

Keywords: factor, GSK-3ß, in vitro, injury, ligase IV, neuroprotection, optic nerve, pathways, protein, repair
Abstract
Glycogen synthase kinase-3β (GSK-3β) has been shown to attenuate DNA damage in nerve cells, thereby enhancing neuronal survival under pathological conditions; however, the underlying mechanism remains unclear. An in vitro serum-starvation retinal neuron model and in vivo ischemia/reperfusion retina injury rat model were established and treated with SB216763, a GSK-3β inhibitor. SB21673 decreased the formation of γ-H2A histone family member X foci and enhanced the viability of ischemic retinal neurons. In addition, SB216763 upregulated expression of phosphorylated-CREB1, a ligase IV transcription factor, and significantly increased the transcriptional activity of ligase IV in ischemic retinal neurons. These results were confirmed in rat retinas following ischemia/reperfusion injury. Furthermore, we found that unlike lithium chlorine (a well-known direct inhibitor of GSK-3β), SB216763 inhibited GSK-3β activity by suppressing its phosphorylation. Taken together, our results suggest that GSK-3β inhibition enhances repair of DNA double-strand breaks by upregulating ligase IV expression in ischemic retinal neurons. This study was approved by the Institutional Animal Care and Use Committee of Zhongshan Ophthalmic Center on February 18, 2018.
Chinese Library Classification No. R456; R741; R774.6
Introduction
As part of the central nervous system, retinal neurons are postmitotic cells that cannot be regenerated under pathological conditions. Accordingly, retinal neurodegenerative blindness, such as glaucoma and optic nerve damage, is a major and refractory ophthalmological blindness (Zhuang et al., 2009; Chen et al., 2018; Smith et al., 2018). An increasing number of studies concerning neuroprotective strategies are currently ongoing, including investigation of DNA repair, antiapoptotic molecular applications, neurotrophic factor delivery, and anti-inflammatory treatments. Among these studies, DNA damage repair is considered to be a promising treatment strategy for therapeutic intervention (Merlo et al., 2005; Martin and Wong, 2017; Ru et al., 2019).
Glycogen synthase kinase-3β (GSK-3β), a multifaceted serine-threonine kinase widely expressed throughout the central nervous system, exhibits its highest expression level during development (Seira and Del Río, 2014). GSK-3β is involved in morphogenesis, axonal polarity, and synaptogenesis of neurons (Guo et al., 2007; Morgan-Smith et al., 2014). Dysregulation of GSK-3β is reportedly associated with diverse neurodegenerative diseases, including Parkinson’s disease and progressive supranuclear palsy (Domínguez et al., 2012; Huang et al., 2018). Numerous studies have shown that GSK-3β activation is closely related to the pathological processes of retinal degenerative diseases, such as glaucoma, retinitis, and diabetic retinopathy (Sun et al., 2014; Wang et al., 2017; Zhu et al., 2018). Several lines of evidence have indicated that under pathological conditions, such as genotoxic or oxidative stress, GSK-3β accelerates cell death by upregulating proapoptotic proteins and inhibiting DNA damage repair (Yang et al., 2011; Biswas et al., 2013; Ahmed et al., 2019; Ding et al., 2019). Both in vivo and in vitro evidence have confirmed that GSK-3 modulation provides cellular and functional neuroprotection in retinitis pigmentosa and retinal neovascularization disease (Sánchez-Cruz et al., 2018; Yu et al., 2020). Moreover, pharmacological inhibition of GSK-3β has been implicated in clinical treatment for diabetes, neurological diseases, and retinal diseases such as glaucoma (Carta et al., 2012; Li et al., 2017). However, mechanisms underlying the neuroprotective effect mediated by GSK-3β inhibition are not well defined.
As a highly conserved kinase, GSK-3β responds to a variety of protein activation and cell signal transductions (Duda et al., 2018; Wang and Wang, 2018; Zakharova et al., 2019). Control of GSK-3β activity is differentially regulated by varied and complex mechanisms, including phosphorylation, protein-protein interactions, priming/substrate specificity, subcellular localization, and proteolytic cleavage (Beurel et al., 2015). Notably, the phospho-serine or -threonine directly bind to the GSK-3 active catalytic site. GSK-3β activity can be directly inhibited by phosphorylation of serine at position 389 (Ser389) or Ser9, or competitively inhibited by phosphorylated peptides patterned after the unique recognition motif of GSK-3 (Plotkin et al., 2003; Thornton et al., 2008; Medina and Wandosell, 2011; Cormier and Woodgett, 2017).
Lithium chloride (LiCl) is a well-known GSK-3β inhibitor that is commonly used for clinical treatment of bipolar disorders (Kurgan et al., 2019; Li et al., 2020; Ng et al., 2020). Investigations of the precise mechanism by which LiCl exerts its neuroprotective effects have revealed the involvement of several cellular signaling pathways and various proteins. Mounting evidence suggests that LiCl protects neurons by directly inhibiting GSK-3β activity using a mechanism that does not alter its phosphorylation level (Seira and Del Río, 2014; Bai et al., 2018; Kurgan et al., 2019). The caspase family, Bax, p53, and other proteins have also been implicated in LiCl-mediated anti-inflammatory and neuroprotective actions (Jacobs et al., 2012). Moreover, our previous study indicated that LiCl promoted DNA damage repair and improved the survival of ischemic retinal neurons by upregulating DNA ligase IV (Zhuang et al., 2009; Yang et al., 2016).
DNA ligase IV is essential for nonhomologous end-joining DNA repair, the main DNA repair pathway for postmitotic neurons (Gatz et al., 2011; Gerodimos et al., 2017; Kaminski et al., 2018), which makes it a promising therapeutic target for neuroprotection. A previous study demonstrated that phosphorylation of the transcription factor CREB1 (p-CREB1) is essential for transcription of ligase IV (Guo et al., 2007). GSK-3β activity has consistently been implicated in p-CREB1 expression in various cells, such as 3T3-L1 cells and lung fibroblasts (Tullai et al., 2011; Park et al., 2016). Another well-known GSK-3β inhibitor, SB216763, also increases CREB1 phosphorylation (Baarsma et al., 2013). Thus, whether GSK-3β inhibition exerts a neuroprotective effect by regulating DNA ligase IV is not well understood.
To address this, an in vitro serum starvation retinal neuron model and in vivo ischemia/reperfusion (I/R) retinal injury rat model were established. SB216763, a specific inhibitor of GSK-3β (Coghlan et al., 2000), was used to evaluate the potential relationship between GSK-3β and DNA ligase IV in ischemic retinal neurons both in vitro and in vivo.
Materials and Methods
Ethics statement
All animal experimental procedures were approved and monitored by the Institutional Animal Care and Use Committee of Zhongshan Ophthalmic Center, China on February 18, 2018, and strictly abided by the Association for Research in Vision and Ophthalmology Standards for the Use of Animals in Ophthalmic and Vision Research. All efforts were made to minimize suffering of rats used in this study. All postnatal 1-day-old Sprague-Dawley rats (n = 30; for in vitro study) and adult female Sprague-Dawley rats (n = 30; aged 6 weeks, weighing 180–220 g; for in vivo study) were provided by the Laboratory Animal Center of Southern Medical University [Guangzhou, China; License No. SCXK (Yue) 2019-075A].
Primary rat retinal neural cells cultures and drug treatment
In rats, retinal ganglion cells are the only differentiated neurons at birth (Okabe et al., 1989; Huang et al., 2016). Briefly, postnatal 1-day-old rats were sacrificed by an intraperitoneal injection of pentobarbital (60 mg/kg; Sigma-Aldrich, St. Louis, MO, USA). Retinas were isolated and dissociated into a single-cell suspension by 0.125% trypsin solution, and then cultured in Dulbecco’s Modified Eagle Medium (Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (Gibco) in accordance with previously described methods (Yang et al., 2016). After 12 hours of cell attachment, 10 μM Ara-C (Sigma-Aldrich) was used to suppress the growth of non-neurons. After 12 hours, the medium was substituted with complete medium (10% fetal bovine serum) containing SB216763 (5 μM) or vehicle control [dimethyl sulfoxide (DMSO); 99.9%, 5 μM; Sigma-Aldrich] for 24 hours. To simulate an ischemic environment in vitro, neurons were nutrient-deprived by serum-free culture media with SB216763 (5 μM) or DMSO for 48 hours. After incubation, cells were harvested for further analysis.
I/R in vivo experiment
Thirty adult rats were randomly divided into control and SB216763 groups (n = 15 rats/group). The SB216763 group received intraperitoneal injections of SB216763 (0.6 mg/kg) for 3 days, while the control group received intraperitoneal injections of 200–400 μL DMSO, the solvent of SB216763.
After 3 days of pretreatment, an I/R experiment was carried on the right eye of each rat, with the left eye serving as a as self-control. Briefly, rats were anesthetized with intraperitoneal injection of pentobarbital (50 mg/kg) and their pupils were dilated with 0.5% tropicamide. Afterwards, a 30-G needle (Kindly Medical Devices Co., Wenzhou, China) connected to a reservoir filled with sterile salt solution was inserted into the anterior chamber of the right eye of rats in both groups. Increasing the intraocular pressure to induce retinal ischemia was achieved by raising the reservoir, yielding a maximum intraocular pressure of 120 mmHg within 2 minutes, which was maintained for 1 hour (Yang et al., 2016). Tono-Pen XL (Mentor, Santa Barbara, CA, USA) was used to monitor intraocular pressure every 10 minutes during experiments. Direct ophthalmoscopy (66 Vision Tech, Suzhou, China) was used to confirm systolic collapse of the central artery in rat eyes. Rats were normothermic with heated jackets throughout the experiments. Eyes were harvested at 1 and 3 days after I/R for subsequent analysis.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
Primary cultured retinal neural cells were seeded in 48-well plates. After nutrient deprivation for 48 hours, retinal neural cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) agent (5 mg/mL) at 37°C for 4 hours, and then 250 μL of DMSO was added into each well. A fluorescence plate reader (Power Wave XS; Bio-Tek, Winooski, VT, USA) was employed to measure the absorbance of each well at 490 nm. Cell viability was assessed by the optical density ratio of treated cultures to untreated controls.
Immunofluorescence analysis
Retinal neural cells were seeded onto slides. After 48 hours of serum deprivation, cells were fixed with 4% paraformaldehyde for 15 minutes, immersed in 0.1% Triton X-100 for 10 minutes, and then blocked with 10% normal goat serum (Boster, Wuhan, China) for 30 minutes. Afterwards, cells were incubated with primary antibodies against rabbit [1:1000; Cell Signaling Technology (CST), Danvers, MA, USA] and mouse anti-microtubule-associated protein-2 (MAP2, 1:100; Boster) overnight at 4°C. Alexa Fluor 488-conjugated goat anti-mouse IgG (1:1000; CST) and Alexa Fluor 555-conjugated goat anti-rabbit IgG (1:1000; CST) were added at room temperature for 1 hour. Next, nuclei were stained with 4',6-diamidino-2-phenylindole for 5 minutes. Images were captured by fluorescence microscopy (Leica, Bensheim, Germany). The amount of γ-H2AX foci was scored in images obtained using a constant exposure time. At least 100 cells from each group were counted.
Real-time reverse transcription-polymerase chain reaction
After 48 hours of serum deprivation, the total RNA of primary retina neural cells was isolated by using TRIzol Reagent (Sigma-Aldrich). Reverse transcription-polymerase chain reaction (RT-PCR) assays were performed with a SYBR Prime Script™ RT-PCR Kit in accordance with the manufacturer’s protocol (Takara, Shiga, Japan). Real-time PCR was employed to measure ligase IV expression using a Roche 480 system (Roche, Indianapolis, IN, USA). Relative target gene expression was quantitated according to the comparative ΔCt method, and normalized to the endogenous control gene β-actin. The results are presented as relative fold change compared with an unstimulated control. The following primer pairs were used: ligase IV (172-bp product size), 5′-GGC ACT TCA AGG AGT TTC TGG A-3′ and 5′-ATG TAA AGC TTA GCC AGC ATG G-3′; and β-actin (166-bp product size), 5′-TCA CCC ACA CTG TGC CCA T-3′ and 5′-TCT TTA ATG TCA CGC ACG ATT-3′.
Western blot assay
Cells were lysed with radioimmunoprecipitation assay buffer containing protease inhibitor cocktail. Next, western blotting was performed using a standard protocol. The following primary antibodies were used overnight at 4°C: ligase IV (1:300; Santa Cruz Biotechnology, Dallas, TX, USA), GSK-3β (1:1000; CST), p-GSK-3β (1:1000; CST), β-catenin (1:2000; BD Biosciences, Franklin Lakes, NJ, USA), CREB1 (1:1000; CST), and p-CREB1 (1:1000; Proteintech, Chicago, IL, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:10,000; CST) served as a loading control. Proteins were visualized following incubation with a horseradish peroxidase-conjugated anti-rabbit or -mouse IgG secondary antibody (1:10,000; CST) for 1 hour at room temperature, and exposured by the enhanced ECL chemiluminescence system (Bio-Rad, Hercules, CA, USA). Relative intensities of bands were quantified by densitometry using ImageJ software (version 1.51k; National Institutes of Health, Bethesda, MD, USA) and normalized to GAPDH levels.
DNA ligase IV promoter-reporter luciferase assay
To investigate whether SB216763 regulates DNA ligase IV through p-CREB1, we constructed a luciferase reporter driven by the ligase IV proximal promoter (–601 to +164), which contains a p-CREB1 binding site in regions –121 and –301. The mutant (GTG ACG TT to GTA TTG TT) was introduced for ligase IV (–601/+164)-luc by digestion and the addition of an inserter. The ligase IV (–601/+164)-luc construct was digested by PvuII and AvrII, and then ligated with the inserter containing the mutation, 5'-CTG GCG GAA ACG CGG GTT TGG CCT CGA CGG TAT TGT TTT CCG GTC GGA ATG AAA GTG GGC GAC TTC TCG GGA GGC TGC-3'. The subclone used in experiments was verified using the specific primers (forward, 5'-GGT TTG GCC TCC GAC GGT ATT G-3'; reverse, 5'-AAC CTG TAA ATC CCA GTC CAG-3').
Primary rat retinal neurons were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) as described previously (Yang et al., 2016). Plasmid transfections contained 2 μg of various reporter plasmids, 2 μg of expression plasmids, or pcDNA3-based vectors, and 100 ng of the Renilla luciferase reporter plasmid pCMV-RL (Promega, Madison, WI, USA). The pCMV-RL plasmid encoding Renilla luciferase was included in all samples to monitor transfection efficiencies. At 24 hours post-transfection, levels of firefly and Renilla luciferase activity were sequentially measured in each sample using the Dual-Glo Luciferase Assay System (Promega). The resulting fluorescence intensity reflects the effect of p-CREB1 on the ligase IV promoter. Levels of firefly luciferase activity were normalized to Renilla luciferase activity.
Small interfering RNA interference
Small interfering RNA (siRNA) sequences used for targeted silencing assay were as follows: ligase IV: 5'-TTG CTC AAT TTA CCA AGA G dTdT-3'; for CREB1: 5'-GCA CTT AAG GAC CTT TAC TDT DT-3'; control, 5'-GGU UUG GCU GGG GUG UUA U dTdT-3'. The siRNA interference assay for primary rat retinal neural cells was performed using Lipofectamine RNAiMAX (Invitrogen).
Statistical analysis
All statistical analyses were performed using GraphPad Prism (version 4.02; GraphPad Software, San Diego, CA, USA) and data are presented as mean ± standard error of mean (SEM). Student’s t-test and analysis of variance were used to evaluate statistical differences between two groups and multiple groups, respectively. A P value less than 0.05 was considered statistically significant for all analyses. All experiments were repeated at least three times.
Results
GSK-3β inhibitor SB216763 decreases DNA double-strand breaks (DSBs) and enhances cell viability in retinal neurons
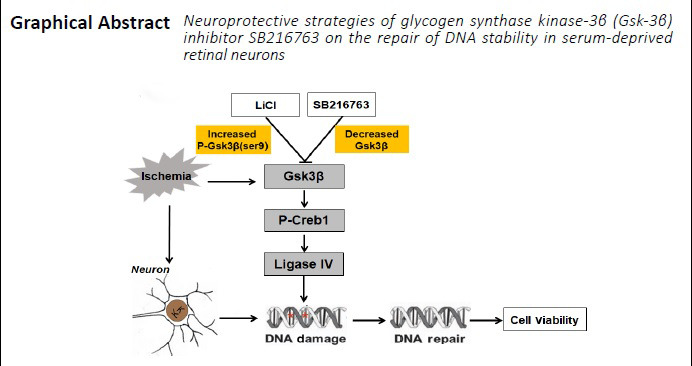
Many studies have indicated that GSK-3β inhibition protects neurons from apoptosis induced by genotoxic and other stresses (Marchena et al., 2017). To explore the underlying mechanism, primary rat retinal neurons were cultured and subjected to serum starvation. Figure 1A shows that 92% of primary cultured retinal cells were positive for MAP2, a well-known neuronal marker (Johnson and Jope, 1992), demonstrating their neuronal characteristics. Pretreatment of primary rat retinal neurons for 24 hours with SB216763, a GSK-3β inhibitor, prior to serum starvation resulted in upregulation of β-catenin, a downstream target negatively regulated by GSK-3β (P < 0.01; Figure 1B and C). These results confirmed that SB216763 mediated GSK-3β inhibition in ischemic retinal neurons. Relative intensities of bands were quantified by densitometry and normalized to GAPDH levels. As shown in Figure 1D, double staining of MAP2 (a neuronal marker) and γ-H2AX [a well-characterized DNA DSB marker (Ivashkevich et al., 2012)] demonstrated that serum deprivation induced pronounced γ-H2AX foci formation in retinal neurons; however, inhibiting GSK-3β with SB216763 pretreatment significantly decreased γ-H2AX expression compared with the control group (P < 0.05; Figure 1E). Accordingly, SB216763 pretreatment significantly improved the cell viability of retinal neurons upon serum deprivation compared with the control treatment (DMSO) (P < 0.001; Figure 1F). These results indicate that GSK-3β inhibition suppressed DNA damage and promoted cell viability in retinal neurons suffering from ischemic injury. However, the mechanism by which GSK-3β inhibition mediates DNA repair is still unknown.
Figure 1.
SB216763 suppresses DNA damage and increases cell viability in serum-deprived retinal neurons.
(A) Retinal neurons were cultured and stained for MAP2 (Alexa Fluor 488, green), and nuclei were stained with DAPI (blue). (B) SB216763 upregulated expression of β-catenin, the substrate of GSK-3β. (C) Relative protein expression of β-catenin is presented as a histogram. (D) Double staining for MAP2 (green staining by Alexa Fluor 488) and γ-H2AX (red staining by Alexa Fluor 555) in serum-deprived retinal neurons. SB216763 significantly inhibited γ-H2AX foci formation in serum-deprived retinal neurons. (E) Relative quantification of γ-H2AX expression was determined by counting foci in 50 randomly selected MAP2-positive cells. Scale bars: 20 μm. (F) SB216763 treatment improved cell viability in serum-deprived retinal neurons, as evidenced by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Data are expressed as the mean ± SEM. n = 3 for each group. *P < 0.05, **P < 0.01, vs. control (Con) group (Student’s t-test). DAPI: 4',6-Diamidino-2-phenylindole; Gapdh: glyceraldehyde 3-phosphate dehydrogenase; GSK-3β: glycogen synthase kinase-3β; MAP2: microtubule-associated protein-2; SB216763: a GSK-3β inhibitor; γ-H2AX: γ-H2A histone family member X.
DNA ligase IV is involved in SB216763-mediated neuroprotection
LiCl has been used as a GSK-3 inhibitor in clinical treatment and scientific research for many years. Our previous study indicated that LiCl increased DNA stability by upregulating DNA ligase IV, thereby facilitating neuroprotective effects (Zhuang et al., 2009; Yang et al., 2011). Therefore, we hypothesized that the GSK-3β inhibitor SB216763 might regulate DNA ligase IV. Accordingly, we measured mRNA and protein levels of DNA ligase IV in primary retinal neurons treated with SB216763 or vehicle control. Consistent with our expectations, 24-hour treatment with SB216763 (5 μM) significantly increased mRNA expression of ligase IV in retinal neurons suffering from ischemic injury compared with control cells pretreated with DMSO vehicle (P < 0.001; Figure 2A). The same effect was observed for protein expression of ligase IV in ischemic retinal neurons (P < 0.01; Figure 2B and C).
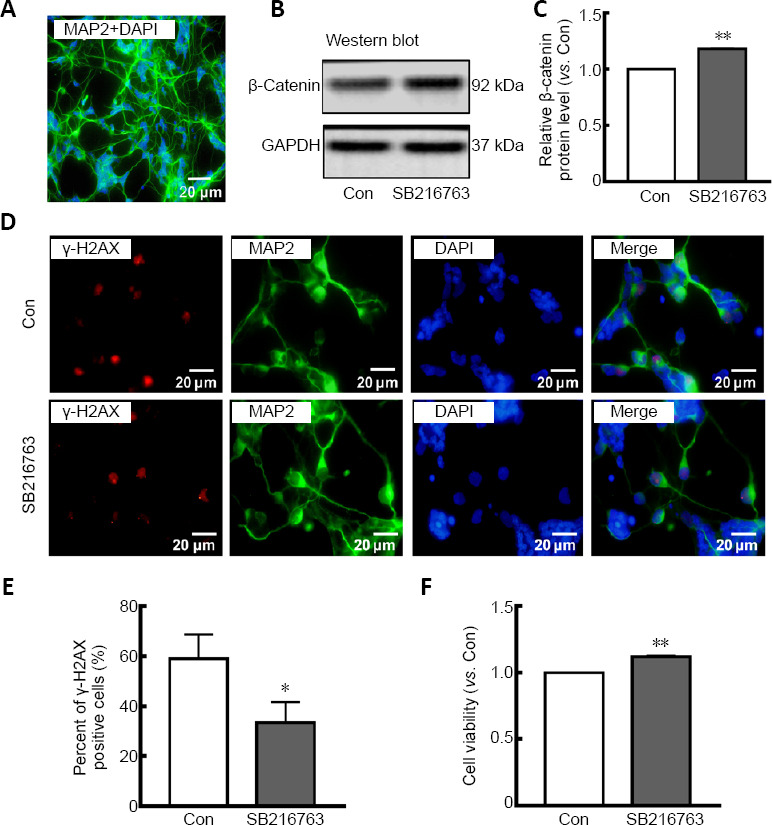
Figure 2.
Ligase IV is involved in the neuroprotective mechanisms of SB216763.
(A) Ligase IV mRNA expression was significantly upregulated in serum-starved retinal neurons pretreated with SB216763 compared with control cells. (B, C) SB216763 pretreatment significantly upregulated ligase IV protein expression in serum-starved retinal neurons, as evidenced by western blot analysis. (D, E) siRNA interference assays were employed to downregulate ligase IV protein expression in serum-starved retinal neurons pretreated with SB216763 or DMSO. (F) SB216763 pretreatment significantly improved cell viability in serum-starved retinal neurons compared with control treatment; however, downregulation of ligase IV by siRNA assay markedly suppressed this effect. Data are expressed as mean ± SEM compared with the control group. n = 3 for each group. *P < 0.05, **P < 0.01, vs. control (Con) group (Student’s t-test). DMSO: Dimethyl sulfoxide; Gapdh: glyceraldehyde 3-phosphate dehydrogenase; GSK-3β: glycogen synthase kinase-3β; SB216763: a GSK-3β inhibitor; siRNA: small interfering RNA.
To further confirm whether ligase IV was involved in SB216763-mediated neuroprotection in retinal neurons suffering from ischemic injury, ligase IV expression was silenced using siRNA interference. As shown in Figure 2D, siRNA interference suppressed the upregulation of ligase IV induced by SB216763 in retinal neurons under serum deprivation conditions (P < 0.05; Figure 2E). Accordingly, SB216763 significantly improved the cell viability of retinal neurons upon serum deprivation, while this increase was abrogated by ligase IV siRNA interference (P < 0.05; Figure 2F). These results support the potential involvement of ligase IV expression in the neuroprotective effect of SB216763.
Inhibition of GSK-3β activity by SB216763 transcriptionally upregulates DNA ligase IV by increasing p-CREB1
Our previous study reported that p-CREB1 may participate in the transcriptional regulation of ligase IV (Yang et al., 2016). Here, we found that SB216763 did not affect CREB1 expression, but instead markedly upregulated p-CREB1 expression in retinal neurons following serum deprivation (Figure 3A). Indeed, SB216763 increased CREB1 phosphorylation by 1.38-fold over the control treatment (P < 0.05; Figure 3B).
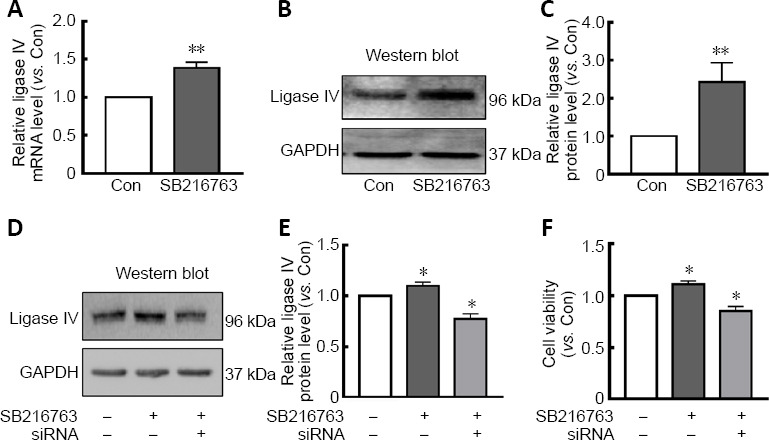
Figure 3.
SB216763 is involved in transcriptional regulation of ligase IV in retinal neurons.
(A) SB216763 pretreatment significantly upregulated p-CREB1 expression in serum-starved retinal neurons, as evidenced by western blot analysis. Additionally, SB216763 did not affect CREB1 expression. (B) Relative protein expression of p-CREB1 is presented as a histogram. (C) One mutation was introduced into ATF sites in the –601/+164 constructs. (D) Levels of luciferase activity were normalized to that of Renilla luciferase. SB216763 pretreatment significantly increased luciferase activity of the ligase IV promotor compared with control cells. p-CREB1 mutations dramatically reduced luciferase activity relative to the WT promoter sequence. Data are expressed as mean ± SEM compared with the control group. n = 3 for each group. *P < 0.05, **P < 0.01, vs. control (Con) group (Student’s t-test). ATF: Activating transcription factor; Gapdh: glyceraldehyde 3-phosphate dehydrogenase; Mt: mutant; p-CREB1: phosphorylated CREB1; SB216763: a glycogen synthase kinase-3β inhibitor; WT: wild type.
The transcriptional mechanism of ligase IV was further investigated by a luciferase reporter assay using a construct driven by the ligase IV proximal promoter (–601 to +164), which contains an activating transcription factor binding site between –121 and –301 (Yang et al., 2016). The transcription factor binding motif of the p-CREB1 DNA-binding sequence (5′-GTG ACG TT-3′) is located at –126. As shown in Figure 3C, the p-CREB1 site was mutated (5′-GTG CCG TT-3′ to 5′-GTA TTG TT-3′) and the entire promoter region from –601 to +164 was assayed in ischemic retinal neurons pretreated with 5 μM SB216763. Twenty-four hours after pretreatment with SB216763, serum-starved retinal neurons were transfected with luciferase reporter constructs containing the ligase IV promoter. At 24 hours post-transfection, levels of firefly and Renilla luciferase activity were measured using the Dual-Glo Luciferase Assay System. The results indicated that SB216763 pretreatment significantly increased luciferase activity compared with control cells. However, p-CREB1 mutations dramatically reduced luciferase activity relative to that of the wild-type promoter sequence (P < 0.05; Figure 3D). These results further suggest that inhibiting GSK-3β elicits neuroprotection by regulating ligase IV expression in ischemic retinal neurons.
Mechanism by which SB216763 inhibits GSK-3β is different from LiCl in retinal neurons
GSK-3β activity is regulated by phosphorylation of Tyr216 or Ser9 (Park et al., 2013). However, SB216763 and LiCl, both well-known GSK-3β inhibitors, have different effects on Ser9 phosphorylation of GSK-3β in various cell lines (Zhang et al., 2003; Noble et al., 2005; Koo et al., 2014; D'Angelo et al., 2016). To explore what occurs in retinal neurons, we further investigated GSK-3β expression and phosphorylation levels in retinal neurons treated with LiCl and SB216763. The relative intensities of bands were quantified by densitometry and normalized to GAPDH levels. As shown in (Figure 4A), LiCl did not affect GSK-3β expression in ischemic retinal neurons compared with the control treatment (P > 0.05; Figure 4A and B). However, LiCl pretreatment significantly upregulated p-GSK-3β expression in ischemic retinal neurons (P < 0.05; Figure 4A and B), indicating that LiCl indirectly inhibited GSK-3β activity by stimulating GSK-3β phosphorylation in retinal neurons. In contrast, SB216763 not only directly decreased GSK-3β expression (P < 0.05), but also dramatically inhibited its phosphorylation (P < 0.001; Figure 4C and D).
Figure 4.
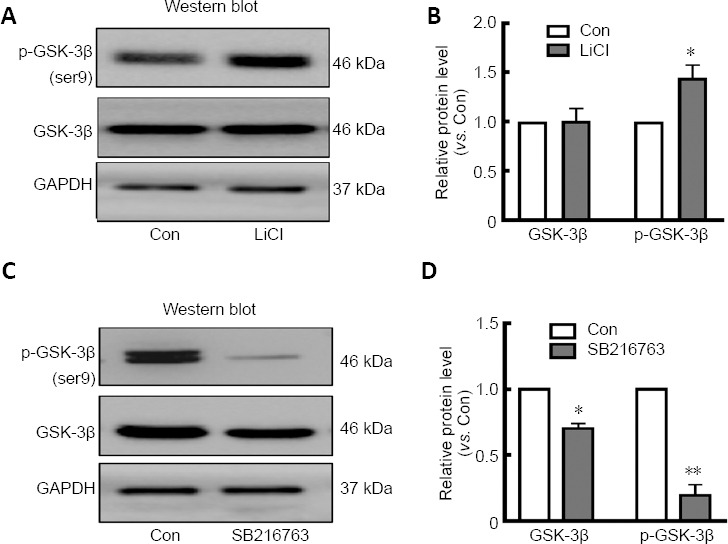
GSK-3β activity is inhibited by different mechanisms in retinal neurons.
(A) LiCl significantly upregulated p-GSK-3β (Ser9) expression in serum-deprived retinal neurons, but did not affect GSK-3β expression. (B) Relative protein expression of p-GSK-3β and GSK-3β are presented as histograms. (C) SB216763 not only directly decreased expression of GSK-3β but also dramatically inhibited its phosphorylation. (D) Relative protein expression of p-GSK-3β and GSK-3β is presented as histograms. Data are expressed as mean ± SEM compared with the control group. n = 3 for each group. *P < 0.05, **P < 0.01, vs. control (Con) group (Student’s t-test). Gapdh: Glyceraldehyde 3-phosphate dehydrogenase; GSK-3β: glycogen synthase kinase-3β; LiCl: lithium chloride; p-GSK-3β: phosphorylated glycogen synthase kinase-3β; SB216763: a GSK-3β inhibitor.
GSK-3β inhibition upregulates DNA ligase IV expression and enhances DNA repair in retinal neurons following ischemic injury in vivo
To further confirm the neuroprotective mechanism of SB216763, a rat I/R model was established to mimic retinal ischemic injury in vivo. As shown in Figure 5A, SB216763 pretreatment significantly upregulated the expression of both ligase IV (P < 0.05) and p-CREB1 (P < 0.01; Figure 5B) in ischemic rat retinas. These observations were consistent with those of our in vitro studies. Moreover, we analyzed γ-H2AX foci formation at the indicated time points (1 and 3 days). Our results showed that γ-H2AX-positive cells were primarily located in the retinal ganglion cell layer after I/R treatment (1 day; Figure 5C). After counting the number of γ-H2AX foci in at least 10 slides, we found that SB216763 pretreatment significantly suppressed γ-H2AX foci formation in the ganglion cell layer at 1 day (P < 0.05) and 3 days (P < 0.001) following I/R surgery (Figure 5D). Thus, these data further indicate that the GSK-3β inhibitor SB216763 enhanced repair of DNA DSBs by upregulating ligase IV expression in serum-deprived retinal neurons.
Figure 5.
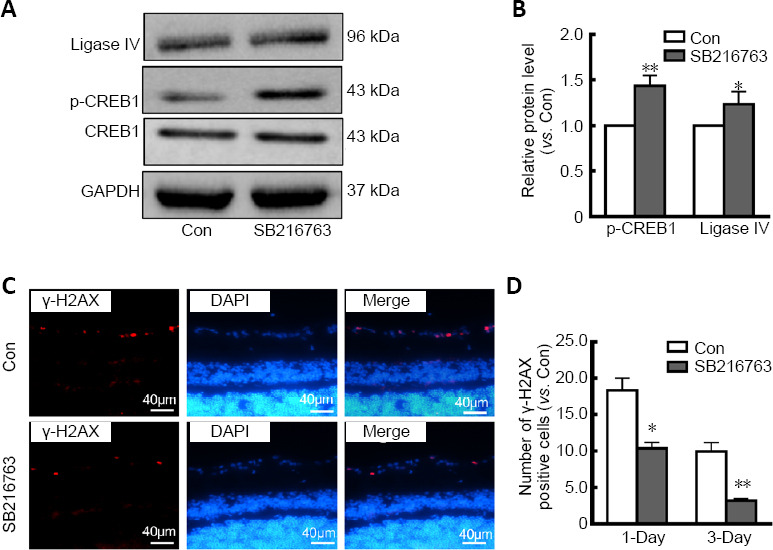
SB216763 protects retinal neurons from ischemic injury in vivo.
(A) SB216763 pretreatment upregulated expression of p-CREB1 and ligase IV in the retinas of rats subjected to I/R injury. (B) Relative expression of p-CREB1 and ligase IV in rat retinas quantified by densitometry is presented as a histogram. (C) γ-H2AX foci labeled double-strand DNA breaks in rat retinas subjected to I/R injury. γ-H2AX-positive cells (red staining by Alexa Fluor 555) were primarily located in the ganglion cell layer of ischemic rat retinas. Scale bars: 40 μm. (D) SB216763 pretreatment significantly suppressed γ-H2AX foci formation at 1 day and 3 days following I/R surgery. Data are expressed mean ± SEM. n = 3 for each group. *P < 0.05, **P < 0.01, vs. control (Con) group (Student’s t-test). DAPI: 4',6-Diamidino-2-phenylindole; I/R: ischemia/reperfusion; p-CREB1: phosphorylated CREB1; SB216763: a glycogen synthase kinase-3β inhibitor; γ-H2AX: γ-H2A histone family member X.
Discussion
Pharmacological inhibition of GSK-3β has been implicated as a promising therapeutic strategy for neurological disease. Here, we demonstrated that the GSK-3β inhibitor SB216763 suppressed DNA damage and promoted cell survival in ischemic retinal neurons by decreasing GSK-3β phosphorylation and upregulating DNA ligase IV expression in vitro. Moreover, DNA ligase IV in retinal neurons was transcriptionally regulated by p-CREB1 after inhibition of GSK-3β. Using a rat I/R injury model, these results were confirmed in vivo. Furthermore, we found that although LiCl and SB216763 are both inhibitors of GSK-3β, LiCl inhibited GSK-3β activity by suppressing its phosphorylation, whereas SB216763 inhibited GSK-3β activity by decreasing its expression. Thus, our findings identify a novel mechanism by which the GSK-3β inhibitor SB216763 exhibits neuroprotective effects, which might shed light on use of GSK-3β inhibition for clinical treatment of ischemia retinal diseases.
GSK-3β is ubiquitously expressed and active in non-stimulated cells, and is involved in various cell functions such as cell division, differentiation, cell migration, and apoptosis (Doble and Woodgett, 2003). Within the nervous system in particular, GSK-3β integrates different signaling pathways from diverse cellular stimuli and has, thus, attracted significant attention as a therapeutic target (Medina et al., 2011). Our results demonstrated that the GSK-3β inhibitor SB216763 improved cell viability in ischemic retinal neurons both in vivo and in vitro, consistent with previous studies. For example, a study by D'Angelo et al. (2016) reported that pharmacological interventions built upon GSK-3β silencing strategies could represent a novel therapy for neonatal brain injury. In addition, some small molecules that target GSK-3 have been shown to protect retinal ganglion cells and photoreceptor cells from various injuries (Marchena et al., 2017). Moreover, SB216763 was shown to block glutamate-induced caspase 3 activation and excitotoxicity in primary rat cortical neurons in vitro (Chuang et al., 2011). Therefore, GSK-3β is an ideal therapeutic target for neuroprotection.
With regard to the mechanism by which GSK-3β mediates neuroprotection, substantial evidence has been reported. For example, GSK-3β inhibition elicited neuroprotection in a neonatal hypoxic-ischemic brain injury, likely by reducing proinflammatory responses through blockade of STAT3 signaling (D'Angelo et al., 2016). As a central module for promoting sensory axon regeneration, PI3K-GSK3-Smad1 signaling plays a critical role in the mammalian nervous system (Saijilafu et al., 2013). Treatment with the GSK-3β inhibitor LiCl could have a dual impact by reducing the accumulation of both amyloid β and pathogenic, insoluble tau proteins (Noble et al., 2005). In the present study, we found that SB216763 significantly decreased DNA DSBs in primary retinal neurons. Moreover, SB216763 not only inhibited GSK-3β activity, but also upregulated mRNA and protein expression of DNA ligase IV in ischemic rat retinal neurons. DNA ligase IV is involved in nonhomologous end-joining repair in postmitotic cells (Zhuang et al., 2009). Upregulation of p-CREB1, a transcriptional regulator of ligase IV, was observed in ischemic retinal neurons treated with SB216763 both in vitro and in vivo. Promoter inducibility assay results further indicated that ligase IV is transcriptionally upregulated in retinal neurons by p-CREB1. Thus, our results suggest that SB216763 offers neuroprotection from ischemia-induced damage by enhancing DNA repair.
Our previous studies demonstrated that LiCl, another well-known GSK-3β inhibitor, promotes DSB repair, thereby improving cell viability in ischemic retinal neurons (Yang et al., 2016). However, we found that SB216763 and LiCl have different effects on GSK-3β phosphorylation, which clearly correlates with GSK-3β kinase activity. Phosphorylation of Ser9 of GSK-3β appears to block its kinase activity, while phosphorylation of Tyr216 has the opposite effect (Hur and Zhou, 2010). However, not all pharmacological inhibitors of GSK-3β suppress its phosphorylation. Indeed, Simón et al. (2008) demonstrated that GSK-3β activation/inactivation is not strictly aligned with its phosphorylation level. Similarly, in our study, the GSK-3β antagonist LiCl inhibited GSK-3β activity in ischemic retinal neurons by promoting its phosphorylation at Ser9, rather than affecting GSK-3β expression. However, another GSK-3β antagonist, SB216763, significantly inhibited GSK-3β expression and phosphorylation in ischemic retinal neurons. It should be noted that other explanations might account for this discrepancy. First, unlike SB216763, LiCl is a nonspecific GSK-3β inhibitor, meaning it may negatively regulate GSK-3β activity by multiple mechanisms. LiCl stabilizes the inactive form of GSK-3β by increasing phosphorylation at Ser9. This modification involves several different kinases, including protein kinase A, protein kinase B (Akt), and protein kinase C (Jope, 2003; Chuang et al., 2011). In addition, the ATP-dependent magnesium-sensitive catalytic activity of GSK-3β is directly suppressed by LiCl. Moreover, as a nonselective inhibitor of GSK-3β, LiCl can exert diverse additional activities and regulate several cell signaling pathways. LiCl reportedly alters expression of CREB, a transcription factor known to regulate GSK-3β expression (Evenson et al., 2005). However, SB216763, a highly selective inhibitor, does not inhibit GSK-3β secondary to the phosphorylation enhancement of GSK-3β. Instead, SB216763 competitively binds to the ATP binding site to inhibit the activity of GSK-3β (Cao et al., 2006). The observed loss of Ser9-phosphorylated GSK-3β induced by SB216763 is consistent with a previous study (Braeuning and Buchmann, 2009) and may be explained by a cellular feedback mechanism involving recruitment of Ser9-phosphorylated GSK-3β when signal transduction is suppressed by SB216763.
There are some limitations of this study. Although we confirmed the neuroprotective effect of SB216763 and its underlying mechanism, the optimal drug administration route and most effective drug concentration were not conclusively explored. Moreover, despite the recent development of novel GSK-3β inhibitors capable of acting by various mechanisms, differences in their neuroprotective actions have not been defined. In our future studies, these questions will be investigated in detail.
In conclusion, neuroprotective strategies are still a worldwide challenge for scientists. The present study provides substantial evidence indicating that GSK-3β inhibition is required for activation of DNA ligase IV, thereby facilitating DNA repair and genetic stability in retinal neurons. This study not only provides new insights into the mechanism by which negatively mediating GSK-3β elicits neuroprotection, but also extends the use of GSK-3β inhibitors in ophthalmic clinical applications.
Footnotes
Conflicts of interest: The authors declare that they have no conflict of interests.
Financial support: This study was supported by the National Natural Science Foundation of China, Nos. 81670848 (to JZhuang) and 81900850 (to YY). The funder had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement: The study was approved by the Institutional Animal Care and Use Committee of Zhongshan Ophthalmic Center, China on February 18, 2019.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This study was supported by the National Natural Science Foundation of China, Nos. 81670848 (to JZhuang) and 81900850 (to YY).
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Deusen AV, Yu J, Song CP; T-Editor: Jia Y
References
- 1.Ahmed Z, Morgan-Warren PJ, Berry M, Scott RAH, Logan A. Effects of siRNA-mediated knockdown of GSK3β on retinal ganglion cell survival and neurite/axon growth. Cells. 2019;8:956. doi: 10.3390/cells8090956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baarsma HA, Engelbertink LH, van Hees LJ, Menzen MH, Meurs H, Timens W, Postma DS, Kerstjens HA, Gosens R. Glycogen synthase kinase-3 (GSK-3) regulates TGF-β1-induced differentiation of pulmonary fibroblasts. Br J Pharmacol. 2013;169:590–603. doi: 10.1111/bph.12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai L, Chang HM, Cheng JC, Chu G, Leung PCK, Yang G. Lithium chloride inhibits StAR and progesterone production through GSK-3β and ERK1/2 signaling pathways in human granulosa-lutein cells. Mol Cell Endocrinol. 2018;461:89–99. doi: 10.1016/j.mce.2017.08.018. [DOI] [PubMed] [Google Scholar]
- 4.Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions and diseases. Pharmacol Ther. 2015;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biswas M, Kwong EK, Park E, Nagra P, Chan JY. Glycogen synthase kinase 3 regulates expression of nuclear factor-erythroid-2 related transcription factor-1 (Nrf1) and inhibits pro-survival function of Nrf1. Exp Cell Res. 2013;319:1922–1931. doi: 10.1016/j.yexcr.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braeuning A, Buchmann A. The glycogen synthase kinase inhibitor 3-(2, 4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2, 5-dione (SB216763) is a partial agonist of the aryl hydrocarbon receptor. Drug Metab Dispos. 2009;37:1576–1580. doi: 10.1124/dmd.109.027821. [DOI] [PubMed] [Google Scholar]
- 7.Cao Q, Lu X, Feng YJ. Glycogen synthase kinase-3beta positively regulates the proliferation of human ovarian cancer cells. Cell Res. 2006;16:671–677. doi: 10.1038/sj.cr.7310078. [DOI] [PubMed] [Google Scholar]
- 8.Carta F, Supuran CT, Scozzafava A. Novel therapies for glaucoma: a patent review 2007 - 2011. Expert Opin Ther Pat. 2012;22:79–88. doi: 10.1517/13543776.2012.649006. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Cho KS, Vu THK, Shen CH, Kaur M, Chen G, Mathew R, McHam ML, Fazelat A, Lashkari K, Au NPB, Tse JKY, Li Y, Yu H, Yang L, Stein-Streilein J, Ma CHE, Woolf CJ, Whary MT, Jager MJ, et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat Commun. 2018;9:3209. doi: 10.1038/s41467-018-05681-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuang DM, Wang Z, Chiu CT. GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front Mol Neurosci. 2011;4:15. doi: 10.3389/fnmol.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- 12.Cormier KW, Woodgett JR. Recent advances in understanding the cellular roles of GSK-3. F1000Res. 2017:6. doi: 10.12688/f1000research.10557.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Angelo B, Ek CJ, Sun Y, Zhu C, Sandberg M, Mallard C. GSK3β inhibition protects the immature brain from hypoxic-ischaemic insult via reduced STAT3 signalling. Neuropharmacology. 2016;101:13–23. doi: 10.1016/j.neuropharm.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding L, Madamsetty VS, Kiers S, Alekhina O, Ugolkov A, Dube J, Zhang Y, Zhang JS, Wang E, Dutta SK, Schmitt DM, Giles FJ, Kozikowski AP, Mazar AP, Mukhopadhyay D, Billadeau DD. Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to chemotherapy by abrogating the TopBP1/ATR-mediated DNA damage response. Clin Cancer Res. 2019;25:6452–6462. doi: 10.1158/1078-0432.CCR-19-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M. Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J Biol Chem. 2012;287:893–904. doi: 10.1074/jbc.M111.306472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duda P, Wiśniewski J, Wójtowicz T, Wójcicka O, Jaśkiewicz M, Drulis-Fajdasz D, Rakus D, McCubrey JA, Gizak A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin Ther Targets. 2018;22:833–848. doi: 10.1080/14728222.2018.1526925. [DOI] [PubMed] [Google Scholar]
- 18.Evenson AR, Fareed MU, Menconi MJ, Mitchell JC, Hasselgren PO. GSK-3beta inhibitors reduce protein degradation in muscles from septic rats and in dexamethasone-treated myotubes. Int J Biochem Cell Biol. 2005;37:2226–2238. doi: 10.1016/j.biocel.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Gatz SA, Ju L, Gruber R, Hoffmann E, Carr AM, Wang ZQ, Liu C, Jeggo PA. Requirement for DNA ligase IV during embryonic neuronal development. J Neurosci. 2011;31:10088–10100. doi: 10.1523/JNEUROSCI.1324-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerodimos CA, Chang HHY, Watanabe G, Lieber MR. Effects of DNA end configuration on XRCC4-DNA ligase IV and its stimulation of Artemis activity. J Biol Chem. 2017;292:13914–13924. doi: 10.1074/jbc.M117.798850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo W, Jiang H, Gray V, Dedhar S, Rao Y. Role of the integrin-linked kinase (ILK) in determining neuronal polarity. Dev Biol. 2007;306:457–468. doi: 10.1016/j.ydbio.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 22.Huang B, Liu J, Meng T, Li Y, He D, Ran X, Chen G, Guo W, Kan X, Fu S, Wang W, Liu D. Polydatin prevents lipopolysaccharide (LPS)-induced parkinson’s disease via regulation of the AKT/GSK3β-Nrf2/NF-κB signaling axis. Front Immunol. 2018;9:2527. doi: 10.3389/fimmu.2018.02527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang GL, Liao D, Chen H, Lu Y, Chen L, Li H, Li B, Liu W, Ye C, Li T, Zhu Z, Wang J, Uchida T, Zou Y, Dong Z, He Z. The protein level and transcription activity of activating transcription factor 1 is regulated by prolyl isomerase Pin1 in nasopharyngeal carcinoma progression. Cell Death Dis. 2016;7:e2571. doi: 10.1038/cddis.2016.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivashkevich A, Redon CE, Nakamura AJ, Martin RF, Martin OA. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012;327:123–133. doi: 10.1016/j.canlet.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D. GSK-3β: A bifunctional role in cell death pathways. Int J Cell Biol. 2012;2012:930710. doi: 10.1155/2012/930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson GV, Jope RS. The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. J Neurosci Res. 1992;33:505–512. doi: 10.1002/jnr.490330402. [DOI] [PubMed] [Google Scholar]
- 28.Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory, actions, multiple outcomes. Trends Pharmacol Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 29.Kaminski AM, Tumbale PP, Schellenberg MJ, Williams RS, Williams JG, Kunkel TA, Pedersen LC, Bebenek K. Structures of DNA-bound human ligase IV catalytic core reveal insights into substrate binding and catalysis. Nat Commun. 2018;9:2642. doi: 10.1038/s41467-018-05024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koo J, Yue P, Gal AA, Khuri FR, Sun SY. Maintaining glycogen synthase kinase-3 activity is critical for mTOR kinase inhibitors to inhibit cancer cell growth. Cancer Res. 2014;74:2555–2568. doi: 10.1158/0008-5472.CAN-13-2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurgan N, Whitley KC, Maddalena LA, Moradi F, Stoikos J, Hamstra SI, Rubie EA, Kumar M, Roy BD, Woodgett JR, Stuart JA, Fajardo VA. A low-therapeutic dose of lithium inhibits GSK3 and enhances myoblast fusion in C2C12 cells. Cells. 2019;8:1340. doi: 10.3390/cells8111340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li M, Xia M, Chen W, Wang J, Yin Y, Guo C, Li C, Tang X, Zhao H, Tan Q, Chen Y, Jia Z, Liu X, Feng H. Lithium treatment mitigates white matter injury after intracerebral hemorrhage through brain-derived neurotrophic factor signaling in mice. Transl Res. 2020;217:61–74. doi: 10.1016/j.trsl.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Li Z, Wu F, Zhang X, Chai Y, Chen D, Yang Y, Xu K, Yin J, Li R, Shi H, Wang Z, Li X, Xiao J, Zhang H. Valproate attenuates endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells via the AKT/GSK3β signaling pathway. Int J Mol Sci. 2017;18:315. doi: 10.3390/ijms18020315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchena M, Villarejo-Zori B, Zaldivar-Diez J, Palomo V, Gil C, Hernández-Sánchez C, Martínez A, de la Rosa EJ. Small molecules targeting glycogen synthase kinase 3 as potential drug candidates for the treatment of retinitis pigmentosa. J Enzyme Inhib Med Chem. 2017;32:522–526. doi: 10.1080/14756366.2016.1265522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin LJ, Wong M. Enforced DNA repair enzymes rescue neurons from apoptosis induced by target deprivation and axotomy in mouse models of neurodegeneration. Mech Ageing Dev. 2017;161:149–162. doi: 10.1016/j.mad.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medina M, Wandosell F. Deconstructing GSK-3: The fine regulation of its activity. Int J Alzheimers Dis. 2011;2011:479249. doi: 10.4061/2011/479249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medina M, Garrido JJ, Wandosell FG. Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Front Mol Neurosci. 2011;4:24. doi: 10.3389/fnmol.2011.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merlo D, Di Stasi AM, Bonini P, Mollinari C, Cardinale A, Cozzolino F, Wisden W, Garaci E. DNA repair in post-mitotic neurons: a gene-trapping strategy. Cell Death Differ. 2005;12:307–309. doi: 10.1038/sj.cdd.4401572. [DOI] [PubMed] [Google Scholar]
- 39.Morgan-Smith M, Wu Y, Zhu X, Pringle J, Snider WD. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. Elife. 2014;3:e02663. doi: 10.7554/eLife.02663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng QX, Yeo WS, Sivalingam V. Lithium-associated renal dysfunction: To stop or not to stop. Bipolar Disord. 2020;22:91–92. doi: 10.1111/bdi.12853. [DOI] [PubMed] [Google Scholar]
- 41.Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okabe S, Shiomura Y, Hirokawa N. Immunocytochemical localization of microtubule-associated proteins 1A and 2 in the rat retina. Brain Res. 1989;483:335–346. doi: 10.1016/0006-8993(89)90178-9. [DOI] [PubMed] [Google Scholar]
- 43.Park CH, Lee BH, Ahn SG, Yoon JH, Oh SH. Serine 9 and tyrosine 216 phosphorylation of GSK-3β differentially regulates autophagy in acquired cadmium resistance. Toxicol Sci. 2013;135:380–389. doi: 10.1093/toxsci/kft158. [DOI] [PubMed] [Google Scholar]
- 44.Park SS, Choi H, Kim SJ, Chang C, Kim E. CREB/GSK-3β signaling pathway regulates the expression of TR4 orphan nuclear receptor gene. Mol Cell Endocrinol. 2016;423:22–29. doi: 10.1016/j.mce.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 45.Plotkin B, Kaidanovich O, Talior I, Eldar-Finkelman H. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J Pharmacol Exp Ther. 2003;305:974–980. doi: 10.1124/jpet.102.047381. [DOI] [PubMed] [Google Scholar]
- 46.Ru Q, Xiong Q, Tian X, Chen L, Zhou M, Li Y, Li C. Tea polyphenols attenuate methamphetamine-induced neuronal damage in PC12 cells by alleviating oxidative stress and promoting DNA repair. Front Physiol. 2019;10:1450. doi: 10.3389/fphys.2019.01450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saijilafu, Hur EM, Liu CM, Jiao Z, Xu WL, Zhou FQ. PI3K-GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nat Commun. 2013;4:2690. doi: 10.1038/ncomms3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sánchez-Cruz A, Villarejo-Zori B, Marchena M, Zaldivar-Díez J, Palomo V, Gil C, Lizasoain I, de la Villa P, Martínez A, de la Rosa EJ, Hernández-Sánchez C. Modulation of GSK-3 provides cellular and functional neuroprotection in the rd10 mouse model of retinitis pigmentosa. Mol Neurodegener. 2018;13:19. doi: 10.1186/s13024-018-0251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seira O, Del Río JA. Glycogen synthase kinase 3 beta (GSK3β) at the tip of neuronal development and regeneration. Mol Neurobiol. 2014;49:931–944. doi: 10.1007/s12035-013-8571-y. [DOI] [PubMed] [Google Scholar]
- 50.Simón D, Benitez MJ, Gimenez-Cassina A, Garrido JJ, Bhat RV, Díaz-Nido J, Wandosell F. Pharmacological inhibition of GSK-3 is not strictly correlated with a decrease in tyrosine phosphorylation of residues 216/279. J Neurosci Res. 2008;86:668–674. doi: 10.1002/jnr.21523. [DOI] [PubMed] [Google Scholar]
- 51.Smith SB, Wang J, Cui X, Mysona BA, Zhao J, Bollinger KE. Sigma 1 receptor: A novel therapeutic target in retinal disease. Prog Retin Eye Res. 2018;67:130–149. doi: 10.1016/j.preteyeres.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun XB, Lu HE, Chen Y, Fan XH, Tong B. Effect of lithium chloride on endoplasmic reticulum stress-related PERK/ROCK signaling in a rat model of glaucoma. Pharmazie. 2014;69:889–893. [PubMed] [Google Scholar]
- 53.Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, Sabio G, Davis RJ, Matthews DE, Doble B, Rincon M. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science. 2008;320:667–670. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tullai JW, Graham JR, Cooper GM. A GSK-3-mediated transcriptional network maintains repression of immediate early genes in quiescent cells. Cell Cycle. 2011;10:3072–3077. doi: 10.4161/cc.10.18.17321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang B, Hu C, Yang X, Du F, Feng Y, Li H, Zhu C, Yu X. Inhibition of GSK-3β activation protects SD rat retina against N-methyl-N-nitrosourea-induced degeneration by modulating the Wnt/β-catenin signaling pathway. J Mol Neurosci. 2017;63:233–242. doi: 10.1007/s12031-017-0973-2. [DOI] [PubMed] [Google Scholar]
- 56.Wang H, Wang X. GSK3-like kinases are a class of positive components in the core ABA signaling pathway. Molecular plant. 2018;11:761–763. doi: 10.1016/j.molp.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 57.Yang ES, Nowsheen S, Wang T, Thotala DK, Xia F. Glycogen synthase kinase 3beta inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro Oncol. 2011;13:459–470. doi: 10.1093/neuonc/nor016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Y, Wu N, Tian S, Li F, Hu H, Chen P, Cai X, Xu L, Zhang J, Chen Z, Ge J, Yu K, Zhuang J. Lithium promotes DNA stability and survival of ischemic retinal neurocytes by upregulating DNA ligase IV. Cell Death Dis. 2016;7:e2473. doi: 10.1038/cddis.2016.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu L, Wu S, Che S, Wu Y, Han N. Inhibitory role of miR-203 in the angiogenesis of mice with pathological retinal neovascularization disease through downregulation of SNAI2. Cell Signal. 2020;71:109570. doi: 10.1016/j.cellsig.2020.109570. [DOI] [PubMed] [Google Scholar]
- 60.Zakharova IO, Sokolova TV, Bayunova LV, Zorina, Rychkova MP, Shpakov AO, Avrova NF. The protective effect of insulin on rat cortical neurons in oxidative stress and its dependence on the modulation of Akt, GSK-3beta, ERK1/2, and AMPK activities. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20153702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]
- 62.Zhu H, Zhang W, Zhao Y, Shu X, Wang W, Wang D, Yang Y, He Z, Wang X, Ying Y. GSK3β-mediated tau hyperphosphorylation triggers diabetic retinal neurodegeneration by disrupting synaptic and mitochondrial functions. Mol Neurodegener. 2018;13:62. doi: 10.1186/s13024-018-0295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhuang J, Li F, Liu X, Liu Z, Lin J, Ge Y, Kaminski JM, Summers JB, Wang Z, Ge J, Yu K. Lithium chloride protects retinal neurocytes from nutrient deprivation by promoting DNA non-homologous end-joining. Biochem Biophys Res Commun. 2009;380:650–654. doi: 10.1016/j.bbrc.2009.01.162. [DOI] [PubMed] [Google Scholar]