

Abstract
OBJECTIVE
To compare efficacy and safety of dulaglutide at doses of 3.0 and 4.5 mg versus 1.5 mg in patients with type 2 diabetes inadequately controlled with metformin.
RESEARCH DESIGN AND METHODS
Patients were randomly assigned to once-weekly dulaglutide 1.5 mg, 3.0 mg, or 4.5 mg for 52 weeks. The primary objective was determining superiority of dulaglutide 3.0 mg and/or 4.5 mg over 1.5 mg in HbA1c reduction at 36 weeks. Secondary superiority objectives included change in body weight. Two estimands addressed efficacy objectives: treatment regimen (regardless of treatment discontinuation or rescue medication) and efficacy (on treatment without rescue medication) in all randomly assigned patients.
RESULTS
Mean baseline HbA1c and BMI in randomly assigned patients (N = 1,842) was 8.6% (70 mmol/mol) and 34.2 kg/m2, respectively. At 36 weeks, dulaglutide 4.5 mg provided superior HbA1c reductions compared with 1.5 mg (treatment-regimen estimand: −1.77 vs. −1.54% [−19.4 vs. −16.8 mmol/mol], estimated treatment difference [ETD] −0.24% (−2.6 mmol/mol), P < 0.001; efficacy estimand: −1.87 vs. −1.53% [−20.4 vs. −16.7 mmol/mol], ETD −0.34% (−3.7 mmol/mol), P < 0.001). Dulaglutide 3.0 mg was superior to 1.5 mg for reducing HbA1c, using the efficacy estimand (ETD −0.17% [−1.9 mmol/mol]; P = 0.003) but not the treatment-regimen estimand (ETD −0.10% [−1.1 mmol/mol]; P = 0.096). Dulaglutide 4.5 mg was superior to 1.5 mg for weight loss at 36 weeks for both estimands (treatment regimen: −4.6 vs. −3.0 kg, ETD −1.6 kg, P < 0.001; efficacy: −4.7 vs. −3.1 kg, ETD −1.6 kg, P < 0.001). Common adverse events through 36 weeks included nausea (1.5 mg, 13.4%; 3 mg, 15.6%; 4.5 mg, 16.4%) and vomiting (1.5 mg, 5.6%; 3 mg, 8.3%; 4.5 mg, 9.3%).
CONCLUSIONS
In patients with type 2 diabetes inadequately controlled by metformin, escalation from dulaglutide 1.5 mg to 3.0 mg or 4.5 mg provided clinically relevant, dose-related reductions in HbA1c and body weight with a similar safety profile.
Introduction
Type 2 diabetes is a chronic disease requiring long-term maintenance of glycemic control to reduce the risk of micro- and macrovascular complications. Treatment intensification is required over time, with most patients eventually requiring two or more drugs to achieve and maintain glycemic goals (1). Current guidance recommends that glucagon-like peptide-1 receptor agonists (GLP-1 RAs) should follow metformin therapy in several clinical situations, because of their effective glucose lowering, low hypoglycemia risk, potential for weight loss, and proven cardiovascular (CV) benefit for most agents in this therapeutic class (2).
Dose selection leading to development of dulaglutide 0.75 mg and 1.5 mg weekly doses was guided by GLP-1 RA class concerns related to increases in heart rate, pancreatic enzyme levels, and gastrointestinal (GI) adverse events (3). Evolution in the understanding of the GLP-1RA benefit-risk profile, especially with respect to CV (4) and pancreatic safety (5,6), and experience with stepwise dose escalation to optimize GI tolerability (7), combined with a persistent need for therapies to maintain glycemic control, prompted study of higher once-weekly doses (3.0 mg and 4.5 mg) of dulaglutide (8). Assessment of Weekly Administration of LY2189265 [dulaglutide] in Diabetes-11 (AWARD-11) was a phase 3 study designed to demonstrate superiority of dulaglutide 3.0 mg and/or 4.5 mg to 1.5 mg for change in HbA1c at 36 weeks and to provide safety data over 52 weeks in patients with type 2 diabetes inadequately controlled by metformin monotherapy.
Research Design and Methods
Study Design and Participants
This randomized, double-blind, parallel-arm study was conducted at 203 sites in 15 countries. The study included three periods: a 2-week lead-in period, followed by a 52-week treatment period (with primary efficacy end point at 36 weeks) and a 4-week safety follow-up period (Supplementary Fig. 1). The study was conducted in accordance with the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, and the International Conference on Harmonization Good Clinical Practices Guidelines. The protocol was approved by local institutional review boards. All patients provided written informed consent.
Eligible adults (aged ≥18 years) had type 2 diabetes for ≥6 months, with HbA1c ≥7.5% (58 mmol/mol) and ≤11.0% (97 mmol/mol) at screening; BMI ≥25 kg/m2; were insulin and GLP-1 RA naïve; and were taking commercially available metformin ≥1,500 mg/day for ≥3 months. A minimum BMI threshold was used to minimize concerns that higher drug exposures, as seen in previous studies with lower-weight patients (9), might predispose patients with lower BMI to GI tolerability limitations during dose escalation. Patients with type 1 diabetes; those using any other glucose-lowering medications (other than metformin) within 3 months before randomization; serum calcitonin level ≥20 ng/L; a history of pancreatitis, ketoacidosis, or hyperosmolar state/coma; recent CV event; or active cancer were excluded. A summary of eligibility criteria is provided in Supplementary Table 1.
Block randomization was used at the country level. Patients were randomly assigned 1:1:1 to dulaglutide 1.5 mg, 3.0 mg, or 4.5 mg, administered once weekly via subcutaneous injection with a single-dose pen. Randomization was stratified by HbA1c (<8.5% [69 mmol/mol], and ≥8.5% [69 mmol/mol]). Consistent with current labeling recommendations in the U.S., treatment was initiated with once-weekly dulaglutide 0.75 mg. After 4 weeks, the dose was escalated every 4 weeks to the randomized dose of 1.5 mg, 3.0 mg, or 4.5 mg (Supplementary Fig. 1).
Patients unable to tolerate dulaglutide during dose escalation were not put in a lower-dose group but could temporarily interrupt and then restart the study drug once. If intolerable symptoms returned, the study drug no longer was administered to the patients. Guidance was provided to study sites regarding treatment of patients with GI symptoms, including advice on dietary behaviors to mitigate nausea symptoms and vomiting, allowance for temporary interruption of study drug, and consideration for the use of oral antiemetic or antidiarrheal medication.
Patients with severe, persistent hyperglycemia during the study could initiate glycemic rescue therapy (other than nonstudy GLP-1 RAs or dipeptidyl peptidase-4 inhibitors, which were not permitted) according to prespecified criteria (Supplementary Appendix 2).
Efficacy Measures and Safety Assessments
The primary efficacy measure was the change in HbA1c from baseline to 36 weeks. Secondary efficacy measures (all assessed at 36 weeks and controlled for type I error) were the proportion of patients achieving HbA1c <7.0% (<53 mmol/mol); change from baseline in fasting serum glucose (FSG) level, determined by the central laboratory; and change from baseline in body weight. All other efficacy measures were exploratory and included comparison of dulaglutide 3.0 mg and 4.5 mg to the 1.5-mg dose at 52 weeks on the primary and secondary efficacy measures, as well as assessment at 36 and 52 weeks of the proportion of patients achieving the HbA1c target of ≤6.5% (48 mmol/mol), six-point self-monitored plasma glucose profile, fasting glucagon level, and measures of insulin resistance and β-cell function.
Adverse events, laboratory parameters, vital signs, and electrocardiograms were assessed for safety. Cases of suspected pancreatitis, CV events, and cause of death were confirmed by independent adjudication. Clinically important hypoglycemia was defined as any episode with a documented blood glucose level <54 mg/dL (<3 mmol/L) (10) or severe hypoglycemia, as defined by the American Diabetes Association (11). An independent data monitoring committee, external to the sponsor, reviewed unblinded safety data through the end of the primary 36-week end point.
Statistical Analysis
Approximately 510 participants per treatment group completing 36 weeks of the study (primary end point) provided at least 80% power to demonstrate superiority of dulaglutide 3.0 mg and/or 4.5 mg relative to 1.5 mg in change from baseline in mean HbA1c. Assumptions for sample-size calculations included a 0.22% difference in HbA1c change between dulaglutide 1.5 mg and at least one of the higher doses, a SD of 1.1%, and a two-sided significance level of 0.05. The primary safety and efficacy analysis population was the intent-to-treat population, defined as all randomly assigned patients who received at least one dose of study drug. Analyses of the 36-week primary and secondary objectives were performed on the basis of a primary database lock after all patients either completed the 36-week primary end-point visit or discontinued the study prior to this visit.
Two primary estimands were defined for analysis of the 36-week primary and secondary efficacy end points: a treatment-regimen estimand and an efficacy estimand (Supplementary Appendix 3). The treatment-regimen estimand included all baseline and end point data (either 36-week or 52-week data) collected regardless of initiation of new antihyperglycemic therapy or premature treatment discontinuation, with imputation of missing end-point data based on measurements from patients in the same treatment group and the same binary status of premature treatment discontinuation. The efficacy estimand included data from all study visits up to either initiation of any new antihyperglycemic medication for >14 days or premature treatment discontinuation, whichever occurred first. The treatment-regimen estimand was prespecified and applied only to the primary and secondary objectives at the 36-week time point in compliance with the U.S. Food and Drug Administration preference for this methodology for regulatory evaluation of safety and efficacy. The efficacy estimand was prespecified as primary for all other purposes and was estimated for all primary, secondary, and exploratory efficacy objectives (including all efficacy analyses through 52 weeks).
For the treatment-regimen estimand, an ANCOVA model was applied to the complete baseline and end-point data using multiple imputation for HbA1c, body weight, and FSG analyses. A logistic regression model was fit to the complete data to analyze the proportions of patients achieving HbA1c target <7.0% (53 mmol/mol), with imputation of missing end-point data as not achieving the target. For the efficacy estimand, a mixed model for repeated measures was used for longitudinal continuous variables and a longitudinal logistic regression model for repeated measures was used for categorical variables. Additional details on the statistical methods are provided in Supplementary Appendix 3.
To control overall type I error, a graphical approach (12) was used to compare the treatment effect among the predefined parameters of interest to address the primary and secondary efficacy objectives at 36 weeks for the intent-to-treat population (Supplementary Appendix 3). This graphical approach was conducted separately for each estimand, and each was tested at the full significance level of 0.05.
Results
Patient Disposition and Baseline Characteristics
A total of 1,842 patients were randomly assigned to a dulaglutide dose (1.5 mg, n = 612; 3.0 mg, n = 616; 4.5 mg, n = 614), of whom 93.1% completed the primary 36-week time point and 89.3% completed 36 weeks of study drug treatment with no difference across dose groups (1.5 mg, 89.7%; 3.0 mg, 88.3%; 4.5 mg, 89.9%; P = 0.623) (Fig. 1). There was no significant difference across dose groups in the proportion of patients completing the study through 52 weeks (1.5 mg, 90.8%; 3.0 mg, 89.1%; 4.5 mg, 91.2%; P = 0.427) or completing the study while taking the study drug (1.5 mg, 87.1%; 3.0 mg, 84.9%; 4.5 mg, 84.7%; P = 0.416) (Fig. 1).
Figure 1.

Patient disposition. *Patient randomly assigned at two different investigator sites, only counted once in randomization total; †Includes patients who discontinued the study.
Baseline characteristics were comparable among treatment groups (Table 1). The proportion of patients receiving new antihyperglycemic medication during the trial for any reason was similar across dose groups at both the primary 36-week time point (1.5 mg, 7.0%; 3.0 mg, 5.5%; 4.5 mg, 6.8%; P = 0.491) and final 52-week time point (1.5 mg, 9.2%; 3.0 mg, 7.1%; 4.5 mg, 9.0%; P = 0.370).
Table 1.
Baseline characteristics and demographics
| Parameter | DU 1.5 mg (n = 612) | DU 3.0 mg (n = 616) | DU 4.5 mg (n = 614) | Total (N = 1,842) |
|---|---|---|---|---|
| Age (years) | 57.8 ± 9.7 | 56.9 ± 10.2 | 56.6 ± 10.2 | 57.1 ± 10.0 |
| ≥65 | 156 (25.5) | 150 (24.4) | 132 (21.5) | 438 (23.8) |
| ≥75 | 20 (3.3) | 14 (2.3) | 21 (3.4) | 55 (3.0) |
| Duration of diabetes (years) | 7.6 ± 5.8 | 7.6 ± 5.5 | 7.7 ± 5.8 | 7.6 ± 5.7 |
| Female sex | 314 (51.3) | 288 (46.8) | 296 (48.2) | 898 (48.8) |
| Race | ||||
| American Indian or Alaska Native | 30 (4.9) | 26 (4.2) | 32 (5.2) | 88 (4.8) |
| Asian | 13 (2.1) | 18 (2.9) | 14 (2.3) | 45 (2.4) |
| Black | 28 (4.6) | 31 (5.0) | 23 (3.7) | 82 (4.5) |
| Native Hawaiian or Pacific Islander | 1 (0.2) | 1 (0.2) | 3 (0.5) | 5 (0.3) |
| White | 529 (86.4) | 521 (84.6) | 530 (86.3) | 1,580 (85.8) |
| Multiple | 11 (1.8) | 19 (3.1) | 12 (2.0) | 42 (2.3) |
| HbA1c (%) | 8.6 ± 0.9 | 8.6 ± 1.0 | 8.6 ± 0.9 | 8.6 ± 1.0 |
| BMI (kg/m2) | 34.4 ± 6.4 | 34.3 ± 6.2 | 34.0 ± 6.2 | 34.2 ± 6.3 |
| Weight (kg) | 95.5 ± 20.2 | 96.3 ± 20.1 | 95.4 ± 20.6 | 95.7 ± 20.3 |
| eGFR (mL/min/1.73 m2) | 93.4 ± 18.2 | 93.3 ± 17.8 | 93.7 ± 18.3 | 93.5 ± 18.1 |
| eGFR category (mL/min/1.73 m2) | ||||
| ≥30–60 | 33 (5.4) | 25 (4.1) | 33 (5.4) | 91 (4.9) |
| ≥60 to <90 | 171 (27.9) | 198 (32.1) | 184 (30.0) | 553 (30.0) |
| ≥90 | 408 (66.7) | 393 (63.8) | 397 (64.7) | 1,198 (65.0) |
| SBP (mmHg) | 132.1 ± 14.2 | 131.1 ± 14.1 | 132.1 ± 14.0 | 131.8 ± 14.1 |
| DBP (mmHg) | 78.8 ± 9.3 | 78.4 ± 8.7 | 79.0 ± 9.0 | 78.7 ± 9.0 |
| HR (bpm) | 75.6 ± 10.1 | 75.3 ± 9.5 | 75.5 ± 10.3 | 75.5 ± 10.0 |
| FSG (mg/dL) | 185.0 ± 52.0 | 184.0 ± 54.4 | 183.4 ± 48.0 | 184.1 ± 51.5 |
All values presented as mean ± SD or n (%). DBP, diastolic blood pressure; DU, dulaglutide; eGFR, estimated glomerular filtration rate; HbA1c, glycated hemoglobin; HR, heart rate; SBP, systolic blood pressure.
Glycemic Control
Treatment-Regimen Estimand
All treatment doses were efficacious using the treatment-regimen estimand (HbA1c least-square mean [LSM] change from baseline at 36 weeks, −1.54% for 1.5 mg, −1.64% for 3.0 mg, and −1.77% for 4.5 mg) (Fig. 2A). Superiority over the 1.5-mg dose for the primary HbA1c end point was observed for the 4.5-mg dose (estimated treatment difference [ETD] −0.24% [95% CI −0.36, −0.11] [−2.6 mmol/mol (95% CI −3.9, −1.3)]; P < 0.001) but not the 3.0-mg dose (ETD −0.10% [95% CI −0.23, 0.02] [−1.1 mmol/mol (−2.5, 0.2)]; P = 0.096) (Fig. 2A). Because superiority was not shown for the 3.0-mg dose, per the graphical testing procedure, subsequent comparisons for prespecified secondary glycemic control measures of the proportion of patients achieving HbA1c <7% (Fig. 2D) and of FSG levels (Supplementary Fig. 2) could not be formally tested for either dose using the treatment-regimen estimand, and all P values displayed for these comparisons are nominal. However, a consistent pattern of dose-related improvement in HbA1c, FSG levels, and the proportion of patients achieving HbA1c <7% was evident for the higher doses at both the primary 36-week time point and final 52-week time point using the treatment-regimen estimand (Fig. 2A and D and Supplementary Fig. 2).
Figure 2.
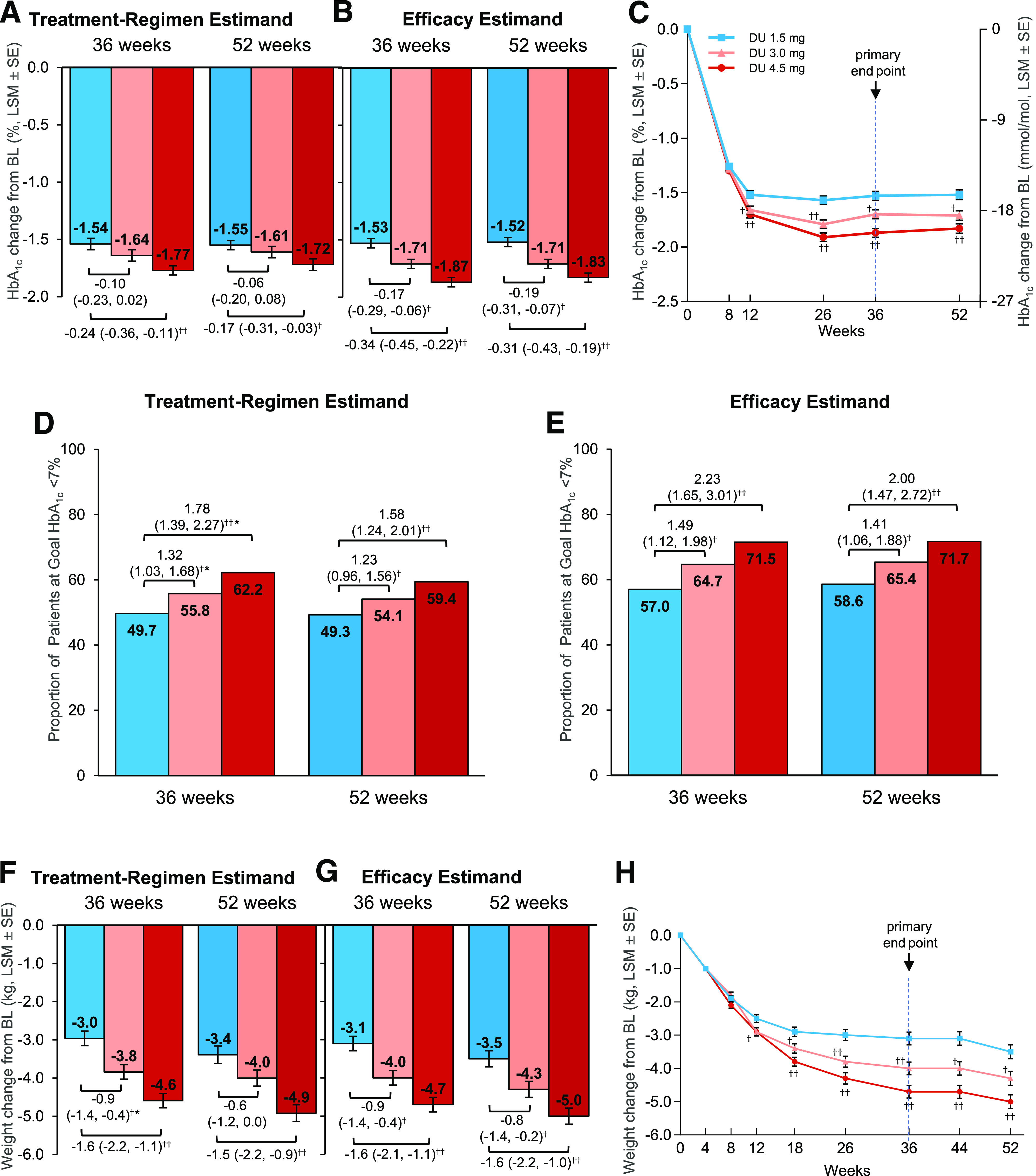
Primary and secondary efficacy outcomes by estimand. A: Change in HbA1c from baseline to 36 weeks and 52 weeks, ANCOVA with multiple imputation (treatment-regimen estimand). B: Change in HbA1c from baseline to 36 weeks (primary time point) and 52 weeks, mixed-model for repeated measure (MMRM; efficacy estimand). C: Change in HbA1c by study week, MMRM (efficacy estimand). D: Proportion of patients achieving HbA1c <7% (53 mmol/mol) at 36 and 52 weeks, logistic regression with imputation of missing end point data as not achieving target (treatment-regimen estimand). E: Proportion of patients achieving HbA1c <7% (53 mmol/mol) at 36 and 52 weeks, longitudinal logistic regression (efficacy estimand). F: Change in body weight from baseline to 36 and 52 weeks, ANCOVA with multiple imputation (treatment-regimen estimand). G: Change in body weight from baseline to 36 and 52 weeks, MMRM (efficacy estimand). H: Change in body weight by study week, MMRM (efficacy estimand). Data presented as LSM ± SE unless otherwise indicated; †P < 0.05, ††P < 0.001 vs. dulaglutide 1.5 mg, respectively. *Nominal P value, not adjusted for multiplicity; comparison vs. dulaglutide 1.5 mg did not achieve statistical superiority using the graphical testing approach. Note that 36-week LSMs in bar graphs for efficacy estimand are from MMRM analysis performed at the 36-week primary database lock, whereas LSMs in the line graphs (C and H) are from MMRM analyses including all data through the final 52-week time point. The efficacy estimand included patients with a nonmissing baseline value and at least one nonmissing postbaseline value of the response variable: n = 1,780 patients for HbA1c (1.5 mg, n = 591; 3.0 mg, n = 595; 4.5 mg, n = 594) and n = 1,809 patients for body weight (1.5 mg, n = 601; 3.0 mg, n = 604; 4.5 mg, n = 604). For the treatment-regimen estimand, data were included from all randomly assigned patients with imputation of missing end point values. BL, baseline; DU, dulaglutide; HbA1c, glycated hemoglobin.
Efficacy Estimand
All treatment doses were efficacious, resulting in significant LSM reductions in HbA1c from baseline to 36 weeks: −1.53% (−16.7 mmol/mol) for 1.5 mg, −1.71% (−18.6 mmol/mol) for 3.0 mg, and −1.87% (−20.4 mmol/mol) for 4.5 mg (Fig. 2B and C). Compared with the 1.5-mg dose, the 3.0- and 4.5-mg doses resulted in significantly greater LSM HbA1c reductions from 12 weeks (Fig. 2C), with ETDs at 36 weeks of −0.17% for 3.0 mg (95% CI −0.29, −0.06 [−1.9 mmol/mol (95% CI −3.2, −0.6)]; P = 0.003) and −0.34% for 4.5 mg (95% CI −0.45, −0.22 [−3.7 mmol/mol (95% CI −4.9, −2.4)]; P < 0.001) (Fig. 2B). Based on prespecified subgroup analysis, escalation to dulaglutide 3.0 mg or 4.5 mg resulted in additional HbA1c reductions versus the 1.5-mg dose regardless of baseline HbA1c (Supplementary Fig. 3). Mean treatment differences in the 4.5-mg group were greater in patients with higher HbA1c at baseline (treatment-by-subgroup interaction P = 0.02) (Supplementary Fig. 3).
Significantly more patients achieved the secondary outcome of HbA1c <7.0% (53 mmol/mol) (Fig. 2E) and exploratory outcome of HbA1c ≤6.5% (48 mmol/mol) (Supplementary Table 2) in the higher dulaglutide dose groups versus the 1.5-mg dose. At the primary 36-week time point, the odds of achieving each HbA1c target (either <7% or ≤6.5%) were approximately two times higher in patients who were escalated to the dulaglutide 4.5-mg dose compared with those maintained at the 1.5-mg dose (odds ratio for achieving HbA1c <7%, 2.2 [95% CI 1.7, 3.0], P < 0.001; odds ratio for achieving HbA1c ≤6.5%, 2.0 [95% CI 1.5, 2.6], P < 0.001) (Supplementary Table 2). Dulaglutide 4.5 mg but not 3.0 mg was superior to the 1.5-mg dose for reduction in FSG at 36 weeks (Supplementary Fig. 2).
Changes from baseline in HbA1c, the proportion of patients achieving HbA1c targets, and FSG levels observed at the primary 36-week time point were sustained through the final 52-week time point using the efficacy estimand (Fig. 2B, C, and E and Supplementary Fig. 2).
Body Weight
For the treatment-regimen estimand, dulaglutide 4.5 mg was superior to 1.5 mg for body weight (ETD, −1.6 kg [95% CI −2.2, −1.1]; P < 0.001) (Fig. 2F). Superiority of the 3.0-mg dose on body weight could not be formally tested using the treatment-regimen estimand in the graphical testing method (Supplementary Appendix 3), because this dose did not achieve superiority on the primary HbA1c end point. However, a pattern of dose-related decrease in body weight was observed at 36 weeks and 52 weeks (Fig. 2F).
For the efficacy estimand, patients whose dulaglutide dose was escalated from 1.5 mg to the 3.0-mg and 4.5-mg doses had greater reductions in body weight compared with those maintained on dulaglutide 1.5 mg from 12 weeks (Fig. 2H), with superior weight loss for 3.0 mg (−4.0 kg; ETD, −0.9 kg [95% CI −1.4, −0.4]; P = 0.001) and 4.5 mg (−4.7 kg; ETD, −1.6 kg [95% CI −2.1, −1.1]; P < 0.001) at 36 weeks (Fig. 2G), with additional weight loss observed in each dose group at week 52 (Fig. 2G and H).
Other Outcomes
Results for the exploratory efficacy outcomes of self-monitored plasma glucose (Supplementary Fig. 4) and markers of glucose metabolism (Supplementary Table 3) were generally consistent with the dose-related improvements in glycemic control discussed and are summarized in the Supplementary Material.
Safety
The proportion of patients reporting at least one treatment-emergent adverse event was similar across dose groups (Table 2). The three most frequently reported adverse events were nausea, diarrhea, and vomiting, although few cases were severe (≤0.5%) (Supplementary Table 4); the incidence of each, although numerically higher in the higher dulaglutide-dose groups compared with the 1.5-mg group, were similar between the 3.0- and 4.5-mg groups (Table 2). Adverse GI events tended to occur early and abate over time, with the incidences of nausea, vomiting, or diarrhea through 52 weeks generally similar to those reported through 36 weeks (Supplementary Table 4). Other GI-related events, such as constipation, dyspepsia, and abdominal pain, were reported in ≤5% of patients overall, with no clinically relevant dose-related differences across groups (Supplementary Table 5).
Table 2.
Summary of adverse events through 52 weeks
| Variable | DU 1.5 mg (n = 612) | DU 3.0 mg (n = 616) | DU 4.5 mg (n = 614) |
|---|---|---|---|
| Patients with ≥1 TEAE | 380 (62.1) | 384 (62.3) | 408 (66.4) |
| TEAEs occurring in ≥5% patients in any group | |||
| Nausea | 87 (14.2) | 99 (16.1) | 106 (17.3) |
| Diarrhea | 47 (7.7) | 74 (12.0) | 71 (11.6) |
| Vomiting | 39 (6.4) | 56 (9.1) | 62 (10.1) |
| Nasopharyngitis | 28 (4.6) | 32 (5.2) | 38 (6.2) |
| Dyspepsia | 17 (2.8) | 31 (5.0) | 17 (2.8) |
| Discontinuation of study drug due to any AE or death | 37 (6.0) | 43 (7.0) | 52 (8.5) |
| Discontinuation of study drug due to common GI events* | 9 (1.5) | 21 (3.4) | 24 (3.9) |
| Nausea | 8 (1.3) | 8 (1.3) | 9 (1.5) |
| Diarrhea | 1 (0.2) | 6 (1.0) | 6 (1.0) |
| Vomiting | 0 (0.0) | 5 (0.8) | 8 (1.3) |
| Dyspepsia | 0 (0.0) | 2 (0.3) | 1 (1.2) |
| Serious adverse events | 51 (8.3) | 42 (6.8) | 38 (6.2) |
| Death | 3 (0.5) | 4 (0.6) | 4 (0.7) |
| Adjudication confirmed | |||
| Acute pancreatitis | 1 (0.2) | 2 (0.3) | 3 (0.5) |
| CV events | 2 (0.3) | 8 (1.3) | 5 (0.8) |
| Gallbladder-related events† | 9 (1.5) | 11 (1.8) | 10 (1.6) |
| Acute renal events‡ | 6 (1.0) | 6 (1.0) | 5 (0.8) |
| Hypoglycemia | |||
| Documented§ (<54 mg/dL) | 8 (1.3) | 2 (0.3) | 7 (1.1) |
| Severe‖ | 1 (0.2) | 0 (0.0) | 1 (0.2) |
All values presented as n (%). Deaths and adjudication-confirmed events include any events reported during the safety follow-up period. AE, adverse event; DU, dulaglutide; PG, plasma glucose; TEAE, treatment-emergent adverse event.
Any AE reported under MedDRA System Organ Class Gastrointestinal Disorders in ≥5% of patients in any dose group.
Defined by Standardized MedDRA Query (SMQ) Acute Gallbladder Disease.
Defined by SMQ Acute Renal Failure.
Any confirmed PG level <54 mg/dL (3.0 mmol/L).
An episode requiring the assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions. These episodes may have been associated with sufficient neuroglycopenia to induce seizure or coma. Plasma glucose measurements may not have been available during such an event, but neurologic recovery attributable to the restoration of PG to normal was considered sufficient evidence that the event was induced by a low PG concentration.
The overall incidence of study drug discontinuation due to common GI events was low (3.9%) but more common in the 3-mg (3.4%) and 4.5-mg (3.9%) groups than in the 1.5-mg group (1.5%) (Table 2). Study drug discontinuation due to nausea was similar across dose groups (1.5 mg, 1.3%; 3.0 mg, 1.3%; 4.5 mg, 1.5%). Study drug discontinuation was more common in the higher-dose groups for vomiting and diarrhea but remained low for these events in the highest 4.5-mg group (1.3% for vomiting and 1.0% for diarrhea) (Table 2).
Fewer serious events were reported in the higher-dose groups than in the 1.5-mg group (Table 2). The incidence and estimated annual rate of documented hypoglycemia (plasma glucose, <54 mg/dL [3.0 mmol/L]) by dose group were as follows: 1.5 mg, 1.3%, n = 0.017 events per year; 3.0 mg, 0.3%, n = 0.003 events per year; 4.5 mg, 1.1%, n = 0.02 events per year. Two patients, one each in the 1.5- and 4.5-mg dose groups, experienced one episode each of severe hypoglycemia.
Six cases of pancreatitis were confirmed by adjudication (Table 2). One of these patients (assigned to dulaglutide 4.5 mg) had a previously undisclosed history of pancreatitis and would have been excluded from enrollment had this been known at screening.
Similar proportions of patients reported gallbladder- or renal-related adverse events (Table 2). Diabetic retinopathy was reported as an adverse event in eight patients (0.4%) overall: 1.5 mg, n = 4 patients (0.7%), 3.0 mg, n = 1 patient (0.2%), and 4.5 mg, n = 3 patients (0.5%). There were no reports of pancreatic cancer, C-cell hyperplasia, or medullary thyroid carcinoma during the treatment period of the study. One patient in the dulaglutide 1.5-mg group presented at the 52-week study visit with elevated serum aminotransferase and lipase concentrations and was subsequently diagnosed with pancreatic adenocarcinoma after completing the study, ∼74 days after receiving the last dose of dulaglutide.
Eleven deaths occurred during the study across the three dose groups: 1.5 mg, n = 3 patients; 3.0 mg, n = 4 patients; 4.5 mg, n = 4 patients. Accounting for the dose-escalation period, the distribution of the 11 deaths according to the dulaglutide dose being taken before or at the time of death was one patient receiving 0.75 mg, three patients receiving 1.5 mg, four patients receiving 3.0 mg, and three patients receiving 4.5 mg.
Fifteen patients had at least one CV event confirmed by adjudication (Table 2). Two of the eight CV events reported in the 3.0-mg dose group occurred during the dose escalation period when the patient was taking the 1.5-mg dose, and one of the five CV events reported in the 4.5-mg group occurred when the patient was taking the 0.75-mg dose.
Significant LSM increases in seated heart rate were observed across all dose groups over time (Supplementary Fig. 5A), including at the 36-week primary time point (1.5 mg, 1.5 bpm; 3.0 mg, 2.7 bpm; 4.5 mg, 2.7 bpm) and the 52 week final time point (1.5 mg, 1.0 bpm; 3.0 mg, 1.9 bpm; 4.5 mg, 1.9 bpm). There were no clinically relevant differences from a safety perspective related to blood pressure (Supplementary Fig. 5), electrocardiogram findings (Supplementary Fig. 6), or serum lipid levels (Supplementary Table 6).
Conclusions
In patients with type 2 diabetes who are taking metformin but experiencing inadequate glycemic control, escalation from dulaglutide 1.5 mg to 3.0 mg or 4.5 mg once weekly provided improved glycemic control and body weight reduction compared with patients taking dulaglutide 1.5 mg at 36 weeks, and these improvements were maintained to 52 weeks. The additional glycemic benefit and weight loss were achieved with a safety profile similar to the 1.5-mg dose of dulaglutide.
Two primary estimands were prespecified, consistent with recent diabetes intervention trials (13). The treatment-regimen estimand included efficacy data for patients no longer taking dulaglutide with or without rescue medication and replaced missing end point data with model-derived values. This estimand provides a conservative estimate of overall effectiveness in populations in which patients may not adhere to treatment or may initiate other therapies. Estimands of this type often ascribe meaningful HbA1c reductions to the placebo arm because patients randomly assigned to placebo therapy are more likely to receive rescue therapies. Similarly, estimates of the active treatment effect can be blunted by inclusion of patients who have discontinued active randomized therapy. The efficacy estimand included data from patients only while they were taking dulaglutide and without initiating another glucose-lowering medication, thus providing an estimate of treatment effects expected in patients adherent to dulaglutide therapy. Neither the treatment-regimen nor the efficacy estimand approaches were applied to safety analyses and therefore do not affect conclusions about safety or tolerability of the higher dulaglutide doses.
Dose-related improvements in glycemic control and body weight were observed using both estimands. Although the statistical inference for superiority using the graphical testing approach was different between estimands, estimates of the absolute changes from baseline in HbA1c and body weight were generally similar between estimands. This is expected on the basis of the high patient retention (90%) in the trial with no significant difference across dose groups in the proportion of patients discontinuing dulaglutide treatment early or receiving rescue therapy. Although the dulaglutide 3.0-mg dose did not achieve statistical superiority to the 1.5-mg dose for the HbA1c primary end point using the treatment-regimen estimand, clinically relevant improvements in HbA1c were seen regardless of estimand used and should have clinical utility in patients requiring treatment intensification. The clinical relevance of the 3.0- and 4.5-mg doses is further supported by the dose-related increase observed using both estimands in the proportion of patients able to achieve the American Diabetes Associated–recommended HbA1c treatment targets of <7% (53 mmol/mol) and ≤6.5% (48 mmol/mol) at 36 weeks and who maintained these targets through 52 weeks, which is particularly important in these patients with poor glycemic control (mean HbA1c, 8.6%) at baseline. On the basis of these results and a favorable risk-benefit balance, both the 3.0- and 4.5-mg dulaglutide doses are considered efficacious and have been approved by the U.S. Food and Drug Administration and the European Medicines Agency in the European Union as additional dosing options for dulaglutide.
The safety of once-weekly dulaglutide 0.75 mg and 1.5 mg, approved in 2014, has been thoroughly characterized (14,15). As with the lower doses, the most common adverse events with the 3.0- and 4.5-mg doses were GI related (primarily nausea, vomiting, and diarrhea) and were predominantly limited in duration and mild in severity. Importantly, among patients whose dose was escalated to 4.5 mg, the incidences of nausea, diarrhea, and vomiting (17.3%, 11.6%, and 10.1%, respectively) were in the range previously reported for lower doses of dulaglutide (14,16), and early treatment discontinuations due to common GI events in AWARD-11 were infrequent (≤4% in each higher-dose group). Thus, escalation of dulaglutide dose from 1.5 mg to 3.0 mg and then to 4.5 mg achieved additional glycemic control while maintaining GI tolerability generally consistent with that previously established for dulaglutide.
Patient retention in the study (>90%) and adherence to assigned treatment (88%–90%) were high and relatively balanced across the treatment groups through 36 weeks, enabling a robust assessment of the primary and secondary objectives of the trial. Generalizability of these results to broader populations requires consideration of the population enrolled in AWARD-11. The study included patients who were overweight or obese (mean BMI, 34.2 kg/m2) with diabetes poorly controlled while taking metformin (mean HbA1c, 8.6%). Although indicative of many patients with chronic hyperglycemia requiring intensification of therapy, the results may not generalize to patients who do not meet these criteria. The dulaglutide 3.0- and 4.5-mg doses were studied here only in combination with metformin. However, the efficacy of dulaglutide 1.5 mg once weekly has been established with a wide variety of background medications (14), and the incremental benefits of the higher doses are expected to be similar when used in combination with other glucose-lowering medications.
In conclusion, the AWARD-11 trial demonstrated glycemic and weight benefits for patients using 3- and 4.5-mg doses of dulaglutide once weekly, without meaningful changes to the safety profile already familiar to clinicians and patients using dulaglutide. Currently approved prescribing information in the U.S. recommends a 0.75-mg starting dose once weekly, which may be increased to 1.5 mg once weekly for additional glycemic control. If additional glycemic control is needed, dose escalation may be performed to 3.0 mg or 4.5 mg at 4-week intervals. The availability of four clinically efficacious dulaglutide doses provides additional tools to individualize patient care, helping patients achieve and maintain glycemic and weight-reduction goals and allowing those already taking 1.5 mg to intensify treatment while continuing to receive a familiar therapy.
Article Information
Acknowledgments. The authors thank the trial investigators, trial staff, and trial participants for their contributions, and Oralee Varnado (Eli Lilly and Company) for writing support. The trial was sponsored by Eli Lilly and Company, which designed the trial, monitored sites, and collected and analyzed the data per a prespecified statistical analysis plan.
Duality of Interest. The trial was funded by Eli Lilly and Company. J.P.F. declares research support from Allergan, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly and Company, Janssen, Madrigal, Merck, Novartis, Novo Nordisk, Pfizer, Sanofi, and Theracos; serves on advisory boards and consults for Boehringer Ingelheim, Coherus Therapeutics, Eli Lilly and Company, Gilead, Merck, Novo Nordisk, and Sanofi; and serves on speaker bureaus for Merck and Sanofi. E.B. declares serving on advisory boards and consulting for Abbott, AstraZeneca, Becton Dickinson, Boehringer Ingelheim, Bristol Myers Squibb, Bruno Farmaceutici, Janssen, Johnson & Johnson, Eli Lilly and Company, Merck Sharp and Dohme, Mundipharma, Novartis, Novo Nordisk, Roche, Sanofi, Servier, and Takeda. Y.G.L., Z.M., R.M., M.A.B., and D.A.C. are employees of Eli Lilly and Company and own stock in the company. Z.Y. is a former employee of Eli Lilly and Company. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. J.P.F., E.B., L.N.R., Y.G.L., Z.Y., Z.M., R.M., M.A.B., and D.A.C. were responsible for the acquisition, analysis, or interpretation of data and drafting or critical revision of the manuscript for important intellectual content. Z.M. and D.A.C. contributed to the conception and design of the trial. Y.G.L. and Z.Y. were responsible for the statistical considerations in the analysis and trial design. All authors had full access to the data related to this study and approved the final version submitted for publication. M.A.B. and D.A.C. are the guarantors of this work and, as such, take responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Some of these data were presented in abstract form at the Endocrine Society annual meeting, held virtually 8–22 June 2020, and the 80th Scientific Sessions of the American Diabetes Association, held virtually 12–16 June 2020.
Footnotes
Clinical trial reg. no. NCT03495102, clinicaltrials.gov
This article contains supplementary material online at https://doi.org/10.2337/figshare.13315403.
This article is featured in a podcast available at https://www.diabetesjournals.org/content/diabetes-core-update-podcasts.
References
- 1.Blonde L, Raccah D, Lew E, et al. Treatment intensification in type 2 diabetes: a real-world study of 2-OAD regimens, GLP-1 RAs, or basal insulin. Diabetes Ther 2018;9:1169–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Diabetes Association . 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020;43(Suppl. 1):S98–S110 [DOI] [PubMed] [Google Scholar]
- 3.Skrivanek Z, Gaydos BL, Chien JY, et al. Dose-finding results in an adaptive, seamless, randomized trial of once-weekly dulaglutide combined with metformin in type 2 diabetes patients (AWARD-5). Diabetes Obes Metab 2014;16:748–756 [DOI] [PubMed] [Google Scholar]
- 4.Kristensen SL, Rørth R, Jhund PS, et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol 2019;7:776–785 [DOI] [PubMed] [Google Scholar]
- 5.Egan AG, Blind E, Dunder K, et al. Pancreatic safety of incretin-based drugs--FDA and EMA assessment. N Engl J Med 2014;370:794–797 [DOI] [PubMed] [Google Scholar]
- 6.Nauck MA, Frossard JL, Barkin JS, et al. Assessment of pancreas safety in the development program of once-weekly GLP-1 receptor agonist dulaglutide. Diabetes Care 2017;40:647–654 [DOI] [PubMed] [Google Scholar]
- 7.Nauck MA, Petrie JR, Sesti G, et al.; Study 1821 Investigators . A phase 2, randomized, dose-finding study of the novel once-weekly human GLP-1 analog, semaglutide, compared with placebo and open-label liraglutide in patients with type 2 diabetes. Diabetes Care 2016;39:231–241 [DOI] [PubMed] [Google Scholar]
- 8.Frias JP, Wynne AG, Matyjaszek-Matuszek B, et al. Efficacy and safety of an expanded dulaglutide dose range: a phase 2, placebo-controlled trial in patients with type 2 diabetes using metformin. Diabetes Obes Metab 2019;21:2048–2057 [DOI] [PubMed] [Google Scholar]
- 9.Geiser JS, Heathman MA, Cui X, et al. Clinical pharmacokinetics of dulaglutide in patients with type 2 diabetes: analyses of data from clinical trials. Clin Pharmacokinet 2016;55:625–634 [DOI] [PubMed] [Google Scholar]
- 10.International Hypoglycaemia Study Group . Glucose concentrations of less than 3.0 mmol/L (54 mg/dL) should be reported in clinical trials: a joint position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2017;40:155–157 [DOI] [PubMed] [Google Scholar]
- 11.Seaquist ER, Anderson J, Childs B, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care 2013;36:1384–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bretz F, Posch M, Glimm E, Klinglmueller F, Maurer W, Rohmeyer K. Graphical approaches for multiple comparison procedures using weighted Bonferroni, Simes, or parametric tests. Biom J 2011;53:894–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aroda VR, Saugstrup T, Buse JB, Donsmark M, Zacho J, Davies MJ. Incorporating and interpreting regulatory guidance on estimands in diabetes clinical trials: the PIONEER 1 randomized clinical trial as an example. Diabetes Obes Metab 2019;21:2203–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jendle J, Grunberger G, Blevins T, Giorgino F, Hietpas RT, Botros FT. Efficacy and safety of dulaglutide in the treatment of type 2 diabetes: a comprehensive review of the dulaglutide clinical data focusing on the AWARD phase 3 clinical trial program. Diabetes Metab Res Rev 2016;32:776–790 [DOI] [PubMed] [Google Scholar]
- 15.Kugler AJ, Thiman ML. Efficacy and safety profile of once-weekly dulaglutide in type 2 diabetes: a report on the emerging new data. Diabetes Metab Syndr Obes 2018;11:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pratley RE, Aroda VR, Lingvay I, et al.; SUSTAIN 7 Investigators . Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol 2018;6:275–286 [DOI] [PubMed] [Google Scholar]