a contribution to a special issue of Neurochemical Research in honor of Dr. White’s career.
References
- 1.Modi AC, Ingerski LM, Rausch JR, Glauser TA, Drotar D (2012) White coat adherence over the first year of therapy in pediatric epilepsy. J Pediatr 161(4):695–699. doi: 10.1016/j.jpeds.2012.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eddy CM, Rickards HE, Cavanna AE (2011) The cognitive impact of antiepileptic drugs. Ther Adv Neurol Disord 4(6):385–407. doi: 10.1177/1756285611417920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Council NANDaSN (2015) Anticonvulsant Screening Program Report.
- 4.Barker-Haliski M, White HS (2015) Antiepileptic drug development and experimental models In: Wyllie E, Gidal BE, Good-kin HP (eds) Wyllie’s treatment of epilepsy. 6th edn Lippencott, Williams & Wilkins, Philadelphia [Google Scholar]
- 5.Putnam TJ, Merritt HH (1937) Experimental determination of the anticonvulsant properties of some phenyl derivatives. Science 85(2213):525–526. doi: 10.1126/science.85.2213.525 [DOI] [PubMed] [Google Scholar]
- 6.Barton ME, Klein BD, Wolf HH, White HS (2001) Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res 47:217–227 [DOI] [PubMed] [Google Scholar]
- 7.Vezzani A, Aronica E, Mazarati A, Pittman QJ (2013) Epilepsy and brain inflammation. Exp Neurol 244:11–21. doi: 10.1016/j.expneurol.2011.09.033 [DOI] [PubMed] [Google Scholar]
- 8.Rowley NM, White HS (2010) Comparative anticonvulsant efficacy in the corneal kindled mouse model of partial epilepsy: correlation with other seizure and epilepsy models. Epilepsy Res 92(2–3):163–169 [DOI] [PubMed] [Google Scholar]
- 9.Matagne A, Klitgaard H (1998) Validation of corneally kindled mice: a sensitive screening model for partial epilepsy in man. Epilepsy Res 31(1):59–71 pii] [DOI] [PubMed] [Google Scholar]
- 10.Loewen JL, Barker-Haliski ML, Dahle EJ, White HS, Wilcox KS (2016) Neuronal injury, gliosis, and glial proliferation in two models of temporal lobe epilepsy. J Neuropathol Exp Neurol. doi: 10.1093/jnen/nlw008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barker-Haliski ML, Vanegas F, Mau MJ, Underwood TK, White HS (2016) Acute cognitive impact of antiseizure drugs in naive rodents and corneal-kindled mice. Epilepsia 57(9):1386–1397. doi: 10.1111/epi.13476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogawski MA (2006) Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res 69(3):273–294. doi: 10.1016/j.eplepsyres.2006.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leclercq K, Matagne A, Kaminski RM (2014) Low potency and limited efficacy of antiepileptic drugs in the mouse 6 Hz corneal kindling model. Epilepsy Res 108(4):675–683. doi: 10.1016/j.eplepsyres.2014.02.013 [DOI] [PubMed] [Google Scholar]
- 14.Smith MD, Adams AC, Saunders GW, White HS, Wilcox KS (2007) Phenytoin- and carbamazepine-resistant spontaneous bursting in rat entorhinal cortex is blocked by retigabine in vitro. Epilepsy Res 74(2–3):97–106. doi: 10.1016/j.eplepsyres.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 15.Suzuki F, Junier MP, Guilhem D, Sorensen JC, Onteniente B (1995) Morphogenetic effect of kainate on adult hippocampal neurons associated with a prolonged expression of brain-derived neurotrophic factor. Neuroscience 64(3):665–674 [DOI] [PubMed] [Google Scholar]
- 16.Bouilleret V, Ridoux V, Depaulis A, Marescaux C, Nehlig A, Le Gal La Salle G (1999) Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience 89(3):717–729 [DOI] [PubMed] [Google Scholar]
- 17.Riban V, Bouilleret V, Pham-Le BT, Fritschy JM, Marescaux C, Depaulis A (2002) Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience 112(1):101–111 [DOI] [PubMed] [Google Scholar]
- 18.Duveau V, Pouyatos B, Bressand K, Bouyssieres C, Chabrol T, Roche Y, Depaulis A, Roucard C (2016) Differential effects of antiepileptic drugs on focal seizures in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. CNS Neurosci Ther 22(6):497–506. doi: 10.1111/cns.12523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, Vezzani A (2011) Interleukin-1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 8(2):304–315. doi: 10.1007/s13311-011-0039-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS (2013) Progress report on new antiepileptic drugs: a summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res 103(1):2–30. doi: 10.1016/j.eplepsyres.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 21.Loscher W (1997) Animal models of intractable epilepsy. Prog Neurobiol 53:239–258 [DOI] [PubMed] [Google Scholar]
- 22.White HS (2003) Preclinical development of antiepileptic drugs: past, present, and future directions. Epilepsia 44(Suppl 7):2–8 [DOI] [PubMed] [Google Scholar]
- 23.Srivastava AK, White HS (2013) Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine-resistant amygdala kindled rats. Epilepsy Res 104(1–2):26–34. doi: 10.1016/j.eplepsyres.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srivastava AK, Alex AB, Wilcox KS, White HS (2013) Rapid loss of efficacy to the antiseizure drugs lamotrigine and carbamazepine: a novel experimental model of pharmacoresistant epilepsy. Epilepsia 54(7):1186–1194. doi: 10.1111/epi.12234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hellier JL, Patrylo PR, Buckmaster PS, Dudek FE (1998) Recurrent spontaneous motor seizures after repeated low-dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Res 31(1):73–84 [DOI] [PubMed] [Google Scholar]
- 26.Thomson KE, Modi A, Glauser TA, White HS (2017) The impact of nonadherence to antiseizure drugs on seizure outcomes in an animal model of epilepsy. Epilepsia In Press, New York: [DOI] [PubMed] [Google Scholar]
- 27.Orban G, Bombardi C, Marino Gammazza A, Colangeli R, Pierucci M, Pomara C, Pessia M, Bucchieri F, Benigno A, Smolders I, De Deurwaerdere P, Di Giovanni G (2014) Role(s) of the 5-HT2C receptor in the development of maximal dentate activation in the hippocampus of anesthetized rats. CNS Neurosci Ther 20(7):651–661. doi: 10.1111/cns.12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamid H, Kanner AM (2013) Should antidepressant drugs of the selective serotonin reuptake inhibitor family be tested as antiepileptic drugs?. Epilepsy Behav 26 (3):261–265. doi: 10.1016/j.yebeh.2012.10.009 [DOI] [PubMed] [Google Scholar]
- 29.Boison D (2012) Adenosine dysfunction in epilepsy. Glia 60(8):1234–1243. doi: 10.1002/glia.22285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS (2017) Progress report on new antiepileptic drugs: a summary of the Thirteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIII). Epilepsia 58(2):181–221. doi: 10.1111/epi.13634 [DOI] [PubMed] [Google Scholar]
- 31.Bialer M, Twyman RE, White HS (2004) Correlation analysis between anticonvulsant ED50 values of antiepileptic drugs in mice and rats and their therapeutic doses and plasma levels. Epilepsy Behav 5 (6):866–872 [DOI] [PubMed] [Google Scholar]
- 32.Finney DJ (1952) Probit analysis A statistical treatment of the sigmoid response curve. University Press, Cambridge [Google Scholar]
- 33.Swinyard EA, Woodhead JH, White HS, Franklin MR (1989) General principles: experimental selection, quantification, and evaluation of anticonvulsants In: Levy RHM RH, Melrum B, Penry JK, Dreifuss FE (eds) Antiepileptic drugs. 3rd edn Raven Press, New York, pp 85–102 [Google Scholar]
- 34.White HS, Woodhead JH, Franklin MR (1995) General principles: experimental selection, quantification, and evaluation of antiepileptic drugs In: Levy RH, Mattson RH, Meldrum BS (eds) Antiepileptic Drugs. 4th edn Raven Press, New York, pp 99–110 [Google Scholar]
- 35.White HS, Johnson M, Wolf HH, Kupferberg HJ (1995) The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital J Neurol Sci 16(1–2):73–77 [DOI] [PubMed] [Google Scholar]
- 36.Racine RJ (1972) Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294 [DOI] [PubMed] [Google Scholar]
- 37.Dunham MS, Miya TA (1957) A note on a simple apparatus for detecting neurological deficit in rats and mice. J Amer Pharm Ass Sci Ed 46:208–209 [DOI] [PubMed] [Google Scholar]
- 38.Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32(3):281–294 [DOI] [PubMed] [Google Scholar]
- 39.Scharfman HE (1997) Hyperexcitability in combined entorhinal/hippocampal slices of adult rat after exposure to brain-derived neurotrophic factor. J Neurophysiol 78(2):1082–1095 [DOI] [PubMed] [Google Scholar]
- 40.Barker-Haliski ML, Heck TD, Dahle EJ, Vanegas F, Pruess TH, Wilcox KS, White HS (2016) Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler’s virus model of temporal lobe epilepsy. Epilepsia 57(12):1958–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barker-Haliski ML, Dahle EJ, Heck TD, Pruess TH, Vanegas F, Wilcox KS, White HS (2015) Evaluating an etiologically-relevant platform for therapy development for temporal lobe epilepsy: effects of carbamazepine and valproic acid on acute seizures and chronic behavioral comorbidities in the theiler’s murine encephalomyelitis virus mouse model. J Pharmacol Exp Ther. doi: 10.1124/jpet.114.222513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomson KE, White HS (2014) A novel open-source drug-delivery system that allows for first-of-kind simulation of nonadherence to pharmacological interventions in animal disease models. J Neurosci Methods. doi: 10.1016/j.jneumeth.2014.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunwiddie TV, Diao L, Kim HO, Jiang JL, Jacobson KA (1997) Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus. J Neurosci 17(2):607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H (2012) Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol 264(3):387–394. doi: 10.1016/j.taap.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boison D (2016) The biochemistry and epigenetics of epilepsy: focus on adenosine and glycine. Front Mol Neurosci 9:26. doi: 10.3389/fnmol.2016.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janusz W, Kleinrok Z (1989) The role of the central serotonergic system in pilocarpine-induced seizures: receptor mechanisms. Neurosci Res 7(2):144–153 [DOI] [PubMed] [Google Scholar]
- 47.Grabenstatter HL, Ferraro DJ, Williams PA, Chapman PL, Dudek FE (2005) Use of chronic epilepsy models in antiepileptic drug discovery: the effect of topiramate on spontaneous motor seizures in rats with kainate-induced epilepsy. Epilepsia 46(1):8–14. doi: 10.1111/j.0013-9580.2005.13404.x [DOI] [PubMed] [Google Scholar]
- 48.Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H (2008) Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol 154(8):1662–1671. doi: 10.1038/bjp.2008.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tosh DK, Paoletta S, Deflorian F, Phan K, Moss SM, Gao ZG, Jiang X, Jacobson KA (2012) Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: receptor docking and potent anticonvulsant activity. J Med Chem 55(18):8075–8090. doi: 10.1021/jm300965a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen HY, Sun H, Hanthorn MM, Zhi Z, Lan JQ, Poulsen DJ, Wang RK, Boison D (2014) Overexpression of adenosine kinase in cortical astrocytes and focal neocortical epilepsy in mice. J Neurosurg 120(3):628–638. doi: 10.3171/2013.10.JNS13918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.During MJ, Spencer DD (1992) Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 32(5):618–624. doi: 10.1002/ana.410320504 [DOI] [PubMed] [Google Scholar]
- 52.Borowicz KK, Piskorska B, Stepniak B, Czuczwar SJ (2012) Effects of fluoxetine on the anticonvulsant action of valproate and ethosuximide in mouse model of myoclonic convulsions. Ann Agric Environ Med 19(3):487–490 [PubMed] [Google Scholar]
- 53.Borowicz KK, Stepien K, Czuczwar SJ (2006) Fluoxetine enhances the anticonvulsant effects of conventional antiepileptic drugs in maximal electroshock seizures in mice. Pharmacol Rep 58(1):83–90 [PubMed] [Google Scholar]
- 54.Deshpande LS, DeLorenzo RJ (2011) Acetaminophen inhibits status epilepticus in cultured hippocampal neurons. Neuroreport 22(1):15–18. doi: 10.1097/WNR.0b013e3283413231 [DOI] [PMC free article] [PubMed] [Google Scholar]