

Summary
The recent increase of immunopeptidomics data, obtained by mass spectrometry or binding assays, opens up possibilities for investigating endogenous antigen presentation by the highly polymorphic human leukocyte antigen class I (HLA-I) protein. State-of-the-art methods predict with high accuracy presentation by HLA alleles that are well represented in databases at the time of release but have a poorer performance for rarer and less characterized alleles. Here, we introduce a method based on Restricted Boltzmann Machines (RBMs) for prediction of antigens presented on the Major Histocompatibility Complex (MHC) encoded by HLA genes—RBM-MHC. RBM-MHC can be trained on custom and newly available samples with no or a small amount of HLA annotations. RBM-MHC ensures improved predictions for rare alleles and matches state-of-the-art performance for well-characterized alleles while being less data demanding. RBM-MHC is shown to be a flexible and easily interpretable method that can be used as a predictor of cancer neoantigens and viral epitopes, as a tool for feature discovery, and to reconstruct peptide motifs presented on specific HLA molecules.
Keywords: Restricted Boltzmann Machines, machine learning, antigen presentation, neoantigen discovery, SARS-CoV-2 epitopes, HLA prediction, immunopeptidomics
Graphical Abstract
Highlights
-
•
Flexible predictor of HLA-presented antigens for custom and newly produced datasets
-
•
Prediction for poorly represented HLA alleles is improved over state-of-the-art tools
-
•
Accurate HLA type prediction when only a few HLA annotations are available
-
•
Lower-dimensional data representation enables feature discovery
Bravi et al. developed a flexible machine-learning method to predict viral and cancer antigens presented to killer T cells by histocompatibility leukocyte antigen (HLA) class I proteins. The method is designed to deliver accurate predictions in newly available samples for which little information on the presenting HLA proteins can be retrieved in existing databases.
Introduction
Recognition of malignant and infected cells by the adaptive immune system requires binding of cytotoxic T cell receptors to antigens, short peptides (typically 8–11-mer) presented by the major histocompatibilty complex (MHC) class I coded by histocompatibility leukocyte antigen class I proteins (HLA-I) alleles (Figure 1A). Tumor-specific neoantigens, i.e., antigens carrying cancer-specific mutations, are currently sought-after targets for improving cancer immunotherapy (Yarchoan et al., 2017; Garcia-Garijo et al., 2019). Computational predictions can help select potential neoantigens and accelerate immunogenicity testing. To be useful, these predictions must be specific to each HLA type.
Figure 1.

RBM Approach to HLA-I Antigen Presentation
(A) Antigens binding to a given HLA match specific “sequence motifs” represented by logos (example of Tax-HLA-A∗02:01 complex structure, PDB-ID:1BD2, Mol∗ image; Sehnal et al., 2018).
(B) RBM model structure, see also Figures S1–S3.
(C) RBM scores for presented versus generic peptides on HLA-A∗02:01 and predictive performance assessed by ROC.
(D) RBM and NetMHC performance at recovering MS-detected peptides from Sarkizova et al., 2020 for 92 HLA-I alleles as measured by AUC and PPV metrics, see also see Figure S5. PPVs and AUCs are plotted as a function of the distance between the corresponding HLA and the closest one in NetMHCpan4.0 training dataset. Bold lines highlight the trend of mean AUC and PPV values (plotted by squares) over subsets of alleles grouped by distance as indicated by vertical dashed lines (63 alleles with distance = 0, 15 alleles with 0 < distance < 0.05, 10 alleles with 0.05 < distance < 0.1, and 4 alleles with distance > 0.1).
(E) Score percentiles on the same allele as (C) of the neoantigens from 5 cancer cell lines validated in Marty et al., 2017: 4 neoantigens validated for cell line SKOV3, 4 for A375, 4 for A2780, 3 for OV90, and 1 for HeLa.
(F) Correlation of experimental stability of SARS-CoV-2 epitopes (94 for each HLA allele; Immunitrack and Intavis, 2020) with scores from NetMHC4.0/NetMHCpan4.0 and scores from RBM trained on BA—not available for HLA-C alleles under consideration—and MS data (with ad hoc sequence re-weighting), see also STAR Methods; Figures S4 and S7; Table S1.
(G) RBM and NetMHC4.0 score percentiles (relative to all SARS-CoV-2 9-mers) of n = 22 homologs of dominant SARS-CoV epitopes identified in Grifoni et al., 2020, see also Figure S7. Boxplots indicate median, upper, and lower quartiles.
State-of-the-art methods (Mei et al., 2020; Abelin et al., 2017; O’Donnell et al., 2018; Sarkizova et al., 2020), such as NetMHC (Andreatta and Nielsen, 2016; Jurtz et al., 2017; Reynisson et al., 2020), are based on artificial neural networks trained in a supervised way to predict peptide presentation from the known peptide-HLA association. They are trained on large datasets at every method release and they provide peptide-scoring schemes that perform best on frequent alleles and well-characterized alleles at the time of release. Their accuracy is degraded for rare or little studied HLA-I alleles, which are poorly represented in databases. In that case, another approach is to train unsupervised models of presentation from custom elution experiments with little or no information about the peptide-HLA association. For instance, MixMHCp (Bassani-Sternberg and Gfeller, 2016; Bassani-Sternberg et al., 2017; Gfeller et al., 2018) can reconstruct, from unannotated peptide sequences, a mixture of generative models, one for each expressed HLA type. However, it makes simplified assumptions about binding specificity and is not designed to leverage available (albeit limited) annotation information from the Immune Epitope Database (Vita et al., 2019) (IEDB) to improve accuracy.
We present an alternative method for predicting peptides presented by specific class I MHCs based on Restricted Boltzmann Machines—RBM-MHC. RBM-MHC is a scoring and classification scheme that can be easily trained “on the fly” on custom datasets, such as patient- or experiment-specific samples, and more generally on newly available data. As such, RBM-MHC enables improved predictions for rare alleles at a fast pace without waiting for new releases of general software like NetMHC (Andreatta and Nielsen, 2016; Jurtz et al., 2017; Reynisson et al., 2020), with which it is not intended to compete on an equal footing.
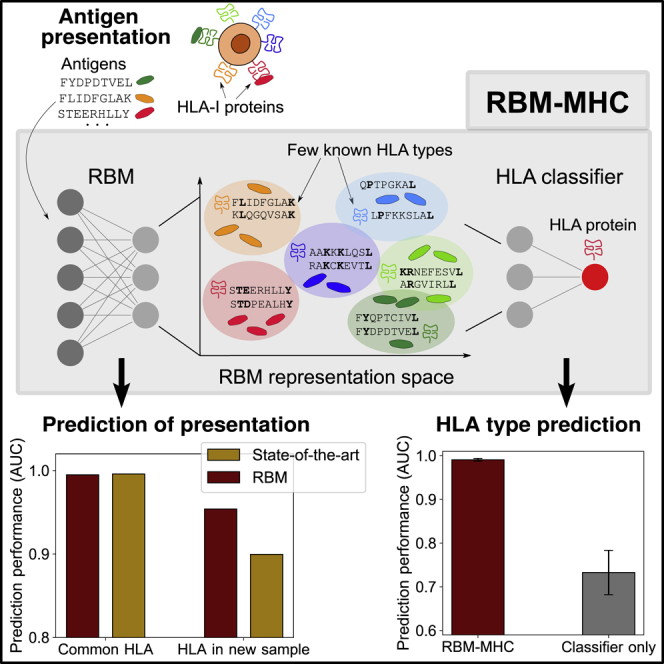
The method consists of two parts. The first component (Figure 1) relies on a RBM, an unsupervised machine-learning scheme that learns probability distributions of sequences given as input (Smolensky, 1986; Hinton, 2002; Tubiana et al., 2019). The RBM estimates presentation scores for each peptide and can generate candidate presentable peptides. The RBM also provides a lower-dimensional representation of peptides with a clear interpretation in terms of associated HLA type. The second component of the method (Figure 2) exploits this efficient representation to classify sequences by HLA restriction in a supervised way, using only a small number of annotations.
Figure 2.

RBM-MHC Approach to HLA-I Classification
(A) RBM-MHC workflow, see also Figures S1–S3. RBM projects sequences onto a representation in terms of “hidden units” (we selected hidden unit 8 and 10 for illustration) through the set of learnt weights. In this representation space, each cluster groups together antigens with the same HLA-binding specificity (given by the color code). Linear classification guided by the knowledge on the HLA-I restriction of a few antigens in each cluster (“labels”) is performed through the HLA-I classifier to predict the HLA-I type of all antigens. Data: IEDB-derived dataset for haplotype 1, Table S2.
(B) RBM distinguishes 2 subclasses in HLA-B∗51:01-binding antigens. The HLA-B∗51:01-peptide bond can be established (Gfeller et al., 2018) via (i) the interaction of the HLA-B∗51:01 residue 62 with peptide position 2, requiring a hydrophilic residue there, typically alanine (A), and a polar or negatively charged residue, typically aspartic acid (D), at position 1; and, (ii) with R62 sidechain facing the solvent, requiring co-occurrence at positions 1–2 of hydrophobic residues, typically proline (P), at position 2. Inspection of the inputs to the 10th hidden unit (), found by projecting peptides predicted by RBM-MHC as HLA-B∗51:01-specific onto the corresponding weights (STAR Methods, Equation 4), reveals a bimodal distribution, enabling the discrimination of the “hydrophilic/charged” pattern (i) from the “hydrophobic” pattern (ii), as recapitulated by the sequence logos.
(C) Performance (AUC) of 6 methods for HLA-I prediction on 9-mer in 10 synthetic-individual samples, each carrying 6 HLA-I covering A, B, and C alleles (see also Figure S8; Table S2). Bars are standard deviations over the 10 datasets.
(D) Sequence logos of clusters found with RBM-MHC trained on melanoma-associated sample 12T (Kalaora et al., 2016, 2018) and IEDB mono-allelic data; ρ gives the Pearson correlation between respective amino acid frequencies.
(E) Performances (average Pearson correlation ρ over clusters) for 3 samples from (Kalaora et al., 2016, 2018; Bassani-Sternberg et al., 2016), see also Figure S9 and Table S3. In C, E the MixMHCp and NetMHC versions used are MixMHCp2.1 and NetMHCpan4.1, respectively.
Results
Restricted Boltzmann Machine for Peptide Presentation
The first building block of the method is a RBM, a probabilistic graphical model defined by a simple structure with one hidden layer and weights connecting the input—the peptide sequence—to the hidden layer (Figure 1B). RBM parameters, i.e., the weights and the biases acting on both input and hidden units, are learned from a training set of presented peptides collected from public repositories or custom samples (STAR Methods; Figures S1A, S2A, and S3). These input peptides may come from mass spectrometry (MS) experiments or binding assays. They may come from several HLA alleles, or a single one. For example, in datasets from single-individual samples, the number of HLAs is at most 6, as each individual inherits 2 alleles of each locus HLA-A, HLA-B, and HLA-C. Another example is given by mono-allelic samples (Abelin et al., 2017; Sarkizova et al., 2020), where peptides are presented by a single HLA protein.
After training, the RBM returns a probability for each sequence, which is interpreted as a global score of antigen presentation by the HLA proteins involved in the training dataset. Since learning the RBM requires fixed-length input sequences, we first reduce peptides of variable length to a reference length by an alignment procedure based on a Hidden Markov Model profile built from aligning subsets of same-length sequences using standard routines (STAR Methods; Figure S1E). We set the reference length to 9-residues, the most common length of peptides presented by class I MHCs (Bassani-Sternberg et al., 2015; Andreatta and Nielsen, 2016; Trolle et al., 2016; Sarkizova et al., 2020).
RBM Performance on Mono-Allelic Data
As a first validity check of RBM-based presentation scores, we built a RBM model to predict presentation by a single common allele, HLA-A∗02:01, from MS data annotated with this HLA restriction in IEDB (Vita et al., 2019). We tested the model score’s ability to discriminate presented peptides from “generic” peptides (randomly drawn from the human proteome). We calculated a receiver operating characteristic (ROC) curve from RBM presentation scores assigned to a test set of presented versus generic peptides (Figure 1C). The area under the ROC, AUC = 0.973, far above the random-expectation value (AUC = 0.5) and close to the maximal value (AUC = 1), proves the RBM predictive power at recovering presented antigens.
We next benchmarked RBM’s predictive power on recent MS datasets from mono-allelic cell lines (Sarkizova et al., 2020). In addition to the AUC, which considers with equal weight presented and generic peptides, we computed the positive predictive value (PPV) (Abelin et al., 2017; Sarkizova et al., 2020), which measures the model’s ability to correctly recognize presented peptides among a 99- or 999-fold excess of generic peptides (equivalent to assuming that only 1%; Yewdell, 2006; Paul et al., 2013; Vitiello and Zanetti, 2017; or 0.1%; Abelin et al., 2017; Sarkizova et al., 2020) of all peptides bind to a given HLA, see STAR Methods). Figure S5B compares the AUC and PPV between RBM and NetMHCpan4.0 (Jurtz et al., 2017) for all the 92 different HLA alleles encompassing A, B, C loci from Sarkizova et al., 2020 (31 HLA-A, 40 HLA-B, and 21 HLA-C). NetMHCpan4.0 is the penultimate version of NetMHCpan, which was trained before the publication of the mono-allelic data (Sarkizova et al., 2020) and is therefore trained on an independent dataset. In contrast, RBM was trained on data presently available in IEDB for all the 92 alleles. To ensure independence from the training set, testing peptides were manually excluded from the training data (STAR Methods). Both methods performed comparably (AUC = 0.97 for RBM versus 0.95 for NetMHC, 1% PPV = 0.52 for RBM versus 0.53 for NetMHC).
When there is limited or no data for a given HLA allele, NetMHCpan extrapolates from the most similar HLA allele for which data are available. Figure 1D shows that NetMHCpan performance degrades with the distance between the queried HLA and nearest neighbor in the training dataset (Nielsen et al., 2007). By contrast, the RBM performs equally well on all HLAs, which is expected as in that case the distance to the training dataset is always zero. This highlights the importance of being able to flexibly and rapidly train models on new data, especially for HLA alleles that are poorly covered in previously available datasets. The four alleles at the largest distance from NetMHCpan4.0 training data, for which RBM outperforms NetMHC, are all HLA-C (HLA-C∗01:02, HLA-C∗02:02, HLA-C∗07:04, and HLA-C∗17:01, see Figure S5B), a locus usually under-represented in existing databases.
RBM Can Predict Cancer Neoantigens
An application of antigen presentability is the identification of neoantigens arising from cancer mutations, which are key to evaluating their potential for immunotherapies. To assess RBM’s ability to predict neoantigens, we looked at missense mutations in 5 ovarian and melanoma cancer cell lines from Marty et al., 2017. We attributed presentation scores to all 8–11-mer peptides harboring these mutations, using the RBM previously trained on HLA∗A:02:01 (known to be expressed in the 5 cell lines), and also computed, for comparison, the corresponding score by NetMHCpan4.1 (Reynisson et al., 2020), the latest version of NetMHCpan. 16 neoantigens were experimentally validated by MS to associate with HLA∗A:02:01 (Marty et al., 2017). Of those, 15 were ranked by RBM in the top 4.1% among generic peptides (mean score percentile 1.5% versus 1.6% for NetMHC) and 4.7% among mutated peptides (mean 1.8% versus 0.9% for NetMHC) for the corresponding cell line. This demonstrates that the RBM reaches state-of-the-art performance at predicting presented neoantigens.
Severe Acute Respiratory Syndrome Coronavirus 2 Epitope Discovery
Predicting which antigens are presented by virus-infected cells is key to the rational design of vaccines targeting these antigens. We tested RBM’s ability to perform this task on the example of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), using recent in vitro measurements of binding stability (Immunitrack and Intavis, 2020) of SARS-CoV-2 candidate epitopes (selected using NetMHC4.0; Andreatta and Nielsen, 2016; and NetMHCpan4.0; Jurtz et al., 2017; see STAR Methods).
We first trained an allele-specific RBM for each HLA-I involved in the experiment. Since the experiment measures binding, we chose our training sets as binding assay (BA) datasets as well as MS datasets that were reweighed to make them comparable to BAs (correcting for amino acid frequency biases, see STAR Methods). We then attributed a RBM score to each peptide and compared it to its experimental binding stability (Figures S7A and S7B). In Figure 1F we report the correlation between the RBM score and measurement for each allele. The performance of the NetMHC version used to select the epitopes (NetMHC4.0/NetMHCpan4.0) is shown for comparison. As before, RBM outperforms NetMHC4.0/NetMHCpan4.0 for rarer HLA-C alleles (HLA-C∗07:01 and HLA-C∗01:02). It is noteworthy that correlations scores for rare alleles are improved using the very recent NetMHCpan4.1 and get comparable to RBM results (see Figure S7C). Supported by the good performance of RBM on rare alleles, we suggest new SARS-CoV-2 epitopes for HLA-C alleles that were not among the top-scoring ones by NetMHC4.0/NetMHCpan4.0. Our predictions, given in Table S1, have been favorably cross-checked with NetMHCpan4.1 scores.
RBM also assigns high presentation scores to SARS-CoV-2 epitopes that are homologous to experimentally validated SARS-CoV cytotoxic T cell epitopes (Grifoni et al., 2020), on par with NetMHC4.0 (Figure 1G) and NetMHCpan4.0/NetMHCpan4.1 (Figure S7E).
RBM Offers Useful Low-Dimension Representations
In addition to providing a presentation score, RBM hidden units allow for mapping peptides onto a lower-dimensional “representation space” given by the inputs to the RBM hidden units (Tubiana et al., 2019) (see STAR Methods). The full potential of this representation is best illustrated on a RBM trained on multi-allelic data, where peptides may be bound to up to 6 different HLA-I proteins. Figure 2A shows an example of such a representation projected in 2 dimensions, on a synthetic dataset obtained by pooling peptides from 6 HLA restrictions in IEDB. The low-dimensional projection organizes peptides into 6 well-defined clusters, reflecting the HLA-binding specificities present in the sample. Each HLA recognizes specific “sequence motifs” (patterns of preferred residues at each position) in the presented peptides. The data-derived RBM parameters underlying that representation play a key role in capturing which amino acids contribute to defining HLA-binding motifs, as illustrated in a biallelic case in Figure S2B.
The representation space can also be useful to reveal new features even in the mono-allelic case. An example is provided by HLA-B∗51:01-restricted peptides derived from a clinical sample (Kalaora et al., 2016, 2018) (see below for details about how restriction was predicted). Projection of HLA-B∗51:01-specific sequences onto a single RBM feature reveals a double-peaked histogram, corresponding to two structurally alternative binding modes, which were validated in Gfeller et al., 2018 (Figure 2B).
HLA Classification with RBM-MHC
This low-dimensional representation of peptides suggests an efficient way to classify them by HLA-I specificity using the annotation of a small number of sequences by their HLA preference. In practice, these annotated sequences may come from other experiments or public databases as IEDB (Vita et al., 2019). Using these annotations, we train a linear classifier to predict the HLA restriction of individual peptides from their RBM representation (see STAR Methods; Figures 2A, S2A, and S3C–S3E). We refer to this architecture, which combines the RBM (trained on unannotated peptides) and the HLA-I classifier (trained on a few annotated ones), as RBM-MHC.
To test performance in a case where the ground truth is known, we trained RBMs on 10 synthetic “single-individual” samples pooling together IEDB 9-mer antigens presented by 6 HLA-I proteins per individual, covering 43 different alleles in total (12 HLA-A, 17 HLA-B, and 14 HLA-C, see STAR Methods and Table S2). We randomly selected 10% of peptides and labeled them with their associated HLA and used them to train the RBM-MHC classifier. The performance of RBM-MHC at predicting HLA association, as measured by AUC = 0.991, is excellent (Figure 2C and STAR Methods). The RBM representation is crucial for achieving high prediction performance. Training a linear HLA-I classifier directly on the annotated sequences (rather than their RBM representation) yields a much poorer performance (AUC=0.733). On the other hand, a completely unsupervised clustering algorithm (K-means; Lloyd, 1982) applied to unlabeled data in RBM representation space (“RBM-km”) performed well (AUC=0.927), while K-means applied directly to sequences did not (AUC = 0.650; Figure 2C and STAR Methods).
The structure of the RBM representation space further allows for setting up a well interpretable protocol to generate new artificial peptides from the model with controlled HLA-binding specificity (STAR Methods, Figures S2C–S2E).
We next extended our HLA type predictions to peptides of variable length (8–11 residues). Multiallelic datasets pose the additional challenge that sequences must be aligned together across different HLA motifs. To optimize this multiple sequence alignment, we resorted to an iterative procedure where the sequence alignment is refined using the HLA assignments from the first iteration of RBM-MHC, after which a second RBM-MHC is trained to obtain the final model. This procedure allows the steps of HLA classification and alignment to inform each other (STAR Methods and Figure S1E). We benchmarked our alignment routine against other well-established alignment strategies (based on the MAFFT software; Katoh and Standley, 2013) and we verified that our routine ensures higher HLA classification performance even without the refinement step (STAR Methods and Figures S1F–S1L).
RBM-MHC Compares Favorably to Existing Methods
RBM-MHC outperforms MixMHCp2.1 (Bassani-Sternberg and Gfeller, 2016; Bassani-Sternberg et al., 2017; Gfeller et al., 2018) in terms of both overall accuracy and stability across the 10 synthetic datasets (Figures 2C and S8). This is in part thanks to the 10% labeled data it exploits. The RBM also captures global sequence correlations that MixMHCp’s independent-site models miss, which are needed to correctly classify antigens across alleles with similar binding motifs. The major drop in MixMHCp2.1 performance occurs precisely in datasets that mix same-supertype alleles (Sidney et al., 2008) (HLA-B∗27:05 and HLA-B∗39:01 belonging to supertype B27; and, HLA-B∗40:01 and HLA-B∗44:03 belonging to B44, see Table S2). RBM-MHC also performs better in terms of AUC than NetMHCpan4.1 (Reynisson et al., 2020), which can be applied here to predict binding to each of the 6 alleles in our datasets and remains comparable to NetMHCpan4.1 when several other performance indicators are considered (Figure S8). The gain in AUC of RBM-MHC over MixMHCp and NetMHC is slightly more pronounced for 9-mer-only samples than for 8–11-mer (Figure S8A), thus suggesting room for further improvement in the alignment.
Application to Single-Patient Cancer Immunopeptidome
To assess the relevance of our approach in a clinical setting, we considered single-patient, melanoma-associated immunopeptidomics datasets (Kalaora et al., 2016, 2018; Bassani-Sternberg et al., 2016), complemented with patient HLA typing and whole-exome sequencing (WES) of tumorous cells, wherein a total 11 neoantigens were identified. We tested the RBM-MHC approach for motif reconstruction. Since in this case, true peptide-HLA associations are unknown, the model performance is evaluated through the correlation between the predicted motifs and motifs reconstructed from IEDB monoallelic data (Figures 2D–2E and S9). RBM-MHC, MixMHCp2.1, and NetMHCpan4.1 perform comparably, with the exception of sample Mel8, where MixMHCp2.1 merges antigens specific to the 2 HLA-C into the same cluster causing a drop in the average ρ (Figures 2E and S9B). In addition, RBM-MHC and its unsupervised version RBM-km trained on the patient dataset (see STAR Methods) systematically predicted the correct HLA association for all neoantigens. They also assigned a top 1% score among all WES mutants to 8 out of the 11 identified neoantigens (Table S3).
Discussion
In this work, we presented RBM-MHC, which is a flexible predictor of antigen sequences presented on specific HLA-I types. It can be broadly used for four tasks. First, the RBM module of the method can be used to score the presentability of (neo)antigens, regardless of HLA information (Figure 1). Second, the low-dimensional representation of peptides provided by the RBM allows one to visualize different HLA-binding motifs (as in Figure 2A), or different binding modes within the same HLA (as in Figure 2B), providing a useful tool for data exploration and feature discovery. Third, RBM-MHC may be used to classify peptides by HLA restriction using only a limited number of annotations, matching state-of-the-art performance (Figure 2C). Finally, the method can generate putative antigens with or without a specific HLA restriction (Figures S2C–S2E). These generated peptides could be experimentally tested in future, providing additional evidence for the validity of the model (Russ et al., 2020).
RBM-MHC’s use will also depend on the choice of the training dataset. Single-allele datasets allow for training allele-specific models to score antigen presentation by a specific HLA allele. In this way, we have proposed new putative SARS-CoV-2 epitopes for HLA-C alleles that could be tested experimentally (Table S1). Multi-allelic datasets (from e.g., peptidome MS of clinical samples) allow for training models to score antigen presentation in a given donor (with all its HLA-I alleles), and to predict the HLA-binding specificity of given peptides. We benchmarked our approach on several examples of datasets. In single-allele datasets, we found that RBM-MHC can perform similarly to established methods, such as NetMHCpan, for frequent, well-characterized alleles (Figures 1D–1G, S5, and S7), despite the fact that NetMHC training is fully supervised, incorporates more information such as binding affinities, and uses positive (binders) as well as negative (non-binders) examples. The number of binders per allele in the version NetMHCpan4.0 ranges between ~50 and ~9,500 (Jurtz et al., 2017), while RBM-MHC is trained on datasets of presented peptides only, making our approach less data demanding. The amount of 8-11-mer peptides per allele considered in this work from IEDB-derived MS records varies from ~100 to 104, with an average (2,350) comparable to NetMHCpan4.0 positive examples. RBM-MHC is a more flexible machine-learning scheme that can be easily trained on newly produced datasets, tracking the fast-growing number of available datasets to improve its predictive power, especially for previously under-represented HLA-I alleles (Figures 1D, 1F, and S5), such as HLA-C alleles.
The latest version of NetMHCpan, NetMHCpan4.1 (Reynisson et al., 2020), was published very recently, three years after the previous release NetMHCpan4.0 (Jurtz et al., 2017). This new version was trained on a total of ~850,000 ligands collected from public (mainly IEDB) and in-house resources (Reynisson et al., 2020), which is a 10-fold increase with respect to NetMHCpan4.0. All our comparisons were done with this latest version, except on the monoallelic data from Sarkizova et al., 2020 (Figure 1D) and on the SARS-CoV-2 epitope data from Immunitrack and Intavis, 2020 (Figure 1F), where we have used version 4.0 for independent validation and consistency. For completeness in Figures S6 and S7C we report the results obtained with NetMHCpan4.1, showing an improvement of performance for rare alleles thanks to the increase of training data. These results support our main conclusion that our approach is especially useful in scenarios where new data become available but current methods are not updated yet to cover the corresponding alleles with good accuracy, as was the case when data from Sarkizova et al., 2020 and Immunitrack and Intavis, 2020 came out while NetMHCpan4.0 was the latest version.
RBM-MHC can also be applied to unannotated, moderate-size multiallelic datasets available from clinical studies, for which there may exist only a limited number of HLA annotations in existing databases (<100) for the patients rarer HLA alleles. We have shown that RBM-MHC efficiently exploits the statistical information in these samples and combines it with limited annotation information to deliver accurate and stable predictions of HLA-I binding (Figures 2C–2E, S8, and S9; Tables S2 and S3).
The flexibility of choice of the training dataset and corresponding applications is even broader. The method is not limited to MS datasets but can be trained on datasets from binding affinity assays to customize presentation score predictions toward the identification of high-affinity ligands (see Figure 1F). If only MS data are available to build a predictor for this task, as is the case for HLA-C alleles in Figure 1F, a commonly acknowledged issue is that biases of MS techniques can potentially affect the amino acid frequency distribution in training datasets and hence the predictor’s results. To address this issue we developed, similarly to Bassani-Sternberg et al., 2017, a sequence re-weighting scheme to efficiently compensate for detection biases in MS and better score ligands tested in vitro (STAR Methods, Figure S4).
Overall, our results show that the approach is useful for systematic applications with newly produced large-scale datasets covering an increasing range of HLA types. Future work will be devoted to also develop an extension to HLA class II presentation. Finally, since our method is designed to be trained on custom samples it could be of relevance to produce sample-specific insights about the complexity of endogenous antigen presentation. As a future direction, we will investigate how the probabilistic framework provided by RBM-MHC can be exploited to develop these insights in a quantitative manner.
STAR★Methods
Key Resources Table
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barbara Bravi (bbravi.bb@gmail.com).
Materials Availability
This study did not generate new materials.
Data and Code Availability
This paper analyzes existing, publicly available data. These datasets’ sources are provided in the Key Resources Table.
RBM-MHC original code and trained models are publicly available at https://github.com/bravib/rbm-mhc.
The scripts used to generate the figures reported in this paper are available at https://github.com/bravib/rbm-mhc.
Any additional information required to reproduce this work is available from the Lead Contact.
Method Details
Schematic Outline of the RBM-MHC Pipeline
The essential steps of the RBM-MHC application pipeline are:
1. Collection of Training Datasets
RBM-MHC can be trained on patient-derived clinical samples or on datasets collected from public repositories - here we constructed training datasets from mass spectrometry and binding affinity assays available in IEDB. See Figure S1A and STAR Methods section “Dataset collection from IEDB and preparation” for a detailed description.
2. Alignment
RBM-MHC works with fixed-length sequences, hence peptide sequences are first reduced to the same length through an alignment routine, see Figure S1E and STAR Methods section “Antigen sequence alignment” for a detailed description.
3. RBM-MHC Algorithm
The full RBM-MHC architecture (Figure 2A) combines a Restricted Boltzmann Machine (RBM) and a HLA-I classifier, trained in successive steps. See Figures S2 and S3 and STAR Methods section “RBM-MHC algorithm” for a detailed description.
Dataset Collection from IEDB and Preparation
The analyses described throughout this work rely on the collection of sets of peptides documented with selected HLA restriction in the Immune Epitope Database (IEDB, last release; Vita et al., 2019).
Mass Spectrometry Datasets
The search in IEDB for immunopeptidomic data from mass spectrometry (MS) was performed as follows. The full set of curated HLA-I ligands was downloaded from IEDB (file mhc_ligand_full.csv from http://www.iedb.org/database_export_v3.php, accessed in July 2020). In this IEDB file, we looked for linear, human peptides eluted from cells and detected by MS techniques (field Assay Group = “ligand presentation”, field Method/Technique = “cellular MHC/mass spectrometry”, “mass spectrometry”, “secreted MHC/mass spectrometry”). Among these data, we prioritized HLA-specific peptides from mono-allelic sources, in such a way that the assignment of HLA-binding specificity is unambiguous and does not rely on additional in silico predictions. To do this in practice, we set the field Allele Evidence Code = “Single allele present”, indicating that antigen presenting cells are known to only express a single HLA-I molecule, as is the case for mono-allelic cell lines (Abelin et al., 2017; Sarkizova et al., 2020). If, for a given allele, we found less than a minimal number (set to 300) of sequences among mono-allelic-source data, we extended the search to peptides with Allele Evidence Code = “Allele Specific Purification”, since this procedure attaches greater confidence to the HLA assignment than its inference by in silico methods. Only for one allele (HLA-B∗39:01) did we extend the search to all other MS data, including the evidence code “Inferred by motif or alleles present”. MS datasets filtered through these steps were used to train allele-specific RBM presentation models (as in Figures 1C–1F) and multi-allelic models (as in Figures 2C–2E). In a multi-allelic setting, we first tested motif reconstruction by considering 10 “synthetic-individual” datasets of antigens with known HLA-I specificity to assess the RBM-MHC classification performance by comparing RBM-MHC predictions against the known HLA-I specificity. These datasets were built by collecting all IEDB antigens (searched as above) associated to 2 haplotypes, i.e., combinations of an A, a B, a C HLA allele observed to co-occur in the human population to preserve linkage (see Table S2). Information on haplotypes co-occurrence was found at allelefrequencies.net (González-Galarza et al., 2015). To apply the RBM-MHC method to multi-allelic, patient-derived immunopeptidomic samples, we sought to have a small amount of labeled peptides, here set to 10%. To this end, we either retrieved a HLA annotation in IEDB for portions of the samples or, when this was not possible, we added to each of them labeled peptides from IEDB (searched as above) for the 6 HLA-I given by the patient’s HLA typing. The RBM-MHC training set was then defined as this minimally extended dataset, to guide the learning of the HLA-I classifier by the labeled peptides, whose predictions are used to reconstruct HLA-I motifs in the original dataset. We did not attempt to identify motifs in the dataset Mel5 from (Bassani-Sternberg et al., 2016) as the patient’s HLA typing was incomplete (lacking the HLA-C alleles).
Binding Assay Datasets
Records of binding affinity (BA) assays from IEDB (as of July 2020) were filtered following (Sarkizova et al., 2020), i.e. selecting peptides annotated with a quantitative measure of binding dissociation constants <500 nM and excluding assay types that led to documented discrepancies between predicted and effective affinity (“purified MHC/direct/radioactivity/dissociation constant KD”, “purified MHC/direct/fluorescence/half maximal effective concentration (EC50)”, “cellular MHC/direct/fluorescence/ half maximal effective concentration (EC50)”). BA datasets were used to train allele-specific models of Figures 1F–1G.
Antigen Sequence Alignment
The typical length of HLA-I peptides, structurally constrained by the MHC-I binding cleft, is generally recognized to be 8 to 11 amino acids (Bassani-Sternberg et al., 2015; Andreatta and Nielsen, 2016; Trolle et al., 2016; Sarkizova et al., 2020), with 9-mers being the most abundant except for very few exceptions (Sarkizova et al., 2020). Hence we focus on datasets containing 8–11-mers (Figures S1B–S1D) and an alignment procedure is implemented to reduce peptide sequences to the typical length of 9 residues. This choice of a 9-mer-centered alignment is also consistent with the treatment of variable-length class I peptides by other algorithms as MixMHCp, which scores their positions based on 9-mer motifs (Gfeller et al., 2018), and NetMHC, which applies to sequences with length different from 9 the insertions and deletions that give optimal predicted scores for presentation by a given allele (Andreatta and Nielsen, 2016).
Our alignment routine is articulated around the construction of a main alignment and an alignment refinement, aimed at improving HLA classification and hence motif reconstruction. The optimal alignment for each peptide is found by separately aligning subsets of peptides sharing the same HLA-binding specificity that best describe the corresponding sequence motif. In turn, optimally aligning a peptide facilitates its correct association the HLA type. However, identifying such subsets in typical samples, which pool together peptides of different specificity, requires first a step of motif reconstruction, i.e. of assignment of putative HLA types to all peptides. The workflow of the alignment procedure, depicted in Figure S1E, is as follows.
Main Alignment
-
•
Progressively align fixed-length subsets of sequences (Step 1 in Figure S1E). We estimate Position Weight Matrix (PWM) profiles of subsets of sequences with the same length and we align these profiles, using respectively the functions seqprofile and profalign (with default options) of the MATLAB Bioinformatics Toolbox (release R2018b). The alignment is made progressively from the minimal length considered (here 8 residues) to the maximal one (here 11 residues) by inserting gaps in shorter profiles, resulting in an alignment of the maximal length considered (11). This alignment is used as seed to learn a Hidden Markov Model (HMM) profile of the reference length = 9 by appeal to the routines hmmprofstruct and hmmprofestimate (with default parameters) of the MATLAB Bioinformatics Toolbox.
-
•
Align sequences to the HMM profile, Step 2 in Figure S1E. Sequences with length different from 9 are re-aligned to the HMM profile relying on the position-specific insertion/deletion probabilities of the HMM (by the hmmprofalign function). This procedure results in a multiple sequence alignment displaying mostly the insertion of a gap in 8-mers and single/double deletions in 10/11-mers.
-
•
Use this first alignment to train RBM-MHC.
Alignment refinement (for HLA classification in multi-allelic samples):
-
•
Build HLA-specific HMM profiles by grouping peptides based on the putative “class” (HLA type) predicted by the HLA-I classifier (Step 3 in Figure S1E). First, for each HLA-I class, we put together the 10% of labeled data and the peptides classified in that class, weighted by the probability of classification, reflecting the degree of confidence of class assignment. We build on this sample a HLA-specific HMM by the routines hmmprofstruct and hmmprofestimate (with default parameters). We use these HMM profiles (essentially capturing the pattern of single-site amino acid frequency for each HLA type) as seeds of each class’s alignment.
-
•
Re-align peptides based on the best HMM alignment scores (Step 4 in Figure S1E). We take every unlabeled peptide and we consider the alignment to each of the classes’ seeds and the corresponding HMM alignment score (both given by the hmmprofalign function). We retain, for each peptide, the alignment with the highest score. Such best scoring alignment can be to a class that is different from the classifier-predicted one, allowing us to re-classify peptides more accurately in the subsequent iteration. This step helps therefore correct classification errors arising from the initial, suboptimal alignment by means of allele-specific HMM alignment scores.
-
•
Repeat the RBM-MHC training after the re-alignment.
In the 10 “synthetic-individual” datasets considered for testing motif reconstruction (Table S2), high classification performance was reached already at the first iteration and we observed a further, systematic improvement after 1 re-alignment step, see Figure S1F. For 2 or more iterations, there are cases in which the classification performance is degraded, therefore in our motif reconstruction applications (Figures 2C–2E) we re-align and re-train once.
The alignment routine is designed in such a way as to be easily tuned to a different reference length and to the inclusion of longer peptides, depending on the particular length preferences of the alleles of interest and on data availability. For instance, in Figure S1H we show that HLA classification performance is rather stable with respect to the choice of the reference length in the range 8–11 residues and could even slightly increase for reference lengths > 9. We recall however that the choice of reference length = 9 is made to reflect the typical length of class I peptides: peptide length distributions of the datasets considered in this work confirm that 9 residues is by far the most abundant length (Figures S1B–S1D).
To test the quality our alignment routine, we compared its performance at HLA classification to that of other common alignment software, MAFFT (latest version 7.471; Katoh and Standley, 2013) and HMMER (http://hmmer.org/). Default options of these methods tend to produce very gapped alignments, that might cause a rather drastic loss of information on the original peptides and hence affect the RBM-MHC classification performance. In contrast, the progressive alignment of profiles obtained from subsets of sequences of the same length (Step 1 in Figure S1E) allows for well controlled gap insertions. We found that large gap penalties are more suitable for applying MAFFT to the type of alignment of interest here, and yield better performance. After a grid search we set in MAFFT the penalty for opening a gap to 8, the one for extending a gap to 12. We implemented MAFFT: (i) with a speed-oriented option, FFT-NS-2, see (Katoh and Standley, 2013); (ii) with an accuracy-oriented option – G-INS-I – recommended for sequences of similar lengths. (We have checked that other accuracy-oriented options, E-INS-I and L-INS-i, lead to rather similar results). MAFFT accuracy-oriented options tend to be slow, hence we ran it only on a subset of the sample. We use this aligned subset as a seed to build a HMM by the HMMER routine hmmbuild (with open-gap penalty and extension-gap penalty respectively increased to 0.49 and 0.99). All the other sequences are then aligned by the routine hmmalign. To get an alignment of length 9, one can adjust accordingly a maximal gap percentage, i.e., columns with more than this percentage of gaps are filtered out to reduce the alignment length. For example, to obtain the alignments of length = 9 used for Figure S1G, columns with in average > 68% of gaps (MAFFT-FFT-NS-2) and > 65% of gaps (MAFFT-G-INS-i + HMMER) were filtered out. Varying this percentage threshold allows one to work with alignments of different reference length, see e.g. Figures S1I–S1L. We did not find evidence that MAFFT-based alignments (i) and (ii) lead to improved performance compared to the alignment routine we adopted, already in the first training iteration (Figure S1G). In particular, the fast, but usually less accurate, FFT-NS-2 option, allowing for a global alignment of the full sample, performs better than the use of a HMM model built from an accurately aligned seed. This might be due to the fact that the initial choice of a unique seed, including peptides of different specificity yet to predict, is inevitably suboptimal.
RBM-MHC Algorithm
Restricted Boltzmann Machine (RBM)
The RBM learns an underlying probability model for the antigen sequences in the training datasets, in our case the HLA-I presented antigens. A RBM (Smolensky, 1986; Hinton, 2002) consists of one layer of observed units and one layer of hidden units connected by weights (see Figure 2A). The observed units stand here for the antigen sequences to model, hence and the number of “symbols”, q, for each observed unit is 21 (20 amino acids and the gap). Mathematically, the model is defined by a joint probability over presented antigen sequences and hidden units:
| (Equation 1) |
where is a matrix of local “potentials” (biases) acting on observed units, are local potentials on hidden units and the weights (arranged in a tensor of size ) couple hidden and observed units. The parametric form of is chosen as a double Rectified Linear Units (dReLu):
| (Equation 2) |
containing parameters () to infer from data during training (see below). The dReLu was shown to outperform other choices of potential such as Gaussian (Tubiana and Monasson, 2017; Tubiana et al., 2019), guaranteeing that correlations beyond pairwise in data are captured. The probability distribution over the presented antigen sequences one is interested in modeling is recovered as the marginal probability over hidden units:
| (Equation 3) |
where . We define , the input to hidden unit μ coming from the observed sequence , as the sum of the weights entering that particular hidden unit:
| (Equation 4) |
During the modeling step called “training”, the weights , the local potentials and the parameters specifying are inferred from data by maximizing the average log-likelihood of sequence data , as previously described (Tubiana and Monasson, 2017; Tubiana et al., 2019). This leads to inferring the RBM probability distribution that optimally reproduces the statistics of the training dataset (see Figure S3H for comparisons of data and model single-site frequency and pairwise connected correlations). During training, the contribution of sequences to can be re-weighted following a re-weighting scheme we have conceived (see STAR Methods section “Sequence re-weighting scheme”) to correct for amino-acid frequency biases in the training dataset, as the ones introduced by mass spectrometry (MS) detection. A regularization, i.e. a penalty term over the weight parameters is introduced to prevent overfitting. The function maximized during training then becomes:
| (Equation 5) |
This regularization, which was first introduced in (Tubiana et al., 2019), plays effectively the role of a regularization, imposing sparsity of weights, with a strength that is adapted to increasing magnitude of weights, hence favoring homogeneity among hidden units (see Tubiana et al., 2019 for more detailed explanations). Examples of inferred sets of weights at different regularization strength are provided in Figures S2B and S3G. The package used for training, evaluating and visualizing RBMs is an updated version of the one described in (Tubiana et al., 2019). The package was ported to Python3 and the optimization algorithm was changed from SGD to RMSprop (i.e., to ADAM without momentum) with learning rate , , , ) (see Kingma and Ba, 2015 for definition of the parameters). The parameter values were chosen for their robust learning performance across a variety of datasets studied in previous works such as MNIST, Ising models, and MSAs of protein domains of various sizes. Overall, the adaptive learning rate of RMSprop/ADAM result in larger updates for the fields and weights attached to rare amino acids, and hence speeds up convergence.
The (hyper-)parametric search for optimal regularization () and number of hidden units () was made from the trend of the RBM log-likelihood on a held-out validation dataset, the aim being to achieve a good fit but to avoid overfitting. Figures S3A and S3B illustrate such (hyper-)parametric search for a multi-allelic RBM. Low regularizations achieve a better fit of training data (see log-likelihood values for the training dataset); when selecting a low regularization (as ), the log-likelihood over the validation dataset starts to decrease beyond , indicating overfitting. Given these trends, we trained the multi-allelic RBM models (Figures 2C–2E) with and . This choice is further supported by considering, in Figure S3C, the accuracy of HLA classification (see below for its definition), which reaches an optimal value for and . The same test with an allele-specific RBM (Figure S3F) shows that already beyond the model could overfit, hence allele-specific RBM presentation models (Figures 1C–1G) were trained with and .
HLA-I Classifier
The HLA-I classifier part of the RBM-MHC takes as input of the peptide sequence and gives a categorical output, i.e. the peptide HLA-I specificity (Figures 2A and S2A). denotes the HLA-I type. Typically for single-individual samples , since each individual displays at most 2 different copies (a maternal and a paternal copy) of HLA-A, HLA-B and HLA-C.
The classifier is trained by minimizing a loss function chosen to be a categorical cross-entropy :
| (Equation 6) |
where the sum runs only over the sequences labeled with their HLA-I association. is the label assigned to for supervised training in one-hot encoding, i.e. only for the c standing for its associated HLA type and zero otherwise so (it is normalized over categories). The choice of one-hot encoding is justified by the fact that, for the sake of discriminating motifs, we select peptides associated to only 1 HLA type in the database (mainly from mono-allelic sources, see STAR Methods section “Dataset collection from IEDB and preparation”). is the categorical output predicted by the HLA-I classifier for , calculated from the softmax activation function:
| (Equation 7) |
where are the classifier weights, connecting input to output layer (see Figure S2A), and are local biases adjusted during training. Element-wise softmax is defined as:
| (Equation 8) |
The activation function softmax has the advantage of giving predictions normalized over categories, thus each element can be interpreted as the probability that sequence belongs to class c. Our numerical implementation relies on Theano and Keras Python libraries. Training is performed in minibatches of 64 sequences, by the ADAM optimizer for 1000 epochs, retaining the model that gives the best accuracy on a held-out partition of the ~30% of the training dataset. The choice of RBM (hyper-)parameters (see above) also ensures a high accuracy of classification (Figure S3C). Accuracy of classification is measured there as an area under the curve (AUC), which is different from the cross-entropy optimized during training (Equation 6) and is defined below, in the section “RBM-MHC performance in multi-allelic samples”. This AUC value is only minimally affected when reducing the RBM training dataset and is close to 1 (indicating the maximal accuracy) already with the small number of labeled sequences used, i.e. 10% of data. (The AUC clearly increases further when increasing this amount, see Figures S3D–S3E)
Combining the probability functions estimated by RBM and HLA-I classifier, we define for each sequence a global score :
| (Equation 9) |
where is the RBM log-likelihood assigned to sequence and is the classifier score, defined from the vector of predicted class probabilities as . is the negative entropy of classification, so it contributes to with higher values when the confidence with which a HLA-I class is predicted is higher.
We shall stress here that we do not attempt to optimize log-likelihood given by the global score Equation 9, as this strategy has the risk of introducing biases in the representation learnt by the RBM in a fully unsupervised way - backpropagating the classifier gradient to the RBM training would have the effect of adapting its hidden-space representation to the prediction of the HLA type. Rather, it is precisely learning the classifier on top of the cluster structure discovered in an unsupervised way in the RBM representation space that ensures robust HLA type prediction in the scenario we envision, i.e., when we have very few labels at our disposal to characterize new datasets. In addition, optimizing Equation 9 could potentially also imply a loss of the feature discovery power of a fully unsupervised approach, as data features not directly contributing to the HLA specificity would not be internally represented.
Unsupervised Clustering
K-Means Algorithm
Given a set of points , K-means (Lloyd, 1982; Forgy, 1965) finds the centroids of clusters and assigns each to the cluster whose centroid has the minimal distance to . If we indicate by the distance between point and a cluster c, we can express the probability that belongs to cluster c as:
| (Equation 10) |
as is a common choice in the “soft” version of K-means. We define RBM-km as the application of K-means to sequence representations in the space of inputs to hidden units (i.e. ) instead of sequences themselves (i.e. ). From such a probabilistic clustering prediction, we define the classification score . Our implementation of K-means relies on the routine available in the Python package Scikit-learn (Pedregosa et al., 2011).
MixMHCp Algorithm
Consistently with the other approaches discussed, we assume that the expected number of clusters is known, and we implement the clustering by MixMHCp2.1 (Bassani-Sternberg and Gfeller, 2016; Bassani-Sternberg et al., 2017; Gfeller et al., 2018). First we build Position Weight Matrices (PWM) for each of the clusters found by MixMHCp; each PWM describes the single-site amino acid frequencies in cluster c, with . For a sequence a set of presentation scores (one per cluster) can be defined from the log-likelihood under the corresponding PWM:
| (Equation 11) |
where the superscript stands for “MixMHCp” and L is the length to which all sequences are reduced in MixMHCp (). The final score of a sequence is taken as the maximal log-likelihood among the clusters, , which is used jointly to predict the HLA association.
Annotation of HLA-I Binding Motifs
The first step in clustering approaches, either by MixMHCp or K-means, is fully unsupervised and consists of optimally assigning peptides to clusters. Clusters then need to be annotated with the corresponding HLA-binding specificity, among the ones known to be expressed in the sample cells from e.g. HLA typing. For this second step, we consider the fraction of data for which we have labels (the same used for training the HLA-I classifier) and we estimate from them a PWM for each HLA type, in such a way as to obtain a set of reference motifs. We next estimate the PWM of each cluster and we label the cluster with the HLA association of the reference motif that minimizes the difference to the cluster PWM. Note that this mapping could give the same HLA type associated to several clusters and other HLA types not associated to any cluster, indicating a poor classification performance.
Generation of HLA-Specific Artificial Peptides
A RBM is a generative model. As it is based on fitting an entire probability distribution to a given dataset, sampling from this distribution allows one to generate new candidate antigens. The binding specificity to HLA-I of such generated sequences can be controlled by conditioning (fixing) the RBM probability on the values of inputs to hidden units coding for the desired specificity. The search for these values is guided by the HLA-I classifier. This procedure directly builds on the idea of sampling while conditioning on structural, functional, phylogenetic features emerging in RBM representations of protein families (Tubiana et al., 2019). Schematically, the steps of this HLA-specific sampling are as follows:
-
1.
we select a HLA-I class c (e.g. c = HLA-A∗01:01) and we find such that is close to 1 (that is, there is high confidence of class c prediction);
-
2.
we estimate from the conditional probability and we sample new sequences from the probability of observed sequences conditioned on , ;
-
3.
to further explore the region encoding for the specificity to HLA-I class c, we randomly move from to (s drawn from a Gaussian );
-
4.
we accept the move with a probability , where and . The parameter β (akin to an inverse temperature in physics) basically controls how stringent the selection for sequences predicted in class c with a high probability is.
-
5.
We set to proceed with new moves as in step 3. Every arbitrary amount of moves, we generate new configurations by conditional sampling as in step 2.
Figure S2C shows examples of values (2 of its components for simplicity) covered in this search with and (dark red points). By this sampling procedure, we generate samples of artificial peptides that would be predicted to be presentable and specifically presented by the selected HLA-I protein complex. Such samples explore a broad diversity of peptides, that are typically 3–4 mutations away from the closest natural ones (Figure S2D) while preserving the profile of amino acid abundances constrained by a given binding specificity (Figure S2E).
Sequence Re-weighting Scheme
RBM presentation models trained on datasets obtained by mass spectrometry (MS) might underestimate the probability of presentation of ligands that are less frequently detected by MS. The most evident case are ligands containing cysteine, an amino acid that can undergo chemical modifications by oxidation typically not included in standard MS searches. We introduced a procedure for re-weighting sequences as a general strategy to correct for biases in the training dataset, such as MS detection biases. We defined a weight for each sequence in the training set:
| (Equation 12) |
where denotes typical amino acid frequencies in MS (as can be estimated from IEDB data) while is a background amino acid frequency distribution that does not suffer from the same biases as the MS one. These weights are incorporated into the average log-likelihood to maximize during RBM training:
| (Equation 13) |
When modeling multiple specificities through mixtures of PWMs (as by MixMHCp and MixMHCpred, see Bassani-Sternberg et al., 2017), an analogous correction can be implemented by rescaling the amino acid frequency composing each PWM by the background frequency. Following Ref. (Bassani-Sternberg et al., 2017), can be chosen as the frequency of amino acids in the human proteome or as the frequency of amino acids in IEDB antigens detected by other techniques than MS (we shall refer to these as ). More precisely, is estimated from frequencies at non-anchor sites (position 4 to position 7 of 9-mers), excluding from the search all alleles that: (i) have fewer than 100 peptides associated to them; (ii) are present in neither MS nor non-MS datasets; (iii) show specificity at positions between 4 and 7.
In Figure S4A, the comparison of and for the 20 amino acids provides a clear indication of the MS detection bias in relation to the amino acids cysteine (C) and tryptophan (W), the frequency difference between MS detection and other techniques being more than 100% of MS frequency itself. The C/W frequency in presented antigens is therefore expected to be underestimated by MS, suggesting that ligands containing C/W, whose binding affinity to HLA-I could be successfully tested in vitro, would be missed by MS. For the purpose of illustration, we test how the re-weighting procedure could correct for such bias on a presentation model built from IEDB, MS data for the allele HLA-A∗01:01, with . We first compute the weights α (Equation 12) for sequences in the training dataset. In Figure S4B, we show separately their distribution for sequences with and without C/W: for the former, α have particularly high values, in such a way as to weight more their statistical information and to compensate for the underestimation of C/W frequencies in MS. The effect of re-weighting can be then assessed on a validation dataset, in particular on its sequences with C/W, looking at the distribution of their presentation scores relative to the average scores over the full validation set (see Figure S4C). When considering a RBM model trained without re-weighting (giving the probability ), these sequences would be assigned a score lower than the average score of the validation set, as a straight consequence of the low amount of ligands with C/W in the training set. Once the RBM model is learned with re-weighting (giving the probability ), the score assigned to these sequences is brought closer to the average value and in particular to overlap with the range of presentation score values that would be assigned by a RBM model () trained on IEDB data for the same HLA allele from techniques different from MS (i.e. data that do not underestimate the occurrence of C/W in presented antigens). The re-weighting scheme can also improve the ability of the RBM model to discriminate antigens containing C/W of validated immunogenicity (Rufer et al., 1998; Amir et al., 2011) from generic peptides, by assigning them presentation scores of higher rank. We show this in Figure S4D, where we consider a re-weighted version of the HLA-A∗02:01-specific model of Figures 1C and 1E. While there (as well as in the analysis of Figures 1D and 2B–2E) the re-weighting was not necessary, as we validated the model on the same type of data (MS-detected) as the training set (MS-based), in Figure S4D we show that the re-weighting is useful for scoring by a MS-based model peptides from binding assays and that contain amino acids underdetected by MS. Similarly, another scenario where we applied the re-weighting scheme is the prediction, by MS models, of SARS-CoV-2 epitopes tested in vitro for binding, as in Figure 1F. The re-weighting procedure just described can be activated as an option in the RBM-MHC software package.
Quantification and Statistical Analysis
Model Predictions in Mono-Allelic MS Datasets
To assign scores of presentation by a specific HLA-I, we resorted simply to the RBM to build HLA-specific presentation models (with , ). To train them, sets of 8–11-mer peptides documented with the corresponding HLA association were collected from IEDB, restricting to mono-allelic-source, mass spectrometry sequence data, as outlined in the section “Dataset collection from IEDB and preparation”. We then used the RBM log-likelihood as probabilistic score of presentation by the HLA under consideration. For the preliminary validity check of the HLA-A∗02:01-specific model, we randomly selected a subset with the 80% of these sequences as training dataset and we kept the remaining 20% as test set to evaluate the model’s predictions in terms of probabilistic scores of presentation (Figure 1C). We randomly selected 5000 peptides from the human proteome (as in UniProt database; UnitProt Consortium, 2019) with length distribution matching the one of presented peptides to serve as a set of peptides predominantly not presented on the cell surface (“generic”).
To test our method on mono-allelic MS datasets from Ref. (Sarkizova et al., 2020), we trained 92 allele-specific RBM models, for all the A, B, C HLAs analyzed in (Sarkizova et al., 2020). We decided to train a series of allele-specific models to minimize the uneven accuracy across alleles, emerging especially in multi-allelic models, due to the different abundances of peptides with different HLA-I preferences available as training data. (The use of multi-allelic models is intended as tool to characterize unannotated samples through motif reconstruction). For each model, we randomly selected in the corresponding mono-allelic dataset 100 peptides to use for model evaluation. Since datasets from (Sarkizova et al., 2020) feature in the latest version of IEDB, we carefully excluded the 100 peptides per model from the IEDB-derived training datasets. We produced, as described above, n-fold excess of generic peptides, choosing n in such a way as to consider a proportion between presented and generic peptides that resembles natural conditions of epitope selection. Large-scale experimental and bioinformatic studies on viral peptidomes (Yewdell, 2006; Paul et al., 2013; Vitiello and Zanetti, 2017) found that ~1% of peptides bind to given MHCs (suggesting the value n=99). Comparing the amount of all 9-long peptides from human proteome (~107) and the average, allele-specific antigen repertoire size (~104 sequences) derived from existing databases as IEDB, Refs. (Abelin et al., 2017; Sarkizova et al., 2020) concluded rather that ~0.1% of peptides is “presentable” (corresponding to n=999). These approximate estimates allowed us to fix a threshold for positive prediction at, respectively, the top-scoring 1% and 0.1% of peptides in order to evaluate model performance by the PPV metric. (The random expectation for the PPV in these conditions is 10−2 and 10−3 for the PPV estimated respectively at the top scoring 1% and 0.1%). AUCs do not vary, apart from noise, with n; the AUC values shown in Figures 1D and S5 are the ones obtained with n=99 (i.e., generic and presented peptides mixed in proportion 100:1). To better rationalize differences between RBM and NetMHCpan4.0 in connection to different HLAs, we monitored performance as a function of the distance between the query HLA and its nearest neighbor in the NetMHCpan4.0 training dataset, where such distance is determined from the similarity between the two HLA-I sequences (more precisely, the 34 residues in contact with the peptide). The distance between sequences X and Y is defined as , where e.g. is a global alignment score between X and Y based on the BLOSUM50 substitution matrix, see (Nielsen et al., 2007; Hoof et al., 2009).
MS-Based Model Validation for Neoantigen Discovery
To evaluate the predictive power of the HLA-A∗02:01-specific RBM model for neoantigen discovery, we acquired the list of missense mutations for ovarian and melanoma cancer cell lines SKOV3, A2780, OV90, HeLa and A375 from the Cosmic Cell Lines Project (Forbes et al., 2016). We further filtered them largely following the steps described in Ref. (Marty et al., 2017), i.e. we retained only mutations in genes with non-zero expression levels in the corresponding cell line (based on expression data from the Genomics of Drug Sensitivity Center; Yang et al., 2013) and we excluded common germline variants documented in the Single Nucleotide Polymorphism Database (Sherry et al., 2001).
As a result, the number of missense mutations considered is as follows: 444 (A375), 521 (SKOV3), 602 (A2780), 280 (OV90), 511 (HeLa). For each cell line, the list of mutated peptides to score results from adding up all the up to 38 peptides of length 8–11 containing each mutation. Since the MS database for the 5 cancer lines produced by Ref. (Marty et al., 2017) was annotated by comparison to the consensus human proteome, it contained only wildtype peptides whose mutated version was in the list of putative neoantigens. As long as a mutation does not affect an anchor site, the mutated version should preserve a high probability of presentation. We followed the authors’ choice of not considering the two peptides mutated at anchor positions as presented and we excluded them from the analysis of Figure 1E. When we consider all neoantigens (including the two neoantigens arising from anchor-site mutations of MS-derived peptides) the mean score percentiles are: RBM-MHC 3.3% vs NetMHCpan4.1 1.6% (among generic peptides) and RBM-MHC 3.6% vs NetMHCpan4.1 0.9% (among all mutated peptides from the same cell line).
The studies from which single-patient samples 12T, Mel8, Mel15, Mel5 were retrieved (Kalaora et al., 2016, 2018; Bassani-Sternberg et al., 2016) listed a total of 11 tumor-presented neoantigens, using techniques that allow for the detection of variants of native proteins by MS. For some of these neoantigens, immunogenicity was also validated in vitro by identification of neoantigen-specific T-cell responses. Their HLA association was predicted by NetMHC software, see (Kalaora et al., 2016, 2018; Bassani-Sternberg et al., 2016). The prediction was to a large extent confirmed by in vitro validation of immunogenicity, which relied on antigen presenting cells that were positive to the predicted HLA-I. As the 11 neoantigens are 9–10 residues long, following Ref. (Bassani-Sternberg et al., 2017) we considered 9-mer and 10-mer peptides overlapping the missense mutations observed in the patient’s WES, which resulted in a list of thousands of putative neoantigens (see Table S3). The RBM-MHC training set per se can thus take into account only 9–10 mers from patients’ samples; patients’ neoantigens to validate were not included in the training dataset. In this multi-allelic case where antigens may be presented by several HLA types (as opposed to the allele-specific case of the previous paragraph) we used the global score accounting for the probability of presentation (by RBM) as well as the confidence of the HLA assignment (either by the HLA-I classifier in RBM-MHC or by K-means clustering in RBM-km) of Equation 9.
Model Predictions for SARS-CoV-2 Epitopes
We downloaded the protein-coding regions of SARS-CoV-2 genome from GenBank: NC_045512.2 (Benson et al., 2013). We extracted all the 9-mers contained in the SARS-CoV-2 proteome, giving a list of 9656 candidate cytotoxic T-cell epitopes (HLA-I antigens). Ref. (Immunitrack and Intavis, 2020) (available at https://www.immunitrack.com/free-coronavirus-report-for-download/) tested in vitro the 94 top scoring epitopes according to NetMHC4.0 (Andreatta and Nielsen, 2016) (a well-known NetMHC version for binding affinity predictions) for each of the HLA-I alleles A∗01:01, A∗02:01, A∗03:01, A∗11:01, A∗24:02, B∗40:01, C∗04:01, C∗07:01, C∗07:02 and according to NetMHCpan4.0 (run with EL option) (Jurtz et al., 2017) for C∗01:02. 159 peptides were identified as high-stability binders (i.e., with stability above the threshold of 60% of the reference peptide value for the corresponding HLA-I allele). Ref. (Immunitrack and Intavis, 2020) also estimated the trend of binding stability (expressed as % of the reference value) vs predicted score (binding affinity in [nM] for NetMHC4.0, rank percentile for NetMHCpan4.0). Here we report and quantify these trends in terms of Pearson correlation for the sake of comparison to RBM (Figures 1F and S7B). To probe our method as a predictor for the high-stability binders found, we learned a series of allele-specific RBM presentation models ( hidden units, ) for the 10 HLA-I alleles considered in this study, prioritizing training datasets from binding assays in IEDB (see STAR Methods section “Dataset collection from IEDB and preparation”). These types of data are almost absent for the 4 HLA-C mentioned, and in these cases models were learned from MS data only but a re-weighting of frequencies, aimed at correcting for MS biases in detection, was applied (see STAR Methods section “Sequence re-weighting scheme”). Final BA datasets used have in average 1269 sequences, MS ones 1678. All peptides tested for binding to a given HLA-I in Ref. (Immunitrack and Intavis, 2020) were scored by the corresponding RBM model (see Figure 1F, where, as a comparison, results achieved by training all RBMs on MS data with re-weighting are also reported). We next estimated their score percentile relative to scores assigned to all candidate epitopes and we assessed that tested binders were predominantly assigned high scores, able to a good extent to discriminate them from the tested non-binders (see Figure S7A).
As an additional test, we considered the SARS-CoV-2 cytotoxic T-cell epitopes identified as potentially associated to high immune responses by Ref. (Grifoni et al., 2020), who mapped the dominant, experimentally validated SARS-CoV-derived epitopes to the corresponding regions in the SARS-CoV-2 proteome. We scored the 22 epitopes in this list with complete (100%) or moderate-high (> 70%) sequence similarity to the homologous SARS-CoV epitope, which should preserve high likelihood of presentation. Since homologous SARS-CoV epitopes were experimentally tested in binding assays, scores were assigned by the same (affinity-trained) models as above covering the HLA restrictions reported in Ref. (Grifoni et al., 2020) (HLA-A∗02:01 to the largest extent, HLA-A∗24:02, HLA-B∗40:01), see Figures 1G and S7D. In addition, we checked that scores estimated by MS-trained RBM models with re-weighting lie in the same range, see Figure S7E.
RBM-MHC Performance in Multi-allelic Samples
We chose the Area Under the Curve (AUC) of the Receiving Operating Characteristic (ROC) as a metric for classification (i.e., HLA assignment) performance, estimated for each approach as follows. RBM-MHC, through the HLA-I classifier, outputs for each peptide a probability to belong to each HLA-I class. MixMHCp2.1 (Bassani-Sternberg and Gfeller, 2016; Bassani-Sternberg et al., 2017; Gfeller et al., 2018) models probabilistically data by a mixture of independent models, thus it describes each sequence by a vector of “responsibilities”, describing the probabilities to belong to each cluster (i.e., HLA-I class). NetMHCpan4.1 (Reynisson et al., 2020) predicts peptide binding values from either the training on eluted data (EL option) or the binding affinity data (BA option) and it estimates from these values presentation scores and percentile rank scores. Low values of percentile rank define binders (following the authors’ recommendations, peptides with percentile rank ¡2% and ¡0.5% are considered HLA-I weak and strong binders respectively). Having built the samples from MS data, we considered NetMHC predictions from the EL option (NetMHCpan4.1-EL). We compare the HLA-I classes by pairs. We consider the probability of belonging to a certain class that each method would assign to “positives” of that class (peptides binding to the respective HLA-I allele) and “negatives” (peptides binding to other alleles): when the classification performance is good, the former has values close to one, the latter has values close to zero. Varying the threshold between false positive and false negative distributions, we build the ROC curve. We take the area under the ROC curve (AUC) as measure of the ability to discriminate the two classes (AUC = 1 means perfect discrimination, AUC = 0.5 means chance). To obtain one cumulative indicator, we average over the AUCs of these pairwise comparisons. For the approaches partially or fully supervised (RBM-MHC and classifier-only), the AUC is measured only from data not in the 10% used for the supervised learning step. To further assess classification performance, we looked also at the HLA type of each peptide predicted based on: the highest responsibility value among the values for MixMHCp2.1; the lowest percentile rank for NetMHCpan4.1-EL; the highest class probability estimated by the HLA-I classifier for RBM-MHC. In this way, when comparing peptides of different classes two by two, we can count true positives of classification (), false positives (), true negatives () and false negatives (). Once these quantities are defined, additional classification performance indicators are: accuracy = , Precision = , Specificity = , Sensitivity = . MixMHCp2.1 was run using default options, which include an additional “trash cluster”. When measuring these classification performance indicators, the assignment to the trash cluster is considered among the false negatives. The comparison of classification performance as measured by these indicators is shown in Figures S8B–S8I.
Acknowledgments
We thank Benjamin Greenbaum for suggesting to look at SARS-CoV-2 and Clément Roussel, Andrea Di Gioacchino, Carlos Olivarez, Hannah Carter, David Gfeller, Shelly Kalaora, Yardena Samuels, David Hoyos, Jayon Lihm, and Anna Paola Muntoni for helpful exchanges. This work was partially supported by the European Research Council consolidator grant number 724208, the European Research Council Marie Curie-Sklodowska ITN QuanTI, the ANR-17 RBMPro and ANR-19 Decrypted CE30-0021-01, the ANR (Agence Nationale de la Recherche) Flash Covid 19 - FRM, PROJET “SARS-Cov-2immunRNAs” grants. J.T. was supported by a postdoctoral fellowship from the Human Frontier Science Program Organization (reference number: LT001058/2019). This material is based upon work supported under a collaboration by Stand Up to Cancer, a program of the Entertainment Industry Foundation, the Society for Immunotherapy of Cancer, and the Lustgarten Foundation.
Author Contributions
Conceptualization, B.B., S.C., R.M., T.M., and A.M.W.; Methodology, B.B., S.C., R.M., T.M., and A.M.W.; Formal Analysis, B.B., S.C., R.M., T.M., and A.M.W.; Investigation, B.B., S.C., R.M., T.M., and A.M.W.; Software, B.B., J.T., S.C., R.M., T.M., and A.M.W.; Writing – Original Draft, B.B., S.C., R.M., T.M., and A.M.W.; Writing – Review & Editing, B.B., S.C., R.M., T.M., and A.M.W.
Declaration of Interests
The authors declare no competing interests.
Published: December 17, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2020.11.005.
Contributor Information
Barbara Bravi, Email: bbravi.bb@gmail.com.
Simona Cocco, Email: simona.cocco@phys.ens.fr.
Rémi Monasson, Email: remi.monasson@phys.ens.fr.
Thierry Mora, Email: thierry.mora@phys.ens.fr.
Aleksandra M. Walczak, Email: aleksandra.walczak@phys.ens.fr.
Supplemental Information
References
- Abelin J.G., Keskin D.B., Sarkizova S., Hartigan C.R., Zhang W., Sidney J., Stevens J., Lane W., Zhang G.L., Eisenhaure T.M. Mass spectrometry profiling of HLA-associated peptidomes in Mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir A.L., van der Steen D.M., Hagedoorn R.S., Kester M.G.D., van Bergen C.A.M., Drijfhout J.W., de Ru A.H., Falkenburg J.H.F., van Veelen P.A., Heemskerk M.H.M. Allo-HLA–reactive T cells inducing graft-versus-host disease are single peptide specific. Blood. 2011;118:6733–6742. doi: 10.1182/blood-2011-05-354787. [DOI] [PubMed] [Google Scholar]
- Andreatta M., Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M., Bräunlein E., Klar R., Engleitner T., Sinitcyn P., Audehm S., Straub M., Weber J., Slotta-Huspenina J., Specht K. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016;7:13404. doi: 10.1038/ncomms13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M., Chong C., Guillaume P., Solleder M., Pak H., Gannon P.O., Kandalaft L.E., Coukos G., Gfeller D. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput. Biol. 2017;13:e1005725. doi: 10.1371/journal.pcbi.1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M., Gfeller D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide–HLA interactions. J. Immunol. 2016;197:2492–2499. doi: 10.4049/jimmunol.1600808. [DOI] [PubMed] [Google Scholar]
- Bassani-Sternberg M., Pletscher-Frankild S., Jensen L.J., Mann M. Mass spectrometry of human leukocyte antigen Class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics. 2015;14:658–673. doi: 10.1074/mcp.M114.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UnitProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes S.A., Beare D., Bindal N., Bamford S., Ward S., Cole C.G., Jia M., Kok C., Boutselakis H., De T. COSMIC: high-resolution cancer genetics using the catalogue of somatic mutations in cancer. Curr. Protoc. Hum. Genet. 2016;91:10.11.1–10.11.37. doi: 10.1002/cphg.21. [DOI] [PubMed] [Google Scholar]
- Forgy E.W. Cluster analysis of multivariate data: efficiency versus interpretability of classifications. Biometrics. 1965;21:768–769. [Google Scholar]
- Garcia-Garijo A., Fajardo C.A., Gros A. Determinants for neoantigen identification. Front. Immunol. 2019;10:1392. doi: 10.3389/fimmu.2019.01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gfeller D., Guillaume P., Michaux J., Pak H.S., Daniel R.T., Racle J., Coukos G., Bassani-Sternberg M. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol. 2018;201:3705–3716. doi: 10.4049/jimmunol.1800914. [DOI] [PubMed] [Google Scholar]
- González-Galarza F.F., Takeshita L.Y., Santos E.J., Kempson F., Maia M.H.T., da Silva A.L.S., Teles e Silva A.L.T., Ghattaoraya G.S., Alfirevic A., Jones A.R. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015;43:D784–D788. doi: 10.1093/nar/gku1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Sidney J., Zhang Y., Scheuermann R.H., Peters B., Sette A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020;27:671–680.e2. doi: 10.1016/j.chom.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton G.E. Training products of experts by minimizing contrastive divergence. Neural Comput. 2002;14:1771–1800. doi: 10.1162/089976602760128018. [DOI] [PubMed] [Google Scholar]
- Hoof I., Peters B.P., Sidney J., Pedersen L.E., Sette A., Lund O., Buus S., Nielsen M. NetMHCpan, a Method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immunitrack, Intavis Covid19 Intavis_Immunitrack stability dataset. 2020. https://www.immunitrack.com/wp/wp-content/uploads/Covid19-Intavis-Immunitrack-datasetV2.xlsx
- Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: improved peptide–MHC Class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaora S., Barnea E., Merhavi-Shoham E., Qutob N., Teer J.K., Shimony N., Schachter J., Rosenberg S.A., Besser M.J., Admon A. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget. 2016;7:5110–5117. doi: 10.18632/oncotarget.6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaora S., Wolf Y., Feferman T., Barnea E., Greenstein E., Reshef D., Tirosh I., Reuben A., Patkar S., Levy R. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in melanoma. Cancer Discov. 2018;8:1366–1375. doi: 10.1158/2159-8290.CD-17-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. software version 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma, D.P., and Ba, L.J. (2015). Adam: A method for stochastic optimization, International Conference on Learning Representations (ICLR).
- Lloyd S.P. Least squares quantization in PCM. IEEE Trans. Inform. Theory. 1982;28:129–137. [Google Scholar]
- Marty R., Kaabinejadian S., Rossell D., Slifker M.J., van de Haar J., Engin H.B., de Prisco N., Ideker T., Hildebrand W.H., Font-Burgada J. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171:1272–1283.e15. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S., Li F., Leier A., Marquez-Lago T.T., Giam K., Croft N.P., Akutsu T., Smith A.I., Li J., Rossjohn J. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief. Bioinform. 2020;21:1119–1135. doi: 10.1093/bib/bbz051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Lundegaard C., Blicher T., Lamberth K., Harndahl M., Justesen S., Røder G., Peters B., Sette A., Lund O. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell T.J., Rubinsteyn A., Bonsack M., Riemer A.B., Laserson U., Hammerbacher J. MHCflurry: open-source Class I MHC binding affinity prediction. Cell Syst. 2018;7:129–132.e4. doi: 10.1016/j.cels.2018.05.014. [DOI] [PubMed] [Google Scholar]
- Paul S., Weiskopf D., Angelo M.A., Sidney J., Peters B., Sette A. HLA Class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J. Immunol. 2013;191:5831–5839. doi: 10.4049/jimmunol.1302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufer N., Wolpert E., Helg C., Tiercy J.M., Gratwohl A., Chapuis B., Jeannet M., Goulmy E., Roosnek E. HA-1 and the SMCY-derived peptide FIDSYICQV (H-Y) are immunodominant minor histocompatibility antigens after bone marrow transplantation. Transplantation. 1998;66:910–916. doi: 10.1097/00007890-199810150-00016. [DOI] [PubMed] [Google Scholar]
- Russ W.P., Figliuzzi M., Stocker C., Barrat-Charlaix P., Socolich M., Kast P., Hilvert D., Monasson R., Cocco S., Weigt M. An evolution-based model for designing chorismate mutase enzymes. Science. 2020;369:440–445. doi: 10.1126/science.aba3304. [DOI] [PubMed] [Google Scholar]
- Sarkizova S., Klaeger S., Le P.M., Li L.W., Oliveira G., Keshishian H., Hartigan C.R., Zhang W., Braun D.A., Ligon K.L. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020;38:199–209. doi: 10.1038/s41587-019-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehnal, D., Rose, A.S., Koča, J., Burley, S.K., and Velankar, S. (2018). Mol∗: Towards a common library and tools for web molecular graphics. Proceedings of the workshop on molecular graphics and visual analysis of molecular data, MolVA ’18, pp. 29–33.
- Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. DbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidney J., Peters B., Frahm N., Brander C., Sette A. HLA class I supertypes: A revised and updated classification. BMC Immunol. 2008;9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolensky P. Information processing in dynamical systems: foundations of harmony theory. In: Rumelhart D.E., McLelland J.L., editors. Vol. 1. MIT Press; 1986. pp. 194–281. (Parallel distributed processing: explorations in the microstructure of cognition: Foundations). [Google Scholar]
- Trolle T., McMurtrey C.P., Sidney J., Bardet W., Osborn S.C., Kaever T., Sette A., Hildebrand W.H., Nielsen M., Peters B. The length distribution of class I–restricted T cell epitopes is determined by both peptide supply and MHC allele–specific binding preference. J. Immunol. 2016;196:1480–1487. doi: 10.4049/jimmunol.1501721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubiana J., Cocco S., Monasson R. Learning protein constitutive motifs from sequence data. eLife. 2019;8:e39397. doi: 10.7554/eLife.39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubiana J., Monasson R. Emergence of compositional representations in restricted Boltzmann machines. Phys. Rev. Lett. 2017;118:138301. doi: 10.1103/PhysRevLett.118.138301. [DOI] [PubMed] [Google Scholar]
- Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello A., Zanetti M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 2017;35:815–817. doi: 10.1038/nbt.3932. [DOI] [PubMed] [Google Scholar]
- Yang W., Soares J., Greninger P., Edelman E.J., Lightfoot H., Forbes S., Bindal N., Beare D., Smith J.A., Thompson I.R. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M., Johnson B.A., Lutz E.R., Laheru D.A., Jaffee E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer. 2017;17:209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell J.W. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity. 2006;25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. These datasets’ sources are provided in the Key Resources Table.
RBM-MHC original code and trained models are publicly available at https://github.com/bravib/rbm-mhc.
The scripts used to generate the figures reported in this paper are available at https://github.com/bravib/rbm-mhc.
Any additional information required to reproduce this work is available from the Lead Contact.