

Systemic inflammation is often accompanied by adaptive responses mediated by the central nervous system, such as lack of motivation and attention, fatigue, malaise, irritation or even depression. This symptoms are summarized under the term “sickness behavior” (Dantzer et al., 2008). The inflammation-induced communication between the body and the brain uses neural as well humoral pathways, likely orchestrated by the major pro-inflammatory cytokines, such as interleukin (IL)-1α and β, tumor necrosis factor-α (TNF-α) and IL-6 (Dantzer et al., 2008). Interestingly, microglia, the main immune cells of the brain, sense peripheral inflammation as early as 5 hours after its induction by means of their intracellular Ca2+ signaling (Riester et al., 2020). Importantly, this change in Ca2+ signaling occurs long before the morphological activation of microglia, which usually takes place 24–48 hours after the induction of inflammation (Kozlowski and Weimer, 2012). Experimentally, the inflammation is often induced by the peripheral injection of lipopolysaccharide (LPS), a major component of the cell wall of Gram-negative bacteria. Both the early LPS-mediated increase in microglial Ca2+ signaling and the delayed LPS-induced morphological activation of microglia were blocked in mutant mice lacking the NACHT-, LRR- and pyrin (PYD)-domain-containing protein 3 (NLRP3) inflammasome (Tejera et al., 2019; Riester et al., 2020). Moreover, the LPS-induced reactive astrocytosis, visualized by an increased expression of glial fibrillary acidic protein, was also absent in Nlrp3–/– mice (Tejera et al., 2019), thus identifying the NLRP3 inflammasome as a key player governing the brain’s immune response to peripheral inflammation.
NLRP3 belongs to a protein family including 22 members in humans and 34 members in mice and the NLRP3 inflammasome is one of the known mediators of the organism’s immune response, causing the activation of caspase-1 and the subsequent maturation and release of IL-1β and IL-18 (He et al., 2016; Tejera et al., 2019). Classically, a two-hit model has being proposed for activation of this inflammasome. The first hit, called priming signal, induces the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB)-mediated expression of the NLRP3 and pro-IL-1β/pro-IL-18 proteins, while the second hit, called activation signal, causes the formation of the inflammasome complex, the cleavage of pro-caspase-1 into active caspase-1 as well as caspase-1-mediated cleavage of pro-IL-1β and pro-IL-18 into their active mature forms (Figure 1). The two-hit model, however, does not seem to apply to monocytes as well as bone marrow-derived dendritic cells, which are able to release mature IL-1β in response to a single pathogenic molecule (hit), such as, for example, LPS (He et al., 2016). Expectedly, the level of IL-1β both in the serum and the brain was significantly reduced in LPS-treated Nlrp3–/– mice, compared to control littermates (Tejera et al., 2019; Riester et al., 2020). This, however, was not the case for TNF-α. Surprisingly, the brain and serum levels of TNF-α more than doubled in Nlrp3–/– mice. Still, the brain’s immune response (see above) was largely absent, thus refuting the widely assumed contribution of TNF-α to this important process (Figure 1B). This conclusion is also consistent with the fact that LPS-induced increase in microglial Ca2+ signaling is preserved in TNF-α–/– mice (Riester et al., 2020).
Figure 1.
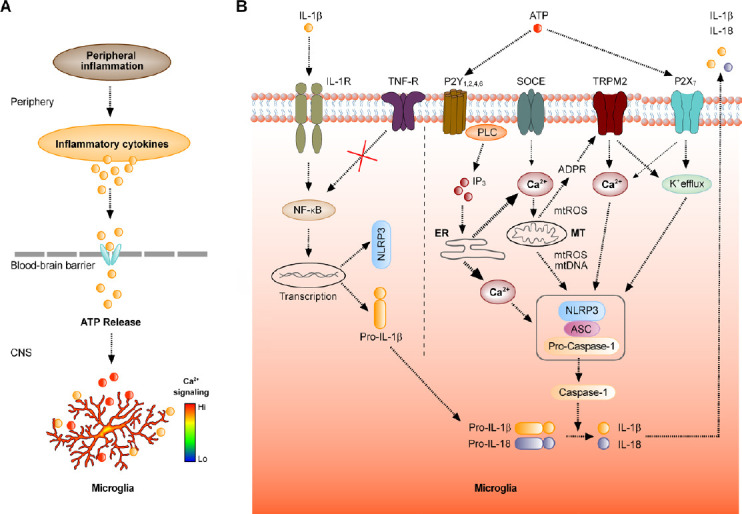
Mechanisms underlying microglial response to peripheral inflammation.
(A) Schematic representation of the signaling pathway underlying changes in microglial Ca2+ signaling, induced by peripheral inflammation. (B) Molecular mechanisms involved into the interplay between the increase in [Ca2+]i and the activation of the NLRP3 inflammasome. The boxed structure consisting of NLRP3, adaptor protein ASC and Pro-Caspase-1 represents the NLRP3 inflammasome. ADPR: ADP-ribose; ER: endoplasmic reticulum; IL: interleukin; IL-1R: IL-1β receptor; IP3: inositol 1,4,5-trisphosphate; MT: mitochondrion; mtDNA: mitochondrial DNA; mtROS: reactive oxygen species of mitochondrial origin; NF-κB: nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells; NLRP3: NACHT-, LRR- and pyrin (PYD)-domain-containing protein 3; P2X7: ionotropic ATP receptor; P2Y1, 2, 4, 6: metabotropic ATP receptors; PLC: phospholipase C; SOCE: store-operated Ca2+ entry channel; TNF-R: tumor necrosis factor-α receptor; TRPM2: transient receptor potential cation channel, subfamily M, member 2.
As the first LPS-induced surge of pro-inflammatory cytokines is produced peripherally, by monocytes, macrophages and dendritic cells, early microglial Ca2+ signals, induced by this stimulus, likely represent the brain’s response to peripheral IL-1β (Figure 1A). How does this response come about? For cortical microglia, the most likely pathway comprises cytokine transporters at the blood-brain barrier (Dantzer et al., 2008). Once in the brain, IL-1β binds to its receptors on the microglial surface to stimulate NF-κB-dependent pathways upregulating, among others, the production of NLRP3 as well pro-IL-1β/pro-IL-18. This provides the first hit for the mentioned above two-hit model of activation of the NLRP3 inflammasome (Figure 1B). At the same time, IL-1β likely stimulates a release of a universal DAMP (damage-associated molecular pattern) molecule ATP. Such a release was documented both in situ (rat hippocampal slices (Sperlagh et al., 2004)) and in vivo (rabbit hypothalamus (Gourine et al., 2007)) and was shown to occur almost without delay. Indeed, in slices the peak of IL-1β-evoked purine release was reached in less than 20 minutes after applying IL-1β, whereas in vivo the release of ATP started 18 minutes and reached its peak 45 minutes after the LPS injection into the ear vein (Gourine et al., 2007). In slices, the ATP release was almost completely blocked by the sodium channel blocker tetrodotoxin as well as by selective blockers of the glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and N-methyl-D-aspartic acid receptors, pointing towards neuronal origin of the released ATP. Why under these conditions the mentioned above signaling cascade is not switched on by the TNF-α remains unclear. This might either be due to the failure to activate NF-κB (as schematically shown in Figure 1B) or due to the insufficient amount of released ATP.
Once released, the ATP binds to ionotropic (P2X7) as well as metabotropic (P2Y1, 2, 4, 6) receptors on microglia (Eichhoff et al., 2011), causing the LPS-induced microglial Ca2+ signals, described above. This would provide the second hit for the two-hit model, causing the assembly of the NLRP3 inflammasome (Figure 1B). Interestingly, however, the in vivo level of ATP, measured by Gourine et al. (2007) reached only 4 μM. This is enough to produce an ATP-mediated Ca2+ signal in microglia caused by Ca2+ release from the endoplasmic reticulum Ca2+ stores (Figure 1B); Eichhoff et al., 2011) but way to low for activation of P2X7 receptors, known to require millimolar ATP concentration (McLarnon, 2005). I hypothesize, therefore, that in case of mild-to-medium peripheral inflammation ATP acts predominantly on metabotropic receptors. Indeed, in vivo DAMP-induced microglial Ca2+ signals are known to rely on Ca2+ release from the intracellular Ca2+ stores (Eichhoff et al., 2011; Brawek et al., 2014) and inhibition of the phospholipase C (Figure 1B) blocks the activation of the NLRP3 inflammasome induced by multiple stimuli, whereas direct activation of phospholipase C induces IL-1β secretion in the absence of any exogenous stimulus (Lee et al., 2012). The intracellular Ca2+ release, in turn, can trigger/activate several key processes, all promoting the assembly of the NLRP3 inflammasome (Figure 1B). Such processes include the activation of the (i) store-operated Ca2+ entry through the cell membrane, (ii) Ca2+-activated K+ channels leading to the K+ efflux, known as potent activator of the NLRP3 inflammasome (Lee et al., 2012; He et al., 2016), and (iii) release of reactive oxygen species as well as oxidized DNA from mitochondria. Intracellular reactive oxygen species, in turn, (iv) increase the production of ADP-ribose thereby activating the Transient Receptor Potential TRPM2 channels. These nonselective cation channels, known for their sensitivity to endogenous reactive oxygen species (Wang et al., 2020), support both Ca2+ influx and K+ efflux, thereby facilitating the activation of the NLRP3 inflammasome (Figure 1B).
The interplay between the increase in the intracellular free Ca2+ concentration ([Ca2+]i) and the activation of the NLRP3 inflammasome marks a qualitative change in the function of microglia, switching from DAMP-sensing sentinel cells surveying their environment to effector cells actively promoting inflammation-induced immune response of the brain. The cells undergo the change in morphology and process motility, proliferate, start to phagocytose and upregulate the production/release of the inflammatory factors (e.g., IL-1β, IL-18, TNF-α, ATP, NO) (Brawek and Garaschuk, 2014; Tejera et al., 2019; Riester et al., 2020). While some of these effects critically depend on the formation of NLRP3 inflammasome (e.g. changes in microglial morphology, phagocytosis, IL-1β/IL-18 and likely also ATP production) the others (e.g., microglial proliferation, production of the TNF-α) do not, thus occurring also in the Nlrp3–/– mice (Tejera et al., 2019). Interestingly, in young wild type mice the microglial phagocytosis is not triggered/upregulated by the mild-to-medium peripheral inflammation (Tejera et al., 2019; Riester et al., 2020). The opposite, however, is true for aged (15-month-old) wild type mice, which show at least a 5x increase in the density of lysosomal marker protein CD68 (Tejera et al., 2019).
Together, these data identify an important role of the NLRP3 inflammasome for governing both the sensor (through the peripheral production of IL-1β) and the effector functions of microglia. The microglia-derived inflammatory factors, in turn, engage the other cell types (e.g., neurons and astrocytes) into the network-wide adaptive response (Brawek and Garaschuk, 2014; Tejera et al., 2019), thus giving the NLRP3 inflammasome a key role in controlling the reaction of central nervous system to peripheral inflammation.
I thank Ariane Kaupp for help with the figure.
This work was supported by the VolkswagenStiftung (grant No. 90233) to OG.
Additional file: Open peer review report 1 (77KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Honghao Wang, Southern Medical University, China.
P-Reviewer: Wang H; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Brawek B, Garaschuk O. Network-wide dysregulation of calcium homeostasis in Alzheimer’s disease. Cell Tissue Res. 2014;357:427–438. doi: 10.1007/s00441-014-1798-8. [DOI] [PubMed] [Google Scholar]
- 2.Brawek B, Schwendele B, Riester K, Kohsaka S, Lerdkrai C, Liang Y, Garaschuk O. Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 2014;127:495–505. doi: 10.1007/s00401-013-1242-2. [DOI] [PubMed] [Google Scholar]
- 3.Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eichhoff G, Brawek B, Garaschuk O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim Biophys Acta. 2011;1813:1014–1024. doi: 10.1016/j.bbamcr.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 5.Gourine AV, Dale N, Llaudet E, Poputnikov DM, Spyer KM, Gourine VN. Release of ATP in the central nervous system during systemic inflammation: real-time measurement in the hypothalamus of conscious rabbits. J Physiol. 2007;585:305–316. doi: 10.1113/jphysiol.2007.143933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 Inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozlowski C, Weimer RM. An automated method to quantify microglia morphology and application to monitor activation state longitudinally in vivo. PLoS One. 2012;7:e31814. doi: 10.1371/journal.pone.0031814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLarnon JG. Purinergic mediated changes in Ca2+ mobilization and functional responses in microglia: effects of low levels of ATP. J Neurosci Res. 2005;81:349–356. doi: 10.1002/jnr.20475. [DOI] [PubMed] [Google Scholar]
- 10.Riester K, Brawek B, Savitska D, Fröhlich N, Zirdum E, Mojtahedi N, Heneka MT, Garaschuk O. In vivo characterization of functional states of cortical microglia during peripheral inflammation. Brain Behav Immun. 2020;87:243–255. doi: 10.1016/j.bbi.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Sperlagh B, Baranyi M, Hasko G, Vizi ES. Potent effect of interleukin-1 beta to evoke ATP and adenosine release from rat hippocampal slices. J Neuroimmunol. 2004;151:33–39. doi: 10.1016/j.jneuroim.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, Latz E, Golenbock D, Heneka MT. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019;38:e101064. doi: 10.15252/embj.2018101064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Negro R, Wu H. TRPM2, linking oxidative stress and Ca2+ permeation to NLRP3 inflammasome activation. Curr Opin Immunol. 2020;62:131–135. doi: 10.1016/j.coi.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.