

Abstract
Aryl diazonium ions have long been used in bioconjugation due to their reactivity towards electron rich aryl residues, such as tyrosine. However, their utility in biological systems have been restricted due to the requirement of harsh conditions for their generation in situ, as well as limited hydrolytic stability. Previous work describing a scaffold known as triazabutadiene (TBD) has shown the ability to protect aryl diazonium ions allowing for increased synthetic utility, as well as triggered release under biologically relevant conditions. Herein, we describe the synthesis and application of a novel TBD, capable of installation of a cyclooctyne on protein surfaces for later use of copper-free click reactions involving functional azides. The probe shows efficient protein labeling across a wide pH range that can be accomplished in a convenient and timely manner. Orthogonality of the cyclooctyne modification was showcased by labeling a model protein in the presence of hen egg proteins, using an azide-containing fluorophore. We further confirmed that the azobenzene modification can be cleaved using sodium dithionite treatment.
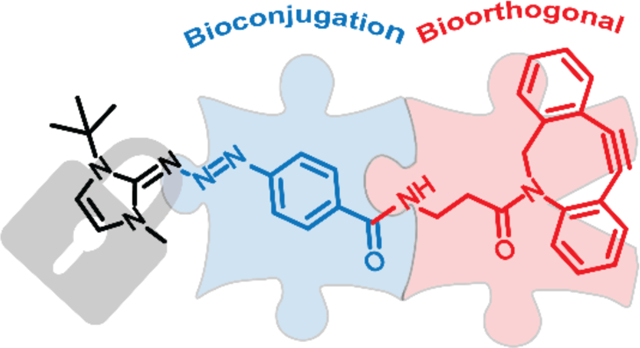
Graphical Abstract
Tyrosine is recognized as an important amino acid in the study of protein-protein interactions as it is often targeted for post-translational modifications including phosphorylation, glycosylation, and oxidation.1 Tyrosine has also been identified as an attractive target in the realm of bioconjugate chemistry due to relatively low abundance on protein surfaces, but high abundance at sites of protein-protein interactions.2 Additionally, tyrosine is uncharged at physiological pH and therefore, tyrosine modifications do not alter the surface charge of proteins in contrast to those that modify charged residues like lysine.1 As such, several new tyrosine conjugation strategies have been developed in the past decade including 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) and sulfur fluoride exchange (SuFeX) analogs, among others.3,4,5 Such strategies have shown utility in a range of applications for protein modification and functionalization including manufacturing of antibody drug conjugates (ADCs).5,6 Amidst the advances, the oldest tyrosine bioconjugation strategy, aryl diazonium ions to form azo-bonds, remains one of the most heavily utilized.7,8 Though effective, aryl diazonium ions generally require generation under highly acidic conditions, and the salts are both prone to degradation and exhibit shock sensitivity, making long term storage a challenge.9,10 Further complicating storage, aryl diazonium ions are susceptible to hydrolysis at biologically relevant pH.11 Despite the limitations, aryl diazonium ions remain a quintessential tool within the realm of bioconjugate chemistry due to their reactivity and relative selectivity.
Diazonium ions can orthogonally label residues such as tyrosine and histidine, but further selectivity can be attained via inclusion of a second, bioorthogonal, moiety. Bioorthogonal modifications are inert towards native biochemical processes and selectively react with functional groups not present in the natural world.12,13 Thus, bioconjugation strategies in conjunction with bioorthogonal chemistry act as powerful tools to achieve selective, modular protein labeling with the potential for a bifurcated workflow (Figure 1a).5 Herein, we describe the synthesis and application of a novel, cyclooctyne containing protected diazonium ion that is bench stable and can be readily added to aqueous environments for protein modification with a bioorthogonal handle.
Figure 1.
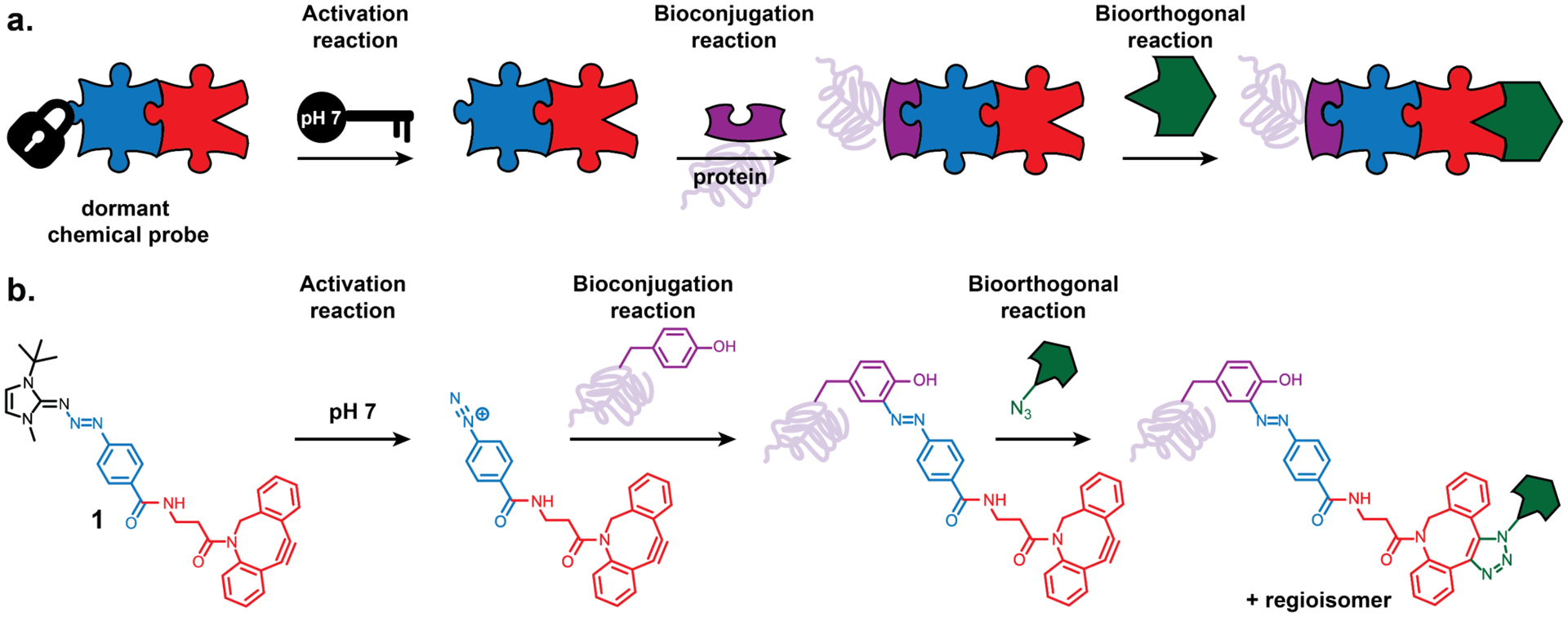
(a) Design workflow showing the activation of the dormant chemical probe, bioconjugation and bioorthogonality. (b) Structural representation of the designed chemical probe and its behavior.
In recent years, our group has utilized a scaffold known as the triazabutadiene (TBD), which acts as a protected diazonium ion. The use of the TBD scaffold incurs the advantage of increased diazonium ion bench stability and more importantly, the ability to release diazonium within controlled environmental conditions via triggers such as low pH, or UV irradiation at physiological pH.14,15 Protonation of the TBD leads to diazonium release, which in turn can be captured by an electron rich aromatic system including residues such as tyrosine and histidine.16 The various building blocks of the TBD scaffold advantageously provide several points of functionalization to steer stability, reactivity, targeted delivery, and has allowed for applications in protein labeling, purification, and crosslinking.14,15,17
Previously, our group developed an alkyne functionalized TBD, capable of bioorthogonal modification under ‘copper click’ conditions.18 Though widely popular in bioconjugate chemistry, copper click reactions are not without their caveats. Not all systems are compatible with copper as it can be toxic, and in some instances reaction conditions can lead to protein degradation.19 Additionally, reaction optimization often includes reducing agents such as ascorbate, which have been found to cause undesired oxidation of histidine and arginine residues, which in turn requires supplementation with novel ligands to avoid.20 Accordingly, copper free click chemistry via cyclooctynes has become an attractive alternative due to their ability to perform strain-promoted alkyne azide cycloadditions (SPAAC) without the need for copper.21 Their thermostability, efficiency, and biocompatibility have spawned an increasing demand for new cyclooctyne probes with numerous applications including bioorthogonal protein labeling, cellular surface functionalization and development of antibody conjugates for novel therapuetics.22 As such, we sought to develop a next generation TBD (1) containing a cyclooctyne reactive handle capable of quick and efficient functionalization of proteins (Figure 1b). Conceptually, the exact nature of the linker chemistry is not important, but modifications adjacent to the aryl diazonium ion would have an impact on its release and the bioconjugation.
The synthesis of compound 1 began with a key coupling between azide 2 (Scheme 1, made in two steps from commercially available 4-aminobenzoic acid), and t-butyl methyl imidazolium iodide (3) in the presence of potassium tert-butoxide (t-BuOK) to provide the triazabutadiene (TBD) core.23,24 The ester on the resulting TBD was hydrolyzed with LiOH, to provide a lithium salt that was coupled to N- hydroxysuccimide (NHS) to form key NHS-ester 4. The NHS-functionalized TBD, 4, readily coupled with dibenzocyclooctyne (DBCO) - amine25,26,27 to provide 1 in reasonable yield over the final three steps. It should be noted that purification prior to final amide bond formation proved to be problematic and accordingly, these final steps were performed on the crude material and the purification was accomplished on the final compound.
Scheme 1.
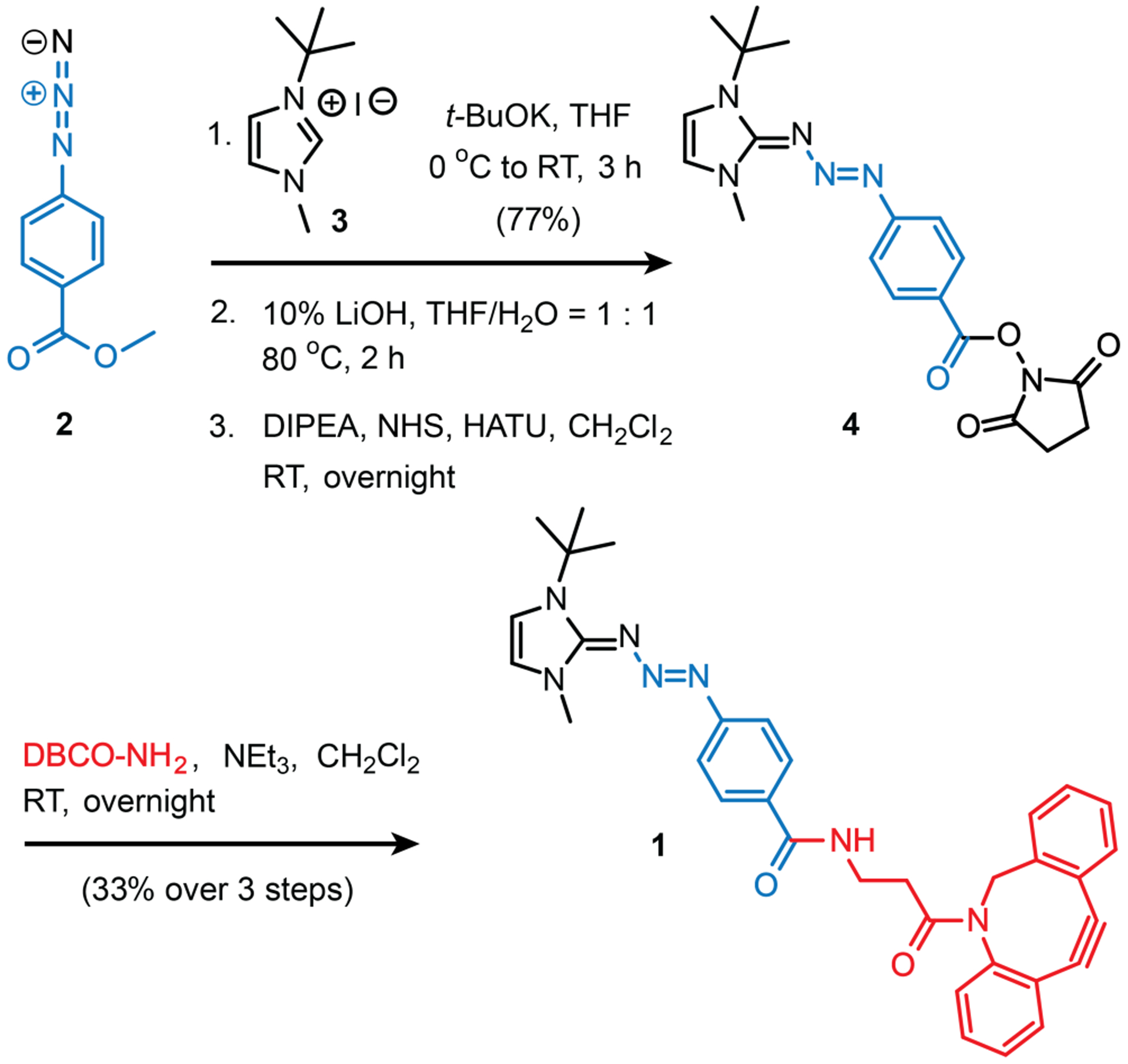
Synthesis of DBCO-functionalized TBD (1)
With compound 1 in hand, we sought to characterize its efficiency in protein labeling. We began these studies by determining the pH range in which 1 was capable of protein labeling (Figure 2a). Bovine Serum Albumin (BSA) was treated with 1 across a range of pH (4–8) for 2 hours and then further incubated with an azide-linked Alexa Fluor™ 488 fluorophore following neutralization. SDS-PAGE analysis indicated that 1 provided robust labeling of BSA at pH 4–7, however, labeling was seen to markedly diminish at pH 8 (Figure 2b). This is consistent with previous TBD literature showing lability at lower pH, but increased stability in alkaline conditions, and is consistent with the proposed mechanism of activation.17 We then sought to confirm that the labeling on BSA was most likely due to desired azo-adduct formation following diazonium release from 1. This was accomplished through both competition experiments, and a reductive cleavage of the azo-bond (Figure 2a, c & d).
Figure 2.
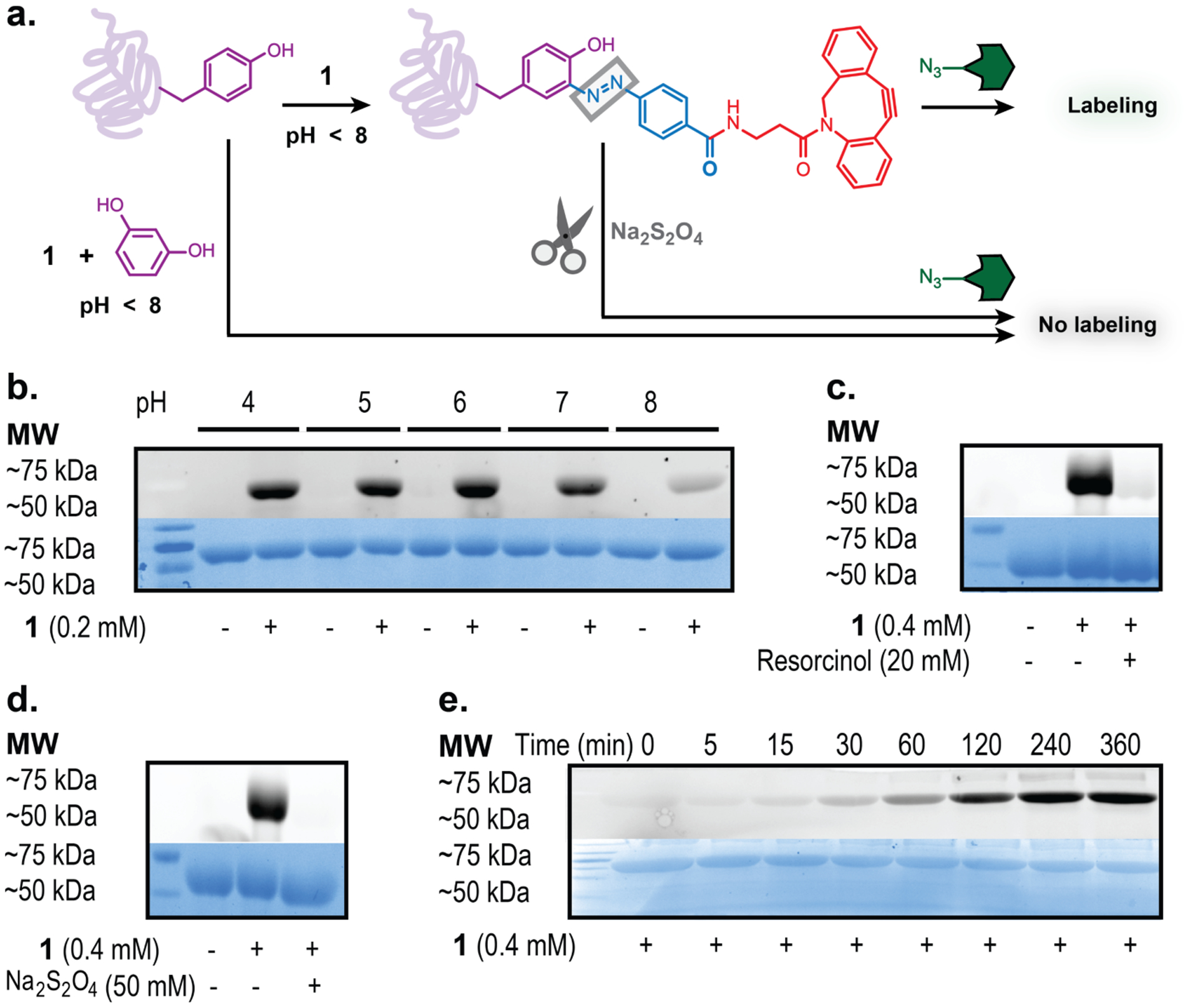
(a) Treatment of protein with 1 at pH < 8 results in azo modification by cyclooctyne diazonium ion. Labeling can be outcompeted by addition of resorcinol or reductively removed with sodium dithionite. All samples, including controls, were treated with AlexaFluor azide in the final step prior to analysis by SDS-PAGE (b-e). (b) BSA treated with 0.2 mM 1 for 2 hours at pH 4–8. (c) BSA treated with 1 with or without excess resorcinol present. (d) BSA treated with 1 with and without a subsequent reaction with 50 mM sodium dithionite at pH 7. (e) Time point assessment of BSA treatment by 1 over the course of 6 hours at pH 7 (0.1 M PBS pH 7).
First, we challenged BSA with 1 in the presence of resorcinol, an aromatic nucleophile that readily reacts with diazonium ions. In the presence of excess resorcinol, fluorescent labeling of BSA was diminished, indicating that resorcinol was able to outcompete aromatic residues on the protein surface (Figure 2c). We further wanted to confirm that azo-modifications on BSA could be removed via reduction by treatment with known azo-reductant sodium dithionite.9,28 BSA (25 μM) was first treated with 0.4 mM 1. Next, an aliquot of the labeled BSA was treated with 50 mM sodium dithionite. After 2 hours, buffer exchange was performed using G-10 SEC resin and the resulting sample was then treated with the fluorescent azide. The loss of fluorescence following sodium dithionite treatment indicated the DBCO moiety had been removed from the BSA prior to azide conjugation (Figure 2d). Taken together, these results are consistent with the observed fluorescence being due to the desired azo modifications resulting from diazonium ion release by 1.
Next, we gauged the rate of the labeling reaction by measuring BSA fluorescence as a function of time incubated with 1 at pH 7. BSA (25 μM) was incubated with 0.4 mM 1 and aliquots were removed and quenched with excess resorcinol at time points ranging from 0–6 hours (Figure 2e). All samples, including controls, were then incubated with the azide-linked fluorophore (0.4 mM) for 2 hours prior to gel imaging. We analyzed the fluorescence intensity of the protein bands as a function of time and observed that after a brief induction time, the fluorescent labeling increased rapidly for the first couple hours before leveling off after 4 hours of incubation at room temperature (see Supporting Information Figure S1).
The next goal for the bioconjugation chemistry was to use mass spectrometry to measure the extent of protein modification through the 2 step process of bioconjugation and bioorthogonal reaction. To accomplish this goal we used MSP1D1T2(−) (MSP), a small model protein with several tyrosine in its sequence.29 First, 25 μM of MSP was treated with 0.5 mM 1 overnight in pH 7 PBS buffer. An aliquot of this experiment was treated with the AlexaFluor azide and analyzed by SDS-PAGE as had been done previously with BSA (Figure 3). The remainder of the first sample was further split into two equal aliquots, one of which was treated with 0.5 mM of a biotin-(PEG)2-azide compound for 4 hours, and the other was left with the cyclooctyne intact. Analysis of these two samples compared with the control apo-MSP using native mass spectrometry confirmed the desired reactivity. The sample treated with 1 clearly showed a distribution of labeled proteins with up to six modifications consistent with the expected mass of the desired azo-adduct. Furthermore, the sample subsequently treated with the biotin-(PEG)2-azide showed all of the modified peaks were further modified, with those of lesser abundance (5 and 6 modifications) no longer visible above the background noise. Together, these data confirm that 1 is capable of robust labeling of several tyrosine residues on MSP, and importantly that the resulting modifications are capable of reacting with multiple azide substrates. This enables the same protein sample to be utilized in several workflows while retaining knowledge of the extent of modification.
Figure 3.
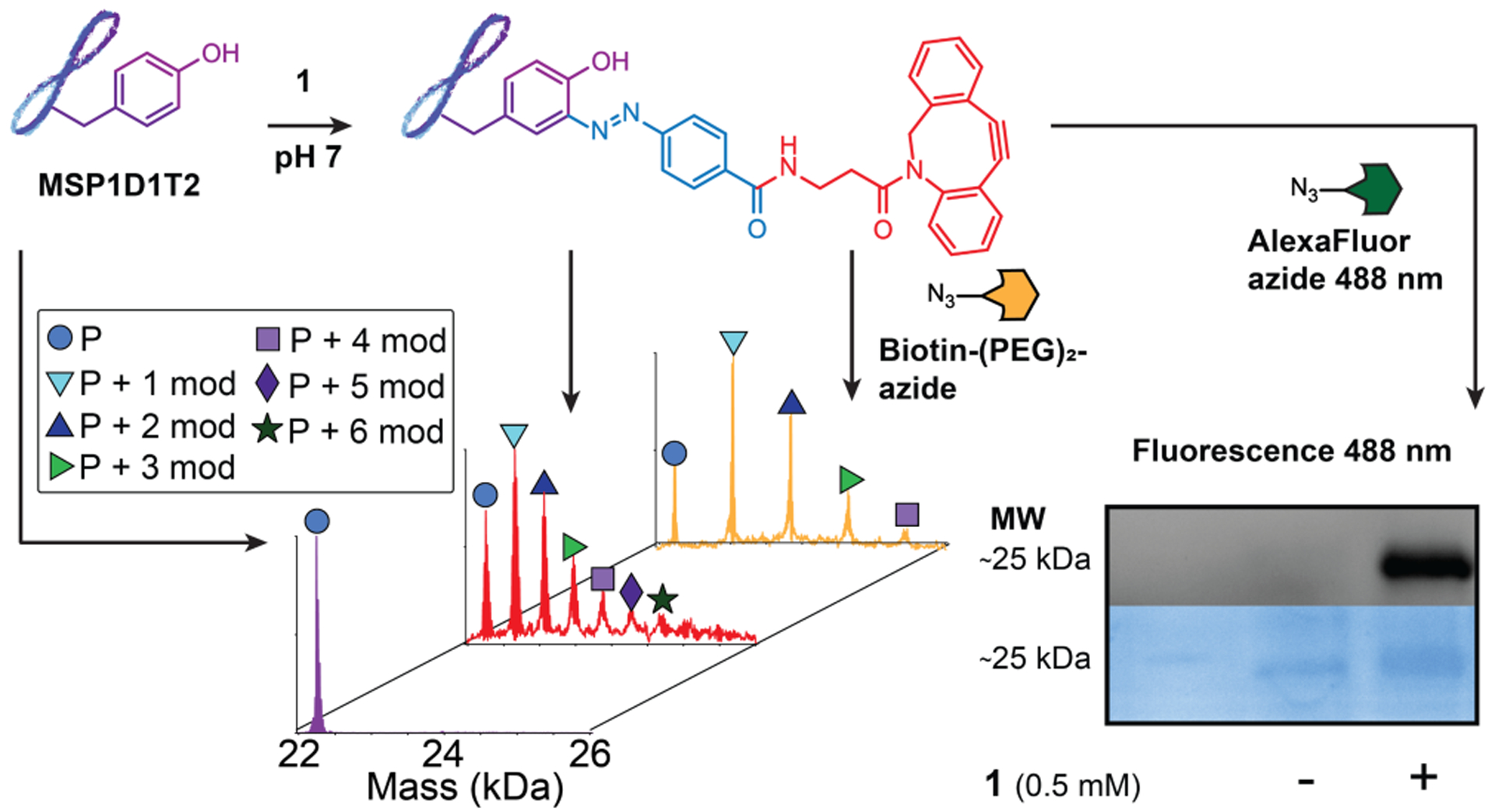
The extent of protein labeling was quantified using a membrane scaffold protein (MSP1D1T2(−)), as a small model protein. MSP (mass spectrum in purple) was first incubated with 1 at pH 7 to provide a cyclooctyne-modified MSP (mass spectrum in red). At this point part of the sample was split. One portion was treated with a fluorescent azide and the modification was observed by SDS-PAGE. The second portion was treated with a biotin-containing azide (mass spectrum shown in orange). Masses corresponding with integer-values of the appropriate modifications were observed and are noted with symbols (P = MSP protein, mod = chemical modification)
After showcasing the versatility and efficiency of 1, we sought to highlight its built-in bioorthogonal reactivity. Functionalizing proteins with cyclooctynes allows for highly specific modifications both in vitro and in vivo, as organic azides will only react with proteins with the complimentary DBCO modification. To illustrate this point, we used hen egg protein extract as a model system. First, we confirmed that 1 was capable of labeling a range of proteins by using it to treat hen egg protein extract (Figure 4a). When comparing untreated hen egg protein extract to that treated with 1, SDS-analysis showed robust fluorescent labeling with minimal background fluorescence attributed to indiscriminate, non-covalent fluorophore labeling (Figure 4a).
Figure 4.
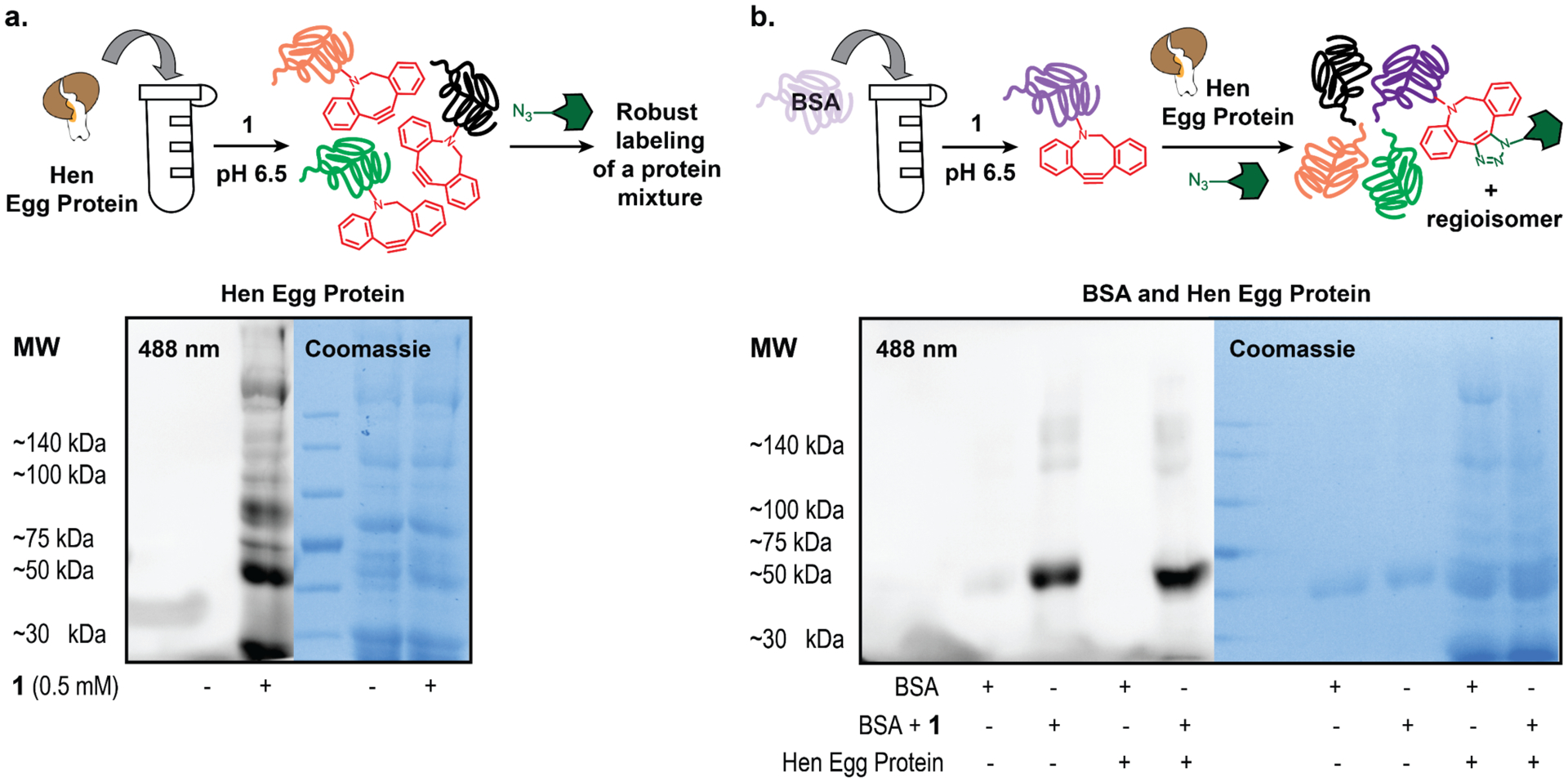
(a) Workflow of hen egg protein treatment by 1 and subsequent fluorescent azide, along with SDS-PAGE of fluorescently labeled hen egg extract. Control hen egg protein shown in the left lane was also treated with the fluorophore. (b) Workflow of BSA treated with 1 and then orthogonally labeled by fluorescent azide in hen egg protein extract, along with SDS-PAGE of BSA treatment with 1 and fluorescent azide with and without the presence of hen egg protein extract. All lanes were treated with the fluorophore.
Having confirmed minimal background labeling within the hen egg protein sample, we moved on to show that BSA modified with 1 could be selectively observed within a complex protein mixture (Figure 4b). We labeled BSA with 1 and then transferred the modified protein to a solution of unlabeled hen egg protein extract. Following incubation with fluorescent azide, we only observed fluorescence of the labeled BSA shown at ~60 kDa (Figure 4b). This band was consistent with the same modified BSA without the presence of the egg protein, confirming it was indeed orthogonally labeled BSA. Finally, we tested the ability for the cyclooctyne to remain intact and reactive in the presence of strong biological nucleophiles, such as cysteine. We observed that 1-modified BSA remained reactive with the fluorescent azide even following treatment with 20 mM cysteine over 24 hours (see Supporting Information Figure S2). While some fluorescence intensity, and by proxy cyclooctyne, was lost, the overall fluorescence of the cysteine treated sample maintained ~60% of the intensity of the non-cysteine-treated sample (see Supporting Information Figure S3).30
Overall, we have shown the ability to synthesize a versatile cyclooctyne containing TBD capable of efficient modification of proteins. Leveraging the physiological compatibility of the novel TBD, we were able to accomplish robust protein labeling across a range of pH, with relatively low reaction times. Importantly, we also displayed that this probe could be used to install a reactive cyclooctyne on the surface of proteins, allowing for quick and selective functionalization of proteins, even in the presence of complex protein mixtures. The triazabutadiene afforded the synthetic opportunity to show the cross-compatibility of cyclooctynes and aryl diazonium ions, two highly reactive and sought after functional groups. Furthermore, the aryl diazonium ion chemistry described can be coupled with other strategies, such as chemoselective rapid azo-coupling reactions (CRACR) with 5-hydroxytryptophan to enhance selectivity.31 Taken together, we believe that this system poses great potential for a wide variety of bioconjugate applications including highly selective and bioorthogonal modification of proteins for purification, imaging, or drug conjugation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NSF-CAREER award, given to J.C.J. (CHE-1552568). Additional funding to J.A.T and M.T.M was provided by the National Institute of General Medical Sciences and National Institutes of Health (T32 GM008804 to J.A.T and R35 GM128624 to M.T.M). All NMR data were collected in the NMR facility at the University of Arizona. The purchase of the Bruker AVANCE III 400 MHz spectrometer was supported by NSF grant 840336 and the University of Arizona. The purchase and upgrade of the Bruker AVANCE DRX 500 MHz spectrometer was partially supported by NSF grant 9214383, the Office of Naval Research, and the University of Arizona. . The purchase of the Bruker NEO 500 MHz spectrometer was supported by NSF grant 1920234 and the University of Arizona. All FTIR spectra were collected in the W.M. Keck Center for Nano-Scale Imaging in the Department of Chemistry and Biochemistry at the University of Arizona. This instrument purchase was supported by Arizona Technology and Research Initiative Fund (A.R.S.§15-1648). We thank Kristen Keck and Yelena Feinstein at the University of Arizona Analytical & Biological Mass Spectrometry Facility for help with the MS analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website . The synthesis and characterization of all new compounds and detailed protocols for bioconjugation are provided as a PDF.
The authors declare no competing financial interest.
REFERENCES
- (1).Dorta DA; Deniaud D; Møvel M; Gouin SG Tyrosine Conjugation Methods for Protein Labelling. Chem. Eur. J 2020, 26 (63), 1–14. [DOI] [PubMed] [Google Scholar]
- (2).Koide S; Sidhu SS The Importance of Being Tyrosine: Lessons in Molecular Recognition from Minimalist Synthetic Binding Proteins. ACS Chem. Biol 2009, 4 (5), 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ban H; Gavrilyuk J; Barbas CF Tyrosine Bioconjugation through Aqueous Ene-Type Reactions: A Click-Like Reaction for Tyrosine. J. Am. Chem. Soc 2010, 132, 1523–1525. [DOI] [PubMed] [Google Scholar]
- (4).Choi EJ; Jung D; Kim J-S; Lee Y; Kim BM Chemoselective Tyrosine Bioconjugation through Sulfate Click Reaction. Chem. - A Eur. J 2018, 24 (43), 10948–10952. [DOI] [PubMed] [Google Scholar]
- (5).Szijj P; Kostadinova K; Spears RJ; Chudasama V Tyrosine Bioconjugation – An Emergent Alternative. Org. Biomol. Chem 2020, 18, 9018–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Park S; Kim SY; Cho J; Jung D; Seo D; Lee J; Lee S; Yun S; Lee H; Park O; et al. Aryl Sulfate Is a Useful Motif for Conjugating and Releasing Phenolic Molecules: Sulfur Fluorine Exchange Click Chemistry Enables Discovery of Ortho-Hydroxy-Protected Aryl Sulfate Linker. Bioconjug. Chem 2019, 30 (7), 1957–1968. [DOI] [PubMed] [Google Scholar]
- (7).Pauly H Uber Die Einwirkung von Diazoniumverbindungen Auf Imidazole. Hoppe-Seyler’s Z. Physiol. Chem 1905, 44, 159–160. [Google Scholar]
- (8).Pauly H Zur Kenntnis Der Diazoreaktion Des Eiwebes. Hoppe-Seyler’s Z. Physiol. Chem 1915, 94, 284–290. [Google Scholar]
- (9).Hooker JM; Kovacs EW; Francis MB Interior Surface Modification of Bacteriophage MS2. J. Am. Chem. Soc 2004, 126, 3718–3719. [DOI] [PubMed] [Google Scholar]
- (10).Schotten C; Leprevost SK; Yong LM; Hughes CE; Harris KDM; Browne DL Comparison of the Thermal Stabilities of Diazonium Salts and Their Corresponding Triazenes. Org. Process Res. Dev 2020, 24, 2336–2341. [Google Scholar]
- (11).Trusova ME; Kutonova KV; Kurtukov VV; Filimonov VD; Postnikov PS Arenediazonium Salts Transformations in Water Media: Coming Round to Origins. Resour. Technol 2016, 2, 36–42. [Google Scholar]
- (12).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Patterson DM; Nazarova LA; Prescher JA Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- (14).Guzman LE; Kimani FW; Jewett JC Protecting Triazabutadienes To Afford Acid Resistance. ChemBioChem 2016, 17, 2220–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jensen SM; Kimani FW; Jewett JC Light-Activated Triazabutadienes for the Modification of a Viral Surface. ChemBioChem 2016, 17, 2216–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Paik CH; Eckelman WC; Reba RC Reactivity of Amino Acids in the Azo Coupling Reaction. I. Dependence of Their Reactivity on PH. Bioorg. Chem 1979, 8, 25–34. [Google Scholar]
- (17).Shadmehr M; Davis GJ; Mehari BT; Jensen SM; Jewett JC Coumarin Triazabutadienes for Fluorescent Labeling of Proteins. ChemBioChem 2018, 19, 2550–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cornali BM; Kimani FW; Jewett JC Cu-Click Compatible Triazabutadienes to Expand the Scope of Aryl Diazonium Ion Chemistry. Org. Lett 2016, 18, 4948–4950. [DOI] [PubMed] [Google Scholar]
- (19).Hong V; Steinmetz NF; Manchester M; Finn MG Labeling Live Cells by Copper-Catalyzed Alkyne-Azide Click Chemistry. Bioconjug. Chem 2010, 21, 1912–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pickens CJ; Johnson SN; Pressnall MM; Leon MA; Berkland CJ Practical Considerations, Challenges, and Limitations of Bioconjugation via Azide-Alkyne Cycloaddition. Bioconjug. Chem 2018, 29, 686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dommerholt J; Rutjes FPJT; van Delft FL Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. Top. Curr. Chem 2016, 374, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jewett JC; Bertozzi CR Cu-Free Click Cycloaddition Reactions in Chemical Biology. Chem. Soc. Rev 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).He J; Kimani FW; Jewett JC A Photobasic Functional Group. J. Am. Chem. Soc 2015, 137, 9764–9767. [DOI] [PubMed] [Google Scholar]
- (24).Khramov DM; Bielawski CW Donor-Acceptor Triazenes: Synthesis, Characterization, and Study of Their Electronic and Thermal Properties. J. Org. Chem 2007, 72, 9407–9417. [DOI] [PubMed] [Google Scholar]
- (25).Debets MF; Van Berkel SS; Schoffelen S; Rutjes FPJT; Van Hest JCM; Van Delft FL Aza-Dibenzocyclooctynes for Fast and Efficient Enzyme PEGylation via Copper-Free (3+2) Cycloaddition. Chem. Commun 2010, 46, 97–99. [DOI] [PubMed] [Google Scholar]
- (26).Kuzmin A; Poloukhtine A; Wolfert MA; Popik VV Surface Functionalization Using Catalyst-Free Azide-Alkyne Cycloaddition. Bioconjug. Chem 2010, 21, 2076–2085. [DOI] [PubMed] [Google Scholar]
- (27).McNelles SA; Pantaleo JL; Adronov A Highly Efficient Multigram Synthesis of Dibenzoazacyclooctyne (DBCO) without Chromatography. Org. Process Res. Dev 2019, 23, 2740–2745. [Google Scholar]
- (28).Jaffe CL; Lis H; Sharon N New Cleavable Photoreactive Heterobifunctional Cross Linking Reagents for Studying Membrane Organization. Biochemistry 1980, 19, 4423–4429. [DOI] [PubMed] [Google Scholar]
- (29).Reid DJ; Keener JE; Wheeler AP; Zambrano DE; Diesing JM; Reinhardt-Szyba M; Makarov A; Marty MT Engineering Nanodisc Scaffold Proteins for Native Mass Spectrometry. Anal. Chem 2017, 89, 11189–11192. [DOI] [PubMed] [Google Scholar]
- (30).Thiol-yne coupling can proceed via radical mechanisms upon UV irradiation, as well as by nucleophilic attack by thiolates, including cysteine. As such, prolonged exposure to reactive thiols could prove detrimental to this technology, see:; Zhang C; Dai P; Vinogradov AA; Gates ZP; Pentelute BL Site-Selective Cysteine–Cyclooctyne Conjugation. Angew. Chem. Int. Ed 2018, 57, 6459–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Addy PS; Erickson SB; Italia JS; Chatterjee A A Chemoselective Rapid Azo-Coupling Reaction (CRACR) for Unclickable Bioconjugation. J. Am. Chem. Soc 2017, 139, 11670–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.