

Decreased neuronal survival-signaling and brain damage: Stroke is a leading cause of death worldwide, the major cause of adult disability and second of dementia. In spite of the social and economic importance of this disorder, and after intense research, no effective drugs have yet reached the clinic. Blood reperfusion with the thrombolytic agent tissue plasminogen activator remains the only pharmacologic treatment currently available for ischemic stroke, the major type of brain infarction (> 85% of total cases). Damage in this situation results from thrombotic or embolic occlusion of a cerebral artery causing a decrease of blood flow to a specific area of the brain parenchyma, neurons being particularly sensitive to a reduction of the supply of glucose and oxygen. It is thus a priority to develop neuroprotective strategies able to preserve neurons from the ischemic injury and, in this way, reduce brain damage and patient disability. A promising approach involves rescue of the area of penumbra surrounding the infarct, a region functionally silent but structurally intact. However, neurons in the penumbra can undergo a process of delayed death known as excitotoxicity, caused by overstimulation of the N-methyl-D-aspartate type of excitatory glutamate receptors (NMDARs). The critical role played by these receptors in synaptic plasticity, learning and memory, together with dual functions in neuronal survival and death (Hardingham et al., 2002), underlies previous failure of NMDAR blockade as a therapeutic target in stroke. Nevertheless, the low-affinity uncompetitive NMDAR antagonist memantine is still able to improve cognitive functions and behavioral disturbances in moderate-to-severe Alzheimer’s disease, a neurodegenerative disorder also associated with excitotoxicity. Anyhow, for stroke treatment, we are currently exploring alternative strategies such as the inhibition of neurotoxic proteins that act downstream overactivated NMDARs or directed to enhance neuronal survival pathways. Concerning the latter, several laboratories have chosen to analyze the promotion of neurotrophin-dependent survival pathways by treatment with brain-derived neurotrophic factor (BDNF) as a possible strategy for neuroprotection in stroke but also other acute or chronic disorders of the central nervous system. However, a potential caveat of this approach is that signaling mediated by BDNF is dramatically subverted by excitotoxicity, a process not only central to stroke but, as mentioned, also associated to many other neurological disorders (Tejeda and Diaz-Guerra, 2017). In models of stroke and human samples, excitotoxicity induces transcriptional and proteolytic mechanisms strongly associated with neurodegeneration that alter the expression of the two major brain isoforms of the BDNF receptor, tropomyosin-related kinase B (TrkB): the catalytically active full-length receptor (TrkB-FL) and a truncated receptor lacking the tyrosine kinase domain (TrkB-T1) (Vidaurre et al., 2012; Tejeda et al., 2016). Nonetheless, recent work from my group has demonstrated that it is possible to interfere TrkB-FL degradation in stroke and, in this way, decrease neuronal death and brain damage (Tejeda et al., 2019). Interestingly, these results have been accomplished by primarily preventing TrkB-FL endocytosis, which is strongly induced by excitotoxicity and precedes receptor processing (Figure 1). In this perspective, we will discuss the prospects of using the modulation of excitotoxicity-induced endocytosis, and the subsequent preservation of membrane survival proteins, as a neuroprotective therapeutic strategy for acute brain insults (stroke, epilepsy, or trauma) and excitotoxicity-associated chronic disorders (e.g., Alzheimer’s, Parkinson´s, Huntington´s diseases).
Figure 1.
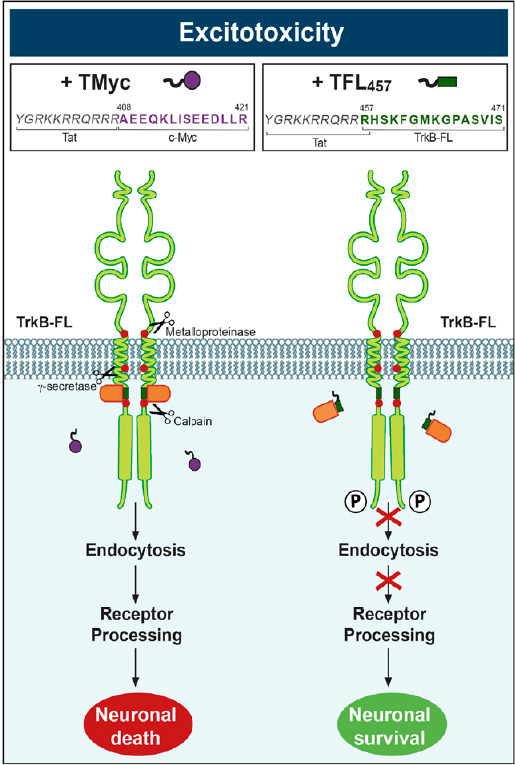
Mechanism of action of neuroprotective peptide TFL457.
The CPP contains a TrkB-FL juxtamembrane sequence (dark green rectangle) that probably competes a TrkB-interacting protein (orange) important for excitotoxicity-induced endocytosis, thus preventing secondary receptor processing and neuronal death. The interaction of TrkB with this unidentified protein is maintained in the presence of the control peptide TMyc (purple circle) and neuronal death is induced by the excitotoxic insult. CPP: Cell-penetrating peptide; TFL457 and TMyc: Tat-derived CPPs; TrkB: tropomyosin-related kinase B; TrkB-FL: the catalytically active full-length receptor.
Dual role of endocytosis in neuronal survival and death: Endocytosis is a ubiquitous physiological process that mediates nutrient uptake, receptor internalization and signaling, essential events for cell growth and survival. In neurons, endocytosis is required at the synaptic cleft after neurotransmitter release for recycling of membranes and surface proteins. Meanwhile, in the postsynaptic membranes of glutaminergic neurons, endocytosis is central for the reduction in long-term depression of surface glutamate receptors, mostly of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid family. NMDARs also undergo internalization in response to ligand binding, synapse maturation or long-term depression. While C-terminal sequences of GluN2A/B subunits direct endocytosed NMDARs to recycling endosomes, conserved motifs near the juxtamembrane region of GluN1 and GluN2A/B drive receptors to late endosomes and degradation (Scott et al., 2004). Notably, trafficking of TrkB isoforms is similarly very important for neurotrophin signaling in physiological conditions. Upon BDNF binding, both isoforms are rapidly and efficiently internalized in a clathrin-dependent way and form signaling endosomes. However, while TrkB-T1 predominantly recycles back to the cell surface by a default mechanism, TrkB-FL recycling is less efficient, relies on its tyrosine kinase activity, and is regulated by binding of the protein Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) to a receptor juxtamembrane region located between the transmembrane and tyrosine kinase domains (Huang et al., 2009).
In addition, endocytosis plays an evolutionarily conserved function in cell destruction by necrosis, a form of death central to stroke and other diseases affecting the central nervous system (Troulinaki and Tavernarakis, 2012). A transient upregulation of the endocytic activity has been observed early after cell death induction which is related to changes in calcium homeostasis taking place along neurodegeneration. The NMDARs are efficient calcium channels and their overactivation, among other mechanisms, profoundly alters the intracellular regulation of this ion. Correspondingly, endocytosis is enhanced in excitotoxicity by a clathrin/dynamin-mediated mechanism that precedes neuronal death in vitro or in models of ischemia, where stressed but viable neurons of the ischemic region have been described as highly endocytic (Vaslin et al., 2009). A crosstalk between endocytosis and the activation by Ca2+ of the protease calpain has been also described. After NMDAR overstimulation, cleavage by calpain of different substrates triggers multiple mechanisms of neurotoxicity, particularly enhancement of calcium overload, disruption of cell structure and promotion of cell-death signaling by degradation of antiapoptotic proteins or, alternatively, those involved in neuroprotective signaling such as TrkB-FL (Vidaurre at al., 2012). The subcellular localization of calpain activation is currently under discussion. Activation is favored near plasma and endosomal membranes but can also occur in microdomains with high local [Ca2+] as well as other cell compartments (mitochondria, nucleus or Golgi membranes). Notably, several components of the clathrin-coated vesicles, including the α- and β2-subunits of the adaptor protein complex 2 (AP-2), are calpain substrates and result hydrolyzed in experimental ischemia and the brain of Alzheimer’s disease patients (Rudinskiy et al., 2009). Cleavage of these AP-2 proteins has been suggested to be a mechanism to reduce clathrin-coated vesicles and moderate endocytosis at late stages of necrotic cell death.
Prevention of TrkB-FL endocytosis is relevant for stroke neuroprotection in vivo: Since endocytosis is a requirement for necrosis, general strategies to downregulate or deplete key proteins mediating different steps of the endocytic process have been devised and shown to actually interfere with neurodegeneration (Troulinaki and Tavernarakis, 2012). Therefore, generic interference of endocytosis might be considered as a therapeutic strategy with potential to reduce cell death due to acute brain insults. However, potential caveats to this approach are the pleiotropic adverse effects it might have considering that essential physiological cell processes would be inhibited as well. As an alternative, we propose the use of molecules able to interfere pathological endocytosis of specific neuronal survival proteins. One candidate to consider is the NMDAR, internalized by its own overstimulation. In fact, endocytosis of GluN2B-containing NMDARs has been shown to mediate excitotoxicity by still unknown mechanisms (Wu et al., 2017). Blockade of this clathrin-dependent endocytic process using a cell-penetrating peptide (CPP) was able to inhibit excitotoxicity in cultured cortical neurons. This CPP (Tat-YEKL) contains a 11 aa domain of the human immunodeficiency virus type 1 Tat protein, which allows attached cargoes to cross the blood-brain barrier and plasma membrane, followed by 12 aa of the GluN2B C-terminus corresponding to the AP-2 binding motif. The selection of CPPs as neuroprotective tools for stroke therapy seems particularly appropriate since the highly endocytic neurons of the ischemic area present an enhanced uptake of Tat-derived peptides (Vaslin et al., 2009) which results in a selective targeting of potential neuroprotective CPPs to damaged neurons. In the future, it will be interesting to establish if this CPP has similar neuroprotective effects when used in vivo and can specifically inhibit pro-death e?ects of NMDAR overactivation while preserving other receptor functions. Another possible candidate to be explored as neuroprotective target is kinase D-interacting substrate of 220 kDa (Kidins220), a downstream effector of neurotrophin receptors and NMDARs essential for neuronal viability. We have demonstrated that Kidins220 processing by calpain in excitotoxicity is secondary to traffic of this protein to the Golgi apparatus and early activation of Rap1-GTPase (López-Menéndez et al., 2019). At later stages of the excitotoxic process, Kidins220 downregulation governs Rap1 inactivation associated to a decrease in ERK activity which compromises neuronal survival. Therefore, prevention of excitotoxicity-induced Kidins220 endocytosis might be investigated as a way to inhibit calpain processing of Rap1 activation complexes and, thus, interfere the shut-off of Kidins220/Rap1/ERK prosurvival cascades.
Finally, we have already demonstrated that interference of TrkB-FL endocytosis is a relevant neuroprotective strategy both in vitro and in vivo. We have found that excitotoxicity decreases the surface levels of this receptor and, sequentially, promotes its intracellular processing by several proteases (Tejeda et al., 2019). The major mechanism of TrkB-FL downregulation is cleavage of its intracellular juxtamembrane region by calpain (Vidaurre et al., 2012; Figure 1). A secondary mechanism for TrkB-FL, but primary for TrkB-T1 regulation, is regulated intramembrane proteolysis by sequential metalloproteinase/γ-secretase action, producing the shedding by these isoforms of identical receptor ectodomains that we have demonstrated act as BDNF-scavengers (Tejeda et al., 2016). Altogether, these mechanisms cause a profound alteration of BDNF signaling by excitotoxicity, observed not only in stroke but also other neurological disorders (Tejeda and Diaz-Guerra, 2017). In order to preserve BDNF-regulated survival pathways, we have designed a Tat-derived CPP (TFL457) containing a short TrkB-FL juxtamembrane sequence (aa 457–471) that we hypothesized might be important for the control of receptor stability and function in excitotoxicity. The selected sequence is part of an intracellular region established as important for regulation of TrkB-FL location and function through interaction with a set of different proteins that include Hrs (Huang et al., 2009). We observed that peptide TFL457 specifically prevented early TrkB-FL endocytosis activated by excitotoxicity, differently from a negative control CPP containing non-related sequences (TMyc). We hypothesized that TFL457 might be competing some protein/s interaction/s established by sequence 457–471 in TrkB-FL which would be required for the promotion of excitotoxicity-induced endocytosis. Since this peptide will only affect interactions specifically established by TrkB-FL, endocytosis of other surface proteins would not be affected. By keeping the unprocessed receptor in the cell surface, TFL457 secondarily interferes TrkB-FL cleavage by calpain and regulated intramembrane proteolysis and, thus, receptor Tyr816 phosphorylation and BDNF/TrkB/PLCγ-signaling are maintained. The result is the preservation of cAMP response-element binding protein and myocyte enhancer factor 2 promoter activities downstream this cascade, which starts a feedback mechanism favoring increased expression of critical prosurvival proteins in neurons, such as BDNF or the TrkB receptor itself, resulting in increased neuronal viability. The developed neuroprotective peptide could be highly relevant for stroke therapy since, in a mouse model of permanent ischemia, it counteracts TrkB-FL downregulation in the infarct and efficiently decreases infarct size by nearly a 30% (Tejeda et al., 2019). Furthermore, TFL457 improves the neurological outcome, as shown by a decrease of 42% in the average number of slips in a test evaluating balance and motor coordination. Therefore, these results unravel endocytosis of TrkB-FL induced by excitotoxicity as a novel and highly relevant target for stroke therapy and open up new avenues for the development of novel drugs specifically directed to other survival proteins.
This work was supported by BFU2016-75973-R (MINECO/AEI/FEDER, EU).
Additional file: Open peer review report 1 (79.7KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Rayudu Gopalakrishna, University of Southern California, USA.
P-Reviewer: Gopalakrishna R; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 2.Huang SH, Zhao L, Sun ZP, Li XZ, Geng Z, Zhang KD, Chao MV, Chen ZY. Essential role of Hrs in endocytic recycling of full-length TrkB receptor but not its isoform TrkB, T1. J Biol Chem. 2009;284:15126–15136. doi: 10.1074/jbc.M809763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.López-Menéndez C, Simón-García A, Gamir-Morralla A, Pose-Utrilla J, Luján R, Mochizuki N, Díaz-Guerra M, Iglesias T. Excitotoxic targeting of Kidins220 to the Golgi apparatus precedes calpain cleavage of Rap1-activation complexes. Cell Death Dis. 2019;10:535. doi: 10.1038/s41419-019-1766-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rudinskiy N, Grishchuk Y, Vaslin A, Puyal J, Delacourte A, Hirling H, Clarke PG, Luthi-Carter R. Calpain hydrolysis of alpha- and beta2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration. J Biol Chem. 2009;284:12447–12458. doi: 10.1074/jbc.M804740200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott DB, Michailidis I, Mu Y, Logothetis D, Ehlers MD. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tejeda GS, Ayuso-Dolado S, Arbeteta R, Esteban-Ortega GM, Vidaurre OG, Diaz-Guerra M. Brain ischaemia induces shedding of a BDNF-scavenger ectodomain from TrkB receptors by excitotoxicity activation of metalloproteinases and gamma-secretases. J Pathol. 2016;238:627–640. doi: 10.1002/path.4684. [DOI] [PubMed] [Google Scholar]
- 7.Tejeda GS, Diaz-Guerra M. Integral characterization of defective BDNF/TrkB signalling in neurological and psychiatric disorders leads the way to new therapies. Int J Mol Sci. 2017;18:268–291. doi: 10.3390/ijms18020268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tejeda GS, Esteban-Ortega GM, San Antonio E, Vidaurre ÓG, Díaz-Guerra M. Prevention of excitotoxicity-induced processing of BDNF receptor TrkB-FL leads to stroke neuroprotection. EMBO Mol Med. 2019;11:e9950. doi: 10.15252/emmm.201809950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Troulinaki K, Tavernarakis N. Endocytosis and intracellular trafficking contribute to necrotic neurodegeneration in C. elegans. EMBO J. 2012;31:654–666. doi: 10.1038/emboj.2011.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaslin A, Puyal J, Clarke PG. Excitotoxicity-induced endocytosis confers drug targeting in cerebral ischemia. Ann Neurol. 2009;65:337–347. doi: 10.1002/ana.21584. [DOI] [PubMed] [Google Scholar]
- 11.Vidaurre OG, Gascón S, Deogracias R, Sobrado M, Cuadrado E, Montaner J, Rodríguez-Peña A, Díaz-Guerra M. Imbalance of neurotrophin receptor isoforms TrkB-FL/TrkB-T1 induces neuronal death in excitotoxicity. Cell Death Dis. 2012;3:e256. doi: 10.1038/cddis.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y, Chen C, Yang Q, Jiao M, Qiu S. Endocytosis of GluN2B-containing NMDA receptor mediates NMDA-induced excitotoxicity. Mol Pain. 2017;13:1744806917701921. doi: 10.1177/1744806917701921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.