

Abstract
Early‐onset Alzheimer's disease (EOAD) is generally known as a dominant disease due to highly penetrant pathogenic mutations in the amyloid precursor protein, presenilin 1 and 2. However, they explain only a fraction of EOAD patients (5% to 10%). Furthermore, only 10% to 15% of EOAD families present with clear autosomal dominant inheritance. Studies showed that only 35% to 60% of EOAD patients have at least one affected first‐degree relative. Parent–offspring concordance in EOAD was estimated to be <10%, indicating that full penetrant dominant alleles are not the sole players in EOAD. We aim to summarize current knowledge of rare variants underlying familial and seemingly sporadic Alzheimer's disease (AD) patients. Genetic findings indicate that in addition to the amyloid beta pathway, other pathways are of importance in AD pathophysiology. We discuss the difficulties in interpreting the influence of rare variants on disease onset and we underline the value of carefully selected ethnicity‐matched cohorts in AD genetic research.
Keywords: Alzheimer's disease, biological pathways of disease, familial AD, genetic etiology, rare variants, sporadic AD
1. INTRODUCTION
Alzheimer's disease (AD; Online Mendelian Inheritance in Man [OMIM]#104300) is a complex, progressive, and irreversible neurodegenerative brain disease (NBD) representing the most common dementia subtype affecting more than 50 million people worldwide. 1 AD is characterized by an insidious onset, progressive loss of memory and additional cognitive functions such as word finding, spatial cognition, and problem solving. 2 While the clinical symptoms of AD display a substantial overlap between multiple other NBDs, neuropathological examination upon brain autopsy can confirm a definite diagnosis of AD. 3 , 4 Neuropathological hallmarks of AD are extracellular depositions of amyloid beta (Aβ) peptides and intracellular neurofibrillary tangles of hyperphosphorylated tau protein, accompanied by gliosis and loss of neurons and synapses. 5
Aging is the most prominent biological risk factor for developing AD at late age with up to 90% of AD patients diagnosed above 65 years (late‐onset Alzheimer's disease [LOAD]). LOAD is a complex and heterogeneous disorder with a genetic etiology of up to 82%. 5 Approximately 10% of AD patients are diagnosed before the age of 65 years (early‐onset Alzheimer's disease [EOAD]) and present with a genetic etiology of up to 100%. 5 , 6 EOAD and LOAD patients are clinically and pathologically similar and both occur in familial and sporadic patients. Approximately 35% to 60% of the EOAD patients have first‐degree relatives with dementia, including 10% to 15% autosomal dominant families with three generation or more. 7 , 8 These multi‐generational EOAD families were essential for the identification of pathogenic mutations in three causal AD genes: amyloid precursor protein (APP) and presenilin 1 and 2 (PSEN1 and PSEN2), which are key players in the Aβ pathway (Table 1, Figure 1). The ε4 allele of the apolipoprotein E (APOE) gene was identified as a major genetic risk factor for LOAD, increasing risk 3 times in heterozygous and 15 times in homozygous carriers. In EOAD patients, risk is increased in homozygous ε4 carriers and in heterozygous ε4 carriers with a positive family history (Box 1). 5 , 9
TABLE 1.
Replicated genes harboring rare variants (MAF < 1%) associated with AD
| Gene | Inheritance pattern | Study | Implicated pathways |
|---|---|---|---|
| APP | Dominant | Alzforum | Aβ pathway 5 |
| Recessive | 78 , 79 | Immune system 131 | |
| de novo/mosaicism | 108 |
BBB integrity 132 Gene expression regulation 134 , 135 |
|
| PSEN1 | Dominant | Alzforum | Aβ pathway 5 |
| Recessive | 74 , 75 , 76 , 77 | Immune system 139 | |
| de novo/mosaicism | 105 , 107 |
BBB integrity 132 Axonal guidance and cytoskeleton function 142 |
|
| PSEN2 | Dominant | Alzforum | Aβ pathway 5 |
| de novo/mosaicism a | 146 |
Immune system 139 Synaptic plasticity 140 Apoptosis, phagocytosis, and autophagy 144 , 145 , 147 , 148 |
|
| SORL1 | Dominant | 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 | Aβ pathway 149 |
| Recessive b | 80 | Lipid metabolism 150 | |
| de novo/mosaicism c | 112 | ||
| ABCA7 | Dominant | 41 , 42 , 43 , 44 , 45 , 46 |
Aβ pathway 151 Immune system 152 Lipid metabolism 153 |
| TREM2 | Dominant | 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 |
Aβ pathway 157 Immune system 157 Lipid metabolism 158 Apoptosis, phagocytosis, and autophagy 159 |
| BIN1 | Dominant | 45 , 65 , 66 |
Aβ pathway 160 Tau pathway 161 Synaptic plasticity 162 |
| UNC5C | Dominant | 67 , 68 , 69 |
Axonal guidance 165 Apoptosis, phagocytosis, and autophagy 165 |
| AKAP9 | Dominant | 67 , 68 | Synaptic plasticity 165 |
| NOTCH3 | Dominant | 70 , 71 , 72 , 73 |
Aβ pathway 166 Gene expression regulation 167 BBB integrity 168 |
| 12S rRNA | Maternal | 98 , 99 , 100 | Mitochondrial cascade hypothesis 171 |
| CLU | Dominant | 101 , 102 , 103 |
Aβ pathway 172 Immune system 173 Lipid metabolism 174 |
| PLCG2 | Dominant | 54 , 56 , 104 |
Immune system 56 |
| ABI3 | Dominant | 54 , 56 , 104 , 105 | Immune system 56 |
| APOE | Modifier | 114 , 115 |
Aβ pathway 178 Immune system 178 Lipid metabolism 178 Synaptic plasticity 178 BBB integrity 179 Axonal guidance and cytoskeleton function 180 , 181 Tau pathway 182 Mitochondrial cascade hypothesis 183 |
Note: Reported inheritance patterns and implicated pathways are mentioned alongside literature references.
Abbreviations: Aβ, amyloid beta; Alzforum, Alzforum mutation database (https://www.alzforum.org/mutations); BBB, blood–brain barrier; MAF, minor allele frequency.
Identified in a Braak II control subject in a single study. 146
Bi‐allelic variants reported in SORL1 in a single study. 80
FIGURE 1.
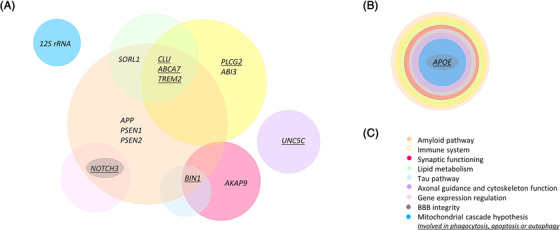
Schematic presentation of Alzheimer's disease (AD) genes and implicated pathways. A, Pathways in which the AD genes are involved. B, Apolipoprotein E (APOE) is a major common AD susceptibility gene involved in nine different pathways and harbors one rare variant modifying onset age. C, Color‐coded legend indicating the different genetic pathways associated with AD
The identification of Aβ plaques in autopsy brains of AD patients and the identification of pathogenic mutations in the three AD genes, which are key in the creation of the Aβ plaques, were at the basis of the Aβ hypothesis. 10 The Aβ hypothesis dominated AD‐related research for more than 25 years by stating that the accumulation of extracellular Aβ plaques in the brain was the central cause of AD pathology, though this was not accepted universally. 10 The hypothesis states that Aβ plaques ultimately trigger a cascade of disease‐causing events including inflammation, formation of tau tangles, and synaptic dysfunction. 10 The observation of Aβ plaques in the brain pf cognitively normal elderly and the absence in some AD patients created a lot of controversy. 11 , 12 Suspected non‐Aβ AD patients, describing AD patients lacking detectable Aβ in cerebrospinal fluid biomarker profiles or brain positron emission tomography imaging provided evidence for the existence of an AD pathology unrelated to the Aβ cascade. 11 Aβ plaque burden in AD brains with Aβ pathology showed only a weak correlation with disease severity and high‐profile clinical trials targeting Aβ failed to deliver functional treatment. 13 , 14 , 15 , 16 Moreover, only 5% to 10% of the EOAD patients can be explained by the pathogenic mutations in the three familial AD genes, suggesting that potential non‐Aβ pathways might also be involved in onset of AD. 5 Concordance studies between parent–offspring and between siblings resulted in <10% parent–offspring concordance and 21.6% concordance among siblings. 6 If AD is caused solely by full penetrant autosomal dominant alleles, concordance between parent–offspring would be estimated ≈50%. 6 These observations indicate that other genes, pathways, and inheritance patterns are involved in the etiology of AD. Here, we summarized the current knowledge of rare variants (minor allele frequency [MAF] < 1%) underlying familial and (seemingly) sporadic AD patients and focus on genes including rare AD associated variants (Figure 2). We hereby highlight the contribution of additional molecular pathways to early disease etiology (Figure 1) and underline the relevance of carefully selected ethnicity‐matched cohorts in AD genetic research.
FIGURE 2.

Presentation of the genes in the familial to sporadic Alzheimer's disease (AD) spectrum. Rare variants (minor allele frequency [MAF] < 1%) associated with familial or sporadic AD by multiple independent studies are shown. All listed genes include heterozygous mutations associated with autosomal dominant AD, except 12S rRNA, which includes heterozygous mtDNA mutations associated with maternal AD. Red box: de novo mutations in PSEN1 and APP have been associated with sporadic AD
RESEARCH IN CONTEXT
Systematic review: PubMed search, meeting abstracts, and presentations were used to collect information concerning the role of rare variants in Alzheimer's disease (AD) genetics.
Interpretation of results: We provide a comprehensive review on the current knowledge of rare variants underlying familial and seemingly sporadic AD.
Future directions: The basis of the current knowledge of AD derived from genetic studies on large early‐onset AD pedigrees in the early 1990s. Here, we underline the value of carefully selected ethnicity‐matched cohorts in AD genetic research to understand the biology and to identify therapeutic targets in the search for medical treatment.
2. FAMILIAL AD
Linkage studies in multi‐generational autosomal dominant EOAD families were crucial for the identification of three causal AD genes, APP on chromosome 21q, PSEN1 on 14q, and PSEN2 on 1q, 4 , 17 and including full penetrant pathogenic mutations. Concordance studies between parent–offspring and between siblings, however, showed that AD is not solely the cause of full penetrant dominant alleles. In addition to the three known causal AD genes, other well‐replicated genes, including rare heterozygous variants with reduced penetrance, are present in familial AD.
2.1. Highly penetrant pathogenic mutations in APP, PSEN1, and PSEN2
Pathogenic missense mutations and whole gene duplications have been identified in APP (https://www.alzforum.org/mutations). Interestingly, most of the pathogenic missense mutations affect APP processing and are located near the β‐ or γ‐secretase cleavage sites or in the Aβ sequence of the APP protein (amino acids 670 to 724). 18 Pathogenic missense mutations appear to result in overproduction of either total Aβ or a shift in the Aβ1‐40/Aβ1‐42 ratio toward the more toxic Aβ1‐42 peptide. 2 , 4 APP duplications of variable size have been reported in AD families and underline the importance of APP gene dosage. 2 , 19 Pathogenic mutations in APP account for <1% of EOAD patients. 5 , 20 Besides pathogenic mutations, an Icelandic protective missense variant p.A673T was also identified in APP. This variant was associated with reduced production of the amyloidogenic Aβ1‐40 and Aβ1‐42 peptides (≈40%). 21 Finally, rare single nucleotide variants (SNVs) in the APP promotor have been associated with increased LOAD susceptibility. 22
PSEN1 and PSEN2 are both essential proteins of the catalytic core of the γ‐secretase complex, which catalyzes the cleavage of membrane proteins including APP. 20 Mutant γ‐secretase increases Aβ1‐42 levels, while decreasing Aβ1‐40 levels, leading to an increased Aβ1‐42/Aβ1‐40 ratio. 17 The majority of pathogenic PSEN mutations are missense mutations; however, pathogenic amino acid insertions and deletions have also been described (https://www.alzforum.org/mutations). Mutations in PSEN1 are the most common cause of familial EOAD and are characterized by the earliest onset ages (on average 8.4 and 14.2 years earlier compared to APP and PSEN2 mutations, respectively 23 ). 18 PSEN1 mutations causing the most severe form of AD explain ≈6% of EOAD patients. 5 In comparison, PSEN2 mutations are rare, explaining <1% of EOAD patients, and may show incomplete penetrance. 5 Carriers present with higher onset ages; however, onset ages are highly variable even between members of the same family. 24 , 25
BOX 1: APOE
A genome‐wide linkage study in familial LOAD patients and subsequent association studies identified APOE as the major susceptibility gene for AD. 5 , 125 , 126 , 127 APOE has three common isoforms, APOE ε2, ‐ε3, and ‐ε4, which have a general frequency of ≈8.4%, 77.9%, and 13.7%, respectively (calculated in 5930 patients and 8607 controls). 128
APOEε3/ε3 is the most common APOE genotype and is neutral concerning AD risk. 114 The APOE ε2 allele is considered to decrease AD risk and is associated with later AD onset ages. 129 The APOE ε4 allele frequency is drastically increased in AD patients to almost 40%, pinpointing a risk increasing effect of this isoform which is associated with earlier disease onset ages. 114 , 128 In comparison to mutations in APP, PSEN1, and PSEN2, the ε4 allele of APOE was neither considered necessary nor sufficient to cause the disease and was therefore categorized as a risk allele for AD. 5 Yet, dependent lifetime risk for AD (assessed in 7351 patients and 10,132 control individuals from Caucasian ancestry) was consistent with semi‐dominant inheritance of a moderately penetrant gene. 130 AD risk at age 85 ranged from 51% in male APOE ε4/ε4 carriers to 60% in female APOE ε4/ε4 carriers. 130 These results urge the consideration of APOE as a gene with semi‐dominant inheritance rather than a risk gene in the genetics of AD. 130
2.2. Familial AD genes including rare mutations with reduced penetrance
Common variants in the sortilin‐related receptor 1 gene (SORL1) were associated with AD for the first time in 2007 26 and several later studies attempted to replicate these findings with mixed results (e.g., Bettens et al. 27 and Li et al. 28 ) (Table 1). Ultimately, a meta‐analysis of 30,393 individuals in 2011 confirmed the association of common SORL1 variants with AD risk. 29 In 2013, genome‐wide association studies (GWAS) also pinpointed SORL1 as a risk gene for AD. 30 Since the association of common SORL1 variants with AD, rare variants in the gene have also received more attention. In 2012, whole‐exome sequencing (WES) in 14 unrelated EOAD probands of families with suggestive autosomal dominant inheritance, identified 5/14 carriers of SORL1 protein truncating variants, suggesting an important role for rare SORL1 variants in familial EOAD. 31 Unfortunately, the lack of DNA of affected family members did not allow segregation analysis of the SORL1 variants in these particular families. Multiple additional studies have described the association of rare SORL1 variants with AD risk. 32 , 33 , 34 Recently, a meta‐analysis of gene based burden association tests was performed, combining data of five studies, 32 , 33 , 35 , 36 , 37 including 9204 cases and 9646 controls of European ancestry. 38 Results showed that estimated AD risk was maximal for premature stop codon (PTC) mutations, which are in line with results of previously performed studies and suggestive for haploinsufficiency of SORL1 (e.g., Verheijen et al. 32 ). Of note, significant association of rare SORL1 PTC variants with AD risk was only reached after pooling data from five large independent studies. A recent WES study identified 19 PTC SORL1 variants among 6965 AD patients and only 1 in 13,252 control individuals of Caucasian ancestry. 39 In addition, SORL1 missense variants were shown to be enriched 1.5‐fold in EOAD patients compared to control individuals. 32 Two SORL1 missense mutations (p.R1303C and p.G1732A) and one splice site variant (c.3050‐2A>G) were shown to segregate with disease in autosomal dominant AD families. 40 SORL1 missense mutations, however, do have a non‐negligible frequency in control individuals, which can in part be explained by reduced penetrance of SORL1 missense variants. 32
Comparable, rare variants in the ATP‐binding cassette, sub‐family A, member 7 (ABCA7) gene are associated with increased AD risk (Table 1). Resequencing studies focusing on the >20 novel AD risk loci (GWAS), 41 , 42 , 43 , 44 , 45 identified an enrichment of rare ABCA7 variants in AD patients, confirming ABCA7 as an AD risk gene. Particularly, PTC variants (observed across the entire ABCA7 transcript) are enriched in AD patients, suggesting ABCA7 haploinsufficiency as the most plausible downstream mechanism. 46 So far, four autosomal dominant AD families have been identified in which rare ABCA7 (PTC and missense) variants co‐segregated with disease. 46 Importantly, the observation of a non‐negligible frequency of missense variants as well as the presence of PTC variants in control individuals, indicate variable penetrance of ABCA7 rare variants. 46 Expression levels of ABCA7 in the brain are highly variable between AD patients (also between PTC carriers). De Roeck et al. 42 observed incomplete nonsense‐mediated decay of ABCA7 transcripts harboring PTC variants. For some variants, the induced PTC could even be removed from the transcript by, for example, the use of cryptic splice sites. Yet, alternative splicing only explains variable expression levels in a handful of patients and is therefore not sufficient to fully explain reduced penetrance of ABCA7 variants in general. 42 Further research remains necessary to fully comprehend the impact of ABCA7 missense and PTC variants in AD pathology. 42
3. FROM FAMILIAL TO SPORADIC AD
There are rare heterozygous variants described to be involved in the genetic etiology of AD. Independent studies replicated many of these associations but seem to explain more isolated patients rather than autosomal dominant families. Most of these genes linked to AD by means of multiple statistical association studies in unrelated case control cohorts and co‐segregating with disease observed in one or two families. Additionally, an underlying recessive genetic aspect or maternal inheritance might in part explain seemingly sporadic AD patients.
3.1. Dominant AD
In 2013, Guerreiro et al. identified significantly more heterozygous variants in exon two of the triggering receptor expressed on myeloid cells 2 (TREM2) gene in Caucasian AD patients (n = 1092) compared to Caucasian control individuals (n = 1107) (Table 1). 47 In particular, the p.R47H rare variant showed the strongest association, which was confirmed by direct genotyping in an extended cohort of 1887 AD patients and 4061 control individuals (Caucasian) and meta‐analysis of three independent GWAS. 47 In the same year, the p.R47H association with AD was also reported in Icelandic, American, German, Dutch, and Norwegian study populations, 48 and was later also confirmed by subsequent studies. 49 , 50 , 51 , 52 , 53 , 54 In a large LOAD family (n = 21 affected individuals), p.R47H was found to co‐segregate completely with disease. 55 Other TREM2 rare variants that are significantly associated with AD were reported, for example, p.R62H, 56 , 57 p.H157Y (initially associated with AD in Han Chinese, 58 and further supported by additional meta‐analyses 52 , 59 ), p.D87N 47 (not replicated in Pottier et al. 53 ), and p.L211P (African‐American populations). 60 However, with the exception of p.R47H and p.R62H, the other associations need to be independently replicated in larger study cohorts. Several additional rare TREM2 variants are enriched in AD patients; however, statistical significance was not reached in single variant analysis. 47 , 48 , 51 , 57 This observation might in part be explained by the frequency of these variants and the investigated cohort sizes. Overall, rare variants in TREM2 are significantly enriched in AD patients (particularly in exon 2) and are indicated to cause a partial loss of TREM2 function. 47 , 61 Of note, the association of p.R47H was mainly observed in Caucasian populations and TREM2 variants in general were not associated with AD in Iranian, 62 Japanese, 63 and Korean 64 populations, hereby indicating that TREM2 influences are most likely population specific.
In the bridging integrator 1 gene (BIN1), the p.K358R rare variant was found to be significantly associated with LOAD in Caribbean Hispanics and segregated in 2/6 Caribbean Hispanic families in which the variant was identified (Table 1). 45 Another BIN1 rare variant, p.P318L, was significantly associated with LOAD in Han Chinese study cohorts including 1133 LOAD patients and 1159 control individuals; however, it was not identified in the study of Vardarajan et al. 66 Additionally, rare variants in BIN1 were associated with AD by an independent study. 66
Rare variants in the Unc‐5 homolog C (UNC5C) and A kinase anchor protein 9 (AKAP9) genes were also associated with AD 67 , 68 (Table 1). A rare UNC5C variant p.T835M, was found to segregate completely with disease in one LOAD family and was associated with disease in four large cohorts of LOAD patients (n = 8050) and control individuals (n = 98,194). 67 The specific p.T835M variant was found to cause increased cell death among different cell types including neurons. 67 Association of p.T835M with AD risk was not replicated in a study in Chinese AD patients (n = 360) and control individuals (n = 400); however, the investigated cohort might be too underpowered to identify significant association of ultra‐rare variants. 69 Of note, four novel variants were identified in exon 15 of UNC5C in the Chinese AD patient cohort. 69
Another study identified two rare AD associated AKAP9 variants (in tight linkage disequilibrium) in African‐American study populations (Table 1). 68 The study performed WES for seven African‐American familial AD patients and 44 rare variants were selected, based on frequency, sequencing quality, and relationship to previously implicated AD genes, for further investigation in 422 cases and 396 control individuals. 68 Two rare AKAP9 variants (rs149979685 and rs144662445) were associated with AD risk and this association was replicated in extended cohorts of 1037 cases and 1869 control individuals from African‐American descent. 68 Another AKAP9 rare variant, p.R434W, was found to segregate with LOAD in two large families. 66
Multiple studies also identified mutations in the notch receptor 3 (NOTCH3) gene in clinically diagnosed AD patients. To start, WES performed in a Turkish patient from a consanguineous AD family identified a rare heterozygous variant in NOTCH3, p.R1231C, as a potential culprit 70 (Table 1). Segregation analysis of the identified NOTCH3 variant p.R1231C was performed in additional family members and was identified in one unaffected at‐risk individual (younger than the reported age at onset in the family and therefore still uninformative). 70 NOTCH3 resequencing in 95 EOAD patients and 95 control individuals did not identify additional rare NOTCH3 variants. 70 Possible incomplete penetrance of the p.R1231C variant in the Turkish family and the complexity of the family (consanguineous) complicate interpretation of the pathogenic nature of this variant. 70 Another study investigated the role of adult‐onset leukodystrophy genes, including NOTCH3, in AD. 71 Adult‐onset leukodystrophies represent a spectrum of rare inherited progressive neurodegenerative disorders affecting the white matter of the central nervous system, which are often misdiagnosed with common sporadic dementing phenotypes. 71 Genetic screening of NOTCH3 was performed in 332 Caucasian AD patients and 676 Caucasian elderly control individuals. Gene‐based analysis was significant for NOTCH3 and this signal was driven by one common synonymous variant (p.P1521P) and three rare coding variants with large effect size (p.V1952M, p.V1183M, and p.H170R). Carrier frequency of the three rare coding variants was two to three times higher in LOAD patients compared to control individuals. 71 Importantly, all three variants had previously been significantly associated with severity of white matter lesions in elderly with hypertension. 72 The most recent study associating NOTCH3 rare variants with AD, applied a strategy focused on rare variants occurring only in cases to identify high penetrant rare variants, using data of the Alzheimer's Disease Sequencing Project (ADSP; 5617 AD patients and 4594 controls individuals). 73 Results identified one rare missense variant in NOTCH3 p.A284T that was present in 10 AD patients and absent from control individuals, and hereby provided the strongest link to date between NOTCH3 and AD. 73
3.2. Recessive AD
An underlying recessive genetic aspect could in part explain sporadic AD patients. Important differences between sporadic AD patients and seemingly isolated patients with underlying recessive AD are observable in the recurrence risk in parent–offspring. A study by Wingo et al. investigated parent–offspring concordance and concordance among siblings in an attempt to identify the likely mechanism of inheritance in the majority of non‐autosomal dominant EOAD cases. 6 A parent–offspring concordance of ≤10% and a concordance among siblings of 21.6% were estimated. These results indicate that autosomal recessive inheritance is the most likely mechanism of inheritance in these particular cases. 7
Two known dominant causal alleles, p.E693∆ and p.A713T in APP, and one p.E280A in PSEN1, were identified in homozygous state in one Japanese, 74 one Italian, 75 and one Columbian 76 AD family, respectively. In all three families, homozygous carriers, however, were not more severely affected with the disease. Another homozygous PSEN1 mutation p.A431E was observed in a 35‐year‐old male with early‐onset dementia, presenting with a relatively aggressive phenotype. 77 These observations demonstrated that homozygous variants in known AD genes are not lethal, as previously assumed. 76 Another homozygous APP variant, p.A673V, was found to cause disease only in homozygous state, whereas heterozygous carriers were unaffected. 78 The APP p.A673V was shown to have two pathogenic effects, shifting APP processing to the amyloidogenic pathway and increasing the aggregation property of the Aβ fibrils. 78 The effect of interaction between wild‐type and mutated alleles was investigated to unravel the seemingly protective effect of heterozygous p.A673V. Results showed that this interaction hinders amyloidogenesis and neurotoxicity, thereby protecting heterozygous carriers. 78 Another mutation at the same APP position, p.A673T, was reported in a patient without clinical signs of dementia and no deposition of Aβ plaques in the brain. 79 Yet, when the mutation was introduced in a synthetic Aβ peptide, the susceptibility to aggregate increased. 78
Both observations underline that benign heterozygous variants in known causal AD genes could harbor pathogenic effects in homozygous state. 78 Finally, bi‐allelic loss‐of‐function of SORL1 was described in one AD patient with maternal and paternal history of dementia in one study. The compound SORL1 variant carrier presented with an earlier onset age (55 years) than the parents, yet, the onset age is in the same range as other heterozygous SORL1 PTC variant carriers. 80 Bi‐allelic variants have also been described by single studies in two novel AD candidate genes: VWA2 and CTSF 81 , 82 (see supporting information). Replication of these findings in larger cohorts is necessary to fully comprehend their contribution to the etiology of AD. Additionally, genome‐wide linkage analysis performed in an extended recessive LOAD family identified a linked region on chromosome 8p22‐p21.2. 83 More than 50 genes are included in the linked region and therefore, further analysis of these genes is necessary (e.g., by means of whole‐genome sequencing in the LOAD family). 83
To identify additional novel loci harboring AD‐associated recessive variants, several studies investigated extended runs of homozygosity (ROHs) in case–control cohorts of different ethnic backgrounds. 84 , 85 , 86 , 87 , 88 , 89 One study performed ROH mapping in 837 LOAD and 550 controls from Northern European and Northern American ancestry and identified excess ROHs in cases versus controls, with the most significant LOAD associated ROH on chromosome 8p11.23. 84 Another study in 1955 AD cases and 955 control individuals with British/Irish ancestry failed to replicate the chromosome 8 finding. 86 The study did not identify an excess of ROHs in cases compared to controls and none of the ROHs showed significant association with AD. 86 Explanations for the discrepancy in results can be found in the study of McQuillan et al., who showed that the genomic location where ROHs could occur can differ significantly between different Caucasian populations. 86 , 88 Another study showed that the burden of ROHs was associated with AD, but the mean length of ROHs per person was significantly larger in AD patients compared to control individuals. 85 Results suggest that recessive risk loci exist in the Caribbean Hispanic population. 85 In addition, in African‐American cohorts including 1917 AD patients and 3858 control individuals, significantly more ROHs > 1, >2, and >3 Mb in AD patients versus control individuals were observed. 87 Smaller ROHs (>0.5 Mb) were also significantly associated with AD in this study population. In addition, in an isolated Arab community from Israel, specific AD‐associated ROHs was observed. 89 Overall, these results suggest that recessive AD risk loci are present in multiple ethnic subgroups; yet, replication studies are needed to prove their involvement in AD.
3.3. Maternal AD
Several studies reported a higher frequency of progressive dementia in mothers compared to fathers of AD patients, 90 , 91 , 92 suggestive for maternal inheritance. Even after correcting for the longer life expectancy of women, higher mother‐to‐father ratios have been observed in affected parents of AD patients. 91 Other studies, however, contradict these observations, with mothers of AD patients not often more affected compared to mothers of control individuals. 93 However, additional evidence for the presence of maternal transmission was found through brain imaging studies showing that maternal AD history predisposes to: reduced brain glucose metabolism, 94 increased atrophy in AD sensitive brain regions, 95 and smaller baseline hippocampi. 96 Additionally, having an AD‐affected mother was associated with poorer cognitive performance in later life and earlier ages at onset in the offspring compared to having an affected father. 97 The involvement of rare homoplasmic variants in mt‐DNA in AD has been suggested by several studies. Two rare variants in 12S rRNA (np 956‐965 and 856 A>G) have been described in AD patient cohorts. The np 956‐965 insertion (3‐4‐5 base pairs) was described in AD patients in three independent studies in European and Japanese cohorts and was absent from control individuals (Table 1). 98 , 99 , 100 Also, the 856 A>G variant was observed in two independent studies in AD patients but not in control individuals. 98 , 99 These studies suggest that variants in 12S rRNA might increase risk of developing AD.
4. SPORADIC AD
Particular genes are linked with AD through significant association in independent case control studies but never described as co‐segregating in families. Hence, these genes seem to be predominantly involved in the genetic etiology of sporadic patients unless there are families segregating extremely rare variants and yet not observed. In this review article, we categorize these genes as non‐familial AD genes. Also, de novo mutations and genetic modifiers have been associated with sporadic AD and are described in this section.
4.1. Non‐familial AD genes
Evidence was provided for a role of rare heterozygous variants in the clusterin (CLU) gene in AD 101 (Table 1). The CLU protein is a multifunctional protein showing striking similarities with the common risk gene APOE (Box 1), and CLU expression is increased in AD‐related brain regions (e.g., hippocampus and entorhinal cortex). Unbiased resequencing of all CLU coding exons in AD patients and control individuals from Flanders‐Belgium (n = 1930), identified 19 rare (MAF < 1%) to intermediate rare (MAF = 1% to 5%) non‐synonymous missense variants and one in‐frame 9 bp deletion p.T445‐D447del, carried by three AD patients. 101 Fourteen out of 19 missense variants were identified in 31 AD patients and eight variants were only in patients. Five variants, present only in controls, were identified in 21 control individuals, but all were labeled as benign by PolyPhen and SIFT prediction tools. 101 While the percentages of CLU rare variants were similar in AD and control cohorts, rare variants in AD patients clustered around exons 5 to 8, which are coding for the CLU β‐chain domain. 101 These observations were replicated in French and Canadian replication cohorts (n = 2755) and a meta‐analysis study including Portuguese, UK, and US Caucasian AD cohorts (n = 11,544). 101 , 102 , 103
In 2017, rare coding variants were identified in the Phospholipase C γ2 (PLCG2) and B3‐domain containing transcription factor ABI3 (ABI3) genes, showing genome‐wide significant association with AD in Caucasian study populations. 56 Results included one protective variant in PLCG2 (p.P522R) and one risk variant in ABI3 (p.S209F) 56 (Table 1). These findings were replicated in 2742 Caucasian AD cases, 3351 Caucasian controls, 181 African‐American AD cases, and 331 African‐American controls, genotyped for both variants. 104 Significant association of both the PLCG2 and ABI3 variants with AD was observed. The association of the p.P522R protective PLCG2 variant with AD was also replicated. 105 The frequencies of PLCG2 p.P522R and ABI3 p.S209F were also investigated in an Argentinian population (419 AD cases and 486 controls). 54 Both variants were observed in similar frequencies as reported by the International Genomics Alzheimer's Project (IGAP) 56 and both modulated susceptibility to AD in populations from Argentina. 54
4.2. De novo alleles
Somatic de novo variants are post‐zygotic variants, which may lead to (somatic and germline) mosaicism, that is, cells with genetic differences in one organism. 106 Pathogenic germline mosaic mutations have already been identified in the known AD genes. So far, 19 de novo PSEN1 mutations (absent from both parents, paternity assessed) with onset ages as early as 23 years old 107 and two de novo APP duplications have been described. 108 Beck et al. reported in 2004 the first confirmed evidence of both a somatic and germline mosaicism in a sporadic EOAD patient. 109 To this date, this is the only proven pathogenic somatic brain mutation ever identified in an AD patient. Nevertheless, involvement of somatic brain variants in AD was further investigated. Several studies described an enrichment of APP recombination and mutation (structural variations as well as SNVs). 110 , 111 However, the exact role of the observed APP recombinants will need further investigation. 110 Based on the current studies, one can conclude that somatic brain variations are not a common cause of sporadic AD, especially not in APP, PSEN1, and PSEN2. 112 Some factors might explain the absence of confirmation of the contribution of somatic variants. Neuropathological AD is characterized by loss of neurons and synapses, including neuronal DNA content. 5 Somatic variants, potentially underlying AD etiology, could be lost in advanced stages of the disease. Also, neuronal DNA is collected post mortem, potentially causing fragmentation of the DNA and complicating somatic variant detection. 106 Follow‐up studies are needed to obtain evidence of the recurrence of somatic brain variants and providing a definite link to AD etiology.
5. GENETIC MODIFIERS AND OLIGOGENIC INHERITANCE
Numerous novel alleles are associated with AD (Table 1). Yet, progress in understanding the effect of the genetic background on the penetrance and expressivity of causal and/or risk alleles is limited. 113 This limited knowledge on the impact of modifiers and oligogenic interactions can be partially explained by the methodological difficulty of identifying interactions of multiple genes on similar phenotypes. 113 Oligogenic interactions could shed light on the network and functioning of AD‐associated genes; however, so far, no conclusive results have been obtained to conclude they are involved in AD etiology.
Identification of modifier genes can act as potential novel therapeutic targets, as they might influence the phenotype of certain pathogenic variants. APOE is probably the most common AD onset‐age modifier (Box 1). 5 In 2019, the first rare modifying AD variant in APOE was described in a woman from the largest autosomal dominant Columbian AD kindred, segregating the pathogenic PSEN1 p.E280A mutation. 114 The mutation carriers are developing disease in their 40s, but this carrier showed the first signs at age 73. 114 WES identified two copies of the rare APOEε3 p.R136S variant (Christchurch 115 ). The woman presented with an unusually high Aβ brain load and limited tau and other NBD measurements. 114 Given its pronounced protective effect on disease onset, this variant could be of high value in development of novel therapeutic approaches.
6. LESSONS LEARNED FROM RARE VARIANTS IN THE GENETIC ETIOLOGY OF AD
In this review article, we focused on replicated rare variants and observed three main features in AD genetic research. First, there is a discrepancy in impact of the variants between ethnic groups. Examples include the TREM2 variants, with strong associations in, for example, Caucasian, African American, and Han Chinese populations and not in, for example, Iranian, Japanese, and Korean populations, suggesting population‐specific influences. Second, it is pivotal to evaluate pathogenicity of each of the variants independently in addition to gene‐based statistical tests taking into account all observed rare variants in one gene. Evidence can be found in the same TREM2 example in which patient‐specific variants were found to be enriched in exon 2. Also in APP, a pathogenic mutation hot spot was observed in exons 16 to 17. Third, the type of variation should also be taken into account. Good examples are SORL1 and ABCA7 in which PTC variants seem to be more penetrant compared to missense variants. Taken together, these observations underline that AD‐associated genes harbor pathogenic variants with variable penetrance as well as benign or even protective variants that cannot be otherwise classified in the absence of functional testing (variants of uncertain significance [VUS]), and that this effect is population specific. The observation of these effects complicates the interpretation of statistical analyses, especially in larger cohorts including individuals of different ethnicities. VUS are in general difficult to interpret. Even the causal AD genes harbor substantial proportions of VUS (>200 known pathogenic variants and more than 90 VUS identified in APP, PSEN1, and PSEN2 [≈45%] 116 ), with unknown contribution to disease onset. 117 Functional profiling of the identified VUS remains crucial to make a link to relevant disease processes. Carefully selected ethnicity‐matched study cohorts with availability of patient‐derived biomaterials are therefore of high value in AD genetic research, especially for ultra‐rare and rare variants that cannot be replicated in large independent studies but might have a clear functional association with AD pathology.
Identification of underlying genetic mechanisms in AD patients is often challenging, especially in seemingly sporadic AD patients. The presence of (ultra‐) rare mutations, de novo alleles, and mosaicisms often make it extra difficult to identify underlying inheritance. Yet, understanding this complexity of the genetic etiology of AD is of high importance, especially to estimate the recurrence risk in patient offspring.
The current knowledge of AD‐associated rare variants provides insights into the pitfalls and difficulties of AD genetic research. In addition, it provides important insights into the molecular pathways that are involved in early disease etiology. As AD is characterized by highly heterogeneous pathological processes, it is not convenient to distinguish disease‐causing pathways from downstream consequences. Genetic research provides evidence for the involvement of the immune system, lipid metabolism, and synaptic functioning in early AD etiology in addition to the Aβ pathway. Also, cytoskeleton function, axonal transport, regulation of gene expression, and post‐translational modification of proteins were found to be implicated in the etiology of AD. 118 Phagocytotic, apoptotic, and autophagic processes have also been associated with AD pathology. 119 , 120 , 121 , 122 , 123 Evidence was also provided for the early involvement of the alternatively proposed tau, dual Aβ‐tau, mitochondrial cascade‐ and neurovascular (blood–brain barrier integrity) pathways 124 (Table 1, Figure 1). Important is that the three AD genes also have biological functions unrelated to the Aβ pathway (Table 1). All these observations can help explain the high heterogeneous etiology of AD, in which apart from the Aβ pathway, other pathological pathways are involved in disease onset and progression and might deliver other potential therapeutic targets. Also, many other rare variants in different genes were associated with AD, which we did not discuss as they had little evidence due to limited number of studies (Table S1 in supporting information). Independent replication studies are essential to unravel the potential role of these rare variants and genes in the heterogeneous genetic etiology of AD.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The research in the authors’ group was funded in part by the Flemish Government–initiated Flanders Impulse Program on Networks for Dementia Research, the Flemish Government–initiated Methusalem Excellence Program, the Research Foundation Flanders (FWO), and the University of Antwerp Research Fund, Belgium. R. Cacace received a postdoctoral fellowship of the FWO.
Hoogmartens J, Cacace R, Van Broeckhoven C. Insight into the genetic etiology of Alzheimer's disease: A comprehensive review of the role of rare variants. Alzheimer's Dement. 2021;13:e12155 10.1002/dad2.12155
REFERENCES
- 1. Alzheimer's Disease International . World Alzheimer Report 2019: Attitudes to dementia. London: Alzheimer's Disease International; 2019.
- 2. Bettens K, Sleegers K, Van Broeckhoven C. Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum Mol Genet. 2010;19:R4‐R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van der Zee J, Sleegers K, Van Broeckhoven C. Invited article: the Alzheimer disease‐frontotemporal lobar degeneration spectrum. Neurology. 2008;71:1191‐1197. [DOI] [PubMed] [Google Scholar]
- 4. Rademakers R, Cruts M, Van Broeckhoven C. Genetics of early‐onset Alzheimer dementia. ScientificWorldJournal. 2003;3:497‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early‐onset Alzheimer's disease revisited. Alzheimers Dement. 2016;12:733‐748. [DOI] [PubMed] [Google Scholar]
- 6. Wingo TS, Lah JJ, Levey AI, Cutler DJ. Autosomal recessive causes likely in early‐onset Alzheimer disease. Arch Neurol. 2012;69:59‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alonso Vilatela ME, Lopez‐Lopez M, Yescas‐Gomez P. Genetics of Alzheimer's disease. Arch Med Res. 2012;43:622‐631. [DOI] [PubMed] [Google Scholar]
- 8. Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van Duijn CM, de Knijff P, Cruts M, et al. Apolipoprotein E4 allele in a population‐based study of early‐onset Alzheimer's disease. Nat Genet. 1994;7:74‐78. [DOI] [PubMed] [Google Scholar]
- 10. Makin S. The amyloid hypothesis on trial. Nature. 2018;559:S4. [DOI] [PubMed] [Google Scholar]
- 11. Chetelat G, La Joie R, Villain N, et al. Amyloid imaging in cognitively normal individuals, at‐risk populations and preclinical Alzheimer's disease. Neuroimage Clin. 2013;2:356‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Serrano‐Pozo A, Qian J, Monsell SE, et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann Neurol. 2014;75:597‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378:321‐330. [DOI] [PubMed] [Google Scholar]
- 14. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370:322‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rygiel K. Novel strategies for Alzheimer's disease treatment: an overview of anti‐amyloid beta monoclonal antibodies. Indian J Pharmacol. 2016;48:629‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Dyck CH. Anti‐amyloid‐beta monoclonal antibodies for Alzheimer's disease: pitfalls and promise. Biol Psychiatry. 2018;83:311‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giri M, Zhang M, Lu Y. Genes associated with Alzheimer's disease: an overview and current status. Clin Interv Aging. 2016;11:665‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dai MH, Zheng H, Zeng LD, Zhang Y. The genes associated with early‐onset Alzheimer's disease. Oncotarget. 2018;9:15132‐15143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sleegers K, Brouwers N, Gijselinck I, et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006;129:2977‐2983. [DOI] [PubMed] [Google Scholar]
- 20. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature. 2012;488:96‐99. [DOI] [PubMed] [Google Scholar]
- 22. Theuns J, Brouwers N, Engelborghs S, et al. Promoter mutations that increase amyloid precursor‐protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006;78:936‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cruts M, Theuns J, Van Broeckhoven C. Locus‐specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33:1340‐1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherrington R, Froelich S, Sorbi S, et al. Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996;5:985‐988. [DOI] [PubMed] [Google Scholar]
- 25. Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer's disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133:1143‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin‐related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bettens K, Brouwers N, Engelborghs S, De Deyn PP, Van Broeckhoven C, Sleegers K. SORL1 is genetically associated with increased risk for late‐onset Alzheimer disease in the Belgian population. Hum Mutat. 2008;29:769‐770. [DOI] [PubMed] [Google Scholar]
- 28. Li Y, Rowland C, Catanese J, et al. SORL1 variants and risk of late‐onset Alzheimer's disease. Neurobiol Dis. 2008;29:293‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reitz C, Cheng R, Rogaeva E, et al. Meta‐analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol. 2011;68:99‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pottier C, Hannequin D, Coutant S, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early‐onset Alzheimer disease. Mol Psychiatry. 2012;17:875‐879. [DOI] [PubMed] [Google Scholar]
- 32. Verheijen J, Van den Bossche T, van der Zee J, et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early‐onset Alzheimer's disease. Acta Neuropathol. 2016;132:213‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bellenguez C, Charbonnier C, Grenier‐Boley B, et al. Contribution to Alzheimer's disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging. 2017;59:220.e1‐e9. [DOI] [PubMed] [Google Scholar]
- 34. Nicolas G, Charbonnier C, Wallon D, et al. SORL1 rare variants: a major risk factor for familial early‐onset Alzheimer's disease. Mol Psychiatry. 2016;21:831‐836. [DOI] [PubMed] [Google Scholar]
- 35. Bis JC, Jian X, Kunkle BW, et al. Whole exome sequencing study identifies novel rare and common Alzheimer's‐associated variants involved in immune response and transcriptional regulation. Mol Psychiatry. 2020;25:1859–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holstege H, van der Lee SJ, Hulsman M, et al. Characterization of pathogenic SORL1 genetic variants for association with Alzheimer's disease: a clinical interpretation strategy. Eur J Hum Genet. 2017;25:973‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sassi C, Ridge PG, Nalls MA, et al. Influence of coding variability in APP‐Abeta metabolism genes in sporadic Alzheimer's disease. PLoS One. 2016;11:e0150079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Campion D, Charbonnier C, Nicolas G. SORL1 genetic variants and Alzheimer disease risk: a literature review and meta‐analysis of sequencing data. Acta Neuropathol. 2019;138:173‐186. [DOI] [PubMed] [Google Scholar]
- 39. Raghavan NS, Brickman AM, Andrews H, et al. Whole‐exome sequencing in 20,197 persons for rare variants in Alzheimer's disease. Ann Clin Transl Neurol. 2018;5:832‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thonberg H, Chiang HH, Lilius L, et al. Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol Commun. 2017;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol. 2015;14:814‐822. [DOI] [PubMed] [Google Scholar]
- 42. De Roeck A, Van den Bossche T, van der Zee J, et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer's disease. Acta Neuropathol. 2017;134:475‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Steinberg S, Stefansson H, Jonsson T, et al. Loss‐of‐function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet. 2015;47:445‐447. [DOI] [PubMed] [Google Scholar]
- 44. Le Guennec K, Nicolas G, Quenez O, et al. ABCA7 rare variants and Alzheimer disease risk. Neurology. 2016;86:2134‐2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome‐wide association studies loci. Ann Neurol. 2015;78:487‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. De Roeck A, Van Broeckhoven C, Sleegers K. The role of ABCA7 in Alzheimer's disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019;138:201‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lill CM, Rengmark A, Pihlstrom L, et al. The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement. 2015;11:1407‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu Y, Liu W, Wang X. TREM2 variants and risk of Alzheimer's disease: a meta‐analysis. Neurol Sci. 2015;36:1881‐1888. [DOI] [PubMed] [Google Scholar]
- 51. Cuyvers E, Bettens K, Philtjens S, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. 2014;35:726.e11‐9. [DOI] [PubMed] [Google Scholar]
- 52. Zhou SL, Tan CC, Hou XH, Cao XP, Tan L, Yu JT. TREM2 variants and neurodegenerative diseases: a systematic review and meta‐analysis. J Alzheimers Dis. 2019;68:1171‐1184. [DOI] [PubMed] [Google Scholar]
- 53. Pottier C, Wallon D, Rousseau S, et al. TREM2 R47H variant as a risk factor for early‐onset Alzheimer's disease. J Alzheimers Dis. 2013;35:45‐49. [DOI] [PubMed] [Google Scholar]
- 54. Dalmasso MC, Brusco LI, Olivar N, et al. Transethnic meta‐analysis of rare coding variants in PLCG2, ABI3, and TREM2 supports their general contribution to Alzheimer's disease. Transl Psychiatry. 2019;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Korvatska O, Leverenz JB, Jayadev S, et al. R47H variant of TREM2 associated with Alzheimer disease in a large late‐onset family: clinical, genetic, and neuropathological study. JAMA Neurol. 2015;72:920‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sims R, van der Lee SJ, Naj AC, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial‐mediated innate immunity in Alzheimer's disease. Nat Genet. 2017;49:1373‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jin SC, Benitez BA, Karch CM, et al. Coding variants in TREM2 increase risk for Alzheimer's disease. Hum Mol Genet. 2014;23:5838‐5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang T, Tan L, Chen Q, et al. A rare coding variant in TREM2 increases risk for Alzheimer's disease in Han Chinese. Neurobiol Aging. 2016;42:217.e1‐e3. [DOI] [PubMed] [Google Scholar]
- 59. Jiang T, Hou JK, Gao Q, et al. TREM2 p.H157Y variant and the risk of Alzheimer's disease: a meta‐analysis involving 14,510 subjects. Curr Neurovasc Res. 2016;13:318‐320. [DOI] [PubMed] [Google Scholar]
- 60. Jin SC, Carrasquillo MM, Benitez BA, et al. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener. 2015;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer's disease and other neurodegenerative disorders. Lancet Neurol. 2018;17:721‐730. [DOI] [PubMed] [Google Scholar]
- 62. Mehrjoo Z, Najmabadi A, Abedini SS, et al. Association study of the TREM2 gene and identification of a novel variant in exon 2 in Iranian patients with late‐onset Alzheimer's disease. Med Princ Pract. 2015;24:351‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miyashita A, Wen Y, Kitamura N, et al. Lack of genetic association between TREM2 and late‐onset Alzheimer's disease in a Japanese population. J Alzheimers Dis. 2014;41:1031‐1038. [DOI] [PubMed] [Google Scholar]
- 64. Chung SJ, Kim MJ, Kim J, et al. Exome array study did not identify novel variants in Alzheimer's disease. Neurobiol Aging. 2014;35:1958.e13‐4. [DOI] [PubMed] [Google Scholar]
- 65. Tan MS, Yu JT, Jiang T, Zhu XC, Guan HS, Tan L. Genetic variation in BIN1 gene and Alzheimer's disease risk in Han Chinese individuals. Neurobiol Aging. 2014;35:1781.e1‐e8. [DOI] [PubMed] [Google Scholar]
- 66. Vardarajan BN, Barral S, Jaworski J, et al. Whole genome sequencing of Caribbean Hispanic families with late‐onset Alzheimer's disease. Ann Clin Transl Neurol. 2018;5:406‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wetzel‐Smith MK, Hunkapiller J, Bhangale TR, et al. A rare mutation in UNC5C predisposes to late‐onset Alzheimer's disease and increases neuronal cell death. Nat Med. 2014;20:1452‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Logue MW, Schu M, Vardarajan BN, et al. Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans. Alzheimers Dement. 2014;10:609‐618.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiao B, Liu X, Tang B, et al. Investigation of TREM2, PLD3, and UNC5C variants in patients with Alzheimer's disease from mainland China. Neurobiol Aging. 2014;35:2422.e9‐e11. [DOI] [PubMed] [Google Scholar]
- 70. Guerreiro RJ, Lohmann E, Kinsella E, et al. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer's disease. Neurobiol Aging. 2012;33:1008.e17‐e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sassi C, Nalls MA, Ridge PG, et al. Mendelian adult‐onset leukodystrophy genes in Alzheimer's disease: critical influence of CSF1R and NOTCH3. Neurobiol Aging. 2018;66:179.e17‐e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schmidt H, Zeginigg M, Wiltgen M, et al. Genetic variants of the NOTCH3 gene in the elderly and magnetic resonance imaging correlates of age‐related cerebral small vessel disease. Brain. 2011;134:3384‐3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Patel D, Mez J, Vardarajan BN, et al. Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Netw Open. 2019;2:e191350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tomiyama T, Nagata T, Shimada H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer's‐type dementia. Ann Neurol. 2008;63:377‐387. [DOI] [PubMed] [Google Scholar]
- 75. Conidi ME, Bernardi L, Puccio G, et al. Homozygous carriers of APP A713T mutation in an autosomal dominant Alzheimer disease family. Neurology. 2015;84:2266‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kosik KS, Munoz C, Lopez L, et al. Homozygosity of the autosomal dominant Alzheimer disease presenilin 1 E280A mutation. Neurology. 2015;84:206‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Parker J, Mozaffar T, Messmore A, Deignan JL, Kimonis VE, Ringman JM. Homozygosity for the A431E mutation in PSEN1 presenting with a relatively aggressive phenotype. Neurosci Lett. 2019;699:195‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Di Fede G, Catania M, Morbin M, et al. A recessive mutation in the APP gene with dominant‐negative effect on amyloidogenesis. Science. 2009;323:1473‐1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Peacock ML, Warren JT Jr, Roses AD, Fink JK. Novel polymorphism in the A4 region of the amyloid precursor protein gene in a patient without Alzheimer's disease. Neurology. 1993;43:1254‐1256. [DOI] [PubMed] [Google Scholar]
- 80. Le Guennec K, Tubeuf H, Hannequin D, et al. Biallelic loss of function of SORL1 in an early onset Alzheimer's disease patient. J Alzheimers Dis. 2018;62:821‐831. [DOI] [PubMed] [Google Scholar]
- 81. Hoogmartens J, Hens E, Engelborghs S, et al. Contribution of homozygous and compound heterozygous missense mutations in VWA2 to Alzheimer's disease. Neurobiol Aging. 2020. [DOI] [PubMed] [Google Scholar]
- 82. Bras J, Djaldetti R, Alves AM, et al. Exome sequencing in a consanguineous family clinically diagnosed with early‐onset Alzheimer's disease identifies a homozygous CTSF mutation. Neurobiol Aging. 2016;46:236.e1‐e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Baron M, Gomez‐Tortosa E, Bochdanovits Z, et al. Extended kindred with recessive late‐onset Alzheimer disease maps to locus 8p22‐p21.2: a genome‐wide linkage analysis. Alzheimer Dis Assoc Disord. 2012;26:91‐95. [DOI] [PubMed] [Google Scholar]
- 84. Nalls MA, Guerreiro RJ, Simon‐Sanchez J, et al. Extended tracts of homozygosity identify novel candidate genes associated with late‐onset Alzheimer's disease. Neurogenetics. 2009;10:183‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ghani M, Sato C, Lee JH, et al. Evidence of recessive Alzheimer disease loci in a Caribbean Hispanic data set: genome‐wide survey of runs of homozygosity. JAMA Neurol. 2013;70:1261‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sims R, Dwyer S, Harold D, et al. No evidence that extended tracts of homozygosity are associated with Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:764‐771. [DOI] [PubMed] [Google Scholar]
- 87. Ghani M, Reitz C, Cheng R, et al. Association of long runs of homozygosity with Alzheimer disease among African American individuals. JAMA Neurol. 2015;72:1313‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. McQuillan R, Leutenegger AL, Abdel‐Rahman R, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sherva R, Baldwin CT, Inzelberg R, et al. Identification of novel candidate genes for Alzheimer's disease by autozygosity mapping using genome wide SNP data. J Alzheimers Dis. 2011;23:349‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Duara R, Lopez‐Alberola RF, Barker WW, et al. A comparison of familial and sporadic Alzheimer's disease. Neurology. 1993;43:1377‐1384. [DOI] [PubMed] [Google Scholar]
- 91. Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer's disease cases: evidence for maternal inheritance. Neurology. 1996;47:254‐256. [DOI] [PubMed] [Google Scholar]
- 92. Gomez‐Tortosa E, Barquero MS, Baron M, et al. Variability of age at onset in siblings with familial Alzheimer disease. Arch Neurol. 2007;64:1743‐1748. [DOI] [PubMed] [Google Scholar]
- 93. Heggeli KA, Crook J, Thomas C, Graff‐Radford N. Maternal transmission of Alzheimer disease. Alzheimer Dis Assoc Disord. 2012;26:364‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mosconi L, Brys M, Switalski R, et al. Maternal family history of Alzheimer's disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A. 2007;104:19067‐19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Honea RA, Swerdlow RH, Vidoni ED, Burns JM. Progressive regional atrophy in normal adults with a maternal history of Alzheimer disease. Neurology. 2011;76:822‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Andrawis JP, Hwang KS, Green AE, et al. Effects of ApoE4 and maternal history of dementia on hippocampal atrophy. Neurobiol Aging. 2012;33:856‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Seshadri S, Wolf PA, Au R, McNulty K, White R, DAgostino RB. Lifetime risk of dementia and Alzheimer's disease: estimates from the Framingham study. Neurology. 1996;46:55002. [DOI] [PubMed] [Google Scholar]
- 98. Tanaka N, Goto Y, Akanuma J, et al. Mitochondrial DNA variants in a Japanese population of patients with Alzheimer's disease. Mitochondrion. 2010;10:32‐37. [DOI] [PubMed] [Google Scholar]
- 99. Tanno Y, Okuizumi K, Tsuji S. mtDNA polymorphisms in Japanese sporadic Alzheimer's disease. Neurobiol Aging. 1998;19:S47‐S51. [DOI] [PubMed] [Google Scholar]
- 100. Shoffner JM, Brown MD, Torroni A, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics. 1993;17:171‐184. [DOI] [PubMed] [Google Scholar]
- 101. Bettens K, Brouwers N, Engelborghs S, et al. Both common variations and rare non‐synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol Neurodegener. 2012;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Guerreiro RJ, Beck J, Gibbs JR, et al. Genetic variability in CLU and its association with Alzheimer's disease. PLoS One. 2010;5:e9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tycko B, Feng L, Nguyen L, et al. Polymorphisms in the human apolipoprotein‐J/clusterin gene: ethnic variation and distribution in Alzheimer's disease. Hum Genet. 1996;98:430‐436. [DOI] [PubMed] [Google Scholar]
- 104. Conway OJ, Carrasquillo MM, Wang X, et al. ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol Neurodegener. 2018;13:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. van der Lee SJ, Conway OJ, Jansen I, et al. A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer's disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. 2019;138:237‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Leija‐Salazar M, Piette C, Proukakis C. Review: somatic mutations in neurodegeneration. Neuropathol Appl Neurobiol. 2018;44:267‐285. [DOI] [PubMed] [Google Scholar]
- 107. Lou F, Luo X, Li M, Ren Y, He Z. Very early‐onset sporadic Alzheimer's disease with a de novo mutation in the PSEN1 gene. Neurobiol Aging. 2017;53:193.e1‐e5. [DOI] [PubMed] [Google Scholar]
- 108. Rovelet‐Lecrux A, Charbonnier C, Wallon D, et al. De novo deleterious genetic variations target a biological network centered on Abeta peptide in early‐onset Alzheimer disease. Mol Psychiatry. 2015;20:1046‐1056. [DOI] [PubMed] [Google Scholar]
- 109. Beck JA, Poulter M, Campbell TA, et al. Somatic and germline mosaicism in sporadic early‐onset Alzheimer's disease. Hum Mol Genet. 2004;13:1219‐1224. [DOI] [PubMed] [Google Scholar]
- 110. Lee MH, Siddoway B, Kaeser GE, et al. Somatic APP gene recombination in Alzheimer's disease and normal neurons. Nature. 2018;563:639‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bushman DM, Kaeser GE, Siddoway B, et al. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer's disease brains. Elife. 2015;4:e05116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Nicolas G, Acuna‐Hidalgo R, Keogh MJ, et al. Somatic variants in autosomal dominant genes are a rare cause of sporadic Alzheimer's disease. Alzheimers Dement. 2018;14:1632‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kousi M, Katsanis N. Genetic modifiers and oligogenic inheritance. Cold Spring Harb Perspect Med. 2015;5:a017145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Arboleda‐Velasquez JF, Lopera F, O'Hare M, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wardell MR, Brennan SO, Janus ED, Fraser R, Carrell RW. Apolipoprotein E2‐Christchurch (136 Arg—‐Ser). New variant of human apolipoprotein E in a patient with type III hyperlipoproteinemia. J Clin Invest. 1987;80:483‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hsu S, Pimenova AA, Hayes K, et al. Systematic validation of variants of unknown significance in APP, PSEN1 and PSEN2. Neurobiol Dis. 2020;139:104817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Perrone F, Cacace R, Van Mossevelde S, et al. Genetic screening in early‐onset dementia patients with unclear phenotype: relevance for clinical diagnosis. Neurobiol Aging. 2018;69:292.e7‐e14. [DOI] [PubMed] [Google Scholar]
- 118. Cuyvers E, Sleegers K. Genetic variations underlying Alzheimer's disease: evidence from genome‐wide association studies and beyond. Lancet Neurol. 2016;15:857‐868. [DOI] [PubMed] [Google Scholar]
- 119. Sweeney MD, Sagare AP, Zlokovic BV. Blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sokolowski JD, Mandell JW. Phagocytic clearance in neurodegeneration. Am J Pathol. 2011;178:1416‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Uddin MS, Stachowiak A, Mamun AA, et al. Autophagy and Alzheimer's disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. 2018;10:04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sole‐Domenech S, Cruz DL, Capetillo‐Zarate E, Maxfield FR. The endocytic pathway in microglia during health, aging and Alzheimer's disease. Ageing Res Rev. 2016;32:89‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liu PP, Xie Y, Meng XY, Kang JS. History and progress of hypotheses and clinical trials for Alzheimer's disease. Signal Transduct Target Ther. 2019;4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pericak‐Vance MA, Bebout JL. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034‐1050. [PMC free article] [PubMed] [Google Scholar]
- 126. Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late‐onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467‐1472. [DOI] [PubMed] [Google Scholar]
- 127. Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high‐avidity binding to beta‐amyloid and increased frequency of type 4 allele in late‐onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977‐1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 129. Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180‐184. [DOI] [PubMed] [Google Scholar]
- 130. Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major gene with semi‐dominant inheritance. Mol Psychiatry. 2011;16:903‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Welikovitch LA, Do Carmo S, Magloczky Z, et al. Early intraneuronal amyloid triggers neuron‐derived inflammatory signaling in APP transgenic rats and human brain. Proc Natl Acad Sci U S A. 2020;117:6844‐6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Montagne A, Zhao Z, Zlokovic BV. Alzheimer's disease: a matter of blood‐brain barrier dysfunction? J Exp Med. 2017;214:3151‐3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hick M, Herrmann U, Weyer SW, et al. Acute function of secreted amyloid precursor protein fragment APPsalpha in synaptic plasticity. Acta Neuropathol. 2015;129:21‐37. [DOI] [PubMed] [Google Scholar]
- 134. Wu Y, Zhang S, Xu Q, et al. Regulation of global gene expression and cell proliferation by. APP Sci Rep. 2016;6:22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Muller T, Meyer HE, Egensperger R, Marcus K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics‐relevance for Alzheimer's disease. Prog Neurobiol. 2008;85:393‐406. [DOI] [PubMed] [Google Scholar]
- 136. Rama N, Goldschneider D, Corset V, Lambert J, Pays L, Mehlen P. Amyloid precursor protein regulates netrin‐1‐mediated commissural axon outgrowth. J Biol Chem. 2012;287:30014‐30023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282‐1288. [DOI] [PubMed] [Google Scholar]
- 138. Cheng N, Jiao S, Gumaste A, Bai L, Belluscio L. APP overexpression causes Abeta‐independent neuronal death through intrinsic apoptosis pathway. eNeuro. 2016;3:ENEURO.0150-16.2016 10.1523/ENEURO.0150-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J. Reduced beta‐amyloid production and increased inflammatory responses in presenilin conditional knock‐out mice. J Biol Chem. 2004;279:46907‐46914. [DOI] [PubMed] [Google Scholar]
- 140. Saura CA, Choi SY, Beglopoulos V, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age‐dependent neurodegeneration. Neuron. 2004;42:23‐36. [DOI] [PubMed] [Google Scholar]
- 141. Wang Y, Greig NH, Yu QS, Mattson MP. Presenilin‐1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol Aging. 2009;30:1061‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lazarov O, Morfini GA, Pigino G, et al. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease‐linked mutant presenilin 1. J Neurosci. 2007;27:7011‐7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wolozin B, Alexander P, Palacino J. Regulation of apoptosis by presenilin 1. Neurobiol Aging. 1998;19:S23‐7. [DOI] [PubMed] [Google Scholar]
- 144. Neely KM, Green KN, LaFerla FM. Presenilin is necessary for efficient proteolysis through the autophagy‐lysosome system in a gamma‐secretase‐independent manner. J Neurosci. 2011;31:2781‐2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Yoon SY, Kim DH. Alzheimer's disease genes and autophagy. Brain Res. 2016;1649:201‐209. [DOI] [PubMed] [Google Scholar]
- 146. Sala Frigerio C, Lau P, Troakes C, et al. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer's disease patients. Alzheimers Dement. 2015;11:1265‐1276. [DOI] [PubMed] [Google Scholar]
- 147. Araki W, Yuasa K, Takeda S, et al. Pro‐apoptotic effect of presenilin 2 (PS2) overexpression is associated with down‐regulation of Bcl‐2 in cultured neurons. J Neurochem. 2001;79:1161‐1168. [DOI] [PubMed] [Google Scholar]
- 148. Gamliel A, Teicher C, Hartmann T, Beyreuther K, Stein R. Overexpression of wild‐type presenilin 2 or its familial Alzheimer's disease‐associated mutant does not induce or increase susceptibility to apoptosis in different cell lines. Neuroscience. 2003;117:19‐28. [DOI] [PubMed] [Google Scholar]
- 149. Cuccaro ML, Carney RM, Zhang Y, et al. SORL1 mutations in early‐ and late‐onset Alzheimer disease. Neurol Genet. 2016;2:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yin RH, Yu JT, Tan L. The role of SORL1 in Alzheimer's disease. Mol Neurobiol. 2015;51:909‐918. [DOI] [PubMed] [Google Scholar]
- 151. Satoh K, Abe‐Dohmae S, Yokoyama S, St George‐Hyslop P, Fraser PE. ATP‐binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J Biol Chem. 2015;290:24152‐24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Abe‐Dohmae S, Ikeda Y, Matsuo M, et al. Human ABCA7 supports apolipoprotein‐mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J Biol Chem. 2004;279:604‐611. [DOI] [PubMed] [Google Scholar]
- 154. Iwamoto N, Abe‐Dohmae S, Sato R, Yokoyama S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J Lipid Res. 2006;47:1915‐1927. [DOI] [PubMed] [Google Scholar]
- 155. Tanaka N, Abe‐Dohmae S, Iwamoto N, Fitzgerald ML, Yokoyama S. Helical apolipoproteins of high‐density lipoprotein enhance phagocytosis by stabilizing ATP‐binding cassette transporter A7. J Lipid Res. 2010;51:2591‐2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Jehle AW, Gardai SJ, Li S, et al. ATP‐binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol. 2006;174:547‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Karanfilian L, Tosto MG, Malki K. The role of TREM2 in Alzheimer's disease; evidence from transgenic mouse models. Neurobiol Aging. 2020;86:39‐53. [DOI] [PubMed] [Google Scholar]
- 158. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid‐beta by microglia. Neuron. 2016;91:328‐340. [DOI] [PubMed] [Google Scholar]
- 159. Kleinberger G, Yamanishi Y, Suarez‐Calvet M, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:243ra86. [DOI] [PubMed] [Google Scholar]
- 160. Miyagawa T, Ebinuma I, Morohashi Y, et al. BIN1 regulates BACE1 intracellular trafficking and amyloid‐beta production. Hum Mol Genet. 2016;25:2948‐2958. [DOI] [PubMed] [Google Scholar]
- 161. Thomas S, Hoxha K, Tran A, Prendergast GC. Bin1 antibody lowers the expression of phosphorylated tau in Alzheimer's disease. J Cell Biochem. 2019;120:18320‐18331. [DOI] [PubMed] [Google Scholar]
- 162. Schurmann B, Bermingham DP, Kopeikina KJ, et al. A novel role for the late‐onset Alzheimer's disease (LOAD)‐associated protein Bin1 in regulating postsynaptic trafficking and glutamatergic signaling. Mol Psychiatry. 2020;25:2000‐2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Nanda SK, Herion D, Liang TJ. The SH3 binding motif of HCV [corrected] NS5A protein interacts with Bin1 and is important for apoptosis and infectivity. Gastroenterology. 2006;130:794‐809. [DOI] [PubMed] [Google Scholar]
- 164. Galderisi U, Di Bernardo G, Cipollaro M, et al. Induction of apoptosis and differentiation in neuroblastoma and astrocytoma cells by the overexpression of Bin1, a novel Myc interacting protein. J Cell Biochem. 1999;74:313‐322. [PubMed] [Google Scholar]
- 165. Del‐Aguila JL, Koboldt DC, Black K, et al. Alzheimer's disease: rare variants with large effect sizes. Current Opinion in Genetics & Development. 2015;33:49‐55. [DOI] [PubMed] [Google Scholar]
- 166. Durrant CS, Ruscher K, Sheppard O, Coleman MP, Ozen I. Beta secretase 1‐dependent amyloid precursor protein processing promotes excessive vascular sprouting through NOTCH3 signalling. Cell Death Dis. 2020;11:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Peters N, Opherk C, Zacherle S, Capell A, Gempel P, Dichgans M. CADASIL‐associated Notch3 mutations have differential effects both on ligand binding and ligand‐induced Notch3 receptor signaling through RBP‐Jk. Exp Cell Res. 2004;299:454‐464. [DOI] [PubMed] [Google Scholar]
- 168. Wang Y, Pan L, Moens CB, Appel B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development. 2014;141:307‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zweidler‐McKay PA, He Y, Xu L, et al. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B‐cell malignancies. Blood. 2005;106:3898‐3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wang T, Baron M, Trump D. An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol. 2008;96:499‐509. [DOI] [PubMed] [Google Scholar]
- 171. Phillips NR, Simpkins JW, Roby RK. Mitochondrial DNA deletions in Alzheimer's brains: a review. Alzheimers Dement. 2014;10:393‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. DeMattos RB, Cirrito JR, Parsadanian M, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193‐202. [DOI] [PubMed] [Google Scholar]
- 173. Kirszbaum L, Bozas SE, Walker ID. SP‐40,40, a protein involved in the control of the complement pathway, possesses a unique array of disulphide bridges. FEBS Lett. 1992;297:70‐76. [DOI] [PubMed] [Google Scholar]
- 174. Jenne DE, Lowin B, Peitsch MC, Bottcher A, Schmitz G, Tschopp J. Clusterin (complement lysis inhibitor) forms a high density lipoprotein complex with apolipoprotein A‐I in human plasma. J Biol Chem. 1991;266:11030‐11036. [PubMed] [Google Scholar]
- 175. Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang CY. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol. 2005;7:909‐915. [DOI] [PubMed] [Google Scholar]
- 176. Zhang B, Wu Q, Ye XF, Liu S, Lin XF, Chen MC. Roles of PLC‐gamma2 and PKCalpha in TPA‐induced apoptosis of gastric cancer cells. World J Gastroenterol. 2003;9:2413‐2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Magno L, Lessard CB, Martins M, et al. Alzheimer's disease phospholipase C‐gamma‐2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 2019;11:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform‐dependent manner in an in vitro blood‐brain barrier model. J Biol Chem. 2011;286:17536‐17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Yin C, Guo ZD, He ZZ, Wang ZY, Sun XC. Apolipoprotein E affects in vitro axonal growth and regeneration via the MAPK signaling pathway. Cell Transplant. 2019;28:691‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Fleming LM, Weisgraber KH, Strittmatter WJ, Troncoso JC, Johnson GV. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp Neurol. 1996;138:252‐260. [DOI] [PubMed] [Google Scholar]
- 182. Wadhwani AR, Affaneh A, Van Gulden S, Kessler JA. Neuronal apolipoprotein E4 increases cell death and phosphorylated tau release in alzheimer disease. Ann Neurol. 2019;85:726‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid‐ and receptor‐binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102:18694‐18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. DeKroon RM, Mihovilovic M, Goodger ZV, et al. ApoE genotype‐specific inhibition of apoptosis. J Lipid Res. 2003;44:1566‐1573. [DOI] [PubMed] [Google Scholar]
- 185. Muth C, Hartmann A, Sepulveda‐Falla D, Glatzel M, Krasemann S. Phagocytosis of apoptotic cells is specifically upregulated in ApoE4 expressing microglia in vitro. Front Cell Neurosci. 2019;13:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Simonovitch S, Schmukler E, Bespalko A, et al. Impaired autophagy in APOE4 astrocytes. J Alzheimers Dis. 2016;51:915‐927. [DOI] [PubMed] [Google Scholar]
- 187. Park JS, Lee J, Jung ES, et al. Brain somatic mutations observed in Alzheimer's disease associated with aging and dysregulation of tau phosphorylation. Nat Commun. 2019;10:3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information