

Abstract
Adaptor proteins modulate substrate selection by AAA+ proteases. The ClpS adaptor delivers N-degron substrates to ClpAP but inhibits degradation of substrates bearing ssrA tags or other related degrons. How ClpS inhibits degradation of such substrates is poorly understood. Here, we demonstrate that ClpS impedes recognition of ssrA-tagged substrates by a non-competitive mechanism and also slows subsequent unfolding/translocation of these substrates as well as of N-degron substrates. This suppression of mechanical activity is largely a consequence of the ability of ClpS to repress ATP hydrolysis by ClpA, but several lines of evidence show that ClpS’s inhibition of substrate binding and its ATPase repression are separable activities. Using ClpS mutants and ClpS-ClpA chimeras, we establish that engagement of the intrinsically disordered N-terminal extension (NTE) of ClpS by ClpA is both necessary and sufficient to inhibit multiple steps of ClpAP-catalyzed degradation. These observations reveal how an adaptor can simultaneously modulate the catalytic activity of a AAA+ enzyme, efficiently promote recognition of some substrates, suppress recognition of other substrates and thereby affect degradation of its menu of substrates in a specific manner. We propose that similar mechanisms are likely to be used by other adaptors to regulate substrate choice and the catalytic activity of molecular machines.
Keywords: AAA+ proteases, Adaptor proteins, Protein degradation, AAA+ unfoldases/translocases, Substrate regulators
INTRODUCTION
Energy-dependent AAA+ proteases, are critical in all domains of life, functioning to maintain proteostasis and to regulate many cellular processes [1, 2]. These proteases consist of a AAA+ ring hexamer that recognizes, unfolds, and translocates protein substrates into the degradation chamber of an associated peptidase [1, 3, 4]. In prokaryotes, protease-associated AAA+ unfoldases recognize small, accessible peptide sequences, called degrons or degradation tags, typically located near the N or C terminus of a protein substrate [5]. For example, the ssrA degron (AANDENYALAA-cooh), which is co-translationally added to the C termini of proteins when translation is compromised, targets ssrA-tagged proteins for degradation by the ClpXP and ClpAP proteases of Escherichia coli [6–8]. N-degrons, in contrast, are single N-terminal amino acids (F, W, Y and L in E. coli) that target substrates for degradation by ClpAP with the assistance of the ClpS adaptor [9, 10].
Adaptor proteins alter the substrate repertoire of AAA+ enzymes and therefore influence many cellular processes [5]. E. coli SspB, one of the best-characterized adaptors, delivers ssrA-tagged substrates to ClpXP. Like SspB, most known AAA+-unfoldase/protease adaptors influence the substrate-recognition step [11, 12]. However, there are more complex adaptor mechanisms. For example, the Bacillus subtilis MecA adaptor regulates both assembly of the ClpCP protease and recognition of the ComK transcription factor [13–15]. ClpS adaptor of the E. coli ClpAP protease represents a unique class of adaptors that regulate ClpA activity either positively or negatively depending on the nature of degron attached to the substrate (Fig 1a). ClpS enhances the degradation of N-degron substrates whereas it inhibits the degradation of ssrA tagged substrates.
Figure 1: The ClpAPS complex.
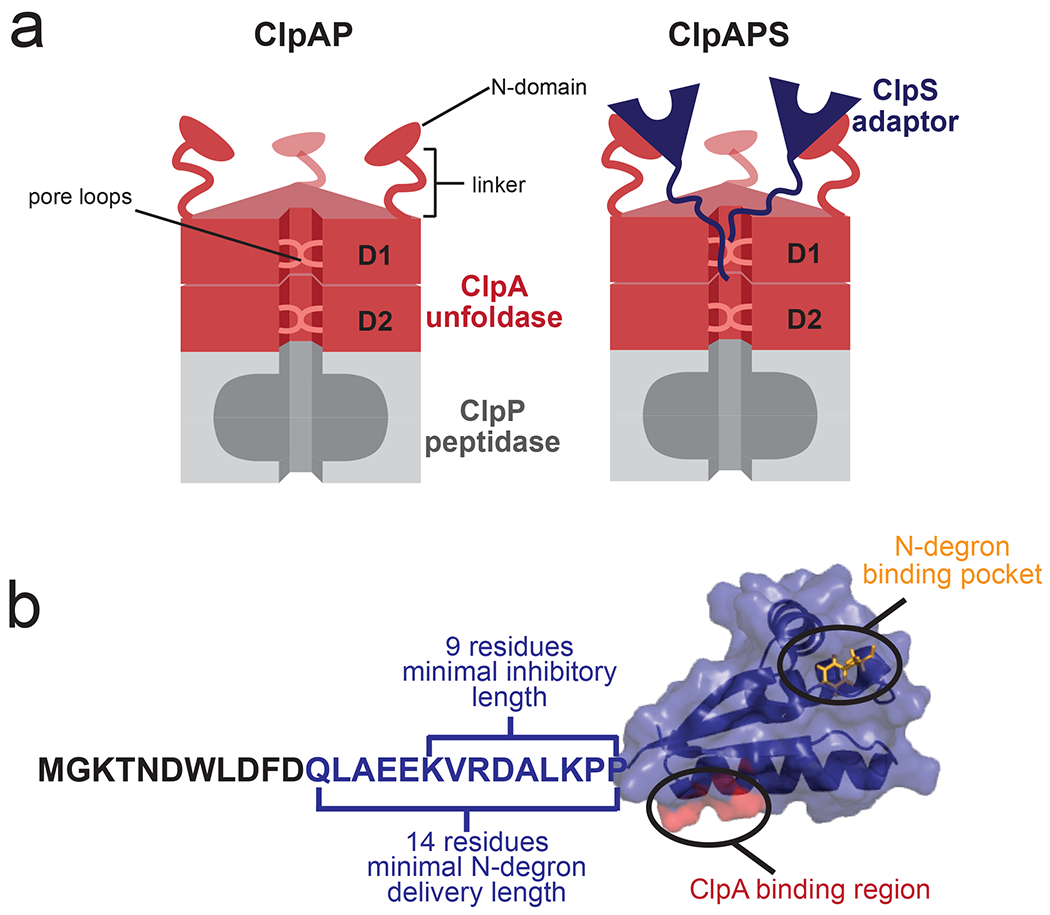
(a) The ClpAP protease. (left) The ClpA hexamer consists of D1 and D2 AAA+ rings, with N-terminal domains connected to the D1 domain of each subunit by a flexible linker. Conserved loops in the ClpA translocation pore grip substrates and mediate translocation and unfolding. (right) The ClpS adaptor binds the N-domain of ClpA. (b) Structure of E. coli ClpS bound to a Phe N-degron (PDB code 3O2B). The ClpS adaptor has an unstructured N-terminal extension (NTE, residues 1-25) and a core domain (residues 26-106), which harbors a binding pocket for N-degrons (orange). A region of ClpS that binds to the N-terminal domain of ClpA is highlighted in red. The portion of the NTE required for ClpS function is color blue.
Here, we dissect the molecular mechanism(s) used by the ClpS adaptor to negatively control degradation of ssrA-tagged substrates by ClpAP, a protease consisting of the hexameric AAA+ ClpA unfoldase and the tetradecameric ClpP peptidase (Fig. 1a). ClpA subunits contain an N-terminal domain and two AAA+ modules (D1 and D2), which assemble into a double-ring homohexamer with an axial translocation pore that aligns with the pore of ClpP (Fig. 1a) [16, 17]. As shown in Fig. 1b, ClpS contains a tightly-folded core domain (residues 26-106), carrying a hydrophobic pocket that binds N-degrons and another surface which binds the N-terminal domain of ClpA. In addition to its folded core, ClpS also has an intrinsically disordered N-terminal extension or NTE (residues 2-25 in E. coli) [18–21]. Current evidence supports an N-degron substrate delivery model in which ClpS enhances degradation by active “handoff” of the N-degron substrate from its binding pocket on ClpS to the translocation pore of ClpA [21, 22]. Prior to this “substrate handoff” from the adaptor to ClpA, ClpS recognizes and binds the N-degron on the substrate, and then a CIpA•ClpS•N-degron-substrate ternary complex assembles. In this complex, the core folded domain of ClpS interacts with the N domain of ClpA, while the ClpS NTE interacts of the ClpA pore. In this three-protein complex, the CIpS•substrate and the ClpS•ClpA binding affinities each are increased 75-fold and 9-fold, respectively, relative to when the third component is missing [21]. Substrate handoff from the adaptor to the enzyme occurs when the NTE of ClpS is engaged, like a degron, by the translocation machinery of the ClpA axial pore. In fact, the NTE is a functional degron in chimeric proteins; for example, NTE-GFP is a good substrate for ClpAP degradation, whereas GFP alone is not [21]. Unlike protein substrates, however, the core domain of ClpS resists ClpA unfolding and thus NTE engagement does not result in ClpS denaturation or degradation [21].
Although ClpS delivery of N-degron substrates to ClpA has been actively studied [10, 21, 23], less is known about the mechanism of ClpS inhibition of ClpAP degradation of ssrA-tagged and related substrates. Some models propose that the NTE directly competes with recognition of ssrA-tagged substrates [18, 24]. Here, we dissect the molecular mechanism by which the ClpS adaptor negatively controls degradation of ssrA-tagged substrates by ClpAP. In contrast to previous reports, we demonstrate that ssrA-tagged substrates do bind, albeit weakly, to the ClpAPS complex but are degraded very slowly as a consequence of reductions in the rates of substrate unfolding and translocation. We also demonstrate that the NTEs from multiple ClpS molecules are needed to efficiently inhibit degradation and show that the ability of the NTE to suppress the maximal rate of degradation parallels its activity in partially repressing ATP hydrolysis by ClpAP. We discuss the ways in which the ClpS NTE acts as a “degron mimic”, compare the inhibitory and stimulatory activities of ClpS, and consider the implications of our results for general strategies of adaptor protein function.
RESULTS
SsrA-tagged substrates bind to ClpAPS but with weakened affinity.
Does ClpS prevent ClpA binding to ssrA-tagged substrates by a competitive mechanism or reduce ClpA-substrate affinity by a non-competitive mechanism? To measure binding using fluorescence anisotropy, we employed fluorescein maleimide to label an ssrA-tagged variant of the DNA-binding domain of λ repressor containing one cysteine (λ*fl-ssrA) [6, 7], a good substrate for ClpAP, or to label a variant of ClpS containing one cysteine (ClpS*fl) [21]. ClpA binds to λ*fl-ssrA with an affinity of ~0.7 μM (Fig. 2a) in the presence of an ATP analog (ATPγS) that is not hydrolyzed [25]. Next, binding of ClpA to ClpS*fl was assayed in the presence of 30 μM λ-ssrA. Under these conditions, ClpA bound ClpS*fl with an affinity of ~0.16 μM (Fig. 2b), whereas an affinity of ~0.18 μM was previously measured for binding of ClpA to ClpS*fl in the absence of ssrA-tagged substrates [21]. These results are inconsistent with a model of strict competition, which predicts that 30 μM λ-ssrA should decrease the apparent affinity of ClpA for ClpS by a factor of approximately 44 (See Methods).
Figure 2: Binding of λ*fl-ssrA to ClpA or ClpAS and degradation by ClpAP or ClpAPS.
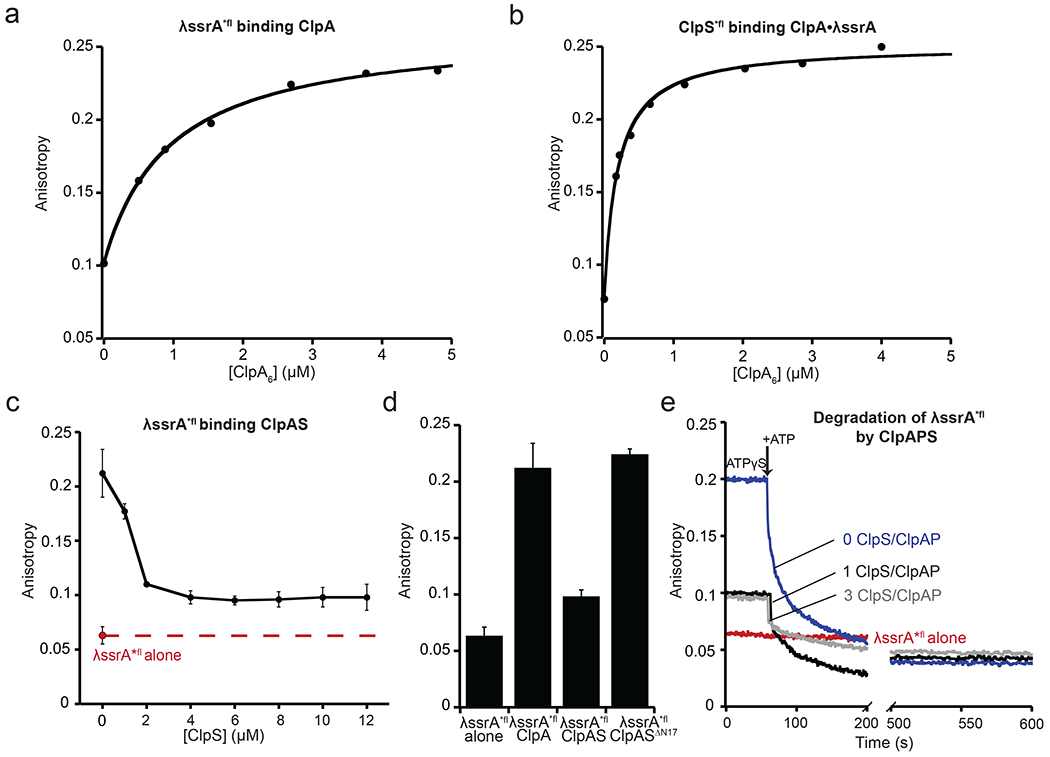
(a) Binding of ClpA to λ*fl-ssrA (0.15 μM) in the presence of 2 mM ATPγS, as assayed by fluorescence anisotropy. The line is fit to a hyperbolic equation with 50% binding (KD) at 740 ± 190 nM. (b) Binding of ClpA to ClpS*fl (0.2 μM) in the presence of λ-ssrA (30 μM) and ATPγS (2 mM), as assayed by fluorescence anisotropy. The line is fit to a quadratic equation for near stoichiometric binding with 50% binding (KD) at 160 ± 51 nM. The KD values in (a) and (b) are averages ± SD (n=3). Data are representative of three independent experiments. (c) Binding of ClpS to ClpA (2 μM) pre-incubated with λ-ssrA*fl (0.15 μM) and ATPγS (2 mM) with increasing concentrations of ClpS. Binding was assayed by equilibrium levels of fluorescence anisotropy. The red dashed line marks the anisotropy of free λ*fl-ssrA. Values are averages (n ≥ 3) ± 1 SD. (d) Fluorescence anisotropy of λ-ssrA*fl (0.15 μM) binding ClpA in the absence or presence of ClpS (2 μM ClpA6, 4 μM ClpS) or ClpSΔN17 (4.5 μM ClpA6, 18 μM ClpSΔN17) and 2 mM ATPγS. (e) Effects of different amounts of ClpS on the kinetics of ClpAP degradation of λ*fl-ssrA, as assayed by fluorescence anisotropy. λ*fl-ssrA was pre-incubated with ClpAP or ClpAPS and ATPγS (2 mM). After ~60s, degradation was initiated by addition of 8 mM ATP. Red trace, λ*fl-ssrA alone. Blue trace, λ*fl-ssrA with ClpAP (2 μM ClpA6, 4 μM ClpP14). Grey trace, λ*fl-ssrA with ClpAPS (2 μM ClpA6, 4 μM ClpP14, 6 μM ClpS). Black trace, λ*fl-ssrA with ClpAPS (2 μM ClpA6, 4 μM ClpP14, 2 μM ClpS).
If λ-ssrA and ClpS compete for ClpA binding by a non-competitive binding mechanism, then excess ClpS should fail to completely displace λ-ssrA from ClpA. To test this prediction, we mixed a small amount of λ*fl-ssrA with a concentration of ClpA sufficient to give ~75% binding of this substrate (2 μM) and then added increasing concentrations of ClpS (Fig. 2c). Importantly, ClpS in two-fold or higher excess over ClpA reduced the anisotropy to a stable plateau that was higher than the anisotropy of λ*fl-ssrA alone. At this plateau ~20% of the λ*fl-ssrA remained bound in a ClpAPS complex while ~80% of this substrate was free, confirming that ClpS and λ*fl-ssrA can bind ClpA at the same time, the hallmark of non-competitive binding. Based on this bound/free ratio, we calculate an affinity (KD) of ~8 μM for the binding of ClpAPS to λ*fl-ssrA under these conditions. Thus, ClpS binding weakens ClpA’s affinity for λ*fl-ssrA ~11-fold. ClpSΔN17, which lacks the 17 N-terminal-most residues of wild-type ClpS, bound ClpA with an affinity of ~0.16 μM (Supplementary Fig. 1) but did not displace λ*fl-ssrA from ClpA (Fig. 2d), establishing that the missing NTE residues are required for non-competitive inhibition.
To test if the intermediate λ*fl-ssrA anisotropy observed in the presence of ClpAPS corresponds to a productive complex, we pre-assembled ClpAP or ClpAPS complexes with λ*fl-ssrA in the presence of ATPγS and monitored anisotropy for ~60 s before adding ATP to initiate degradation. Both with no ClpS and with ClpS in three-fold excess over ClpAP, the anisotropy decreased to a value lower than that of free λ*fl-ssrA following addition of ATP (Fig. 2e), as expected if λ*fl-ssrA was degraded into peptides. Following addition of ATP, the loss of anisotropy was biphasic, likely because ATP hydrolysis causes λ*fl-ssrA dissociation in addition to supporting degradation. We note that non-competitive inhibition of λ*fl-ssrA binding to ClpA saturated at a 2:1 ratio of ClpS:ClpA (Fig. 2c), whereas a 4:1 ratio of ClpS:ClpA is required for strong degradation inhibition [18, 19]. Thus, the mechanisms by which ClpS weakens substrate binding and inhibits overall degradation have distinctive features.
ClpS increases KM and decreases Vmax for SFGFP-ssrA degradation.
To analyze ClpS inhibition of enzyme function, we assayed the effects of ClpS on the steady-state kinetics of ClpAP degradation of super-folder GFP with an ssrA tag (SFGFP-ssrA) [26, 27]. Rates of initial degradation of different concentrations of SFGFP-ssrA were determined by loss of native fluorescence and fit to the Michaelis-Menten equation. With ClpS present in six-fold excess over ClpAP, SFGFP-ssrA was degraded with a 12-fold weaker KM and 4-fold slower Vmax compared to degradation by ClpAP alone (Fig. 3a). Thus, ClpS decreased Vmax/KM, the second-order rate constant for degradation, by a factor of ~50-fold. Addition of Phe-Val, an N-degron dipeptide that stabilizes ClpAPS complexes [21], did not result in substantially stronger inhibition (Fig. 3a), suggesting that ClpA is already saturated with ClpS under the conditions of this experiment. The observed changes in KM and Vmax parameters support a classical mixed-inhibition model that is fully consistent with non-competitive binding. Moreover, the Vmax decrease suggests that ClpS negatively affects one or more mechanical activities of ClpAP.
Figure 3: ClpS inhibits recognition and degradation of an ssrA-tagged substrate.
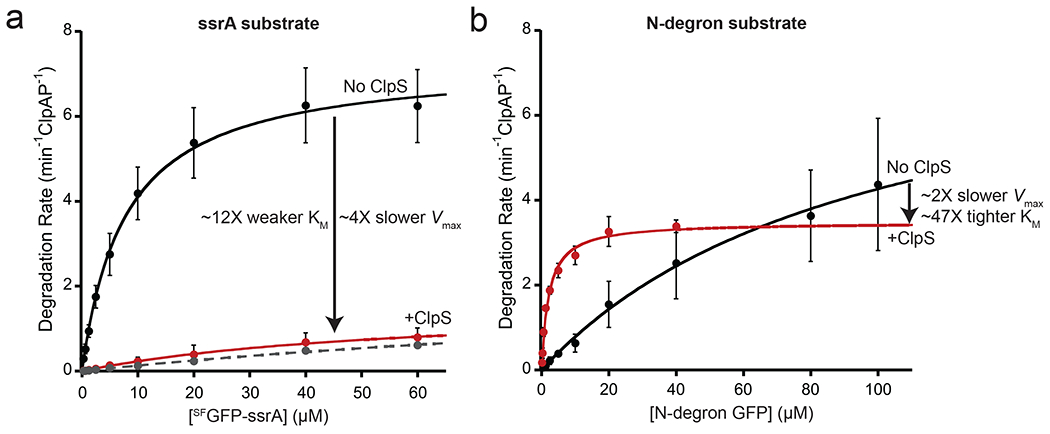
(a) Michaelis-Menten analysis of steady-state degradation of SFGFP-ssrA by ClpAP (black circles) or ClpAPS (red circles). ClpAP (0.4 μM ClpA6, 0.8 μM ClpP14) degraded SFGFP-ssrA with a KM of 7.8 μM and a Vmax of 7.3 min−1 ClpAP−1. ClpS (2.4 μM) weakened KM to 93 μM and reduced Vmax to 1.8 min−1 ClpAPS−1. In the presence of the N-degron dipeptide Phe-Val (10 μM), ClpAPS degraded SFGFP-ssrA with a KM of 167 μM and a Vmax of 2.4 min−1 ClpAPS−1 (Gray dotted line) (b) Degradation of the N-degron substrate ylfvq-GFP by ClpAP (black circles) or ClpAPS (red circles). ClpAP (0.2 μM ClpA6, 0.4 μM ClpP14) degraded ylfvq-GFP with a KM of 98 μM and Vmax of 8.4 min−1 ClpAP−1. ClpS (1.2 μM) tightened the KM to 2.1 μM (47-fold) but slowed Vmax to 3.5 min−1 ClpAPS−1. In both panels, values are average ± 1 SD (n = 3), and solid lines are fits to the Michaelis-Menten equation.
ClpS depression of Vmax has also been observed for ClpAP degradation of N-degron-tagged variants of the I27 domain of human titin (N-titinI27) [10]. Likewise, compared to ClpAP, we found that ClpAPS displayed a reduced Vmax for degradation of the N-degron substrate ylfvq-GFP, even as it caused enhanced N-degron recognition, as revealed by the tighter KM (Fig. 3b). In combination, these results support a model in which ClpS reprograms ClpAP to alter substrate specificity but at a cost of slowing mechanical unfolding and/or translocation of the substrate. For both ssrA-tagged and N-degron tagged substrates, the tags are the first part of the substrates to be degraded by ClpAP. As tag binding provides only the initial enzyme-substrate contact, it seems unlikely that the tags themselves rather than ClpS are responsible for reprogramming translocation of the entire substrate.
ClpS inhibits substrate unfolding and translocation.
Following ClpAP binding and engagement, substrates must be unfolded and translocated through the axial pore of ClpA to allow entry into ClpP for degradation. We sought to determine if ClpS affects the two mechanical activities of ClpA, both of which require ATP hydrolysis. To measure any impact of ClpS on ClpAP machine functions, we studied degradation of a previously described multi-domain substrate, CFP-GFP-titin I27V15P-ssrA (Fig. 4a), in which the CFP and GFP domains have comparable time constants for enzymatic unfolding and translocation [28]. Because degradation of this substrate proceeds from the C-terminus to the N-terminus, GFP fluorescence is lost before CFP fluorescence, and the lag between the GFP and CFP curves depends on the rate at which unfolded GFP (~240 amino acids) is translocated and the CFP domain is unfolded. Notably, the lag for ClpAPS degradation was approximately twice as long as the lag for ClpAP degradation (Fig. 4b), suggesting that ClpS slows translocation and/or unfolding. Indeed, the solid lines in Fig. 4b represent a simulation in which ClpAPS both unfolded and translocated the GFP and CFP domains at half of the rate of ClpAP. These results support the idea that ClpS slows the mechanical unfolding and translocation of ssrA-tagged protein substrates. Moreover, single-molecule estimates of the average times for GFP unfolding and translocation by ClpAP indicate that the translocation is the rate-determining step and hence the majority of the lag in both of the Fig. 4b experiments is likely to represent translocation [29]. Consistent with these data, when we assayed ClpAP and ClpAPS degradation of a stable native substrate (titin I27-ssrA) compared with the same protein unfolded by carboxymethylation of cysteines normally buried in the hydrophobic core (titin I27CM-ssrA), ClpS reduced the rate of degradation of the native substrate (~7-fold) as well as the unfolded substrate (~2-fold) (Figs. 4c, 4d). Thus, both the mechanical processes of ClpA: protein unfolding and protein translocation are negatively regulated by ClpS.
Figure 4: ClpS inhibits post-engagement mechanical steps during ClpAP degradation.
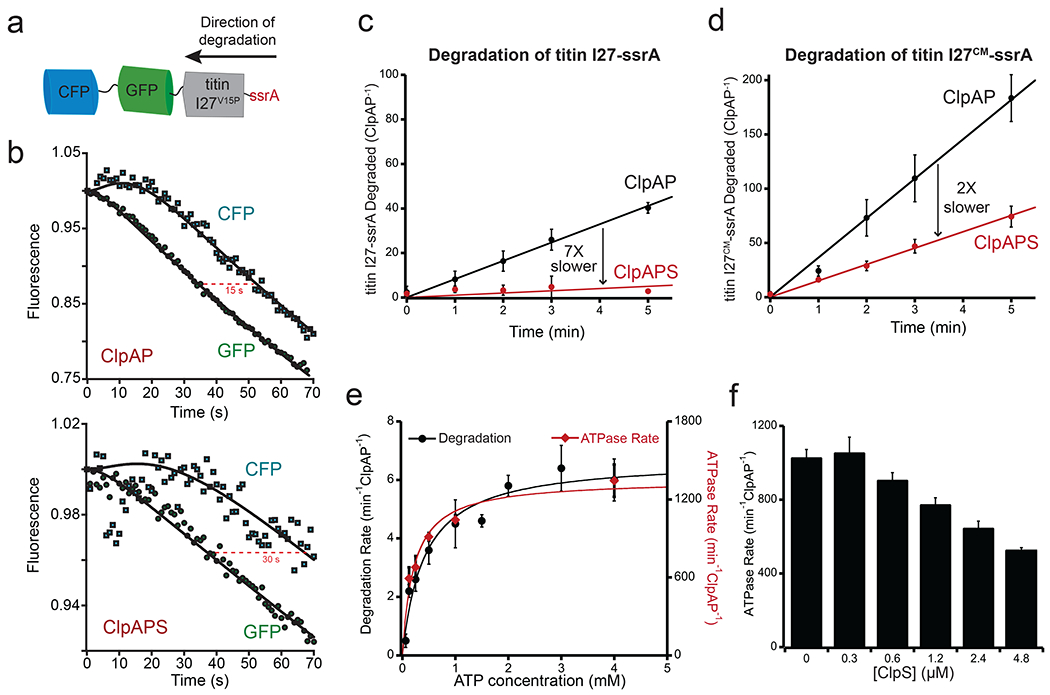
(a) Cartoon of the multi-domain substrate CFP-GFP-titin I27V15P-ssrA. (b) Degradation of the GFP and CFP domains of CFP-GFP-titin I27V15P-ssrA (0.5 μM) by ClpAP (4.5 μM ClpA6, 9 μM ClpP14) in the absence (top) or presence (bottom) of ClpS (27 μM). The curves shown are representative of three independent experiments. The lines are kinetic simulations for a model with first-order rate constants for binding, engagement, and degradation of the titin I27V15P domain (k1), for unfolding of the GFP/CFP domains (k2), and for translocation of the GFP/CFP domains (k3). For the ClpAP simulation, the values of k1, k2, and k3 were 0.00435 s−1, 0.25 s−1, and 0.15 s−1, respectively. For the ClpAPS simulations, these constants were 0.0012 s−1, 0.125 s−1, and 0.075 s−1, respectively. The initial increase in CFP fluorescence results from loss of FRET upon unfolding of the GFP domain. (c) Degradation of [35S]-titin I27-ssrA (40 μM) by ClpAP (0.2 μM ClpA6, 0.4 μM ClpP14) in the absence (black circles) or presence (red circles) of ClpS (1.2 μM), as measured by release of TCA-soluble peptides. ClpAP degraded [35S]-titin I27-ssrA at 7.3 ± 0.39 min−1ClpAP−1. ClpS slowed degradation to 1 ± 0.56 min−1ClpAP−1. (d) Degradation of [35S]-titin I27CM-ssrA (40 μM) by ClpAP in the absence (black circles) or presence (red circles) of ClpS. See (c) for experimental conditions. ClpAP degraded [35S]-titin I27CM-ssrA at 34 ± 5 min−1ClpAP−1. ClpS slowed degradation to 15 ± 1.6 min−1ClpAP−1. Values in (c) and (d) are averages (n = 3) ± 1 SD. (e) Covariation of rates of substrate degradation and ATP hydrolysis. Rates of SFGFP-ssrA degradation (black circles) and ATP hydrolysis (red diamonds) by ClpAP (0.4 μM ClpA6, 0.8 μM ClpP14) were determined at different ATP concentrations. Values are averages (n = 3) ± 1 SD. (f) Suppression of ClpAP ATPase rate by ClpS. ATP hydrolysis rates by ClpAP (0.4 μM ClpA6, 0.8 μM ClpP14) were determined in the presence of 30 μM λ-ssrA at increasing ClpS concentrations. Values are averages (n = 3) ± 1 SD.
ClpS slowing of ClpAP’s mechanical activities is likely to result, at least in part, from suppression of the rate of ATP hydrolysis of ClpAP [19]. In support of this model, we found that the rates of ATP hydrolysis and degradation of SFGFP-ssrA were well correlated over a wide range of ATP concentrations (Fig. 4e). Moreover, when we assayed ATP hydrolysis by ClpAP in the presence of high concentrations of λ-ssrA, increasing concentrations of ClpS reduced ATPase activity ~2-fold (Fig. 4f). ClpA has two ATPase rings, D1 and D2. The top (or N terminal) D1 ring is the slower ATPase and plays important, but auxiliary roles in the mechanical processing of protein substrates. The C terminal D2 ATPase is, in contrast, responsible for ~85% of ClpA’s total ATPase activity and is the major motor involved in protein unfolding and translocation [30]. To investigate whether ClpS was able to regulate this critical D2 motor, we assayed ATP hydrolysis by ClpAP, a variant that carries a WT D2 ring but an ATPase-defective mutation (E286Q) in the D1 ring [31]. ATPase activity of ClpAP was determined in the presence of λ-ssrA at different concentrations of ClpS. ClpA showed a very similar response to ClpS addition as did ClpA, with ATPase activity ~50% inhibited at saturating levels of ClpS (compare Fig. 4f and Supplementary Fig. 2). These data establish that ClpS can inhibit ClpA’s major ATPase motor, likely by contacts between the NTE and the D2 ring. Interestingly, concentrations of ClpS that did not lead to additional weakening of λ-ssrA binding did cause additional reduction in ATPase activity (compare Figs. 2c and 4f), suggesting that these activities of ClpS are distinct.
Inhibition requires ClpA access to the ClpS NTE.
The length but not the sequence of the ClpS NTE is critical for inhibiting ClpAP degradation of ssrA-tagged substrates and for delivering N-degron substrates to ClpAP, with the latter activity requiring engagement of the NTE by the ClpA translocation machinery [18, 19, 21, 22, 32]. To test the importance of NTE access during inhibition, we fused H6-tagged mouse dihydrofolate reductase to ClpS (H6-DHFR-ClpS) [22]. The H6 tag of this protein serves as a degron for ClpA, but access to the full NTE is impeded unless ClpAP can unfold and degrade DHFR, which occurs slowly in the presence of methotrexate (MTX) [33]. Fig. 5a shows ClpAP degradation of SFGFP-ssrA with and without wild-type ClpS (open and closed black symbols, respectively) and with H6-DHFR-ClpS in the absence or presence of MTX (open and closed red symbols, respectively). H6-DHFR-ClpS + MTX inhibited SFGFP-ssrA degradation only marginally, yielding a curve similar to the no ClpS control. In contrast, when H6-DHFR-ClpS was present in the absence of MTX, we observed a biphasic inhibition curve, with the rate of SFGFP-ssrA degradation between the no ClpS (uninhibited) and +ClpS (fully inhibited) conditions. During the initial phase of ~300 s, SFGFP-ssrA was clearly degraded at approximately half the rate observed in the absence of any inhibitor. However, during the remainder of the time course, degradation was much slower, reflecting enhanced inhibition. An attractive interpretation of this time course is that the first phase, when inhibition is poor, corresponds to the time required for most or all of the H6-DHFR portion of the adaptor-chimera to be degraded. This degradation releases protein that is identical to WT ClpS, with a small region of DHFR (estimated to be approximately 15 amino acids) N-terminal to the ClpS NTE. This proteolytically trimmed chimera then functions as a good inhibitor of SFGFP-ssrA degradation, although perhaps not as strongly inhibitory as native ClpS.
Figure 5: The NTE is critical for inhibition.
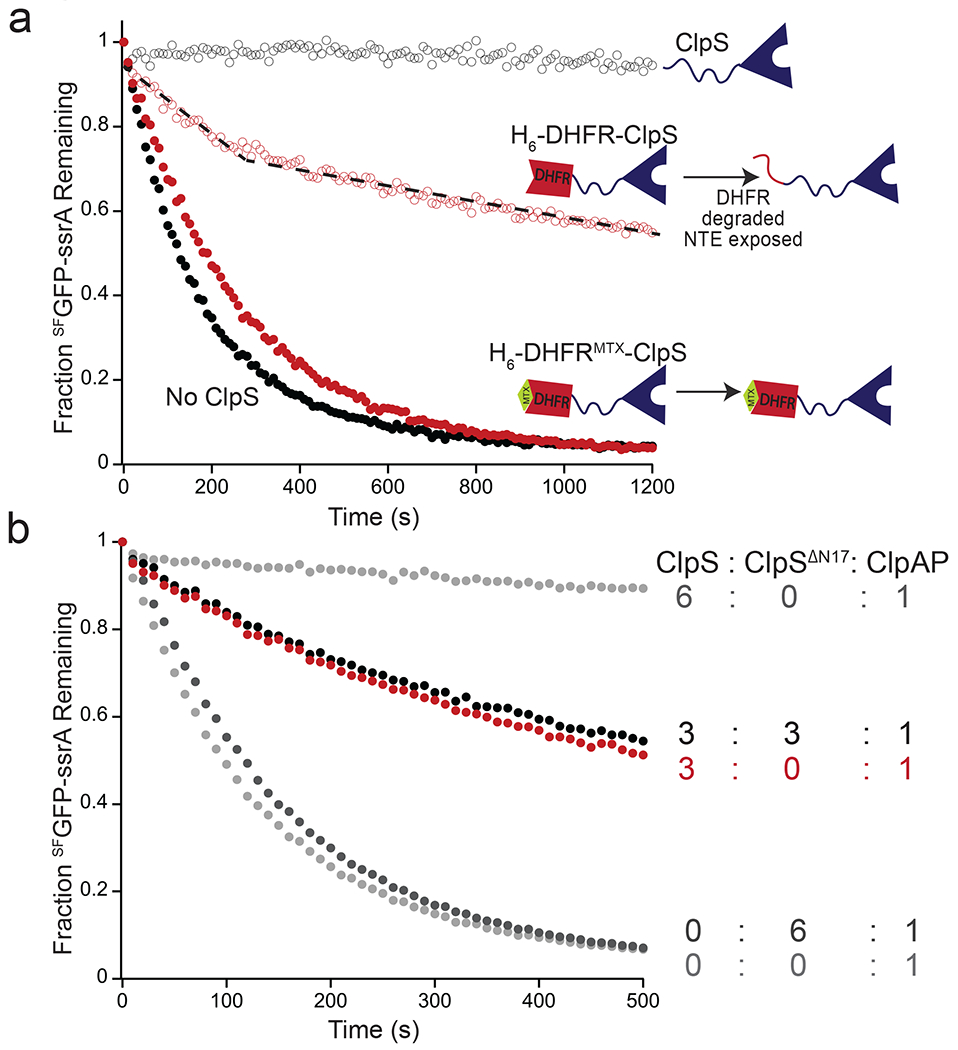
(a) Degradation of SFGFP-ssrA (5 μM) by ClpAP (0.2 μM ClpA6, 0.4 μM ClpP14) alone (solid black circles) or in the presence of 1 μM ClpS (empty black circles), 1 μM H6-DHFR-ClpS (empty red circles), or 1 μM H6-DHFR-ClpS and 10 μM MTX (solid red circles). Data are representative of 3 independent experiments. The dotted line is a visual cue to indicate the biphasic nature of the degradation kinetics in presence of H6-DHFR-ClpS. (b) Inhibition of degradation of SFGFP-ssrA (5 μM) by ClpAP (1 μM ClpA6, 2 μM ClpP14) with ClpS alone (3 μM or 6 μM), ClpSΔN17 alone (6 μM), or a mixture of ClpS (3 μM) and ClpSΔN17 (3 μM). Curves are representative of 3 independent experiments.
Four to six molar equivalents of ClpS per ClpA hexamer are required for maximum inhibition [19]. To test if the same number of NTEs are required for inhibition, we performed SFGFP-ssrA degradation assays with mixtures of ClpS and the truncated ClpSΔN17 variant, which binds ClpAP but fails to inhibit (Fig. 5b). As observed previously, three ClpS molecules per ClpA6 provided ~50% of the inhibition achieved with six ClpS molecules per ClpA hexamer [19]. Strikingly, inhibition did not improve when three ClpSΔN17 molar equivalents and three ClpS molar equivalents per ClpA hexamer were present. This result strongly suggests that binding of the ClpS cores to the N-domains of ClpA is important only because they bring along their attached NTEs, which directly mediate inhibition.
The NTE is sufficient for inhibition.
To test if the ClpS NTE is sufficient for inhibition, we initially constructed an NTE-ClpAΔN fusion but this protein was subject to severe “autodegradation” in the presence of ClpP. To minimize this problem, we generated NTE-DHFR-ClpAΔN and DHFR-ClpAΔN chimeras, variants that were much more slowly removed by autodegradation when MTX was present (Figs. 6a, 6b). When NTE-DHFR-ClpAΔN and DHFR-ClpAΔN were each incubated with ClpP, SFGFP-ssrA, ATP, and MTX, DHFR-ClpAΔN-ClpP degraded SFGFP-ssrA efficiently whereas NTE-DHFR-ClpAΔN-ClpP did not (Fig. 6c). Thus, the NTE can suppress degradation of an ssrA-tagged substrate in the absence of both the core domain of ClpS and the N-terminal domain of ClpA. We also assayed the steady-state kinetics of SFGFP-ssrA degradation under the same conditions over the first 10 minutes of the reaction, when autodegradation is minimal. The fusion enzyme containing the NTE displayed a 10-fold weaker KM and 2.5-fold lower Vmax for degradation compared to the enzyme lacking the NTE (Fig. 6d). These results parallel the inhibitory effects of ClpS and support a model in which the NTE is largely responsible for inhibition, with ClpS binding to the ClpA N-terminal domain simply positioning it properly for engagement by ClpA whereas NTE interactions with the translocation machinery suppress the rate of ATP hydrolysis. As predicted by this model, NTE-DHFR-ClpAΔN hydrolyzed ATP with a substantially (~3-fold) slower rate than the ClpA variant lacking the NTE (DHFR-ClpAΔN) (Supplementary Figure 3).
Figure 6: The NTE is sufficient for inhibition.
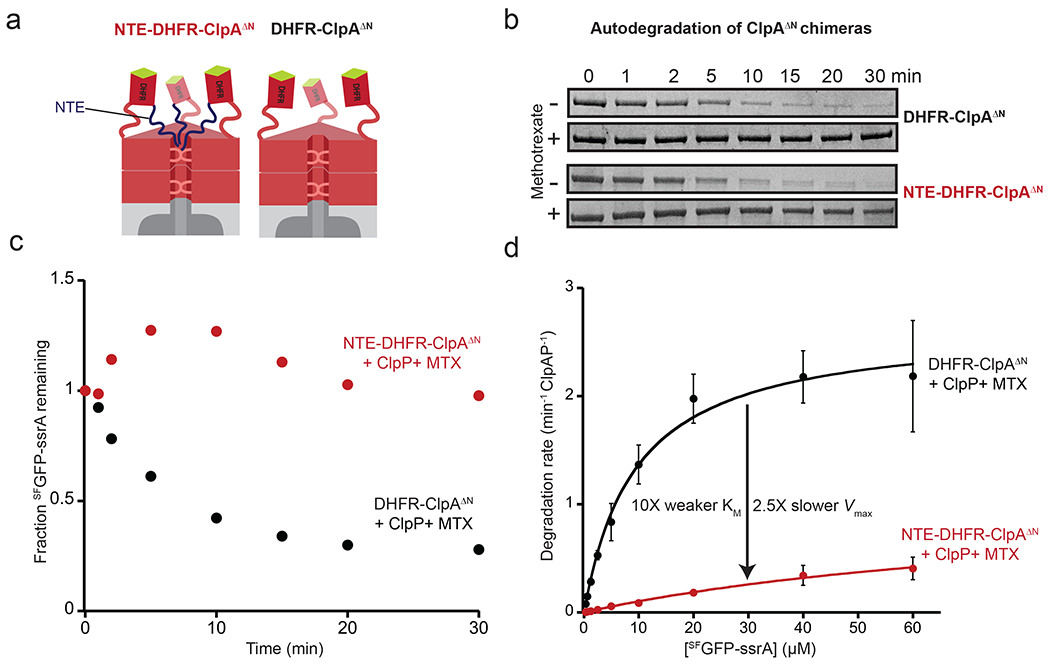
(a) Cartoons NTE-DHFR-ClpAΔNP (left), or DHFR-ClpAΔNP (right). ΔN carries a deletion of ClpA residues 1-168, which are not required for degradation of ssrA-tagged proteins [59, 60] (b) Autodegradation of NTE-DHFR-ClpAΔN or DHFR-ClpAΔN in the presence and absence of MTX (10 μM) assayed by SDS-PAGE. Experiments contained a ClpA variant (0.5 μM), ClpP (1 μM), and SFGFP-ssrA (20 μM) but only the ClpA variant band is shown. (c) Quantification kinetics of SFGFP-ssrA degradation from the plus MTX experiments described in panel b by densitometry of the SDS gels. Data in (b) and (c) are representative of 3 independent experiments. (d) Steady-state kinetics of ClpP (0.8 μM) degradation of SFGFP-ssrA supported by DHFR-ClpAΔN or NTE-DHFR-ClpAΔN (0.4 μM each) with MTX (10 μM). For DHFR-ClpAΔN supported degradation, KM was 9.5 μM and Vmax was 2.7 min−1 enz−1. For NTE-DHFR-ClpAΔN supported degradation, KM was 94 μM and Vmax was 1.1 min−1 enz−1. Values are average ± 1 SD (n = 3).
NTE length and the mechanism of inhibition.
Previous studies established that ClpS bearing an NTE of nine residues (ClpSΔN16) inhibits degradation of GFP-ssrA as efficiently as wild-type ClpS, whereas deletion of one additional residue (ClpSΔN17) results in essentially no inhibition [19]. These experiments were performed under conditions where weakened substrate recognition was the major cause of ClpS inhibition. Using a set of ClpS variants with truncated NTEs of different lengths [19], we determined Vmax values for ClpAPS* degradation of SFGFP-ssrA. As the length of the NTE increased from nine to 14 residues, there was an almost linear decrease in Vmax (Fig. 7). These results support a model in which inhibition of substrate recognition and inhibition of substrate unfolding and translocation via changes in the ATP-hydrolysis rate are mediated by different, although possibly overlapping regions of the ClpS NTE. Furthermore, by plotting the normalized average ClpAPS* ATP hydrolysis rates reported by Roman-Hernandez et. al. [21] together with Vmax values for ClpAPS* degradation of SFGFP-ssrA it was clear that these rates followed a very similar trend as a function of NTE length, with inflection points in both curves occurring between 11 and 14 NTE residues.
Figure 7: Effect of NTE length on Vmax for degradation.
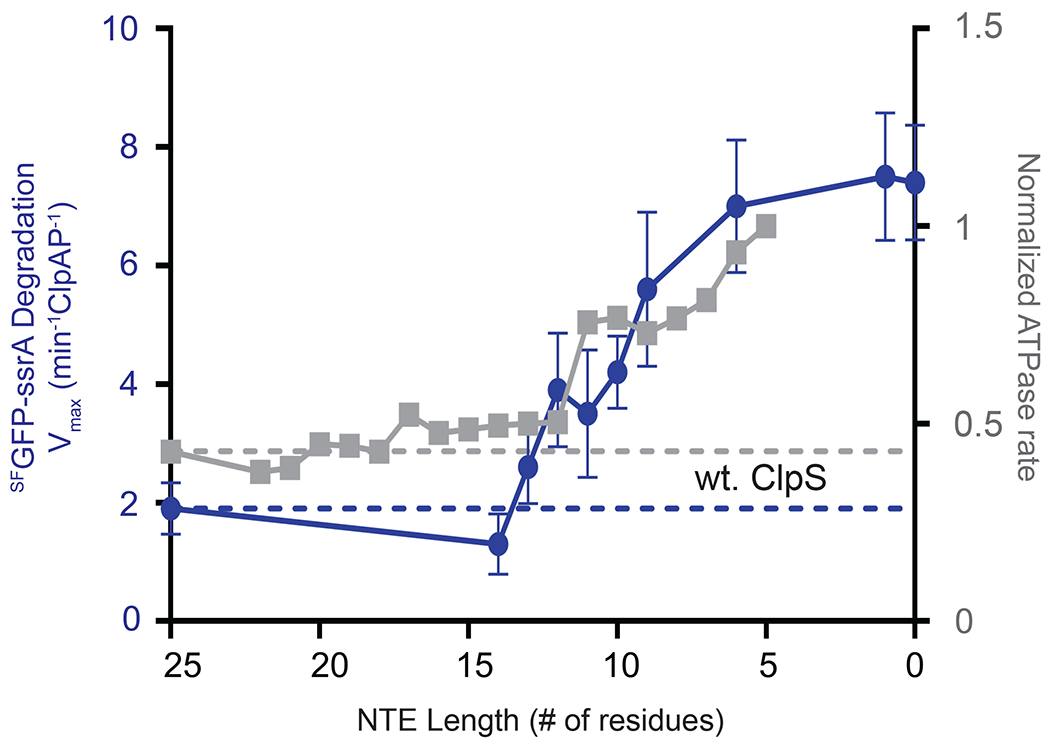
Vmax (blue circles) for ClpAP (0.4 μM ClpA6, 0.8 μM ClpP14) degradation of different concentrations of SFGFP-ssrA were determined by Michaelis-Menten analysis of experiments performed in the presence of NTE truncation variants of ClpS (2.4 μM). Values are average ± 1 SD (n = 3). ATPase rates (grey squares) for ClpAP in presence of ClpS NTE truncation variants from Roman-Hernandez et al [21]. Values were normalized to the wt. ClpA and plotted allowing direct comparison of the changes in Vmax and ATPase rates resulting from varying the ClpS NTE length. The dotted lines represent the values obtained in the presence of wt. ClpS.
DISCUSSION
AAA+ adaptors are typically described as facilitators of substrate recognition by their partner ATPase machines [3, 5, 11, 34]. Although this paradigm holds for many adaptors, ClpS represents an interesting exception. In fact, ClpS is a multifaceted adaptor, as it acts as an efficient stimulator of recognition of one class of substrates whereas strongly inhibiting degradation of other substrate classes [18, 19]. Most previous studies have focused on elucidating how ClpS acts as an enhancer of N-degron substrate degradation [10, 19, 21–23, 35–38]. Our current work reveals new mechanistic aspects of the strategy that ClpS employs to modulate substrate degradation by ClpAP. We find that, in addition to modulating substrate recognition, ClpS affects catalytic steps of the ClpAP degradation cycle. Based on these results, our current view on how ClpS regulates ClpAP activity is depicted in the model shown in Fig. 8. This model describes two general mechanisms that ClpS employs to reprogram the ClpAP protease. In the absence of ClpS, ClpAP preferentially degrades ssrA-tagged substrates compared to N-degron substrates [19]. When ClpS binds the N-terminal domain of ClpA, it positions its unstructured NTE for recognition and engagement by the ClpA pore, much like ClpA would engage a substrate’s degron; ClpS therefore can be considered an “undegradable” substrate mimic [21]. As ClpA attempts to unfold and translocate ClpS, which resists degradation, ATP hydrolysis is slowed, delivery of N-degron substrates is enhanced markedly, but degradation of these substrates is also slowed because of ClpA’s reduced ATPase rate. The ClpAPS complex both binds and degrades ssrA-tagged substrates more slowly than ClpAP does. Thus, as in cases of kinetic proofreading [39–42], ClpAPS sacrifices efficiency to obtain higher specificity.
Figure 8: Model for reprogramming of ClpAP by ClpS.
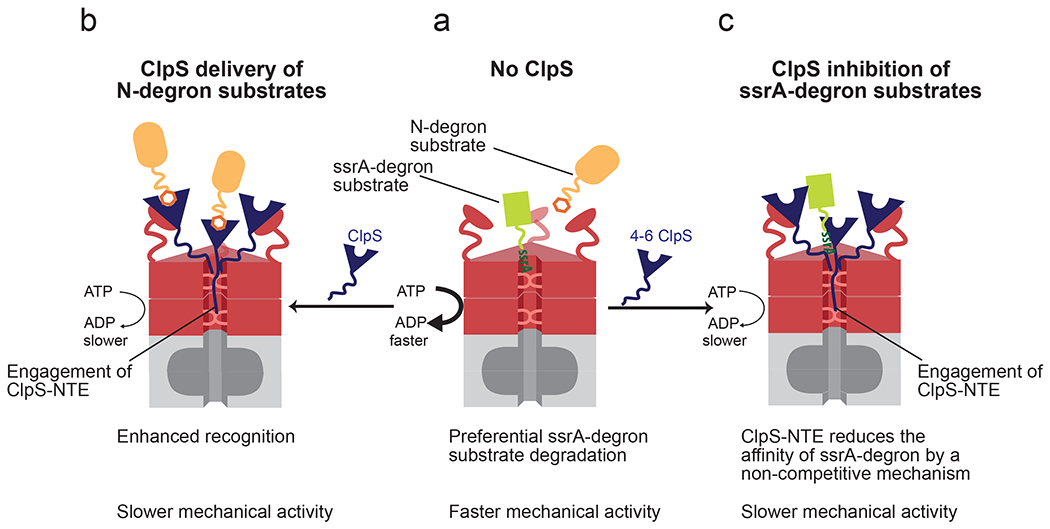
(a) In the absence of ClpS, ClpAP preferentially degrades ssrA-tagged substrates (green) relative to N-degron substrates (orange). (b) When the ClpS core binds the ClpA N-terminal domain, it positions the NTE for engagement by ClpA pore loops. ClpS NTE interactions with the translocation machinery suppress the rate of ATP hydrolysis by ClpA, slowing its mechanical activities. When N-degron substrates are present, ClpS also markedly enhances their recognition by ClpAP. (c) When ClpS and ssrA degron substrates are present, ClpS decreases the affinity for this substrate class by a non-competitive binding mechanism. Under these conditions the ClpS NTE also interacts with, and slows the translocation machinery of ClpA.
Our experiments reveal that ClpS is more than a simple binding switch, functioning in part as a rheostat for ClpAP substrate preference. Kinetic analysis of SFGFP-ssrA substrate degradation, as well as solution binding assays using λ-ssrA, demonstrate that ClpS weakens but does not prevent the binding of ssrA-tagged substrates to ClpA. This aspect of inhibition, along with the observation that inhibition of ssrA-tagged substrate recognition progressively increases as more ClpS molecules bind to the ClpA hexamer [19], suggests that ClpS can “tune” (like a rheostat) substrate recognition. For example, an increased ratio of ClpS to ClpAP in the cell could temporarily favor degradation of N-degron substrates without completely halting the degradation of ssrA-tagged substrates. In E. coli, the ClpS:ClpA6 ratio is ~6:1 during exponential growth but shifts to ~2:1 during stationary phase, resulting in an increased capacity for degradation of ssrA-tagged proteins and other non-N-degron substrates [19, 43].
Previous studies have highlighted the role of the ClpS NTE for both the mechanisms of delivery and inhibition [18, 19, 21, 22, 24]. The ClpS NTE, which lacks sequence conservation among orthologs, must be at least 14 residues long for efficient N-degron delivery, suppression of ClpA ATP hydrolysis [21], and efficient inhibition of Vmax for degradation of ssrA-tagged substrates.
Importantly, we find that the NTE must be actively engaged by ClpA for inhibition, as it is the case for N-degron substrate delivery [22]. By fusing the NTE to ClpA, we established that the NTE is sufficient for raising KM and decreasing Vmax for degradation of ssrA-tagged substrates. Notably, an NTE-DHFR protein is a poor inhibitor in trans (data not shown), suggesting that the ClpS core plays two functions: (i) binding the ClpA N-terminal domain with tight affinity, and (ii) positioning the ClpS NTE for engagement by ClpA. Importantly, the ClpS core resists unfolding and degradation by ClpAP [22]. Thus, we propose that ClpS acts as a substrate mimic. In fact, the NTE has been shown to act as a degradation signal when appended at the N-terminus of GFP [21], and mutations at the junction of the NTE and ClpS core can render ClpS susceptible to ClpAP degradation with the NTE functioning as the degron (Izarys Rivera-Rivera, personal communication).
A second striking aspect of ClpS inhibition is the slowing of substrate processing. We found that ClpS decreases the maximal degradation rate of ssrA-tagged substrates. Importantly, this and previous studies also show that the maximal degradation rate of N-degron substrates is also slower in the presence of ClpS [10]. The ability of ClpS to suppress the rate of ATP hydrolysis by ClpAP ~2-fold is probably responsible for the general slowing of substrate unfolding and translocation [21]. Indeed, when we adjusted the ClpAP ATPase rate to 50% of the maximal rate by changing the ATP concentration, SFGFP-ssrA was also degraded at ~50% of the normal rate. Importantly, this same pattern of ATPase suppression by ClpS was also observed with a ClpA variant in which only the D2 ATPase, the major unfolding and translocation motor, was active further connecting ClpS’s ATPase inhibition with the slowing of mechanical protein processing. Lastly, we also demonstrated that inhibition of the degradation Vmax and suppression of the ATPase rate had very similar dependences on the length of the ClpS NTE [21]. This collection of evidence strongly argues that ClpS slows substrate processing by suppressing the ATPase rate of ClpAP. PinA, an adaptor that non-competitively inhibits substrate degradation by the Lon protease, also suppresses Lon ATPase activity [44]. Interestingly, adaptors like SspB, MecA and α -SNAP – which enhance substrate recognition by ClpX, ClpC and NSF, respectively – stimulate ATP hydrolysis of their partner AAA+ enzymes [45–47]. Thus, modulation of ATP-hydrolysis rates appears to be a common strategy that adaptors can employ to regulate their cognate AAA+ partners.
ClpS slowing the substrate translocation rate, as observed for CFP-GFP-ssrA, is consistent with the idea that it also slows the conformational changes derived from the ATPase cycles that drive translocation [48, 49]. Furthermore, saturating ClpS suppresses ATP hydrolysis and the degradation of an unfolded substrate ~2-fold. However, we also observe more than a 2-fold inhibition of Vmax for degradation of a natively folded ssrA-tagged substrate by ClpS, suggesting that the full range of ClpS inhibition strategies involves additional effects on substrate engagement and/or unfolding. The recent use of single-molecule optical-trapping methods to study ClpXP and ClpAP provides an opportunity to probe these steps [29, 50]. These experiments reveal distinct phases of degradation reactions including pre-unfolding dwell times, translocation velocities, step dwells and step sizes. Thus, analyzing the multiple features of ClpAP unfolding, translocation and degradation in the presence of ClpS, or some of the ClpS chimeras/variants designed here, holds promise for further elucidating mechanism by which ClpS controls the mechanical workings of ClpAP. Furthermore, although numerous AAA+ unfoldases and unfoldase-substrate structures have been recently elucidated by cryo EM [16, 51, 52], these studies leave unanswered questions regarding regulation of AAA+ unfoldases, and particularly do not yet address the steps of initial substrate docking and irreversible engagement, steps where adaptor proteins are often especially important. The ClpAPS•N-degron substrate assembly, as a biologically important and highly stable complex is an especially attractive candidate for the next phase of structural studies.
Although the simplest and most commonly considered type of adaptor for AAA+ machines are direct “molecular matchmakers” that prioritize substrate choice by forming bipartite protein-protein contacts between the machine and a specific substrate (e.g. cargo or client), it’s clear that the ClpS adaptor functions using a more complex mechanism involving at least three features: (i) recognizing N-degron substrates and tethering them near the enzyme pore; (ii) weakening enzyme interaction with other substrate classes, and (iii) modulating the machine activity of the ClpAP enzyme by controlling its ATPase activity. Importantly, as analysis of the adaptors of AAA+ machines has matured, it is becoming evident that many, perhaps most adaptors work using multi-faceted mechanisms. For example, The Caulobacter crescentus CpdR adaptor binds to the N-terminal domain of the Caulobacter ClpX unfoldase, activating enhanced degradation without interacting with specific substrates; this adaptor also recruits new co-adaptors by protein-protein interactions, which in turn deliver new substrates [53]. CpdR also passively inhibits recognition of some substrates by controlling access to the enzyme’s N-terminal domains needed for their efficient recognition. Looking more broadly, the AAA+ motor dynein, which transports multiple types of cellular cargo (its multiple substrates) along microtubules in eukaryotic cells depends in large part on adaptor proteins to match each type of cargo with the dynein machine [54]. Participation of numerous different adaptor proteins have been reported and interestingly, a large fraction of these are known as “activating adaptors” that both recruit a specific cargo to dynein and directly stimulate the mechanical functions of dynein, promoting motility of that adaptor’s specifically-loaded cargo [54]. Thus, with ClpS protein as one important example, it appears judicious to consider both the binding/matchmaking properties and the ability to modify the cognate enzyme’s activities when characterizing novel biological adaptor proteins.
METHODS
Strains and plasmids
H6-SUMO-λ(1-93)A21C-ssrA was generated using the QuickChange Site-Directed Mutagenesis Kit protocol (Agilent). The cloned construct was inserted into a pET23b vector at the C-terminus of H6-SUMO. To generate the NTE-DHFR-ClpAΔN1-168 chimera, residues 1-26 of the ClpS NTE, followed by mouse dihydrofolate reductase (DHFR) were fused to the N-terminus of ClpAΔN1-168 in a pET9a vector using standard cloning techniques. To generate DHFR-ClpAΔN1-168, residues 1-26 of the ClpS NTE were deleted from NTE-DHFR-ClpAΔN1-168.
Protein expression and purification.
All proteins were expressed in E. coli strain BL21 (DE3) pLysS that had been transformed with appropriate plasmid vectors. 35S-labelled titin I27-ssrA was expressed and purified as described [38, 55]. Cysteines in 35S-titin I27-ssrA were carboxymethylated by incubation for 2 h with a 200-fold molar excess of iodoacetic acid in the presence of 5 M GuHCl (pH 8.9) at 25 °C. ClpA, NTE-DHFR-ClpAΔN1-168, and DHFR-ClpAΔN1-168 were purified as described [19]. Briefly, after cell lysis, the cleared lysate was brought to 40% (w/v) saturated ammonium sulfate and centrifuged. The pellet was resuspended in S-Sepharose buffer (25 mM HEPES, pH 7.5, 2 mM DTT, 0.1 mM EDTA, 10% (v/v) glycerol) and centrifuged again. The supernatant was loaded onto an S-Sepharose column (GE Healthcare) and the protein was eluted in a gradient from 0.2 to 1 M KCl in S-Sepharose buffer. Peak fractions were combined and dialyzed into 50 mM HEPES, pH 7.5, 20 mM MgCl2, 0.3 M NaCl, 10% (v/v) glycerol and 0.5 mM DTT. ClpA, ClpP and ClpS were purified as described [21, 31, 56]. After expression, H6-SUMO-ClpS was purified by Ni-NTA affinity chromatography (Qiagen) and then cleaved with Ulp1 protease. A second round of Ni-NTA chromatography removed the H6-SUMO fragment. ClpS was purified by gel filtration on a Superdex 75 column (GE Healthcare). ClpS was concentrated and stored in 20 mM HEPES (pH 7.5), 150 mM KCl, 1 mM DTT, and 10% glycerol (v/v). H6-DHFR-ClpS was a gift from Izarys Rivera-Rivera (MIT). ClpS NTE deletions variants were a gift from Jennifer Hou (MIT).
After expression, H6-SUMO-λ-ssrA fusion protein was purified by Ni-NTA chromatography (Qiagen) in the presence of 8 M urea. Urea was removed and the protein was cleaved with Ulp1 protease. A second round of Ni-NTA chromatography removed the H6-SUMO fragment. λ-ssrA was concentrated and stored in 10 mM Tris (pH 8), 100 mM NaCl, 1 mM DTT, and 10% glycerol. SFGFP-ssrA and CFP-GFP-titin I27V15PssrA were purified as described 49]. YLFVQ-GFP was a gift from Benjamin Stein (MIT).
Fluorescent Labeling.
λ-ssrA or ClpS variants containing a single cysteine were labeled with fluorescein as described [21]. Briefly, λ-ssrA or ClpS (25 μM) was incubated with 50 mM DTT in 100 mM TrisCl (pH 8) for 1 h at 4 °C, and then buffer-exchanged into 100 mM Sodium Phosphate (pH 8) and 1 mM EDTA. λ-ssrA was labeled with 0.3 mg/mL fluorescein-5-maleimide (Invitrogen) for 2 h at room temperature in the dark. Excess fluorescein maleimide was removed by size-exclusion chromatography. Fluorescently labeled λ-ssrA was stored in 20 mM Tris (pH 8), 150 mM NaCl, 1 mM DTT, and 10% glycerol. Fluorescently labeled ClpS was stored in 10 mM HEPES (pH 7.5), 200 mM KCl, and 1 mM DTT.
Biochemical assays.
ClpAP and ClpAPS degradation assays were performed as described [21]. Briefly, ClpA6 (0.4 μM), ClpP14 (0.8 μM), and ClpS or variants (2.4 μM) were preincubated in reaction buffer (50 mM HEPES, pH 7.5, 300 mM NaCl, 20 mM MgCl2, 0.5 mM DTT, and 10% glycerol (v/v) with substrate for 10 min at 30 °C before adding 16 mM ATP and a regeneration system (200 μg/ml creatine kinase, 20 mM creatine phosphate) to initiate degradation. For the ylfvq-GFP degradation experiments, 0.2 μM ClpA6, 0.4 μM ClpP14, and 1 μM ClpS were used. Initial rates of degradation of different concentrations of SFGFP-ssrA or ylfvq-GFP were assayed by loss of fluorescence (420 nm excitation; 540 nm emission), and data were fitted to the Michaelis-Menten equation to obtain KM and Vmax. ATP-hydrolysis rates were monitored using a coupled assay by following loss of NADH absorbance at 340 nm as described [57] under similar conditions used for the protein degradation assays. Reported values of kinetic parameters were averages (n = 3) ± 1 SD.
Solution binding.
Binding assays were monitored by fluorescence anisotropy using a Photon Technology International Fluorimeter. λ*fl-ssrA (0.15 μM) was incubated with different concentrations of ClpA and 2 mM ATPγS in the presence or absence of ClpS until equilibrium was reached. Similarly, ClpS*fl (0.2 μM) was incubated with different concentrations of ClpA in the presence of 30 μM λ-ssrA until equilibrium was reached. Data were fitted to a hyperbolic binding isotherm using a non-linear least-squares algorithm. For anisotropy degradation assays, λ*fl-ssrA was incubated with ClpAP and 2 mM ATPγS in the presence or absence of ClpS. Degradation was initiated by the addition of ATP and the regeneration mix.
Calculation of apparent affinity of ClpS.
According to modified Michaelis-Menten equation for competitive inhibition, the apparent affinity of the substrate (ClpS) for the enzyme (ClpA) in the presence of inhibitor (λ-ssrA) is:
λfl-ssrA binds to ClpA with an affinity (KI) of 0.7 μM; hence at a concentration of 30 μM of λ-ssrA, the apparent affinity of ClpS is
Simulations.
To simulate the decrease in GFP fluorescence during degradation of CFP-GFP-titin I27V15P-ssrA (CGT), we used a two-step CGT→CG→CU model. The first step has a rate constant (k1) for pseudo first-order binding of the substrate by excess ClpAP and degradation of most of the titin I27V15P domain to generate CG, which retains native CFP and GFP fluorescence. The second step has a rate constant (k2) for unfolding of the GFP domain to generate CU, which retains native CFP fluorescence. Values of k1 and k2 for ClpAP and ClpAPS degradation were determined by fitting the decrease in GFP fluorescence using KinTek Explorer [58], constraining k2 for ClpAPS to 0.5•k2 for ClpAP. To model the decrease in CFP fluorescence, a four-step CGT→CG→CU→C→U mechanism was used, with k1 and k2 defined as above, k3 representing translocation of the unfolded GFP domain, and k4 representing unfolding of the CFP domain. To simulate the CFP data using Tenua (bililite.com), we increased CFP fluorescence by a factor of 1.7 upon unfolding of GFP, used k1 and k2 from the GFP fitting, set k4 = k2, and varied k3 (with the value for ClpAPS constrained to 0.5•k3 for ClpAP) by trial-and-error until the experimental and modeled trajectories for ClpAP and ClpAPS degradation were similar.
Supplementary Material
Highlights:
ClpS adaptor enhances and inhibits degradation by ClpAP, tuning substrate choice.
ClpS impedes degradation of ssrA-substrates, however the mechanism has been unclear.
In one mechanism, ClpS acts non-competitively to decrease ssrA-tag affinity to ClpA.
ClpS also reduces the ClpA ATPase, thereby slowing protein unfolding/translocation.
To inhibit, ClpS’s intrinsically disordered “domain” is necessary and sufficient.
ClpS is multi-faceted, controlling both substrate binding and enzyme activity.
Acknowledgements.
We thank D. Finley, J. Hou, J. Kardon, A. Keating, A. Olivares, I. Rivera-Rivera, F. Solomon, B. Stein for discussions, materials, and advice. Supported in part by National Institutes of Health Pre-Doctoral Training Grant T32GM007287, the Howard Hughes Medical Institute, and National Institutes of Health Grants GM-49224 and Al-16892
REFERENCES:
- [1].Olivares AO, Baker TA, Sauer RT. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat Rev Microbiol (2016);14:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol (2005);6:519–29. [DOI] [PubMed] [Google Scholar]
- [3].Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem (2011);80:587–612. [DOI] [PubMed] [Google Scholar]
- [4].Striebel F, Kress W, Weber-Ban E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol (2009);19:209–17. [DOI] [PubMed] [Google Scholar]
- [5].Baker TA, Sauer RT. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci (2006);31:647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gottesman S, Roche E, Zhou Y, Sauer RT. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev (1998);12:1338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Keiler KC, Waller PR, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science (1996);271:990–3. [DOI] [PubMed] [Google Scholar]
- [8].Karzai AW, Roche ED, Sauer RT. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nature structural biology (2000);7:449–55. [DOI] [PubMed] [Google Scholar]
- [9].Tobias JW, Shrader TE, Rocap G, Varshavsky A. The N-end rule in bacteria. Science (1991);254:1374–7. [DOI] [PubMed] [Google Scholar]
- [10].Wang KH, Sauer RT, Baker TA. ClpS modulates but is not essential for bacterial N-end rule degradation. Genes Dev (2007);21:403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dougan DA, Mogk A, Zeth K, Turgay K, Bukau B. AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett (2002);529:6–10. [DOI] [PubMed] [Google Scholar]
- [12].Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol (2005);7:742–9. [DOI] [PubMed] [Google Scholar]
- [13].Turgay K, Hahn J, Burghoorn J, Dubnau D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J (1998);17:6730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kirstein J, Schlothauer T, Dougan DA, Lilie H, Tischendorf G, Mogk A, et al. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J (2006);25:1481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mei Z, Wang F, Qi Y, Zhou Z, Hu Q, Li H, et al. Molecular determinants of MecA as a degradation tag for the ClpCP protease. The Journal of biological chemistry (2009);284:34366–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lopez KE, Rizo AN, Tse E, Lin J, Scull NW, Thwin AC, et al. Conformational Plasticity of the ClpAP AAA+ Protease Couples Protein Unfolding and Proteolysis. bioRxiv 820209 [Preprint], (2019) doi: 10.1101/820209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kessel M, Maurizi MR, Kim B, Kocsis E, Trus BL, Singh SK, et al. Homology in structural organization between E. coli ClpAP protease and the eukaryotic 26 S proteasome. J Mol Biol (1995);250:587–94. [DOI] [PubMed] [Google Scholar]
- [18].Dougan DA, Reid BG, Horwich AL, Bukau B. ClpS, a substrate modulator of the ClpAP machine. Mol Cell (2002);9:673–83. [DOI] [PubMed] [Google Scholar]
- [19].Hou JY, Sauer RT, Baker TA. Distinct structural elements of the adaptor ClpS are required for regulating degradation by ClpAP. Nat Struct Mol Biol (2008);15:288–94. [DOI] [PubMed] [Google Scholar]
- [20].Zeth K, Ravelli RB, Paal K, Cusack S, Bukau B, Dougan DA. Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nature structural biology (2002);9:906–11. [DOI] [PubMed] [Google Scholar]
- [21].Roman-Hernandez G, Hou JY, Grant RA, Sauer RT, Baker TA. The ClpS adaptor mediates staged delivery of N-end rule substrates to the AAA+ ClpAP protease. Mol Cell (2011);43:217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rivera-Rivera I, Roman-Hernandez G, Sauer RT, Baker TA. Remodeling of a delivery complex allows ClpS-mediated degradation of N-degron substrates. Proc Natl Acad Sci U S A (2014);111:E3853–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Erbse A, Schmidt R, Bornemann T, Schneider-Mergener J, Mogk A, Zahn R, et al. ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature (2006);439:753–6. [DOI] [PubMed] [Google Scholar]
- [24].De Donatis GM, Singh SK, Viswanathan S, Maurizi MR. A single ClpS monomer is sufficient to direct the activity of the ClpA hexamer. The Journal of biological chemistry (2010);285:8771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Reid BG, Fenton WA, Horwich AL, Weber-Ban EU. ClpA mediates directional translocation of substrate proteins into the ClpP protease. Proc Natl Acad Sci U S A (2001);98:3768–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol (2006);24:79–88. [DOI] [PubMed] [Google Scholar]
- [27].Nager AR, Baker TA, Sauer RT. Stepwise unfolding of a beta barrel protein by the AAA+ ClpXP protease. J Mol Biol (2011);413:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Martin A, Baker TA, Sauer RT. Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat Struct Mol Biol (2008);15:139–45. [DOI] [PubMed] [Google Scholar]
- [29].Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA. Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol (2014);21:871–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kress W, Mutschler H, Weber-Ban E. Both ATPase domains of ClpA are critical for processing of stable protein structures. The Journal of biological chemistry (2009);284:31441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kotamarthi HC, Sauer RT, Baker TA. The Non-dominant AAA+ Ring in the ClpAP Protease Functions as an Anti-stalling Motor to Accelerate Protein Unfolding and Translocation. Cell Rep (2020);30:2644–54 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guo F, Maurizi MR, Esser L, Xia D. Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. The Journal of biological chemistry (2002);277:46743–52. [DOI] [PubMed] [Google Scholar]
- [33].Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell (2001);7:627–37. [DOI] [PubMed] [Google Scholar]
- [34].Kirstein J, Moliere N, Dougan DA, Turgay K. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol (2009);7:589–99. [DOI] [PubMed] [Google Scholar]
- [35].Wang KH, Roman-Hernandez G, Grant RA, Sauer RT, Baker TA. The molecular basis of N-end rule recognition. Mol Cell (2008);32:406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Roman-Hernandez G, Grant RA, Sauer RT, Baker TA. Molecular basis of substrate selection by the N-end rule adaptor protein ClpS. Proc Natl Acad Sci U S A (2009);106:8888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang KH, Oakes ES, Sauer RT, Baker TA. Tuning the strength of a bacterial N-end rule degradation signal. The Journal of biological chemistry (2008);283:24600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stein BJ, Grant RA, Sauer RT, Baker TA. Structural Basis of an N-Degron Adaptor with More Stringent Specificity. Structure (2016);24:232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Blossey R, Schiessel H. Kinetic proofreading of gene activation by chromatin remodeling. HFSP J (2008);2:167–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yamane T, Hopfield JJ. Experimental evidence for kinetic proofreading in the aminoacylation of tRNA by synthetase. Proc Natl Acad Sci U S A (1977);74:2246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Swain PS, Siggia ED. The role of proofreading in signal transduction specificity. Biophys J (2002);82:2928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci U S A (1974);71:4135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Farrell CM, Grossman AD, Sauer RT. Cytoplasmic degradation of ssrA-tagged proteins. Mol Microbiol (2005);57:1750–61. [DOI] [PubMed] [Google Scholar]
- [44].Hilliard JJ, Simon LD, Van Melderen L, Maurizi MR. PinA inhibits ATP hydrolysis and energy-dependent protein degradation by Lon protease. The Journal of biological chemistry (1998);273:524–7. [DOI] [PubMed] [Google Scholar]
- [45].Wah DA, Levchenko I, Rieckhof GE, Bolon DN, Baker TA, Sauer RT. Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease. Mol Cell (2003);12:355–63. [DOI] [PubMed] [Google Scholar]
- [46].Barnard RJ, Morgan A, Burgoyne RD. Stimulation of NSF ATPase activity by alpha-SNAP is required for SNARE complex disassembly and exocytosis. J Cell Biol (1997);139:875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K. MecA, an adaptor protein necessary for ClpC chaperone activity. Proc Natl Acad Sci U S A (2003);100:2306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Martin A, Baker TA, Sauer RT. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell (2008);29:441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Stinson BM, Nager AR, Glynn SE, Schmitz KR, Baker TA, Sauer RT. Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell (2013); 153:628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cordova JC, Olivares AO, Shin Y, Stinson BM, Calmat S, Schmitz KR, et al. Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell (2014);158:647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Puchades C, Sandate CR, Lander GC. The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat Rev Mol Cell Biol (2020);21:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fei X, Bell TA, Jenni S, Stinson BM, Baker TA, Harrison SC, et al. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Elife (2020);9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lau J, Hernandez-Alicea L, Vass RH, Chien P. A Phosphosignaling Adaptor Primes the AAA+ Protease ClpXP to Drive Cell Cycle-Regulated Proteolysis. Mol Cell (2015);59:104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Reck-Peterson SL, Redwine WB, Vale RD, Carter AP. The cytoplasmic dynein transport machinery and its many cargoes. Nat Rev Mol Cell Biol (2018);19:382–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell (2003);114:511–20. [DOI] [PubMed] [Google Scholar]
- [56].Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell (2000);5:639–48. [DOI] [PubMed] [Google Scholar]
- [57].Burton RE, Siddiqui Sm, Kim YI, Baker TA, Sauer RT. Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J (2001);20:3092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Johnson KA. Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods Enzymol (2009);467:601–26. [DOI] [PubMed] [Google Scholar]
- [59].Lo JH, Baker TA, Sauer RT. Characterization of the N-terminal repeat domain of Escherichia coli ClpA-A class I Clp/HSP100 ATPase. Protein Sci (2001);10:551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Singh SK, Rozycki J, Ortega J, Ishikawa T, Lo J, Steven AC, et al. Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. The Journal of biological chemistry (2001);276:29420–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.