

Alzheimer’s disease (AD), a common neurodegenerative disease, afflicts 26 million people worldwide currently with projection of a fourfold increase in this figure by the year 2050 (Brookmeyer et al., 2018). The majority of AD cases (95%) are sporadic, having the late-onset affecting those over 65 years old. About 15% among those 65 years and older suffer from AD, and the incidence of AD is close to 50% for those aged over 85 years (Brookmeyer et al., 2018). There are a number of changes in the ageing brain, such as reduced cerebral blood flow, white matter changes, iron overload, and neuroinflammation (Chen et al., 2010). The reduction of cerebral blood flow leads to hypoperfusion, thus causing cerebral hypoxia, which is a common vascular component among the AD risk factors. Prolonged and severe hypoxia can cause neuronal loss and memory impairment. It has been understood that patients with stroke are at risk of AD. Up to 1/3 of stroke survivors suffer from post stroke dementia (Mijajlovic et al., 2017). The most common cause of post stroke dementia are vascular dementia, AD and mixed dementia.
Hypoxia is a condition where oxygen tensions are below the normal level of tissue oxygen tension (Chen et al., 2018). Hypoxia can be classified as acute, intermittent, chronic according to the duration; mild, moderate, severe according to the extent. The duration and extent of hypoxia in different cells dictate their beneficial or detrimental effects on the cells. Severe and/or chronic hypoxia, impair delivery of oxygen, affect cellular metabolism, ATP production, Ca2+ homeostasis, and lead to reactive oxygen species (ROS) formation and inflammation (Chen et al., 2018). While mild, moderate and/or intermittent hypoxia have been found to induce protective adaptations in the brain (Lall et al., 2019).
In the AD process, hypoxia enhances a shift in amyloid precursor protein processing toward the amyloidogenic pathway and down-regulates the function of α-secretase. Hypoxia inhibits the expression and activity of an amyloid-degrading peptidase ‘neprilysin’ in cortical neurons of rat, increasing the accumulation of amyloid beta (Aβ) peptides in the affected regions. It has been shown that hypoxia interplays with Aβ peptide aggravating the neuronal death. There is an increase vulnerability of hippocampal neurons to Aβ peptide toxicity during hypoxia. Calcium dyshomeostasis is a fundamental mechanism in AD pathogenesis. Aβ interaction with the plasma membrane leads to elevated cytoplasmic Ca2+ concentrations and enhances excitation of neuron. Chronic hypoxia enhances Ca2+ entry and mitochondrial Ca2+ content, potentiates posttranscriptional trafficking of L-type Ca2+ channels. Both hypoxia and Aβ can trigger the activation of microglia, leading to a maladaptive neuroinflammatory response. Whilst, neuroinflammation itself could be another initiating pathological trigger of AD (Lall et al., 2019). Neuroinflammation plays a detrimental role in AD pathogenesis, as microglia depletion by colony stimulating factor receptor 1 inhibitors improves AD symptoms in in vivo (Rawlinson et al., 2020).
Cells respond to hypoxia by stabilizing hypoxia inducible factor (HIF), a key transcription factor regulating oxygen homeostasis. The HIF levels in cells are directly regulated by four oxygen-sensitive hydroxylases: 3 prolyl hydroxylases (PHD1-3) and 1 asparaginyl hydroxylase, factor inhibiting HIF (Chen et al., 2018). Both oxygen and 2 oxoglutarate are substrates of these enzymes, while iron and ascorbate are co-factors. In a normal oxygen environment, HIF hydroxylase is active and degrades the HIF-α subunit. Yet, in a reduced oxygen environment, the absence of oxygen as a substrate, renders the HIF hydroxylases inactive, thus the HIF-α subunit accumulates and binds to the HIF-α subunit, forming the HIF molecule (Figure 1). The stabilization of HIF upregulates the expression of hundreds of HIF targeted genes that help cells and tissue survival in hypoxia, as well as a number of apoptotic genes, such as Bax, BNIP3 (Figure 1). HIF-1α is ubiquitously expressed, and appears the most active isoform during short periods (2–24 hours) of intense hypoxia or anoxia (< 0.1% O2) in some cell lines. HIF-2α is more tissue specific and is emerging as a distinct entity in target gene induction in the vascular endothelia cells, and is known as an endothelium specific HIF-α isoform. HIF-2α is active under mild or physiological hypoxia, and continues to be active even after 48–72 hours of hypoxia. HIF-2α shares 48% amino acid homology with HIF-1α, and binds to similar promoter sites but differs in the cofactors it recruits. HIF-1 and HIF-2 have largely overlapping but also some non-redundant functions. In some contexts, HIF-1α plays a key role in initial response to hypoxia whereas HIF-2α drives the hypoxic response during chronic hypoxic exposure.
Figure 1.
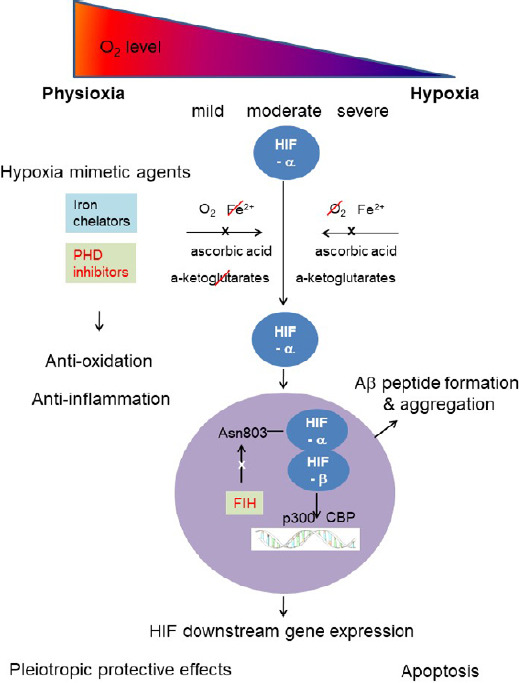
Schematic diagram describing effects of hypoxia and hypoxia mimetic agents on neurons in Alzheimer’s disease (AD) process.
Severe and/or chronic hypoxia can leads to amyloid beta (Aβ) peptide formation and aggregation, Ca2+ dyshomeostasis, reactive oxygen species (ROS) formation in neurons as well as neuroinflammation (Chen et al., 2018). While mild, moderate and/or intermittent hypoxia have been found to induce protective adaptations in the brain (Lall et al., 2019). Hypoxia inducible factor (HIF), a key mediator of oxygen homeostasis, generates numerous pleiotropic protective effects, but also participates in Aβ peptide formation and aggregation. Hypoxia mimetic agents, both iron chelators and HIF prolyl hydroxylase (PHD) inhibitors, can remove ROS and reduce neuroinflammation, in addition to activating HIF. Pharmacological stabilization of HIF can be neuroprotective and be explored as an adjunctive therapy for AD.
It has been revealed by sequence analysis and gel shift studies that HIF-1 binds to β-secretase 1 (BACE1) promoter, and that overexpression of HIF-1α in neuronal cells increases BACE1 mRNA and protein levels, and down-regulation of HIF-1α reduces the levels of BACE1. It has been observed that HIF-1 binds to the promoter of anterior pharynx-defective phenotype (APH-1) to up-regulate its expression, leading to an increase in γ-cleavage of amyloid precursor protein and Notch. HIF-1 also binds to gene promoter of neprilysin and suppresses its transcription (Lall et al., 2019).
On the other hand, HIF-1 has been proposed as a neuroprotective factor, which has ability to suppress neuronal cell death caused by hypoxia or oxidative stress, and to protect against Aβ peptide toxicity (Zheng et al., 2015; Ashok et al., 2017; Merelli et al., 2018). Recombinant adeno-associated virus vector expressing the human HIF-1α gene recombinant adeno-associated virus-HIF-1α inhibits neuronal apoptosis of the hippocampus induced by Aβ peptides. HIF-1 increases glycolysis and the hexose monophosphate shunt, maintains the mitochondrial membrane potential and cytosolic accumulation of cytochrome C, thereby inactivating caspase-9 and caspase-3, and thus prevents neuronal death in the AD brain. Oxidative damage, caused by Aβ peptide induces mitochondrial dysfunction, which is a major characteristic of neuronal apoptosis. Additional pathological features of AD are astrocyte activation and reduced glucose metabolism in some selected brain areas. Maintenance of HIF-1α levels reverses Aβ peptide-induced glial activation and glycolytic changes, thus mediating a neuroprotective response to Aβ peptide by maintaining metabolic integrity. HIF-1 is the major transcription factor that increases capillary network density and improves blood circulation in living tissue by regulating proteins expression such as erythropoietin, glucose transporter 1, 3 and vascular endothelial growth factor. Erythropoietin is able to block the Aβ generated neuronal apoptosis, while glucose transporter 1, 3 increase glucose transport into brain nerve cells. All in all, HIF-1 participates in hypoxia-induced adaptive reactions to restore cellular homeostasis, and postpone the progression of AD (Figure 1).
A group of agents which can upregulate the HIF levels in normoxia are termed as hypoxia mimetic agents (Chen et al., 2018). These agents include HIF hydroxylase inhibitors and iron chelators. The inhibition of HIF hydroxylase not only exerts pleiotropic neuroprotective effects as a consequence of HIF induction, but also has anti-oxidant and anti-inflammatory effects (Figure 1). HIF hydroxylase inhibition, which engages multiple downstream effector pathways, is a promising therapeutic intervention that can challenge the heterogeneity in AD pathophysiology present in humans. Because the HIF hydroxylase inhibition leverages endogenous adaptive programs, the breadth of the response will not lead to an increased likelihood of toxicity. It is important to emphasize that HIF hydroxylase inhibition does not equal HIF activation. HIFs are only one of a number of growing substrates known to modulate via HIF hydroxylase (Chen et al., 2018). To our best knowledge, there are no studies applying PHD inhibitors for AD treatment. Li et al. (2018) applied FG4592 for the treatment of Parkinson’s disease both in vitro and in vivo. FG4592, a HIF PHD inhibitor, is currently used for anaemia treatment in patients with chronic kidney disease. Li et al. (2018) concluded that FG4592 could be potentially used for treating Parkinson’s disease by improving the neuronal mitochondrial function under oxidative stress.
Iron chelation has been widely studied to treat neurodegenerative diseases, including AD and Parkinson’s disease, as iron accumulation is common in ageing and in neurodegenerative diseases (Devos et al., 2020). Abnormal iron metabolism generates hydroxyl radicals through the Fenton reaction, triggers oxidative stress reactions, causes lipid peroxidation and damages in cell protein and DNA, and ultimately leads to cell death. Iron promotes Aβ aggregation and induces aggregation of hyperphosphorylated tau. Iron-Aβ interaction exhibits toxic effects through ROS. Iron chelators, such as desferoxamine, deferiprone, deferasirox, have generated some promising results in both preclinical studies and some clinical trials for the AD treatment. However, the potential role of HIF stabilization by these agents has not been documented (Merelli et al., 2018). Iron chelators can cause iron chelation and HIF activation simultaneously, as iron is one of the cofactors of HIF PHD (Figure 1). M30, a multi-target iron chelator, has been reported to increase the HIF-1 protein level and to upregulate expression of the HIF-1 downstream genes, such as vascular endothelial growth factor and erythropoietin. It has been reported M30 simultaneously attenuates tau phosphorylation and protects cortical neurons against Aβ25–35 toxicity (Merelli et al., 2018). In a series of single blinded clinical trials, deferiprone is found to be feasible to treat the mild cognitive impairment of AD patients (NCT02878538).
However, there was no evidence for the cognitive efficacy of two of metal protein attenuating compounds – clioquinol and PBT2, in AD patients (Sampson et al., 2014). Large clinical trials are now requested to demonstrate cognitive efficacy of PBT2.
A new generation of iron chelators with multifunction is needed for treating AD. Ideally, the new agents will have the capacity to interfere with another signaling pathway in addition to iron chelation, to stop or reverse the AD process. Iron chelators can lead to both iron chelation and HIF activation, but the latter is not well studied when treating the neurodegenartive disease with iron chelators. Activation of HIF can add an advantage for iron chelation therapy to activate endogenous neuroregenerative pathways, in addition to inhibition of ROS production (Figure 1). Since the expression and function of PHDs have been shown to vary among tissues, the distinct role of HIF alpha isoforms should be considered. Attention need to be pay on the ability of the novel iron chelators to induce different response of HIF-1α and HIF-2α.
In conclusion, cerebral hypoxia is associated with AD. The effects of hypoxia on the brain depends on its duration and extent. Mild, moderate and/or intermittent hypoxia can be protective while severe and/or chronic hypoxia is detrimental. Chronic hypoxia not only promotes Aβ formation and accumulation, but also aggravates Aβ toxicity by increasing ROS and causing calcium dyshomeostasis. Furthermore, both chronic hypoxia and Aβ activate microglia leading to a neuroinflammatory response, which has a detrimental role in AD pathogenesis. The restoration of normal oxygen tension in the brain would reduce or converse the neurodegeneration. The molecules targeted by hypoxia can provide therapeutic strategies and interventions against some common neurodegenerative diseases, including the AD. HIF, an important physiological response mechanism to hypoxia, enhances neuroprotective compensatory pathways involved in many physiological processes. HIF stabilization is not the same as hypoxia, and is only a part of hypoxia-induced changes in the cells.
Pharmacological stabilization of HIF can be neuroprotective and explored as an adjunctive therapy for AD. Iron chelators have the potential to activate HIF in addition to iron chelation. The neuroprotective effects of iron chelators are acting against the generation of free radicals derived from iron, but could also be due to upregulation of the hypoxia rescue genes via HIF stabilization (Merelli et al., 2018). The amount of HIF can be adjusted with doses of hypoxia mimetic agents, and the specific HIF-1 or HIF-2 activation can be achievable (Chen et al., 2018). Carefully monitoring the HIF-1 and HIF-2 activation by some novel iron chelators will help translate iron chelation therapy for neurodegenerative diseases, including the AD. The tissue specific HIF alpha isoform activation by iron chelators merits further investigation.
The authors thank Prof. Christopher Exley, Keele University, UK, for critical comments.
This work was supported by a funding from the Petroleum Technology Development Fund, Nigeria.
Additional file: Open peer review report 1 (81.1KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Ivan Fernandez-Vega, Universidad de Oviedo, Spain.
P-Reviewer: Fernandez-Vega I; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Ashok BS, Ajith TA, Sivanesan S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin Exp Pharmacol Physiol. 2017;44:327–334. doi: 10.1111/1440-1681.12717. [DOI] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Abdalla N, Kawas CH, Corrada MM. Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimers Dement. 2018;14:121–129. doi: 10.1016/j.jalz.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen RL, Balami J, Esiri M, Chen LH, Buchan AM. Ischaemic stroke in the elderly: an overview of evidence. Nat Rev Neurol. 2010;6:256–265. doi: 10.1038/nrneurol.2010.36. [DOI] [PubMed] [Google Scholar]
- 4.Chen RL, Lai UH, Zhu LL, Singh A, Ahmed M, Forsyth NR. Reactive Oxygen Species (ROS) formation in the brain at different oxygen levels: role of hypoxia inducible factors. Front Cell Dev Biol. 2018;6:132. doi: 10.3389/fcell.2018.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devos D, Cabantchik ZI, Moreau C, Danel V, Mahoney-Sanchez L, Bouchaoui H, Gouel F, Rolland AS, Duce JA, Devedjian JC. FAIRPARK-II and FAIRALS-II studygroups (2020) Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J Neural Transm (Vienna) 127:189–203. doi: 10.1007/s00702-019-02138-1. [DOI] [PubMed] [Google Scholar]
- 6.Lall R, Mohammed R, Ojha U. What are the links between hypoxia and Alzheimer’s disease. Neuropsychiatr Dis Treat. 2019;15:1343–1354. doi: 10.2147/NDT.S203103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Cui X, Chen Y, Chen T, Xu H, Yin H, Wu Y. Therapeutic potential of a prolyl hydroxylase inhibitor FG-4592 for Parkinson’s diseases in vitro and in vivo: regulation of redox biology and mitochondrial function. Front Aging Neurosci. 2018;10:121. doi: 10.3389/fnagi.2018.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merelli A, Rodríguez JCG, Folch J, Regueiro MR, Camins A, Lazarowski A. Understanding the role of hypoxia inducible factor during neurodegeneration for new therapeutics opportunities. Curr Neuropharmacol. 2018;16:1484–1498. doi: 10.2174/1570159X16666180110130253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mijajlović MD, Pavlović A, Brainin M, Heiss WD, Quinn TJ, Ihle-Hansen HB, Hermann DM, Assayag EB, Richard E, Thiel A, Kliper E, Shin Y, Kim YH, Choi S, Jung S, Lee YB, Sinanović O, Levine DA, Schlesinger I, Mead G, et al. Post-stroke dementia - a comprehensive review. BMC Med. 2017;15:11. doi: 10.1186/s12916-017-0779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rawlinson C, Jenkins SI, Thei L, Dallas ML, Chen RL. Post-ischaemic immunological response in the brain: targeting microglia in ischaemic stroke therapy. Brain Sci. 2020;10:159. doi: 10.3390/brainsci10030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampson EL, Jenagaratnam L, McShane R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst Rev. 2014;2 doi: 10.1002/14651858.CD005380.pub5. CD005380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng H, Fridkin M, Youdim M. New approaches to treating Alzheimer’s disease. Perspect Medicin Chem. 2015;7:1–8. doi: 10.4137/PMC.S13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.