

Abstract
Traumatic brain injury (TBI) can lead to significant neuropsychiatric problems and neurodegenerative pathologies, which develop and persist years after injury. Neuroinflammatory processes evolve over this same period. Therefore, we aimed to determine the contribution of microglia to neuropathology at acute [1 d postinjury (dpi)], subacute (7 dpi), and chronic (30 dpi) time points. Microglia were depleted with PLX5622, a CSF1R antagonist, before midline fluid percussion injury (FPI) in male mice and cortical neuropathology/inflammation was assessed using a neuropathology mRNA panel. Gene expression associated with inflammation and neuropathology were robustly increased acutely after injury (1 dpi) and the majority of this expression was microglia independent. At 7 and 30 dpi, however, microglial depletion reversed TBI-related expression of genes associated with inflammation, interferon signaling, and neuropathology. Myriad suppressed genes at subacute and chronic endpoints were attributed to neurons. To understand the relationship between microglia, neurons, and other glia, single-cell RNA sequencing was completed 7 dpi, a critical time point in the evolution from acute to chronic pathogenesis. Cortical microglia exhibited distinct TBI-associated clustering with increased type-1 interferon and neurodegenerative/damage-related genes. In cortical neurons, genes associated with dopamine signaling, long-term potentiation, calcium signaling, and synaptogenesis were suppressed. Microglial depletion reversed the majority of these neuronal alterations. Furthermore, there was reduced cortical dendritic complexity 7 dpi, reduced neuronal connectively 30 dpi, and cognitive impairment 30 dpi. All of these TBI-associated functional and behavioral impairments were prevented by microglial depletion. Collectively, these studies indicate that microglia promote persistent neuropathology and long-term functional impairments in neuronal homeostasis after TBI.
SIGNIFICANCE STATEMENT Millions of traumatic brain injuries (TBIs) occur in the United States alone each year. Survivors face elevated rates of cognitive and psychiatric complications long after the inciting injury. Recent studies of human brain injury link chronic neuroinflammation to adverse neurologic outcomes, suggesting that evolving inflammatory processes may be an opportunity for intervention. Here, we eliminate microglia to compare the effects of diffuse TBI on neurons in the presence and absence of microglia and microglia-mediated inflammation. In the absence of microglia, neurons do not undergo TBI-induced changes in gene transcription or structure. Microglial elimination prevented TBI-induced cognitive changes 30 d postinjury (dpi). Therefore, microglia have a critical role in disrupting neuronal homeostasis after TBI, particularly at subacute and chronic timepoints.
Keywords: CSF1R antagonist, microglia, neuroinflammation, neurotrauma, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a leading cause of neurologic and neuropsychiatric disability in the United States (Gualtieri and Cox, 1991; Millis et al., 2001; Silver et al., 2009). For instance, TBI patients are 5–10 times more likely to develop symptoms of depression compared with the general population (Kumar et al., 2018; Singh et al., 2018) and 30% of TBI patients show detectable cognitive decline (Himanen et al., 2006; Salmond et al., 2006; Till et al., 2008; Ramlackhansingh et al., 2011). Recent development of PET-detectable ligands that bind to activated microglia allowed for in vivo approximation of inflammation after TBI. Elevated binding of PK11195 at benzodiazepine receptors on microglia was detected in TBI patients up to 17 years postinjury and was associated with impaired cognitive processing speed (Ramlackhansingh et al., 2011). Coughlin et al. (2017) studied former National Football League players and found chronically activated microglia/macrophages evident up to 10 years after retirement and detectable before cognitive impairment, which is common in former professional football players. There are no targeted pharmacologic therapies to prevent TBI-related neuropsychiatric sequelae, however aberrant changes in microglial homeostasis persist months to years after injury in humans and may represent an opportunity for therapeutic intervention.
Chronic derangements in microglial structure and CNS inflammation are modeled in rodent models of TBI. Repeated closed-head injury in mice resulted in increased Iba1 (microglia) labeling and white matter damage that corresponded with deficits in hippocampal-dependent learning 12–18 months after injury (Mouzon et al., 2014). CD68+ microglia were detected in the lesion site one year after focal controlled cortical impact (CCI) injury (Loane et al., 2014) and increased MHCII and CD68 labeling was detected 14–30 d after diffuse brain injury in both rats (Ziebell et al., 2012) and mice (Fenn et al., 2014). Inflammatory/primed MHC-II+ microglia were evident 30 d postinjury (dpi); lipopolysaccharide (LPS) challenge 30 dpi led to exaggerated microglial production of IL-1β, impaired cognitive performance, and protracted sickness and depressive-like behaviors (Fenn et al., 2014; Muccigrosso et al., 2016). Microglia are involved in several homeostatic processes including synaptic pruning (Schafer et al., 2012), immune surveillance (Nimmerjahn et al., 2005), and debris clearance (Neumann et al., 2009); therefore, chronic disruption of microglial homeostasis may underlie neuropsychiatric complications and neurodegenerative diseases following injury.
Myriad histologic evidence suggests dynamic structural associations between neurons and microglia after TBI. Microglia associate with axons immediately following TBI in pigs (Wofford et al., 2017) and dynamically interact with neurons in a species-dependent manner (Lafrenaye et al., 2015; Gorse and Lafrenaye, 2018). In addition to process convergence, a subset of microglia undergoes a structural transition and become rod-shaped in the cortex of mice and rats following midline fluid percussion injury (FPI; Ziebell et al., 2012). Rod microglia align closely with apical dendrites of Layer V pyramidal neurons following FPI (Witcher et al., 2018). This morphology appears in other contexts, including chronic viral infection (pigs) and in normal human aging (Ji et al., 2016; Bachstetter et al., 2017). The functional significance of microglia-neuron structural interactions after TBI is unknown.
The overarching goal of this study was to determine whether microglia drive neuropathology after diffuse TBI. We previously showed that eliminating microglia via CSF1R antagonism before injury and up to 7 dpi prevented diverse immune and inflammatory gene expression after midline FPI (mFPI; Witcher et al., 2018). The aims of the current study were 2-fold: (1) to understand the role of microglia in progression of neuropathological gene expression from acute (1 dpi) and subacute (7 dpi) to chronic (30 dpi) phases of injury; and (2) to understand whether microglia mediate neuronal transcriptional, structural, and physiological responses to injury. CSF1R antagonism was used to deplete CNS microglia before injury and bulk and single-cell cortical gene expression, neuronal plasticity, electrophysiology, and cognitive function were assessed. Our resulting data indicate that microglia are the critical mediators of chronic inflammation and suppress neuronal homeostasis at transcriptional, structural, physiological, and functional levels.
Materials and Methods
Mice
Adult (8- to 10-week-old) male C57BL/6 mice were purchased from Charles River Breeding Laboratories. For neuronal tracing and dendritic spine analysis, Thy1-YFP-H mice were purchased from The Jackson Laboratory (B6.Cg-Tg-Thy1-YFP-HJrs/J; stock number 003782). Mice were housed in groups of four and kept under a 12/12 h light/dark cycle with ad libitum access to food and water. All procedures were performed in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and the Public Health Service's Policy on Humane Care and Use of Laboratory Animals and Guide for the Care use of Laboratory Animals and were approved by The Ohio State University Institutional Laboratory Animal Care and Use Committee.
PLX5622 administration
PLX5622 was provided by Plexxikon Inc. and formulated in AIN-76A rodent chow by Research Diets at a concentration of 1200 mg/kg. Standard AIN-76A diet was provided as a vehicle control. Mice were provided ad libitum access to PLX5622 or vehicle diet for 14 d to deplete microglia before TBI. Mice were maintained on the experimental diets for the duration of the study. This dose and time was validated in our lab to deplete ∼96% of microglia in C57BL/6 mice (McKim et al., 2018).
mFPI
Mice received a midline diffuse TBI using a FPI apparatus (Custom Design & Fabrication) as we previously described (Fenn et al., 2014, 2015; Rowe et al., 2016; Witcher et al., 2018). This diffuse injury occurs in the absence of a contusion or tissue cavitation and causes diffuse axonal injury in the neocortex, hippocampus, and dorsolateral thalamus (Kelley et al., 2006, 2007; Bachstetter et al., 2013; Fenn et al., 2014). In brief, a midline craniectomy was performed with a 3 mm outer diameter trephine and a rigid Luer-loc needle hub was secured over the craniectomy. After recovery, injury was induced by filling the injury hub with saline and imposing a 10-ms pulse of saline (1.2 atmospheres; 670–720 mV) onto the dura through the hub (Witgen et al., 2006; Lifshitz et al., 2007b; Fenn et al., 2014; Rowe et al., 2016). Immediately after injury, the hub was removed, dural integrity was confirmed, and mice were evaluated for injury severity using the self-righting test (Lifshitz et al., 2007a). Mice were euthanized immediately if dural integrity was compromised. Self-righting inclusion criteria were based on our previous work with C57BL/6 mice (Witcher et al., 2018). Only mice with a moderate TBI were used (200–540 s). In these studies, control mice were naive and uninjured.
Iba1, P2RY12, and GFAP immunofluorescence
Iba1 and GFAP were labeled as previously described (Witcher et al., 2018). In brief, mice were perfused with PBS followed by 4% formaldehyde. Brains were removed and postfixed for 24 h and dehydrated in 30% sucrose for 48 h. Tissue was snap-frozen in isopentane, cryosectioned at 30 µm, and sections were stored in cryoprotectant. Somatosensory cortical sections were washed in PBS and blocked (0.1% Triton X-100, 5% BSA, and 5% NDS). Next, sections were incubated overnight with primary antibodies for anti-Iba1 (rabbit anti-Iba1, 1:1000, Wako catalog #019-19471, RRID:AB_2665520), anti-P2RY12 (rabbit anti-P2RY12, 1:1000, AnaSpec, EGT Group catalog #55043A, RRID:AB_2298886), or anti-GFAP (goat anti-GFAP, 1:500, Abcam catalog #ab53554, RRID:AB_880202) diluted in 0.1% Triton X-100, 1% BSA, and 1% NDS in PBS. Next, sections were washed and incubated with an appropriate fluorochrome-conjugated secondary antibody (donkey anti-rabbit or goat; Alexa Fluor 488/594/647; Invitrogen). Sections were mounted on charged slides and cover-slipped with Fluoromount (Beckman Coulter).
Microscopy and analysis
Fluorescent labeling within the somatosensory cortex was visualized and imaged using an EVOS FL Auto 2 imaging system (Thermo Fisher Scientific). Imaging parameters were kept constant throughout each experiment. To determine percent-area of Iba1 or GFAP labeling, single-channel images were converted to eight-bit TIFF format and constant thresholds were used to quantify positively labeled pixels (ImageJ Software). To count number of microglia, images of Iba1 and P2RY12 labeling were merged and double-positive cells were counted. Values from four to six images per mouse were averaged, and these values were used to calculate group averages and variance for each injury or treatment group (P2RY12 labeling not shown; over 99% of cells were double-positive). Investigators were blinded to injury/treatment group before all microscopy and throughout image analysis.
Cortical microdissociation for NanoString
RNA was extracted from microdissected regions of cortex containing hypertrophied and rod-shaped microglia (Witcher et al., 2018). At time of killing, brains were rapidly removed and snap-frozen in dry ice-cooled isopentane. Fresh-frozen tissue was cryosectioned at alternating thicknesses of 30 µm and 200 µm onto charged slides. Thin sections, or guide slides, were labeled for CD11b (1:300, biotinylated) and visualized using an ABC solution (Vector Laboratories) and a diaminobenzidine (DAB) peroxidase substrate kit (Vector Laboratories). Labeled guide slides were used to identify regions of interest in the frozen thick sections, which were then microdissected, immediately lysed, and RNA was extracted (PicoPure RNA extraction kit, Thermo Fisher Scientific). RNA integrity was confirmed by Agilent BioAnalyzer and preamplification cycles were determined using expression of housekeeping genes (Gapdh, Actb); n = 5–9 mice per experimental group.
NanoString gene expression analysis
Gene expression was quantified using the NanoString neuropathology panel, with 30 added custom genes (NanoString Technologies). Technical normalization was first performed to positive and negative controls per manufacturer's protocols. Housekeeping genes were validated based on strong correlation with total counts, and seven were used to normalize data (Aars, Asb7, Ccdc127, Cnot10, Csnk2a2, Lars, Mto1). Data were normalized and differential expression testing was performed using DESeq2 in R (Love et al., 2014). Pathway analysis was performed using ingenuity pathway analysis (IPA; QIAGEN). Raw RCC files and normalized count matrix are available in NCBI's Gene Expression Omnibus (GEO; GSE160651); raw and normalized counts are provided in Extended Data Figure 1-2; p values below 0.05 were considered statistically significant. Genes were determined to be PLX dependent if p < 0.05 for Veh-Con versus Veh-TBI comparison but p > 0.05 (not significantly different) for Veh-Con versus PLX-TBI. If p < 0.05 for both Veh-Con versus Veh-TBI and Veh-Con versus PLX-TBI comparisons, the gene was determined to be PLX independent. Two outliers were removed from this analysis because of technical errors during the NanoString nCounter assay (one oversaturation, one undersaturation).
Cortical dissociation for single-cell sequencing
Mice were perfused with ice-cold PBS, brains were rapidly removed, and the cortex was microdissected. The cortex was dissociated using the Adult Mouse Brain Dissociation kit for mouse and rat per manufacturer's instructions with minor modifications (Miltenyi Biotec). Artificial CSF (aCSF; as described below) was used to make up enzyme solutions. Dissected cortex was immediately placed into prewarmed (37°C) Enzyme P solution, Enzyme A solution was added, and tissue was dissociated in C-tubes at 37°C using a gentleMACS dissociator (Miltenyi Biotec). Density centrifugation was used to remove myelin debris, remaining red blood cells were lysed, and cell viability and concentration was determined using a hemocytometer. Cortices from three mice per group (Veh-Con, Veh-TBI, PLX5622-Con, PLX5622-TBI) were pooled into each sample. This experiment was performed in two replicates, such that six mice were used for each experimental group. Approximately 3000–7000 cells were recovered from each cortical homogenate.
10× Genomics single-cell RNA sequencing
Single-cell cortical suspension was loaded onto a 10× chip and run on a Chromium Controller to generate gel-bead emulsions (10× Genomics). Protocol for UMI barcoding, cDNA amplification, and library construction followed the 10× Genomics Single Cell 3′ v2 reagent kit protocol. Library quality was determined by Agilent High Sensitivity DNA BioAnalyzer chip and was sequenced using an Illumina HiSeq. Sequencing reads were aligned to the mm10 mouse genome and count matrices were generated using CellRanger (10× Genomics). Raw FASTQ files and count matrices are available in the NCBI GEO (GSE160763). Low-quality cells (doublets, low counts, high counts) were filtered using Seurat in R (Butler et al., 2018). Cell types were assigned to each cluster based on significantly increased gene expression. Tmem119+ microglia, Gja1+ astrocytes, Plp1+ oligodendrocytes, and Meg3+ neurons and were extracted from the full dataset and further analyzed. Cluster-specific expression and differential expression between experimental groups were determined using Seurat (p-adj < 0.05). Extended Data Figure 3-2 contains raw output from analyses determining cluster-specific markers and differential expression between Veh-Con and Veh-TBI groups for each cell type.
To determine reversal by PLX5622, genes affected by TBI were determined to be (1) reversed if expression returned to baseline (Veh-Con) levels in the PLX-TBI group and if Veh-TBI versus PLX-TBI were significantly different from one another; (2) partially reversed if expression returned to baseline (Veh-Con), but was not significant between Veh-TBI and PLX-TBI; (3) and unaffected if PLX-TBI was significantly altered from baseline in the same direction as Veh-TBI.
Compound action potential (CAP) recording
CAPs were recorded from the corpus callosum in ex vivo slice preparations as previously described (Reeves et al., 2005). In brief, mice were anesthetized with isoflurane, euthanized, and brains were rapidly dissected into ice-cold cutting solution containing the following: 250 mm sucrose, 25 mm D-glucose, 2.5 mm KCl, 24 mm NaHCO3, 1.25 mm NaH2PO4, 2.0 mm CaCl2, 1.5 mm MgSO4, and 1.0 mm kynurenic acid (pH 7.3–7.4). Whole-brain coronal slices (400 µm) were prepared using a Vibratome (VT1200S, Leica). Brain slices with intact corpus callosum were transferred to chambers filled with aCSF (bubbled with 95% O2 and 5% CO2) containing the following: 124 mm NaCl, 3 mm KCl, 24 mm NaHCO3, 1.25 mm NaH2PO4, 2 mm CaCl2, 1.0 mm MgSO4, and 10 mm D-glucose (pH 7.3–7.4), allowed to recover at 37C for 30 min, then moved to room temperature for at least 1 h. Slices were transferred to a submerged chamber at room temperature and a gravity perfusion system was used for solution exchange (2–3 ml/min) with oxygenated aCSF during recording. Borosilicate glass electrodes filled with aCSF (1.5–3 MΩ) were positioned in the center of the corpus callosum on one hemphisphere to record CAPs evoked by electrical stimulation through a custom-made twisted nichrome wire stimulating electrode placed in the corpus callosum of the other hemisphere. The distance between the electrodes was ∼1.5 mm. Differing stimulation intensities (0–2 mA, 21 steps, 200 µS, every 5 S) were applied to evoke CAPs and to create intensity-response plots. CAPs were low-pass filtered at 1 kHz and digitally sampled at 50 kHz. All data were digitized using an Axopatch 200B amplifier, Digidata 1440A, and pClamp 10.6 software (Molecular Devices). Data were monitored on-line and analyzed off-line from typical traces using Clampfit 10.6 software; n = 6 mice per group; peak values from three to four slices per mouse were averaged to generate N1, N2, and N2/N1 ratios. Mouse averages were then used to calculate group means and error. The representative traces were evoked with maximum stimulation.
Neuronal tracing and dendritic spine analysis
Dendritic complexity was determined as previously described with minor modifications (Hao et al., 2016; Chunchai et al., 2018). In brief, Thy1-YFP-H mice were used because 10–12% of Layer V cortical neurons express YFP and allow for visualization of small neuronal structures (Feng et al., 2000). Mice were injured, perfused, fixed, and brains were sectioned through the cortex at 30 μm thickness. A series of z-stacks were taken from cortex Layer IV/V using a Leica SP8 confocal microscope (Leica Biosystems). Neuronal morphology was analyzed by Imaris software 7.0 (Oxford Instruments). Three neuronal cells per brain section and three brain sections per mouse with n = 6 mice per group were assessed. Whole neurons were traced and Sholl analysis of process branching/complexity was performed using Imaris software. The number of intersections were plotted against the Sholl radius and the area under the curve was calculated from the 3D construction. In addition, to determine dendritic spine density, the three tertiary segments were used to randomly measure dendritic spine density. YFP+ dendrites were quantified per 10-μm dendritic segment and further classified as mushroom or stubby spines using Imaris.
Rotarod and wire hang assessments of locomotor activity
Accelerating Rotarod testing was performed as previously described (Fenn et al., 2014). In brief, latency to fall off of the accelerating Jrotarod was determined. Rotation speed started at 4 rpm and accelerated to 40 rpm over 200 s. Mice had three attempts per day, which were averaged. Training was 3 d before mFPI and the final day counted as baseline performance. Mice were tested 1 dpi and weekly following TBI. The horizontal bar-hang test was performed as previously described (Deacon, 2013). In brief, the front paws were placed on a 6-mm diameter stiff wire elevated over a pad and latency for mouse to escape onto a platform 20 cm away was determined. No mice fell from the wire during this test; n = 6 mice per experimental group.
Novel object location (NOL) and recognition
NOL and recognition were determined as previously described (Denninger et al., 2018) with minor modifications. These tests involved four 10-min phases each separated by 24 h: habituation (no objects), acclimation (two objects), location (two objects, one new location), and recognition (two objects, with one new object). Trials were videotaped and time spent investigating each object was measured by a blinded reviewer. Discrimination index was calculated for both location and recognition trials as follows: [(timenovel – timefamiliar)/timetotal] × 100; n = 10 per experimental group.
Experimental design and statistical analysis
Number of mice per experiment including number of slices/cells/images analyzed are included in the methods for each experiment above. Statistical analysis for NanoString and scRNA-sequencing (scRNAseq) using DESeq2 or CellRanger respectively are described above. IBM SPSS Statistics (version 24) was used for ANOVA of histologic, behavioral, and electrophysiological data. One-way, two-way, or repeated-measures ANOVA was used as-appropriate to determine main effects and interactions between factors. Least squares difference (LSD) was used for post hoc analysis when main effects and/or interactions were determined and p < 0.05 was considered statistically significant.
Results
Microglia-dependent and microglia-independent differential gene expression in the cortex changes over 1, 7, and 30 dpi
We previously reported that depletion of microglia with the CSF1R antagonist PLX5622 reversed mRNA expression of inflammatory-related genes and attenuated the formation of unique rod-shaped microglia in the cortex 7 dpi (Witcher et al., 2018). Here, we sought to determine the contribution of microglia to cortical neuropathology at acute (1 dpi), subacute (7 dpi), and chronic timepoints (30 dpi). Here, microglia were depleted using oral administration of PLX5622 for 14 d before TBI (Fig. 1A) and mice were maintained on experimental diet (Veh or PLX5622) for the duration of the experiment. At each endpoint, the brain was collected, sectioned, and labeled with anti-Iba1. As expected (Elmore et al., 2014; Witcher et al., 2018), there was a robust reduction in the number of Iba1+ microglia with PLX5622 administration at baseline (F(1,31) = 1658, p < 0.001; Fig. 1B,C) and at 1, 7, or 30 dpi (F(3,31) = 4.3, p = 0.015). Sections were co-labeled with P2RY12 and over 99% of Iba1+ cells were also P2RY12+ (data not shown). Overall, microglia were depleted for the duration of PLX5622 administration.
Figure 1.
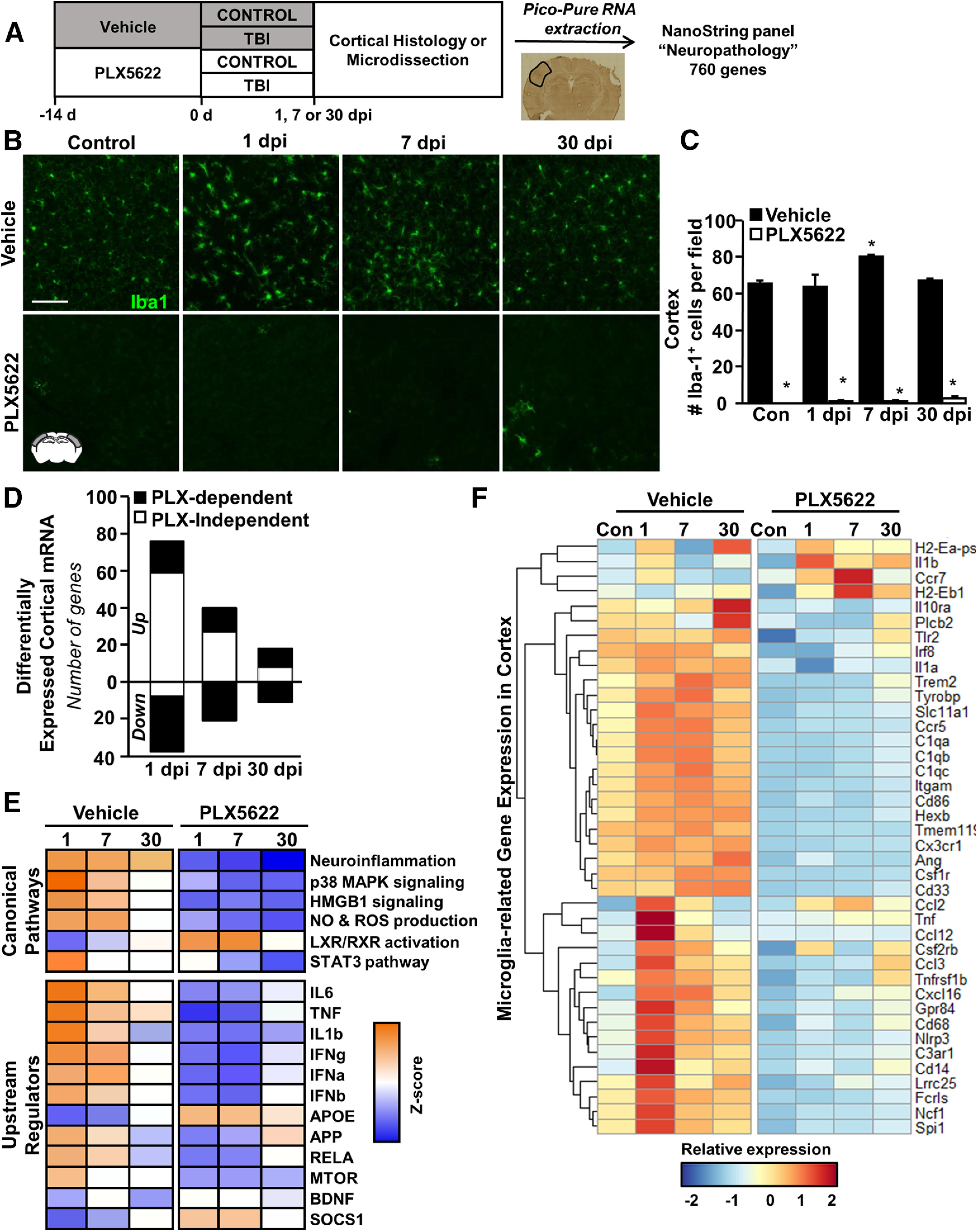
Microglia-dependent and microglia-independent differential gene expression in the cortex changes over 1, 7, and 30 dpi. A, Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or a CSF1R antagonist (PLX5622) for 14 d. Next, mice were uninjured (Con) or were subjected to mFPI (TB1; n = 4). Mice were maintained on vehicle or PLX diet for the duration of the experiment. At each endpoint (1, 7, or 30 dpi), mice were perfused, fixed, and cortical tissue was cryosectioned and labeled for Iba1 (microglia). B, Representative images of Iba1 labeling in the lateral cortex. Scale bar: 50 μm. C, Quantification of average number of Iba1+ microglia per 20× field. In a separate experiment, mice were provided diets formulated as above for 14 d before TBI (n = 5–9). Mice were killed 1, 7, or 30 dpi. Brains were flash-frozen fresh and cryosectioned at either 30 or 200 µm. The 30-µm sections were labeled for CD11b and used to identify cortical regions of interest. The 200-µm sections were dissected while frozen and immediately lysed for RNA extraction. RNA copy number was determined using NanoString Neuropathology panel plus. D, The number of differentially expressed genes (increased or decreased) by TBI and influenced by time or PLX5622. E, IPA comparison analysis of canonical pathways and upstream regulators of the differentially expressed genes based on z score (p-adj < 0.05). Orange indicates high relative z score while blue indicates low relative z score. F, Heatmap shows average z score of microglial signature genes that were differentially expressed following PLX5622 administration. Extended Data Figure 1-1 shows (1) representative images of GFAP labeling in the lateral cortex (scale bar: 50 μm); and (2) quantification of average GFAP+ percent-area. Raw and normalized counts are provided in Extended Data Figure 1-2, and IPA results are available in Extended Data Figure 1-3. Bars represent the mean ± SEM. Means with (*) are significantly different from Veh-Con (p < 0.05).
Time course of GFAP expression in the cortex after TB1 with and without microglia. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or a CSF1R antagonist (PLX5622) for 14 d. Next, mice were uninjured (Con) or were subjected to mFPI (TBI; n = 4). Mice were maintained on vehicle or PLX diet for the duration of the experiment. At each endpoint (1, 7, or 30 dpi), mice were perfused, fixed, and cortical tissue was cryosectioned and labeled for GFAP (subset of astrocytes). A, Representative images of GFAP labeling in the lateral cortex. Scale bar: 50 μm. B, Quantification of average GFAP+ percent-area labeled. Bars represent the mean ± SEM. Means with * are significantly different from Veh-Con (p < 0.05). Download Figure 1-1, TIF file (1.3MB, tif) .
Table of NanoString neuropathology raw and normalized data. Table contains raw and normalized count data for full NanoString Neuropathology dataset. Download Figure 1-2, XLSX file (845.9KB, xlsx) .
Table of IPA results from NanoString Neuropathology analysis. Table contains output from IPA for each differential expression comparison. Download Figure 1-3, XLSX file (271.6KB, xlsx) .
In a separate experiment, the brain was collected and fresh-frozen guide sections were labeled with anti-CD11b to visualize microglia. Based on the identification of reactive or rod-shaped microglia on guide sections, corresponding cortical regions from adjacent thick sections were microdissected while frozen. Analysis of neuropathology-associated gene expression (NanoString Neuropathology nCounter panel, 760 genes plus 30 custom genes) in these microdissected cortices postinjury showed that differential gene expression was influenced by injury, time, and microglial depletion (Fig. 1D). For example, there were 114 differentially expressed genes (76 increased and 38 decreased) at 1 dpi. At 7 dpi there were 61 differentially expressed genes (40 increased and 21 decreased) and at 30 dpi there were 29 differentially expressed genes (18 increased and 11 decreased). Figure 1D highlights the TBI-induced genes that were PLX independent (white bars) or PLX dependent (p < 0.05, black bars). At 1 dpi, only 33% of the genes influenced by TBI were reversed by microglial depletion. The contribution of TBI-induced genes that were influenced by microglial depletion (PLX5622) was increased to 66% at 7 dpi and to 80% by 30 dpi. Taken together, acute gene expression after TBI was predominately microglia independent, while persistent changes in neuropathological mRNA expression were dependent on microglia.
Next, IPA was used to identify canonical signaling pathways and upstream regulators for these differentially expressed genes (Krämer et al., 2014) Raw outputs from IPA are included in Extended Data Figure 1-3. IPA comparison analysis was then used to compare pathway changes across postinjury time points (1, 7, 30 dpi) and treatment conditions (Veh, PLX5622; Fig. 1E). As expected, canonical pathways associated with neuroinflammation were maintained 1, 7, and 30 dpi. Other significant canonical pathways that were increased at the acute and subacute time points (1 and 7 dpi) included p38 signaling, NO and ROS production, and HMGB1 signaling. There was a suppression of LXR/RXR pathways after TBI in both the acute and subacute phases of injury. These pathway-level effects of TBI were ameliorated by microglial depletion.
IPA analysis of upstream regulators (p < 0.05; absolute z score > 2) revealed similar effects of TBI and microglial depletion. This analysis predicts signaling pathways that may explain observed patterns in gene expression. Expression patterns at acute and subacute time points (1 and 7 dpi) were consistent with elevated cytokine (IL-6, TNF, IL-1β) and interferon (IFNα, IFNβ, IFNγ) signaling. Of these, TNF-associated genes remained elevated 30 dpi compared with controls. There was a TBI-associated suppression of APOE and SOCS1 pathways at 1 and 7 dpi. Overall, these expression patterns after TBI were not present in mice without microglia and showed opposite expression patterns relative to controls (Fig. 1E, bottom panel).
Gene expression patterns with and without PLX5622 confirm the histologic evidence of microglial depletion. Figure 1F shows a heat map of microglia-related genes affected by TBI and PLX at 1, 7, or 30 dpi. For instance, there was a main effect of PLX5622 (p < 0.05) on the majority of microglia-related genes including Tmem119, Cx3cr1, Csf1r, C1qc, Trem2, and Itgam (CD11b). Of the microglia-related genes, Il10ra, Gpr84, Cd68, Cd14, Ccr5, Cd33, and Nlrp3 were increased by TBI (p < 0.05) and attenuated in mice in which microglia were depleted before TBI. Of note, some other microglia/myeloid genes, Ccr7, Il1b, H2-Eb1, and Ccl2, were increased by TBI (p < 0.05) independent of PLX5622. Some microglia-related genes increased in the PLX-TBI mice compared with PLX-Con mice by 30 dpi, including Cd68, Cd14, and Trem2. This may be related to a relative expansion of microglia in the brain of PLX-TBI mice by 30 dpi (Fig. 1C). Collectively, the majority of inflammatory responses attributed to microglia were blocked 1 dpi and remained low 7 and 30 dpi when microglia were depleted before and after TBI.
Inflammatory mRNA expression in the cortex evolves over 1, 7, and 30 dpi
Related to the data presented in Figure 1, the heat map in Figure 2A highlights the interaction between TBI and time on genes associated with the neuropathology with sections reflecting genes significantly (p < 0.05) differentially expressed at 1 (top), 7 (middle), and 30 (bottom) dpi. Figure 2B reflects genes differentially expressed at multiple timepoints. Overall, there are effects of injury, time, and microglial depletion (PLX5622). The highest level of differential mRNA expression in the cortex was at 1 dpi. Acutely, TBI increased several inflammation-related (Cd14, Cd68, Gpr84, Myd88, Tnf), interferon-related (Irf7, Ifi2712a), and chemokine-related (Ccl12, Ccl3, Cxcl10, Cxcl16) transcripts compared with controls (p < 0.05). These TBI-associated increases 1 dpi were attenuated by PLX5622 (p < 0.05). Nonetheless, a higher level of gene expression (66%; Fig. 1D) was unaffected by PLX. For instance, TBI increased Icam1, Ptgs2, Ccl2, Cd44, Gfap, Atf3, Osmr, Nex, collagen components (Col4a1, Col4a2, and Col6a), and matrix-metalloproteinases (Mmp12, Mmp14, and Mmp19). The increases in these genes at 1 dpi were unaffected by microglial depletion. Thus, much of the differential expression of mRNA in the cortex 1 dpi was independent of microglia.
Figure 2.
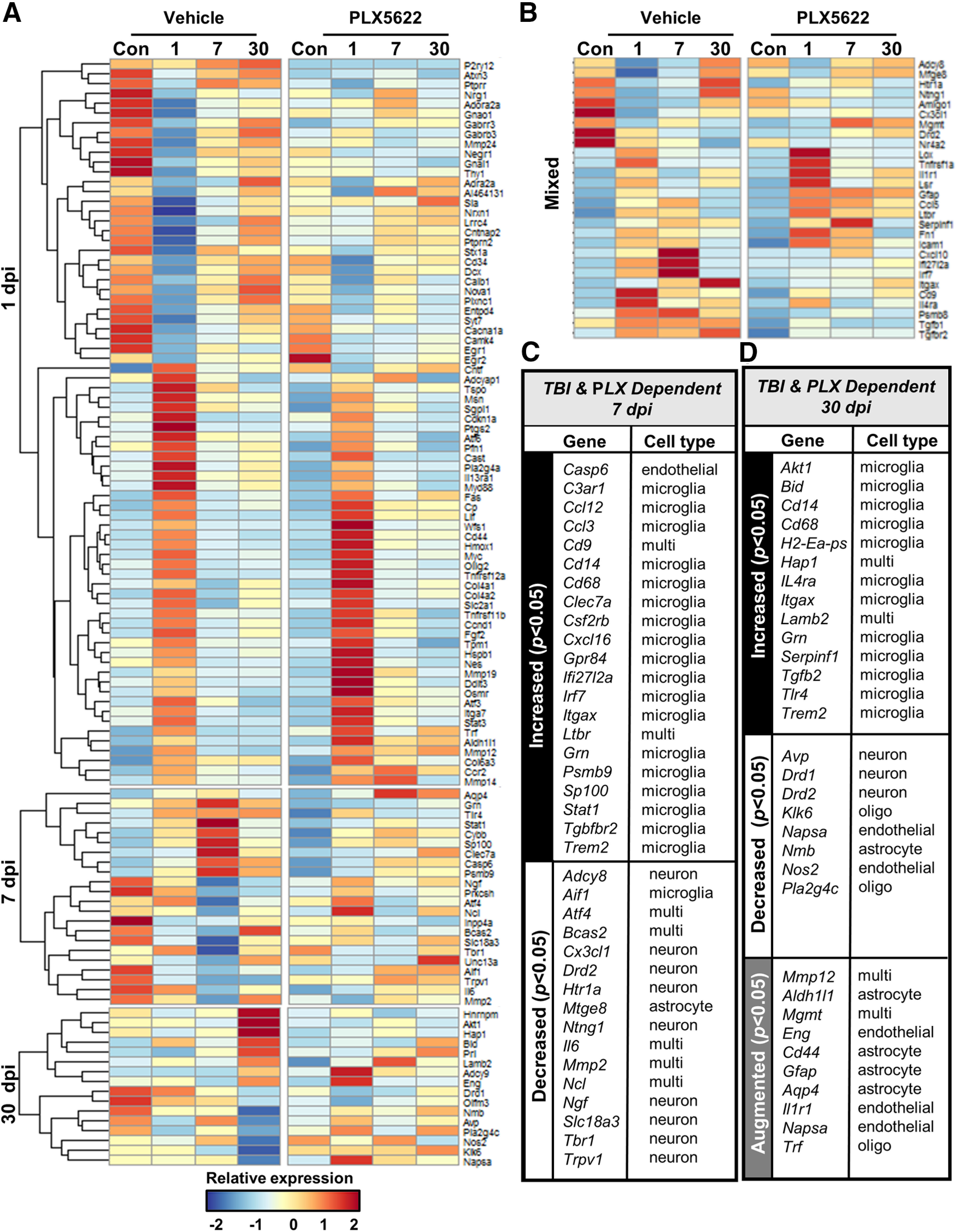
Evolving inflammatory mRNA expression in the cortex 1, 7, and 30 dpi. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d before TBI (n = 5–9). Mice were maintained on vehicle or PLX diet for the duration of the experiment. Mice were killed 1, 7, or 30 dpi. Brains were flash-frozen fresh and cryosectioned at either 30 or 200 µm. The 30-µm sections were labeled for CD11b and used to identify cortical regions of interest. The 200-µm sections were dissected while frozen and immediately lysed for RNA extraction. RNA copy number was assessed using the NanoString Neuropathology panel. A, Heatmap of average z scores of differentially expressed genes influenced by TBI, time, or PLX5622. B, Table of genes increased or decreased 7 dpi and influenced by PLX5622. C, Table of genes increased or decreased 7 dpi and influenced by PLX5622. D, Table of genes augmented by TBI-PLX compared with TBI-vehicle at 30 dpi. All genes in the tables were differentially expressed (TBI and PLX, p < 0.05).
Over time, however, the number of genes that were influenced by TBI and microglia were increased. This was apparent in the heat map, which was organized by time (Fig. 2A). Genes differentially expressed 7 and 30 dpi are summarized in Figure 2C,D and the Barres Laboratory Brain RNA-seq Mus musculus dataset (http://www.brainrnaseq.org) was used to approximate which cell type expressed each gene (Zhang et al., 2014). At 7 dpi, several inflammation-related (Cd14, Cd68, Gpr84, Clec7a, Itgax, Tlr4, Trem2), interferon-related (Irf7, Ifi2712a), and chemokine-related (Ccl12, Ccl3, Cxcl10, Cxcl16) transcripts were increased compared with controls (p < 0.05) and reversed by the depletion of microglia (p > 0.05). There were also several genes decreased in the cortex 7 dpi (p < 0.05) including genes associated with neurons (Adcy8, Cxc3rl, Drd2, Htr1a, Ngf, Slc18a3, and Trpv1), which were prevented by the depletion of microglia (p > 0.05).
At 30 dpi, several transcripts related to innate immunity (Cd14, Cd68, Gpr84, Itgax, Tlr4, Trem2) were increased by TBI compared with controls (p < 0.05) and reversed by the depletion of microglia (p > 0.05). There were also several genes decreased in the cortex 30 dpi (p < 0.05) including neuron-associated transcripts (Avp, Drd1, Drd2). These reductions were again reversed by the depletion of microglia before TBI (p > 0.05).
Another notable observation at 30 dpi was that there were several genes augmented in the TBI-PLX group compared with Veh-TBI. These data may indicate there was compensation/expansion of astrocytes after TBI when microglia were depleted. In support of this notion, there was higher relative mRNA expression of Aldh1l1, Cd44, Gfap, and Aqp4 (p < 0.05; Fig. 2D) in the PLX-TBI group compared with the Veh-TBI group. Extended Data also shows increased GFAP labeling in the TBI-PLX group compared with the TBI-Con group 30 dpi (Extended Data Fig. 1-1).
Single-cell sequencing of cortical cells after TBI. Cortical mRNA analysis showed a robust contribution of microglia to neuroinflammatory pathways that persisted over time postinjury
These data also indicate that persistent inflammation in the subacute and chronic time points after TBI, which may affect surrounding neurons, oligodendrocytes and endothelial cells. To investigate this hypothesis, a single-cell sequencing approach was used to determine cell type-specific gene expression within the cortex 7 dpi with and without microglial depletion (Fig. 3A). Pooled single-cell suspensions were generated from microdissected cortices and the 10× Genomics pipeline was used to perform single-cell mRNA sequencing. Distinct CNS cell types were evident by clustering based on cell-selective gene expression (Fig. 3B,C). For instance, microglia (Tmem119), astrocytes (Gja1), oligodendrocytes (Plp1), neurons (Meg3), and endothelial cells (Ly6c1) all formed distinct clusters (Fig. 3C). The distribution of all 36 000 sequenced cells across all four experimental groups (Veh-Con, Veh-TBI, PLX-Con, PLX-TBI) is represented in the dot plot in Figure 3D and Extended Data Figure 3-1. Extended Data Figure 3-1 highlights the difference in number of microglia between Veh-treated and PLX-treated groups. In addition, microglia (12,181 cells) and neuron (2878 cells) populations were subclustered for further analysis. Dot plots of subclusters show distribution of cells by experimental group for microglia (Fig. 3E) and neurons (Fig. 3F).
Figure 3.
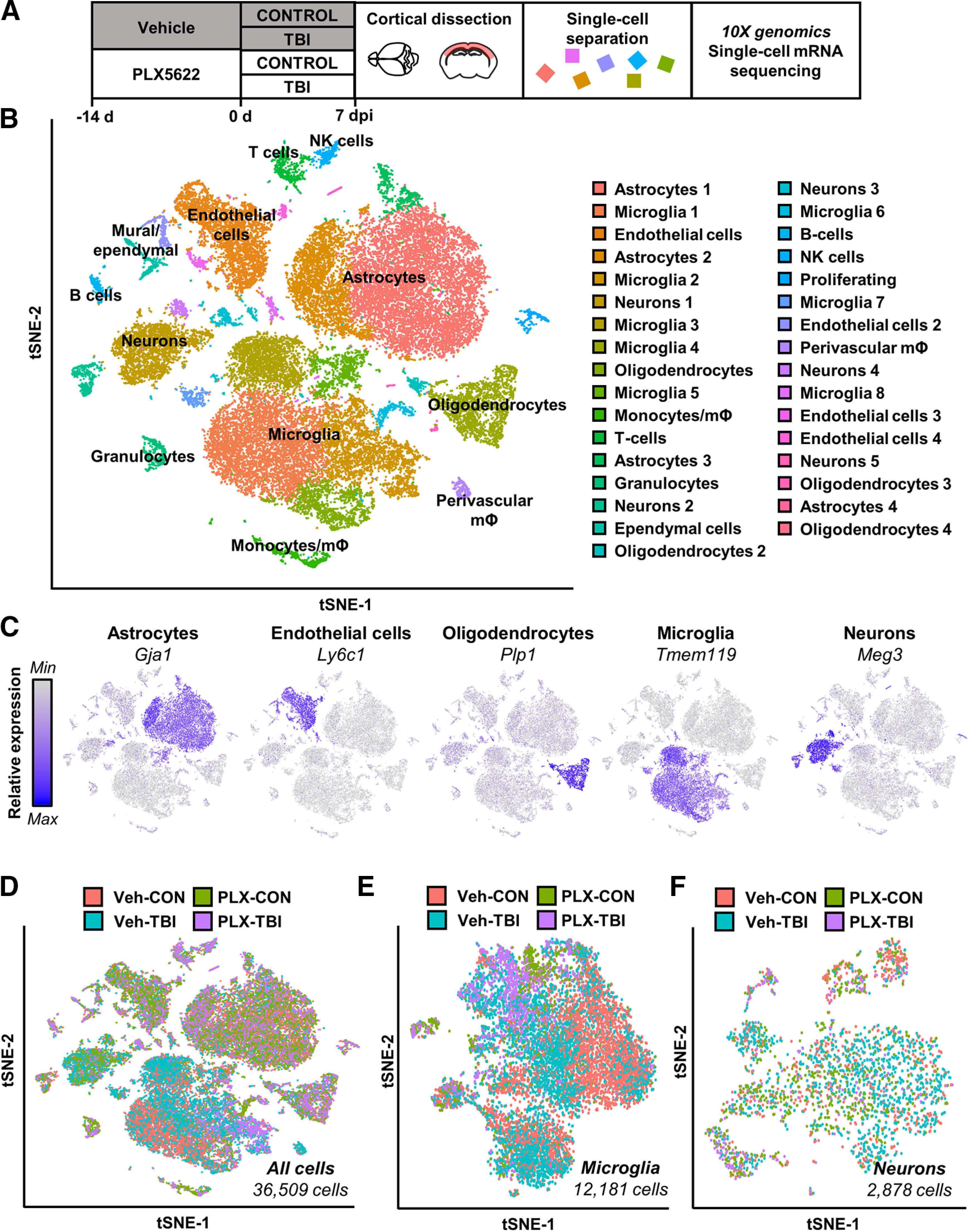
Single-cell RNA sequencing of cortex 7 dpi. A, Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (Con) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. Mice were killed 7 dpi, and the cortex was microdissected and enzymatically digested into a single-cell suspension. Cortices from three mice were pooled from each group; data reflect two replicate experiments (n = 6 mice per group). Single cells were run on a 10× genomics chromium controller, mRNA from individual cells was barcoded, and cDNA libraries were generated and sequenced. Sequencing results were aligned to the mouse mm10 genome and doublets and low-quality cells were removed. Clustering and differential expression was generated using Seurat in R. B, tSNE clusters show individual cell types identified based on marker gene expression. C, tSNE plots highlight primary CNS cell types by marker gene expression: astrocytes (Gja1), endothelial cells (Ly6c1), oligodendrocytes (Plp1), microglia (Tmem119), and neurons (Meg3). D, tSNE highlights distribution of all cells across experimental groups. Subclusters of microglia (E) and neurons (F) were selected based on expression of Tmem119 and Meg3, respectively. tSNE plots highlight distribution of experimental groups within subclusters. tSNE plots in Extended Data Figure 3-1 represent distribution of cells across the four experimental groups: (1) Veh-CON, (2) PLX-CON, (3) Veh-TB1, and (4) PLX-TB1. Results from cluster expression analysis and differential expression for cell type subsets is available in Extended Data Figure 3-2.
Distribution of experimental groups across cell-type clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. tSNE plots represent distribution of cells across the four experimental groups: (1) Veh-CON, (2) PLX-CON, (3) Veh-TB1, and (4) PLX-TB1. Download Figure 3-1, TIF file (1.5MB, tif) .
Table of cluster markers and differential expression results. Table contains output from overall cluster marker analysis, cluster marker analysis for individual cell types, and differential expression between Veh-CON and Veh-TB1 groups for each cell type. Download Figure 3-2, XLSX file (426.4KB, xlsx) .
Trauma-associated mRNA signatures and increased type-1 interferon signaling evident in cortical microglia after TBI
First, subclustering of microglia revealed 10 (0–9) distinct microglial clusters determined by each cell's transcriptome (Fig. 4A). Clusters are presented in Figure 4B with the total number of cells per cluster and the proportion of cells derived from each experimental group (Veh-Con, Veh-TBI, PLX-Con, PLX-TBI). Here, PLX5622 and TBI -dependent gene expression patterns were evident. A representative heat map of the gene expression defining each cluster is provided (Extended Data Fig. 4-1). The pie charts (Fig. 4C) reflect the proportion of microglia from each experimental condition per cluster. Genes from overrepresented clusters in each experimental group are listed and were expressed significantly higher in those microglia compared with all other microglia (p-adj < 0.05). For instance, cluster 3 (green) was predominantly represented in microglia from the cortex of Veh-Con mice (Fig. 4B,C). Cluster three microglia expressed higher levels of the homeostatic microglial genes including Cx3cr1 and Tgfbr1. Cluster 2 microglia (army green) accounted for ∼50% of PLX-Con and PLX-TBI microglia and were enriched in the Veh-TBI group. Cluster 2 highly expressed mRNA encoding ribosomal proteins, which may suggest increased proliferative or metabolic burden. Cluster 7 was also enriched by PLX treatment. Cluster 7 expressed proliferative-related genes including ubiquitin-conjugating enzyme E2C (Ube2c) and centromere protein A (Cenpa), and cytoskeletal components including tubulin-related and actin-related genes. Thus, clusters 2 and 7 may represent active turnover of microglia.
Figure 4.
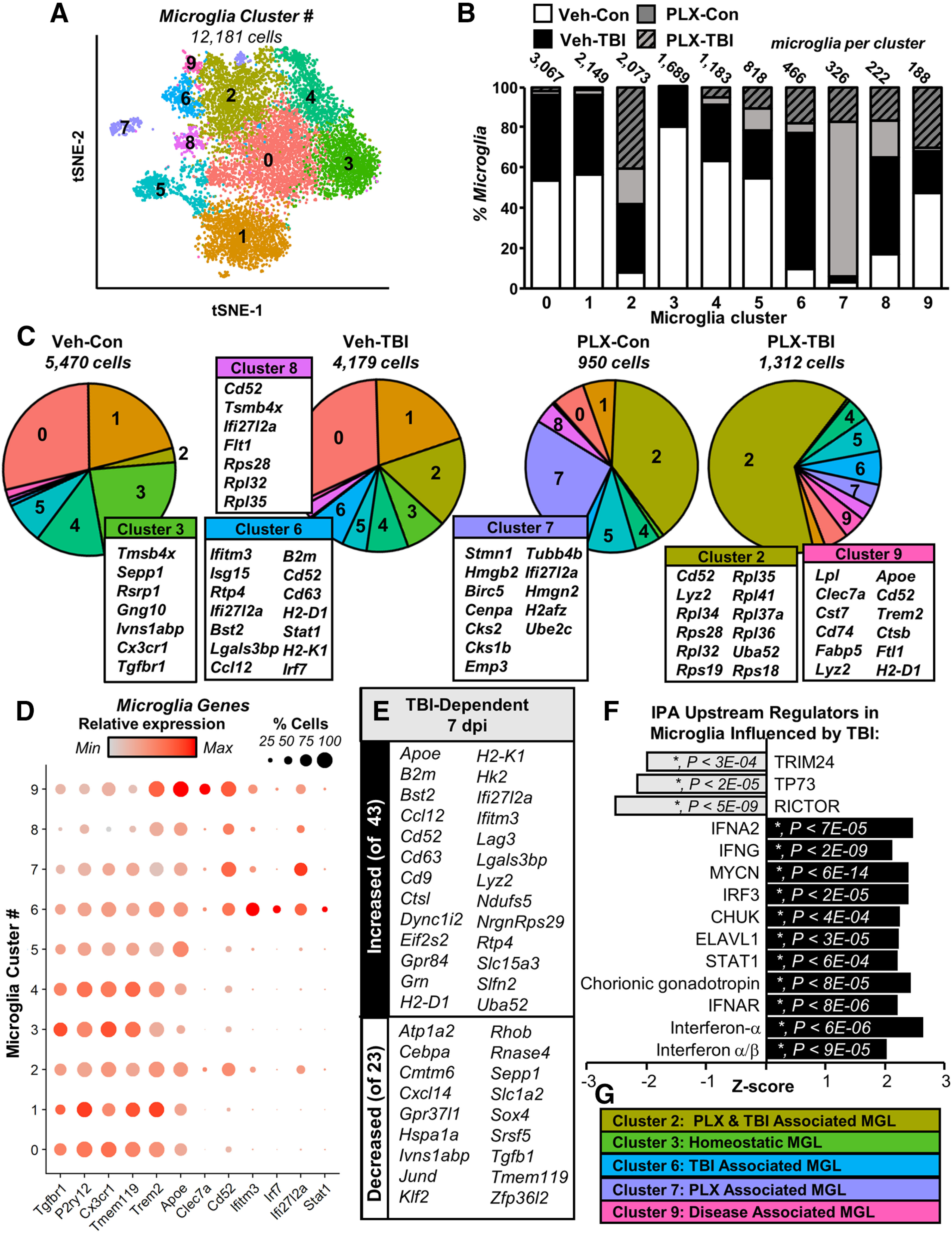
Trauma-associated mRNA signatures and increased type-1 interferon signaling evident in cortical microglia after TB1. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. A, tSNE reflects subclustering of Tmem119+ microglia (12,181 cells) from all samples. B, Distribution of microglia from each experimental condition across the 10 microglial clusters. C, Pie-graphs reflect proportion of each experimental condition in each microglial cluster. Top genes expressed significantly more in a given cluster than in all other microglia are shown. D, Dot plot shows relative and proportional expression of microglial signature (Tgfbr1, P2ry12, Cx3cr1, Tmem119), neurodegenerative-associated (Trem2, Apoe, Clec7a, Cd52), and interferon-related genes (Ifitm3, Irf7, Ifi27l2a, Stat1). E, Subset of genes increased (top) and decreased (bottom) in Veh-TB1 compared with Veh-Con (p-adj < 0.05). F, IPA (upstream regulators) of significantly differentially expressed genes between Con-Veh and TB1-Veh (p-adj < 0.05). G, Clusters enriched in specific experimental groups. Heatmap in Extended Data Figure 4-1 reflects top genes expressed by each Tmem119+ microglia cluster.
Heatmap of top genes expressed in microglial clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. Heatmap reflects top genes expressed by each Tmem119+ microglia cluster. Download Figure 4-1, TIF file (16.8MB, tif) .
The effect of TBI on the microglial subclusters was associated with increased inflammation and type-1 interferon signaling. For instance, cluster 6 (blue) was predominately derived from microglia from Veh-TBI cortices, which highly expressed interferon-responsive genes (Ifitm3, Isg15, Ifi27l2a) and other immune-related genes (Ccl12, Cd63, Cd52, H2-D1, and H2-K1). Furthermore, cluster 8 (purple) was also enriched in the Veh-TBI group and contained increased interferon-related (Ifi27l2a) and damage-associated genes (Cd52, Flt1, Tsmb4x, Apoe). In addition, cluster 9 (pink) was enriched in the Veh-TBI and the PLX-TBI groups and had increased expression of Cd52, Clec7a, Apoe, and Trem2. As these profiles increased with TBI (7 dpi), clusters with more homeostatic genes (clusters 3 and 4) that were evident in the Veh-Con group were proportionally reduced after TBI. These data are consistent with the increased interferon pathways evident in the bulk cortical mRNA analysis (Fig. 1E).
Cluster-specific expression patterns are further highlighted in Figure 4D, which shows genes related to microglial homeostasis (Tgfbr1, P2ry12, Cx3cr1, Tmem119), genes involved in microglial responses to damage (Trem2, Apoe, Clec7a, Cd52), and interferon-responsive genes (Ifitm3, Irf7, Ifi27l2a, Stat1). Dot plots show relative gene expression by cluster (color intensity) and proportion of cells per cluster expressing a gene (dot size). Clusters with more inflammatory gene expression had corresponding reduced expression of homeostatic genes as previously described (Krasemann et al., 2017). For example, the TBI-enriched cluster, cluster 6, had relative increases in expression of Cd52, Ifitm3, Irf7, Ifi27l2a and Stat1, with corresponding reduction in homeostatic genes including Tgfb1, Cx3cr1, and Tmem119 (Fig. 4D). In addition, the disease-associated cluster (Krasemann et al., 2017; Deczkowska et al., 2018), cluster 9, had enriched Trem2, Apoe, Clec7a, Cd52 with corresponding reductions in homeostatic genes including Tgfb1, Cx3cr1, and Tmem119 (Fig. 4D).
When Veh-Con and Veh-TBI groups were compared across all clusters, 66 genes were significantly differentially expressed (p-adj < 0.05; 43 increased and 23 decreased). A selection of these TBI-dependent genes is shown in Figure 4E. IPA upstream regulator analysis of the 66 differentially expressed in microglia between Veh-Con and Veh-TBI samples was consistent with increased type-1 interferon signaling at 7 dpi as a function of TBI. For instance, affected upstream regulators 7 dpi in microglia included increased IFNA, IRF3, IFNAR, and STAT1. Thus, 7 dpi there was a robust interferon-associated pattern of expression in microglia. More specifically, the trauma-enriched cluster 6 was highly responsive to type-1 interferon signaling. This may reflect persistent inflammation and cellular damage in the cortex 7 dpi. Overall, there were 5 clusters of microglia that were enriched within specific treatment groups (Fig. 4G).
TBI induced inflammatory and reactive astrocyte signature that was partially prevented by microglial depletion
Further analysis of Gja+ astrocytes revealed seven (0–6) clusters of cells across all four experimental groups (Fig. 5A). Figure 5B,C highlights genes strongly expressed in specific astrocyte clusters. For instance, cluster 5 was associated with high Gfap expression consistent with reactive astrocytes while Spry2 is implicated in BDNF signaling and neurite outgrowth. These findings were consistent with previous reports that astrocytes maintain heterogeneous cell surface markers and have myriad functions within the adult CNS (Morel et al., 2019; Pestana et al., 2020; Götz et al., 2021).
Figure 5.
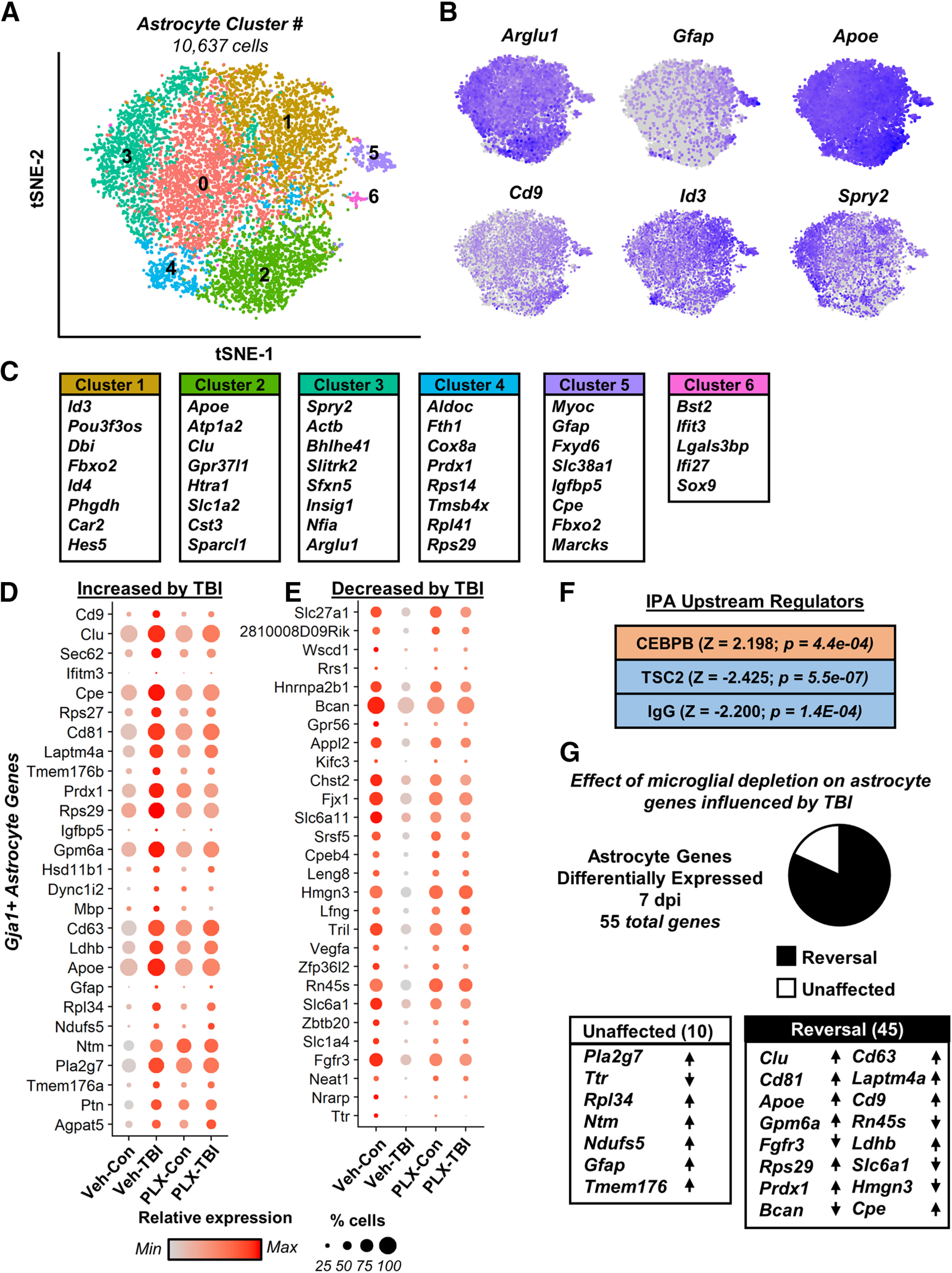
TB1 induced inflammatory and reactive astrocyte signature that was partially prevented by microglial depletion. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. A, tSNE reflects subclustering of Gja+ astrocytes (10,637 cells) from all samples. B, tSNE plots highlight gene expression of astrocytic genes across subclusters, with darker color reflecting higher expression. C, Top genes significantly expressed in a given cluster than in all other astrocytes. Differential gene expression between neurons from Veh-Con and Veh-TBI was determined (p-adj < 0.05). Dot plots reflect genes increased (D) and decreased (E) in Veh-TBI compared with Veh-CON. Differentially expressed genes between Veh-CON and Veh-TBI were used to determine IPA upstream regulators (F). G, Pie-graph reflects the proportion of these genes whose expression pattern was reversed (black) or unaffected (white) by PLX5622 administration. Lists reflect genes with lowest p-adj between Veh-Con and Veh-TBI and arrows reflect expression change after TBI (increased or decreased).
There were 55 differentially expressed genes in Gja1+ astrocytes 7 dpi (Veh-Con vs Veh-TBI, p-adj < 0.05). Of these, 27 were increased (Fig. 5D) and 28 were decreased (Fig. 5E) following TBI. All differentially expressed genes are represented in Figure 5D,E where dot plots reflect relative gene expression by treatment group (color intensity) and proportion of cells per cluster expressing a gene (dot size). Increased genes included Gfap, Apoe, and Clu, which are associated with astrocyte reactivity in injury/disease. Furthermore, expression of genes related to growth-factor signaling (Vegfa, Fgfr3) and extracellular matrix remodeling (Bcan) and astrocytic Notch signaling (Lfng, Nrarp) were decreased. Differentially expressed genes were used for IPA and three upstream regulators were significantly different following injury (p < 0.05; Fig. 5F). Notably, TBI induced gene expression was consistent with increased CEBPB signaling, a transcription factor that regulates inflammatory gene expression in astrocytes. Of the 55 differentially expressed genes 7 dpi, 82% (45 genes) of this signature was reversed in the PLX-TBI group compared with Veh-TBI (p < 0.05; Fig. 5G). Collectively, these data demonstrated that astrocytes adopted an inflammatory and reactive signature characterized by increased inflammatory and reduced homeostatic gene expression that was attenuated by microglial depletion.
Myelinating oligodendrocytes had transcriptional changes consistent with myelination and CNS-damage response after TBI that was attenuated by microglial depletion
Plp1+ cells were further analyzed revealing nine (0–8) clusters (Fig. 6A). Figure 6B,C highlights oligodendrocyte genes increased in specific populations relative to other Plp1+ cells. For instance, Mobp and Mog were highly expressed in myelinating oligodendrocyte clusters (0–4, 6, 8), while Pdgfra was highly expressed in oligodendrocyte precursor cells (OPCs; cluster 5). Cluster 7 was comprised of cells expressing genes implicated in oligodendrocyte maturation (Slc1a1, Gpr17, Bcan) with reduced expression of myelination genes Mog and Mobp.
Figure 6.
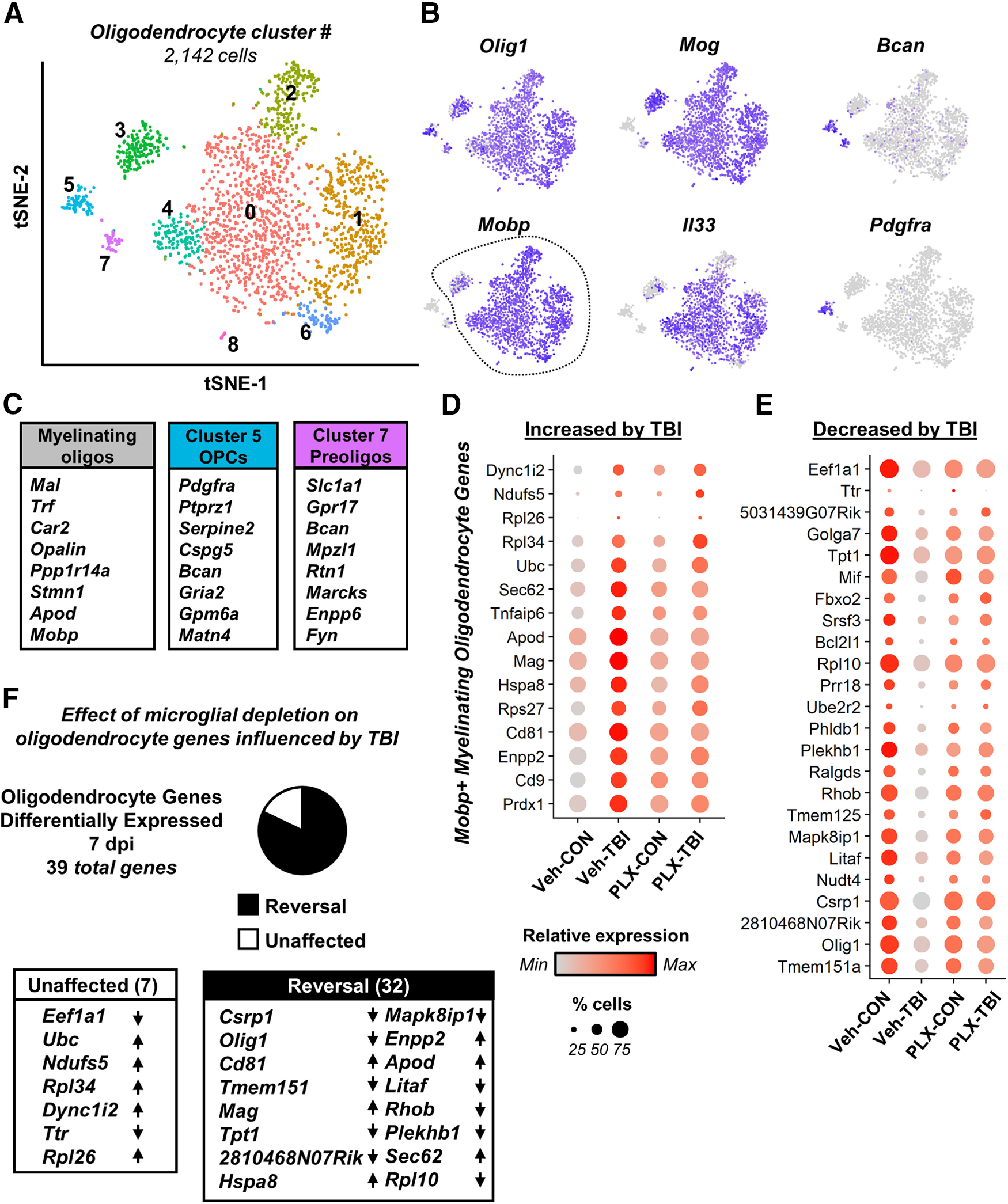
Myelinating oligodendrocytes underwent transcriptional changes consistent with myelination and CNS-damage response after TBI that was attenuated by microglial depletion. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. A, tSNE reflects subclustering of Plp1+ oligodendrocytes (2142 cells) from all samples. B, tSNE plots highlight gene expression of oligodendrocyte genes across subclusters, with darker color reflecting higher expression. C, Shows top genes significantly expressed by myelinating oligodendrocytes (Mobp+, clusters 0–4, 6, 8), OPCs (cluster 5), and preoligodendrocytes (cluster 7). Differential gene expression between myelinating oligodendrocytes from Veh-Con and Veh-TB1 was determined (p-adj < 0.05). Dot plots reflect genes increased (D) and decreased (E) in Veh-TB1 compared with Veh-CON. F, Pie-graph reflects the proportion of these genes whose expression pattern was reversed (black) or unaffected (white) by PLX5622 administration. Lists reflect genes with lowest p-adj between Veh-Con and Veh-TB1 and arrows reflect expression change after TB1 (increased or decreased).
Following TBI, there were 39 differentially expressed genes in myelinating oligodendrocytes (Veh-Con vs Veh-TBI, p-adj < 0.05). Of these, 15 were increased (Fig. 6D) and 24 were decreased 7 dpi (Fig. 6E). All differentially expressed genes are represented in Figure 6D,E where dot plots reflect relative gene expression by treatment group (color intensity) and proportion of cells per cluster expressing a gene (dot size). Increased genes included those encoding tetraspanins (Cd9, Cd81), which are implicated in oligodendrocyte development, and genes associated with oligodendrocyte responses to CNS injury/stress (Apod, Hspa8). Of the 39 differentially expressed genes, 32 were reversed by microglial depletion (Fig. 6F). While the number of differentially expressed genes in oligodendrocytes was relatively modest, there were detectable changes following TBI that were dependent on microglia.
TBI-associated reduction of neuronal homeostasis 7 dpi was ameliorated by microglial depletion
Next, Meg3+ cortical neurons (2878) were analyzed and eleven (0–10) distinct clusters were identified (Fig. 7A; expression heatmap shown in Extended Data Fig. 7-1). Figure 7B highlights neuron-specific genes (Meg3, Camk2n1) and genes associated with projection neurons (Thy1, App), inhibitory neurons (Gad1), or axonal transport (Kif5a). The proportion of each cluster derived from the four experimental groups was relatively consistent across clusters (Extended Data Fig. 7-2). Overall, there were 233 differentially expressed genes in Meg3+ cortical neurons 7 dpi (Veh-Con vs Veh-TBI). Of these, 95% (221 genes) were reduced and 5% (12 genes) were increased 7 dpi (p-adj < 0.05; Fig. 7C).
Figure 7.
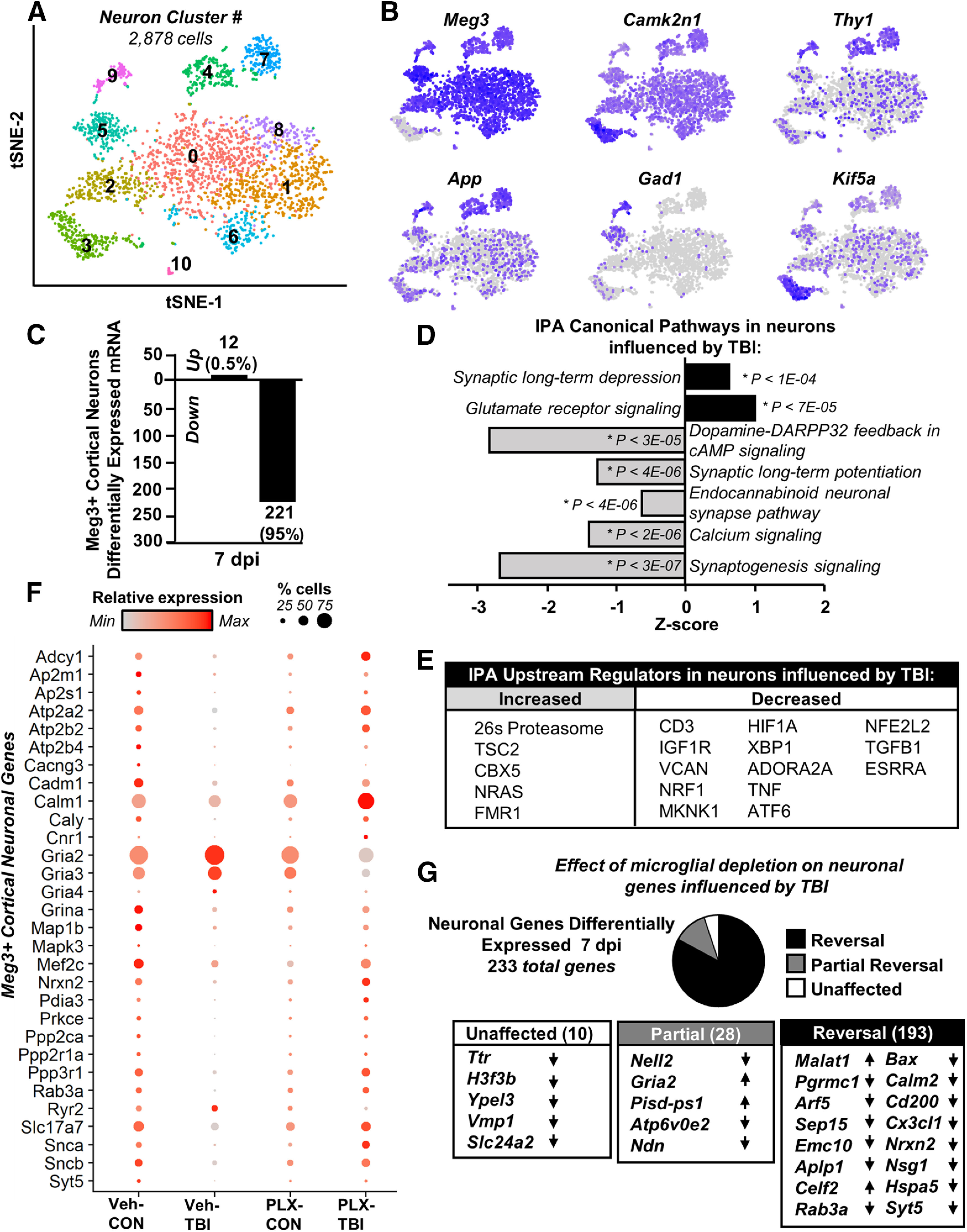
TB1-associated reduction of neuronal homeostatic gene expression 7 dpi was ameliorated by microglial depletion. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. A, tSNE reflects subclustering of Meg3+ neurons (2878 cells) from all samples. B, tSNE plots highlight gene expression of neuronal markers across subclusters, with darker color reflecting higher expression. Differential gene expression between neurons from Veh-Con and Veh-TB1 was determined (p-adj < 0.05). C, Number of increased and decreased genes. IPA (D, canonical pathways; E, upstream regulators) of differentially expressed genes between Con-Veh and TB1-Veh (p-adj < 0.05). F, Dot plot reflects expression of genes used in top pathways affected by TB1 in neurons. G, Pie-graph reflects the proportion of these genes whose expression pattern was reversed (black), partially reversed (gray), or unaffected (white) by PLX5622 administration. Lists reflect genes with lowest p-adj between Veh-Con and Veh-TB1 and arrows reflect expression change after TB1 (increased or decreased). Heatmap in Extended Data Figure 7-1 reflects top genes expressed by each Meg3+ neuron cluster. Graph in Extended Data Figure 7-2 reflects distribution of Meg3+ neurons from each experimental condition across the 11 neuronal clusters.
Heatmap of top genes expressed in neuronal clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. Heatmap reflects top genes expressed by each Meg3+ microglia cluster. Download Figure 7-1, TIF file (15.4MB, tif) .
Distribution of experimental groups across neuronal clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. Graph reflects distribution of Meg3+ neurons from each experimental condition across the 11 neuronal clusters. Download Figure 7-2, TIF file (618.3KB, tif) .
Differentially expressed genes were used for IPA and the suppression of several canonical pathways was evident (Fig. 7D). For instance, there was suppression of calcium signaling, synaptogenesis, and synaptic long-term potentiation 7 dpi (p < 0.00,003, for each). Furthermore, there was increased synaptic long-term depression and glutamate receptor signaling 7 dpi (p < 0.0001). Upstream regulator analysis in cortical neurons is shown in Figure 7E (p < 0.05; absolute z score > 2). Neuronal gene expression after TBI was consistent with reduced expression in regulators associated with growth and survival (TGFB, MKNK1, IGF1), neurite outgrowth (NRF1), and regulation of cellular stress (XBP1, ATF6, NFE2L2). Key genes driving these pathway-level changes are shown (Fig. 7F), with relative gene expression by treatment group (color intensity) and proportion of cells per cluster expressing a gene (dot size). Genes encoding glutamate AMPA receptor subunits (Gria2, Gria3, Gria4) were all increased in Veh-TBI compared with Veh-Con (p-adj < 0.05; Fig. 7E). All other neuronal genes were suppressed. For example, genes related to calcium homeostasis and signaling (Ryr2, Calm1, Caly) were decreased in cortical neurons after TBI.
The majority of gene expression changes were reversed in the PLX-TBI group compared with Veh-TBI (p < 0.05). For instance, the depletion of microglia with PLX5622 attenuated 95% of the TBI-dependent transcriptional changes within cortical neurons (Fig. 7G). Of the 233 genes differentially expressed following TBI, the expression pattern of 193 genes were returned to Veh-Con levels, and 28 were partially reversed. Select genes from each category are shown (Fig. 7G). Genes related to neuronal plasticity and vesicular trafficking (Nsg1, Syt5, Arf5) and neuronal regulation of microglia (Cx3cl1, Cd200) were suppressed following TBI (p-adj < 0.05) and reversed by microglial depletion (p < 0.05). Collectively these data indicate that genes supporting homeostatic neuronal function were suppressed in a microglia-dependent manner 7 d after TBI.
Reduced dendritic complexity after TBI is attenuated by microglial elimination
Our current and previous RNA and scRNAseq profiling studies indicate that microglia mediate chronic inflammation and potential pathology (Witcher et al., 2018) and that pathways associated with neuronal homeostasis were reduced. Other studies show evidence that microglia are involved in dendritic remodeling and modulating synapses (Schafer et al., 2012). Therefore, neuronal morphology and spine density were assessed in the cortex of Thy1-YFP-H mice after TBI (7 dpi) with or without microglial depletion (Fig. 8A). Here, we show that neuronal complexity (Sholl intersections) was decreased in the cortex 7 dpi (F(3,80) = 2.912, p = 0.0273; Fig. 8B–D). Moreover, this effect was absent in mice in which microglia were eliminated before TBI. Dendritic spine density in Thy1-YFP-H mice was also assessed (Fig. 8E) and there was no difference in total dendritic spine density after TBI (Fig. 8F). There was, however, a significant reduction in mushroom (or mature) spines 7 dpi (F(3,57) = 0.4471, p = 0.0169; Fig. 8G) with a corresponding increase in stubby (immature) spines compared with controls (F(3,57) = 0.4471, p = 0.0245; Fig. 8G). The reduction in mushroom (mature spines) with TBI (Veh-TBI) and increase in stubby spines (immature) was not detected in the PLX-TBI mice with depleted microglia. Taken together, microglial depletion prevented the TBI-associated remodeling of cortical dendrites 7 dpi.
Figure 8.
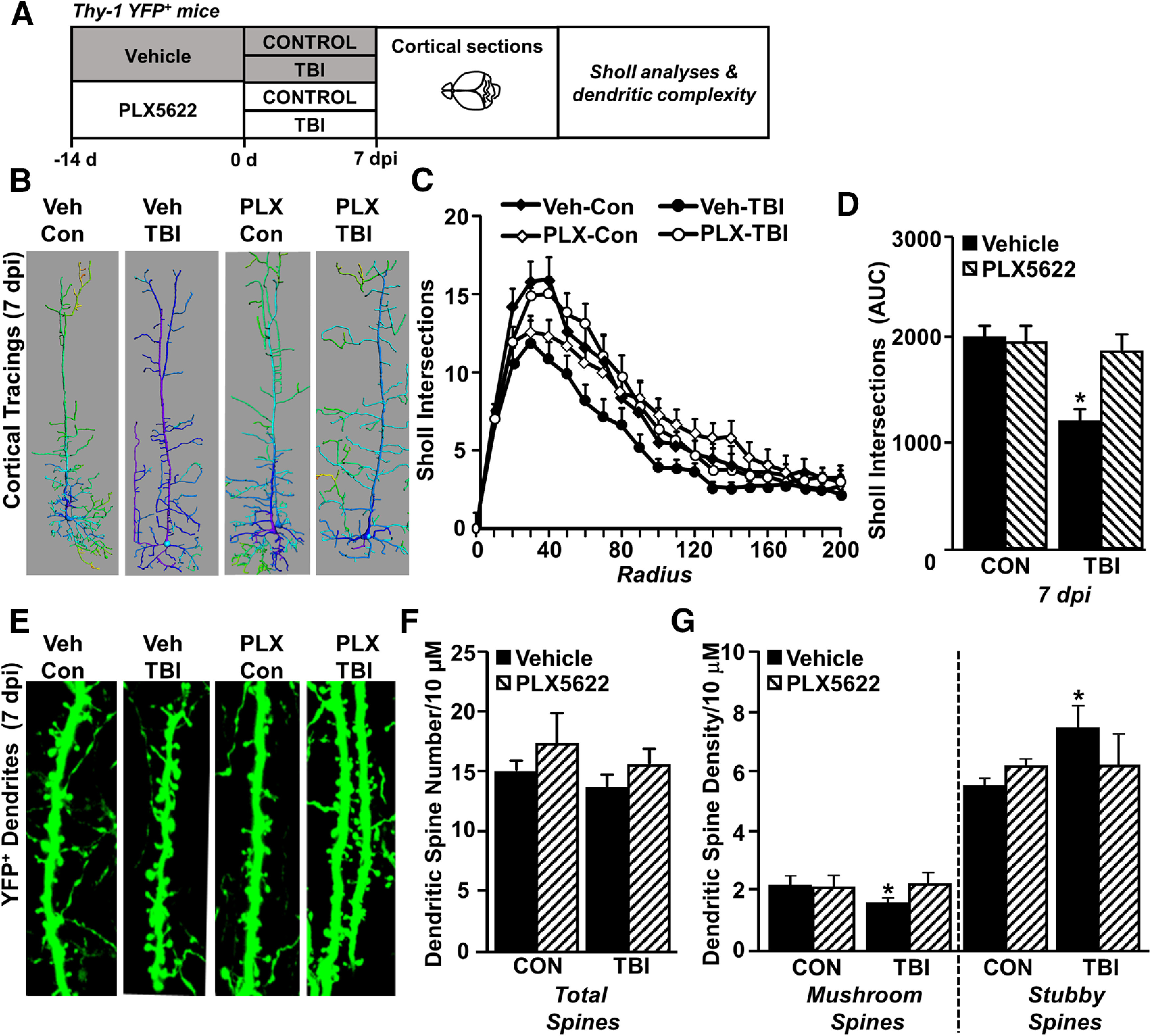
Increased dendritic remodeling 7 dpi was dependent on microglia. A, Adult Thy-1 GFP+ mice were provided diets formulated with either vehicle (Veh) or a CSF1R antagonist (PLX5622) for 14 d. Next, mice were uninjured (Con) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. At 7 dpi, mice were perfused, fixed, and brains were cryosectioned and mounted on slides for microscopy analysis of dendritic density and morphology (n = 6). B, Representative images of cortical neuron reconstructions (generated in Imaris) at 7 dpi from each treatment group. C, Sholl analysis of intersections by radius in the cortex. D, Sholl intersections area under the curve. E, Images of Thy-1 GFP + dendrites 7 dpi. F, Cortical dendritic spine number. G, Dendritic spine density classified by mature mushroom or immature stubby spines. Bars represent mean ± SEM. Means with * are significantly different from Veh-Con group (p < 0.05).
TBI-associated deficits in CAPs 30 dpi were dependent on microglia
We provide novel mRNA evidence of inflammation and neuronal pathology 7 and 30 dpi and demonstrate that neuronal RNA expression is influenced by microglia after TBI. The corpus callosum contains axons from neurons in cortical layers that are injured following diffuse TBI (Ramos et al., 2008). Thus, we examined neuronal connectivity through the corpus callosum 7 and 30 dpi in the presence or absence of microglia (Fig. 9A). CAPs were stimulated and recorded from ex vivo brain slices from the corpus callosum (Fig. 9A). Representative tracings show distinct N1 and N2 components of the action potential, with control and TBI. These data were interpreted to indicate that N1 represents fast-conducting fibers (large, myelinated) and N2 represents slow-conducting fibers (small, unmyelinated).
Figure 9.

TB1-associated deficits in neuronal connectivity 30 dpi were microglia dependent. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. A, At 7 and 30 dpi, mice were killed and CAP was determined from ex vivo preparation of the corpus callosum (n = 6). Representative N1 and N2 tracings of CAP from control and TB1 mice 7 dpi are shown. Graphs reflect average recording amplitude across range of stimulus intensities for (B) N1, (C) N2, and (D) N2/N1 ratio. Recorded values for maximum stimulus intensity (2 mA) are shown for (E) N2/N1 ratio. Graphs reflect averages ± SEM. Means with * are significantly different from Veh-Con group (p < 0.05). Means with # tend to be significantly different (p = 0.06) from Veh-Con group.
The N1 and N2 components of the action potential are shown for each treatment group at 7 and 30 dpi (Fig. 9B,C). The N2 CAPs tended to be reduced by TBI 7 and 30 dpi (p = 0.08, for each; Fig. 9C). The main effect of TBI was a suppression of the N2/N1 ratio 7 dpi and 30 dpi (F(4,32) = 5.5, p = 0.002; Fig. 9D). Post hoc analyses confirmed that the N2/N1 ratio was decreased in mice subjected to TBI 7 dpi and 30 dpi (Veh-TBI groups) compared with controls (p < 0.001 for 7 dpi; p = 0.018 for 30 dpi). At 7 dpi, the suppression of the N2/N1 ratio was unaffected by microglial depletion (PLX-TBI 7 dpi vs control p < 0.001). By 30 dpi, the TBI-induced reduction in N2/N1 ratio was no longer evident in PLX-treated mice (PLX-TBI 30 dpi vs control p = 0.712). The effect of TBI and the reversal by PLX 30 dpi were evident at the maximum stimulus (2 mA) and the N2/N1 ratio was at control levels in PLX-TBI mice, but not Veh-TBI mice (p = 0.02; Fig. 9E). Thus, impaired CAP at a chronic time point after TBI (30 dpi) was dependent on microglia.
TBI-associated deficits in memory 30 dpi were dependent on microglia. We provide evidence of microglia-mediated inflammation in the cortex suppresses neuronal homeostatic gene expression and structural/electrophysiologic function. We next sought to determine whether cortex-dependent sensorimotor and cognitive behaviors were affected by microglia up to 30 dpi (Fig. 10A). To determine sensorimotor function, the accelerating Rotarod and horizontal bar tests were used. These tests require integration of sensory and motor inputs and depend on intact cortical function (Cao et al., 2015). There were, however, no influences of either TBI or PLX5622 on sensorimotor function in the accelerating rotarod (Fig. 10B) or latency to escape the horizontal bar test (Fig. 10C) after mFPI. Other sensorimotor behaviors were also examined, but no long-lasting effects of either TBI or PLX were detected (data not shown). Thus, there were no major long-term deficits in locomotion or balance after diffuse TBI.
Figure 10.

TB1-associated deficits in NOR 30 dpi were microglia dependent. A, Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were maintained on vehicle or PLX diet for the duration of the experiment. In the first study (n = 6), mice were subjected to the accelerating Rotarod and wire-hang test at multiple time points (1, 7, 14, 21, and 28 dpi). In the second study (n = 10), mice were treated as above and NOL and recognition was assessed 28–31 dpi. B, Latency to fall from the accelerating Rotarod and (C) latency to escape from horizontal bar were determined. In a separate cohort, mice were used in the object location and recognition tests. At 30 dpi, (D) total time spent exploring, (E) percent of time spent investigating the novel location, and (F) discrimination index for the novel location were determined. At 31 dpi, (G) total time spent exploring, (H) percent of time spent investigating the novel object, and (I) discrimination index for the novel object were determined. Graphs represent mean ± SEM. Means with * are significantly different from vehicle controls (p < 0.05).
NOL and novel object recognition (NOR) were determined at 30 and 31 dpi, respectively. NOL/NOR are cognitive assessments that depend on intact hippocampal and cortical function (Antunes and Biala, 2012). The first phase of the test examined object location (hippocampal) and the second phase examined NOR (cortical; Fig. 10A). There were no significant differences in total time spent exploring in either phases of testing between the four experimental groups (Fig. 10D,G). In the “location” phase of testing, there was a tendency for an interaction between TBI and PLX intervention (F(1,40) = 3.65, p = 0.063). Post hoc analysis confirmed that Veh-TBI mice investigated the novel location for proportionally less time compared with all other groups, including the PLX-TBI mice (vs Veh-Con p < 0.001; vs PLX-Con p = 0.001; vs PLX-TBI p = 0.026; Fig. 10E). These effects were also evident in the discrimination index for the NOL (Fig. 10F), indicating that the Veh-TBI mice spent significantly more time with the object in familiar location. Similar to the novel location trials, there was an interaction between TBI and PLX administration (F(1,39) = 18.7, p = 0.0001) for NOR aspect of testing. Post hoc analysis confirmed that Veh-TBI mice investigated the novel object for less time compared with all other groups, including the PLX-TBI group (vs Veh-Con p < 0.001; vs PLX-Con p < 0.001; vs PLX-TBI p < 0.001; Fig. 10H). These differences were reflected in a suppressed discrimination index for the novel object in Veh-TBI mice compared with all other groups (Fig. 10I). PLX-TBI mice did not have these reductions in time spent investigating the novel object or discrimination index. Collectively, these data indicate there was TBI related decline in cortical/hippocampal memory 30 dpi, which was dependent on microglia.
Discussion
The goals of this study were to determine how microglial contributions to postinjury inflammation and pathology evolved over time and how microglia influence neuronal dysfunction after diffuse TBI. Novel mRNA data showed that cortical inflammation evolved from acute (1 dpi) to subacute (7 dpi), then to chronic (30 dpi) time points after mFPI. Single-cell RNA sequencing of cortical cells 7 dpi revealed distinct clusters of microglia associated with inflammation, robust type-1 interferon signaling, and damage/neuropathology. In addition to characterizing the inflammatory response, we provide direct evidence of microglia-mediated inflammation causing neuronal dysfunction after TBI. Analysis of scRNA-sequenced cortical neurons showed suppression of genes associated with dopamine signaling, long-term potentiation, calcium signaling, and synaptogenesis. Microglial depletion with PLX5622 prevented the majority of these transcripts from being suppressed after TBI. There were functional deficits in neuronal plasticity 7 dpi and neuronal connectivity 30 dpi. These TBI-associated impairments were blocked by microglial depletion and corroborated by behavioral studies showing that chronic postinjury cognitive impairment was prevented in mice without microglia.
Microglial depletion prevented inflammatory and neuropathology-associated gene expression at subacute (7 dpi) and chronic (30 dpi) time points after injury, while many acute (1 dpi) changes were microglia independent. This suggests the acute transcriptional response to injury may depend on the physical forces of injury while evolving inflammatory and immune processes drive progression from subacute to chronic injury. Critically, microglial depletion with PLX5622 not only influenced genes expressed by microglia, but also genes largely expressed by other CNS cells including neurons and oligodendrocytes. We used the Barres Laboratory Brain RNA-seq Mus musculus dataset (http://www.brainrnaseq.org) to code genes by predominant cell type of expression. Increased transcripts were predominantly expressed by microglia (i.e., Clec7a, Trem2, Tlr4, Ifi27l2a, Stat1) while suppressed transcripts were attributed to neurons (i.e., Cx3cl1, Drd1, Drd2, Trpv1) at both 7 and 30 dpi. One limitation of these data is that use of a predetermined neuropathology panel by NanoString can bias these findings, thus our confirmatory studies using scRNAseq are an important complement. Nonetheless, these data corroborate previous evidence showing microglia become “primed” after TBI and express higher levels of innate immune markers (Fenn et al., 2014; Muccigrosso et al., 2016) and suggest that development of primed microglia occurs alongside suppression of neuronal transcriptional homeostasis.
Single-cell RNA sequencing demonstrated that interferon-responsive gene expression was (Sen et al., 2020) a critical pathway within microglia 7 dpi. Consistent with IPA, trauma-related clusters 6 and 8 were defined by high expression of genes related to interferon signaling or CNS injury. Microglia respond to type-1 interferons via IFNα receptors (IFNAR1, IFNAR2) and express myriad interferon response factors (IRFs). Sen et al. (2020) recently demonstrated that neuronal stress after contusion TBI causes increased stimulator of interferon genes (STING) expression in damaged neurons, which in turn promotes neuronal type-1 interferon release and subsequent microglial cytokine/chemokine release. STING was separately shown to be expressed in postmortem human tissue at late postinjury time points (Abdullah et al., 2018). IFNβ knock-out reduced lesion volume, prevented hippocampal neuron loss, and ameliorated chronic behavioral impairments (Barrett et al., 2020). Importantly, these studies used CCI, which is associated with cortical tissue loss and blood brain barrier disruption, unlike mFPI, which models diffuse injury and is not associated with lesion formation. Nonetheless, similar STING upregulation may occur in the cortex after diffuse TBI, where neurons are injured (Greer et al., 2011) and there is evidence of cell-membrane permeability (Lafrenaye et al., 2012) and mitochondrial dysfunction (Harris et al., 2001). In our scRNAseq dataset we were unable to identify specific cell types that produced type-1 interferons 7 dpi. More acute timepoints may better capture interferon expression if it is transient after injury. Nonetheless, our previous findings showed that IRF expression was microglia dependent (Witcher et al., 2018). Here, we show that subsets of microglia were robustly responsive to IFN signals 7 dpi, which are likely initiated by neurons and glia in response to cellular and mechanical damage following TBI.
PLX5622-mediated depletion was used to determine the global contribution of microglia to postinjury neuropathology and neuronal dysfunction; however, the paradigm was not designed to be therapeutic and microglia remaining after PLX5622-mediated depletion have a distinct mRNA profile. For instance, cluster 2 and 7 microglia were enriched in PLX5622-treated groups and expressed an overabundance of mRNA encoding ribosomal proteins (Rpl34, Rpl 32, Rps28, Rps19) or genes involved in cellular proliferation (Ube2c, Cenpa, tubulin, and actin components). Ribosomal proteins are implicated in p53-mediated programmed cell death, tumorigenesis, and abnormal cell proliferation (Guimaraes and Zavolan, 2016). Our data indicate that PLX causes a state of constant depletion and proliferation of microglia and may explain how microglia can repopulate the brain within 1–3 d after PLX5622 has been removed (Elmore et al., 2014; Rice et al., 2017; Weber et al., 2019). While consistent PLX5622 administration before and postinjury was a valuable tool in our studies, other groups have used microglial depletion and rapid repopulation to reverse microglial dysregulation after murine TBI. Willis et al. (2020) recently demonstrated in a mouse contusion TBI model that microglial depletion and subsequent repopulation shifted microglia to a neuroprotective phenotype, improving learning deficits in an IL6-dependent manner. In a separate study, microglial depletion and repopulation promoted motor and cognitive behavioral recovery at chronic postinjury time points (Henry et al., 2020). Therefore, promoting microglial turnover may have therapeutic benefit.
Single-cell RNA sequencing allows for unprecedented detection of CNS heterogeneity, but is insufficient to answer all relevant questions. To detect changes in single-cell gene expression, expression levels must be robust. While we detected differential expression in major CNS cell types, we were unable to perform in-depth analysis of small cell populations. Relative cell-type distribution is skewed based on ease of dissociation: microglia and astrocytes are overrepresented while neurons and oligodendrocytes are underrepresented. Despite these limitations, our primary conclusions of scRNAseq data are consistent with our NanoString datasets previously presented and included here (Witcher et al., 2018).
TBI and microglia-mediated inflammation suppressed over 200 genes in cortical neurons. Genes with reduced expression included neuronal regulators of microglia (Cd200, Cx3cl1), calcium signaling (Rab3, Calm2, Syt5, Adcyl, Caly), redox signaling (Prrmcl, SpeXV), and synaptic/dendritic plasticity (Mef2c, Sncb, Map1b, Nsg1, Aplp, Ndnm, Nell2, Maphs). Microglial depletion reversed 95% of these neuronal alterations 7 dpi. Furthermore, the TBI effect in neurons showed evidence of increased protein degradation (26s proteasome and glutamate signaling; FMR1) and suppressed redox regulation (ATF6, HIF1A, XBO1, NFE2l2), growth/survival (IGFR1, MKNK1, TGFB1, ESRRA), and plasticity (NRF1, ADORA2A). This neuronal suppression pattern is in contrast to astrocytes and oligodendrocytes both in magnitude and directionality: far fewer genes were differentially expressed compared with neurons and both glial populations had mixed increased/decreased expression changes in genes related to reactivity or homeostasis. TBI-induced changes in neuronal genes and pathways were interpreted to represent a microenvironment that has higher glutamate signaling with reduced calcium signaling, synaptogenesis, and dendritic restructuring.
TBI-induced changes in neuronal transcripts were corroborated by structural, electrophysiological, and behavioral studies. There was evidence of cortical dendritic restructuring 7 dpi, with a reduction in overall dendrite complexity and reduction in mature spines. This time point and location is also when microglia become rod-shaped and align with apical pyramidal dendrites after mFPI (Witcher et al., 2018). Dendritic complexity and maturity were not altered by TBI in the absence of microglia. Expression of complement ligands (C1qa, C1qb) and receptors (Cd11b) were increased in the NanoString and scRNAseq datasets. While we found that spine morphology changes after TBI is microglia-dependent, it is unclear whether this is complement-mediated in the context of TBI (Schafer et al., 2012; Hong et al., 2016; Vasek et al., 2016). We observed a persistent reduction in N2/N1 ratio following injury, which was in part driven by reduction in N2 amplitude (conduction of small diameter and/or unmyelinated fibers). These findings were consistent with previous data collected in rats following mFPI (Reeves et al., 2005). The extension here is that microglial elimination reversed these CAP impairments 30 dpi, indicating that microglia-mediated inflammation disrupts either fiber integrity or conduction.
A final important discussion point is that the RNA and functional profiles of reduced neuronal plasticity corresponded with functional deficits in memory. NOL and recognition depend on both cortical and hippocampal memory (Vogel-Ciernia and Wood, 2014). Thus, the changes in the gene level and in dendritic processes were associated with cortical and hippocampal cognitive impairment 30 dpi. While we anticipated that the cortical inflammation would also affect somatosensory parameters, this was not the case. Cognitive impairments, however, are well established in rodent models of TBI and there is mild cognitive decline after diffuse brain injury by 30 dpi (Bachstetter et al., 2015; Muccigrosso et al., 2016). Here, we show that microglial depletion was protective in preventing TBI-associated cognitive deficits 30 dpi.
Collectively, these studies provide novel evidence that microglia not only respond to and mediate inflammatory signaling but influence the neuronal response to TBI. These studies present a comprehensive picture of microglia as critical mediators of chronic inflammation that corresponded with suppression of neuronal homeostasis at the transcriptional, structural, physiological, and functional level.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) Grant R56-NS-090311 and the National Institute of Aging (NIA) Grant R01-AG-051902 (to J.P.G.). In addition, this work was supported by a P30 Core Grant (NINDS P30-NS-045758). K.G.W. and S.M.O. were supported by the National Institute of Dental and Craniofacial Research Training Grant T32-DE-014320 and K.G.W. by the Ohio State University Presidential Fellowship. T.C. was supported by the Thailand Research Fund-Royal Golden Jubilee Program Grant PHD/0146/2558 TC&SCC. We thank Plexxikon, Inc. for the use of PLX5622, Dr. Paolo Fadda at the Ohio State University Comprehensive Cancer Center Genomics Shared Resource (P30-CA-016058), and the Chronic Brain Injury Discovery Themes Initiative at Ohio State University. Our scRNA-sequencing (scRNAseq) analysis was made possible by an allotment of resources from the Ohio Supercomputing Center.
The authors declare no competing financial interests.
References
- Abdullah A, Zhang M, Frugier T, Bedoui S, Taylor JM, Crack PJ (2018) STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J Neuroinflammation 15:323. 10.1186/s12974-018-1354-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes M, Biala G (2012) The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process 13:93–110. 10.1007/s10339-011-0430-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ (2013) The p38α MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci 33:6143–6153. 10.1523/JNEUROSCI.5399-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Webster SJ, Goulding DS, Morton JE, Watterson DM, Van Eldik LJ (2015) Attenuation of traumatic brain injury-induced cognitive impairment in mice by targeting increased cytokine levels with a small molecule experimental therapeutic. J Neuroinflammation 12:69. 10.1186/s12974-015-0289-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Ighodaro ET, Hassoun Y, Aldeiri D, Neltner JH, Patel E, Abner EL, Nelson PT (2017) Rod-shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol Aging 52:98–105. 10.1016/j.neurobiolaging.2016.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JP, Henry RJ A, Shirey K, Doran SJ, Makarevich OD, Ritzel RR, Meadows VA, Vogel SN, Faden AI, Stoica BA, Loane DJ (2020) Interferon-β plays a detrimental role in experimental traumatic brain injury by enhancing neuroinflammation that drives chronic neurodegeneration. J Neurosci 40:2357–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36:411–420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao VY, Ye Y, Mastwal S, Ren M, Coon M, Liu Q, Costa RM, Wang KH (2015) Motor learning consolidates Arc-expressing neuronal ensembles in secondary motor cortex. Neuron 86:1385–1392. 10.1016/j.neuron.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chunchai T, Thunapong W, Yasom S, Wanchai K, Eaimworawuthikul S, Metzler G, Lungkaphin A, Pongchaidecha A, Sirilun S, Chaiyasut C, Pratchayasakul W, Thiennimitr P, Chattipakorn N, Chattipakorn SC (2018) Decreased microglial activation through gut-brain axis by prebiotics, probiotics, or synbiotics effectively restored cognitive function in obese-insulin resistant rats. J Neuroinflammation 15:11. 10.1186/s12974-018-1055-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin JM, Wang Y, Minn I, Bienko N, Ambinder EB, Xu X, Peters ME, Dougherty JW, Vranesic M, Koo SM, Ahn HH, Lee M, Cottrell C, Sair HI, Sawa A, Munro CA, Nowinski CJ, Dannals RF, Lyketsos CG, Kassiou M, Smith Get al. (2017) Imaging of Glial Cell Activation and White Matter Integrity in Brains of Active and Recently Retired National Football League Players. JAMA neurology 74:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM (2013) Measuring motor coordination in mice. J Vis Exp. Advance online publication. Retrieved May 29, 2013. doi: 10.3791/2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I (2018) Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173:1073–1081. 10.1016/j.cell.2018.05.003 [DOI] [PubMed] [Google Scholar]
- Denninger JK, Smith BM, Kirby ED (2018) Novel object recognition and object location behavioral testing in mice on a budget. J Vis Exp (141):10.3791/58593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. 10.1016/s0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP (2014) Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 76:575–584. 10.1016/j.biopsych.2013.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Skendelas JP, Moussa DN, Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP (2015) Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J Neurotrauma 32:127–138. 10.1089/neu.2014.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorse KM, Lafrenaye AD (2018) The importance of inter-species variation in traumatic brain injury-induced alterations of microglial-axonal interactions. Front Neurol 9:778. 10.3389/fneur.2018.00778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz S, Bribian A, López-Mascaraque L, Götz M, Grothe B, Kunz L (2021) Heterogeneity of astrocytes: electrophysiological properties of juxtavascular astrocytes before and after brain injury. Glia 69:346–361. 10.1002/glia.23900 [DOI] [PubMed] [Google Scholar]
- Greer JE, McGinn MJ, Povlishock JT (2011) Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci 31:5089–5105. 10.1523/JNEUROSCI.5103-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualtieri T, Cox DR (1991) The delayed neurobehavioural sequelae of traumatic brain injury. Brain Inj 5:219–232. 10.3109/02699059109008093 [DOI] [PubMed] [Google Scholar]
- Guimaraes JC, Zavolan M (2016) Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol 17:236. 10.1186/s13059-016-1104-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Dey A, Yu X, Stranahan AM (2016) Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav Immun 51:230–239. 10.1016/j.bbi.2015.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LK, Black RT, Golden KM, Reeves TM, Povlishock JT, Phillips LL (2001) Traumatic brain injury-induced changes in gene expression and functional activity of mitochondrial cytochrome C oxidase. J Neurotrauma 18:993–1009. 10.1089/08977150152693692 [DOI] [PubMed] [Google Scholar]
- Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, Szeto GL, Wu J, Stoica BA, Faden AI, Loane DJ (2020) Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J Neurosci 40:2960–2974. 10.1523/JNEUROSCI.2402-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen L, Portin R, Isoniemi H, Helenius H, Kurki T, Tenovuo O (2006) Longitudinal cognitive changes in traumatic brain injury: a 30-year follow-up study. Neurology 66:187–192. 10.1212/01.wnl.0000194264.60150.d3 [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji P, Schachtschneider KM, Schook LB, Walker FR, Johnson RW (2016) Peripheral viral infection induced microglial sensome genes and enhanced microglial cell activity in the hippocampus of neonatal piglets. Brain Behav Immun 54:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BJ, Farkas O, Lifshitz J, Povlishock JT (2006) Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp Neurol 198:350–360. 10.1016/j.expneurol.2005.12.017 [DOI] [PubMed] [Google Scholar]
- Kelley BJ, Lifshitz J, Povlishock JT (2007) Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol 66:989–1001. 10.1097/NEN.0b013e3181588245 [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr, Tugendreich S (2014) Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30:523–530. 10.1093/bioinformatics/btt703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, et al. (2017) The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47:566–581.e9. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RG, Gao S, Juengst SB, Wagner AK, Fabio A (2018) The effects of post-traumatic depression on cognition, pain, fatigue, and headache after moderate-to-severe traumatic brain injury: a thematic review. Brain Inj 32:383–394. 10.1080/02699052.2018.1427888 [DOI] [PubMed] [Google Scholar]
- Lafrenaye AD, McGinn MJ, Povlishock JT (2012) Increased intracranial pressure after diffuse traumatic brain injury exacerbates neuronal somatic membrane poration but not axonal injury: evidence for primary intracranial pressure-induced neuronal perturbation. J Cereb Blood Flow Metab 32:1919–1932. 10.1038/jcbfm.2012.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrenaye AD, Todani M, Walker SA, Povlishock JT (2015) Microglia processes associate with diffusely injured axons following mild traumatic brain injury in the micro pig. J Neuroinflammation 12:186. 10.1186/s12974-015-0405-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz J, Kelley BJ, Povlishock JT (2007a) Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J Neuropathol Exp Neurol 66:218–229. 10.1097/01.jnen.0000248558.75950.4d [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Witgen B, Grady M (2007b) Acute cognitive impairment after lateral fluid percussion brain injury recovers by 1 month: evaluation by conditioned fear response. Behav Brain Res 177:347–357. 10.1016/j.bbr.2006.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI (2014) Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol 73:14–29. 10.1097/NEN.0000000000000021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, Ramirez-Chan K, Wang Y, Roeth RM, Sucaldito AD, Sobol CG, Quan N, Sheridan JF, Godbout JP (2018) Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry 23:1421–1431. 10.1038/mp.2017.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millis SR, Rosenthal M, Novack TA, Sherer M, Nick TG, Kreutzer JS, High WM Jr, Ricker JH (2001) Long-term neuropsychological outcome after traumatic brain injury. J Head Trauma Rehabil 16:343–355. 10.1097/00001199-200108000-00005 [DOI] [PubMed] [Google Scholar]
- Morel L, Men Y, Chiang MSR, Tian Y, Jin S, Yelick J, Higashimori H, Yang Y (2019) Intracortical astrocyte subpopulations defined by astrocyte reporter Mice in the adult brain. Glia 67:171–181. 10.1002/glia.23545 [DOI] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F (2014) Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol 75:241–254. 10.1002/ana.24064 [DOI] [PubMed] [Google Scholar]
- Muccigrosso MM, Ford J, Benner B, Moussa D, Burnsides C, Fenn AM, Popovich PG, Lifshitz J, Walker FR, Eiferman DS, Godbout JP (2016) Cognitive deficits develop 1 month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun 54:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ (2009) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132:288–295. 10.1093/brain/awn109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Pestana F, Edwards-Faret G, Belgard TG, Martirosyan A, Holt MG (2020) No longer underappreciated: the emerging concept of astrocyte heterogeneity in neuroscience. Brain Sci 10:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ (2011) Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70:374–383. 10.1002/ana.22455 [DOI] [PubMed] [Google Scholar]
- Ramos RL, Tam DM, Brumberg JC (2008) Physiology and morphology of callosal projection neurons in mouse. Neuroscience 153:654–663. 10.1016/j.neuroscience.2008.02.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Povlishock JT (2005) Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol 196:126–137. 10.1016/j.expneurol.2005.07.014 [DOI] [PubMed] [Google Scholar]
- Rice RA, Pham J, Lee RJ, Najafi AR, West BL, Green KN (2017) Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 65:931–944. 10.1002/glia.23135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RK, Griffiths DR, Lifshitz J (2016) Midline (central) fluid percussion model of traumatic brain injury. Methods Mol Biol 1462:211–230. 10.1007/978-1-4939-3816-2_13 [DOI] [PubMed] [Google Scholar]
- Salmond CH, Menon DK, Chatfield DA, Pickard JD, Sahakian BJ (2006) Changes over time in cognitive and structural profiles of head injury survivors. Neuropsychologia 44:1995–1998. 10.1016/j.neuropsychologia.2006.03.013 [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Saha P, Gupta R, Foley LM, Jiang T, Abakumova OS, Hitchens TK, Sen N (2020) Aberrant ER stress induced neuronal-IFNβ elicits white matter injury due to microglial activation and T-cell infiltration after TBI. J Neurosci 40:424–446. 10.1523/JNEUROSCI.0718-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver JM, McAllister TW, Arciniegas DB (2009) Depression and cognitive complaints following mild traumatic brain injury. Am J Psychiatry 166:653–661. 10.1176/appi.ajp.2009.08111676 [DOI] [PubMed] [Google Scholar]
- Singh R, Mason S, Lecky F, Dawson J (2018) Prevalence of depression after TBI in a prospective cohort: the SHEFBIT study. Brain Inj 32:84–90. 10.1080/02699052.2017.1376756 [DOI] [PubMed] [Google Scholar]
- Till C, Colella B, Verwegen J, Green RE (2008) Postrecovery cognitive decline in adults with traumatic brain injury. Arch Phys Med Rehabil 89:S25–S34. 10.1016/j.apmr.2008.07.004 [DOI] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS, Schmidt RE, et al. (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534:538–543. 10.1038/nature18283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Wood MA (2014) Examining object location and object recognition memory in mice. Curr Protoc Neurosci 69:31.1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MD, McKim DB, Niraula A, Witcher KG, Yin WY, Sobol CG, Wang YF, Sawicki CM, Sheridan JF, Godbout JP (2019) The influence of microglial elimination and repopulation on stress sensitization induced by repeated social defeat. Biol Psychiatry 85:667–678. 10.1016/j.biopsych.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis EF, MacDonald KPA, Nguyen QH, Garrido AL, Gillespie ER, Harley SBR, Bartlett PF, Schroder WA, Yates AG, Anthony DC, Rose-John S, Ruitenberg MJ, Vukovic J (2020) Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell 180:833–846.e16. 10.1016/j.cell.2020.02.013 [DOI] [PubMed] [Google Scholar]
- Witcher KG, Bray CE, Dziabis JE, McKim DB, Benner BN, Rowe RK, Kokiko-Cochran ON, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP (2018) Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia 66:2719–2736. 10.1002/glia.23523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witgen B, Lifshitz J, Grady M (2006) Inbred mouse strains as a tool to analyze hippocampal neuronal loss after brain injury: a stereological study. J Neurotrauma 23:1320–1329. 10.1089/neu.2006.23.1320 [DOI] [PubMed] [Google Scholar]
- Wofford KL, Harris JP, Browne KD, Brown DP, Grovola MR, Mietus CJ, Wolf JA, Duda JE, Putt ME, Spiller KL, Cullen DK (2017) Rapid neuroinflammatory response localized to injured neurons after diffuse traumatic brain injury in swine. Exp Neurol 290:85–94. 10.1016/j.expneurol.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J (2012) Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation 9:247. 10.1186/1742-2094-9-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time course of GFAP expression in the cortex after TB1 with and without microglia. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or a CSF1R antagonist (PLX5622) for 14 d. Next, mice were uninjured (Con) or were subjected to mFPI (TBI; n = 4). Mice were maintained on vehicle or PLX diet for the duration of the experiment. At each endpoint (1, 7, or 30 dpi), mice were perfused, fixed, and cortical tissue was cryosectioned and labeled for GFAP (subset of astrocytes). A, Representative images of GFAP labeling in the lateral cortex. Scale bar: 50 μm. B, Quantification of average GFAP+ percent-area labeled. Bars represent the mean ± SEM. Means with * are significantly different from Veh-Con (p < 0.05). Download Figure 1-1, TIF file (1.3MB, tif) .
Table of NanoString neuropathology raw and normalized data. Table contains raw and normalized count data for full NanoString Neuropathology dataset. Download Figure 1-2, XLSX file (845.9KB, xlsx) .
Table of IPA results from NanoString Neuropathology analysis. Table contains output from IPA for each differential expression comparison. Download Figure 1-3, XLSX file (271.6KB, xlsx) .
Distribution of experimental groups across cell-type clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. tSNE plots represent distribution of cells across the four experimental groups: (1) Veh-CON, (2) PLX-CON, (3) Veh-TB1, and (4) PLX-TB1. Download Figure 3-1, TIF file (1.5MB, tif) .
Table of cluster markers and differential expression results. Table contains output from overall cluster marker analysis, cluster marker analysis for individual cell types, and differential expression between Veh-CON and Veh-TB1 groups for each cell type. Download Figure 3-2, XLSX file (426.4KB, xlsx) .
Heatmap of top genes expressed in microglial clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. Heatmap reflects top genes expressed by each Tmem119+ microglia cluster. Download Figure 4-1, TIF file (16.8MB, tif) .
Heatmap of top genes expressed in neuronal clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. Heatmap reflects top genes expressed by each Meg3+ microglia cluster. Download Figure 7-1, TIF file (15.4MB, tif) .
Distribution of experimental groups across neuronal clusters. Adult C57BL/6 mice were provided diets formulated with either vehicle (Veh) or PLX5622 (PLX) for 14 d. Next, mice were uninjured (control) or were subjected to mFPI (TB1). Mice were killed 7 dpi, and single cells were isolated and sequenced as described in Figure 3. Graph reflects distribution of Meg3+ neurons from each experimental condition across the 11 neuronal clusters. Download Figure 7-2, TIF file (618.3KB, tif) .