

Abstract
Coronary artery disease (CAD) is one of the most common causes of death worldwide. The introduction of percutaneous revascularization has revolutionized the therapy of patients with CAD. Despite the advent of drug-eluting stents, restenosis remains the main challenge in treating patients with CAD. In-stent restenosis (ISR) indicates the reduction in lumen diameter after percutaneous coronary intervention, in which the vessel’s lumen re-narrowing is attributed to the aberrant proliferation and migration of vascular smooth muscle cells (VSMCs) and dysregulation of endothelial cells (ECs). Increasing evidence has demonstrated that epigenetics is involved in the occurrence and progression of ISR. In this review, we provide the latest and comprehensive analysis of three separate but related epigenetic mechanisms regulating ISR, namely, DNA methylation, histone modification, and non-coding RNAs. Initially, we discuss the mechanism of restenosis. Furthermore, we discuss the biological mechanism underlying the diverse epigenetic modifications modulating gene expression and functions of VSMCs, as well as ECs in ISR. Finally, we discuss potential therapeutic targets of the small molecule inhibitors of cardiovascular epigenetic factors. A more detailed understanding of epigenetic regulation is essential for elucidating this complex biological process, which will assist in developing and improving ISR therapy.
Keywords: in-stent restenosis, epigenetics, DNA methylation, histone modification, non-coding RNAs, target therapy
Graphical Abstract
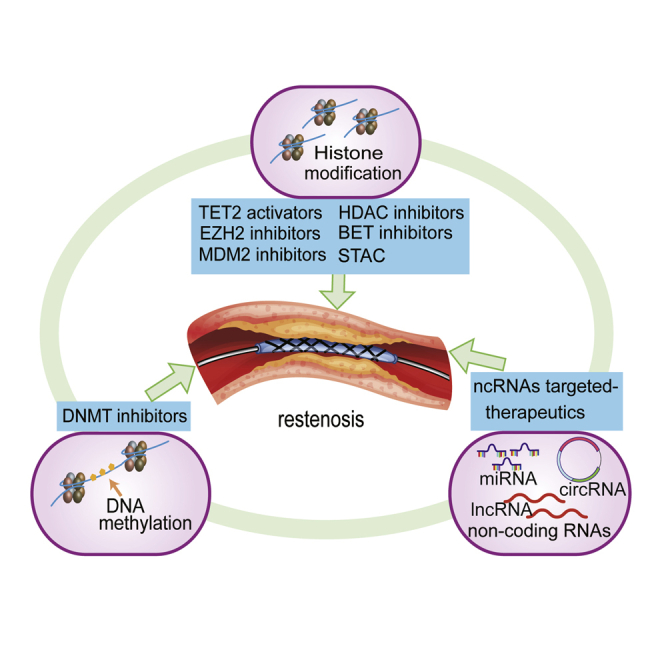
Yang et al. review the mechanism of epigenetic regulation of restenosis and discuss molecule regulators for epigenetic modification. This review provides important insights into the underlying mechanisms of restenosis, and it provides potential regulatory therapeutic targets for proliferative vascular diseases, especially for restenosis.
Main text
Coronary artery disease (CAD) represents the most significant cause of morbidity and mortality and poses a major health and economic burden worldwide.1 The 2017 Global Burden of Disease (GBD) study noted that cardiovascular diseases were responsible for most deaths due to non-communicable diseases globally, with approximately 178,000 deaths, and the estimated cost of CAD treatment is expected to increase by 100% from 2015 to 2030.2 Currently, percutaneous coronary intervention (PCI) is used for treating patients with CAD, which remains the focus of intensive research and development.3 In the past, PCI has reduced the mortality accompanying CAD, particularly those cases related to acute myocardial infarction (AMI).4 However, restenosis due to stent implantation is one of the major drawbacks of this therapy, which may lead to vascular injury and neointima formation, subsequently causing stent implantation failure, necessitating repeat intervention.
Restenosis, defined as the development of luminal narrowing at previously stented regions, usually happens several months after PCI and is accompanied by arterial injury and neointimal formation.5 Initially, the restenosis rate of balloon angioplasty alone was approximately 40%, which was mostly associated with elastic recoil and neointimal hyperplasia.6 Therefore, coronary stents were introduced to overcome these limitations. Subsequently, the introduction of bare metal stents (BMSs) minimized dissections and elastic recoil. Nevertheless, the application of BMSs is limited by the high rate of in-stent restenosis (ISR), which is mainly caused by the generation of a neointima.6,7 The emergence of drug-eluting stents (DESs) (especially second-generation) and drug-coated balloons (DCBs) has further reduced the incidence of restenosis by <10%.8 However, current DESs cannot effectively repress the proliferation and migration of vascular smooth muscle cells (VSMCs) and cannot induce adequate re-endothelialization within the vasculature, which increases the risk of restenosis.4 Therefore, a better understanding of the underlying mechanisms and regulatory targets are required and warrant detailed investigation.
Epigenetic regulation has emerged as a novel direction to study human disease. Epigenetic modification is regulated on multiple sophisticated levels via various mechanisms, including DNA methylation, post-transcriptional histone modification, and non-coding RNAs (ncRNAs). Unlike genetic mutations, epigenetic alterations are largely reversible and vulnerable to nutritional and environmental factors, and they can return to their normal state via modifications and/or drug targeting.9 Hence, their functions, both as potential biomarkers and as novel therapeutic targets, have been extensively studied in recent years. In particular, several aspects of the vascular response to injury are regulated via epigenetic mechanisms, which modulate not only cell proliferation, differentiation, apoptosis, and inflammation, but also thrombosis and vascular remodeling (Figure 1). Interestingly, changes in gene expression levels have been observed in cardiovascular disease, so these abnormal expression patterns are thought to be associated with restenosis.10
Figure 1.

Overview of risk factors, pathogenesis, epigenetic regulation, and currently available targeted inhibitors associated with restenosis
Targeted inhibitors are shown may address pathophysiological pathways associated with the formation and development of restenosis following stenting in coronary artery disease.
In this review, we provide a new perspective regarding the various epigenetic mechanisms regulating ISR and therapies on epigenetic factors targeting ISR. We expect that our observations will contribute to clarify the biological processes of epigenetic regulation of restenosis, as well as provide new ideas for the treatment of restenosis.
Mechanisms of restenosis
Continuous efforts have been made to delineate the mechanism of ISR after stent implantation. Restenosis is a progressive phenomenon that begins after balloon dilation (barotrauma) is determined by PCI. The pathogenesis of restenosis can be subdivided mainly into three parts: (1) early elastic return (recoil) (ER), (2) vascular remodeling, and (3) neointimal hyperplasia. In addition, the presence of metallic struts leads to neointimal hyperplasia.8
ER and vascular remodeling
The internal and external elastic laminae of the human epicardial coronary arteries contain elastin fibers, which may cause ER after balloon hyperextension. ER usually occurs between a few seconds and a few minutes after the balloon is vented, resulting in a 40% reduction in the lumen area. Although angioplasty and stents themselves can cause overstretching injuries, stents can reduce acute recoil. Vascular remodeling is a process that occurs after acute arterial injury, constriction of the local arterial wall, and narrowing of the lumen in the injured vascular segment. However, the exact mechanism of arterial remodeling is not yet fully understood. There are several mechanisms that may explain arterial remodeling, including (1) changes in shear force, (2) oxidative stress, (3) endothelial dysfunction, (4) collagen metabolism, and (5) secretion of growth factors.11 Studies have demonstrated that blood vessels self-remodel in response to long-term flow changes, thus changing the lumen area to maintain a predetermined level of shear stress (OS).12 Interestingly, the presence of stents almost eliminates the two mechanisms of ER and vascular remodeling, while the presence of metal struts promotes neointimal hyperplasia.
Neointimal hyperplasia
The key pathophysiological phenomenon of restenosis after stent implantation is neointimal hyperplasia, which is predominantly due to VSMC proliferation and subsequent abluminal migration.13 Under physiological conditions, VSMCs are a highly specialized, almost fixed cell population whose primary function is to maintain vascular tension and ensure the ability for vasoconstriction. VSMCs are contractile in adult blood vessels and are characterized by a low proliferation rate (about 5%) and a group of specific contractile proteins, including smooth muscle myosin heavy chain (SMMHC), α-smooth actin (α-SMA), and calponin, among others. However, VSMCs have high plasticity and can easily change the phenotype from highly contractile to synthetic under specific stimuli, such as transforming growth factor (TGF)-β, endothelin-1 (ET-1), hyperhomocysteinemia, fibroblast growth factor (FGF), monocyte chemokine protein 1 (MCP-1), prostaglandin D2, and cyclic stretch,14 thus promoting abnormal proliferation and migration while reducing the expression level of contractile proteins, resulting in lumen reduction.15
ISR is primarily a nonspecific inflammatory response to vascular wall damage due to the continued “insult” of foreign elements (metal struts serving as a stent). The stent implantation causes a lesion in the endothelium, which is accompanied by consecutive endothelial denudation, re-endothelialization, and subsequent generation of the neo-endothelium.16 Meanwhile, the endothelial cells (ECs) are denuded, resulting in a series of inflammatory responses, such as platelet activation and circulating leukocyte recruitment, and the release of cytokines and growth factors, including FGF-2, platelet-derived growth factors (PDGFs), TGF-β, vascular endothelial growth factor (VEGF), and insulin-like growth factor-1 (IGF-1).17 These cytokines regulate the phenotypic transformation of VSMCs and change from contractile to synthetic, resulting in proliferation and migration, following which they enter the cell cycle and migrate to the intimal and extracellular matrix (ECM) to then form neointima.7,18
At present, paclitaxel, sirolimus, and zotarolimus are widely used as targeted drugs for restenosis.19 Paclitaxel has a cytostatic effect. The mode of action is the α and β unit polymerization of tubulin, thus stabilizing the microtubules needed for G2 conversion to the M phase. Paclitaxel inhibits the proliferation and migration of VSMCs by modifying the structure of the cytoskeleton.20 Sirolimus, also known as rapamycin, is a macrocyclic antibiotic with strong immunosuppressive properties. Zotarolimus is an analog of rapamycin. Sirolimus and its analog could inhibit the proliferation and migration of VSMCs by blocking the cell cycle from the G1 to S phase21,22 through suppressing rapamycin mammalian targets. These drugs reduce the proliferation of VSMCs but harm the endothelialization of scaffold strut support. Also, delivery of these drugs does not eradicate ISR completely, and more beneficial therapeutic targets should be explored.
Epigenetic mechanisms in ISR
Epigenetic regulation, which differs from genetic events in that the underlying DNA sequence is not changed, can have far-reaching effects on the transcription of genes and hence on cell lineage, function, and fate. In epigenetic regulation, DNA and histones are enzymatically modified by proteins known as “writers” (add modifying groups), “erasers” (remove modifying groups), and “readers” (recognize modifying groups).23 Writers include DNA methyltransferases (DNMTs), histone acetyltransferases (HATs), histone methyltransferases (HMTs), and E3 ligases, among others.23 Erasers constitute ten-eleven translocation (TET) enzymes, histone demethylases (HDMs), histone deacetylases (HDACs), and deubiquitinases (DUBs).23 Readers include ubiquitin (Ub)-like PHD and RING finger domain-containing 1 (UHRF1) and the bromodomain and extra-terminal (BET) domain-containing family.24 This multilateral regulation ensures robust execution of cellular functions, including VSMC proliferation and migration, and EC dysfunction.
DNA methylation
DNA methylation is one of the main epigenetic modulations involving direct chemical modification of DNA. The extent of DNA methylation is regulated by the balance in the activities of DNA-modifying enzymes, which either add (DNMTs) or remove methyl marks (DNA demethylases) from the DNA. In the following sections, we discuss the regulation of VSMCs and ECs via DNA methylation both in vivo and in vitro.
DNA methylation is maintained by the DNMT family, which is classified into two types: DNMT1 mainly maintains the DNA methylation pattern, whereas de novo DNA methylation is typically mediated by DNMT3A and DNMT3B.25 DNMTs are responsible for transferring methyl groups from S-adenosyl methionine (SAM) to the fifth carbon of a cytosine residue to form 5-methylcytosine (5mC). DNA methylation or hypermethylation inhibits transcription, whereas hypomethylation is associated with transcriptional activation.25
Aberrant DNA methylation affects the function of different cell types, such as VSMCs and ECs. Previous studies have reported that increased methylation of the promoter regions of Src homology 2-containing protein tyrosine phosphatase 1 (SHP-1) in oxidative low-density lipoprotein (ox-LDL)-treated VSMCs inhibited SHP-1 expression. The methylation level of the SHP-1 promoter was positively related to the risk of restenosis, suggesting that methylated SHP-1 is a novel target for preventing restenosis.26
Homocysteine (Hcy) is an independent risk factor for coronary and peripheral atherosclerosis and plays a key role in the regulation of vascular cell function. Hcy-induced VSMCs showed increased DNA methylation at the promoter of mitofusin-2 (MFN2), encoding a key transmembrane GTPase widely distributed in the mammalian mitochondrial outer membrane, which inhibited MFN2 transcription, resulting in VSMC proliferation. These studies provided novel insights regarding DNMT1-regulated VSMC proliferation during restenosis.27 In addition, DNA methylation coordinates with paracrine mechanisms to regulate cell function. Previous studies have shown that PDGFs modulate numerous pathophysiological events, including cell proliferation and migration, ECM accumulation, and production of pro- and anti-inflammatory regulators. Hcy-induced DNA hypomethylation is mediated mainly via inhibition of DNMT1 expression, which may increase the expression and paracrine secretion of PDGFs from ECs and further enhance the proliferation and migration of VSMCs. As discussed above, Hcy upregulates PDGF expression via DNA demethylation in ECs, affecting the crosstalk between ECs and VSMCs and thereby activating VSMCs.28 Previous studies have shown that VSMC contraction is dependent on ATP derived from mitochondrial respiration in animal arteries. DNMT1 translocates to the mitochondria and induces mitochondrial DNA (mtDNA) hypermethylation, resulting in repression of mtDNA transcription and mitochondrial dysfunction, and subsequently impairing VSMC contractility during restenosis. These results filled a knowledge gap between mtDNA methylation and VSMC function and may provide a molecular mechanism for further understanding of vascular homeostasis and dysfunction. In addition, they provide a potential therapeutic direction for the treatment of vascular disease.29
Furthermore, several studies have emphasized the importance of microRNAs (miRNAs) in the complex regulation of gene expression. Proteins harboring Uhrf1 induce VSMC proliferation, bind directly to hemimethylated DNA, and play a key role in maintaining DNA methylation by recruiting DNMT1 to these DNA sites. A study has demonstrated that miR-145 overexpression attenuates Uhrf1 expression. In a mouse model, intimal hyperplasia in the mouse carotid artery was attenuated by Uhrf1 short hairpin RNAs (shRNAs) or Uhrf1 VSMC-specific knockout. This clearly indicates that Uhrf1 may be a therapeutic target in vascular pathologies.30 In addition, the findings of this study provided insights regarding the miRNA-mediated regulation of DNA methylation, adding a new layer to the mechanisms regulating VSMC phenotype regulation.
Zhang et al.31 observed that the expression of miR-143 is negatively correlated with both DNMT3a mRNA and protein levels in Hcy-induced VSMCs. The effects of DNMT3a on the methylation status of miR-143 further confirmed the correlation between the expression of miR-143 and DNMT3a. In addition, studies have reported mutual regulation between miR-125b and DNMT3b, which can mediate Hcy-induced VSMC proliferation. On the one hand, DNMT3b is a target of miR-125b. On the other hand, Hcy-induced reduction in miR-125b increased the DNMT3b level and eventually caused the proliferation of VSMCs.32 Taken together, these studies indicated that complicated mechanisms of epigenetic crosstalk between DNA methylation and miRNA coordinately regulate VSMC proliferation.
A large body of evidence has shown that hemodynamic forces, including OS, regulate EC function, which is dysregulated by activation of mechanotransduction. Zhou et al.33 observed that OS induced hypermethylation of ECs by upregulating the expression and promoting the nuclear translocation of DNMT-1. This was reversed by 5-aza-2′-deoxycytidine treatment, which inhibits DNMT-1 and thereby hypermethylation. Consistent with these observations, the DNMT1 level and methylation were upregulated in the OS regions of the partially ligated carotid artery of model rats.34 This was the first study to demonstrate the disturbed blood flow-induced changes in ECs, which were regulated at the epigenomic level in a DNMT- and DNA methylation-dependent manner. Furthermore, this suggests that flow regulates gene expression in a DNMT-dependent manner and reveals new targets for regulating EC function. In addition, Zhang et al.35 also showed that OS-mediated DNMT1 overexpression and mammalian target of rapamycin (mTOR) were involved in this signaling pathway both in vitro and in vivo. DNMT1 regulates proliferative and inflammation molecules including proliferating cell nuclear antigen (PCNA), vascular cell adhesion molecule 1 (VCAM-1), and intercellular cell adhesion molecule 1 (ICAM-1). Furthermore, DNMT1 deficiency improved the dysregulation of ECs caused by disruption of EC proliferation and pro-inflammation. However, the detailed mechanism of OS inducing site-specific binding and distribution of DNMT1 in the genome remains to be elucidated.
Krüppel-like factor 4 (KLF4) is a transcription factor that is extensively expressed in various vascular cells, such as VSMCs, ECs, and other vascular cells. Jiang et al.36 reported that OS promoted hypermethylation of the KLF4 promoter by increasing the expression of DNMT3A and inhibiting KLF4 transcription in ECs. Furthermore, in accordance with in vitro data, the hypermethylation of the KLF4 promoter and the KLF4 and NOS3 gene bodies decreased in the OS region of aortic ECs. The above studies showed that OS increased DNMT expression, resulting in alterations in DNA methylation, which further regulated EC function both in vitro and in vivo.
Lipopolysaccharide (LPS) was also involved in regulating EC function. Endothelial KLF2 is a key transcription factor involved in endothelium-dependent vascular homeostasis. LPS, which is a proinflammatory stimulus, increased hypermethylation of the KLF2 promoter and subsequently downregulated KLF2 and its downstream genes, including that encoding endothelial nitric oxide (NO) synthase (eNOS), in ECs. The loss of DNMT1 reversed KLF2 downregulation in LPS-induced ECs, suggesting that KLF2 methylation is associated with DNMT1 in mediating the inflammatory function of ECs.37
Overall, abnormal DNA methylation altered gene expression in VSMCs and ECs in ISR, leading to disease development. Furthermore, DNMT expression determines the state of DNA methylation, and miRNAs also play a key role in this process.
DNA demethylation
The Tet family, which includes Tet1, Tet2, and Tet3, is a set of enzymes capable of activating DNA demethylation. Tet enzymes promote DNA demethylation and gene transcription by catalyzing the conversion 5mC to 5-hydroxymethylcytosine (5hmC). Recently, the roles of TET in various diseases, particularly in cancer, were investigated extensively.38 In addition, downregulation of TET2 was observed in human atherosclerotic samples and in a mouse femoral artery wire injury model. Importantly, TET2 overexpression dramatically improves intimal hyperplasia in vivo following vascular injury.39
TET2, a key enzyme in the DNA demethylation pathway, is highly expressed in VSMCs and is a key regulator of VSMCs.39 TET2 and 5hmC are upregulated during VSMC differentiation, whereas they are downregulated during VSMCs dedifferentiation.39 Liu et al.39 observed that TET2 is a master epigenetic regulator of VSMC plasticity, as it activates the key transcription factors such as myocardin (MYOCD), SRF, and MYH11 and inhibits KLF4. In addition, the loss of TET2 hampers the rapamycin-induced VSMC phenotype, whereas TET2 overexpression induces contractile differentiation. These findings showed that TET2 is a novel epigenetic regulator of the phenotypic transformation of VSMCs.
Emerging evidence indicates the importance of the relationship between DNA methylation and EC function. Peng et al.40 demonstrated that elevated levels of TET2 repressed nuclear factor κB (NF-κB) activation in ox-LDL-induced ECs, downregulating pro-inflammatory chemotactic molecules, including ICAM-1 and VCAM-1, and protecting EC function, whereas TET2 knockdown induced the opposite effect. Interestingly, consistent with these results, previous studies have suggested that TET2 ameliorated low OS-mediated EC dysregulation,41 increased autophagy, and decreased the expression of inflammatory factors in an ApoE−/− mouse model to hinder the development of atherosclerosis.42 However, the exact molecular mechanisms of the TET2 effect and its role in signal transduction pathways have not been elucidated, which may help prevent or treat endothelial dysfunction in restenosis. A recent study investigated the relationships between TET2 and miRNA in modulating the abnormal function of ECs. miR-101-3p overexpression induced EC-mediated generation of reactive oxygen species (ROS), activated NF-κB, and subsequently dysregulated the ECs. Mechanistically, miR-101-3p contributed to EC dysfunction by repressing TET2 expression at the post-transcriptional level. Furthermore, elevated levels of TET2 attenuated cell death, ROS production, and inflammation caused by the miR-101-3p-induced ECs. This study presented a new approach to improving ISR lesions by targeting miR-101-3p and its downstream targets via the TET2-NF-κB signaling pathway.43 Further studies should shed light on the other signaling pathways related to TET2 and EC function.
Considering that TET2 plays a critical role in EC dysregulation and phenotype transformation of VSMCs, these studies further suggest the possibility of TET2 as a new target for restenosis treatment.
Histone modification
Histone modification can be subdivided into the following categories: (1) methylation, (2) acetylation, (3) ubiquitination, (4) sumoylation, and (5) phosphorylation.44 Histone modifications compass a complex set of multiple modifications that regulate chromatin by “writing” and “erasing” epigenetic marks.45 In the eukaryote nucleus, 147 bp of DNA are wrapped around the octameric core of histones H2A, H2B, H3, and H4, called the nucleosome (Figure 2).46 Each major globular (core) histone has a characteristic side chain or tail, rich in lysine and arginine residues.47 The histone tail undergoes extensive covalent post-translational modifications (PTMs), which collaboratively regulate the chromatin state. A large body of evidence shows that histone PTMs can directly modify chromatin structure.48 In addition, histone modifications also interact with DNMTs/TETs to drive or suppress transcription.
Figure 2.

Illustrations of various epigenetic posttranslational modifications
The nucleosome is comprised of approximately 147 bp of DNA comprising an octamer of two copies of each of the four core histone proteins H2A, H2B, H3, and H4. The histone tail is specifically modified by writing an enzyme that adds or erases the PTM shown.
Histone methylation
Detailed studies are required to understand the effect of histone methylation/demethylation on VSMCs and ECs in the pathological process of ISR. Activation or inhibition of transcription by different histone lysine methylations depends on the site, degree of methylation, genomic location, and other co-existing PTM states.49 It is generally thought that methylated histone H3 lysine 4, 36, and 79 (H3K4, H3K36, and H3K79) are closely related to transcriptional activation, while methylated histone H3 lysine 9 and 27 (H3K9 and H3K27) and histone H4 lysine 20 (H4K20) are involved in transcriptional repression.50 In addition to transcriptional regulation, histone lysine methylation is associated with many other cellular processes, such as DNA damage repair, DNA replication, and mRNA splicing. The functions of HMTs in ISR are discussed in the following section.
To date, more than 50 lysine HMTs have been identified,51 which are highly selective of the target histone lysine residue. Furthermore, histone lysine methylation states depend on activation or repression of HMTs. In the following sections, we discuss the role of HMTs in vascular function and ISR development.
EZH2 is a member of the HMT family that plays important catalytic roles in the polycomb repressor complex 2 (PRC2). Studies have shown that PRC2 methylates H3K27, thereby causing transcriptional repression of the target genes.52
The long ncRNA (lncRNA) taurine-upregulated gene 1 (TUG1) was upregulated in synthetic VSMCs, accompanied by an increase in EZH2 expression. However, the level of lysine methylation was high in differentiated VSMCs. Further studies are required to understand the effects of methylation sites and amounts of lysine on VSMC biology.53 Kolliputi and colleagues54 and Liang et al.55 observed that overexpression of EZH2 contributed to the proliferation and migration of SMCs and inhibited apoptosis, resulting in intima thickening. Mechanistically, an upregulated EZH2 level increased H3K27me3 levels, thereby inducing VSMC proliferation.56 Conversely, Li et al.57 concluded that elevated EZH2 facilitated VSMC growth, which depended on autophagy rather than proliferation. A reduced EZH2 level promoted autophagosome formation and led to aortic dissection. Furthermore, Hcy elevated the expression of EZH2 in the aorta in an ApoE−/− mode mouse.58 Repression of EZH2 ameliorated thoracic aortic aneurysm in the mouse model of Marfan syndrome.59
In addition to regulating various VSMC functions, EZH2 is also associated with EC function. Previous studies by Smits et al.,60 Floris et al.,61 and Xu et al.62 have shown that miR-101 expression led to pro-angiogenic effects by partially diminishing the EZH2 level, thereby promoting hypermethylation of H3K27 and altering the transcriptome. Furthermore, EZH2 acts not only as a target but also as a transcriptional inhibitor of miR-101 in ECs. Suppression of EZH2 reduces EC migration in vitro, indicating that EZH2 inhibitors can be used for treating abnormal vascularization. Xu et al.62 and Maleszewska et al.63 observed that fluid OS decreased the mRNA and protein levels of EZH2, leading to endothelial anti-inflammatory phenotype. Further research should focus on the precise role of EZH2 in endothelium-specific knockout mice using laminar flow induction. A recent study indicated that under ox-LDL treatment, lncRNA OIP5-AS1 promoted EC apoptosis by targeting GSK-3β via EZH2.64 In addition, Tsou et al.65 demonstrated that overexpression of EZH2 in ECs inhibited angiogenesis and restored normal angiogenesis in scleroderma.
These results implied that EZH2 plays a central role in vascular cells may be useful as a new therapeutic target for treating ISR.
HDMs
To date, more than 20 HDMs have been identified and characterized.66 Most importantly, aberrant expression of HDMs has been correlated with many diseases, including cardiovascular disease. The subsequent sections highlight the key HDMs that play crucial roles in the pathologic process of ISR.
JMJD3 (also known as KDM6B), a member of the JmjC-domain protein demethylase family, demethylates H3K27me2/3.67 Activation of NF-κB is a feature of inflammation. JMJD3 increases the level of inflammatory cytokines (interleukin [IL]-1β, tumor necrosis factor [TNF]-α, IL-6) and adherence factors (ICAM-1, matrix metalloprotease [MMP]-9) and nuclear translocation of NF-κB/p65 in LPS-mediated ECs, subsequently leading to vascular cell inflammation.68,69 Mechanistically, LPS promoted JMJD3 nuclear accumulation and assembly of JMJD3 and NF-κB at the promoter of target genes, indicating that both JMJD3 and NF-κB can activate the expression of target genes.69 These findings revealed the epigenetic regulation of the LPS-induced NF-κB pathway in the endothelium and suggested Jmjd3 as a key regulator of the NF-κB pathway and potential therapeutic targets for NF-κB-related diseases. In addition, anti-inflammatory agents have been used to regulate JMJD3. Na et al.70 reported that JMJD3 expression was inhibited in dexamethasone-induced VSMCs by negative glucocorticoid response element binding in the upstream region of the gene, subsequently resulting in suppression of MMP-2, MMP-3, and MMP-9. Furthermore, dexamethasone activates the expression of claudin 5 and occluding-encoding genes. This suggests that dexamethasone can maintain the integrity of ECs under inflammatory conditions. In addition, Lee et al.71 demonstrated that JMJD3 is upregulated in ECs under oxygen-glucose deprivation/reperfusion injury. JMJD3-induced demethylation results in activation of IL6.
Recent studies have also reported the association of JMJD3 with NF-κB, resulting in the proliferation of bladder SMCs and collagen accumulation, which promotes bladder fibrosis.72 Luo et al.73 demonstrated that PDGF induced reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) expression and elevated the JMJD3 level, which subsequently reduced H3K27me3 marks at the NOX4 promoter and activated autophagy in VSMCs. An inhibitor of JMJD3 inhibited NOX4 expression owing to H3K27me3 enrichment at the gene promoter. Furthermore, loss of JMJD3 and NOX4 inhibited autophagic activation, thereby alleviating vascular remodeling after carotid injury. Notably, elevations in JMJD3 and NOX4 levels are observed during the formation of human atherosclerotic plaques. Lack of JMJD3 blunts neointima formation after vascular injury by inhibiting the activation of Nox4 autophagy signals, demonstrating that JMJD3 may be a prospective target for the prevention and treatment of vascular diseases. Overall, although increasing evidence shows JMJD3 to be associated with vascular cell function, the mechanism responsible for JMJD3 regulation remains to be completely elucidated.
JMJD1 (KDM3a) is another HDM. Previous studies have reported that elevation of KDM3a expression promoted high glucose and angiotensin II (AngII)-induced proliferation and migration of VSMCs, which were reduced considerably under conditions of KDM3a deficiency. Furthermore, consistent with this in vitro result, elevated levels of KDM3a were promoted, whereas loss of KDM3a decreased neointima formation after vascular injury in a balloon injury model. Furthermore, JMJD1 expression was upregulated in vessels of a diabetic rat. We observed that KDM3a may act as a novel target for regulating VSMC function and vascular neointimal hyperplasia.74
Histone acetylation
Histone acetylation is a conserved reversible modification modulated by HATs and HDACs, which is associated with various cellular functions. Previous studies in animal models have shown that reversible acetylation plays a crucial role in cardiovascular disorders, such as vascular diseases, heart failure, hypertension, arrhythmia, and angiogenesis.75 HATs are classified into three major families: (1) GCN5-related N-acetyltransferases (GNATs), (2) p300/CREB-binding protein (p300/CBP), and (3) and the MOZ, Ybf2, Sas2, and Tip60 (MYST) family.76 Histone acetylation increases gene expression by “opening” the chromatin structure. In contrast, HDACs “close” the chromatin structure and inhibit gene expression. HDACs not only deacetylate histone, but they also bind to non-histone proteins to inhibit transcription. HDACs have been classified into four groups based on their sequence similarity: type I (HDAC1, 2, 3, and 8), type II (IIa, HDAC4, 5, 7, and 9; IIb, HDAC6 and 10), type III (NAD+-dependent sirtuin [SIRT]1–7), and type IV (HDAC11).77 In the next section we focus on the role of HATs and HDACs in restenosis.
p300 is a major acetyltransferase and a transcriptional co-activator, which is closely related to many diseases, including cardiovascular disorder. A recent study reported that MFN2 exerts an anti-proliferative effect in VSMCs both in vivo and in vitro.27 All-trans retinoic acid promotes KLF4 acetylation by promoting the interaction of p300 with KLF4. Furthermore, the co-expression of p300 and KLF4 activates the MFN2 promoter to induce MFN2 expression in VSMCs.78 In addition, He et al.79 showed that KLF4 modulated the association between TGF-β1-induced gene transcription and H3 acetylation in VSMCs. These results describe a novel mechanism wherein all-trans retinoic acid upregulated KLF4 expression and phosphorylation to eventually activate the MFN2 promoter and further expand the KLF4 acting network in VSMCs. KLFs also regulate inflammation in VSMCs. Lu et al.80 demonstrated that the loss of KLF15 activated the inflammatory pathway in VSMCs and promoted atherogenesis. Mechanistically, KLF15 deletion enhanced the acetylation status and activity of the NF-κB pathway due to interaction with p300. Furthermore, the marked decrease in the expression of KLF15 in human atherosclerotic specimens supported this observation. de Jong et al.81 demonstrated that p300/CBP-associated factor (PCAF) accelerated NF-κB-induced inflammation and played a key role in regulating subsequent VSMC proliferation after injury. Interestingly, three independent large prospective studies have shown an association between mutations in the −2481C allele of the PCAF gene and reduced vascular mortality, suggesting that PCAF is a possible diagnostic marker for restenosis.82
lncRNAs are also related to histone acetylation. lncRNA growth arrest-specific 5 (GAS5) inhibits proliferation, induces apoptosis, and promotes cell cycle arrest of VSMCs. Mechanistically, GAS5 directly binds to p300 and p53, stabilizing the interaction of p53 with p300 and regulating VSMC function.83 Shah et al.84 showed that atherosclerotic plaques predominantly exhibit oxidative DNA damage, mainly as 8-oxoguanine (8oxoG) lesions owing to decreased acetylation of OGG1. p300 was identified as the OGG1 acetyltransferase, which modulates the endogenous expression of OGG1 via acetylation. p300 was downregulated in the VSMCs of the human atherosclerosis plaque response to oxidative stress. Furthermore, p300 regulated both OGG1 stability and activity.
Shali et al.85 showed that glucose upregulated p300 and caused nuclear DNA damage in ECs. p300 induced histone acetylation and activated various transcription factors by binding to the promoters of genes encoding angioactive factors and ECM proteins, subsequently leading to upregulation of vasoactive factors and ECM proteins. Data from these studies show that p300 upregulation is an important upstream epigenetic mechanism for regulating the expression of vasoactive factors and ECM protein genes in ECs. Altogether, p300 plays an important role in regulating inflammation, proliferation, and autophagy. Hence, understanding these mechanisms may contribute to the development of better strategies for treating restenosis.
HDACs
HDAC5 is a class IIa HDAC. Previous studies have indicated that AngII-mediated HDAC5 phosphorylation and nuclear export lead to enhanced MEF2 transcriptional activity and VSMC hypertrophy.86 This research demonstrates new roles for HDAC5 in AngII signaling in VSMCs. Tsou et al.87 showed that HDAC5 knockdown enhanced the formation of the EC tube, thereby improving scleroderma. Furthermore, fluid OS also induced HDAC5 phosphorylation, and hence it downregulated HDAC5-induced KLF2 and eNOS expression, with consequent anti-inflammatory effects.88,89 Previous experiments have also indicated that HDAC5 plays a key regulatory role in EC proliferation and migration in a gene-deficient mouse model.90 Taken together, the results presented in this section support the view that HDAC5 silencing can be a therapeutic target against neointimal hyperplasia and restenosis.
HDAC9 is a member of class IIa HDAC. Genome-wide association studies have shown that several genetic variants of HDAC991 are associated with large vessel ischemic stroke92 and myocardial infarction.93 Recently, Malhotra et al.94 observed that loss of HDAC9 reduced VSMC proliferation and increased contractility. Furthermore, mutations in the VSMC cytoskeleton led to the formation of ternary complexes containing HDAC9, chromatin-remodeling enzyme BRG1, and lncRNA MALAT1. Knockdown of HDAC9 can restore contractility protein expression and improve aortic wall structure.95 In ECs, HDAC9 expression was increased with ox-LDL exposure to apoptosis-inducing doses.96 Han et al.96 and Shi et al.97 have observed that HDAC9 depletion significantly antagonizes ox-LDL-induced apoptosis and inflammatory response. Taken together, this evidence suggests that targeted inhibition of HDAC9 is a promising strategy to prevent restenosis development.
Knockdown of HDAC1 and HDAC3 (type I HDAC) decreased SMC proliferation and neointima formation in murine models of vascular injury.98 Arginase 2 (Arg2) is a key target in the progression of atherosclerosis because it controls endothelial NO, proliferation, fibrosis, and inflammation. HDAC2 (type I HDAC) overexpression inhibited Arg2 expression in ECs, while knockdown of HDAC2 increased Arg2 expression. Furthermore, HDAC2 overexpression improves ox-LDL-induced vascular dysfunction.99 These studies provide a novel therapy for endothelial dysfunction and restenosis by activation of HDAC2. HDAC3 is also critical for maintaining endothelial integrity through the phosphatidylinositol 3-kinase (PI3K)/Akt mechanism.100 Zhang et al.101 and Usui et al.102 found that HDAC4 (type II HDAC) promoted the proliferation and migration of VSMCs mediated by PDGF-BB.102 In vivo, the HDAC inhibitor MC1568 attenuated neointimal hyperplasia in a mouse model of carotid artery ligation.103 These findings suggest that HDAC4 regulates VSMC proliferation and migration, and HDAC4 inhibitors are expected to be novel targets for the prevention of restenosis. HDAC6 is a member of the type IIb HDAC family. Knockdown of HDAC6 attenuates the proliferation and migration of VSMCs and reduces neointima formation after vascular injury.103,104 In addition, Zhang et al.104 observed that loss of HDAC6 prevents dedifferentiation of VSMCs in vitro and reduces the formation of new intima and restenosis in vivo. The mechanism is that inhibition of HDAC6 enhances SRF nuclear activity and maintains SMC contractile levels. These findings suggest that targeting HDAC6 represents an attractive therapeutic approach to treat proliferative vascular diseases, including restenosis. The role of HDAC in restenosis progression deserves further study in experimental animal models.
The SIRTs belong to the family of class III NAD+-dependent HDACs, which catalyze deacetylation/deacylation on histones and protein substrates.105 Seven closely related SIRT family members have been identified, that is, SIRT1–SIRT7. SIRTs regulates numerous pathophysiological processes, for instance, energy metabolism, inflammation, genomic stability, and aging.106 SIRT1 is the most widely studied member of SIRTs, and accumulating studies have found that SIRT1 plays a protective role in CADs.105,107 In the vascular system, SIRT1 negatively regulates vascular inflammation,108 endothelial dysfunctions,109 and VSMC proliferation and migration,110 thereby preventing vascular aging, intimal hyperplasia, and vascular proliferative disease. The expression of SIRT1 protein is decreased in the plaque area of atherosclerotic patients.111,112 In addition, expression of the SIRT1 gene in mononuclear cells in patients with coronary atherosclerosis was reduced,113 suggesting a systemic decline in SIRT1 levels in the disease. However, the question as to whether the impaired SIRT1 level in patients with coronary atherosclerosis is a result of the atherosclerotic process itself, or vice versa, remains to be investigated. SIRT1 expression was also decreased in ApoE−/− mice.114 In addition, SIRT1 EC-115 and VSMC-specific112 overexpression can prevent the development of atherosclerosis in mice. These pieces of evidence suggest that SIRT1 has a protective effect on the cardiovascular system in general. Despite some controversy, overexpression of SIRT1 may be a new therapeutic target for cardiovascular diseases.
In human VSMCs, endogenous SIRT1 is gradually lost with aging, leading to cellular functional defects, including impaired stress response and reduced cell migration and proliferation ability.116 The neointima formation was related to the decreased expression of SIRT1, while the overexpression of SIRT1 in VSMCs weakened the neointima formation after vascular injury.117 Overexpression of SIRT1 in VSMCs inhibited AngII-118 as well as injury-induced119 VSMC hypertrophy and neointimal formation. Additionally, Li et al.117 showed that SIRT inhibited the proliferation and migration of VSMCs through G1/S cell cycle stagnation, thus weakening the formation of neointimal formation. Furthermore, SIRT1 inhibits the uptake of ox-LDL in VSMCs by inhibiting expression levels of LOX-1 and NF-κB signaling pathways.112 These findings offer a new view that the inhibitory effect of SIRT1 on VSMC proliferation and migration is the basis of neointima formation and suggest that SIRT1 may be a potential target for vascular disease intervention. Increasing SIRT1 activity should be further investigated as a promising treatment option for restenosis.
SIRT1 also exerts a protective role in ECs. SIRT1 is highly expressed in the growing vascular endothelium.120 However, the expression level and activity of SIRT1 in inflammatory ECs were significantly decreased.121 SIRT1 binds to the calmodulin of eNOS and thus enhances the production of endothelial NO. However, inhibition of SIRT1 results in decreased bioavailability of NO, suggesting that SIRT1 plays a key role in endothelium-dependent eNOS-mediated vascular homeostasis.122 However, Guo et al.123 found that no improvement in the bioavailability of NO was observed in SIRT1-overexpressed eNOS gene knockout mice. This suggests that eNOS plays an important role together with SIRT1, which has a beneficial effect on the bioavailability of NO. Besides NO, ET-1 (vasoconstrictor) and other factors may also play a key role in endothelial function. The reduction of SIRT1 was related to endothelial injury and increased expression of ET-1.124 Although ET-1 is primarily a vasoconstrictor, it induces NO and prostaglandins to promote vasodilation. SIRT1 is very important to the regulation of NO and ET-1, respectively; thus, the question occurs whether SIRT1 can regulate the conversion between NO and ET-1. If so, how will SIRT1 regulate this process? Hcy induced EC injury, which is caused by downregulation of SIRT1 in the coronary artery and increased oxidative stress, in a mouse model of hyperhomocysteinemia.125 Lu et al.126 found that the high-fat diet-induced EC damage in rats was accompanied by an increased oxidative stress response and a decrease in eNOS and SIRT1 mRNA. Similarly, SIRT1 inhibition increases the expression of p53 and plasminogen activator inhibitor 1, leading to damage-induced neointimal formation and remodeling.127 These studies demonstrated that the downregulation of SIRT1 may be a key factor mediating the endothelial inflammatory response and oxidative stress, leading to endothelial dysfunction. Therefore, activation of SIRT1 may provide a promising therapeutic strategy for cardiovascular diseases.
SIRT6 is a site-specific HDAC that regulates chromatin structure.128 Similarly, in ApoE−/− mice129 and human atherosclerotic plaques,130 the expression of SIRT6 was decreased. Furthermore, Yao et al.131 observed that SIRT6 mediated by the TGF-1/Smad signaling pathway regulates VSMC differentiation. SIRT6 is an important regulator of EC function. The protective effect of SIRT6 on endothelial function is attributed to promoting endothelial-dependent vasodilation and bioavailability of vascular NO, reducing cell permeability, improving endothelial senescence and apoptosis, and promoting autophagy.132 Accumulating research has demonstrated that SIRT6 deficiency enhances the expression of endothelial proinflammatory cytokines and the transcriptional activity of NF-κB.130,133,134 Collectively, these studies suggest that SIRT6 plays a crucial role in regulating vascular cells, especially endothelial dysfunction, by regulating a variety of pathophysiological pathways.
AngII leads to cell migration, whereas depletion of SIRT2 inhibits cell migration. AngII induces microtubule redistribution and deacetylation through SIRT2 in ECs, implying that SIRT2 plays a new role in vascular remodeling induced by hypertension.135 SIRT3 was stably expressed in mice VSMCs, while AngII increased SIRT3 in VSMCs.136 SIRT3 elevations significantly inhibit VSMC proliferation and neointimal hyperplasia.137 Yet, SIRT3 silencing will further increase cell proliferation.136 However, the role of SIRT in restenosis needs to be studied more extensively in experimental animal models.
Histone ubiquitination
Ubiquitin is a small highly conserved globular protein of 76 aa, which can be covalently attached to proteins by ubiquitin ligases. Ubiquitination is an ATP-dependent three-stage enzymatic cascade. Ubiquitin is activated by the ubiquitin E1 enzyme, bound by the E2 enzyme, and ligated to the target protein by the E3 enzyme.46 While evaluating the effect of histone ubiquitination and deubiquitination in ISR, Zhao et al.138 reported that interferon (IFN)-γ exacerbates neointimal hyperplasia by facilitating ubiquitin-dependent liver X receptor-a (LXR-a) degradation. Consistent with the in vitro results, neointimal hyperplasia was observed after 4 weeks of carotid ligation in ApoE−/− mice after treatment with LXR agonist (T0901317). Furthermore, IFN-γ promoted inflammation by inducing ubiquitination. Ubiquitination is closely regulated by E3 ligases, which participate in the regulation of various cellular functions. In subsequent sections, we discuss the specific roles of E3 ligases in ISR.
The E3 ubiquitin ligase mouse double minute 2 homolog (MDM2) is the main negative regulator of p53, as it induces p53 ubiquitination and degradation and impedes p53 transcriptional activity.139 A large number of studies have revealed the correlation between MDM2 and the p53 signaling pathways in vascular ECs and VSMCs.
The function of MDM2 in VSMCs has received considerable attention. In particular, although MDM2 is a vital regulator of p53 activity, the transcription of MDM2 itself also depends on p53, forming a p53/MDM2 negative feedback loop. Hashimoto et al.140 have indicated that disruption of MDM2-p53 interaction inhibited neointimal hyperplasia after vascular injury. Furthermore, Li et al.141 reported that p55γ, a regulatory subunit of PI3Ks, clearly blunted the MDM2-mediated ubiquitination of p53 in VSMCs. In vitro, overexpression of p55γ inhibited VSMC proliferation, increased the p53 protein level, and reduced the amount of ubiquitinated p53. This study indicated that p55γ stabilizes p53 by hindering the ability of MDM2 to interact with p53, thereby hampering p53 ubiquitination. In vivo, loss of p55γ increases neointimal thickening following injury. Furthermore, lncRNAs are also involved in ubiquitination. Long intergenic non-coding (linc)RNA-p21 inhibits VSMC proliferation and induces apoptosis in vitro. Mechanistic studies have indicated that lincRNA-p21 and p53 compete for binding to MDM2 in VSMCs. lincRNA-p21 directly binds to MDM2 and promotes p53-p300 interaction, enhancing p53 transcriptional activity. Reduced lincRNA-p21 expression was observed in the atherosclerotic plaque of ApoE−/− mice and in patients with CAD.142 Thus, these studies demonstrated that disruption of the MDM2-p53 interaction impedes VSMC proliferation and neointimal hyperplasia.
MDM2 also plays a significant role in ECs. OS and VEGF-A stimulation upregulated MDM2 phosphorylation on serine 166.143,144 Furthermore, overexpression of a Ser166-MDM2 phosphorylation mimetic increased the binding of MDM2 to FoxO1, decreased the FoxO1 level (known to promote an angiostasis gene), and promoted EC migration.143 These results highlighted the importance of MDM2 in maintaining survival and responsiveness to pro-angiogenic stimuli in ECs.
Overall, E3 ubiquitin ligases are regulated in VSMCs and ECs and may act as “druggable” targets for the treatment of ISR.
Histone deubiquitination
DUBs are peptidases that reverse ubiquitination by removing ubiquitin molecules from target proteins.145 The human genome encodes approximately 100 DUBs, which can be divided into two main groups according to the mechanism of enzymatic cleavage: (1) cysteine proteases, and (2) zinc metalloproteases. Furthermore, cysteine proteases can be classified into four groups: ubiquitin-specific proteases (USPs), ubiquitin carboxyl-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), and Machado-Joseph disease protein domain proteases (MJDs).145 In the next section, we describe DUBs, particularly USPs, in the context of vascular cell function and ISR development.
USPs are the most abundant DUBs, with approximately 60 proteases in humans.146 USP20 inhibits NF-κB activation via Toll-like receptor (TLR)4,147 IL-1R, and the TNF148 inflammatory pathway in VSMCs. Intriguingly, USP20 impedes TLR4-induced NF-κB activation by deubiquitinating TNF receptor-associated factor 6,147 whereas it inhibits IL-1R and TNF-triggered NF-κB activation by deubiquitinating receptor-interacting protein kinase 1.148 Consistent with the results of the in vitro experiments, USP20 decreased neointimal hyperplasia caused by carotid endothelial denudation in a transgenic mice model.147 In addition, USP20 attenuated atherosclerosis in an LDLr−/− mouse model by inhibiting NF-κB activation in vivo.148 Therefore, increased expression/activity of USP20 may offer protection against inflammatory pathways in patients with ISR.
BET
The BET protein family consists of two tandem domains (reader for lysine acetylation) and one extra-terminal domain, including BRD2, BRD3, BRD4, and BRDT. The BET family proteins use bromodomain-targeted modifications of histones and regulate transcription through interactions with transcription mechanisms.24 Mechanical force, injury, and cytokine response to VSMC stimulation can lead to (re)stenosis and other proliferative vascular lesions, all involving BET, especially BRD4.24 Wang et al.149 found that BRD4 is dramatically elevated in the neointima of injured rat carotid arteries as well as human vein and artery grafts. However, (+)-JQ1 (BET-specific inhibitor) can reduce the proliferation and migration of SMCs in vitro, thus effectively mitigating the neointima formation.149,150 Further studies suggest that (+)-JQ1 induces G0/G1 cell cycle arrest and prevents VSMC proliferation.150 In ECs, VEGF stimulation enhances the acetylation of ETS-1 (v-ETS avian erythroblastosis virus E26 oncogene homolog 1), causing its binding to BRD4, resulting in angiogenesis.151 Thus, decreasing BRD4 expression/activity may provide protection against restenosis patients.
ncRNAs
The relationship between ncRNAs and epigenetics has opened up new domains of research, as they have been shown to modulate gene expression. In addition to DNA methylation and histone modifications, accumulating evidence has highlighted the role of ncRNAs in modulating ISR.152 ncRNAs are mainly categorized into small RNAs (<200 bp) (small ncRNAs) or long RNAs (>200 bp) (lncRNAs) based on transcript size. Small ncRNAs consist of miRNAs, small nucleic RNAs (snRNAs), small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs), QDE-2-interacting RNAs (qiRNAs), and piwi-interacting RNAs (piRNAs).153 lncRNAs consist of lincRNA, circular RNA (circRNAs), natural antisense transcripts (NATs), and enhancer RNAs (eRNAs).154 Interestingly, increasing evidence proved that ncRNAs are critically involved in atherosclerosis, particularly ISR.155, 156, 157, 158, 159 The human ncRNAs circulate in the bloodstream and can act as ISR biomarkers for the diagnosis and prediction of disease outcomes. Several studies have shown the critical role of ncRNAs in ISR regulation. The following section discusses the relationship between ISR and ncRNAs, primarily miRNAs, lncRNAs, and circRNAs.
miRNAs are small endogenous RNAs 19–25 nt in length, which modulate gene expression by targeting the 3′ untranslated region of specific mRNAs, resulting in post-transcriptional gene silencing.153,160 miRNAs, for example, miR-125b1 and miR-124, are involved in epigenetic regulation in combination with the incorporation of DNA methylation, inhibitory histone marks, or deficiency of transcription factors.161,162 Furthermore, various miRNAs mediate the expression of epigenetic elements, such as DNMTs (regulated by miR-152, miR-30, and miR-148a/b), HDACs (regulated by miR-140, miR-1, and miR-449a), and a polycomb group of genes (regulated by miR-101 and miR-26a).163, 164, 165, 166, 167, 168, 169, 170, 171 Previous studies have indicated that miRNAs are expressed in the vascular system. As shown in Table 1, numerous studies have indicated that miRNAs impinge on a wide variety of cellular activity networks, related to the ISR biological pathway, including VSMC and EC function. miRNAs regulate VSMC proliferation, migration, phenotypic switch, calcification, inflammation, and apoptosis. In ECs, miRNAs modulate inflammation, leukocyte adhesion, and eNOS-dependent NO production. Several recent studies have shown that miRNAs can be detected in multiple body fluids, such as cerebrospinal fluid, saliva, and urine; furthermore, miRNAs are stable in blood circulation,190 indicating the potential of miRNAs as biomarkers (Table 2) of restenosis diagnosis, prognosis, and treatment. Thus, the regulation of miRNA levels represents an exciting opportunity for the development of new treatments for ISR.
Table 1.
Role of miRNAs in vascular pathophysiology
| miRNA | Mechanism | Cell type | Function | Animal models | Ref. |
|---|---|---|---|---|---|
| miR-145 | KLF5 | VSMC | phenotypic switch | rat carotid artery balloon injury model | 172 |
| miR-100 | mTOR | EC, VSMC | EC proliferation, VSMC migration | femoral artery occlusion in mice model | 173 |
| miR-143 | EC, VSMC | ↑EC migration, angiogenesis, ↑VSMC proliferation, ↓VSMC apoptosis | calf models of PAH | 174 | |
| miR-221/222 | p27 (Kip1)/p57 (Kip2) | VSMC | ↑proliferation | rat carotid artery balloon injury model | 175 |
| miR-24 | Chi3l1 | VSMC | ↓proliferation | murine models of AAA | 176 |
| miR-92b-3p | TSC1 | VSMC | ↑proliferation | 177 | |
| miR-22 | EVI1 | VSMC | phenotypic switch | mouse femoral arterial injury models | 178 |
| miR-663 | JunB/Myl9 | VSMC | phenotypic switch, ↓proliferation, ↓migration, ↓neointimal formation | rat carotid artery ligation injury | 179 |
| miR-133 | Sp-1 | VSMC | phenotypic switch, ↓proliferation, ↓neointimal formation | rat carotid artery balloon injury model | 180 |
| miR-128-3p | KLF4 | VSMC | phenotypic switch, ↓proliferation, ↓migration | mice model of stenosis | 181 |
| miR-223 | PDGFRβ | VSMC | phenotypic switch, ↓neointimal formation | mice femoral artery wire injury model | 182 |
| miR-10 | FLT1 | EC | angiogenic | zebrafish | 183 |
| miR-30 | DLL4 | EC | angiogenic | zebrafish | 184 |
| miR-133a | GCH1 | EC | endothelial dysfunction | mice or rat models of hyperglycemia, hyperhomocysteinemia, and dyslipidemia | 185 |
| miR-92a | SOCS5 | EC | endothelial dysfunction | LDLR−/− mice | 186 |
| miR-126-5p | DLK1 | EC | ↑proliferation | 187 | |
| miR-424 | CUL2 | EC | angiogenic | rat myocardial infarction models | 188 |
| miR-181b | NF-κB | EC | inflammation | APOE−/− mice | 189 |
Ref., reference; KLF5, Krüppel-like factor-5; VSMC, vascular smooth muscle cell; EC, endothelial cell; mTOR, mammalian target of rapamycin; Chi3l1, chitinase 3-like 1; AAA, abdominal aortic aneurysm; TSC1, consisting of tuberous sclerosis 1; EVI1, ecotropic virus integration site 1 protein homolog; Myl9, myosin light chain 9; FLT1, fms-related tyrosine kinase 1; DLL4, Delta-like 4; GCH1, GTP cyclohydrolase 1; SOCS5, suppressor of cytokine signaling 5; DLK1, Delta-like 1; CUL2, cullin 2; NF-κB, nuclear factor κB.
Table 2.
Biomarkers of miRNA in the development of restenosis
| miRNA | Expression ISR patients | Stent type | ISR | Sample | Reference |
|---|---|---|---|---|---|
| miR-21 | ↑ | DES | coronary | plasma | 191 |
| miR-100 | ↓ | DES | coronary | plasma | 191 |
| miR-143 | ↓ | DES | coronary | plasma | 191 |
| miR-145 | ↓ | DES | coronary | plasma | 191 |
| miR-19a | ↓ | DES | coronary | plasma | 192 |
| miR-126 | ↓ | DES | coronary | plasma | 192 |
| miR-210 | ↓ | DES | coronary | plasma | 192 |
| miR-378 | ↓ | DES | coronary | plasma | 192 |
| miR-93-5p | ↑ | DES/BMS | coronary | plasma | 193 |
| miR-146a | ↑ | DES | coronary | plasma | 194 |
| miR-146b | ↑ | DES | coronary | plasma | 194 |
ISR, in-stent restenosis; DES, drug-eluting stent; BMS, bare metal stent; ↑, upregulated; ↓, downregulated.
Until now, studies have shown that lncRNAs play a vital role in gene regulation during the development of cardiovascular diseases. lncRNAs can indirectly regulate DNA methylation at CpG sites, thereby modulating gene expression. For instance, lncRNA HOXB13-AS1 recruits DNMT3b to methylate and increase transcription from the HOXB13 promoter.195 Similarly, lncRNA SPRY4-IT1 recruits the de novo DNA demethylase DNMT1 to regulate cholangiocarcinoma development.196 lncRNAs can be used as a scaffold to promote interactions among proteins, RNA, and DNA, leading to gene activation or inhibition.197 Similarly, the expression of many VSMC- and EC-related genes involved in the ISR process is modulated via lncRNAs (for example, MALAT1, MANTIS, and NEAT1).95,198,199 lncRNAs are not only involved in epigenetic regulation such as DNA methylation and histone modification, but also in the regulation of gene expression in vascular systems. Similar to miRNAs, lncRNAs such as MYOSLID and NEAT1 regulate the VSMC phenotype switch,199,200 XR007793 regulates proliferation and migration,201 and GAS5 regulates apoptosis.83 lncRNAs modulate endothelial function and dysfunction, including proliferation (by CRNDE),202 migration (by HOTTIP),203 apoptosis (by OIP5-AS1),64 and inflammation (by TGFB2-OT1).204
In addition to these classic linear RNAs, recent studies have indicated that some lncRNAs can generate circRNAs. Interestingly, circRNAs are more stable than lncRNAs and are relatively tissue-specific.205, 206, 207 Owing to their stability and tissue specificity, circRNAs might become a research hotspot for CAD—in particular, ISR—in the future.208 Currently, the role of circRNAs in ISR has attracted considerable attention. Microarray chip analysis has revealed a specific circRNA expression profile in rat carotid artery after balloon injury. Studies have shown the presence of circDiaph3 in injured carotid artery tissues, which regulates VSMC differentiation, proliferation, and migration by targeting Igf1r.209 Furthermore, numerous studies have reported that alterations in DNMTs and the methylation status of some genes that host circRNAs can lead to circRNA silencing, suggesting that circRNAs are involved in epigenetic regulation.210 Furthermore, circRNAs can act as sponges for miRNAs and protect mRNAs from degradation, serve as scaffolds, and interact directly with proteins.205 Similarly, circRNAs also regulate vascular cells in ISR. circRNAs mediate EC proliferation (hsa_circ_0003575)211 and migration (circ_0003204),212 as well as VSMC proliferation (circ_Lrp6),213 migration (circTET3),214 and phenotypic transformation (circ-SATB2).215 In the future, ISR may be treated by targeting circRNAs.
Epigenetic drugs in treating ISR
Despite our understanding of the pathophysiological mechanisms underlying ISR, the incidence of ISR remains high, indicating that extensive efforts are required for developing a new treatment approach. The advent of epigenetics not only helps us to understand the mechanisms underlying restenosis but also provides new targets for ISR drug development. However, it is uncertain whether evidence exists that epigenetic molecular regulators are suitable for treating diseases. At present, several molecular regulators have been approved by a regulatory authority or are undergoing evaluation in clinical trials (Table 3). In most cases, these drugs are currently under evaluation or have been approved for the treatment of various malignancies.
Table 3.
Drugs that target epigenetic regulation that have been approved or are in clinical trials
| Drug | Epigenetic mechanisms | Phase | Disease | Reference or ClinicalTrials.gov identifier |
|---|---|---|---|---|
| Azacitidine | DNMTi | FDA approved | MDS | 216 |
| Guadecitabine | DNMTi | III | AML | NCT02920008 |
| Entinostat | HDACi | III | breast adenocarcinoma | NCT02115282 |
| Vorinostat | HDACi | FDA approved | CTCL | 217 |
| Romidepsin | HDACi | FDA approved | CTCL | 216 |
| Panobinostat | HDACi | FDA approved | multiple myeloma | 218 |
| Givinostat | HDACi | II | chronic myeloproliferative neoplasms | NCT01761968 |
| Mocetinostat | HDACi | II | non-small cell lung cancer | NCT02954991 |
| Valproic acid | HDACi | I | CLL | NCT00810680 |
| Belinostat | HDACi | FDA approved | PTCL | 219 |
| CXD101 | HDACi | I | advanced cancer | NCT01977638 |
| MPT0E028 | HDACi | I | advanced solid malignancies | NCT02350868 |
| CPI-1205 | EZH2 inhibitor | I | B cell lymphoma | NCT02395601 |
| Tazemetostat | EZH2 inhibitor | II | diffuse large B cell lymphoma | NCT02875548 |
DNMTi, DNA methyltransferases inhibitor; FDA, US Food and Drug Administration; MDS, myelodysplastic syndrome; AML, acute myeloid leukemia; HDACi, histone deacetylases inhibitor; CTCL, cutaneous T cell lymphoma; CLL, chronic lymphocytic leukemia; PTCL, peripheral T cell lymphoma; EZH2, enhancer of zeste 2.
As the epigenome is reversible and vulnerable to environmental factors, it may become a new avenue for ISR-related research. Designing clinically meaningful targets for treating ISR now appears feasible, as the interplay between epigenetic regulatory pathways is being revealed. Recent studies on epigenetic regulation have confirmed their potential as druggable targets in clinical medicine. To date, several epigenetic regulators have been considered effective in regulating vascular cells, raising the possibility of treating ISR by targeting epigenetic regulation in humans (Figure 3; Table 4). The subsequent sections discuss in detail the potential of using epigenetic drugs.
Figure 3.

Image representing the hypothetical structure of a stent capable of eluting a targeted inhibitor
The scaffold surface is coated with polylactic acid-glycolic acid copolymer (PLGA). At the bottom of the figure are listed targeted regulatory inhibitors that have the potential to be regulated for therapeutic use.
Table 4.
Drugs that target the epigenetic process of restenosis and associated vascular diseases
| Compound | Type | Animal models | Reference |
|---|---|---|---|
| 5-Aza-2′-deoxycytidine | DNMTi | (1) ApoE−/− mice | 220 |
| (2) LDLr−/− mice | 221 | ||
| Quercetin | DNMTi | (1) LDLr−/− mice | 222 |
| (2) ApoE−/− mice | 223,224 | ||
| (3) mice with a carotid injury model | 225 | ||
| Vitamin C | TET2 activator | (1) rabbits+HCD | 226 |
| (2) ApoE−/− mice | 227,228 | ||
| (3) pig coronary balloon injury | 229 | ||
| Scriptaid | HDACi | murine model of endovascular endothelial denudation | 98 |
| SAHA | HDACi | ApoE−/− mice | 230 |
| Valproic acid | HDACi | (1) LDLr−/− mice | 231 |
| (2) ApoE−/− mice | 232 | ||
| Phenylbutyrate | HDACi | LDLr−/− mice | 231 |
| (R)-trichostatin A | HDACi | (1) LDLr−/− mice | 233 |
| (2) neonatal rat | 234 | ||
| (3) murine carotid ligation model | 235 | ||
| Resveratrol | SIRT1 activator | (1) wire-injured carotid artery model | 236,237 |
| (2) rat carotid artery balloon angioplasty model | 238 | ||
| (3) ApoE−/−/LDLr−/− mice | 239 | ||
| (4) Marfan mouse model | 240 | ||
| (5) ApoE−/− mice | 241 | ||
| SRT1720 | SIRT1 activator | (1) HFHS diet | 242 |
| (2) ApoE−/− mice+AngII infusion ApoE−/− mice | 243 | ||
| SRT3025 | SIRT1 activator | (1) ApoE−/− mice | 244 |
| (2) rabbit iliac artery in-stent restenosis model | 245, 246, 247 | ||
| Fucoidan | SIRT6 activator | ApoE−/− mice | 248 |
| Cyanidin | SIRT6 activator | ApoE−/− mice | 249 |
| Icariin | SIRT6 activator | rabbits+HFD | 250,251 |
| Statins | EZH2 inhibitors | (1) ApoE−/− mice | 251 |
| (2) ApoE∗3-Leiden mice | 252,253 | ||
| (3) LDLr−/− mice | 254 | ||
| (4) rat carotid artery balloon angioplasty model | 255 | ||
| 256 | |||
| GSK343 | EZH2 inhibitors | Marfan syndrome mouse | 59 |
| UNC1999 | EZH2 inhibitors | murine carotid ligation model |
55 |
| (+)-JQ1 | BET inhibitor | (1) rat carotid artery balloon angioplasty | 256 |
| (2) AngII-infused mice | 257 | ||
| RVX-208 (apabetalone) | BET inhibitor | (1) ApoE−/− mice | 258 |
| (2) CAD patients | 259 | ||
| Nutlin-3 | MDM2 inhibitors | murine carotid ligation model |
140 |
ApoE, apolipoprotein E; LDLr−/−, LDL receptor-deficient; TET2, Tet methylcytosine dioxygenase 2; HCD, high-cholesterol diet; SAHA, suberoylanilide hydroxamic acid; HFHS, high-fat, high-sucrose; AngII, angiotensin II; HFD, high-fat diet; CAD, coronary artery disease; MDM2, mouse double minute 2 homolog.
DNMT inhibitors (DNMTis)
DNMTis are of two types, that is, nucleoside-based and non-nucleoside inhibitors.260 DNMTi, which includes 5-Aza and 5-aza-2′-deoxycytidine (decitabine, DAC), has been approved by the US Food and Drug Administration (FDA) for the treatment of myelodysplastic syndrome and acute myeloid leukemia (AML).261 Dunn et al.220 reported that 5-Aza decreased OS-mediated EC inflammation. Furthermore, 5-Aza inhibited atherosclerosis in an ApoE−/− mouse model. However, nucleoside inhibitors, including 5-Aza, are incorporated into DNA and are hence cytotoxic.262 Non-nucleoside inhibitors, including hydralazine, procaine,263 and mithramycin A,264 as well as RG108,263 also suppress the activity of DNMTs. Notably, some natural products have also been indicated to inhibit the activities of DNMTs, such as (−)-epigallocatechin-3-gallate (EGCG),265 curcumin,252 and genistein.266 These findings provide further support regarding the possibility of treating vascular diseases with DNMTi.
TET2 activators
Recent studies have indicated that vitamin C is not only an antioxidant but also a cofactor for dioxygenases of DNA methylation. Several studies have shown that vitamin C decreased EC barrier permeability to protect the integrity and function of ECs.267 Furthermore, Barabutis et al.268 reported that the combination of vitamin C and hydrocortisone prevented and repaired the LPS-mediated dysregulation of the pulmonary endothelial barrier. Altogether, the above results are due to vitamin C’s ability to preserve NO and scavenge reactive oxygen. Furthermore, vitamin C enhances DNA methylation by promoting TET2 expression.269 Nevertheless, the precise mechanism of action of vitamin C on vascular cells remains to be elucidated. In addition to vitamin C, evidence indicates the involvement of several factors related to the expression and activity of TET2, including Fe2+,270 vicenin-2,271 α-ketoglutarate,272 VprBP,273 andnd p300.274 In particular, p300 acetylates TET2, which consequently partners with DNMT1.274 Collectively, an increase in TET2 level protects vascular cells.
HDAC inhibitors (HDACis)
HDACis are divided into five main types according to their structure: hydroxamic acids, benzamides, depsipeptides, short-chain fatty acids, and cyclic peptides.275 Findeisen et al.98 showed that Scriptaid, an HDACi, caused mediation of cell cycle arrest, suppressed proliferation of VSMCs, and repressed neointima formation following vascular injury in a murine model of endovascular endothelial denudation. (R)-trichostatin A (TSA) is a natural compound, which belongs to the class of hydroxamate antibiotics.276 Previous studies have indicated that TSA exacerbates neointima formation in the LDLr−/− mouse model,233 and it furthermore facilitates myocardial hypertrophy in neonatal rats234 and neointimal thickening after vascular injury in a murine model.235 The HDACi activity of butyrate was first identified in HeLa and Friend erythroleukemia cell lines in 1977.277 A recent study has demonstrated that butyrate inhibits proliferation via the HDAC and PI3K/Akt signaling network.278
In addition, miRNAs also inversely regulate HDACi. Zhang et al.101 reported that miR-149-5p inhibits VSMC proliferation and migration by targeting HDAC4, suggesting that miR-149-5p also suppresses HDAC4. These studies clearly indicate that HDACi can be potentially used for prevention of vascular cell damage as a bioactive component of dietary fiber and in therapeutic intervention of restenosis.
SIRT-activating compounds (STACs)
According to the overall protection of SIRT against VSMCs and ECs, several natural and synthetic SIRT activators have been developed and have shown protective effects against cardiovascular disease. Currently, the best studied activator of SITR1 is resveratrol, a plant polyphenol plant antitoxin.132 In a wire-injured artery mouse model, oral administration of resveratrol significantly inhibited intimal hyperplasia.225,279 However, oral resveratrol is highly absorbable in the human body, but bioavailability is low. This raises the question of whether other mechanisms are available to mediate indirect SIRT1 activation.280 In a rat wire-injured artery model, intraperitoneal injection of the phytochemical resveratrol suppresses the development of intimal hyperplasia.236,238 Yet, recent studies have shown that relatively high concentrations of resveratrol exhibit arginase inhibitory activity in VSMCs, which can enhance the vasoconstriction response.281 However, recent studies have shown that relatively higher concentrations of resveratrol enhance vasoconstriction by inhibiting arginase activity in VSMCs. Thus, the optimal delivery method of resveratrol in vivo should be fully studied. In addition, multiple SIRT6 activators have been reported to have inhibited VSMC proliferation, including fluvastatin282 and fucoidan283 (a rat wire-injured artery model). However, the control of SIRT1 and SIRT6 activity in restenosis by specific activators remains a challenge, and although SIRT1 has made some progress, in vitro experiments on SIRT6 are still limited.
HMTis (EZH2 inhibitors)
A series of inhibitors of EZH2, including 3-deazaneplanocin A (DZNep), EPZ005687, EI1, GSK126, GSK343, or UNC1999,284 have been developed and studied.285 GSK126 inhibits monocyte adhesion to ECs and thus protects from endothelial inflammation.62 Furthermore, treatment with GSK343 recovered SM22α expression and subsequently improved aortic performance in the Marfan mouse model.59 Liang et al.55 indicated that application of UNC1999 protected against the neointimal formation and VSMC proliferation in a carotid wire-guided injury model. Overall, these results indicate that EZH2 inhibitors can be used potentially as candidate drugs for vascular diseases.
E3 inhibitors (MDM2 inhibitors)
The effect of E3 ubiquitin ligase inhibitors, such as MDM2 inhibitors, has been extensively investigated. MDM2 and the MDM2-p53 pathway are targeted for preventing and/or treating cancer. Nutlins, one of the earliest MDM2 antagonists, were identified in the early 21st century. The nutlin-3 treatment alleviates neointimal hyperplasia and the intimal/media area ratio following vascular injury in mice.140 A phase 1 trial experiment of another MDM2 antagonist, DS-3032b, in combination with quizartinib, has been conducted for patients with AML (ClinicalTrials.gov: NCT03552029). Similarly, the MDM2 antagonist, SAR405838, is under evaluation in a phase 1 clinical trial in combination with pimasertib for the treatment of malignant neoplasms (ClinicalTrials.gov: NCT01985191).286 So far, several natural products, namely, genistein,287 oroxylin,288 apigenin,289 ginsenosides290 (25-OCH3-PPD, 25-OH-PPD, and 20(S)-ginsenoside Rg3291), matrine,291 and gambogic acid,292 have been shown to suppress MDM2 expression in cancer cells. Despite the anti-cancer activities of these natural products, the use of MDM2 inhibitors in ISR remains to be assessed. However, these findings offer an important opportunity to advance current understanding regarding the role of MDM2 inhibitors as ISR protection agents in clinical practice.
BET inhibitor (BETi)
(+)-JQ1, a BET bromodomain inhibitor, displays antiproliferative effects in several animal models, including intimal hyperplasia of the carotid artery in rats with balloon injury149 and in the mouse carotid artery model of reendothelialization.150 In addition, intraperitoneal injection of (+)-JQ1 ameliorated hypertension, hypertrophy, and inflammation in mice induced by AngII.257 Rvx-208 (also known as apabetalone) is another BETi with cardiovascular benefits.293 Oral administration of RVX-208 reduces LDL levels, thereby lowing atherosclerosis in ApoE−/− mice. Furthermore, RVX-208 suppresses TNF-induced increase of MCP-1 and VCAM-1 in human ECs.258 In addition, the BRD4-specific inhibitor ZL0513 suppressed angiogenesis by regulating activating protein-1 expression.294 Interfering with the function of BET through the use of such inhibitors, which are already clinically used to treat different diseases, may be an attractive approach with the great translational potential to support current interventional therapy by minimizing the negative vascular remodeling process.
ncRNA targeted therapeutics
As reviewed above, ISR is a complicated disease that includes diverse mechanisms and multiple cell types. The emergence of ncRNAs opens a new avenue for ISR treatment. A considerable number of miRNAs have been successfully demonstrated to act as biomarkers or therapeutic targets. Similar evidence for lncRNA is also available. On the basis of therapeutic purpose, two approaches to changing ncRNA levels in diseases have been developed.295 Depending on their activity (deleterious or advantageous), ncRNA function can be enhanced or mimicked via delivery strategies, or their function can be suppressed using antisense oligonucleotides (ASOs) targeting RNA transcripts.295
Wang et al.296 have indicated the merit of locally delivering the miR-21 sponge for inhibiting VSMC proliferation and neointima formation in vivo.296 AntimiR therapies have some advantages, including the coordinated regulation of multiple gene targets via single miRNAs.297 Nevertheless, a major hurdle in individual miRNA targeting is the off-target effect of antimiRs.297 Use of stable, synthetic, and biodegradable miRNA carriers, and optimal delivery approaches, are being assessed to ensure the efficiency of miRNA targeting without toxicity.298
lncRNA is regulated by injecting oligonucleotides that inhibit their expression.299 Furthermore, two main approaches are followed for inhibiting lncRNA expression in preclinical models,300 namely, the RNA interference (RNAi)-based methods301 and the clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) system.302 CRISPR represses the expression of miRNA and lncRNA, such as GAS5, MALAT1, UCA1, lncRNA-21A, and miRNA-21.303 In the context of ISR, antisense oligonucleotides or CRISPR-Cas9 can be used to remove or activate/inhibit the expression of lncRNA to establish a restenosis therapeutic target.
However, certain unanswered problems in ncRNA-based treatment persist, including safe and effective means of administration, the long-term effectiveness of their effects, and side effects after long-term treatment. Future studies addressing these problems are therefore recommended. We anticipate that in the coming years, advancements in biological research will improve our understanding and provide more insights regarding ncRNA-based therapies.
Discussion
Although the progress of the current generation of DESs has solved several problems associated with the use of DESs, restenosis is still an evolving clinical problem. Even with the latest advances in coronary angioplasty (e.g., the introduction of bioabsorbable scaffolds), the problems of restenosis have not been solved. Whereas the understanding of the mechanisms of restenosis is positive in itself, the increasing incidence of CAD and the grim prognosis of those who develop restenosis in CAD require a serious effort to identify new treatment opportunities.
Ideally, PCI should eliminate restenosis by preventing VSMC proliferation and enhance EC regeneration. However, there is no effective therapeutic approach at this time. The advent of epigenetics may help us to understand the mechanism underlying restenosis and provide new targets for ISR drug development.
Restenosis pathogenesis can be mainly attributed to complex interactions between aberrant proliferation and migration of VSMCs and dysfunction of ECs. Epigenetic mechanisms, consisting of DNA methylation, histone modifications, and ncRNAs, may integrate these interactions. Hence, epigenetics may provide new insights for understanding ISR. Several histone modifications, DNA methylation, and numerous ncRNAs have been shown to be involved in regulating the expression of genes responsible for proliferation, migration, and inflammation in ISR. In this review, we have discussed the mechanism underlying the pathological process of ISR and the diverse epigenetic changes that regulate VSMC gene expression and function (migration, proliferation, inflammation, hypertrophy, and phenotypic switch), as well as EC function (vascular homeostasis, angiogenesis, inflammation, and mechanotransduction). Furthermore, epigenetics enzymes regulating vascular cells and the major mechanisms via which they respond to varying stimuli have been described. Based on their significance, the epigenetic enzymes are considered as potential targets in the treatment of ISR. For instance, DNMT1 maintains the synthetic state of VSMCs through hypermethylation, thus promoting the proliferation and migration of VSMCs,304 whereas TET2 has the opposite effect by producing 5hmC demethylation.39 Interestingly, the DNMT inhibitor 5-Aza can reverse DNA hypermethylation in the atherosclerotic aorta of ApoE−/− mice by not only inhibiting DNMT activity but also by restoring the TET2 expression. Similarly, several studies have shown that EZH2 promotes not only VSMC proliferation and migration,53, 54, 55,57 but also Es migration and angiogenesis.57,61 Consistent with these reports, administering the EZH2 inhibitor (such as UNC1999) orally or perivascularly in the model of carotid wire-guided injury, the neointima formation in rats was significantly reduced.55 Thus, it has the potential to be used as a stent-coating drug for restenosis. Moreover, p300 plays an epigenetic role mainly through the interaction with a variety of pro-inflammatory genes in vascular cells and the regulation of NF-κB-dependent expression.77 In HDACs, depletion of HDAC590 and HDAC994 inhibits VSMC proliferation and migration, and inhibition of HDAC996,97 reverses ox-LDL-induced EC inflammatory responses both in vivo and in vitro. These studies supported that HDAC inhibitors are promising potential targets for the treatment of restenosis. In ubiquitination, MDM2, by interacting with p53, not only inhibits neointimal hyperplasia after vascular injury in mice,140 but it also supports the response of spinal cord cells to angiogenic stimulation.144 Targeting MDM2-p53 interaction is expected to provide the potential target for the treatment of restenosis. In addition to DNA methylation and histone modification, non-RNAs also play significant roles, mainly including miRNAs, lncRNAs, circRNAs, and others. Changes in the expression of different epigenetic factors, particularly miRNAs,305 can be detected from their concentrations in body fluids such as the bloodstream, and hence they can be used as biomarkers for ISR in the future. Interestingly, we found that ncRNAs could act as both epigenetic enzyme regulators and epigenetic enzyme targets. For example, the downregulation of miR-101 in ECs promotes angiogenesis by reducing EZH2 inhibition.60 Studies have demonstrated that miR-145 can attenuate the proliferation and migration of VSMCs and neointimal hyperplasia by inhibiting the expression of UHRF1.30 These data provided a cross-regulating network in vascular cells and may unveil a novel dimension of ISR biology.
Considering the importance of epigenetic modification in restenosis, the enormous therapeutic potential of molecular regulators with epigenetic alterations have been reported recently. Consequently, we have discussed several promising molecules herein, including DNMTi, TET2 activators, HDACi, STAC, HMTi, MDM2 inhibitors, BETi, and ncRNA-targeted therapeutics, which might contribute to the prevention or reversal of restenosis. As mentioned earlier, we summarized these targeted therapeutic agents in Table 3, including natural products and chemical compounds.
However, there are still some unanswered questions that deserve further deep studies, such as safety, effective delivery systems, off-target effects, the dose and duration of action, and side effects after long-term treatment. First, the application of ncRNAs is hindered by insufficient targeting specificity, insufficient site-specific delivery, and possible off-targets.306 How can miRNA be delivered to the target in a stable and precise manner? Whether miRNA or miRNA inhibitors can be delivered to the required targeted vascular cells through the development of new vectors (e.g., nanoparticles, ultrasound-targeted microbubbles) is critical for the implementation of location-specific drug delivery and therapeutic effects.307 Second, the optimal dose or treatment time also needs to be considered. Low doses of DNMT drugs have persistent antitumor effects on blood and epithelial tumor cells.308 However, epigenetic regulation takes time, especially if the drug targets the writer protein: epigenetic markers are lost as a result of passive demethylation or deacetylation.309 Does this mean the patient needs to be treated for life? Finally, these drugs may have adverse effects and, although tolerated in the context of cancer, are still unacceptable for the treatment of chronic diseases. For example, the most common adverse reactions of the HDAC inhibitor (vorinostat) are diarrhea, nausea, anorexia, fatigue, thrombocytopenia, and dysgeusia.310 Furthermore, some adverse reactions to HDACi may result in the impairment of other important biological processes. For example, specificity class I HDACis show a strong antitumor effect, but inhibition of class I HDACs leads to a DNA damage response.311 Other major problems include cardiotoxicity312 of some HDACis and cellular resistance to some HDACis. Hence, it is crucial to develop drugs that are tolerated and specific enough to treat complex chronic diseases.
Although the problems of restenosis have not been fully solved, the prevailing mood in the field of epigenetics for restenosis is one of optimism. Clearly, the path to effective restenosis treatment is long and tortuous. Nevertheless, what we may now have is a promising road to follow.
Conclusions
In conclusion, a better understanding of the relationship between vascular cells and epigenetics will not only highlight their role in ISR but will also offer new targets for the prevention and treatment of ISR. Therefore, epigenetic drugs for cardiovascular diseases such as ISR may complement the standard DES treatment in clinical practice. Hence, understanding epigenetics and ISR is expected to yield new insights into the pathogenesis of ISR that, in turn, can be translated into novel biomarkers and therapeutic modalities.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant nos. 81870331, 31701208, and 81800238); the Key R&D Project of Shandong Province (2018GSF118241); the Natural Science Foundation of Shandong Province (grant no. ZR2017MC067); the China International Medical Foundation (Z-2016-23-2001-25); and by the Qingdao Municipal Science And Technology Bureau Project (grant no. 18-2-2-65-jch).
Author contributions
X.Y., Y.Y., J.G., X.L., T.Y., and Y.L. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Tao Yu, Email: yutao0112@qdu.edu.cn.
Yonghong Li, Email: liyonghong-66@163.com.
References
- 1.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee Heart Disease and Stroke Statistics–2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Roth G.A., Abate D., Abate K.H., Abay S.M., Abbafati C., Abbasi N., Abbastabar H., Allah F.A., Abdela J., Abdelalim A., GBD 2017 Causes of Death Collaborators Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1736–1788. doi: 10.1016/S0140-6736(18)32203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canfield J., Totary-Jain H. 40 years of percutaneous coronary intervention: history and future directions. J. Pers. Med. 2018;8:33. doi: 10.3390/jpm8040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borovac J.A., D’Amario D., Vergallo R., Porto I., Bisignani A., Galli M., Annibali G., Montone R.A., Leone A.M., Niccoli G., Crea F. Neoatherosclerosis after drug-eluting stent implantation: a novel clinical and therapeutic challenge. Eur. Heart J. Cardiovasc. Pharmacother. 2019;5:105–116. doi: 10.1093/ehjcvp/pvy036. [DOI] [PubMed] [Google Scholar]
- 5.Park S.J., Kang S.J., Virmani R., Nakano M., Ueda Y. In-stent neoatherosclerosis: a final common pathway of late stent failure. J. Am. Coll. Cardiol. 2012;59:2051–2057. doi: 10.1016/j.jacc.2011.10.909. [DOI] [PubMed] [Google Scholar]
- 6.Jukema J.W., Verschuren J.J., Ahmed T.A., Quax P.H. Restenosis after PCI. Part 1: pathophysiology and risk factors. Nat. Rev. Cardiol. 2011;9:53–62. doi: 10.1038/nrcardio.2011.132. [DOI] [PubMed] [Google Scholar]
- 7.Varela N., Lanas F., Salazar L.A., Zambrano T. The current state of microRNAs as restenosis biomarkers. Front. Genet. 2020;10:1247. doi: 10.3389/fgene.2019.01247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buccheri D., Piraino D., Andolina G., Cortese B. Understanding and managing in-stent restenosis: a review of clinical data, from pathogenesis to treatment. J. Thorac. Dis. 2016;8:E1150–E1162. doi: 10.21037/jtd.2016.10.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stratton M.S., Farina F.M., Elia L. Epigenetics and vascular diseases. J. Mol. Cell. Cardiol. 2019;133:148–163. doi: 10.1016/j.yjmcc.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pons D., de Vries F.R., van den Elsen P.J., Heijmans B.T., Quax P.H., Jukema J.W. Epigenetic histone acetylation modifiers in vascular remodelling: new targets for therapy in cardiovascular disease. Eur. Heart J. 2009;30:266–277. doi: 10.1093/eurheartj/ehn603. [DOI] [PubMed] [Google Scholar]
- 11.Pasterkamp G., de Kleijn D.P., Borst C. Arterial remodeling in atherosclerosis, restenosis and after alteration of blood flow: potential mechanisms and clinical implications. Cardiovasc. Res. 2000;45:843–852. doi: 10.1016/s0008-6363(99)00377-6. [DOI] [PubMed] [Google Scholar]
- 12.Kamiya A., Togawa T. Adaptive regulation of wall shear stress to flow change in the canine carotid artery. Am. J. Physiol. 1980;239:H14–H21. doi: 10.1152/ajpheart.1980.239.1.H14. [DOI] [PubMed] [Google Scholar]
- 13.Farb A., Weber D.K., Kolodgie F.D., Burke A.P., Virmani R. Morphological predictors of restenosis after coronary stenting in humans. Circulation. 2002;105:2974–2980. doi: 10.1161/01.cir.0000019071.72887.bd. [DOI] [PubMed] [Google Scholar]
- 14.Badran A., Nasser S.A., Mesmar J., El-Yazbi A.F., Bitto A., Fardoun M.M., Baydoun E., Eid A.H. Reactive oxygen species: modulators of phenotypic switch of vascular smooth muscle cells. Int. J. Mol. Sci. 2020;21:8764. doi: 10.3390/ijms21228764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gareri C., De Rosa S., Indolfi C. MicroRNAs for restenosis and thrombosis after vascular injury. Circ. Res. 2016;118:1170–1184. doi: 10.1161/CIRCRESAHA.115.308237. [DOI] [PubMed] [Google Scholar]
- 16.Grewe P.H., Deneke T., Machraoui A., Barmeyer J., Müller K.M. Acute and chronic tissue response to coronary stent implantation: pathologic findings in human specimen. J. Am. Coll. Cardiol. 2000;35:157–163. doi: 10.1016/s0735-1097(99)00486-6. [DOI] [PubMed] [Google Scholar]
- 17.Wang J., Jin X., Huang Y., Ran X., Luo D., Yang D., Jia D., Zhang K., Tong J., Deng X., Wang G. Endovascular stent-induced alterations in host artery mechanical environments and their roles in stent restenosis and late thrombosis. Regen. Biomater. 2018;5:177–187. doi: 10.1093/rb/rby006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welt F.G., Rogers C. Inflammation and restenosis in the stent era. Arterioscler. Thromb. Vasc. Biol. 2002;22:1769–1776. doi: 10.1161/01.atv.0000037100.44766.5b. [DOI] [PubMed] [Google Scholar]
- 19.Ang H., Koppara T.R., Cassese S., Ng J., Joner M., Foin N. Drug-coated balloons: technical and clinical progress. Vasc. Med. 2020;25:577–587. doi: 10.1177/1358863X20927791. [DOI] [PubMed] [Google Scholar]
- 20.Lansky A., Grubman D., Scheller B. Paclitaxel-coated balloons: a safe alternative to drug-eluting stents for coronary in-stent restenosis. Eur. Heart J. 2020;41:3729–3731. doi: 10.1093/eurheartj/ehz731. [DOI] [PubMed] [Google Scholar]
- 21.Kobo O., Saada M., Meisel S.R., Hellou E., Frimerman A., Fanne R.A., Mohsen J., Danon A., Roguin A. Modern stents: where are we going? Rambam Maimonides Med. J. 2020;11:e0017. doi: 10.5041/RMMJ.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dangas G.D., Claessen B.E., Caixeta A., Sanidas E.A., Mintz G.S., Mehran R. In-stent restenosis in the drug-eluting stent era. J. Am. Coll. Cardiol. 2010;56:1897–1907. doi: 10.1016/j.jacc.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 23.Yang A.Y., Kim H., Li W., Kong A.N. Natural compound-derived epigenetic regulators targeting epigenetic readers, writers and erasers. Curr. Top. Med. Chem. 2016;16:697–713. doi: 10.2174/1568026615666150826114359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borck P.C., Guo L.W., Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ. Res. 2020;126:1190–1208. doi: 10.1161/CIRCRESAHA.120.315929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Zeng C. Role of DNA methylation in cardiovascular diseases. Clin. Exp. Hypertens. 2016;38:261–267. doi: 10.3109/10641963.2015.1107087. [DOI] [PubMed] [Google Scholar]
- 26.Qi W., Li Q., Liew C.W., Rask-Madsen C., Lockhart S.M., Rasmussen L.M., Xia Y., Wang X., Khamaisi M., Croce K., King G.L. SHP-1 activation inhibits vascular smooth muscle cell proliferation and intimal hyperplasia in a rodent model of insulin resistance and diabetes. Diabetologia. 2017;60:585–596. doi: 10.1007/s00125-016-4159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L., Hao H., Hao Y., Wei G., Li G., Ma P., Xu L., Ding N., Ma S., Chen A.F., Jiang Y. Aberrant MFN2 transcription facilitates homocysteine-induced VSMCs proliferation via the increased binding of c-Myc to DNMT1 in atherosclerosis. J. Cell. Mol. Med. 2019;23:4611–4626. doi: 10.1111/jcmm.14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D., Chen Y., Xie X., Liu J., Wang Q., Kong W., Zhu Y. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J. Mol. Cell. Cardiol. 2012;53:487–496. doi: 10.1016/j.yjmcc.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y.F., Zhu J.J., Yu Tian X., Liu H., Zhang T., Zhang Y.P., Xie S.A., Zheng M., Kong W., Yao W.J. Hypermethylation of mitochondrial DNA in vascular smooth muscle cells impairs cell contractility. Cell Death Dis. 2020;11:35. doi: 10.1038/s41419-020-2240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elia L., Kunderfranco P., Carullo P., Vacchiano M., Farina F.M., Hall I.F., Mantero S., Panico C., Papait R., Condorelli G., Quintavalle M. UHRF1 epigenetically orchestrates smooth muscle cell plasticity in arterial disease. J. Clin. Invest. 2018;128:2473–2486. doi: 10.1172/JCI96121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H.P., Wang Y.H., Cao C.J., Yang X.M., Ma S.C., Han X.B., Yang X.L., Yang A.N., Tian J., Xu H. A regulatory circuit involving miR-143 and DNMT3a mediates vascular smooth muscle cell proliferation induced by homocysteine. Mol. Med. Rep. 2016;13:483–490. doi: 10.3892/mmr.2015.4558. [DOI] [PubMed] [Google Scholar]
- 32.Cao C., Zhang H., Zhao L., Zhou L., Zhang M., Xu H., Han X., Li G., Yang X., Jiang Y. miR-125b targets DNMT3b and mediates p53 DNA methylation involving in the vascular smooth muscle cells proliferation induced by homocysteine. Exp. Cell Res. 2016;347:95–104. doi: 10.1016/j.yexcr.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Li Y.S., Wang K.C., Chien S., please add this new reference Epigenetic Mechanism in Regulation of Endothelial Function by Disturbed Flow: Induction of DNA Hypermethylation by DNMT1. Cell. Mol. Bioeng. 2014;7:218–224. doi: 10.1007/s12195-014-0325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunn J., Qiu H., Kim S., Jjingo D., Hoffman R., Kim C.W., Jang I., Son D.J., Kim D., Pan C. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J. Clin. Invest. 2014;124:3187–3199. doi: 10.1172/JCI74792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y.P., Huang Y.T., Huang T.S., Pang W., Zhu J.J., Liu Y.F., Tang R.Z., Zhao C.R., Yao W.J., Li Y.S. The mammalian target of rapamycin and DNA methyltransferase 1 axis mediates vascular endothelial dysfunction in response to disturbed flow. Sci. Rep. 2017;7:14996. doi: 10.1038/s41598-017-15387-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang Y.Z., Jiménez J.M., Ou K., McCormick M.E., Zhang L.D., Davies P.F. Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ. Res. 2014;115:32–43. doi: 10.1161/CIRCRESAHA.115.303883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan Z., Deng Y., Jiao F., Guo J., Ou H. Lipopolysaccharide downregulates Kruppel-like factor 2 (KLF2) via inducing DNMT1-mediated hypermethylation in endothelial cells. Inflammation. 2017;40:1589–1598. doi: 10.1007/s10753-017-0599-0. [DOI] [PubMed] [Google Scholar]
- 38.Rasmussen K.D., Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733–750. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu R., Jin Y., Tang W.H., Qin L., Zhang X., Tellides G., Hwa J., Yu J., Martin K.A. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013;128:2047–2057. doi: 10.1161/CIRCULATIONAHA.113.002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng J., Tang Z.H., Ren Z., He B., Zeng Y., Liu L.S., Wang Z., Wei D.H., Zheng X.L., Jiang Z.S. TET2 protects against oxLDL-induced HUVEC dysfunction by upregulating the CSE/H2S system. Front. Pharmacol. 2017;8:486. doi: 10.3389/fphar.2017.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Q., Li X., Li R., Peng J., Wang Z., Jiang Z., Tang X., Peng Z., Wang Y., Wei D. Low shear stress inhibited endothelial cell autophagy through TET2 downregulation. Ann. Biomed. Eng. 2016;44:2218–2227. doi: 10.1007/s10439-015-1491-4. [DOI] [PubMed] [Google Scholar]
- 42.Peng J., Yang Q., Li A.F., Li R.Q., Wang Z., Liu L.S., Ren Z., Zheng X.L., Tang X.Q., Li G.H. Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE−/− mice. Oncotarget. 2016;7:76423–76436. doi: 10.18632/oncotarget.13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Q., Li X., Kong L., Xu Q., Wang Z., Lv Q. miR-101-3p induces vascular endothelial cell dysfunction by targeting tet methylcytosine dioxygenase 2. Acta Biochim. Biophys. Sin. (Shanghai) 2020;52:180–191. doi: 10.1093/abbs/gmz154. [DOI] [PubMed] [Google Scholar]
- 44.Hulshoff M.S., Xu X., Krenning G., Zeisberg E.M. Epigenetic regulation of endothelial-to-mesenchymal transition in chronic heart disease. Arterioscler. Thromb. Vasc. Biol. 2018;38:1986–1996. doi: 10.1161/ATVBAHA.118.311276. [DOI] [PubMed] [Google Scholar]
- 45.Uckelmann M., Sixma T.K. Histone ubiquitination in the DNA damage response. DNA Repair (Amst.) 2017;56:92–101. doi: 10.1016/j.dnarep.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 46.Du H.N. Transcription, DNA damage and beyond: the roles of histone ubiquitination and deubiquitination. Curr. Protein Pept. Sci. 2012;13:447–466. doi: 10.2174/138920312802430617. [DOI] [PubMed] [Google Scholar]
- 47.Audia J.E., Campbell R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016;8:a019521. doi: 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J., Guo Y., Zeng W., Huang L., Pang Q., Nie L., Mu J., Yuan F., Feng B. ER stress triggers MCP-1 expression through SET7/9-induced histone methylation in the kidneys of db/db mice. Am. J. Physiol. Renal Physiol. 2014;306:F916–F925. doi: 10.1152/ajprenal.00697.2012. [DOI] [PubMed] [Google Scholar]
- 49.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Chi P., Allis C.D., Wang G.G. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng Q., Wang H., Ng H.H., Erdjument-Bromage H., Tempst P., Struhl K., Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 52.Morera L., Lübbert M., Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics. 2016;8:57. doi: 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen R., Kong P., Zhang F., Shu Y.N., Nie X., Dong L.H., Lin Y.L., Xie X.L., Zhao L.L., Zhang X.J., Han M. EZH2-mediated α-actin methylation needs lncRNA TUG1, and promotes the cortex cytoskeleton formation in VSMCs. Gene. 2017;616:52–57. doi: 10.1016/j.gene.2017.03.028. [DOI] [PubMed] [Google Scholar]
- 54.Aljubran S.A., Cox R., Jr., Tamarapu Parthasarathy P., Kollongod Ramanathan G., Rajanbabu V., Bao H., Mohapatra S.S., Lockey R., Kolliputi N. Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS ONE. 2012;7:e37712. doi: 10.1371/journal.pone.0037712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang J., Li Q., Cai W., Zhang X., Yang B., Li X., Jiang S., Tian S., Zhang K., Song H. Inhibition of polycomb repressor complex 2 ameliorates neointimal hyperplasia by suppressing trimethylation of H3K27 in vascular smooth muscle cells. Br. J. Pharmacol. 2019;176:3206–3219. doi: 10.1111/bph.14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Margueron R., Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li R., Yi X., Wei X., Huo B., Guo X., Cheng C., Fang Z.M., Wang J., Feng X., Zheng P. EZH2 inhibits autophagic cell death of aortic vascular smooth muscle cells to affect aortic dissection. Cell Death Dis. 2018;9:180. doi: 10.1038/s41419-017-0213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiaoling Y., Li Z., ShuQiang L., Shengchao M., Anning Y., Ning D., Nan L., Yuexia J., Xiaoming Y., Guizhong L., Yideng J. Hyperhomocysteinemia in ApoE−/− mice leads to overexpression of enhancer of zeste homolog 2 via miR-92a regulation. PLoS ONE. 2016;11:e0167744. doi: 10.1371/journal.pone.0167744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lino Cardenas C.L., Kessinger C.W., MacDonald C., Jassar A.S., Isselbacher E.M., Jaffer F.A., Lindsay M.E. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight. 2018;3:e97493. doi: 10.1172/jci.insight.97493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smits M., Mir S.E., Nilsson R.J., van der Stoop P.M., Niers J.M., Marquez V.E., Cloos J., Breakefield X.O., Krichevsky A.M., Noske D.P. Down-regulation of miR-101 in endothelial cells promotes blood vessel formation through reduced repression of EZH2. PLoS ONE. 2011;6:e16282. doi: 10.1371/journal.pone.0016282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Floris I., Descamps B., Vardeu A., Mitić T., Posadino A.M., Shantikumar S., Sala-Newby G., Capobianco G., Mangialardi G., Howard L. Gestational diabetes mellitus impairs fetal endothelial cell functions through a mechanism involving microRNA-101 and histone methyltransferase enhancer of zester homolog-2. Arterioscler. Thromb. Vasc. Biol. 2015;35:664–674. doi: 10.1161/ATVBAHA.114.304730. [DOI] [PubMed] [Google Scholar]
- 62.Xu S., Xu Y., Yin M., Zhang S., Liu P., Koroleva M., Si S., Little P.J., Pelisek J., Jin Z.G. Flow-dependent epigenetic regulation of IGFBP5 expression by H3K27me3 contributes to endothelial anti-inflammatory effects. Theranostics. 2018;8:3007–3021. doi: 10.7150/thno.21966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maleszewska M., Vanchin B., Harmsen M.C., Krenning G. The decrease in histone methyltransferase EZH2 in response to fluid shear stress alters endothelial gene expression and promotes quiescence. Angiogenesis. 2016;19:9–24. doi: 10.1007/s10456-015-9485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang M., Liu Y., Li C., Zhang Y., Zhou X., Lu C. Long noncoding RNA OIP5-AS1 accelerates the ox-LDL mediated vascular endothelial cells apoptosis through targeting GSK-3β via recruiting EZH2. Am. J. Transl. Res. 2019;11:1827–1834. [PMC free article] [PubMed] [Google Scholar]
- 65.Tsou P.S., Campbell P., Amin M.A., Coit P., Miller S., Fox D.A., Khanna D., Sawalha A.H. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc. Natl. Acad. Sci. USA. 2019;116:3695–3702. doi: 10.1073/pnas.1813006116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi Y.G., Tsukada Y. The discovery of histone demethylases. Cold Spring Harb. Perspect. Biol. 2013;5:e017947. doi: 10.1101/cshperspect.a017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang X., Liu L., Yuan X., Wei Y., Wei X. JMJD3 in the regulation of human diseases. Protein Cell. 2019;10:864–882. doi: 10.1007/s13238-019-0653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen X., Xiu M., Xing J., Yu S., Min D., Guo F. Lanthanum chloride inhibits LPS mediated expressions of pro-inflammatory cytokines and adhesion molecules in HUVECs: involvement of NF-κB-Jmjd3 signaling. Cell. Physiol. Biochem. 2017;42:1713–1724. doi: 10.1159/000479439. [DOI] [PubMed] [Google Scholar]
- 69.Yu S., Chen X., Xiu M., He F., Xing J., Min D., Guo F. The regulation of Jmjd3 upon the expression of NF-κB downstream inflammatory genes in LPS activated vascular endothelial cells. Biochem. Biophys. Res. Commun. 2017;485:62–68. doi: 10.1016/j.bbrc.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 70.Na W., Shin J.Y., Lee J.Y., Jeong S., Kim W.S., Yune T.Y., Ju B.G. Dexamethasone suppresses JMJD3 gene activation via a putative negative glucocorticoid response element and maintains integrity of tight junctions in brain microvascular endothelial cells. J. Cereb. Blood Flow Metab. 2017;37:3695–3708. doi: 10.1177/0271678X17701156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee K., Na W., Lee J.Y., Na J., Cho H., Wu H., Yune T.Y., Kim W.S., Ju B.G. Molecular mechanism of Jmjd3-mediated interleukin-6 gene regulation in endothelial cells underlying spinal cord injury. J. Neurochem. 2012;122:272–282. doi: 10.1111/j.1471-4159.2012.07786.x. [DOI] [PubMed] [Google Scholar]
- 72.Lai J., Ge M., Shen S., Yang L., Jin T., Cao D., Xu H., Zheng X., Qiu S., Wang K. Activation of NFKB-JMJD3 signaling promotes bladder fibrosis via boosting bladder smooth muscle cell proliferation and collagen accumulation. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:2403–2410. doi: 10.1016/j.bbadis.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 73.Luo X., Yang D., Wu W., Long F., Xiao C., Qin M., Law B.Y., Suguro R., Xu X., Qu L. Critical role of histone demethylase Jumonji domain-containing protein 3 in the regulation of neointima formation following vascular injury. Cardiovasc. Res. 2018;114:1894–1906. doi: 10.1093/cvr/cvy176. [DOI] [PubMed] [Google Scholar]
- 74.Chen J., Zhang J., Yang J., Xu L., Hu Q., Xu C., Yang S., Jiang H. Histone demethylase KDM3a, a novel regulator of vascular smooth muscle cells, controls vascular neointimal hyperplasia in diabetic rats. Atherosclerosis. 2017;257:152–163. doi: 10.1016/j.atherosclerosis.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 75.Yiew K.H., Chatterjee T.K., Hui D.Y., Weintraub N.L. Histone deacetylases and cardiometabolic diseases. Arterioscler. Thromb. Vasc. Biol. 2015;35:1914–1919. doi: 10.1161/ATVBAHA.115.305046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ali I., Conrad R.J., Verdin E., Ott M. Lysine acetylation goes global: from epigenetics to metabolism and therapeutics. chem. rev. 2018;118:1216–1252. doi: 10.1021/acs.chemrev.7b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu S., Kamato D., Little P.J., Nakagawa S., Pelisek J., Jin Z.G. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol. Ther. 2019;196:15–43. doi: 10.1016/j.pharmthera.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang R., Han M., Zheng B., Li Y.J., Shu Y.N., Wen J.K. Krüppel-like factor 4 interacts with p300 to activate mitofusin 2 gene expression induced by all-trans retinoic acid in VSMCs. Acta Pharmacol. Sin. 2010;31:1293–1302. doi: 10.1038/aps.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He M., Zheng B., Zhang Y., Zhang X.H., Wang C., Yang Z., Sun Y., Wu X.L., Wen J.K. KLF4 mediates the link between TGF-β1-induced gene transcription and H3 acetylation in vascular smooth muscle cells. FASEB J. 2015;29:4059–4070. doi: 10.1096/fj.15-272658. [DOI] [PubMed] [Google Scholar]
- 80.Lu Y., Zhang L., Liao X., Sangwung P., Prosdocimo D.A., Zhou G., Votruba A.R., Brian L., Han Y.J., Gao H. Kruppel-like factor 15 is critical for vascular inflammation. J. Clin. Invest. 2013;123:4232–4241. doi: 10.1172/JCI68552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Jong R.C.M., Ewing M.M., de Vries M.R., Karper J.C., Bastiaansen A.J.N.M., Peters H.A.B., Baghana F., van den Elsen P.J., Gongora C., Jukema J.W., Quax P.H.A. The epigenetic factor PCAF regulates vascular inflammation and is essential for intimal hyperplasia development. PLoS ONE. 2017;12:e0185820. doi: 10.1371/journal.pone.0185820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pons D., Trompet S., de Craen A.J., Thijssen P.E., Quax P.H., de Vries M.R., Wierda R.J., van den Elsen P.J., Monraats P.S., Ewing M.M., PROSPER study group. WOSCOPS study group. GENDER study group Genetic variation in PCAF, a key mediator in epigenetics, is associated with reduced vascular morbidity and mortality: evidence for a new concept from three independent prospective studies. Heart. 2011;97:143–150. doi: 10.1136/hrt.2010.199927. [DOI] [PubMed] [Google Scholar]
- 83.Tang R., Mei X., Wang Y.C., Cui X.B., Zhang G., Li W., Chen S.Y. lncRNA GAS5 regulates vascular smooth muscle cell cycle arrest and apoptosis via p53 pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:2516–2525. doi: 10.1016/j.bbadis.2019.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shah A., Gray K., Figg N., Finigan A., Starks L., Bennett M. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. 2018;138:1446–1462. doi: 10.1161/CIRCULATIONAHA.117.033249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen S., Feng B., George B., Chakrabarti R., Chen M., Chakrabarti S. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2010;298:E127–E137. doi: 10.1152/ajpendo.00432.2009. [DOI] [PubMed] [Google Scholar]
- 86.Pang J., Yan C., Natarajan K., Cavet M.E., Massett M.P., Yin G., Berk B.C. GIT1 mediates HDAC5 activation by angiotensin II in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2008;28:892–898. doi: 10.1161/ATVBAHA.107.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsou P.S., Wren J.D., Amin M.A., Schiopu E., Fox D.A., Khanna D., Sawalha A.H. Histone deacetylase 5 is overexpressed in scleroderma endothelial cells and impairs angiogenesis via repression of proangiogenic factors. Arthritis Rheumatol. 2016;68:2975–2985. doi: 10.1002/art.39828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kwon I.S., Wang W., Xu S., Jin Z.G. Histone deacetylase 5 interacts with Krüppel-like factor 2 and inhibits its transcriptional activity in endothelium. Cardiovasc. Res. 2014;104:127–137. doi: 10.1093/cvr/cvu183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang W., Ha C.H., Jhun B.S., Wong C., Jain M.K., Jin Z.G. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. 2010;115:2971–2979. doi: 10.1182/blood-2009-05-224824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vega R.B., Harrison B.C., Meadows E., Roberts C.R., Papst P.J., Olson E.N., McKinsey T.A. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nikpay M., Goel A., Won H.H., Hall L.M., Willenborg C., Kanoni S., Saleheen D., Kyriakou T., Nelson C.P., Hopewell J.C. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bellenguez C., Bevan S., Gschwendtner A., Spencer C.C., Burgess A.I., Pirinen M., Jackson C.A., Traylor M., Strange A., Su Z., International Stroke Genetics Consortium (ISGC) Wellcome Trust Case Control Consortium 2 (WTCCC2) Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat. Genet. 2012;44:328–333. doi: 10.1038/ng.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Foroud T., Lai D., Koller D., Van’t Hof F., Kurki M.I., Anderson C.S., Brown R.D., Jr., Connolly E.S., Eriksson J.G., Flaherty M., Familial Intracranial Aneurysm Study Investigators Genome-wide association study of intracranial aneurysm identifies a new association on chromosome 7. Stroke. 2014;45:3194–3199. doi: 10.1161/STROKEAHA.114.006096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malhotra R., Mauer A.C., Lino Cardenas C.L., Guo X., Yao J., Zhang X., Wunderer F., Smith A.V., Wong Q., Pechlivanis S. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 2019;51:1580–1587. doi: 10.1038/s41588-019-0514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lino Cardenas C.L., Kessinger C.W., Cheng Y., MacDonald C., MacGillivray T., Ghoshhajra B., Huleihel L., Nuri S., Yeri A.S., Jaffer F.A. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018;9:1009. doi: 10.1038/s41467-018-03394-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Han X., Han X., Wang Z., Shen J., Dong Q. HDAC9 regulates ox-LDL-induced endothelial cell apoptosis by participating in inflammatory reactions. Front. Biosci. 2016;21:907–917. doi: 10.2741/4428. [DOI] [PubMed] [Google Scholar]
- 97.Shi W., Wei X., Wang Z., Han H., Fu Y., Liu J., Zhang Y., Guo J., Dong C., Zhou D. HDAC9 exacerbates endothelial injury in cerebral ischaemia/reperfusion injury. J. Cell. Mol. Med. 2016;20:1139–1149. doi: 10.1111/jcmm.12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Findeisen H.M., Gizard F., Zhao Y., Qing H., Heywood E.B., Jones K.L., Cohn D., Bruemmer D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler. Thromb. Vasc. Biol. 2011;31:851–860. doi: 10.1161/ATVBAHA.110.221952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chamorro-Jorganes A., Lee M.Y., Araldi E., Landskroner-Eiger S., Fernández-Fuertes M., Sahraei M., Quiles Del Rey M., van Solingen C., Yu J., Fernández-Hernando C. VEGF-induced expression of miR-17-92 cluster in endothelial cells is mediated by ERK/ELK1 activation and regulates angiogenesis. Circ. Res. 2016;118:38–47. doi: 10.1161/CIRCRESAHA.115.307408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeng L., Xiao Q., Margariti A., Zhang Z., Zampetaki A., Patel S., Capogrossi M.C., Hu Y., Xu Q. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J. Cell Biol. 2006;174:1059–1069. doi: 10.1083/jcb.200605113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang B., Dong Y., Liu M., Yang L., Zhao Z. miR-149-5p inhibits vascular smooth muscle cells proliferation, invasion, and migration by targeting histone deacetylase 4 (HDAC4) Med. Sci. Monit. 2019;25:7581–7590. doi: 10.12659/MSM.916522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Usui T., Morita T., Okada M., Yamawaki H. Histone deacetylase 4 controls neointimal hyperplasia via stimulating proliferation and migration of vascular smooth muscle cells. Hypertension. 2014;63:397–403. doi: 10.1161/HYPERTENSIONAHA.113.01843. [DOI] [PubMed] [Google Scholar]
- 103.Lin X., Cheng C., Zhong J., Liu B., Luo C., Ou W., Mo P., Huang Q., Liu S. Resveratrol inhibits angiotensin II-induced proliferation of A7r5 cells and decreases neointimal hyperplasia by inhibiting the CaMKII-HDAC4 signaling pathway. Mol. Med. Rep. 2018;18:1007–1014. doi: 10.3892/mmr.2018.9056. [DOI] [PubMed] [Google Scholar]
- 104.Zhang M., Urabe G., Little C., Wang B., Kent A.M., Huang Y., Kent K.C., Guo L.W. HDAC6 regulates the MRTF-A/SRF axis and vascular smooth muscle cell plasticity. JACC Basic Transl. Sci. 2018;3:782–795. doi: 10.1016/j.jacbts.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Winnik S., Auwerx J., Sinclair D.A., Matter C.M. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur. Heart J. 2015;36:3404–3412. doi: 10.1093/eurheartj/ehv290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haigis M.C., Guarente L.P. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 107.Ma L., Li Y. SIRT1: role in cardiovascular biology. Clin. Chim. Acta. 2015;440:8–15. doi: 10.1016/j.cca.2014.10.036. [DOI] [PubMed] [Google Scholar]
- 108.Yan W., Sun W., Fan J., Wang H., Han S., Li J., Yin Y. Sirt1-ROS-TRAF6 signaling-induced pyroptosis contributes to early injury in ischemic mice. Neurosci. Bull. 2020;36:845–859. doi: 10.1007/s12264-020-00489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang Z., Zhang M., Wang Z., Guo Z., Wang Z., Chen Q. Cyanidin-3-O-glucoside attenuates endothelial cell dysfunction by modulating miR-204-5p/SIRT1-mediated inflammation and apoptosis. BioFactors. 2020;46:803–812. doi: 10.1002/biof.1660. [DOI] [PubMed] [Google Scholar]
- 110.Xia L., Sun C., Zhu H., Zhai M., Zhang L., Jiang L., Hou P., Li J., Li K., Liu Z. Melatonin protects against thoracic aortic aneurysm and dissection through SIRT1-dependent regulation of oxidative stress and vascular smooth muscle cell loss. J. Pineal Res. 2020;69:e12661. doi: 10.1111/jpi.12661. [DOI] [PubMed] [Google Scholar]
- 111.Thompson A.M., Wagner R., Rzucidlo E.M. Age-related loss of SirT1 expression results in dysregulated human vascular smooth muscle cell function. Am. J. Physiol. Heart Circ. Physiol. 2014;307:H533–H541. doi: 10.1152/ajpheart.00871.2013. [DOI] [PubMed] [Google Scholar]
- 112.Gorenne I., Kumar S., Gray K., Figg N., Yu H., Mercer J., Bennett M. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. 2013;127:386–396. doi: 10.1161/CIRCULATIONAHA.112.124404. [DOI] [PubMed] [Google Scholar]
- 113.Breitenstein A., Wyss C.A., Spescha R.D., Franzeck F.C., Hof D., Riwanto M., Hasun M., Akhmedov A., von Eckardstein A., Maier W. Peripheral blood monocyte Sirt1 expression is reduced in patients with coronary artery disease. PLoS ONE. 2013;8:e53106. doi: 10.1371/journal.pone.0053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rodella L.F., Favero G., Rossini C., Foglio E., Bonomini F., Reiter R.J., Rezzani R. Aging and vascular dysfunction: beneficial melatonin effects. Age (Dordr.) 2013;35:103–115. doi: 10.1007/s11357-011-9336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang Q.J., Wang Z., Chen H.Z., Zhou S., Zheng W., Liu G., Wei Y.S., Cai H., Liu D.P., Liang C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sung J.Y., Kim S.G., Cho D.H., Kim J.R., Choi H.C. SRT1720-induced activation of SIRT1 alleviates vascular smooth muscle cell senescence through PKA-dependent phosphorylation of AMPKα at Ser485. FEBS Open Bio. 2020;10:1316–1325. doi: 10.1002/2211-5463.12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li L., Zhang H.N., Chen H.Z., Gao P., Zhu L.H., Li H.L., Lv X., Zhang Q.J., Zhang R., Wang Z. SIRT1 acts as a modulator of neointima formation following vascular injury in mice. Circ. Res. 2011;108:1180–1189. doi: 10.1161/CIRCRESAHA.110.237875. [DOI] [PubMed] [Google Scholar]
- 118.Li L., Gao P., Zhang H., Chen H., Zheng W., Lv X., Xu T., Wei Y., Liu D., Liang C. SIRT1 inhibits angiotensin II-induced vascular smooth muscle cell hypertrophy. Acta Biochim. Biophys. Sin. (Shanghai) 2011;43:103–109. doi: 10.1093/abbs/gmq104. [DOI] [PubMed] [Google Scholar]
- 119.Kong P., Yu Y., Wang L., Dou Y.Q., Zhang X.H., Cui Y., Wang H.Y., Yong Y.T., Liu Y.B., Hu H.J. circ-Sirt1 controls NF-κB activation via sequence-specific interaction and enhancement of SIRT1 expression by binding to miR-132/212 in vascular smooth muscle cells. Nucleic Acids Res. 2019;47:3580–3593. doi: 10.1093/nar/gkz141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Potente M., Ghaeni L., Baldessari D., Mostoslavsky R., Rossig L., Dequiedt F., Haendeler J., Mione M., Dejana E., Alt F.W. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kao C.L., Chen L.K., Chang Y.L., Yung M.C., Hsu C.C., Chen Y.C., Lo W.L., Chen S.J., Ku H.H., Hwang S.J. Resveratrol protects human endothelium from H2O2-induced oxidative stress and senescence via SirT1 activation. J. Atheroscler. Thromb. 2010;17:970–979. doi: 10.5551/jat.4333. [DOI] [PubMed] [Google Scholar]
- 122.Mattagajasingh I., Kim C.S., Naqvi A., Yamamori T., Hoffman T.A., Jung S.B., DeRicco J., Kasuno K., Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guo Y., Xu C., Man A.W.C., Bai B., Luo C., Huang Y., Xu A., Vanhoutte P.M., Wang Y. Endothelial SIRT1 prevents age-induced impairment of vasodilator responses by enhancing the expression and activity of soluble guanylyl cyclase in smooth muscle cells. Cardiovasc. Res. 2019;115:678–690. doi: 10.1093/cvr/cvy212. [DOI] [PubMed] [Google Scholar]
- 124.Mortuza R., Feng B., Chakrabarti S. SIRT1 reduction causes renal and retinal injury in diabetes through endothelin 1 and transforming growth factor β1. J. Cell. Mol. Med. 2015;19:1857–1867. doi: 10.1111/jcmm.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chan S.H., Hung C.H., Shih J.Y., Chu P.M., Cheng Y.H., Lin H.C., Hsieh P.L., Tsai K.L. Exercise intervention attenuates hyperhomocysteinemia-induced aortic endothelial oxidative injury by regulating SIRT1 through mitigating NADPH oxidase/LOX-1 signaling. Redox Biol. 2018;14:116–125. doi: 10.1016/j.redox.2017.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lu Y., Chen Y., Li R., Liu Q., Wang N., Zhang Y., Li B., Fang Z. Protective effects of Danzhi jiangtang capsule on vascular endothelial damages induced by high-fat diet and palmitic acid. Biomed. Pharmacother. 2018;107:1631–1640. doi: 10.1016/j.biopha.2018.08.129. [DOI] [PubMed] [Google Scholar]
- 127.Wan Y.Z., Gao P., Zhou S., Zhang Z.Q., Hao D.L., Lian L.S., Li Y.J., Chen H.Z., Liu D.P. SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell. 2014;13:890–899. doi: 10.1111/acel.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang H.N., Dai Y., Zhang C.H., Omondi A.M., Ghosh A., Khanra I., Chakraborty M., Yu X.B., Liang J. Sirtuins family as a target in endothelial cell dysfunction: implications for vascular ageing. Biogerontology. 2020;21:495–516. doi: 10.1007/s10522-020-09873-z. [DOI] [PubMed] [Google Scholar]
- 129.Liu Z., Wang J., Huang X., Li Z., Liu P. Deletion of sirtuin 6 accelerates endothelial dysfunction and atherosclerosis in apolipoprotein E-deficient mice. Transl. Res. 2016;172:18–29.e2. doi: 10.1016/j.trsl.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 130.Balestrieri M.L., Rizzo M.R., Barbieri M., Paolisso P., D’Onofrio N., Giovane A., Siniscalchi M., Minicucci F., Sardu C., D’Andrea D. Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: effects of incretin treatment. Diabetes. 2015;64:1395–1406. doi: 10.2337/db14-1149. [DOI] [PubMed] [Google Scholar]
- 131.Yao Q.P., Zhang P., Qi Y.X., Chen S.G., Shen B.R., Han Y., Yan Z.Q., Jiang Z.L. The role of SIRT6 in the differentiation of vascular smooth muscle cells in response to cyclic strain. Int. J. Biochem. Cell Biol. 2014;49:98–104. doi: 10.1016/j.biocel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 132.Guo J., Wang Z., Wu J., Liu M., Li M., Sun Y., Huang W., Li Y., Zhang Y., Tang W. Endothelial SIRT6 is vital to prevent hypertension and associated cardiorenal injury through targeting Nkx3.2-GATA5 signaling. Circ. Res. 2019;124:1448–1461. doi: 10.1161/CIRCRESAHA.118.314032. [DOI] [PubMed] [Google Scholar]
- 133.D’Onofrio N., Servillo L., Giovane A., Casale R., Vitiello M., Marfella R., Paolisso G., Balestrieri M.L. Ergothioneine oxidation in the protection against high-glucose induced endothelial senescence: Involvement of SIRT1 and SIRT6. Free Radic. Biol. Med. 2016;96:211–222. doi: 10.1016/j.freeradbiomed.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 134.Liu R., Liu H., Ha Y., Tilton R.G., Zhang W. Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. BioMed Res. Int. 2014;2014:902842. doi: 10.1155/2014/902842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hashimoto-Komatsu A., Hirase T., Asaka M., Node K. Angiotensin II induces microtubule reorganization mediated by a deacetylase SIRT2 in endothelial cells. Hypertens. Res. 2011;34:949–956. doi: 10.1038/hr.2011.64. [DOI] [PubMed] [Google Scholar]
- 136.Wu, X., Bu, P., Liu, J., Zhao, L., Wang, X., and Li, N. (2013). [Inhibitory effect of Sirt3 on proliferation of vascular smooth muscle cells induced by angiotensin II]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 29, 237–241. [PubMed]
- 137.Lu H., Sun L., Chen W., Zhou Y., Liu K., Chen J., Zhang Z., Zhang C., Tian H. Sirtuin 3 therapy attenuates aging expression, oxidative stress parameters, and neointimal hyperplasia formation in vein grafts. Ann. Vasc. Surg. 2020;64:303–317. doi: 10.1016/j.avsg.2019.05.044. [DOI] [PubMed] [Google Scholar]
- 138.Zhao Q., Zhou D., You H., Lou B., Zhang Y., Tian Y., Guo N., Chen X., Liu Y., Wu Y. IFN-γ aggravates neointimal hyperplasia by inducing endoplasmic reticulum stress and apoptosis in macrophages by promoting ubiquitin-dependent liver X receptor-α degradation. FASEB J. 2017;31:5321–5331. doi: 10.1096/fj.201700327R. [DOI] [PubMed] [Google Scholar]
- 139.Li S., Zhao J., Shang D., Kass D.J., Zhao Y. Ubiquitination and deubiquitination emerge as players in idiopathic pulmonary fibrosis pathogenesis and treatment. JCI Insight. 2018;3:e120362. doi: 10.1172/jci.insight.120362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hashimoto T., Ichiki T., Ikeda J., Narabayashi E., Matsuura H., Miyazaki R., Inanaga K., Takeda K., Sunagawa K. Inhibition of MDM2 attenuates neointimal hyperplasia via suppression of vascular proliferation and inflammation. Cardiovasc. Res. 2011;91:711–719. doi: 10.1093/cvr/cvr108. [DOI] [PubMed] [Google Scholar]
- 141.Li G., Xie N., Yao Y., Zhang Y., Guo J., Feng Y., Lv F., Xiao R.P., Cao C.M. Identification of PI3K regulatory subunit p55γ as a novel inhibitor of vascular smooth muscle cell proliferation and neointimal formation. Cardiovasc. Res. 2015;105:75–85. doi: 10.1093/cvr/cvu235. [DOI] [PubMed] [Google Scholar]
- 142.Wu G., Cai J., Han Y., Chen J., Huang Z.P., Chen C., Cai Y., Huang H., Yang Y., Liu Y. lincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation. 2014;130:1452–1465. doi: 10.1161/CIRCULATIONAHA.114.011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aiken J., Roudier E., Ciccone J., Drouin G., Stromberg A., Vojnovic J., Olfert I.M., Haas T., Gustafsson T., Grenier G., Birot O. Phosphorylation of murine double minute-2 on Ser166 is downstream of VEGF-A in exercised skeletal muscle and regulates primary endothelial cell migration and FoxO gene expression. FASEB J. 2016;30:1120–1134. doi: 10.1096/fj.15-276964. [DOI] [PubMed] [Google Scholar]
- 144.Milkiewicz M., Roudier E., Doyle J.L., Trifonova A., Birot O., Haas T.L. Identification of a mechanism underlying regulation of the anti-angiogenic forkhead transcription factor FoxO1 in cultured endothelial cells and ischemic muscle. Am. J. Pathol. 2011;178:935–944. doi: 10.1016/j.ajpath.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cai J., Culley M.K., Zhao Y., Zhao J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell. 2018;9:754–769. doi: 10.1007/s13238-017-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pfoh R., Lacdao I.K., Saridakis V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr. Relat. Cancer. 2015;22:T35–T54. doi: 10.1530/ERC-14-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jean-Charles P.Y., Zhang L., Wu J.H., Han S.O., Brian L., Freedman N.J., Shenoy S.K. Ubiquitin-specific protease 20 regulates the reciprocal functions of β-arrestin2 in Toll-like receptor 4-promoted nuclear factor κB (NFκB) activation. J. Biol. Chem. 2016;291:7450–7464. doi: 10.1074/jbc.M115.687129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jean-Charles P.Y., Wu J.H., Zhang L., Kaur S., Nepliouev I., Stiber J.A., Brian L., Qi R., Wertman V., Shenoy S.K., Freedman N.J. USP20 (ubiquitin-specific protease 20) inhibits TNF (tumor necrosis factor)-triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018;38:2295–2305. doi: 10.1161/ATVBAHA.118.311071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang B., Zhang M., Takayama T., Shi X., Roenneburg D.A., Kent K.C., Guo L.W. BET bromodomain blockade mitigates intimal hyperplasia in rat carotid arteries. EBioMedicine. 2015;2:1650–1661. doi: 10.1016/j.ebiom.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dutzmann J., Haertle M., Daniel J.M., Kloss F., Musmann R.J., Kalies K., Knöpp K., Pilowski C., Sirisko M., Sieweke J.T. BET bromodomain containing epigenetic reader proteins regulate vascular smooth muscle cell proliferation and neointima formation. Cardiovasc. Res. 2020 doi: 10.1093/cvr/cvaa121. Published online April 30, 2020. [DOI] [PubMed] [Google Scholar]
- 151.Chen J., Fu Y., Day D.S., Sun Y., Wang S., Liang X., Gu F., Zhang F., Stevens S.M., Zhou P. VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis. Nat. Commun. 2017;8:383. doi: 10.1038/s41467-017-00405-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu S., Yang Y., Jiang S., Tang N., Tian J., Ponnusamy M., Tariq M.A., Lian Z., Xin H., Yu T. Understanding the role of non-coding RNA (ncRNA) in stent restenosis. Atherosclerosis. 2018;272:153–161. doi: 10.1016/j.atherosclerosis.2018.03.036. [DOI] [PubMed] [Google Scholar]
- 153.Romano G., Veneziano D., Acunzo M., Croce C.M. Small non-coding RNA and cancer. Carcinogenesis. 2017;38:485–491. doi: 10.1093/carcin/bgx026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Boon R.A., Jaé N., Holdt L., Dimmeler S. Long noncoding RNAs: from clinical genetics to therapeutic targets? J. Am. Coll. Cardiol. 2016;67:1214–1226. doi: 10.1016/j.jacc.2015.12.051. [DOI] [PubMed] [Google Scholar]
- 155.Wang Q., Yang Y., Fu X., Wang Z., Liu Y., Li M., Zhang Y., Li Y., Li P.F., Yu T., Chu X.M. Long noncoding RNA XXYLT1-AS2 regulates proliferation and adhesion by targeting the RNA binding protein FUS in HUVEC. Atherosclerosis. 2020;298:58–69. doi: 10.1016/j.atherosclerosis.2020.02.018. [DOI] [PubMed] [Google Scholar]
- 156.Liu Y., Yang Y., Wang Z., Fu X., Chu X.M., Li Y., Wang Q., He X., Li M., Wang K. Insights into the regulatory role of circRNA in angiogenesis and clinical implications. Atherosclerosis. 2020;298:14–26. doi: 10.1016/j.atherosclerosis.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 157.Liu S., Yang Y., Jiang S., Xu H., Tang N., Lobo A., Zhang R., Liu S., Yu T., Xin H. miR-378a-5p regulates proliferation and migration in vascular smooth muscle cell by targeting CDK1. Front. Genet. 2019;10:22. doi: 10.3389/fgene.2019.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tang N., Jiang S., Yang Y., Liu S., Ponnusamy M., Xin H., Yu T. Noncoding RNAs as therapeutic targets in atherosclerosis with diabetes mellitus. Cardiovasc. Ther. 2018;36:e12436. doi: 10.1111/1755-5922.12436. [DOI] [PubMed] [Google Scholar]
- 159.Yang Y., Yu T., Jiang S., Zhang Y., Li M., Tang N., Ponnusamy M., Wang J.X., Li P.F. miRNAs as potential therapeutic targets and diagnostic biomarkers for cardiovascular disease with a particular focus on WO2010091204. Expert Opin. Ther. Pat. 2017;27:1021–1029. doi: 10.1080/13543776.2017.1344217. [DOI] [PubMed] [Google Scholar]
- 160.Barros-Viegas A.T., Carmona V., Ferreiro E., Guedes J., Cardoso A.M., Cunha P., Pereira de Almeida L., Resende de Oliveira C., Pedro de Magalhães J., Peça J., Cardoso A.L. miRNA-31 improves cognition and abolishes amyloid-β pathology by targeting APP and BACE1 in an animal model of Alzheimer’s disease. Mol. Ther. Nucleic Acids. 2020;19:1219–1236. doi: 10.1016/j.omtn.2020.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Soto-Reyes E., González-Barrios R., Cisneros-Soberanis F., Herrera-Goepfert R., Pérez V., Cantú D., Prada D., Castro C., Recillas-Targa F., Herrera L.A. Disruption of CTCF at the miR-125b1 locus in gynecological cancers. BMC Cancer. 2012;12:40. doi: 10.1186/1471-2407-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lujambio A., Ropero S., Ballestar E., Fraga M.F., Cerrato C., Setién F., Casado S., Suarez-Gauthier A., Sanchez-Cespedes M., Git A. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 163.Amodio N., Rossi M., Raimondi L., Pitari M.R., Botta C., Tagliaferri P., Tassone P. miR-29s: a family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget. 2015;6:12837–12861. doi: 10.18632/oncotarget.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen Z., Wang X., Liu R., Chen L., Yi J., Qi B., Shuang Z., Liu M., Li X., Li S., Tang H. KDM4B-mediated epigenetic silencing of miRNA-615-5p augments RAB24 to facilitate malignancy of hepatoma cells. Oncotarget. 2017;8:17712–17725. doi: 10.18632/oncotarget.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Garzon R., Liu S., Fabbri M., Liu Z., Heaphy C.E., Callegari E., Schwind S., Pang J., Yu J., Muthusamy N. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Malumbres M. miRNAs and cancer: an epigenetics view. Mol. Aspects Med. 2013;34:863–874. doi: 10.1016/j.mam.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tuddenham L., Wheeler G., Ntounia-Fousara S., Waters J., Hajihosseini M.K., Clark I., Dalmay T. The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett. 2006;580:4214–4217. doi: 10.1016/j.febslet.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 168.Varambally S., Cao Q., Mani R.S., Shankar S., Wang X., Ateeq B., Laxman B., Cao X., Jing X., Ramnarayanan K. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Verrando P., Capovilla M., Rahmani R. Trans-nonachlor decreases miR-141-3p levels in human melanocytes in vitro promoting melanoma cell characteristics and shows a multigenerational impact on miR-8 levels in Drosophila. Toxicology. 2016;368–369:129–141. doi: 10.1016/j.tox.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 170.Wang X., Wu Q., Xu B., Wang P., Fan W., Cai Y., Gu X., Meng F. miR-124 exerts tumor suppressive functions on the cell proliferation, motility and angiogenesis of bladder cancer by fine-tuning UHRF1. FEBS J. 2015;282:4376–4388. doi: 10.1111/febs.13502. [DOI] [PubMed] [Google Scholar]
- 171.Hernández-Romero I.A., Guerra-Calderas L., Salgado-Albarrán M., Maldonado-Huerta T., Soto-Reyes E. The regulatory roles of non-coding RNAs in angiogenesis and neovascularization from an epigenetic perspective. Front. Oncol. 2019;9:1091. doi: 10.3389/fonc.2019.01091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cheng Y., Liu X., Yang J., Lin Y., Xu D.Z., Lu Q., Deitch E.A., Huo Y., Delphin E.S., Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res. 2009;105:158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Grundmann S., Hans F.P., Kinniry S., Heinke J., Helbing T., Bluhm F., Sluijter J.P., Hoefer I., Pasterkamp G., Bode C., Moser M. MicroRNA-100 regulates neovascularization by suppression of mammalian target of rapamycin in endothelial and vascular smooth muscle cells. Circulation. 2011;123:999–1009. doi: 10.1161/CIRCULATIONAHA.110.000323. [DOI] [PubMed] [Google Scholar]
- 174.Deng L., Blanco F.J., Stevens H., Lu R., Caudrillier A., McBride M., McClure J.D., Grant J., Thomas M., Frid M. MicroRNA-143 activation regulates smooth muscle and endothelial cell crosstalk in pulmonary arterial hypertension. Circ. Res. 2015;117:870–883. doi: 10.1161/CIRCRESAHA.115.306806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu X., Cheng Y., Zhang S., Lin Y., Yang J., Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2009;104:476–487. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Maegdefessel L., Spin J.M., Raaz U., Eken S.M., Toh R., Azuma J., Adam M., Nakagami F., Heymann H.M., Chernogubova E. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat. Commun. 2014;5:5214. doi: 10.1038/ncomms6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lee J., Heo J., Kang H. miR-92b-3p-TSC1 axis is critical for mTOR signaling-mediated vascular smooth muscle cell proliferation induced by hypoxia. Cell Death Differ. 2019;26:1782–1795. doi: 10.1038/s41418-018-0243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yang F., Chen Q., He S., Yang M., Maguire E.M., An W., Afzal T.A., Luong L.A., Zhang L., Xiao Q. miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation. 2018;137:1824–1841. doi: 10.1161/CIRCULATIONAHA.117.027799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li P., Zhu N., Yi B., Wang N., Chen M., You X., Zhao X., Solomides C.C., Qin Y., Sun J. MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ. Res. 2013;113:1117–1127. doi: 10.1161/CIRCRESAHA.113.301306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Torella D., Iaconetti C., Catalucci D., Ellison G.M., Leone A., Waring C.D., Bochicchio A., Vicinanza C., Aquila I., Curcio A. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res. 2011;109:880–893. doi: 10.1161/CIRCRESAHA.111.240150. [DOI] [PubMed] [Google Scholar]
- 181.Farina F.M., Hall I.F., Serio S., Zani S.M., Climent M., Salvarani N., Carullo P., Civilini E., Condorelli G., Elia L., Quintavalle M. miR-128-3p is a novel regulator of vascular smooth muscle cell phenotypic switch and vascular diseases. Circ. Res. 2020;126:e120–e135. doi: 10.1161/CIRCRESAHA.120.316489. [DOI] [PubMed] [Google Scholar]
- 182.Zeng Z., Xia L., Fan X., Ostriker A.C., Yarovinsky T., Su M., Zhang Y., Peng X., Xie Y., Pi L. Platelet-derived miR-223 promotes a phenotypic switch in arterial injury repair. J. Clin. Invest. 2019;129:1372–1386. doi: 10.1172/JCI124508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hassel D., Cheng P., White M.P., Ivey K.N., Kroll J., Augustin H.G., Katus H.A., Stainier D.Y., Srivastava D. MicroRNA-10 regulates the angiogenic behavior of zebrafish and human endothelial cells by promoting vascular endothelial growth factor signaling. Circ. Res. 2012;111:1421–1433. doi: 10.1161/CIRCRESAHA.112.279711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bridge G., Monteiro R., Henderson S., Emuss V., Lagos D., Georgopoulou D., Patient R., Boshoff C. The microRNA-30 family targets DLL4 to modulate endothelial cell behavior during angiogenesis. Blood. 2012;120:5063–5072. doi: 10.1182/blood-2012-04-423004. [DOI] [PubMed] [Google Scholar]
- 185.Li P., Yin Y.L., Guo T., Sun X.Y., Ma H., Zhu M.L., Zhao F.R., Xu P., Chen Y., Wan G.R. Inhibition of aberrant microRNA-133a expression in endothelial cells by statin prevents endothelial dysfunction by targeting GTP cyclohydrolase 1 in vivo. Circulation. 2016;134:1752–1765. doi: 10.1161/CIRCULATIONAHA.116.017949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Loyer X., Potteaux S., Vion A.C., Guérin C.L., Boulkroun S., Rautou P.E., Ramkhelawon B., Esposito B., Dalloz M., Paul J.L. Inhibition of microRNA-92a prevents endothelial dysfunction and atherosclerosis in mice. Circ. Res. 2014;114:434–443. doi: 10.1161/CIRCRESAHA.114.302213. [DOI] [PubMed] [Google Scholar]
- 187.Schober A., Nazari-Jahantigh M., Wei Y., Bidzhekov K., Gremse F., Grommes J., Megens R.T., Heyll K., Noels H., Hristov M. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 2014;20:368–376. doi: 10.1038/nm.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ghosh G., Subramanian I.V., Adhikari N., Zhang X., Joshi H.P., Basi D., Chandrashekhar Y.S., Hall J.L., Roy S., Zeng Y., Ramakrishnan S. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J. Clin. Invest. 2010;120:4141–4154. doi: 10.1172/JCI42980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sun X., He S., Wara A.K.M., Icli B., Shvartz E., Tesmenitsky Y., Belkin N., Li D., Blackwell T.S., Sukhova G.K. Systemic delivery of microRNA-181b inhibits nuclear factor-κB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ. Res. 2014;114:32–40. doi: 10.1161/CIRCRESAHA.113.302089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gupta S.K., Bang C., Thum T. Circulating microRNAs as biomarkers and potential paracrine mediators of cardiovascular disease. Circ. Cardiovasc. Genet. 2010;3:484–488. doi: 10.1161/CIRCGENETICS.110.958363. [DOI] [PubMed] [Google Scholar]
- 191.He M., Gong Y., Shi J., Pan Z., Zou H., Sun D., Tu X., Tan X., Li J., Li W. Plasma microRNAs as potential noninvasive biomarkers for in-stent restenosis. PLoS ONE. 2014;9:e112043. doi: 10.1371/journal.pone.0112043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dai R., Liu Y., Zhou Y., Xiong X., Zhou W., Li W., Zhou W., Chen M. Potential of circulating pro-angiogenic microRNA expressions as biomarkers for rapid angiographic stenotic progression and restenosis risks in coronary artery disease patients underwent percutaneous coronary intervention. J. Clin. Lab. Anal. 2020;34:e23013. doi: 10.1002/jcla.23013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.O’Sullivan J.F., Neylon A., Fahy E.F., Yang P., McGorrian C., Blake G.J. miR-93-5p is a novel predictor of coronary in-stent restenosis. Heart Asia. 2019;11:e011134. doi: 10.1136/heartasia-2018-011134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang H., Zhang Q., Liu Y., Xue T. miR-146a and miR-146b predict increased restenosis and rapid angiographic stenotic progression risk in coronary heart disease patients who underwent percutaneous coronary intervention. Ir. J. Med. Sci. 2020;189:467–474. doi: 10.1007/s11845-019-02101-9. [DOI] [PubMed] [Google Scholar]
- 195.Xiong Y., Kuang W., Lu S., Guo H., Wu M., Ye M., Wu L. Long noncoding RNA HOXB13-AS1 regulates HOXB13 gene methylation by interacting with EZH2 in glioma. Cancer Med. 2018;7:4718–4728. doi: 10.1002/cam4.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Xu Y., Yao Y., Jiang X., Zhong X., Wang Z., Li C., Kang P., Leng K., Ji D., Li Z. SP1-induced upregulation of lncRNA SPRY4-IT1 exerts oncogenic properties by scaffolding EZH2/LSD1/DNMT1 and sponging miR-101-3p in cholangiocarcinoma. J. Exp. Clin. Cancer Res. 2018;37:81. doi: 10.1186/s13046-018-0747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kuo C.C., Hänzelmann S., Sentürk Cetin N., Frank S., Zajzon B., Derks J.P., Akhade V.S., Ahuja G., Kanduri C., Grummt I. Detection of RNA-DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019;47:e32. doi: 10.1093/nar/gkz037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Leisegang M.S., Fork C., Josipovic I., Richter F.M., Preussner J., Hu J., Miller M.J., Epah J., Hofmann P., Günther S. Long noncoding RNA MANTIS facilitates endothelial angiogenic function. Circulation. 2017;136:65–79. doi: 10.1161/CIRCULATIONAHA.116.026991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ahmed A.S.I., Dong K., Liu J., Wen T., Yu L., Xu F., Kang X., Osman I., Hu G., Bunting K.M. Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA. 2018;115:E8660–E8667. doi: 10.1073/pnas.1803725115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhao J., Zhang W., Lin M., Wu W., Jiang P., Tou E., Xue M., Richards A., Jourd’heuil D., Asif A. MYOSLID is a novel serum response factor-dependent long noncoding RNA that amplifies the vascular smooth muscle differentiation program. Arterioscler. Thromb. Vasc. Biol. 2016;36:2088–2099. doi: 10.1161/ATVBAHA.116.307879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wu Y.X., Zhang S.H., Cui J., Liu F.T. Long noncoding RNA XR007793 regulates proliferation and migration of vascular smooth muscle cell via suppressing miR-23b. Med. Sci. Monit. 2018;24:5895–5903. doi: 10.12659/MSM.908902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Moran M., Cheng X., Shihabudeen Haider Ali M.S., Wase N., Nguyen N., Yang W., Zhang C., DiRusso C., Sun X. Transcriptome analysis-identified long noncoding RNA CRNDE in maintaining endothelial cell proliferation, migration, and tube formation. Sci. Rep. 2019;9:19548. doi: 10.1038/s41598-019-56030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liao B., Chen R., Lin F., Mai A., Chen J., Li H., Xu Z., Dong S. Long noncoding RNA HOTTIP promotes endothelial cell proliferation and migration via activation of the Wnt/β-catenin pathway. J. Cell. Biochem. 2018;119:2797–2805. doi: 10.1002/jcb.26448. [DOI] [PubMed] [Google Scholar]
- 204.Huang S., Lu W., Ge D., Meng N., Li Y., Su L., Zhang S., Zhang Y., Zhao B., Miao J. A new microRNA signal pathway regulated by long noncoding RNA TGFB2-OT1 in autophagy and inflammation of vascular endothelial cells. Autophagy. 2015;11:2172–2183. doi: 10.1080/15548627.2015.1106663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Leeper N.J., Maegdefessel L. Non-coding RNAs: key regulators of smooth muscle cell fate in vascular disease. Cardiovasc. Res. 2018;114:611–621. doi: 10.1093/cvr/cvx249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Santer L., Bär C., Thum T. Circular RNAs: a novel class of functional rna molecules with a therapeutic perspective. Mol. Ther. 2019;27:1350–1363. doi: 10.1016/j.ymthe.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhang C., Huo S.T., Wu Z., Chen L., Wen C., Chen H., Du W.W., Wu N., Guan D., Lian S., Yang B.B. Rapid development of targeting circRNAs in cardiovascular diseases. Mol. Ther. Nucleic Acids. 2020;21:568–576. doi: 10.1016/j.omtn.2020.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zhang S., Wang W., Wu X., Zhou X. Regulatory roles of circular RNAs in coronary artery disease. Mol. Ther. Nucleic Acids. 2020;21:172–179. doi: 10.1016/j.omtn.2020.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Xu J.Y., Chang N.B., Rong Z.H., Li T., Xiao L., Yao Q.P., Jiang R., Jiang J. circDiaph3 regulates rat vascular smooth muscle cell differentiation, proliferation, and migration. FASEB J. 2019;33:2659–2668. doi: 10.1096/fj.201800243RRR. [DOI] [PubMed] [Google Scholar]
- 210.Ferreira H.J., Davalos V., de Moura M.C., Soler M., Perez-Salvia M., Bueno-Costa A., Setien F., Moran S., Villanueva A., Esteller M. Circular RNA CpG island hypermethylation-associated silencing in human cancer. Oncotarget. 2018;9:29208–29219. doi: 10.18632/oncotarget.25673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li C.Y., Ma L., Yu B. Circular RNA hsa_circ_0003575 regulates oxLDL induced vascular endothelial cells proliferation and angiogenesis. Biomed. Pharmacother. 2017;95:1514–1519. doi: 10.1016/j.biopha.2017.09.064. [DOI] [PubMed] [Google Scholar]
- 212.Zhang S., Song G., Yuan J., Qiao S., Xu S., Si Z., Yang Y., Xu X., Wang A. Circular RNA circ_0003204 inhibits proliferation, migration and tube formation of endothelial cell in atherosclerosis via miR-370-3p/TGFβR2/phosph-SMAD3 axis. J. Biomed. Sci. 2020;27:11. doi: 10.1186/s12929-019-0595-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 213.Hall I.F., Climent M., Quintavalle M., Farina F.M., Schorn T., Zani S., Carullo P., Kunderfranco P., Civilini E., Condorelli G., Elia L. circ_Lrp6, a circular RNA enriched in vascular smooth muscle cells, acts as a sponge regulating miRNA-145 function. Circ. Res. 2019;124:498–510. doi: 10.1161/CIRCRESAHA.118.314240. [DOI] [PubMed] [Google Scholar]
- 214.Yao Q.P., Liu Z., Yao A.H., Liu J.T., Jiang J., Chen Y., Li S.S., Han Y., Jiang Z.L., Qi Y.X. Circular RNA circTET3 mediates migration of rat vascular smooth muscle cells by targeting miR-351-5p. J. Cell. Physiol. 2020;35:6831–6842. doi: 10.1002/jcp.29577. [DOI] [PubMed] [Google Scholar]
- 215.Mao Y.Y., Wang J.Q., Guo X.X., Bi Y., Wang C.X. circ-SATB2 upregulates STIM1 expression and regulates vascular smooth muscle cell proliferation and differentiation through miR-939. Biochem. Biophys. Res. Commun. 2018;505:119–125. doi: 10.1016/j.bbrc.2018.09.069. [DOI] [PubMed] [Google Scholar]
- 216.Ganesan A., Arimondo P.B., Rots M.G., Jeronimo C., Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin. Epigenetics. 2019;11:174. doi: 10.1186/s13148-019-0776-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Marks P.A., Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 218.Issa M.E., Takhsha F.S., Chirumamilla C.S., Perez-Novo C., Vanden Berghe W., Cuendet M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics. 2017;9:17. doi: 10.1186/s13148-017-0319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.O’Connor O.A., Horwitz S., Masszi T., Van Hoof A., Brown P., Doorduijn J., Hess G., Jurczak W., Knoblauch P., Chawla S. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J. Clin. Oncol. 2015;33:2492–2499. doi: 10.1200/JCO.2014.59.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dunn J., Simmons R., Thabet S., Jo H. The role of epigenetics in the endothelial cell shear stress response and atherosclerosis. Int. J. Biochem. Cell Biol. 2015;67:167–176. doi: 10.1016/j.biocel.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cao Q., Wang X., Jia L., Mondal A.K., Diallo A., Hawkins G.A., Das S.K., Parks J.S., Yu L., Shi H. Inhibiting DNA methylation by 5-aza-2′-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology. 2014;155:4925–4938. doi: 10.1210/en.2014-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Leckey L.C., Garige M., Varatharajalu R., Gong M., Nagata T., Spurney C.F., Lakshman R.M. Quercetin and ethanol attenuate the progression of atherosclerotic plaques with concomitant up regulation of paraoxonase1 (PON1) gene expression and PON1 activity in LDLR−/− mice. Alcohol. Clin. Exp. Res. 2010;34:1535–1542. doi: 10.1111/j.1530-0277.2010.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Shen Y., Ward N.C., Hodgson J.M., Puddey I.B., Wang Y., Zhang D., Maghzal G.J., Stocker R., Croft K.D. Dietary quercetin attenuates oxidant-induced endothelial dysfunction and atherosclerosis in apolipoprotein E knockout mice fed a high-fat diet: a critical role for heme oxygenase-1. Free Radic. Biol. Med. 2013;65:908–915. doi: 10.1016/j.freeradbiomed.2013.08.185. [DOI] [PubMed] [Google Scholar]
- 224.Jiang Y.H., Jiang L.Y., Wang Y.C., Ma D.F., Li X. Quercetin attenuates atherosclerosis via modulating oxidized LDL-induced endothelial cellular senescence. Front. Pharmacol. 2020;11:512. doi: 10.3389/fphar.2020.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Khandelwal A.R., Hebert V.Y., Kleinedler J.J., Rogers L.K., Ullevig S.L., Asmis R., Shi R., Dugas T.R. Resveratrol and quercetin interact to inhibit neointimal hyperplasia in mice with a carotid injury. J. Nutr. 2012;142:1487–1494. doi: 10.3945/jn.112.162628. [DOI] [PubMed] [Google Scholar]
- 226.Das S., Ray R., Snehlata, Das N., Srivastava L.M. Effect of ascorbic acid on prevention of hypercholesterolemia induced atherosclerosis. Mol. Cell. Biochem. 2006;285:143–147. doi: 10.1007/s11010-005-9070-x. [DOI] [PubMed] [Google Scholar]
- 227.Nespereira B., Pérez-Ilzarbe M., Fernández P., Fuentes A.M., Páramo J.A., Rodríguez J.A. Vitamins C and E downregulate vascular VEGF and VEGFR-2 expression in apolipoprotein-E-deficient mice. Atherosclerosis. 2003;171:67–73. doi: 10.1016/j.atherosclerosis.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 228.Qu K., Ma X.F., Li G.H., Zhang H., Liu Y.M., Zhang K., Zeng J.F., Lei J.J., Wei D.H., Wang Z. Vitamin C down-regulate apo(a) expression via Tet2-dependent DNA demethylation in HepG2 cells. Int. J. Biol. Macromol. 2017;98:637–645. doi: 10.1016/j.ijbiomac.2017.02.025. [DOI] [PubMed] [Google Scholar]
- 229.Nunes G.L., Sgoutas D.S., Redden R.A., Sigman S.R., Gravanis M.B., King S.B., 3rd, Berk B.C. Combination of vitamins C and E alters the response to coronary balloon injury in the pig. Arterioscler. Thromb. Vasc. Biol. 1995;15:156–165. doi: 10.1161/01.atv.15.1.156. [DOI] [PubMed] [Google Scholar]
- 230.Xu Y., Xu S., Liu P., Koroleva M., Zhang S., Si S., Jin Z.G. Suberanilohydroxamic acid as a pharmacological Kruppel-like factor 2 activator that represses vascular inflammation and atherosclerosis. J. Am. Heart Assoc. 2017;6:e007134. doi: 10.1161/JAHA.117.007134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Huang A., Young T.L., Dang V.T., Shi Y., McAlpine C.S., Werstuck G.H. 4-Phenylbutyrate and valproate treatment attenuates the progression of atherosclerosis and stabilizes existing plaques. Atherosclerosis. 2017;266:103–112. doi: 10.1016/j.atherosclerosis.2017.09.034. [DOI] [PubMed] [Google Scholar]
- 232.Bowes A.J., Khan M.I., Shi Y., Robertson L., Werstuck G.H. Valproate attenuates accelerated atherosclerosis in hyperglycemic apoE-deficient mice: evidence in support of a role for endoplasmic reticulum stress and glycogen synthase kinase-3 in lesion development and hepatic steatosis. Am. J. Pathol. 2009;174:330–342. doi: 10.2353/ajpath.2009.080385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Choi J.H., Nam K.H., Kim J., Baek M.W., Park J.E., Park H.Y., Kwon H.J., Kwon O.S., Kim D.Y., Oh G.T. Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005;25:2404–2409. doi: 10.1161/01.ATV.0000184758.07257.88. [DOI] [PubMed] [Google Scholar]
- 234.Davidson S.M., Townsend P.A., Carroll C., Yurek-George A., Balasubramanyam K., Kundu T.K., Stephanou A., Packham G., Ganesan A., Latchman D.S. The transcriptional coactivator p300 plays a critical role in the hypertrophic and protective pathways induced by phenylephrine in cardiac cells but is specific to the hypertrophic effect of urocortin. ChemBioChem. 2005;6:162–170. doi: 10.1002/cbic.200400246. [DOI] [PubMed] [Google Scholar]
- 235.Song S., Kang S.W., Choi C. Trichostatin A enhances proliferation and migration of vascular smooth muscle cells by downregulating thioredoxin 1. Cardiovasc. Res. 2010;85:241–249. doi: 10.1093/cvr/cvp263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Orozco-Sevilla V., Naftalovich R., Hoffmann T., London D., Czernizer E., Yang C., Dardik A., Dardik H. Epigallocatechin-3-gallate is a potent phytochemical inhibitor of intimal hyperplasia in the wire-injured carotid artery. J. Vasc. Surg. 2013;58:1360–1365. doi: 10.1016/j.jvs.2012.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lu Y.J., Jan Y.J., Ko B.S., Liang S.M., Chen L., Wu C.C., Chin C.H., Kuo C.C., Yet S.F., Liou J.Y. Expression of Nik-related kinase in smooth muscle cells attenuates vascular inflammation and intimal hyperplasia. Aging (Albany NY) 2020;12:7511–7533. doi: 10.18632/aging.103104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhu Y., Takayama T., Wang B., Kent A., Zhang M., Binder B.Y., Urabe G., Shi Y., DiRenzo D., Goel S.A. Restenosis inhibition and re-differentiation of TGFβ/Smad3-activated smooth muscle cells by resveratrol. Sci. Rep. 2017;7:41916. doi: 10.1038/srep41916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Fukao H., Ijiri Y., Miura M., Hashimoto M., Yamashita T., Fukunaga C., Oiwa K., Kawai Y., Suwa M., Yamamoto J. Effect of trans-resveratrol on the thrombogenicity and atherogenicity in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Blood Coagul. Fibrinolysis. 2004;15:441–446. doi: 10.1097/00001721-200408000-00001. [DOI] [PubMed] [Google Scholar]
- 240.Hibender S., Franken R., van Roomen C., Ter Braake A., van der Made I., Schermer E.E., Gunst Q., van den Hoff M.J., Lutgens E., Pinto Y.M. Resveratrol inhibits aortic root dilatation in the Fbn1C1039G/+ Marfan mouse model. Arterioscler. Thromb. Vasc. Biol. 2016;36:1618–1626. doi: 10.1161/ATVBAHA.116.307841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Norata G.D., Marchesi P., Passamonti S., Pirillo A., Violi F., Catapano A.L. Anti-inflammatory and anti-atherogenic effects of cathechin, caffeic acid and trans-resveratrol in apolipoprotein E deficient mice. Atherosclerosis. 2007;191:265–271. doi: 10.1016/j.atherosclerosis.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 242.Fry J.L., Al Sayah L., Weisbrod R.M., Van Roy I., Weng X., Cohen R.A., Bachschmid M.M., Seta F. Vascular smooth muscle sirtuin-1 protects against diet-induced aortic stiffness. Hypertension. 2016;68:775–784. doi: 10.1161/HYPERTENSIONAHA.116.07622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chen Y.X., Zhang M., Cai Y., Zhao Q., Dai W. The Sirt1 activator SRT1720 attenuates angiotensin II-induced atherosclerosis in apoE−/− mice through inhibiting vascular inflammatory response. Biochem. Biophys. Res. Commun. 2015;465:732–738. doi: 10.1016/j.bbrc.2015.08.066. [DOI] [PubMed] [Google Scholar]
- 244.Miranda M.X., van Tits L.J., Lohmann C., Arsiwala T., Winnik S., Tailleux A., Stein S., Gomes A.P., Suri V., Ellis J.L. The Sirt1 activator SRT3025 provides atheroprotection in Apoe−/− mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur. Heart J. 2015;36:51–59. doi: 10.1093/eurheartj/ehu095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Yokota T., Nomura K., Nagashima M., Kamimura N. Fucoidan alleviates high-fat diet-induced dyslipidemia and atherosclerosis in ApoEshl mice deficient in apolipoprotein E expression. J. Nutr. Biochem. 2016;32:46–54. doi: 10.1016/j.jnutbio.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 246.Yin J., Wang J., Li F., Yang Z., Yang X., Sun W., Xia B., Li T., Song W., Guo S. The fucoidan from the brown seaweed Ascophyllum nodosum ameliorates atherosclerosis in apolipoprotein E-deficient mice. Food Funct. 2019;10:5124–5139. doi: 10.1039/c9fo00619b. [DOI] [PubMed] [Google Scholar]
- 247.Xu Y., Zhu W., Wang T., Jin L., Liu T., Li X., Guan Z., Jiang Z., Meng X., Wang J., Guo Y. Low molecule weight fucoidan mitigates atherosclerosis in ApoE (-/-) mouse model through activating multiple signal pathway. Carbohydr. Polym. 2019;206:110–120. doi: 10.1016/j.carbpol.2018.10.097. [DOI] [PubMed] [Google Scholar]
- 248.Deux J.F., Meddahi-Pellé A., Le Blanche A.F., Feldman L.J., Colliec-Jouault S., Brée F., Boudghène F., Michel J.B., Letourneur D. Low molecular weight fucoidan prevents neointimal hyperplasia in rabbit iliac artery in-stent restenosis model. Arterioscler. Thromb. Vasc. Biol. 2002;22:1604–1609. doi: 10.1161/01.atv.0000032034.91020.0a. [DOI] [PubMed] [Google Scholar]
- 249.Zhang Y., Wang X., Wang Y., Liu Y., Xia M. Supplementation of cyanidin-3-O-β-glucoside promotes endothelial repair and prevents enhanced atherogenesis in diabetic apolipoprotein E-deficient mice. J. Nutr. 2013;143:1248–1253. doi: 10.3945/jn.113.177451. [DOI] [PubMed] [Google Scholar]
- 250.Zhang Y., Ma X., Li X., Zhang T., Qin M., Ren L. Effects of Icariin on atherosclerosis and predicted function regulatory network in ApoE deficient mice. BioMed Res. Int. 2018;2018:9424186. doi: 10.1155/2018/9424186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Xiao H.B., Sui G.G., Lu X.Y. Icariin improves eNOS/NO pathway to prohibit the atherogenesis of apolipoprotein E-null mice. Can. J. Physiol. Pharmacol. 2017;95:625–633. doi: 10.1139/cjpp-2016-0367. [DOI] [PubMed] [Google Scholar]
- 252.Zhang S., Zou J., Li P., Zheng X., Feng D. Curcumin protects against atherosclerosis in apolipoprotein E-knockout mice by inhibiting Toll-like receptor 4 expression. J. Agric. Food Chem. 2018;66:449–456. doi: 10.1021/acs.jafc.7b04260. [DOI] [PubMed] [Google Scholar]
- 253.Bea F., Blessing E., Shelley M.I., Shultz J.M., Rosenfeld M.E. Simvastatin inhibits expression of tissue factor in advanced atherosclerotic lesions of apolipoprotein E deficient mice independently of lipid lowering: potential role of simvastatin-mediated inhibition of Egr-1 expression and activation. Atherosclerosis. 2003;167:187–194. doi: 10.1016/s0021-9150(02)00387-8. [DOI] [PubMed] [Google Scholar]
- 254.Kleemann R., Princen H.M., Emeis J.J., Jukema J.W., Fontijn R.D., Horrevoets A.J., Kooistra T., Havekes L.M. Rosuvastatin reduces atherosclerosis development beyond and independent of its plasma cholesterol-lowering effect in APOE∗3-Leiden transgenic mice: evidence for antiinflammatory effects of rosuvastatin. Circulation. 2003;108:1368–1374. doi: 10.1161/01.CIR.0000086460.55494.AF. [DOI] [PubMed] [Google Scholar]
- 255.Lin C.P., Huang P.H., Lai C.F., Chen J.W., Lin S.J., Chen J.S. Simvastatin attenuates oxidative stress, NF-κB activation, and artery calcification in LDLR-/- mice fed with high fat diet via down-regulation of tumor necrosis factor-α and TNF receptor 1. PLoS ONE. 2015;10:e0143686. doi: 10.1371/journal.pone.0143686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Yang N., Dong B., Yang J., Li Y., Kou L., Liu Y., Qin Q. Effects of rosuvastatin on apolipoprotein J in balloon-injured carotid artery in rats. Arq. Bras. Cardiol. 2018;111:562–568. doi: 10.5935/abc.20180163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Das S., Senapati P., Chen Z., Reddy M.A., Ganguly R., Lanting L., Mandi V., Bansal A., Leung A., Zhang S. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017;8:1467. doi: 10.1038/s41467-017-01629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jahagirdar R., Zhang H., Azhar S., Tobin J., Attwell S., Yu R., Wu J., McLure K.G., Hansen H.C., Wagner G.S. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis. 2014;236:91–100. doi: 10.1016/j.atherosclerosis.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 259.Nicholls S.J., Ray K.K., Johansson J.O., Gordon A., Sweeney M., Halliday C., Kulikowski E., Wong N., Kim S.W., Schwartz G.G. Selective BET protein inhibition with apabetalone and cardiovascular events: a pooled analysis of trials in patients with coronary artery disease. Am. J. Cardiovasc. Drugs. 2018;18:109–115. doi: 10.1007/s40256-017-0250-3. [DOI] [PubMed] [Google Scholar]
- 260.Liu K., Liu Y., Lau J.L., Min J. Epigenetic targets and drug discovery. Part 2: histone demethylation and DNA methylation. Pharmacol. Ther. 2015;151:121–140. doi: 10.1016/j.pharmthera.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 261.Bryan J., Kantarjian H., Garcia-Manero G., Jabbour E. Pharmacokinetic evaluation of decitabine for the treatment of leukemia. Expert Opin. Drug Metab. Toxicol. 2011;7:661–672. doi: 10.1517/17425255.2011.575062. [DOI] [PubMed] [Google Scholar]
- 262.Sharma S., Kelly T.K., Jones P.A. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Brueckner B., Kuck D., Lyko F. DNA methyltransferase inhibitors for cancer therapy. Cancer J. 2007;13:17–22. doi: 10.1097/PPO.0b013e31803c7245. [DOI] [PubMed] [Google Scholar]
- 264.Lin R.K., Hsu C.H., Wang Y.C. Mithramycin A inhibits DNA methyltransferase and metastasis potential of lung cancer cells. Anticancer Drugs. 2007;18:1157–1164. doi: 10.1097/CAD.0b013e3282a215e9. [DOI] [PubMed] [Google Scholar]
- 265.Wang W., Zhang Z.Z., Wu Y., Wang R.Q., Chen J.W., Chen J., Zhang Y., Chen Y.J., Geng M., Xu Z.D. (−)-Epigallocatechin-3-gallate ameliorates atherosclerosis and modulates hepatic lipid metabolic gene expression in apolipoprotein E knockout mice: involvement of TTC39B. Front. Pharmacol. 2018;9:195. doi: 10.3389/fphar.2018.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Paul B., Li Y., Tollefsbol T.O. The effects of combinatorial genistein and sulforaphane in breast tumor inhibition: role in epigenetic regulation. Int. J. Mol. Sci. 2018;19:1754. doi: 10.3390/ijms19061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ulker E., Parker W.H., Raj A., Qu Z.C., May J.M. Ascorbic acid prevents VEGF-induced increases in endothelial barrier permeability. Mol. Cell. Biochem. 2016;412:73–79. doi: 10.1007/s11010-015-2609-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Barabutis N., Khangoora V., Marik P.E., Catravas J.D. Hydrocortisone and ascorbic acid synergistically prevent and repair lipopolysaccharide-induced pulmonary endothelial barrier dysfunction. Chest. 2017;152:954–962. doi: 10.1016/j.chest.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Yin R., Mao S.Q., Zhao B., Chong Z., Yang Y., Zhao C., Zhang D., Huang H., Gao J., Li Z. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013;135:10396–10403. doi: 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- 270.Cimmino L., Dolgalev I., Wang Y., Yoshimi A., Martin G.H., Wang J., Ng V., Xia B., Witkowski M.T., Mitchell-Flack M. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 2017;170:1079–1095.e20. doi: 10.1016/j.cell.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Hassan N., Ali A., Withycombe C., Ahluwalia M., Al-Nasseri R.H., Tonks A., Morris K. TET-2 up-regulation is associated with the anti-inflammatory action of Vicenin-2. Cytokine. 2018;108:37–42. doi: 10.1016/j.cyto.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 272.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Nakagawa T., Lv L., Nakagawa M., Yu Y., Yu C., D’Alessio A.C., Nakayama K., Fan H.Y., Chen X., Xiong Y. CRL4VprBP E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol. Cell. 2015;57:247–260. doi: 10.1016/j.molcel.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Zhang Y.W., Wang Z., Xie W., Cai Y., Xia L., Easwaran H., Luo J., Yen R.C., Li Y., Baylin S.B. Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol. Cell. 2017;65:323–335. doi: 10.1016/j.molcel.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Seidel C., Schnekenburger M., Dicato M., Diederich M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012;7:357–367. doi: 10.1007/s12263-012-0283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Tsuji N., Kobayashi M., Nagashima K., Wakisaka Y., Koizumi K. A new antifungal antibiotic, trichostatin. J. Antibiot. (Tokyo) 1976;29:1–6. doi: 10.7164/antibiotics.29.1. [DOI] [PubMed] [Google Scholar]
- 277.Riggs M.G., Whittaker R.G., Neumann J.R., Ingram V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 278.Mathew O.P., Ranganna K., Mathew J., Zhu M., Yousefipour Z., Selvam C., Milton S.G. Cellular effects of butyrate on vascular smooth muscle cells are mediated through disparate actions on dual targets, histone deacetylase (HDAC) activity and PI3K/Akt signaling network. Int. J. Mol. Sci. 2019;20:2902. doi: 10.3390/ijms20122902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kim J.W., Lim S.C., Lee M.Y., Lee J.W., Oh W.K., Kim S.K., Kang K.W. Inhibition of neointimal formation by trans-resveratrol: role of phosphatidyl inositol 3-kinase-dependent Nrf2 activation in heme oxygenase-1 induction. Mol. Nutr. Food Res. 2010;54:1497–1505. doi: 10.1002/mnfr.201000016. [DOI] [PubMed] [Google Scholar]
- 280.Walle T., Hsieh F., DeLegge M.H., Oatis J.E., Jr., Walle U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 281.Choi C.I., Koo B.H., Hong D., Kwon H.J., Hoe K.L., Won M.H., Kim Y.M., Lim H.K., Ryoo S. Resveratrol is an arginase inhibitor contributing to vascular smooth muscle cell vasoconstriction via increasing cytosolic calcium. Mol. Med. Rep. 2019;19:3767–3774. doi: 10.3892/mmr.2019.10035. [DOI] [PubMed] [Google Scholar]
- 282.Sanyour H.J., Li N., Rickel A.P., Torres H.M., Anderson R.H., Miles M.R., Childs J.D., Francis K.R., Tao J., Hong Z. Statin-mediated cholesterol depletion exerts coordinated effects on the alterations in rat vascular smooth muscle cell biomechanics and migration. J. Physiol. 2020;598:1505–1522. doi: 10.1113/JP279528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Hlawaty H., Suffee N., Sutton A., Oudar O., Haddad O., Ollivier V., Laguillier-Morizot C., Gattegno L., Letourneur D., Charnaux N. Low molecular weight fucoidan prevents intimal hyperplasia in rat injured thoracic aorta through the modulation of matrix metalloproteinase-2 expression. Biochem. Pharmacol. 2011;81:233–243. doi: 10.1016/j.bcp.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 284.Papale M., Ferretti E., Battaglia G., Bellavia D., Mai A., Tafani M. EZH2, HIF-1, and their inhibitors: an overview on pediatric cancers. Front Pediatr. 2018;6:328. doi: 10.3389/fped.2018.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Gulati N., Béguelin W., Giulino-Roth L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk. Lymphoma. 2018;59:1574–1585. doi: 10.1080/10428194.2018.1430795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Kocik J., Machula M., Wisniewska A., Surmiak E., Holak T.A., Skalniak L. Helping the released guardian: drug combinations for supporting the anticancer activity of HDM2 (MDM2) antagonists. Cancers (Basel) 2019;11:1014. doi: 10.3390/cancers11071014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Li M., Zhang Z., Hill D.L., Chen X., Wang H., Zhang R. Genistein, a dietary isoflavone, down-regulates the MDM2 oncogene at both transcriptional and posttranslational levels. Cancer Res. 2005;65:8200–8208. doi: 10.1158/0008-5472.CAN-05-1302. [DOI] [PubMed] [Google Scholar]
- 288.Mu R., Qi Q., Gu H., Wang J., Yang Y., Rong J., Liu W., Lu N., You Q., Guo Q. Involvement of p53 in oroxylin A-induced apoptosis in cancer cells. Mol. Carcinog. 2009;48:1159–1169. doi: 10.1002/mc.20570. [DOI] [PubMed] [Google Scholar]
- 289.Fang J., Xia C., Cao Z., Zheng J.Z., Reed E., Jiang B.H. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005;19:342–353. doi: 10.1096/fj.04-2175com. [DOI] [PubMed] [Google Scholar]
- 290.Qin J.J., Li X., Hunt C., Wang W., Wang H., Zhang R. Natural products targeting the p53-MDM2 pathway and mutant p53: recent advances and implications in cancer medicine. Genes Dis. 2018;5:204–219. doi: 10.1016/j.gendis.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wang W., Rayburn E.R., Zhao Y., Wang H., Zhang R. Novel ginsenosides 25-OH-PPD and 25-OCH3-PPD as experimental therapy for pancreatic cancer: anticancer activity and mechanisms of action. Cancer Lett. 2009;278:241–248. doi: 10.1016/j.canlet.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Rong J.J., Hu R., Qi Q., Gu H.Y., Zhao Q., Wang J., Mu R., You Q.D., Guo Q.L. Gambogic acid down-regulates MDM2 oncogene and induces p21Waf1/CIP1 expression independent of p53. Cancer Lett. 2009;284:102–112. doi: 10.1016/j.canlet.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 293.Schooling C.M., Zhao J.V. How might bromodomain and extra-terminal (BET) inhibitors operate in cardiovascular disease? Am. J. Cardiovasc. Drugs. 2019;19:107–111. doi: 10.1007/s40256-018-00315-3. [DOI] [PubMed] [Google Scholar]
- 294.Zhou Z., Li X., Liu Z., Huang L., Yao Y., Li L., Chen J., Zhang R., Zhou J., Wang L., Zhang Q.Q. A bromodomain-containing protein 4 (BRD4) inhibitor suppresses angiogenesis by regulating AP-1 expression. Front. Pharmacol. 2020;11:1043. doi: 10.3389/fphar.2020.01043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.van der Harst P., de Windt L.J., Chambers J.C. Translational perspective on epigenetics in cardiovascular disease. J. Am. Coll. Cardiol. 2017;70:590–606. doi: 10.1016/j.jacc.2017.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wang X.W., Zhang C., Lee K.C., He X.J., Lu Z.Q., Huang C., Wu Q.C. Adenovirus-mediated gene transfer of microRNA-21 sponge inhibits neointimal hyperplasia in rat vein grafts. Int. J. Biol. Sci. 2017;13:1309–1319. doi: 10.7150/ijbs.20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Hamburg N.M., Leeper N.J. Therapeutic potential of modulating microRNA in peripheral artery disease. Curr. Vasc. Pharmacol. 2015;13:316–323. [Google Scholar]
- 298.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 299.Poller W., Dimmeler S., Heymans S., Zeller T., Haas J., Karakas M., Leistner D.M., Jakob P., Nakagawa S., Blankenberg S. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur. Heart J. 2018;39:2704–2716. doi: 10.1093/eurheartj/ehx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Skuratovskaia D., Vulf M., Komar A., Kirienkova E., Litvinova L. Promising directions in atherosclerosis treatment based on epigenetic regulation using microRNAs and long noncoding RNAs. Biomolecules. 2019;9:226. doi: 10.3390/biom9060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Kole R., Krainer A.R., Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Chery J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016;4:35–50. doi: 10.14304/surya.jpr.v4n7.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Ho T.T., Zhou N., Huang J., Koirala P., Xu M., Fung R., Wu F., Mo Y.Y. Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 2015;43:e17. doi: 10.1093/nar/gku1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zhuang J., Luan P., Li H., Wang K., Zhang P., Xu Y., Peng W. The Yin-Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2017;37:84–97. doi: 10.1161/ATVBAHA.116.307923. [DOI] [PubMed] [Google Scholar]
- 305.Valihrach L., Androvic P., Kubista M. Circulating miRNA analysis for cancer diagnostics and therapy. Mol. Aspects Med. 2020;72:100825. doi: 10.1016/j.mam.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 306.To K.K.W., Fong W., Tong C.W.S., Wu M., Yan W., Cho W.C.S. Advances in the discovery of microRNA-based anticancer therapeutics: latest tools and developments. Expert Opin. Drug Discov. 2020;15:63–83. doi: 10.1080/17460441.2020.1690449. [DOI] [PubMed] [Google Scholar]
- 307.Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 308.Tsai H.C., Li H., Van Neste L., Cai Y., Robert C., Rassool F.V., Shin J.J., Harbom K.M., Beaty R., Pappou E. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Erdmann A., Halby L., Fahy J., Arimondo P.B. Targeting DNA methylation with small molecules: what’s next? J. Med. Chem. 2015;58:2569–2583. doi: 10.1021/jm500843d. [DOI] [PubMed] [Google Scholar]
- 310.Moj D., Britz H., Burhenne J., Stewart C.F., Egerer G., Haefeli W.E., Lehr T. A physiologically based pharmacokinetic and pharmacodynamic (PBPK/PD) model of the histone deacetylase (HDAC) inhibitor vorinostat for pediatric and adult patients and its application for dose specification. Cancer Chemother. Pharmacol. 2017;80:1013–1026. doi: 10.1007/s00280-017-3447-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Khabele D. The therapeutic potential of class I selective histone deacetylase inhibitors in ovarian cancer. Front. Oncol. 2014;4:111. doi: 10.3389/fonc.2014.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Shultz M.D., Cao X., Chen C.H., Cho Y.S., Davis N.R., Eckman J., Fan J., Fekete A., Firestone B., Flynn J. Optimization of the in vitro cardiac safety of hydroxamate-based histone deacetylase inhibitors. J. Med. Chem. 2011;54:4752–4772. doi: 10.1021/jm200388e. [DOI] [PubMed] [Google Scholar]