

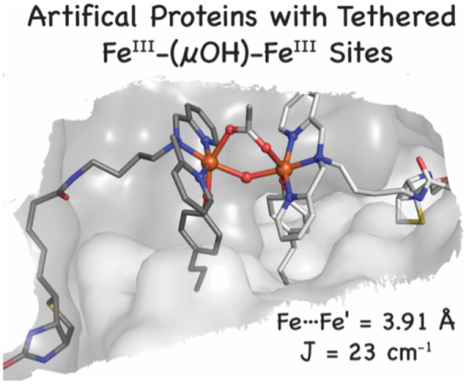
Abstract
Dinuclear iron centers with a bridging hydroxido or oxido ligand form active sites within a variety of metalloproteins. A key feature of these sites is the ability of the protein to control the structures around the Fe centers, which leads to entatic states that are essential for function. To simulate this controlled environment, artificial proteins have been engineered using biotin-streptavidin (Sav) technology in which Fe complexes from adjacent subunits can assemble to form [FeIII-(μ-OH)-FeIII] cores. The assembly process is promoted by the site-specific localization of the Fe complexes within a subunit through the designed mutation of a tyrosinate side chain to coordinate the Fe centers. An important outcome is that the Sav host can regulate the Fe⋯Fe separation, which is known to be important for function in natural metalloproteins. Spectroscopic and structural studies from X-ray diffraction methods revealed uncommonly long Fe⋯Fe separations that change by less than 0.3 Å upon the binding of additional bridging ligands. The structural constraints imposed by the protein host on the di-Fe cores are unique and create examples of active sites having entatic states within engineered artificial metalloproteins.
Graphical Abstract
Introduction
We describe a method for producing artificial metalloproteins (ArMs) that utilizes a protein-assisted assembly process to generate di-nuclear Fe cores. Such Fe species have been shown to be essential for function in a range of metalloproteins that includes hemerythrin for O2 binding,1 soluble methane monooxygenase hydroxylase (MMOH)2 and the R2 subunit of class I ribonucleotide reductases (RNR R2)3–6 for O2 activation, flavin diiron proteins (FDP) for NO and O2 reduction,7,8 and purple acid phosphatases for hydrolysis.9–11 A common structural feature among these metalloproteins is the ability of the protein host to regulate the coordination environments around the Fe centers, which includes managing the Fe⋯Fe separation during turnover.12–16 Regulation of the di-Fe core is necessary to achieve the desired function and, equally critically, it prevents the formation of unwanted structures that could lead to inactivity. Attempts to model di-Fe centers have been dominated by synthetic constructs that utilize either self-assembly approaches or dinucleating ligands.17–25 In both cases, it is difficult to control the Fe⋯Fe separation in the way that is found in proteins and to further allow exogenous ligands to bind.
To reproduce these naturally occurring di-Fe cores, we have turned to the use of a protein host. Many approaches are utilized to develop ArMs, and most aim to leverage the rich functional chemistry of synthetic metal complexes with the structural diversity of protein active sites.26–42 For instance, DeGrado elegantly showed how to design a four-helix bundle to reproduce the triply-bridged di-Fe core found in hemerythrin.43 We have taken a different approach in which we employ the existing biotin-streptavidin (Sav) technology, which has been shown to effectively produce ArMs that catalyze an array of transformations.44–49 Streptavidin is a homotetramer of eight-stranded β-barrels that assemble into a dimer of dimers (Figures 1A, S1) in which the biotin binding pockets face one another within a dimer.50–53 In previous examples of ArMs made with a Sav host, the metal complexes embedded in each subunit function independently.32,48,54 Based on the structure of the Sav dimer, however, it should be possible to assemble di-metallic species from monomeric complexes housed within individual subunits. To demonstrate this concept, we re-engineered Sav to assemble an ArM with a unique [FeIII–(μ-OH)-FeIII] core in which the Fe complexes reside in adjacent subunits of the dimer. Structural and spectroscopic studies support the formation of this complex and its ability to bind additional bridging ligands, such as acetate, azide, and cyanide ions. An important outcome of our work is the demonstration of how variants of Sav impose structural constraints on the assembly of the di-Fe complexes to produce unusually long Fe···Fe separations that cannot be achieved without a protein host. These structures are rare examples of entatic states engineered in an ArM.
Figure 1.
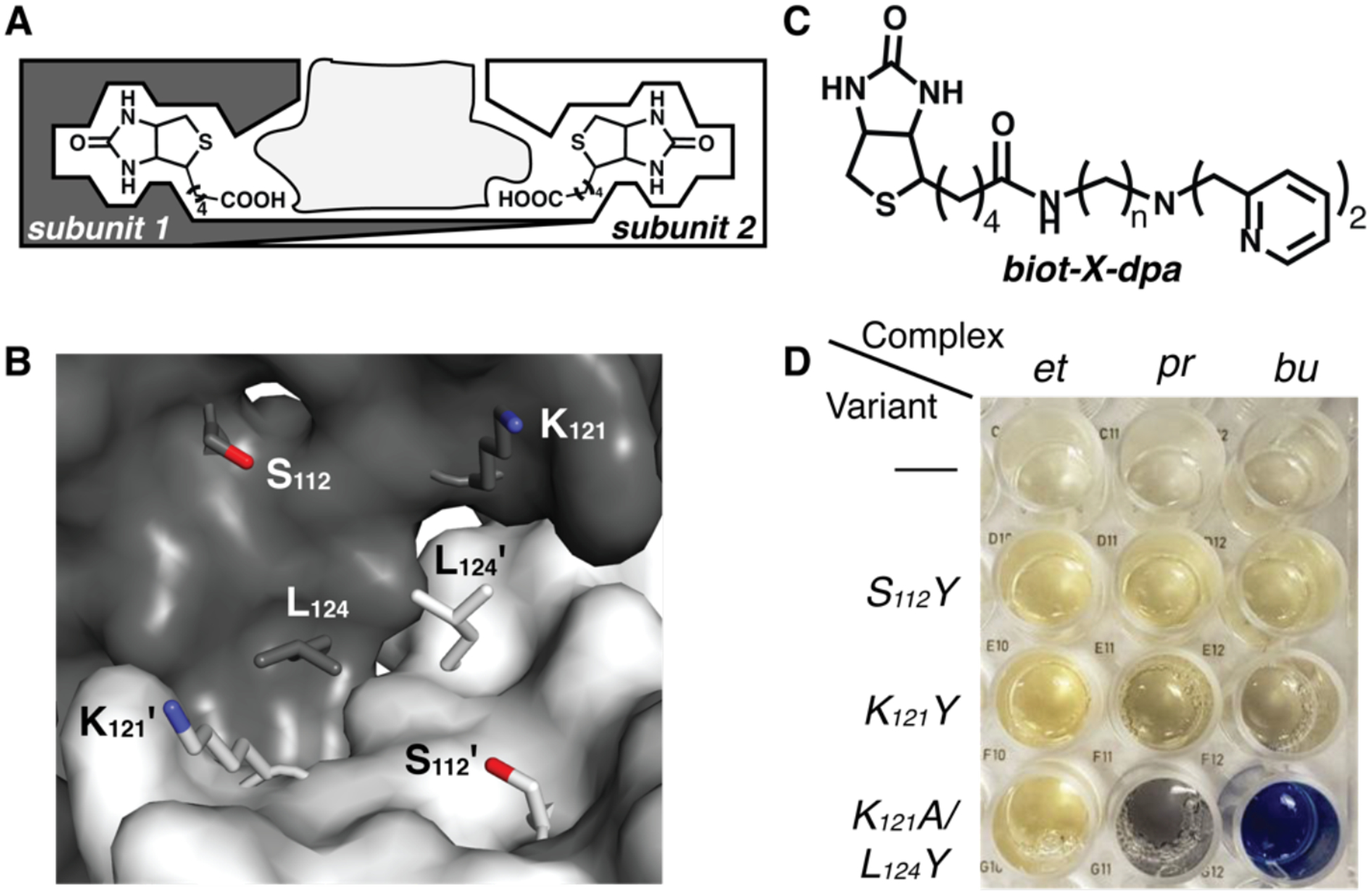
Schematic of a Sav dimer with the space for embedding metal complexes indicated in light gray (A), a close-up view of a Sav dimer showing the amino acids that were mutated to Y (B), the biotinylated ligand used to generate the di-Fe ArM (C), and a picture of the optical assay used to determine the positional match that generated the di-Fe ArM. Key in C: n=2, X=et; n=3, X=pr; n=4, X=bu. Abbreviations of the linkers are used to denote the complexes in D. The first row in D is of the Fe complexes in the absence of a Sav variant.
Results and Discussion
Design Considerations and Preparation of Di-Fe Proteins.
The insertion of biotinylated metallocofactors into Sav variants is controlled by the strong, non-covalent affinity that Sav has for biotin (Ka > 1014 M−1),55 which ensures the reproducible binding of biotinylated species within the dimers. To achieve the site-specific localization of a metallocofactor, additional binding to Sav is needed – either by covalent bonding to an amino acid side chain or through non-covalent interactions, such as hydrogen bonds (H-bonds).56–59 However, it is still difficult to predict the exact placement of a metallocofactor within a Sav dimer. To ensure that these types of bonding interactions are favored, we have found that a positional match between the embedded metal complex and specific amino acid side chains is needed. Positional matching can be accomplished by finding the correct linker between the biotin moiety and ligand that situates a metal complex in a specific region of the Sav dimer (Figure 1). Because Sav is a relatively rigid protein, especially within the interior of the Sav dimer, the linker thus provides a tool for positioning an artificial metallocofactor proximal to an amino acid side chain to promote bonding interactions.
The importance of these design concepts is highlighted in our efforts to prepare ArMs with di-Fe cores. Our initial attempts using wild-type Sav and biotinylated Fe complexes with different linkers were unsuccessful, with both spectroscopic and structural data indicating that only monomeric species were present. We reasoned that the binding of an additional amino acid side chain to the Fe complexes was necessary to localize the Fe complexes and induce the formation of the desired di-Fe species. To accomplish this localization, Sav needed to be re-engineered because there are no amino acid side chains within the dimer of wild-type Sav that can bind covalently to an Fe complex. We therefore developed an optical assay with Sav variants to test the ability of mutated amino acids within the dimer to bind to the Fe centers. This assay used tyrosine mutations because the intense phenolate-to-iron charge transfer transition that occurs when an FeIII center covalently binds to a tyrosine side chain provides a ready visual indicator of Fe–OY interactions within Sav.11,60,61 In addition, such bonding interactions are known to occur in natural oxygenases.11,60
We did not know prior to our studies which combination of biotinylated Fe complex and Sav mutation would produce the desired Fe–OY coordination. Therefore, we prepared three Sav variants with the mutations S112Y, K121Y, and K121A/L124Y. Positions 112 and 121 were selected based on evidence from prior studies that side chains from amino acids other than Y at these positions could interact with metal complexes through either covalent or non-covalent interactions.48,56,57,59 The close proximity of position 124 to the dimer interface led us to prepare the K121A/L124Y variant; we found that the double mutation was necessary to achieve better expression, and the K121A mutation did not play a role in cofactor binding. In addition to the Sav variants, three biotinylated [FeIIIdpa]3+ complexes59 (dpa, dipyridylmethylamine) that differed by only the choice of either an ethyl, propyl, or butyl linker were prepared (Figure 1C). Each biotinylated dpa ligand and its corresponding FeIII complex ([FeIII(biot-X-dpa)(OH2)3]Cl3 (X=et, pr, bu)) were independently prepared and had the expected physical properties (see SI). For instance, each FeIII complex exhibited an electron paramagnetic resonance (EPR) spectrum that is indicative of a high-spin complex and an electronic absorbance spectrum with absorption features at λmax ~ 260, 320, and 480 nm (Figures S2 & S3).59 From these three Fe complexes and three Sav variants, a nine-membered assay was created to determine which combination gave rise to Fe–OY binding. Each ArM had an ~4:1 cofactor:variant stoichiometry, as determined by solution titration studies (Figure S4). The optical assay provided an immediate visual assessment of Fe–Tyr binding, with only [FeIII-bu-dpa] ⊂ K121A/L124Y-Sav (1) producing a strong absorption to indicate the formation of an FeIII–OY bond (Figure 1D). The assay was done in unbuffered, nanopure water – we found that the addition of buffers prevented interactions between the Fe complex and the Tyr side chains. The formation of ArMs with a pentyl linker using [FeIII(biot-pent-dpa)(OH2)3]Cl3 and the K121A/L124Y-Sav variant was also explored but no visible features were observed to indicate that an FeIII–OY bond was formed. These optical results indicated that just the Fe complex with the butyl linker gave the correct positional match for Fe–OY binding and only the solution properties of 1 were studied further in H2O to determine whether a di-Fe core was generated.
Spectroscopic Studies on 1.
As illustrated in the optical assay, 1 had a distinct blue color, which arises from an absorption band at λmax = 605 nm (εM = 2800 M−1cm−1, Figure 2A).11,60,61 Resonance Raman (RR) spectra obtained with 647-nm laser excitation exhibited intense bands at 1172, 1292, 1500, and 1600 cm−1 that constitute the signature of phenol ring vibrations of Fe(III)-coordinated tyrosinate chromophores. The low-frequency modes at 590 and 888 cm−1 are assigned to ν(Fe-OY) and ν(C-OY) modes, as previously reported for other Fe–tyrosine containing proteins (Figure 2B).62–64 The ⊥-mode EPR spectrum of 1 was featureless, which implies that the FeIII centers are magnetically coupled. The Mössbauer spectrum of 57Fe-1 conclusively supports this magnetic coupling, revealing parameters indicative of an antiferromagnetically coupled [FeIII -(μ-OH)-FeIII] species (Figure 2C). A single quadrupole doublet with an isomer shift (δ) of 0.52 mm/s and a quadrupole splitting (ΔEQ) of −0.83 mm/s were observed at low magnetic field; these data are consistent with a coupled system of equivalent FeIII sites (see below for more details).18
Figure 2.
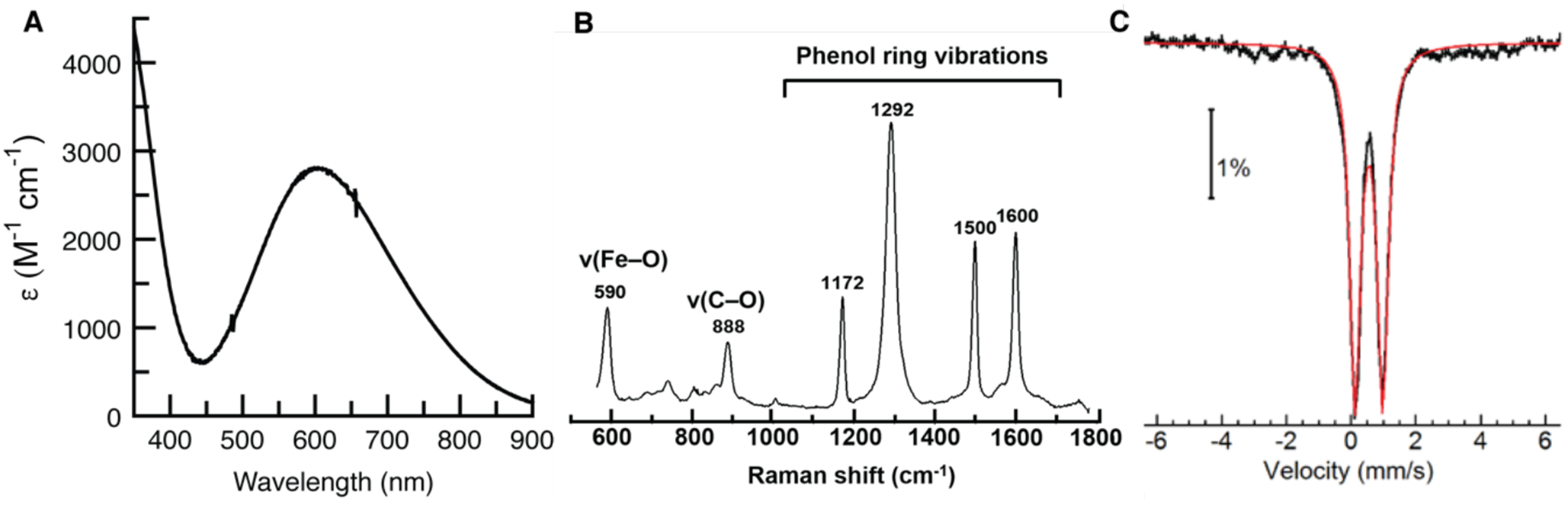
Absorption spectrum of 1 (A), RR spectrum of 1 with λexc = 647 nm (B), and Mössbauer spectrum of 57Fe-1. All samples were prepared in nanopure H2O. The spectra in A and B were measured at room temperature, and that in C was recorded at 4 K.
To gain a better understanding of the coordination environment of 1, X-ray absorption spectroscopic (XAS) studies were done on frozen samples of 1. An X-ray absorption near-edge structure (XANES) analysis provided a K-edge energy of 7124.4 eV for 1, which falls well within the range found for other di-FeIII enzymes (Figure S5).65–67 Additional analysis of the XANES region revealed a pre-edge area value of 8.1 units for 1 that is consistent with those found in hydroxido-bridged [FeIII -(μ-OH)-FeIII] species, which have an average area of ~7 units.68,69 This experimentally determined value is significantly below the average value of ~14 units for oxido-bridged [FeIII -(μ-O)-FeIII] species.68 In addition to agreeing with our Mössbauer results, this finding is consistent with data from our RR experiments. For oxido-bridged [FeIII -(μ-O)-FeIII] chromophores, resonance enhancement of νs(Fe-O-Fe) and νas(Fe-O-Fe) is typically observed when using 407-nm laser excitation.18,70 However, these peaks were not observed for 1, and instead, only non-resonant Raman vibrations from the protein amide backbone and aromatic sidechains were found, thereby supporting the formation of a hydroxido-bridged [FeIII -(μ-OH)-FeIII] species (see SI, Figure S6).
Data from extended X-ray absorption fine structure (EXAFS) analysis of 1 were fit with two sets of scatterers in the primary coordination sphere: Fe–N/O at 1.89 Å and 2.10 Å (Figure S7A; Tables S1&S2). The longer distance was fit with four scatterers and had a relatively high mean-squared deviation (σ2) value of ~8 • 10−3 Å2. Attempts to split the shell resulted in a set of shorter (~2.05 Å) and longer (~2.15 Å) scatterers with reasonable σ2 values, but the resolution of the data (ΔR = 0.13 Å) did not justify the inclusion of the split shell in the best fit. The Fe–N/O bond distance at 1.89 Å is consistent with the binding of the tyrosinate residue to the FeIII centers. In addition, our analysis found an Fe⋯Fe’ distance at 4.02 Å (see below). Carbon scatterers were required at 3.05 Å; these scatterers are consistent with the alkyl linkages between the pyridyl arms of the ligand as well as the carbon atoms adjacent to the N atoms of the pyridyl groups.
Structural Characterization of 1 and 1-OAc.
To further probe the molecular structure of the Fe species within the Sav dimer of 1, single crystals were prepared by soaking crystals of apo-K121A-L124Y-Sav with [FeIII(biot-bu-dpa)(OH2)3]Cl3 (see SI for details). The blue crystals were solved to a resolution of 1.30 Å using X-ray diffraction (XRD) methods to reveal a dinuclear Fe complex immobilized within each Sav dimer (Figures 3A&B, S8A&S9; Tables 1&S3–S8). The Fe centers have distorted six-coordinate primary coordination spheres with N1, N2, and N3 from dpa bound in a meridional fashion with an average Fe–N bond distance of 1.97 Å. In support of the solution studies and color of the crystals, the O-atom donor (O1) from the phenolate group of Y124 also binds to each Fe center with an Fe–O1 bond length of 1.78 Å. Our analysis found additional density between the two Fe centers at 100% occupancy that was modeled to an O-atom bridge (O2) with an Fe/Fe’–O2 bond length of 2.16 Å and an Fe–O2–Fe’ bond angle of 133°. The O1–Fe–O2 angle is small at 65°, which is a consequence of the constrained coordination geometry around the Fe centers imposed by the Sav host (see below). Additional electron density was found between the Fe centers and was difficult to model. Our best fit suggests the possibility of a disordered acetate ion that could originate from the crystallization buffer used to form single crystals of the K121A/L124Y-Sav variant (SI). The acetate ion interacts with the di-Fe unit but with significantly different Fe–O distances of 2.17 Å and 3.39 Å. Finally, the Fe⋯Fe’ distance is 3.96 Å, which agrees with the results found by EXAFS (Figure S7B; Tables S1&S10).
Figure 3.
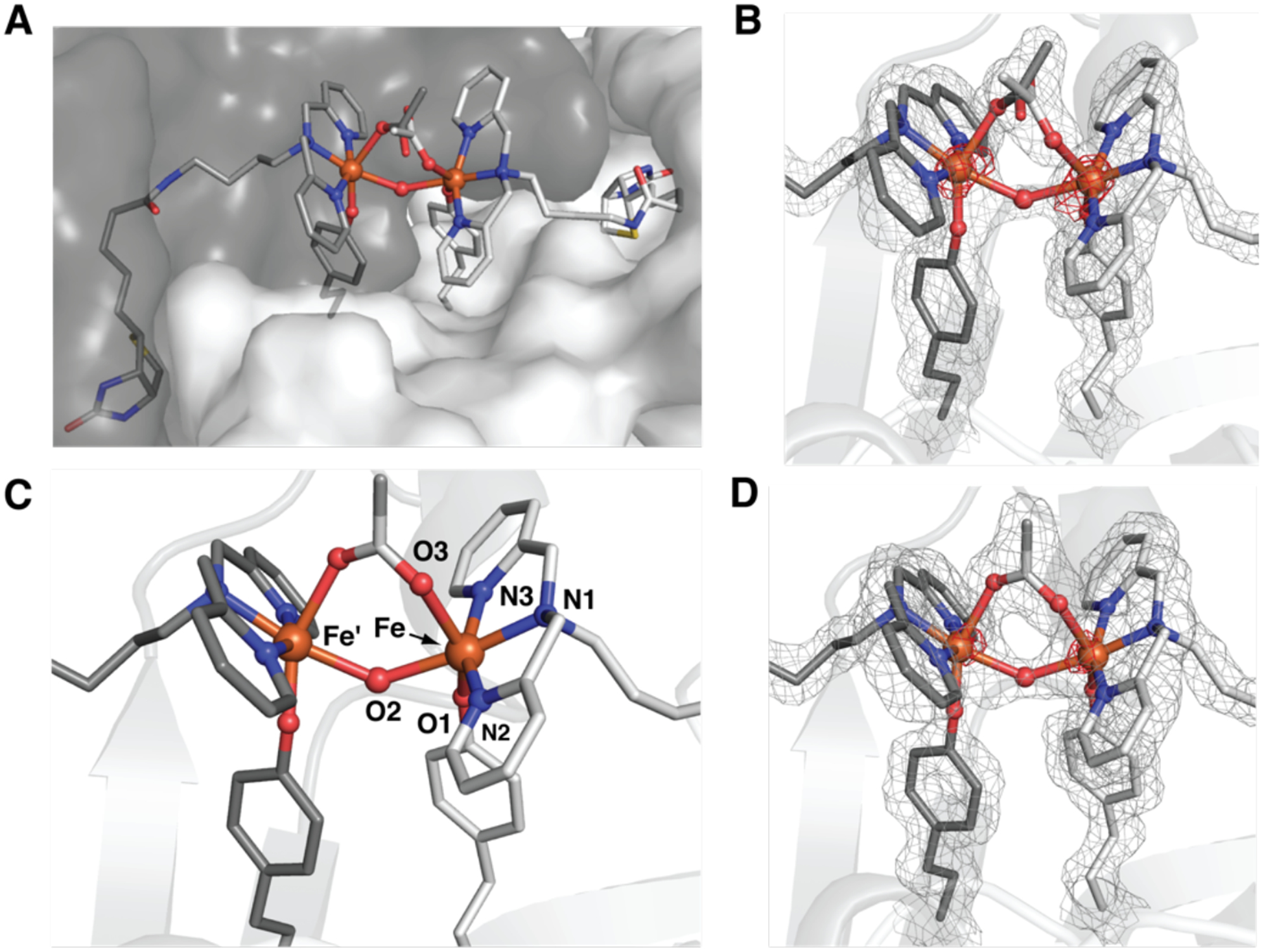
Structure of the biotinylated Fe complexes in a Sav dimer of 1 (A) (PDB 6VOZ), structure of the di-Fe complex in 1 (B), and structure of the di-Fe complex in 1-OAc (C, D) (PDB 6VO9). The gray mesh represents the 2F0-FC electron density map (contoured at 1σ), and the red mesh is the anomalous difference density (contoured at 4 σ). Fe ions are represented as orange spheres, N-atoms as blue spheres, and O-atoms as red spheres.
Table 1.
| Bond length/angle | 1 | 1-OAc | 1-N3 | 1-CN |
|---|---|---|---|---|
| Fe–O1 | 1.78 | 1.82 | 1.81 | 1.83 |
| Fe–O2 | 2.16 | 2.12 | 2.14 | 2.04 |
| Fe–O3 | 2.17 | 2.20 | – | – |
| Fe–Ndpac | 1.97 | 1.99 | 2.03 | 2.01 |
| Fe–N/C | – | – | 2.45 | 2.47 |
| Fe---Fe’ | 3.96 | 3.91 | 3.82 | 3.73 |
| Fe–O2–Fe’ | 133 | 134 | 126 | 132 |
| O1–Fe–O2 | 65 | 70 | 78 | 74 |
bond distances and angles reported in Å and deg, respectively;
solved in I4122 space group,
average values
The possibility of binding an additional exogenous bridging ligand to the di-FeIII center prompted us to examine the molecular structure of 1-OAc, which is the ArM produced after acetate ions are added intentionally to 1. Single crystals of 1-OAc were prepared by soaking crystals of 1 in a 100 mM solution of NaOAc. Analysis of a crystal that diffracted to a 1.50 Å resolution again revealed the formation of a di-Fe active site within each dimer of Sav, where the dpa ligand, the phenolate side chain of L124Y, and the O-atom bridge are coordinated in the same positions as those found in 1 (Figures 3C&D, S8B, & S10; Tables 1, S3–S7, & S9). For 1-OAc, new electron density was observed between the Fe centers and fit to an ordered μ−1,3-acetato ligand at 100 % occupancy, in which the acetato ligand is skewed from the Fe–Fe axis by ~46°. The Fe–O3 bond distance is 2.20 Å with an Fe⋯Fe’ distance of 3.91 Å and O1–Fe–O2 and Fe–O2–Fe’ angles of 70° and 134°, respectively, which are similar to those in 1. These values agree with those obtained from XAS measurements on solution of 1-OAc. For instance, EXAFS analyses were best fit to four Fe–N/O bonds at 2.08 Å, a single Fe–O/N bond at 1.86 Å, and Fe⋯Fe’ distance of 4.00 Å (Figures S5A, S7B & Tables S1 and S10).
The metrical parameters of the di-FeIII centers within crystals of 1 and 1-OAc suggest that the single-atom bridges between the Fe centers are hydroxido ligands, as was the case in solution. However, the resolution of the XRD data did not allow us to conclusively determine whether the O atoms were protonated. We addressed this issue by measuring the Mössbauer properties of 57Fe-enriched crystals of 1 and 1-OAc that were prepared in batches using [57FeIII(biot-bu-dpa)(OH2)3]Cl3. The Mössbauer spectra of 1 and 1-OAc crystals were essentially the same at low-field and gave parameters similar to those measured for solution samples. We collected high-field (7 T) Mössbauer spectra of 57Fe-1-OAc crystals at various temperatures (Figure 4), and simulations revealed an exchange coupling J value of 27(5) cm−1 (J S1.S2) – this value is within the range observed for [FeIII -(μ-OH)-FeIII] species.68 The spectra recorded at higher temperatures (≥50 K) were broad and featureless owing to intermediate electronic relaxation relative to the 57Fe nuclear Larmor frequency.
Figure 4.
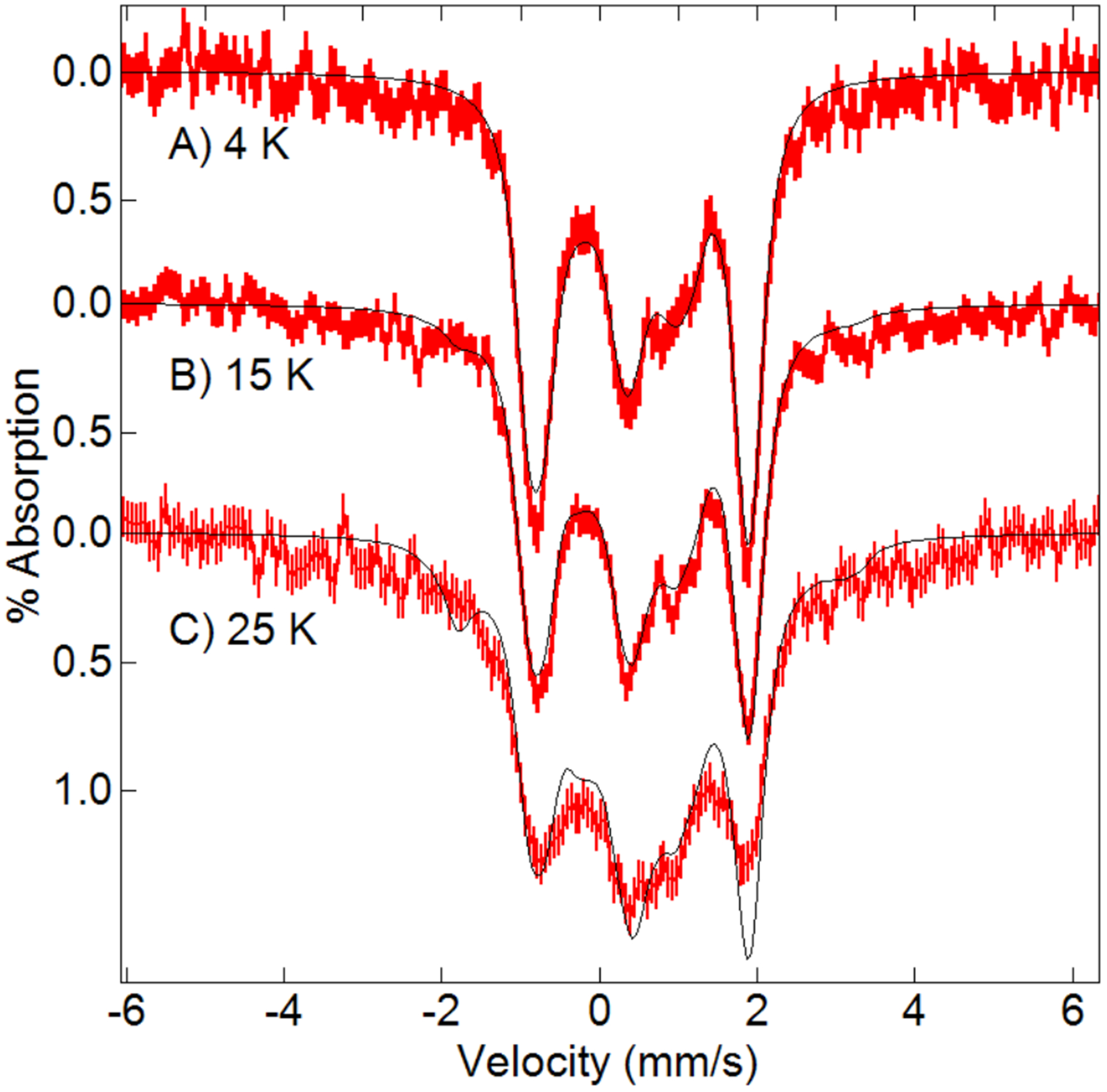
Mössbauer spectra (red) and simulations (black) of 57Fe-1–OAc crystals recorded in a magnetic field of 7 T parallel to the γ-ray direction at the temperatures listed on the figure. The simulations, which were performed in slow relaxation, are for two equivalent exchange-coupled S=5/2 FeIII sites with δ = 0.52 mm/sec, ΔEQ = −0.83 mm/s, η = 0.5, Γ = 0.35 mm/s, A(isotropic) = −20 T, ⊺ = 27 cm−1.
Structural Comparisons and Entatic States.
The structures of the di-Fe cores in 1 and 1-OAc are unusual, and the separations between Fe centers are amongst the longest found for a di-FeIII system with a bridging hydroxido/oxido ligand. There are synthetic systems examples with comparable Fe⋯Fe separations but these only contain a single hydroxido bridging ligand and cannot bind additional ligands to the iron centers.71–74 The closest biological example are found in the FDP proteins, which catalyze the reduction of O2 to water or NO to N2O.75 The crystal structures of FDPs from several organisms show a di-Fe core having μ-OH and μ-carboxylate bridges (Figure 5A).75–77 These structures show a range of Fe⋯Fe distances of 3.3 to 3.5 Å, which are significantly shorter than that in 1, and an Fe–O–Fe angle of ~125°, which is smaller than the angle in 1. However, spectroscopic studies of the diiron core of FDP indicate that the di-FeIII core has two OH bridges and that upon reduction, the di-FeII core retains a single OH bridge.78 In related systems with single oxido bridges, the di-Fe ArM of DeGrado has an [FeIII-(μ-O)(μ-carboxylate)-FeIII] core but a shorter Fe⋯Fe’ distance of 3.6 Å than that in 1-OAc.43 Met RNR R2 also has oxido and carboxylato ions as bridging ligands between two FeIII centers, and its Fe⋯Fe separation of 3.3(5) Å and Fe1–O–Fe2 angle of 120° are smaller than those in 1-OAc (Figure 5B).79 In contrast, the synthetic complex of Que, [Fe2L2(μ-O)(μ−1,3-OBz)]+ (where L is N-(o-hydroxybenzyl)-N,N-bis(2-pyridylmethyl)amine and [OBz]− is the benzoate ion), is similar to 1-OAc (Figure 5C).80 This complex was prepared by a self-assembly method from two monomeric FeIII complexes but without the influence of a protein host and has the same set of ligands to the di-FeIII unit as 1-OAc with the exception of a bridging oxido ligand. Its molecular structure contains an [FeIII-(μ-O)(μ−1,3-carboxylato)-FeIII] core but with significantly different metrical parameters than 1-OAc, which include an Fe⋯Fe separation of 3.218(2) Å and a contracted Fe1–O–Fe2 angle of 128.3 (3)°.
Figure 5.
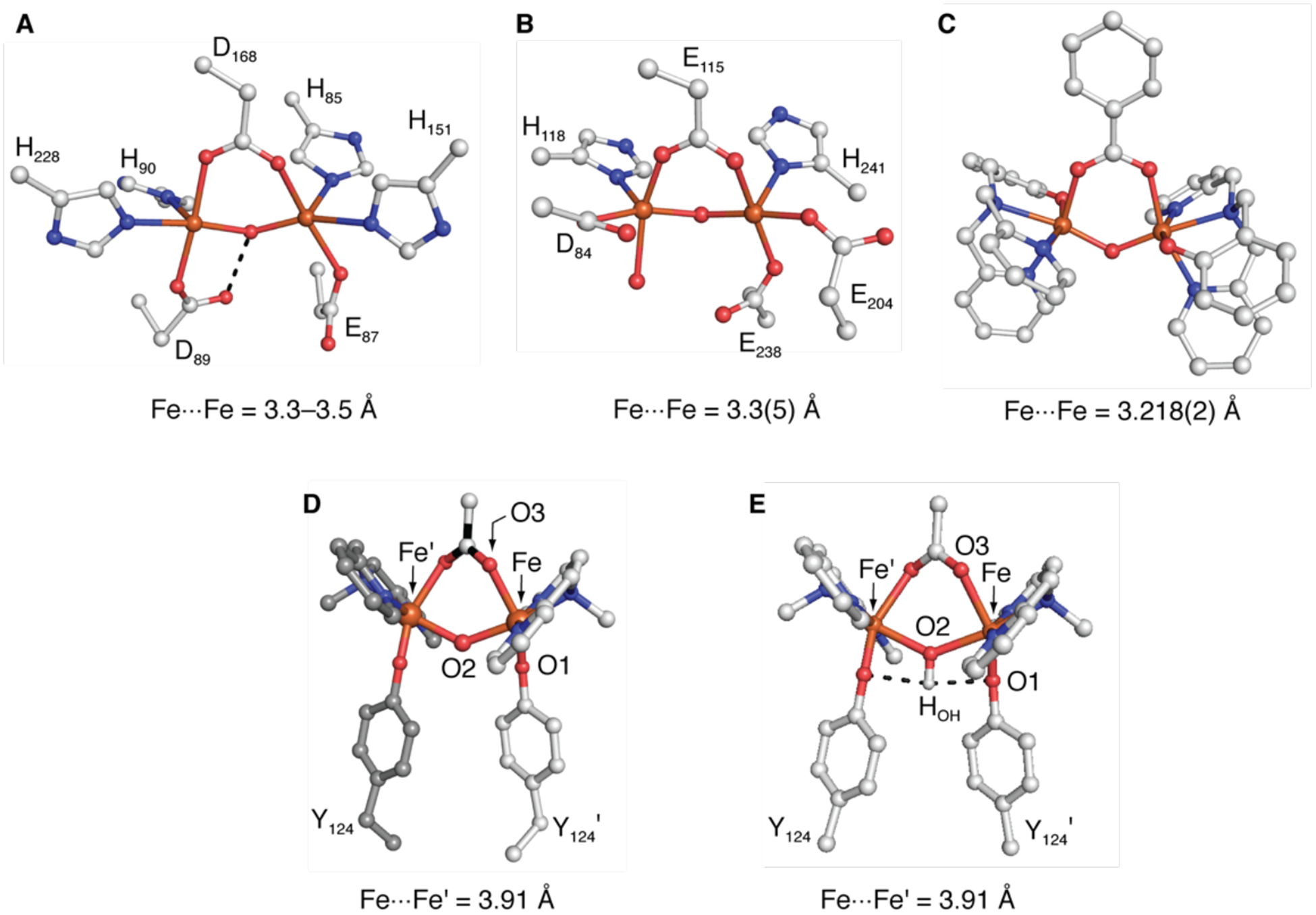
Comparison of the di-FeIII cores of FDP (1VME) (A), met RNR R2 (1MXR) (B), [Fe2L2(μ-O)(μ−1,3-OBz)]+ (C), 1-OAc (D), and the DFT-optimized structure of 1-OAc calculated using XRD coordinates with an unconstrained hydrogen atom placed onto the bridging ligand that shows H-bonds (dashed lines) (E).
The differences between the di-Fe structures in the above examples and that in 1-OAc (and 1) can be attributed to the structural constraints imposed by the Sav host. As mentioned above, the space between the biotin binding sites within a Sav dimer is relatively inflexible, and once bound, biotin is fixed within its binding site. We suggest that the linker within the FeIII cofactor and the position of the mutation at L124Y also impact the structure of the di-Fe core. The optical assay revealed that the butyl linker was necessary to match the position of the Fe complex to that of Y124. Our structural investigations further indicated that the linker also functions to tether the Fe centers within the biotin binding cavity after Fe center coordinates to Y124. With biotin secured in its binding site in Sav, the butyl linkers are unable to move and can only position the Fe complexes near Y124, resulting in practically inflexible di-Fe cores in which the movement of the Fe centers toward each other is restricted. The limited movement of the butyl linker is reinforced by constraints imposed by amino acid side chains that line the interior of Sav subunits and are positioned near the linker. There are close contacts (less then 4.5 Å) between the side chains at S112 and L110 and the carbon atoms of the linker that appears to restrict the conformations of the butyl chain because of steric interactions (Figure S11). Moreover, the side chains at Y124/Y’124 are predisposed to binding: our structural studies found that the tyrosine residue at position 124 has the same conformation in apo-K121A-L124Y-Sav as in the Fe ArMs (Figure S12, Tables S11–S12, PDB 7KNL). The conformation of Y124 appears to be regulated by non-covalent interactions from neighboring side chain at L110 and a weak π-π stacking interaction between the phenolate rings within a dimer. Taken together, these results indicate that the Fe centers within Sav are constrained to produce dinuclear complexes with Fe⋯Fe separations that are significantly longer than those normally observed in [FeIII-(μ-O(H))-FeIII] species.
Proteins can control the assembly and structure of metallocofactors and induce molecular structures that have no analogs in synthetic chemistry. These structures, or entatic states, arise because of structural constraints imposed on the metal ion(s) and its ligands by the protein.25,81,82 We argue that the atypical di-FeIII structures in 1 and 1-OAc are examples of an entatic state, as these [FeIII-(μ-OH)-FeIII] cores cannot be readily achieved within synthetic constructs.20,25 Even with long Fe⋯Fe separations, these ArMs are nevertheless magnetically coupled and, as shown for 1, are capable of binding an exogenous ligand (also see below). Moreover, the Fe⋯Fe separations in either ArM are incapable of collapsing into a thermodynamic sink that would normally produce shorter separations because of the structural constraint imposed by the Sav host. We point to the structure of [Fe2L2(μ-O)(μ−1,3-OBz)]+ (Figure 5C),80 which has the same bridging carboxylate ancillary ligand to the FeIII centers as 1-OAc but a significantly shorter Fe⋯Fe separation and an oxido bridge instead of the hydroxido bridge of 1-OAc (see above). Moreover, the Fe–Ooxide bond lengths in [Fe2L2(μ-O)(μ−1,3-OBz)]+ are ~1.78 Å, which is over 0.4 Å shorter than the analogous Fe–O2 bond lengths in 1-OAc. These metrical parameters of [Fe2L2(μ-O)(μ−1,3-OBz)]+ favor the formation of oxido-bridged di-FeIII cores. For 1-OAc, the K121A/L124Y-Sav host controls the assembly of the di-FeIII species to produce a new [FeIII-(μ-OH)(μ−1,3-OAc)-FeIII] cofactor that stabilizes a bridging hydroxido ligand.
DFT Calculations.
To further understand the properties of the di-Fe species within 1-OAc, we examined several DFT-derived structures (see SI). We first considered a species with an [FeIII-(μ-OH)(μ−1,3-carboxylato)-FeIII] core using a fragment from the structure obtained from XRD measurements (Figure 5D) in which all the atoms were constrained except for the movement of the added proton on the O-atom bridge. The optimized structure exhibited H-bonds between the hydroxido ligand and the O atoms of Y124 with O1⋯HOH and O1⋯O2 distances of 2.14 and 3.21 Å, respectively (Figures 5E&S13A, denoted DFTOH in Table 2). This calculation gave a J value of 15 cm−1, which is close to the value determined experimentally. We also considered the analogous aqua- and oxido-bridged species and computed their exchange coupling constants (Table S13). The aqua complex gave J = 2 cm−1, which was much lower than the experimental value, whereas the oxido complex gave a value of J which was an order of magnitude larger than the experimental value.18 These DFT results support our experimental findings of an [FeIII-(μ-OH)(μ−1,3-carboxylato)-FeIII] core within 1-OAc and prompted us to focus additional DFT calculations on di-Fe systems with a hydroxido bridge. An unconstrained DFT optimization of the [(dpa)FeIII-(μ-OH)(μ−1,3-carboxylato)-FeIII(dpa)] species (Figure S13B, denoted DFT-1 in Table 2) was performed, and the resultant structure had different metrical parameters than the protein-bound species determined by XRD methods. For instance, the Fe–O2 and Fe–O3 bond distances contracted by 0.10 and 0.14 Å, respectively, in the optimized structure to 2.02 and 2.09 Å (Table 2) to give a shorter Fe⋯Fe’ distance of 3.81 Å. In addition, the O1⋯HOH distance lengthened substantially to 2.53 Å with a concomitant change in the O1–Fe1–O2 angle from 70° in the XRD-determined structure to nearly 90° in the DFT-optimized structure. To achieve an O1–Fe1–O2 bond angle of ~90°, the DFT optimization moved O1 away from the hydroxido bridge to a position that is nearly trans to O3 of the bridging acetato ligand. Similar movements are not possible in 1-OAc because of the significant protein constraints imposed on Y124, as described above. The computed J value of 23 cm−1 for this structure is within experimental error of the value measured experimentally. For comparison, the unconstrained DFT-optimized structure for the oxido-bridged species gave an Fe⋯Fe’ distance of 3.49 Å and an exchange coupling of J = +185 cm−1, which is typical of oxido-bridged species.
Table 2.
DFT-calculated geometrical and exchange parameters obtained for the [(dpa)FeIII-(μ-OH)(μ−1,3-carboxylato)-FeIII(dpa)] species.a
| DFTOHb | DFT-1c | DFT-2d | |
|---|---|---|---|
| Fe···Fe’ | 3.91 | 3.81 | 3.91 |
| Fe–O1 | 1.82 | 1.84 | 1.85 |
| Fe–O2 | 2.12 | 2.02 | 2.01 |
| Fe–O3 | 2.23 | 2.09 | 2.11 |
| O1···HO2 | 2.14 | 2.53 | 2.16 |
| O1-Fe-O2 | 70 | 90 | 82 |
| Fe-O2-Fe’ | 134 | 142 | 154 |
| JDFT (cm−1) | 15 | 23 | 34 |
bond distances and angles reported in Å and deg,
the position of the hydroxyl proton has been optimized in the XRD structure,
unconstrained optimized structure,
Y124 residues frozen at the XRD position and the iron centers to the XRD distance.
An additional partially constrained DFT calculation (Figure S13C, denoted DFT-2 in Table 2) was performed to further analyze the influence of Y124 on the exchange coupling. For DFT-2, the Y124 residues within the dimer were frozen to their positions determined in the XRD structure, and the Fe⋯Fe’ distance was also frozen to that in the XRD structure. The optimization was performed with the remaining atoms in the [FeIII-(μ-OH)(μ−1,3-carboxylato)-FeIII] fragment unconstrained. The J value for this structure is larger than those calculated for both the XRD structure and the structure optimized without constraints (Table 2). We attribute the increase in J value to a combination of shorter Fe–O2 and O1⋯HOH distances in this partially constrained optimized structure. The shorter O1⋯HOH distance would weaken the O–H bond of the hydroxido ligand and confer some oxido character to the bridge, resulting in an increased J value. We also note that the structures obtained from DFT optimizations did not reproduce the acute O1–Fe–O2 angle of ~ 70° found by XRD (Table 2), suggesting it to be a consequence of protein constraints rather than hydrogen bonding between the hydroxido bridge and the O1 atoms. Attempts to duplicate some protein constraints included freezing the Cβ atom of the Y124 residues and the tertiary N atoms of the dpa ligands to the positions determined from XRD methods (Table S13, denoted DFT-3). Even with these constraints, the increased freedom during optimization allowed the OY124 atoms to move and revert the O1⋯HOH distance and O1-Fe-O2 angle to those in the unconstrained DFT structure.
Structural Characterization of 1-N3 and 1-CN.
To explore the contribution of the tethering effect to the unusually long separation between Fe centers in the di-Fe units, we prepared ArMs 1-N3 and 1-CN with bound azide and cyanide ions, respectively. These ArMs allowed us to examine how the binding of different external ligands influenced the structure of the di-Fe core and whether the tethering effect was the primary factor responsible for the long Fe⋯Fe’ distance. The molecular structures of the resulting complexes were solved to a resolution of 1.70 Å and revealed a similar primary coordination sphere as that in 1-OAc, including Fe–OY bonds and an FeIII -(μ-OH)-FeIII core. New electron density in each structure was found between the Fe centers and was modeled as a second bridging ligand. For 1-N3, the density fit an azido ligand that was coordinated in μ−1,3 fashion to the two Fe centers (Figures 6A, S8C, & S14, Tables 1, S3–S7, & S14). The Fe–Nazido bond distances are 2.45 Å, the Fe⋯Fe’ distance is 3.82 Å, and the Fe–O2–Fe’ bond angle is 126°. This Fe⋯Fe’ distance is only 0.14 Å shorter than that of 1-OAc. For 1-CN, the limitations of crystallography prevented us from determining the coordination of the C/N atoms to specific Fe centers: both conformations were modeled, and no differences in the Fe–C/N bond metrics were found (Figure 6B, S15, & S16; Tables 1, S3 & S15–S17). Nevertheless, we determined that the cyanido ligand bound to the di-Fe core through a μ-(1,2) coordination mode, and the Fe⋯Fe’ distance shortened to 3.73 Å (a difference of 0.23 Å) with an Fe–O2–Fe’ bond angle of 132°. We found no structural evidence of any other azide or cyanide binding modes, including a μ-(1,1) coordination mode that often occurs in less-restrained di-Fe systems. The small change in the Fe⋯Fe’ distance of at most 0.23 Å between 1 and the 1-X ArMs indicates that the tethering effect plays a dominate role in determining the separation between the two Fe centers.
Figure 6.
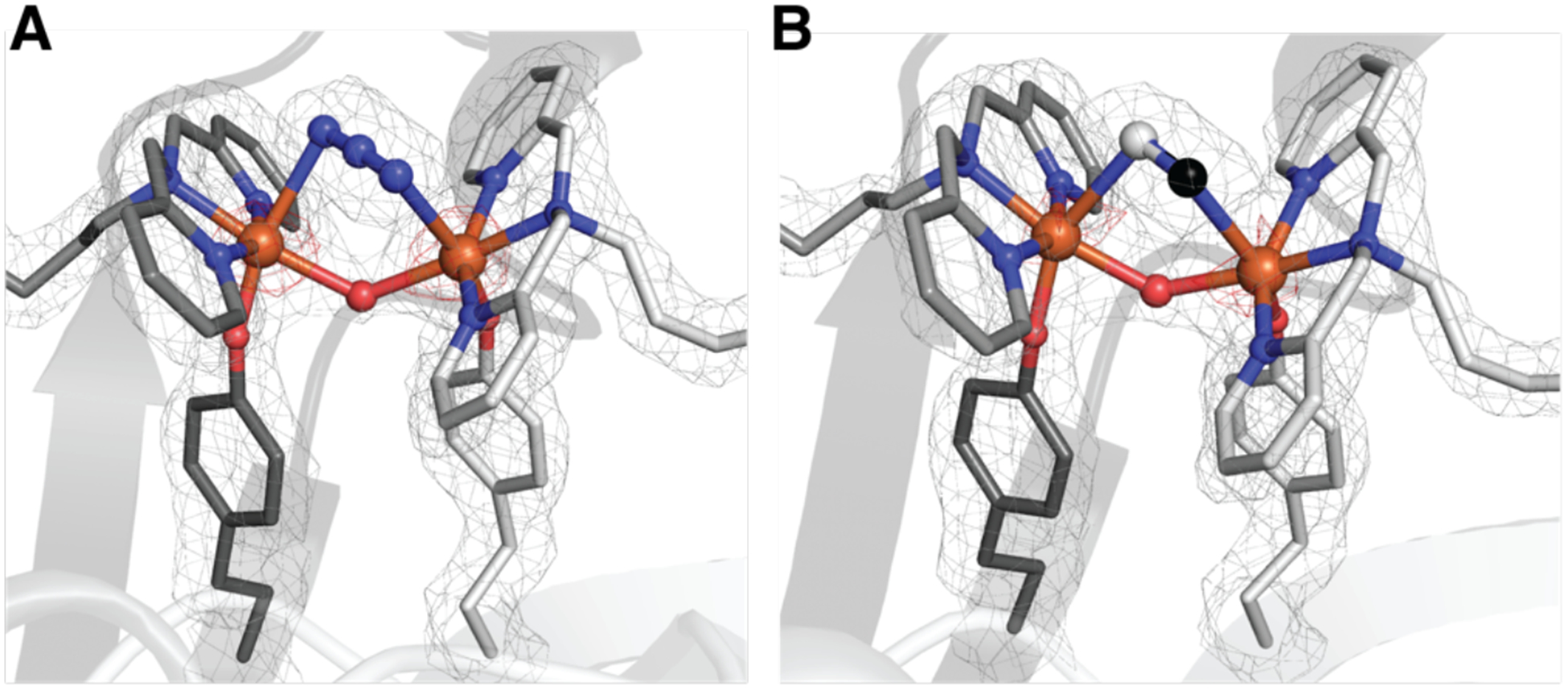
Structures of the di-Fe complex in 1-N3 (A) and 1-CN (B) (PDB 6VOB and 7KBY). The gray mesh represents the 2F0-FC electron density map (contoured at 1σ) and red mesh is the anomalous difference density (contoured at 4 σ). Atom colors and labels are the same as in Figure 3.
The 1-N3 ArM was studied by RR spectroscopy to evaluate if the μ−1,3-N3 binding mode found by XRD is also present in solution. Treating 1 with NaN3 in water resulted in an apparent blue shift of the visible absorption band to λmax ~ 490 nm, which is assigned to an azide-to-iron(III) charge transfer transition (Figure 7A).83 RR spectra obtained with 488-nm excitation showed significant decreases in the resonance enhancement of the tyrosinate vibrations at 587, 740, 887, 1501, and 1602 cm−1 and the appearance of new bands at 396, 2060, and 2088 cm−1 (Figure 7B&C). These new bands downshifted with 15N-labeled azide ions and are therefore assigned to ν(Fe-N3) and νas(NNN) vibrations. The ν(Fe-N3) vibration at 396 cm−1 compares well with frequencies observed in other nonheme di-FeIII azido proteins (Table S18). The observation of two νas(NNN) bands at 2088 and 2060 cm−1, which are 43 and 15 cm−1 lower than the asymmetric stretch of the free azide ion in solution, suggests that two different azido complex conformers exist in solution. Upon 15N labeling of 1-N3 with 15NNN−, both νas(NNN) bands downshifted by 13 cm−1 with no evidence of splitting. If the di-Fe species had either η1-N3 or μ−1,1-azido ligands, the νas(15NNN) bands would have been split by 10 to 20 cm−1, respectively (Table S18).83 Thus, the RR data are consistent with μ−1,3-N3 binding for both conformers in solution. The identity of the two conformers is currently not known.
Figure 7.
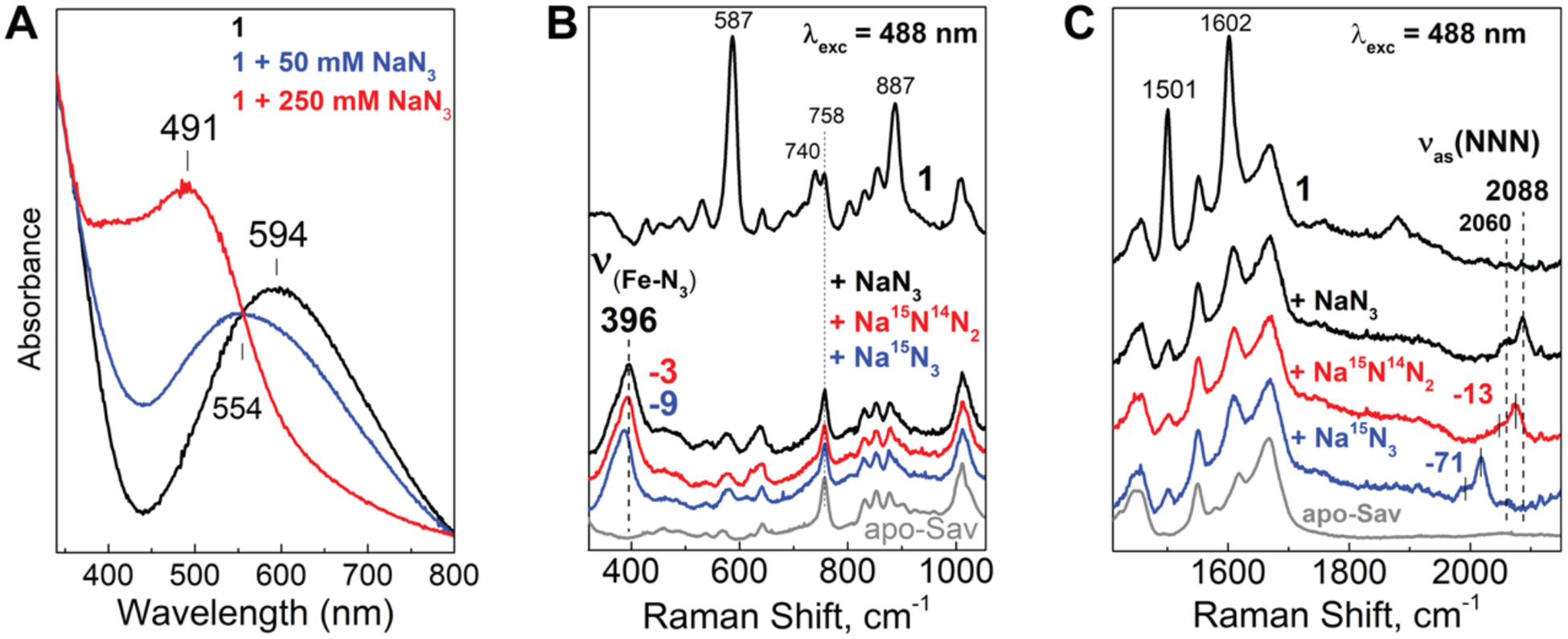
Optical and RR spectra of 1-N3: absorption changes upon the addition of N3− ions to 1 (A) and RR spectra in the low-energy region (B) and high-energy region (C).
Summary and Conclusions
There are now many different approaches to the development of ArMs, and all share a common prerequisite of site-specific insertion of a metallocofactor within a protein host. This requirement is necessary to avoid random cofactor binding, which can lead to heterogenous active sites, but site-specific binding is difficult to achieve and verify. Our engineered di-Fe protein, 1, demonstrated an effective approach that used biotin-Sav technology to ensure the reproducible formation of biomimetic active sites, in which an optical assay clearly indicated the proper match between artificial biotinylated metallocofactors and Sav variants. This assay relied on the formation of FeIII–OY bonds and demonstrated how the intrinsic properties of metallocofactor-protein interactions can be used to discover sites capable of binding metal ions within an ArM. The localization of the Fe complexes within Sav depended on the correct positional match between the linker of the artificial metallocofactors and the location of the tyrosine within the Sav dimer: in our system, only the butyl linker produced an ArM with FeIII–OY bonds.
The spectroscopic and structural investigations of 1 and its analogs, 1-X (X = OAc−, N3−, and CN−), revealed the formation of new di-Fe complexes within each Sav dimer. In solution, RR, XAS, and Mössbauer data agreed that [FeIII-(μ-OH)-FeIII] cores were present for 1 and 1-OAc. XRD experiments on single crystals revealed that the di-Fe complexes span the Sav dimer and function as crosslinks between the two subunits. The metrical parameters around the Fe centers in 1 and 1-OAc suggested the formation of hydroxido-bridged [FeIII-(μ-OH)-FeIII] cores based on the relatively long Fe–O2 bond lengths and Fe⋯Fe’ distances, which are inconsistent with those found in oxido-bridged species. However, our data did not allow us to assign with certainty the protonation states of the oxido ligand, which is a common problem with XRD studies on metalloproteins. We thus turned to Mössbauer studies on 57Fe-enriched crystals of 1 and 1-OAc, and the results further supported the presence of [FeIII-(μ-OH)-FeIII] cores: these studies were possible because our approach permits the generation of the large number of crystals needed for spectroscopic characterization. The ability to perform spectroscopic measurements on crystals of ArMs is an obvious advantage of the biotin-Sav approach, and as we illustrated here, it can offer a powerful complement to XRD experiments for investigating the molecular structures of active sites in ArMs.
The tethering of Fe complexes within adjacent subunits limits the movement of the Fe centers – we observed a small change in the Fe⋯Fe’ distance of only 0.23 Å from 1 to 1-CN that can be attributed to binding of the external bridging ligands. However, the predominate structural constraints on the di-Fe centers arise from the Sav host in which the Fe⋯Fe’ distances in the ArMs developed here are significantly longer than those observed in comparable di-Fe systems. These effects are best observed in 1-OAc, in which the Fe⋯Fe separation is 0.5 Å longer than in similar synthetic systems. The longer Fe⋯Fe separations represent examples of entatic states, whereby the protein exerts significant structural effects on the metallocofactor to produce a di-Fe core with an unusual molecular structure. ArMs like 1, and its derivatives, offer the opportunity to search for new types of reactivity because most biological and synthetic di-Fe systems carry out chemical transformations with Fe⋯Fe’ distances of less than 3.4 Å distances. Our work thus demonstrates the ability to engineer active sites that combine structural constraints with the ability to bind external species, offering a powerful approach in developing ArMs with bimetallic cores with enhanced function.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the NIH (GM120349 to A.S.B., GM077387 to M.P.H., and GM074785 to P.M.L.) for funding. The use of the Advanced Light Source at Lawrence Berkeley National Laboratory was supported through grant ALS-09183 to ASB. Use of the Stanford Synchrotron Radiation Light Source, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393). We thank Jonathan Paretsky for helpful advice.
Footnotes
Supporting Information
The following file is available free of charge.
Experimental descriptions for all new compounds and proteins, a full account of crystallographic data collection and refinements, explanations of RR, XAS, and Mössbauer spectral measurements, and details of DFT calculations. Also, Figures S1–S15 and Tables S1–S18 (PDF).
Reference
- (1).Klotz IM; Kurtz DM Binuclear Oxygen Carriers: Hemerythrin. Acc. Chem. Res 1984, 17, 16–22. [Google Scholar]
- (2).Tinberg CE; Lippard SJ Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res 2011, 44, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nordlund P; Reichard P Ribonucleotide Reductases. Annu. Rev. Biochem 2006, 75, 681–706. [DOI] [PubMed] [Google Scholar]
- (4).Minnihan EC; Nocera DG; Stubbe JA Reversible, Long-Range Radical Transfer in E. Coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res 2013, 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kang G; Taguchi AT; Stubbe JA; Drennan CL Structure of a Trapped Radical Transfer Pathway within a Ribonucleotide Reductase Holocomplex. Science (80-. ) 2020, 368, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Greene BL; Greene BL; Greene BL; Kang G; Cui C; Cui C; Bennati M; Bennati M; Nocera DG; Drennan CL; Drennan CL; Drennan CL; Stubbe JA; Stubbe JA Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem 2020, 89, 45–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Caranto JD; Weitz A; Giri N; Hendrich MP; Kurtz DM A Diferrous-Dinitrosyl Intermediate in the N2O-Generating Pathway of a Deflavinated Flavo-Diiron Protein. Biochemistry 2014, 53, 5631–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hayashi T; Caranto JD; Wampler DA; Kurtz DM; Moënne-Loccoz P Insights into the Nitric Oxide Reductase Mechanism of Flavodiiron Proteins from a Flavin-Free Enzyme. Biochemistry 2010, 49, 7040–7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Uppenberg J; Lindqvist F; Svensson C; Ek-Rylander B; Andersson G Crystal Structure of Mammalian Purple Acid Phosphatase. J. Mol. Biol 1999, 290, 201–211. [DOI] [PubMed] [Google Scholar]
- (10).Guddat LW; McAlpine AS; Hume D; Hamilton S; De Jersey J; Martin JL Crystal Structure of Mammalian Purple Acid Phosphatase. Structure 1999, 7, 757–767. [DOI] [PubMed] [Google Scholar]
- (11).Schenk G; Mitić NŠ; Hanson GR; Comba P Purple Acid Phosphatase: A Journey into the Function and Mechanism of a Colorful Enzyme. Coord. Chem. Rev 2013, 257, 473–482. [Google Scholar]
- (12).Whittington DA; Lippard SJ Crystal Structures of the Soluble Methane Monooxygenase Hydroxylase from Methylococcus Capsulatus (Bath) Demonstrating Geometrical Variability at the Dinuclear Iron Active Site. J. Am. Chem. Soc 2001, 123, 827–838. [DOI] [PubMed] [Google Scholar]
- (13).Rardin RL; Tolman WB; Lippard SJ Monodentate Carboxylate Complexes and the Carboxylate Shift : Implications for Polymetalloprotein Structure and Function. Nouv. J. Chim 1991, 15, 417–430. [Google Scholar]
- (14).Tolman WB; Liu S; Bentsen JG; Lippard SJ Models of the Reduced Forms of Polyiron-Oxo Proteins: An Asymmetric, Triply Carboxylate Bridged Diiron(II) Complex and Its Reaction with Dioxygen. J. Am. Chem. Soc 1991, 113, 152–164. [Google Scholar]
- (15).Shu L; Nesheim JC; Kauffmann K; Münck E; Lipscomb JD; Que LJ An Fe2IVO2 Diamond Core Structure for the Key Intermediate Q of Methane Monooxygenase. Science (80-. ) 1997, 275, 515–518. [DOI] [PubMed] [Google Scholar]
- (16).Cutsail GE; Banerjee R; Zhou A; Que L; Lipscomb JD; Debeer S High-Resolution Extended X-Ray Absorption Fine Structure Analysis Provides Evidence for a Longer Fe⋯Fe Distance in the Q Intermediate of Methane Monooxygenase. J. Am. Chem. Soc 2018, 140, 16807–16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Que L; Dong Y Modeling the Oxygen Activation Chemistry of Methane Monooxygenase and Ribonucleotide Reductase. Acc. Chem. Res 1996, 29, 190–196. [Google Scholar]
- (18).Kurtz DM Oxo- and Hydroxo-Bridged Diiron Complexes: A Chemical Perspective on a Biological Unit. Chem. Rev 1990, 90, 585–606. [Google Scholar]
- (19).Tshuva EY; Lippard SJ Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem. Rev 2004, 104, 987–1012. [DOI] [PubMed] [Google Scholar]
- (20).Do LH; Lippard SJ Evolution of Strategies to Prepare Synthetic Mimics of Carboxylate-Bridged Diiron Protein Active Sites. J. Inorg. Biochem 2011, 105, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Friedle S; Reisner E; Lippard SJ Current Challenges of Modeling Diiron Enzyme Active Sites for Dioxygen Activation by Biomimetic Synthetic Complexes. Chem. Soc. Rev 2010, 39, 2768–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kodera M; Itoh M; Kano K; Funabiki T; Reglier M A Diiron Center Stabilized by a Bis-TPA Ligand as a Model of Soluble Methane Monooxygenase: Predominant Alkene Epoxidation with H2O2. Angew. Chemie - Int. Ed 2005, 117, 7266–7268. [DOI] [PubMed] [Google Scholar]
- (23).Dong Y; Ménage S; Brennan BA; Elgren TE; Jang HG; Pearce LL; Que L Dioxygen Binding to Diferrous Centers. Models for Diiron-Oxo Proteins. J. Am. Chem. Soc 1993, 115, 1851–1859. [Google Scholar]
- (24).Fontecave M; Ménage S; Duboc-Toia C Functional Models of Non-Heme Diiron Enzymes. Coord. Chem. Rev 1998, 178–180, 1555–1572. [Google Scholar]
- (25).Jasniewski AJ; Que L Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev 2018, 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Arnold FH Directed Evolution: Bringing New Chemistry to Life. Angew. Chemie - Int. Ed 2018, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rosati F; Roelfes G Artificial Metalloenzymes. ChemCatChem 2010, 2, 916–927. [Google Scholar]
- (28).Delapierre CM; Rondot L Oxidation Catalysis by Rationally Designed Artificial Metalloenzymes. Isr. J. Chem 2015. 55, 61–75. [Google Scholar]
- (29).Oohora K; Onoda A; Hayashi T Hemoproteins Reconstituted with Artificial Metal Complexes as Biohybrid Catalysts. Acc. Chem. Res 2019, 52, 945–954. [DOI] [PubMed] [Google Scholar]
- (30).Lu Y; Yeung N; Sieracki N; Marshall NM Design of Functional Metalloproteins. Nature 2009, 460, 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ueno T; Abe S; Yokoi N; Watanabe Y Coordination Design of Artificial Metalloproteins Utilizing Protein Vacant Space. Coord. Chem. Rev 2007, 251, 2717–2731. [Google Scholar]
- (32).Heinisch T; Ward TR Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Challenges and Opportunities. Acc. Chem. Res 2016, 49, 1711–1721. [DOI] [PubMed] [Google Scholar]
- (33).Hartwig JF; Ward TR New “Cats” in the House: Chemistry Meets Biology in Artificial Metalloenzymes and Repurposed Metalloenzymes. Acc. Chem. Res 2019, 52, 1145. [DOI] [PubMed] [Google Scholar]
- (34).Nastri F; Chino M; Maglio O; Bhagi-Damodaran A; Lu Y; Lombardi A Design and Engineering of Artificial Oxygen-Activating Metalloenzymes. Chem. Soc. Rev 2016, 45, 5020–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yu F; Cangelosi VM; Zastrow ML; Tegoni M; Plegaria JS; Tebo AG; Mocny CS; Ruckthong L; Qayyum H; Pecoraro VL Protein Design: Toward Functional Metalloenzymes. Chem. Rev 2014, 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Simmons TR; Berggren G; Bacchi M; Fontecave M; Artero V Mimicking Hydrogenases: From Biomimetics to Artificial Enzymes. Coord. Chem. Rev 2014, 270–271, 127–150. [Google Scholar]
- (37).Reetz MT Directed Evolution of Artificial Metalloenzymes: A Universal Means to Tune the Selectivity of Transition Metal Catalysts? Acc. Chem. Res 2019, 52, 336–344. [DOI] [PubMed] [Google Scholar]
- (38).Lombardi A; Pirro F; Maglio O; Chino M; DeGrado WF De Novo Design of Four-Helix Bundle Metalloproteins: One Scaffold, Diverse Reactivities. Acc. Chem. Res 2019, 52, 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Calhoun JR; Nastri F; Maglio O; Pavone V; Lombardi A; DeGrado WF Artificial Diiron Proteins: From Structure to Function. Biopolym. - Pept. Sci. Sect 2005, 80, 264–278. [DOI] [PubMed] [Google Scholar]
- (40).Maglio O; Nastri F; Martin de Rosales RT; Faiella M; Pavone V; DeGrado WF; Lombardi A Diiron-Containing Metalloproteins: Developing Functional Models. Comptes Rendus Chim 2007, 10, 703–720. [Google Scholar]
- (41).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (42).Churchfield LA; Tezcan FA Design and Construction of Functional Supramolecular Metalloprotein Assemblies. Acc. Chem. Res 2019, 52, 345–355. [DOI] [PubMed] [Google Scholar]
- (43).Wade H; Stayrook SE; DeGrado WF The Structure of a Designed Diiron(III) Protein: Implications for Cofactor Stabilization and Catalysis. Angew. Chemie - Int. Ed 2006, 45, 4951–4954. [DOI] [PubMed] [Google Scholar]
- (44).Liang AD; Serrano-Plana J; Peterson RL; Ward TR Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Enzymatic Cascades and Directed Evolution. Acc. Chem. Res 2019, 52, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ward TR Artificial Metalloenzymes Based on the Biotin - Avidin Technology: Enantioselective Catalysis and Beyond. Acc. Chem. Res 2011, 44, 47–57. [DOI] [PubMed] [Google Scholar]
- (46).Wilson ME; Whitesides GM Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety. J. Am. Chem. Soc 1978, 100, 306–307. [Google Scholar]
- (47).Roy A; Vaughn MD; Tomlin J; Booher GJ; Kodis G; Simmons CR; Allen JP; Ghirlanda G Enhanced Photocatalytic Hydrogen Production by Hybrid Streptavidin-Diiron Catalysts. Chem. - A Eur. J 2020, 26, 6240–6246. [DOI] [PubMed] [Google Scholar]
- (48).Hyster TK; Knörr L; Ward TR; Rovis T Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C-H Activation. Science (80-. ) 2012, 338, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Serrano-Plana J; Rumo C; Rebelein JG; Peterson RL; Barnet M; Ward TR Enantioselective Hydroxylation of Benzylic C(Sp3)-H Bonds by an Artificial Iron Hydroxylase Based on the Biotin-Streptavidin Technology. J. Am. Chem. Soc 2020, 142, 10617–10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).DeChancie J; Houk KN The Origins of Femtomolar Protein-Ligand Binding: Hydrogen-Bond Cooperativity and Desolvation Energetics in the Biotin-(Strept)Avidin Binding Site. J. Am. Chem. Soc 2007, 129, 5419–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Freitag S; Trong ILE; Klumb L; Stayton PS; Stenkamp RE Structural Studies of the Streptavidin Binding Loop. Protein Sci. 1997, 6, 1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hendrickson WA; Pähler A; Smith JL; Satow Y; Merritt EA; Phizackerley RP Crystal Structure of Core Streptavidin Determined from Multiwavelength Anomalous Diffraction of Synchrotron Radiation. Proc. Natl. Acad. Sci. U. S. A 1989, 86, 2190–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hyre DE Cooperative Hydrogen Bond Interactions in the Streptavidin-Biotin System. Protein Sci. 2006, 15, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wu S; Zhou Y; Rebelein JG; Kuhn M; Mallin H; Zhao J; Igareta NV; Ward TR Breaking Symmetry: Engineering Single-Chain Dimeric Streptavidin as Host for Artificial Metalloenzymes. J. Am. Chem. Soc 2019, 141, 15869–15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Weber PC; Ohlendorf DH; Wendoloski JJ; Salemme FR Structural Origins of High-Affinity Biotin Binding to Streptavidin. Science (80-. ) 1989, 243, 85–88. [DOI] [PubMed] [Google Scholar]
- (56).Mann SI; Heinisch T; Weitz AC; Hendrich MP; Ward TR; Borovik AS Modular Artificial Cupredoxins. J. Am. Chem. Soc 2016, 138, 9073–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Mann SI; Heinisch T; Ward TR; Borovik AS Peroxide Activation Regulated by Hydrogen Bonds within Artificial Cu Proteins. J. Am. Chem. Soc 2017, 139, 17289–17292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Olshansky L; Huerta-Lavorie R; Nguyen AI; Vallapurackal J; Furst A; Tilley TD; Borovik AS Artificial Metalloproteins Containing Co4O4 Cubane Active Sites. J. Am. Chem. Soc 2018, 140, 2739–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Miller KR; Paretsky JD; Follmer AH; Heinisch T; Mittra K; Gul S; Kim IS; Fuller FD; Batyuk A; Sutherlin KD; Brewster AS; Bhowmick A; Sauter NK; Kern J; Yano J; Green MT; Ward TR; Borovik AS Artificial Iron Proteins: Modeling the Active Sites in Non-Heme Dioxygenases. Inorg. Chem 2020, 59, 6000–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Que L Metalloproteins with Phenolate Coordination. Coord. Chem. Rev 1983, 50, 73–108. [Google Scholar]
- (61).Davis JC; Lin SS; Averill BA Kinetics and Optical Spectroscopic Studies on the Purple Acid Phosphatase from Beef Spleen. Biochemistry 1981, 20, 4062–4067. [DOI] [PubMed] [Google Scholar]
- (62).Siu DCT; Que L; Orville AM; Lipscomb JD; Ohlendorf DH Resonance Raman Studies of the Protocatechuate 3, 4-Dioxygenase from Brevibacterium Fuscum. Biochemistry 1992, 31, 10443–10448. [DOI] [PubMed] [Google Scholar]
- (63).Averill BA; Davis JC; Burman S; Zirino T; Sanders-loehr J; Loehr TM; Sage JT; Debrunner PG Spectroscopic and Magnetic Studies of the Purple Acid Phosphatase from Bovine Spleen. J. Am. Chem. Soc 1987, 109, 3760–3767. [Google Scholar]
- (64).Que L; Mayer R; Lawrence AR; Robert H Resonance Raman Studies of Pyrocatechase-Inhibitor Complexes. Biochemistry 1980, 19, 2588–2593. [DOI] [PubMed] [Google Scholar]
- (65).Penner-Hahn JE A Short Fe-Fe Distance in Peroxodiferric Ferritin: Control of Fe Substrate versus Cofactor Decay? Science (80-. ) 2000, 287, 122–125. [DOI] [PubMed] [Google Scholar]
- (66).Griese JJ; Kositzki R; Schrapers P; Branca RMM; Nordström XA; Lehtiö J; Haumann M; Högbom XM Structural Basis for Oxygen Activation at a Heterodinuclear Manganese/Iron Cofactor. J. Biol. Chem 2015, 290, 25254–25272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Jasniewski AJ; Komor AJ; Lipscomb JD; Que L Unprecedented (μ−1,1-Peroxo)Diferric Structure for the Ambiphilic Orange Peroxo Intermediate of the Nonheme N-Oxygenase CmlI. J. Am. Chem. Soc 2017, 139, 10472–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Westre TE; Kennepohl P; DeWitt JG; Hedman B; Hodgson KO; Solomon EI A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes. J. Am. Chem. Soc 1997, 119, 6297–6314. [Google Scholar]
- (69).Roe AL; Schneider DJ; Mayer RJ; Pyrz JW; Widom J; Que L X-Ray Absorption Spectroscopy of Iron-Tyrosinate Proteins. J. Am. Chem. Soc 1984, 106, 1676–1681. [Google Scholar]
- (70).Sanders-Loehr J; Wheeler WD; Shiemke AK; Averill BA; Loehr TM Electronic and Raman Spectroscopic Properties of Oxo-Bridged Dinuclear Iron Centers in Proteins and Model Compounds. J. Am. Chem. Soc 1989, 111, 8084–8093. [Google Scholar]
- (71).Jullien J; Juhász G; Mialane P; Dumas E; Mayer CR; Marrot J; Rivière E; Bominaar EL; Münck E; Sécheresse F Structure and Magnetic Properties of a Non-Heme Diiron Complex Singly Bridged by a Hydroxo Group. Inorg. Chem 2006, 45, 6922–6927. [DOI] [PubMed] [Google Scholar]
- (72).Alam MA; Nethaji M; Ray M Structural Characterization of an Enantiopure Hydroxo-Bridged Binuclear Iron(III) Complex with Empty One-Dimensional Helical Channels. Inorg. Chem 2005, 44, 1302–1308. [DOI] [PubMed] [Google Scholar]
- (73).Jozwiuk A; Ingram AL; Powell DR; Moubaraki B; Chilton NF; Murray KS; Houser RP Redox and Acid–Base Properties of Asymmetric Non-Heme(Hydr)Oxo-Bridged Diiron Complexes. J. Chem. Soc. Dalt. Trans 2014, 43, 9740–9753. [DOI] [PubMed] [Google Scholar]
- (74).Turowski PN; Armstrong WH; Liu S; Brown SN; Lippard SJ Synthesis and Characterization of Hydroxo-Bridged Diiron(III) Complexes Containing Carboxylate or Phosphate Ester Bridges: Comparisons to Diiron(III) Proteins. Inorg. Chem 1994, 33, 636–645. [Google Scholar]
- (75).Kurtz DM Flavo-Diiron Enzymes: Nitric Oxide or Dioxygen Reductases? J. Chem. Soc. Dalt. Trans 2007, No. 37, 4115–4121. [Google Scholar]
- (76).Romão CV; Vicente JB; Borges PT; Victor BL; Lamosa P; Silva E; Pereira L; Bandeiras TM; Soares CM; Carrondo MA; Turner D; Teixeira M; Frazão C Structure of Escherichia Coli Flavodiiron Nitric Oxide Reductase. J. Mol. Biol 2016, 428, 4686–4707. [DOI] [PubMed] [Google Scholar]
- (77).Di Matteo A; Scandurra FM; Testa F; Forte E; Sarti P; Brunori M; Giuffrè A The O2-Scavenging Flavodiiron Protein in the Human Parasite Giardia Intestinalis. J. Biol. Chem 2008, 283, 4061–4068. [DOI] [PubMed] [Google Scholar]
- (78).Weitz AC; Giri N; Caranto JD; Kurtz DM; Bominaar EL; Hendrich MP Spectroscopy and DFT Calculations of a Flavo-Diiron Enzyme Implicate New Diiron Site Structures. J. Am. Chem. Soc 2017, 139, 12009–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Högbom M; Galander M; Andersson M; Kolberg M; Hofbauer W; Lassmann G; Nordlund P; Lendzian F Displacement of the Tyrosyl Radical Cofactor in Ribonucleotide Reductase Obtained by Single-Crystal High-Field EPR and 1.4-Å x-Ray Data. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3209–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Yan S; Que L; Taylor LF; Anderson OP A Model for the Chromophoric Site of Purple Acid Phosphatases. J. Am. Chem. Soc 1988, 110, 5222–5224. [Google Scholar]
- (81).Wallar BJ; Lipscomb JD Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem. Rev 1996, 96, 2625–2658. [DOI] [PubMed] [Google Scholar]
- (82).Vallee BL; Williams RJ Metalloenzymes: The Entatic Nature of Their Active Sites. Proc. Natl. Acad. Sci. U. S. A 1968, 59, 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Berry JF; Bill E; Bothe E; Weyhermüller T; Wieghardt K Octahedral Non-Heme Non-Oxo Fe(IV) Species Stabilized by a Redox-Innocent N-Methylated Cyclam-Acetate Ligand. J. Am. Chem. Soc 2005, 127, 11550–11551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.