

Abstract
Despite recent therapeutic advances in cancer treatment, metastasis remains the principal cause of cancer death. Recent work has uncovered the unique biology of metastasis-initiating cells that results in tumor growth in distant organs, evasion of immune surveillance and co-option of metastatic microenvironments. Here we review recent progress that is enabling therapeutic advances in treating both micro- and macrometastases. Such insights were gained from cancer sequencing, mechanistic studies and clinical trials, including of immunotherapy. These studies reveal both the origins and nature of metastases and identify new opportunities for developing more effective strategies to target metastatic relapse and improve patient outcomes.
Metastasis causes greater than 90% of cancer death. Unlike primary tumors, which can often be cured using local surgery or radiation, metastasis is a systemic disease. Systemic approaches, including screening, chemotherapy, targeted therapy and immunotherapy, are therefore the mainstay of metastasis prevention and treatment. Concerted efforts to improve cancer therapeutics in recent years are bearing fruit. The US cancer mortality rate declined by 29% from 1991 to 2017, with an average decline of 1.5% per year between 2013 and 2017. The steepest declines have been observed in metastatic melanoma (−6.4%) and lung cancer (−4.3%), largely owing to the transformative impact of immunotherapy1. In metastatic breast cancer, for which checkpoint immunotherapy was less widely effective but for which several new targeted therapies have been approved, the median 5-year survival for patients diagnosed with recurrent disease increased from 18.4% (95% confidence interval (CI), 13.6–24.8%) in 2000 to 32.6% (95% CI, 20.6–51.4%) in 2010 (ref.2). Despite these advances, mortality rates have stagnated or risen for several cancers, including those of the pancreas, liver, uterus and sarcomas, and the vast majority of patients with recurrent or de novo metastatic cancer of any type still die within 5 years of their diagnosis1,3. Treating metastasis therefore remains a challenge.
Progress in both basic cancer science and clinical oncology is critical to further improving the treatment of metastatic cancer. The last two decades have witnessed unprecedented collaboration between cancer biologists and clinical investigators. Technological advances have allowed the rapid accumulation of tumor genomic data annotated with disease progression and drug response information. Clinical trials increasingly include extensive real-time biospecimen collection and patient-specific model generation, such as patient-derived xenografts and organoids, before and during treatment and following the development of drug resistance. Innovative trial designs such as basket, umbrella and platform trials have shortened the time needed to bring a drug to the clinic4.
Such approaches enable investigators to nimbly identify biomarkers of therapeutic response, validate resistance mechanisms in ex vivo models and develop next-generation drugs. Rich datasets derived from this process lead to hypotheses on the underlying mechanisms of metastasis, which can then be tested in functional assays. Thus, the interplay between preclinical and postclinical studies is accelerating understanding of the biology of metastasis, allowing the development of new treatments. The goal of current research efforts is to develop new treatments targeting the singular biology of metastatic seeding, dormancy and micrometastatic growth during the dormant phase of metastasis, as well as to augment the efficacy of current therapies against overt metastasis. Here we focus on a selection of recent biological insights and how these advances point to new therapeutic opportunities to improve outcomes in patients with cancer.
The origins and progression of metastasis
Although cancer cell dissemination can start early during tumor progression5–7, most cells leaving a tumor fail to colonize distant organs and instead succumb to various stresses8. To form metastases, cancer cells must negotiate a series of steps previously termed the ‘metastatic cascade’, with each step requiring specific functions9,10 (Fig. 1). By acting on heterogeneous cancer cell populations, these pressures select for clones with fitness to colonize distant organs.
Fig. 1 |. Steps, biological functions and cancer cell vulnerabilities in the metastasis cascade.
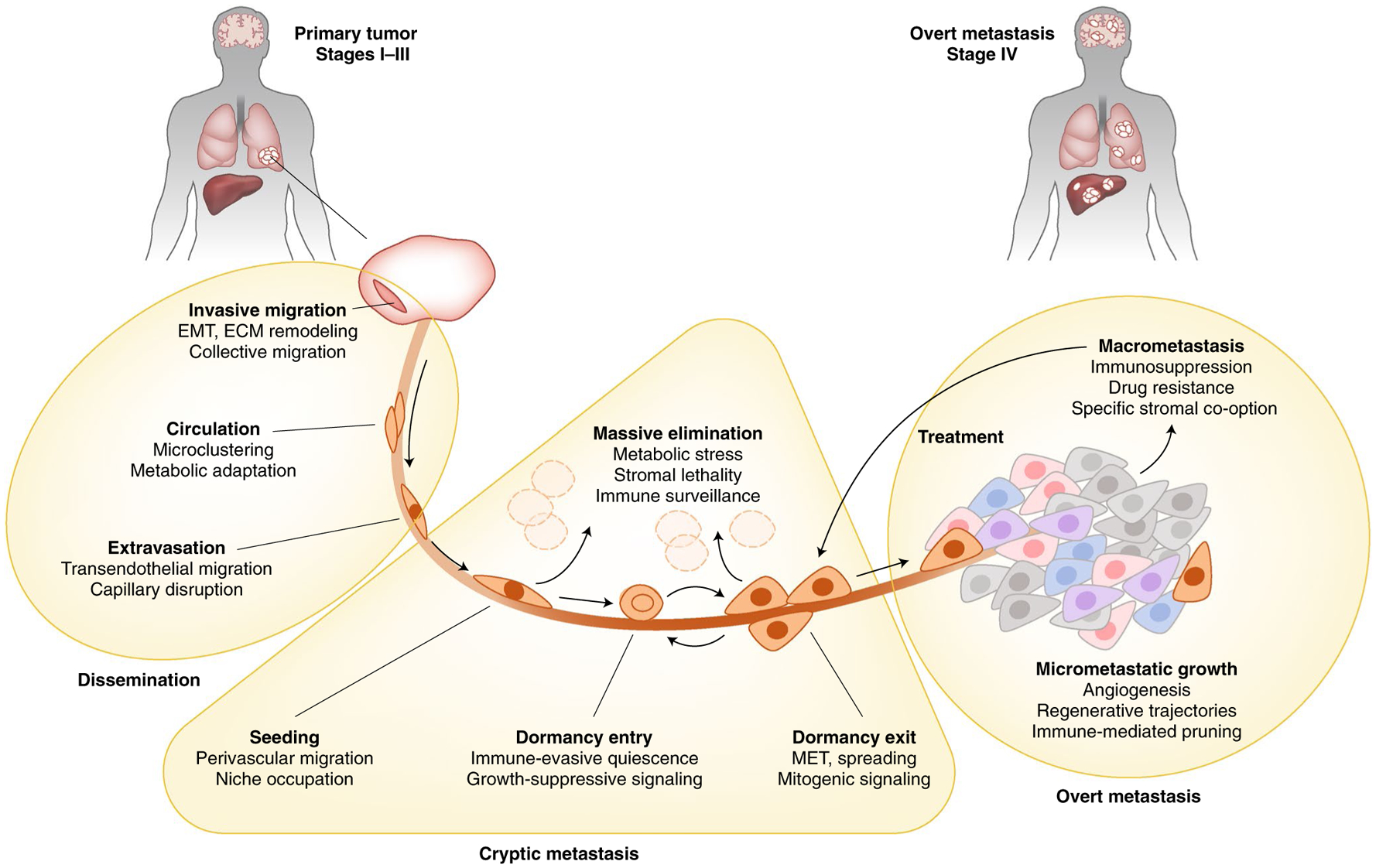
Local surgery or radiation and systemic approaches including chemotherapy, targeted therapy and immunotherapy are currently the mainstay of metastasis prevention and treatment and are frequently effective at reducing metastatic tumor mass. However, these treatments do not specifically target the cryptic phase of metastasis or regenerative progenitors that persist following therapeutic debulking of macrometastatic disease. Cancer cells disseminating from a primary tumor via the blood or lymphatic system require specific functions (as listed under each boldface step) to adapt to a number of stresses in order to invade vessels, survive the loss of niche factors from the originating organ and survive in the circulation. On reaching distant organs (gray area), cancer cells enter and exit proliferative dormancy, evade immunity and acquire mitogenic signals by co-opting the stroma of the distant organs. The majority of cancer cells leaving a primary tumor are unable to survive these stresses and are cleared. Cancer cells that survive and retain the ability to regenerate the tumor during the cryptic phase of metastasis are called metastasis-initiating cells (MICs). MICs launch overt metastatic growth in distant organs, develop along tissue-regenerative trajectories and deploy organ-specific stromal co-option functions. Clinically overt macrometastases may be effectively debulked by classic therapies, but resistance and relapse are driven by the plasticity and persistence of MIC states within macrometastases. ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; MET, mesenchymal-epithelial transition.
Sources of intratumoral heterogeneity.
The heterogeneity of cancer cell populations is rooted not only in genomic instability and genetic variation within a tumor but also in the capacity of malignant progenitor cells for extensive phenotypic variation. Stem-like malignant progenitors have the ability to fluidly adopt diverse phenotypic states in response to cell-intrinsic developmental programs as well as external stromal signals. This property, called phenotypic plasticity, enables cancer cells to adapt to specific microenvironments, overcome metastasis barriers and resist therapy11–13. Phenotypic plasticity exacerbates the heterogeneity of genomically diverse cancer cell populations14. Changes in the heterogeneity of cancer cell populations may in turn influence the composition of the tumor stroma and, with that, the emergence of cancer cells with the propensity to establish metastatic relapse.
Metastasis-promoting genes.
Studies combining experimental models of metastasis and patient-derived tumor gene expression data have identified genes whose expression in cancer cells promotes metastasis in mouse models and is associated with relapse in the clinic (reviewed in refs.10,15–17). A large array of genes are known to facilitate cancer cell dissemination steps such as invasion, circulation, extravasation, resistance to stromal and metabolic stresses, formation of metastatic niches, co-option of organ-specific stromal components and other pro-metastatic functions9,10. Some of these genes are expressed by cancer cells in the primary tumor, priming the cells for metastasis upon dissemination to particular organs. The expression of other metastasis-promoting genes becomes manifest as disseminated cancer cells adapt to a specific host tissue environment. Metastasis-promoting genes are candidate targets of treatments against metastasis, and several are the object of ongoing clinical trials (reviewed in ref.18).
Metastasis-driving mutations.
Other studies have leveraged large cancer genomics datasets to identify pro-metastatic mutations. This work is guided by the principle that cancer involves the evolution of malignant cell populations under selective pressure to survive the stresses of tumor progression19. The mutation patterns and overall mutational burden in primary and metastatic cancers are largely concordant, as demonstrated in comparisons of large cohorts of colorectal, non-small-cell lung, pancreatic and renal cell cancers, among others, although discrete subclonal genetic alterations suggest the sequential dominance of certain clones19–24. Cancer genomics studies have identified few recurrent metastasis-associated mutations, and recurrent mutations may be associated with resistance to specific therapies in metastatic disease, not mediators of metastatic cascade progression per se. Genes that regulate DNA methylation and chromatin modification are frequently mutated in aggressive tumors24, and studies of clinical and experimental metastasis have shown that acquired epigenetic and transcriptional changes are critical drivers of metastasis25–31. Alterations at the epigenetic level could favor phenotypes that are adept at dissemination and organ colonization.
Metastasis and cancer stem cells.
The concept that metastasis may be driven by metastasis-initiating cells (MICs) is rooted in stem cell biology. The cancer stem cell (CSC) hypothesis posits that only certain cells have the ability to initiate and propagate tumors32,33. Studies on leukemia, intestinal cancers and other cancers showed that tumor-initiating CSCs are homeostatic stem cells with acquired oncogenic mutations34–38. In principle, CSCs could directly become MICs by activating an additional set of metastasis-promoting genes while retaining their original stem cell phenotype. Indeed, metastatic lesions frequently mimic the histology, and hence the differentiation trajectory, of the tumor of origin39. However, experimental evidence from studies on colorectal cancer (CRC) suggests that MICs are phenotypically distinct from CSCs and are more akin to regenerative progenitors than to homeostatic stem cells40,12.
Metastasis and phenotypic plasticity.
MICs are endowed with marked phenotypic plasticity and undergo dynamic phenotypic changes (Fig. 2). For example, in CRC, mutations that hyperactivate the WNT signaling pathway, which is essential for the proliferation of intestinal stem cells, turn intestinal LGR5+ cells into tumor-initiating CSCs37. While tumors are initiated by LGR5+ cells, the cells that disseminate from the primary tumor invasion front, circulate in the blood and seed liver metastasis in mouse models are predominantly LGR5− (ref.13). Once established in the liver, some proliferating metastatic cells reacquire LGR5 expression as they go on to establish macrometastatic colonies13.
Fig. 2 |. Model of metastasis subverting normal regenerative processes.
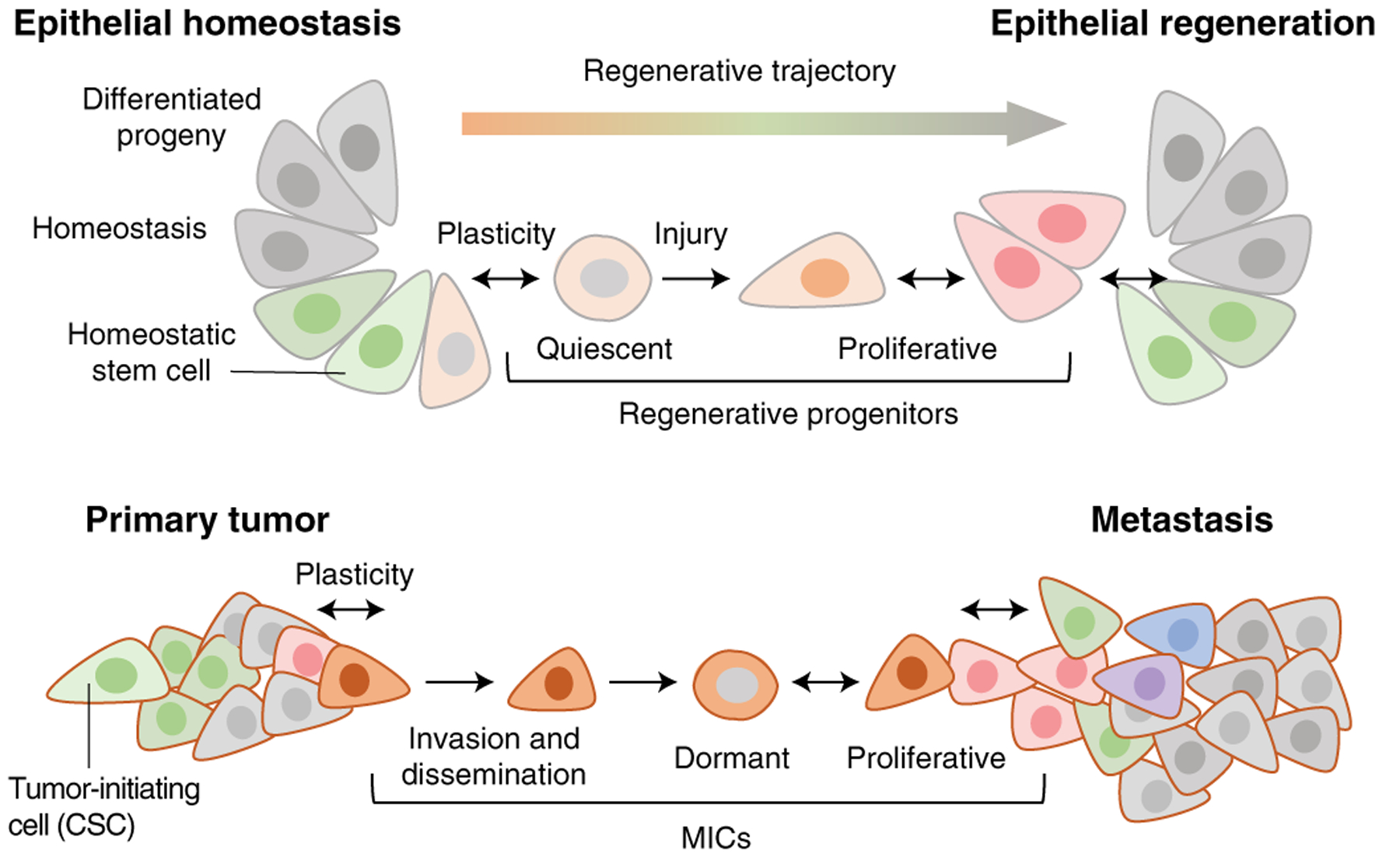
In normal epithelia, stem cells continuously generate differentiated progeny and maintain tissue homeostasis. Upon injury, quiescent progenitors emerge and give rise to proliferative daughter cells, which restore the epithelial barrier and lead to the regeneration of a homeostatic epithelium. Metastasis co-opts these regenerative processes. Oncogenic mutations in homeostatic stem cells generate primary tumor-initiating cells (CSCs). During tumor progression, cancer cells emerge that have a distinct regenerative progenitor phenotype. These cells can invade, disseminate and enter reversible quiescence to seed distant organs and eventually regrow a tumor, thus acting as MICs. Under appropriate environmental cues, these cells can regenerate heterogeneous tumors that recapitulate the developmental trajectories of the originating organ. Phenotypic plasticity (two-headed arrows) is prominent throughout these normal and tumorigenic regenerative processes.
Additional insights into the role of plasticity in metastasis come from studies on MICs expressing L1CAM, a cell adhesion molecule whose expression in primary tumors is associated with poor outcome in many types of cancer41. L1CAM is expressed by intestinal progenitors after injury and is required for regeneration of the epithelium40. In invasive carcinomas, dissociation of epithelial structures induces malignant progenitors to adopt a highly plastic regenerative phenotype endowed with migration, anoikis evasion and L1CAM-dependent growth reinitiation capacity40,42. In metastatic cells derived from CRC, lung, breast and renal carcinomas, L1CAM expression mediates initiation of metastatic growth in the brain, lungs, liver and bone marrow42,43. Thus, L1CAM+ MICs have the phenotype of regenerative progenitors that emerge upon disruption of epithelial integrity and drive the distant regrowth of tumors as metastases40 (Fig. 2).
Epithelial-mesenchymal transition.
Another phenotypic plasticity process relevant to metastasis is the epithelial-mesenchymal transition (EMT), in which epithelial cells lose polarity and cell-cell adhesions, migrate and invade stroma to generate tissue. EMT occurs during gastrulation, in several other developmental events and in wound healing, and it frequently involves a continuum of ‘partial EMT’ states between the mesenchymal and epithelial endpoints44–46. Cells with features of EMT are present at the invasion front of carcinomas, and EMT enables migration, invasion and metastatic dissemination of cancer cells47 (Fig. 1). At metastatic sites, cancer cells undergo a reverse process of mesenchymal-epithelial transition (MET) as a step toward the initiation of metastatic growth48,49. EMTs are driven by the SNAIL and ZEB transcriptional repressors of epithelial genes. Transforming growth factor (TGF)-β is a potent inducer of EMT in cooperation with other pathways, particularly the RAS-MAPK signaling pathway50.
These studies on regenerative progenitors and their phenotypic plasticity are illuminating a multi-stage process that mirrors epithelial repair reenacted in carcinoma metastasis (Fig. 2). Such lineage plasticity is often found in very aggressive end-stage metastatic cancers and is associated with resistance to standard tissue-lineage-specific therapies11,51. Understanding the mechanisms and functional consequences of plasticity is therefore vital to improving cancer therapeutics52,53.
Circulating tumor cells.
Cancer cells disseminate from tumors by invading blood and lymphatic vessels (Fig. 1). Cutaneous melanoma cells can also disperse by migrating on the abluminal surface of lymphatic vessels, a phenomenon known as extravascular migratory metastasis54. Perineural invasion of cancer cells that migrate along nerves has also been documented55. However, hematogenous dissemination is considered to be the main form of metastatic spread to distant organs. Cancer cells in the blood circulation, referred to as circulating tumor cells (CTCs), are a focus of intense research. CTCs express progenitor and EMT markers, suggesting that these cells are primed to grow as metastatic tumors56,57.
The study of CTCs provides a unique window into the biology of cancer cells that are on the way to seed metastasis58,59. CTCs have a short half-life in circulation and largely disappear upon removal of the primary tumor. The vast majority of CTCs are eliminated and never go on to form metastases. The extent to which some CTCs persist or reappear after the primary tumor is eliminated likely reflects the existence of active metastases. Therefore, treatment will still require targeting of metastatic cells that have already established residence in distant organs.
Detection of CTCs in untreated patients with localized disease as identified by imaging may help identify patients with subclinical metastatic disease, potentially sparing these patients invasive surgeries that are unlikely to be curative or driving more aggressive therapy. Specific molecular features may identify CTCs that have a greater propensity for eventual metastatic relapse and thus may serve as clinically relevant biomarkers. In this regard, it is note-worthy that cancer cells circulate singly as well as in clusters60, and CTC clusters have a superior ability to seed metastasis in experimental models61. CTC clusters are enriched in genome methylation patterns that denote a stem-like cancer cell state57. It remains to be determined whether stem-like cancer cells seed metastasis because they form clusters or form clusters and seed metastasis because they are stem-like.
While the sensitivity of current CTC detection assays remains limiting, improved technology for CTC and tumor cell-free DNA (cfDNA) detection is enabling early detection and may potentially guide early treatment of metastatic relapse62,63. In contrast, patients with established metastatic disease typically have higher CTC and cfDNA burdens than can readily be detected using current assays, which have utility in enabling serial noninvasive ‘liquid biopsy’ monitoring of therapy response and the emergence of resistant tumor subclones64–66. Importantly, unlike a tissue biopsy of one tumor, liquid biopsies integrate information from multiple, spatially discrete metastatic tumors, capturing intertumoral heterogeneity and maximizing the chance of detecting subclonal resistance-associated mutations that may be specific to individual tumors67.
Protective dormancy.
In many cancers, surgical resection of the primary tumor is followed by a period lasting from months to years without clinical evidence of disease but ultimately followed by aggressive outgrowth of cryptic metastases (Fig. 1). MICs that were disseminated before the removal of the primary tumor can remain latent, dynamically fluctuating between dormant and proliferative states until conditions allow these cells to evade clearance by the immune system and grow as a metastatic outbreak. It is thought that there are two mechanisms for this: cellular dormancy, in which disseminated cancer cells fail to form a colony by entering proliferative quiescence, and tumor mass dormancy, in which tumor growth becomes limited by the failure to activate angiogenesis or reaching an equilibrium with other constraints imposed by the host stroma68,69.
Disseminated cancer cells in mouse models of metastasis are frequently located near capillaries, where they enter growth arrest70–72. TGF-β, a potent growth inhibitor for epithelial progenitors73, is present in perivascular locations and inhibits the growth of disseminated cancer cells70,74,75. Although metastatic dormancy is commonly attributed to the effect of stromal growth-inibitory signals on MICs, MICs can proactively suppress local growth-promoting signals in order to enter a quiescent state71, which protects MICs form immune surveillance76–79. The relevance of the immune system in metastatic dormancy is clearly implied by cases in which immunosuppressed organ recipients developed metastasis in organs transplanted from donors who had been deemed cured of melanoma80.
Establishing a niche.
Normal adult stem cells depend on cues that balance their proliferation, self-renewal and differentiation. These cues are provided by contacts with neighboring cells, extracellular matrix and diffusible factors that form a local tissue structure or composition called the ‘stem cell niche’ (refs.81,82). The survival, quiescence and outbreak of disseminated metastatic cells similarly depend on stromal signals, cell contacts, extracellular matrix and metabolic cues collectively referred to as the ‘metastasis niche’ (refs.18,39,83). The interactions between MICs and their niches are bidirectional, with cancer cell-derived factors making the host tissue more conducive to MIC survival.
Disseminated cancer cells are initially unwelcome in the host tissue10. Tissue-resident macrophages and natural killer (NK) cells are an important line of defense against disseminated cancer cells84–86. In addition, bone marrow-derived and peripheral immune cells are recruited to newly seeded metastases. Depletion of T cells and NK cells increases metastasis in experimental models71,87,88. In line with experimental observations, tumor lymphocyte infiltration as measured by an ‘immunoscore’ was inversely correlated with metastasis size in patients with CRC89. Non-immune cells, such as astrocytes in the brain, also mediate killing of infiltrated cancer cells43. However, these mediators of anti-metastatic defense may ultimately be co-opted by MICs to provide a supportive niche. Recent work is rapidly expanding the list of stromal cell types capable of supporting metastatic tumor growth, which may in turn be potential targets for therapeutic intervention: these include osteoblasts and osteoclasts in bone metastasis16,90–92; monocytes93, neutrophils94–96 and normal epithelial cells97 in pulmonary metastasis; astrocytes98, microglia99 and neurons100 in brain metastasis; and endothelial cells in multiple sites42,43,70. The composition of metastatic niches may vary depending on the colonization step, from niches that regulate MIC dormancy to those that support aggressive metastasis growth.
Work with mouse models of metastatic disease showed that factors released from primary tumors into the circulation can prime distant organs to become supportive of the growth of arriving cancer cells, thus creating a ‘pre-metastatic niche’ (ref.101). A number of tumor-derived factors have shown activity in this context, including chemokines, cytokines and hormones released by tumors in soluble form or packed in a variety of extracellular vesicles94,102. How tumor-derived cytokines could affect metastatic relapse years after the removal of a primary tumor remains unknown. While these data are intriguing, clinical evidence for the targetability of niche-priming factors is presently lacking.
Treating metastatic cancer
The growing knowledge about the biology of metastasis provides opportunities to improve clinical outcomes in patients with cancer. Discerning the biological differences between micro- and macrometastases, targeting the vulnerabilities of metastatic cancer cells and exploiting the properties of metastatic tumor microenvironments provide a basis for present and future treatments of metastasis.
Developing therapies for micro- and macrometastases.
Patients with overt metastatic disease present as having stage IV cancer or develop a distant recurrence following previous removal of the primary tumor. These patients have metastases of at least 1 cm3 that can be detected by cross-sectional imaging. Such overt metastasis may be clinically manageable by systemic therapy but is typically incurable. The other context in which patients receive systemic therapy is early-stage cancer, where no metastasis is apparent but micrometastasis is assumed to be present. Therapy might be administered either before surgery (neoadjuvant therapy) or after surgery (adjuvant therapy) and is applied to high-risk early-stage cancer, in which a visible tumor is localized to the originating organ and can be completely surgically removed. In most cancers, adjuvant and/or neoadjuvant therapy substantially reduces recurrence rates and prolongs overall survival for subsets of patients with stage II or stage III cancer, demonstrating the feasibility of eliminating disseminated tumor cells or their metastasis-initiating capacity.
In cases of early-stage cancer where the primary tumor is entirely removed by surgery but the patient later relapses with macrometastatic disease, patients must have already harbored disseminated cancer cells in distant organs before tumor removal. While targeting the early steps of tumor invasion and survival in the circulation are, in our opinion, unlikely to be effective at preventing or treating metastasis, targeting micrometastatic cells could prevent metastasis. However, the majority of therapeutic effort has focused on the final steps of the metastasis cascade: colonization of distant organs. New drugs are typically first evaluated in clinical trials in patients with advanced, therapy-resistant metastatic disease, primarily because these are the patients for whom no effective standard treatments exist. This provides an ethical rationale for testing untried new agents in these patients. Second, the effectiveness of therapy can be evaluated in a relatively short time frame of a few months, given the rapid rate of growth of advanced metastatic disease103. Only when drugs have been shown to effectively control advanced metastatic disease are they then tested in patients who may have other standard treatment options, first in the treatment of patients with newly diagnosed metastatic disease and, only then, in the adjuvant/neoadjuvant setting, in patients with surgically removed disease, where a fraction of patients remain at risk of later macrometastatic relapse due to the proliferation of pre-existing micrometastatic cancer cells. Because many patients with early-stage disease will be cured by surgery alone and may not require anti-metastatic therapy, the threshold for testing a new drug, which may have unknown and potentially lifelong side effects, is very high. Further, the cost and administrative burden of performing an adjuvant/neoadjuvant study are substantially greater than those for a study in the advanced metastatic setting, because several years of follow-up of large patient cohorts is needed to establish treatment efficacy.
To date, no drugs have been approved exclusively for use in the adjuvant setting and not for macrometastatic disease, although some dormancy-specific trials in the adjuvant setting are in progress (reviewed in ref.104). Indeed, several drugs that have been demonstrated to provide a survival benefit in treating macrometastasis fail to show benefit in the adjuvant setting against micrometastasis. In colon, breast and non-small-cell lung (NSCLC) cancers, only one in three agents approved to treat macrometastatic cancer have been approved for perioperative treatment when no visible metastases are present105. While such attrition could be due to inadequate trial sample size or insufficient depth of pathway inhibition, it could also be due to the intrinsic differences in the biology of dormant and indolent micrometastases in the adjuvant setting versus growing, radiologically apparent macrometastases.
Most preclinical studies assessing novel therapeutic approaches aim to prevent metastasis or slow tumor growth in animal models, not to shrink large tumors. Thus, there is a disconnect between preclinical studies, which aim to control micrometastases, and first-in-human clinical studies, which assess shrinkage of macrometastases. This disconnect may explain why several drugs that showed promise in controlling micrometastatic tumor growth in preclinical studies failed to achieve shrinkage of macrometastatic tumors in human clinical trials. As a corollary, drugs that may in fact control micrometastatic disease fail to reach the patients who may most benefit from them106.
Targeting metastatic cancer cells.
Both micrometastatic and macrometastatic disease are currently treated with three broad systemic approaches: chemotherapy, targeted therapy and immunotherapy, sometimes in combination. While cytotoxic chemotherapy remains the backbone of metastasis treatment and is currently the only option for many cancer subtypes, drugs that target tumor-driving oncoproteins, or ‘targeted therapy’, are improving outcomes in many cancers107 (Fig. 3). However, mutation-specific therapy can elicit dramatic tumor responses that are often short-lived, and these therapies often select for the expansion of tumor subclones that harbor drug-resistance mutations or bypass targeted pathways and secretomes107–109.
Fig. 3 |. Classic and new opportunities for the treatment of metastatic cancer.
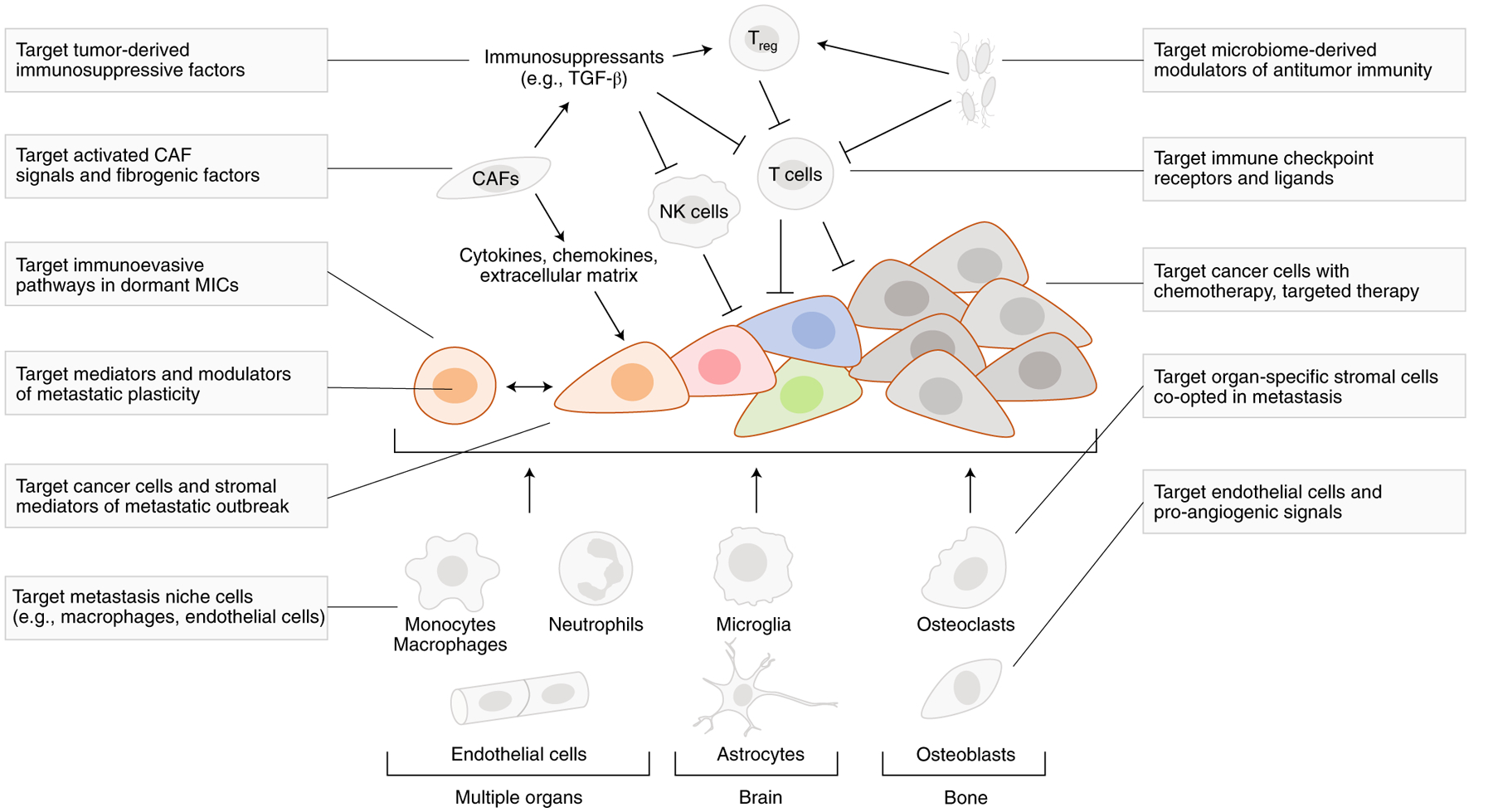
Targeting cancer cells with chemotherapy and targeted therapies is a mainstay of metastasis prevention and treatment. However, the recent success of ICI therapy demonstrates the value of targeting specific components of the tumor stroma (T cells) to treat metastasis. Leveraging recent insights into the regenerative origins, phenotypic plasticity, immune evasion and organ-specific colonization strategies of MICs could yield more potent approaches to prevent metastasis by targeting its cryptic phase during dormancy and micrometastasis and to augment the efficacy of ICI and other therapies by more effective elimination of drug-resistant macrometastatic disease. CAFs, cancer-associated fibroblasts; Treg, regulatory T cells; NK, natural killer cells; TGF-β, transforming growth factor-β.
Patient biospecimen-based studies are rapidly revealing resistance mechanisms and suggesting routes to improvement. In NSCLC, for example, first-generation tyrosine kinase inhibitors (TKIs) against activating epidermal growth factor receptor (EGFR) mutations, gefitinib and erlotinib, improved overall survival in metastatic disease but not in the adjuvant setting110,111. In contrast, the third-generation TKI osimertinib, which targets the drug-resistant EGFRT790M mutant, improved overall survival in both the metastatic and adjuvant settings112–114, underscoring the need for improving drugs in the pipeline to attack subclonal disease-resistance mutations earlier in the course of disease and more effectively.
Other strategies to overcome resistance include combination or sequential therapies to target resistance pathways, often guided by synthetic lethality screens, where cancer cells may compensate for the genetic or pharmacological inhibition of an individual signaling pathway by becoming dependent on a complementary signaling pathway for their survival. Targeting both pathways simultaneously may thus enable selective killing of cancer cells while sparing normal cells that have not become dependent on the second pathway. Recent successes of the synthetic lethality approach include poly(ADP-ribose) polymerase (PARP) inhibitors for cancers with DNA damage repair defects115,116 and cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors with estrogen receptor antagonists for hormone receptor-positive breast cancer117. Several investigational approaches aim to deliver higher doses of biologically active drug more specifically to the tumor while limiting the toxicity of inhibiting the same pathway in normal cells, thus maximizing the therapeutic window (the difference between the drug dose needed to kill cancer cells and the dose needed to kill normal cells), for example, by using antibody-drug conjugates, nanoparticle delivery systems or bispecific antibodies.
Targeting the metastasis microenvironment.
In 1889, Stephen Paget described metastasis as a fertile “seed” landing on a receptive “soil” (ref.118). While cancer treatments have historically focused on tumor cell-intrinsic properties, the advent of immunotherapy has underscored the importance of the tumor microenvironment in controlling metastasis119 (Fig. 3). Tumor neovasculature was an early therapeutic target in the tumor microenvironment120, and, indeed, anti-vascular endothelial growth factor (VEGF) TKIs and the anti-VEGF antibody bevacizumab have shown efficacy in the metastatic setting in several cancers121. In contrast, in renal cell cancer and CRC, VEGF-targeted therapy was not effective in the adjuvant setting122,123, in line with dormant micrometastases not having activated angiogenesis for proliferation.
The past decade has witnessed a revolution in metastasis treatment in the form of immune checkpoint inhibition (ICI). For the reasons discussed above, the majority of immunotherapy clinical trials to date have been conducted in patients with macrometastatic cancer that has become resistant to standard therapy. Unlike other drugs for metastatic cancer, antibodies that block the receptor-ligand interactions of CTLA-4 and PD-1 can induce long-term, durable responses, including complete tumor regression in some patients124,125 (Fig. 3). At present, only a limited portion of patients with cancer benefit from ICI126, including those with metastatic melanoma116 and lung127, bladder or renal cell128 carcinomas and those with mismatch repair-deficient cancers129,130. The types of metastatic cancer that respond to current ICI have a high tumor mutational burden, thus generating more mutated peptides that are processed, displayed on tumor cell-surface major histocompatibility complex (MHC) class I molecules and recognized as foreign ‘neoantigens’ by the immune system131–135. To understand further why there are differing responses, researchers are applying diverse technologies, including single-cell mRNA sequencing, spatial transcriptomics and mass cytometry approaches, to gain insight into the mechanisms of tumor cell recognition by immune cells and immune cell priming and infiltration. The goal is to use this information to induce more effective antitumor ICI responses. Strategies targeting the immunosuppressive cross-talk among immune cells, cancer cells and other components of the metastatic tumor microenvironment may be particularly beneficial136,137 (Fig. 3). The recent success of combination multi-kinase inhibitor therapy together with ICI in metastatic cancers that were otherwise immune unresponsive illustrates the promise of this approach138–140.
To what extent do the principles of immunotherapy of established macrometastasis apply to micrometastasis? ICI in melanoma in the adjuvant setting in patients with completely resected stage III and stage IV disease results in macrometastatic relapse-free survival and, in some cases, an overall survival benefit over placebo control141–144. Given the typically short relapse-free survival (<2 years) in these patients with melanoma, melanoma micrometastases are likely to be rapidly proliferating, albeit too small to detect with current imaging technology at the time of surgery in these individuals. Patients with melanoma who have a greater time lag from removal of the primary tumor to eventual radiological detection of macrometastasis may harbor micrometastases with a distinct biology, potentially with a greater tendency to enter dormancy, or that use distinct mechanisms of immune evasion. Ongoing clinical studies will determine the extent to which ICI is effective in treating micrometastases in surgically resectable cancers beyond melanoma.
Intriguingly, in syngeneic mouse models of triple-negative breast cancer, neoadjuvant anti-PD-1 and anti-CD137 treatment, with the primary tumor still present and subsequently surgically removed after ICI, increased tumor-specific T cell expansion, enhanced eradication of micrometastasis and prolonged survival in comparison to adjuvant ICI treatment administered after primary tumor removal145. In patients with melanoma or NSCLC, early neoadjuvant ICI trials have also demonstrated increased T cell expansion, an enhanced BATF3+ dendritic cell signature associated with increased antigen presentation, a broader T cell repertoire and an excellent primary tumor response, in comparison to adjuvant treatment, with survival data not yet mature146–150. One interpretation of these data is that neoantigens derived from the primary tumor may help prime the immune system during neoadjuvant checkpoint therapy, driving a more robust immune response against distant micrometastases that share these neoantigens. Once the tumor is surgically removed, this immune priming effect may be blunted because fewer tumor cells, and hence neoantigens, remain when ICI therapy is administered. Seeking to leverage the immune-priming effect of tumor neoantigens to boost anti-metastatic immune reactions, several groups are attempting to vaccinate patients using neoantigen peptides that are either patient specific or based on common mutations shared by many patients131.
Treating organ-specific metastasis.
While most anti-metastatic drugs are appropriately systemic, there are opportunities to substantially improve patient outcomes by targeting organ-specific mediators of metastasis. Some cancers metastasize to a single site and remain exclusively in that organ for the remainder of the patient’s life or spread to other organs only after a prolonged period of time. Such phenotypes offer opportunities to improve survival via regional therapy. CRC metastasis to the liver is the classic example. All blood draining the intestine flows first into the liver via the hepatic portal vein. This anatomical constraint means that the vast majority of CTCs disseminating from an intestinal cancer must first traverse the hepatic sinusoids, where they become trapped and initiate metastases with far higher frequency than in other sites such as the lung or bone. Surgical resection of small-volume CRC liver metastases is an established standard of care and is curative in approximately 20% of patients151. Liver metastasis-directed therapies are increasingly being used and include hepatic artery infusion chemotherapy, radiofrequency ablation and embolization152. Beyond CRC liver metastasis, recent randomized phase 2 studies have demonstrated improved overall survival for patients with oligometastatic cancers with consolidative radiotherapy administered to kill residual cancer cells in the tumor bed following surgical resection of the primary tumor153,154.
Bone is typically an early site, and sometimes the only site, of metastasis for hormonally driven breast and prostate cancers. The alpha emitter radium-223, which selectively binds to areas of increased bone turnover, improves overall survival in patients with bone-metastatic prostate cancer155. There is also promise in targeting host organ-specific stromal components (Fig. 3). In the adjuvant setting, meta-analyses of studies in thousands of women showed a small but statistically significant overall survival benefit from bisphosphonates, which inhibit osteoclast-mediated bone resorption156,157.
Another case for organ-specific therapy is the brain, which is typically the last site of metastasis relapse. Because space-occupying lesions in the constrained physical parameters of the skull can cause rapid demise, any disease control here can improve survival in patients with end-stage cancer158. Indeed, prophylactic cranial irradiation increases overall survival and is a standard of care in the adjuvant treatment of small-cell lung cancer, which invariably metastasizes to the brain159. Along with the brain, other metastatic sites that are separated from the circulation by tissue barriers or have immunosuppressive microenvironments include the leptomeningeal space and the peritoneal cavity. Improved understanding and targeting of metastasis mechanisms in these unique microenvironments are likely to improve outcomes for cancers that disseminate to these clinically challenging sites43,98,99,160–163.
Future directions
While current approaches to treat metastatic cancer target well-known vulnerabilities such as the high proliferative activity, oncogene dependence and immune susceptibility of cancer cells, recent insights into the biology of metastasis have revealed new types of potential targets that could be exploited for therapeutic advantage.
Targeting plasticity.
Basic interrogations of cancer mechanisms and insights from the application of novel technologies such as single-cell RNA sequencing to clinical specimens are revealing more fully the complexity of targeting highly plastic and heterogeneous cancers164,165. At the same time, the curative potential of immunotherapy and steady progress made by improvements in mechanism-driven targeted therapy are driving progress. The very presence of tumor cell plasticity suggests numerous routes to therapy resistance; however, MICs across multiple cancer types share conserved regenerative phenotypes. Identification of key molecular determinants of such states, such as L1CAM40 and CD36 (ref.166), is likely to yield improved therapeutic targets (Fig. 3). Defining mechanisms of plasticity using transcriptional and epigenetic approaches may enable manipulation of regenerative pathways, for example, by blocking phenotypic transitions that provide MICs with adaptability or by triggering conversion of MICs into immunosensitive states53. Novel phenotypes, such as neuroendocrine differentiation, that arise as a consequence of lineage plasticity could be preempted using drugs that target the new cell states11,167–169. Targeting chromatin remodeling using selective inhibitors of DNA methylation or histone-modifying enzymes is likely to be a particularly beneficial therapeutic approach to limiting plasticity or enforcing terminal differentiation of regenerative progenitor states170.
Targeting metastatic dormancy.
Improved mouse models of metastasis and rapid autopsy programs in patients are enabling novel insights into the maintenance and outgrowth of dormant MICs. The role of the immune system in maintaining tumor dormancy is increasingly being appreciated. The results of ongoing neoadjuvant ICI studies, anticipated in the next few years, will provide valuable insight into the extent to which such therapy can eliminate micrometastasis, identifying novel response biomarkers as well as molecules and cell types of interest for mechanistic interrogation. In addition to immunotherapy approaches, knowledge gleaned from mouse modeling of metastasis could be leveraged to inhibit the reawakening of quiescent MICs or to accelerate their elimination. Defining whether MICs in patients, in particular, non-cycling cells, have distinct immune evasion mechanisms will be crucial to designing strategies to eliminate such cells.
While there is a dearth of specific therapy for dormant micrometastasis, it is also evident that large numbers of patients are over-treated with drugs in the adjuvant setting that are ineffective at, or unnecessary for, prevention of metastatic recurrence but cause lifelong toxicity. Large randomized clinical trials in common cancers, including TAILORx in breast cancer and IDEA in CRC, have demonstrated that adjuvant therapy can safely be de-escalated in many patients171–173. Several planned and ongoing adjuvant studies are exploring novel biomarkers, notably, circulating tumor DNA and immune correlates, to enable appropriate stratification of patients’ probabilities of developing metastasis and selection of the patients most likely to benefit from adjuvant therapy174–178. There is a need to develop clinically actionable, sensitive biomarkers that can distinguish between the two regenerative stages of metastasis discussed above, as distinct treatments are likely to be needed for cells in different phenotypic states.
The microbiome environment.
The microbiome, both mucosal and intratumoral, is emerging as a potentially important determinant of metastasis progression and therapy response179,180. A recent study demonstrated that both epithelial cells and immune cells in many cancers not traditionally associated with microbial contact, including those of the breast, ovary, bone and brain, have a rich and distinctive intratumoral microbiome181. Fusobacterium strains have been demonstrated to reside in both primary and metastatic CRC and breast cancer and to drive metastasis182,183. Antibiotic treatment to reduce the Fusobacterium load reduced metastatic burden in mouse xenografts182 (Fig. 3). The composition and diversity of the gut microbiome have been shown to correlate with ICI response in metastatic cancers184,185. The majority of metabolites, mechanisms and therapeutic targets in the human microbiome remain to be uncovered. Microbes that target specific tissue types or bind to cancer cell-specific receptors can also be used as drug delivery vehicles186.
Patient-derived models.
Recent advances have underscored the importance of plasticity in metastatic cancers. Thus, preclinical models that may be used to inform future therapeutic targeting should ideally recapitulate dynamic plasticity between the two regenerative states that emerge during metastasis and therapy. Patient-derived organoid systems, which can be generated with high efficiency from the majority of patients and recapitulate developmental trajectories and regenerative states, are well suited to meet this need and complement existing cell lines, genetically engineered mouse models and patient-derived xenograft models187. Patient-derived organoids have been demonstrated to faithfully reproduce the drug sensitivities observed in patients, underscoring the validity of this approach188,189. One limitation of most organoid modeling to date has been the lack of the tumor microenvironment, but this is increasingly being addressed using sophisticated co-culture systems, tumor fragment cultures and bioengineering approaches190,191. The advent of immunotherapy has highlighted the need for syngeneic immunocompetent genetically engineered mouse models that are immunogenic. Metastatic human cancers typically have a higher mutational burden and are more aneuploid than genetically engineered mouse tumors. The extent to which even the most complex mouse models recapitulate the neoantigen landscape and immune microenvironment of advanced metastatic cancer in patients is unknown. The development of humanized mice enables patient-specific tumor organoid and immune cell reconstitution in an orthotopic in vivo environment192. It remains to be seen whether such models, which are currently only sparingly used owing to their complexity and expense, may prove to be better mimics of cancer in humans.
Molecular profiling of patients.
Clinical trials for metastatic cancer are increasingly seeing more biomarker-based patient selection and biospecimen collection and monitoring on trial to enable rapid discovery and validation of resistance mechanisms and populations most likely to benefit from specific interventions193. Rapid bidirectional translation between the clinic and the laboratory is becoming the norm rather than the exception, to the benefit of patients with cancer. Interrogation of novel MIC biomarkers and targets, emerging from preclinical studies, in clinical trials is likely to bear fruit. Deep genetic and immunological characterization of ‘exceptional responders’ to therapy is providing novel insights into mechanisms of metastatic control194,195. Basket trials, which allow drugs targeting a specific mutant allele to be tested in ‘baskets’ of patients from different cancer types, are an emerging trend and have led to the pan-cancer approval of pembrolizumab for microsatellite-high/mismatch repair-deficient cancers and tumor mutation burden-high metastatic cancers129 and entrectinib for metastatic cancers harboring an NTRK fusion196. However, the tissue-lineage-agnostic approach has its limitations: tissue-specific resistance mechanisms have been described (for example, single-agent BRAFV600E inhibitors are effective in melanoma but not in the colon owing to adaptive resistance from feedback activation of EGFR in the latter197), and results must be interpreted with caution.
Conclusions
Effective therapeutic targeting of metastatic cancer must address not only the dynamic plasticity of the cancer cells themselves as they progress through the metastatic cascade but also the mechanisms used by dormant and growing metastases to corrupt and co-opt their niches and evade immune surveillance. Converging lines of evidence from close observation of patient phenotypes, analysis of clinical samples and experimental studies in improved preclinical models are illuminating the properties of metastatic cancers as regenerative states distinct from primary tumors. It has long been appreciated that cancer is akin to a wound that never heals198, and now we know more about why this is so.
Acknowledgements
The authors’ work in the subject area of this Review is supported by NIH grants R35CA252978 (J.M.), K08CA23021 (K.G.), P30-CA008748 (MSKCC) and the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center at MSKCC (J.M.).
Footnotes
Competing interests
J.M. owns stock of Scholar Rock, Inc. J.M. is an inventor on the following patents and patent applications: Assay for anti-metastatic agents (inventors: J.M. and L. Norton; no. 7,829,066; issued 9 November 2010), S100A8/A9 as a diagnostic marker and a therapeutic agent (inventor: J.M.; no. 2,831,593; granted 30 June 2018), Inhibiting cancer metastasis (inventors: J.M. and M. Valiente Cortes; no. 3,047,039; granted 31 July 2019), Methods for treating brain metastasis (inventors: J.M., Q. Chen and A. Boire; no. 10,413,522; issued 17 September 2019), Modulating permeability of the blood cerebrospinal fluid barrier (inventors: J.M. and A. A. Boire; application no. PCT/US2016/062880; published 18 November 2016). K.G. and J.M. are inventors on the following patent application: Treating metastatic cancer and model systems for metastatic disease (inventors: J.M., M. Valiente Cortes and K.G.; application no. PCT/US2017/045145; published 2 August 2017).
Peer review information Hannah Stower was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
References
- 1.Siegel RL, Miller KD & Jemal A Cancer statistics, 2020. CA Cancer J. Clin 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Caswell-Jin JL et al. Change in survival in metastatic breast cancer with treatment advances: meta-analysis and systematic review. JNCI Cancer Spectr. 2, pky062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howlader N et al. (eds.) in SEER Cancer Statistics Review, 1975–2016 1423–1437 (National Cancer Institute, 2019). [Google Scholar]
- 4.May M Twenty-five ways clinical trials have changed in the last 25 years. Nat. Med 25, 2–5 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Hosseini H et al. Early dissemination seeds metastasis in breast cancer. Nature 540, 552–558 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harper KL et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 540, 588–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Z & Curtis C Looking backward in time to define the chronology of metastasis. Nat. Commun 11, 3213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chambers AF, Groom AC & MacDonald IC Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563–572 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Lambert AW, Pattabiraman DR & Weinberg RA Emerging biological principles of metastasis. Cell 168, 670–691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massagué J & Obenauf AC Metastatic colonization by circulating tumour cells. Nature 529, 298–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quintanal-Villalonga A et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol 17, 360–371 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laughney AM et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med 26, 259–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fumagalli AO et al. Plasticity of Lgr5-negative cancer cells drives metastasis in colorectal cancer. Cell Stem Cell 26, 569–578 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswas D et al. A clonal expression biomarker associates with lung cancer mortality. Nat. Med 25, 1540–1548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obenauf AC & Massagué J Surviving at a distance: organ-specific metastasis. Trends Cancer 1, 76–91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esposito M, Guise T & Kang Y The biology of bone metastasis. Cold Spring Harb. Perspect. Med 10.1101/cshperspect.a031252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boire A, Brastianos PK, Garzia L & Valiente M Brain metastasis. Nat. Rev. Cancer 20, 4–11 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Celià-Terrassa T & Kang Y Metastatic niche functions and therapeutic opportunities. Nat. Cell Biol 20, 868–877 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Turajlic S & Swanton C Metastasis as an evolutionary process. Science 352, 169–175 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Reiter JG et al. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 19, 639–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Razavi P et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 34, 427–438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiter JG et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 361, 1033–1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Vega F et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173, 321–337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Z, Li Z, Ma Z & Curtis C Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet 52, 701–708 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacob LS et al. Metastatic competence can emerge with selection of preexisting oncogenic alleles without a need of new mutations. Cancer Res 75, 3713–3719 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denny SK et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 166, 328–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roe JS et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 170, 875–888 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanharanta S et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat. Med 19, 50–56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavazoie SF et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomes AP et al. Dynamic incorporation of histone H3 variants into chromatin is essential for acquisition of aggressive traits and metastatic colonization. Cancer Cell 36, 402–417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald OG et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet 49, 367–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reya T, Morrison SJ, Clarke MF & Weissman IL Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Batlle E & Clevers H Cancer stem cells revisited. Nat. Med 23, 1124–1134 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Lapidot T et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Bonnet D & Dick JE Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 3, 730–737 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Shimokawa M et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 545, 187–192 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Barker N et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Visvader JE Cells of origin in cancer. Nature 469, 314–322 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Oskarsson T, Batlle E & Massagué J Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 14, 306–321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganesh K et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 1, 28–45 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altevogt P, Doberstein K & Fogel M L1CAM in human cancer. Int. J. Cancer 138, 1565–1576 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Er EE et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol 20, 966–978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valiente M et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nieto MA, Huang RY, Jackson RA & Thiery JP Emt: 2016. Cell 166, 21–45 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Yang J et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol 21, 341–352 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pastushenko I et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Shibue T & Weinberg RA EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol 14, 611–629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsai JH, Donaher JL, Murphy DA, Chau S & Yang J Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ocana OH et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Su J et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 577, 566–571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rickman DS, Beltran H, Demichelis F & Rubin MA Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med 23, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Yuan S, Norgard RJ & Stanger BZ Cellular plasticity in cancer. Cancer Discov 9, 837–851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boumahdi S & de Sauvage FJ The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov 19, 39–56 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Lugassy C, Kleinman HK, Vermeulen PB & Barnhill RL Angiotropism, pericytic mimicry and extravascular migratory metastasis: an embryogenesis-derived program of tumor spread. Angiogenesis 23, 27–41 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Kuol N, Stojanovska L, Apostolopoulos V & Nurgali K Role of the nervous system in cancer metastasis. J. Exp. Clin. Cancer Res 37, 5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu M et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gkountela S et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 176, 98–112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pantel K & Speicher MR The biology of circulating tumor cells. Oncogene 35, 1216–1224 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Micalizzi DS, Maheswaran S & Haber DA A conduit to metastasis: circulating tumor cell biology. Genes Dev 31, 1827–1840 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fidler IJ The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur. J. Cancer 9, 223–227 (1973). [DOI] [PubMed] [Google Scholar]
- 61.Aceto N, Toner M, Maheswaran S & Haber DA En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer 1, 44–52 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Chemi F et al. Pulmonary venous circulating tumor cell dissemination before tumor resection and disease relapse. Nat. Med 25, 1534–1539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heller G et al. Circulating tumor cell number as a response measure of prolonged survival for metastatic castration-resistant prostate cancer: a comparison with prostate-specific antigen across five randomized phase III clinical trials. J. Clin. Oncol 36, 572–580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alix-Panabieres C, Schwarzenbach H & Pantel K Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med 63, 199–215 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Cristofanilli M et al. The clinical use of circulating tumor cells (CTCs) enumeration for staging of metastatic breast cancer (MBC): international expert consensus paper. Crit. Rev. Oncol. Hematol 134, 39–45 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Pantel K, Hille C & Scher HI Circulating tumor cells in prostate cancer: from discovery to clinical utility. Clin. Chem 65, 87–99 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Parikh AR et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med 25, 1415–1421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sosa MS, Bragado P & Aguirre-Ghiso JA Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer 14, 611–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Phan TG & Croucher PI The dormant cancer cell life cycle. Nat. Rev. Cancer 20, 398–411 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Ghajar CM et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol 15, 807–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malladi S et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165, 45–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carlson P et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol 21, 238–250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.David CJ & Massagué J Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol 19, 419–435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bragado P et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38ɑ/β signalling. Nat. Cell Biol 15, 1351–1361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prunier C, Baker D, Ten Dijke P & Ritsma L TGF-β family signaling pathways in cellular dormancy. Trends Cancer 5, 66–78 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Pommier A et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 10.1126/science.aao4908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pantel K et al. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 51, 4712–4715 (1991). [PubMed] [Google Scholar]
- 78.Koebel CM et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Eyob H et al. Inhibition of Ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 3, 751–760 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ali FR & Lear JT Melanoma in organ transplant recipients: incidence, outcomes and management considerations. J. Skin Cancer 2012, 404615 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tikhonova AN, Lasry A, Austin R & Aifantis I Cell-by-cell deconstruction of stem cell niches. Cell Stem Cell 27, 19–34 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scadden DT Nice neighborhood: emerging concepts of the stem cell niche. Cell 157, 41–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Plaks V, Kong N & Werb Z The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16, 225–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mohme M, Riethdorf S & Pantel K Circulating and disseminated tumour cells—mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol 14, 155–167 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Garner H & de Visser KE Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat. Rev. Immunol 10.1038/s41577-019-0271-z (2020). [DOI] [PubMed] [Google Scholar]
- 86.Quail DF & Joyce JA Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eyles J et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Invest 120, 2030–2039 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bidwell BN et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med 18, 1224–1231 (2012). [DOI] [PubMed] [Google Scholar]
- 89.Van den Eynde M et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell 34, 1012–1026 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Kang Y et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003). [DOI] [PubMed] [Google Scholar]
- 91.Zheng H et al. Therapeutic antibody targeting tumor- and osteoblastic niche-derived Jagged1 sensitizes bone metastasis to chemotherapy. Cancer Cell 32, 731–747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang J et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J. Clin. Invest 107, 1235–1244 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen Q, Zhang XH & Massagué J Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 20, 538–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Acharyya S et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Albrengues J et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 10.1126/science.aao4227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Teijeira A et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity 52, 856–871 (2020). [DOI] [PubMed] [Google Scholar]
- 97.Ombrato L et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 572, 603–608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen Q et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sevenich L et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol 16, 876–888 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeng Q et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kaplan RN et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peinado H et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017). [DOI] [PubMed] [Google Scholar]
- 103.Fontebasso Y & Dubinett SM Drug development for metastasis prevention. Crit. Rev. Oncog 20, 449–473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Risson E, Nobre AR, Maguer-Satta V & Aguirre-Ghiso JA The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 1, 672–680 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Parsons S, Maldonado EB & Prasad V Comparison of drugs used for adjuvant and metastatic therapy of colon, breast, and non-small cell lung cancers. JAMA Netw. Open 3, e202488 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Steeg PS Perspective: The right trials. Nature 485, S58–59 (2012). [DOI] [PubMed] [Google Scholar]
- 107.Vasan N, Baselga J & Hyman DM A view on drug resistance in cancer. Nature 575, 299–309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lito P, Rosen N & Solit DB Tumor adaptation and resistance to RAF inhibitors. Nat. Med 19, 1401–1409 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Sabnis AJ & Bivona TG Principles of resistance to targeted cancer therapy: lessons from basic and translational cancer biology. Trends Mol. Med 25, 185–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu Y-L et al. CTONG1104: adjuvant gefitinib versus chemotherapy for resected N1-N2 NSCLC with EGFR mutation—final overall survival analysis of the randomized phase III trial 1 analysis of the randomized phase III trial. J. Clin. Oncol 38, 9005 (2020). [Google Scholar]
- 111.Jassem J Adjuvant EGFR tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: still an investigational approach. Transl. Lung Cancer Res 8, S387–S390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mok TS et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med 376, 629–640 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Herbst RS et al. Osimertinib as adjuvant therapy in patients (pts) with stage IB–IIIA EGFR mutation positive (EGFRm) NSCLC after complete tumor resection: ADAURA. J. Clin. Oncol 38, abstract LBA5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Soria JC et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med 378, 113–125 (2018). [DOI] [PubMed] [Google Scholar]
- 115.Mateo J et al. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol 30, 1437–1447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Farmer H et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 117.Spring LM et al. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet 395, 817–827 (2020). [DOI] [PubMed] [Google Scholar]
- 118.Paget S The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 8, 98–101 (1989). [PubMed] [Google Scholar]
- 119.Binnewies M et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Folkman J Tumor angiogenesis: therapeutic implications. N. Engl. J. Med 285, 1182–1186 (1971). [DOI] [PubMed] [Google Scholar]
- 121.Haibe Y et al. Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol 10, 221 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Iheanacho K & Vaishampayan U Perioperative approaches to kidney cancer. Clin. Adv. Hematol. Oncol 18, 56–65 (2020). [PubMed] [Google Scholar]
- 123.Allegra CJ et al. Bevacizumab in stage II–III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J. Clin. Oncol 31, 359–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ribas A & Wolchok JD Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Larkin J et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med 381, 1535–1546 (2019). [DOI] [PubMed] [Google Scholar]
- 126.Haslam A, Gill J & Prasad V Estimation of the percentage of US patients with cancer who are eligible for immune checkpoint inhibitor drugs. JAMA Netw. Open 3, e200423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hellmann MD et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 18, 31–41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Motzer RJ et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med 378, 1277–1290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Long GV et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol. 19, 672–681 (2018). [DOI] [PubMed] [Google Scholar]
- 131.Schumacher TN, Scheper W & Kvistborg P Cancer neoantigens. Annu. Rev. Immunol 37, 173–200 (2019). [DOI] [PubMed] [Google Scholar]
- 132.Hellmann MD et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med 378, 2093–2104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marabelle A et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 Study. J. Clin. Oncol 38, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Snyder A et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rizvi NA et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tauriello DVF et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Mariathasan S et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Makker V et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 20, 711–718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rini BI et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med 380, 1116–1127 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Fukuoka S et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J. Clin. Oncol 38, 2053–2061 (2020). [DOI] [PubMed] [Google Scholar]
- 141.Eggermont AMM et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med 378, 1789–1801 (2018). [DOI] [PubMed] [Google Scholar]
- 142.Weber J et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med 377, 1824–1835 (2017). [DOI] [PubMed] [Google Scholar]
- 143.Eggermont AM et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N. Engl. J. Med 375, 1845–1855 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zimmer L et al. Adjuvant nivolumab plus ipilimumab or nivolumab monotherapy versus placebo in patients with resected stage IV melanoma with no evidence of disease (IMMUNED): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 395, 1558–1568 (2020). [DOI] [PubMed] [Google Scholar]
- 145.Liu J et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 6, 1382–1399 (2016). [DOI] [PubMed] [Google Scholar]
- 146.Versluis JM, Long GV & Blank CU Learning from clinical trials of neoadjuvant checkpoint blockade. Nat. Med 26, 475–484 (2020). [DOI] [PubMed] [Google Scholar]
- 147.Blank CU et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med 24, 1655–1661 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Amaria RN et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med 24, 1649–1654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Forde PM et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med 378, 1976–1986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Spranger S, Dai D, Horton B & Gajewski TF Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Creasy JM et al. Actual 10-year survival after hepatic resection of colorectal liver metastases: what factors preclude cure? Surgery 163, 1238–1244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Clark ME & Smith RR Liver-directed therapies in metastatic colorectal cancer. J. Gastrointest. Oncol 5, 374–387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gomez DR et al. Local consolidative therapy vs. maintenance therapy or observation for patients with oligometastatic non-small-cell lung cancer: long-term results of a multi-institutional, phase II, randomized study. J. Clin. Oncol 37, 1558–1565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Palma DA et al. Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (SABR-COMET): a randomised, phase 2, open-label trial. Lancet 393, 2051–2058 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Parker C et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med 369, 213–223 (2013). [DOI] [PubMed] [Google Scholar]
- 156.Early Breast Cancer Trialists’ Collaborative Group. Adjuvant bisphosphonate treatment in early breast cancer: meta-analyses of individual patient data from randomised trials. Lancet 386, 1353–1361 (2015). [DOI] [PubMed] [Google Scholar]
- 157.O’Carrigan B et al. Bisphosphonates and other bone agents for breast cancer. Cochrane Database of Systematic Reviews 10.1002/14651858.CD003474.pub4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lin X & DeAngelis LM Treatment of brain metastases. J. Clin. Oncol 33, 3475–3484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Auperin A et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N. Engl. J. Med 341, 476–484 (1999). [DOI] [PubMed] [Google Scholar]
- 160.Boire A et al. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell 168, 1101–1113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Haro MA, Dyevoich AM, Phipps JP & Haas KM Activation of B-1 cells promotes tumor cell killing in the peritoneal cavity. Cancer Res. 79, 159–170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Priego N et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med 24, 1024–1035 (2018). [DOI] [PubMed] [Google Scholar]
- 163.Chi Y et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 369, 276–282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Suva ML & Tirosh I Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol. Cell 75, 7–12 (2019). [DOI] [PubMed] [Google Scholar]
- 165.Rozenblatt-Rosen O et al. The Human Tumor Atlas Network: charting tumor transitions across space and time at single-cell resolution. Cell 181, 236–249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pascual G et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017). [DOI] [PubMed] [Google Scholar]
- 167.Rekhtman N et al. Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin. Cancer Res 22, 3618–3629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hamilton K et al. Prevalence and prognostic significance of neuroendocrine cells in esophageal adenocarcinoma. Mod. Pathol 13, 475–481 (2000). [DOI] [PubMed] [Google Scholar]
- 169.Adams EJ et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature 571, 408–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kaur J, Daoud A & Eblen ST Targeting chromatin remodeling for cancer therapy. Curr. Mol. Pharm 12, 215–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sparano JA et al. Clinical and genomic risk to guide the use of adjuvant therapy for breast cancer. N. Engl. J. Med 380, 2395–2405 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sobrero AF et al. Overall survival (OS) and long-term disease-free survival (DFS) of three versus six months of adjuvant (adj) oxaliplatin and fluoropyrimidine-based therapy for patients (pts) with stage III colon cancer (CC): final results from the IDEA (International Duration Evaluation of Adj chemotherapy) collaboration. J. Clin. Oncol 38, 4004–4004 (2020). [Google Scholar]
- 173.Grothey A et al. Duration of adjuvant chemotherapy for stage III colon cancer. N. Engl. J. Med 378, 1177–1188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Coakley M, Garcia-Murillas I & Turner NC Molecular residual disease and adjuvant trial design in solid tumors. Clin. Cancer Res 25, 6026–6034 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Pantel K & Alix-Panabières C Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat. Rev. Clin. Oncol 16, 409–424 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Phallen J et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med 10.1126/scitranslmed.aan2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cohen JD et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Razavi P et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med 25, 1928–1937 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V & Wargo JA The microbiome, cancer, and cancer therapy. Nat. Med 25, 377–388 (2019). [DOI] [PubMed] [Google Scholar]
- 180.Geller LT et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nejman D et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bullman S et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Parhi L et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat. Commun 11, 3259 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Routy B et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 185.Gopalakrishnan V et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Forbes NS et al. White paper on microbial anti-cancer therapy and prevention. J. Immunother. Cancer 6, 78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tuveson D & Clevers H Cancer modeling meets human organoid technology. Science 364, 952–955 (2019). [DOI] [PubMed] [Google Scholar]
- 188.Vlachogiannis G et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ganesh K et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med 25, 1607–1614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hapach LA, Mosier JA, Wang W & Reinhart-King CA Engineered models to parse apart the metastatic cascade. NPJ Precis Oncol 3, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yuki K, Cheng N, Nakano M & Kuo CJ Organoid models of tumor immunology. Trends Immunol 10.1016/j.it.2020.06.010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.De La Rochere P et al. Humanized mice for the study of immuno-oncology. Trends Immunol 39, 748–763 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Priestley P et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Balachandran VP et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Iyer G et al. Genome sequencing identifies a basis for everolimus sensitivity. Science 338, 221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Doebele RC et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol 21, 271–282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Prahallad A et al. Unresponsiveness of colon cancer to BRAFV600E inhibition through feedback activation of EGFR. Nature 483, 100–103 (2012). [DOI] [PubMed] [Google Scholar]
- 198.Dvorak HF Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med 315, 1650–1659 (1986). [DOI] [PubMed] [Google Scholar]