

Abstract
Understanding the processes shaping population structure and reproductive isolation of marine organisms can improve their management and conservation. Using genomic markers combined with estimation of individual ancestries, assignment tests, spatial ecology, and demographic modeling, we (i) characterized the contemporary population structure, (ii) assessed the influence of space, fishing depth, and sampling years on contemporary distribution, and (iii) reconstructed the speciation history of two cryptic redfish species, Sebastes mentella and S. fasciatus. We genotyped 860 individuals in the Northwest Atlantic Ocean using 24,603 filtered single nucleotide polymorphisms (SNPs). Our results confirmed the clear genetic distinctiveness of the two species and identified three ecotypes within S. mentella and five populations in S. fasciatus. Multivariate analyses highlighted the influence of spatial distribution and depth on the overall genomic variation, while demographic modeling revealed that secondary contact models best explained inter‐ and intragenomic divergence. These species, ecotypes, and populations can be considered as a rare and wide continuum of genomic divergence in the marine environment. This acquired knowledge pertaining to the evolutionary processes driving population divergence and reproductive isolation will help optimizing the assessment of demographic units and possibly to refine fishery management units.
Keywords: demographic models, fishery management, population genomics, related species, Sebastes, spatial ecology
1. INTRODUCTION
Delineating sustainable management boundaries through the characterization of genomic variation among and within species can provide an indication of reproductive isolation and ultimately help to reduce the risk of depletion of marine resources (Laikre et al., 2009; Ovenden et al., 2015; Reiss et al., 2009). Yet, this represents a challenging endeavor since it reflects both contemporary and historical processes (Waples and Gaggiotti, 2006). It has now become feasible to document these two processes occurring at different timescales by combining population genomics, spatial ecology, and demographic model approaches. Together, these three approaches can identify how species, ecotypes, and populations diverged through space and time, which ultimately lead to more suitable recommendations for the management and conservation of commercially valuable stocks. Population genomics allows detecting and separating locus specific from genome‐wide effects to improve our understanding of evolution (Black et al., 2001). Population genomics information can then be used in spatial ecology to quantify the drivers that may explain genomic distance between individuals from different areas, by using multivariate methods to describe the association between explanatory (i.e., environmental variables) and response (i.e., genomic distances) variables (summarized in Riginos et al., 2016). Both population genomics and spatial ecology have their limitations, but combining these approaches allows these potential weaknesses to be better addressed and reinforces the interpretation of the observed marine connectivity scheme. Furthermore, coupling population genomics and spatial ecology approaches with information about the demographic history of species and populations can deliver a broader portrait by delineating the moment and the geographic conditions (i.e., allopatry vs sympatry) under which the genetic clusters identified in the population genomics approach formed.
Genetic clusters can be formed following habitat fragmentation and climatic/environmental change, for instance as it occurred during the Quaternary period (Hewitt, 2000; Rougeux et al., 2017). This period characterized by change in important environmental driver has shaped the connectivity of populations and the dynamic distribution of marine species in the North Atlantic (Maggs et al., 2008). Demographic modeling is fundamental toward determining whether species diverge in the face of gene flow under the action of divergent ecological selection alone (i.e., ecological speciation, Rundle and Nosil, 2005) or if an initial isolation period precedes subsequent divergence (Endler, 1977; Roux et al., 2016). Methods, either using coalescent simulations (Roux et al., 2014; Roux et al., 2016) or solving the site frequency spectrum through a diffusion approach (Gutenkunst et al., 2009), are suitable to test for these scenarios and also to model local effects in the genome, which can improve demographic inferences (Cruickshank and Hahn, 2014; Ewing and Jensen, 2016; Rougeux et al., 2017; Roux et al., 2014; Schrider et al., 2016; more details provided in the methods).
With more than 110 closely related species worldwide (Hyde and Vetter, 2007), the marine genus Sebastes has attracted the attention of evolutionary biologists that aim to study their speciation history (Johns and Avise, 1998). Redfish are ovoviviparous (i.e., internal fertilization), resulting in lecithotrophic larvae feeding exclusively on the yolk of the egg (Johns and Avise, 1998). In the North Atlantic Ocean, Sebastes mentella Travin 1951 and S. fasciatus Storer 1954 are two well‐suited species to study speciation that have overlapping (i.e., sympatric) and nonoverlapping (i.e., allopatric) distributions. Both species co‐occur in the Northwest Atlantic and inhabit at different depth intervals (Atkinson, 1987). The barrier to gene flow among Sebastes spp. in the North Atlantic Ocean is partially porous. In the Gulf of St. Lawrence (GSL) where S. mentella and S. fasciatus live in sympatry, S. mentella releases its larvae approximately three to four weeks earlier than S. fasciatus (Sévigny et al., 2000). Yet, evidence of introgression between S. mentella and S. fasciatus is present across their overlapping distribution but also between S. mentella and S. norvegicus or S. viviparous in the Northeast Arctic (Roques et al., 2002; Saha et al., 2017). Both S. mentella and S. fasciatus also show some intraspecific population structure.
For S. mentella, two ecotypes have been characterized across the North Atlantic Ocean, namely S. mentella shallow and S. mentella deep, which inhabit specific depths between 300 and 500 m and greater than 500 m, respectively (Cadrin et al., 2010; Saha et al., 2017; Valentin et al., 2014). Environmental gradients related to depth were suggested to play a role in the diversification of S. mentella ecotypes (Shum et al., 2014). S. fasciatus inhabits depths between 100 and 400 m and was previously characterized with weak population structure in the Northwest Atlantic using 13 microsatellite loci (Valentin et al., 2014). Along the Canadian coast, four populations were possibly present, namely (1) Gulf of Maine, (2) Gulf of St. Lawrence—Laurentian Channel, (3) Bonne Bay, and (4) Grand Banks—northward areas.
Redfish populations in the Northwest Atlantic have been extensively fished since the late 1950, with the fishery initially managed as three stocks established by NAFO (Northwest Atlantic Fisheries Organization). In 1993, this management plan was redefined based on a stronger biological basis, resulting in six Canadian management units (i.e., Unit 1, Unit 2, Unit 3, and NAFO divisions 2 + 3K, 3LN, and 3O, see Figure 1). A rapid decrease in landings since 1993 led to a moratorium on fishing in Unit 1 starting in 1995. In 2010, the Committee on the Status of Endangered Wildlife in Canada (COSEWIC) designated Unit 1 S. mentella as endangered and S. fasciatus as threatened. Recently, however, the abundance of Sebastes spp. juveniles located in the northern GSL was reported to be 230 times and 4 times higher in 2017 than their respective average abundances for the period 1993‒2012 (Senay et al., 2019). The stronger recruitment levels indicating new cohorts in the GSL may be a precursor to stock recovery and present hope for a potential future re‐opening of the redfish fishery.
FIGURE 1.
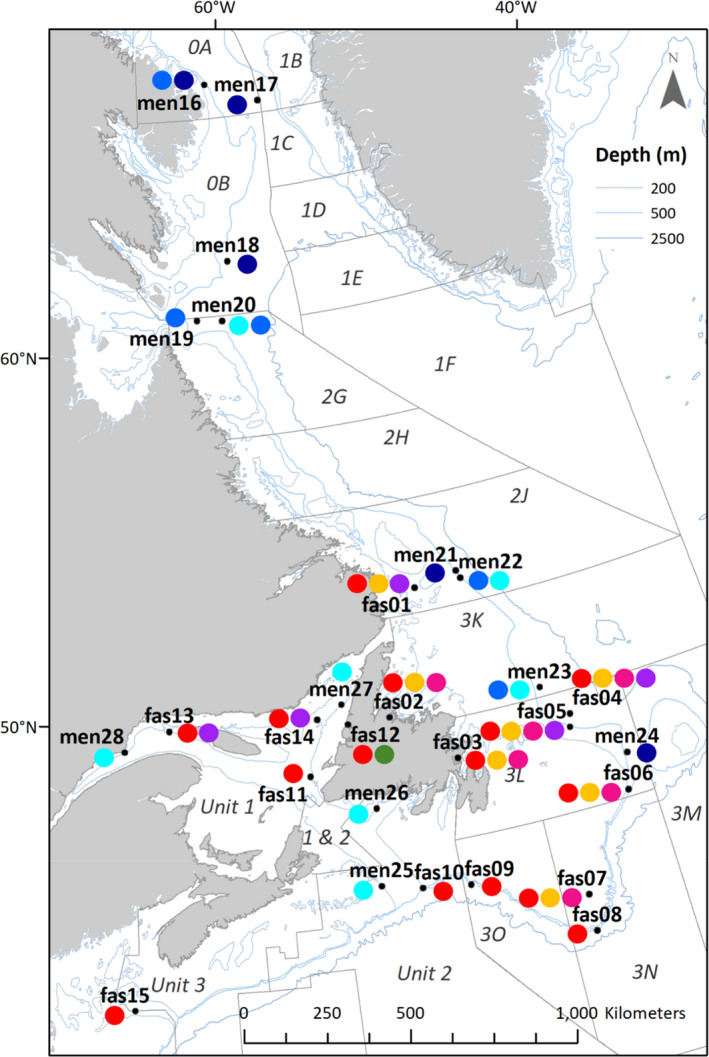
Map of the 28 sampling sites of Sebastes mentella and S. fasciatus along the Northwest Atlantic. Delineation of the current management units specific to redfish species (i.e., Units 1 to 3) and NAFO divisions are presented. Each sampling site is represented by a black point, and adjacent colored circles represent genetic clusters detected at this site. A genetic cluster was indicated as detected when at least an individual shows at least 50% of ancestry to this genetic cluster. There are three ecotypes described in S. mentella: GSL (cyan), shallow (blue), and deep (dark blue). The unknown S. mentella genetic cluster is identified in light gray. There are five populations observed in S. fasciatus: I (purple), II (orange), III (red), IV (green), and IV (pink)
While population genetic structure in S. mentella and S. fasciatus has been intensively investigated using microsatellites, no study has yet used a genome‐wide dataset to assess population genomic structure, spatial ecology, and demographic history to infer how contemporaneous and historical three‐dimensional geographic drivers played a role in the formation of distinct genetic clusters, at the species, ecotype, and population levels. Here, our aim was to genotype S. mentella and S. fasciatus individuals sampled at different depths using genotype by sequencing (GBS) to (i) delineate the contemporary population genomic structure in Sebastes spp. located in the Northwest Atlantic Ocean, including the recent GSL cohorts observed, (ii) assess how space, fishing depth, and sampling years explain the contemporary distribution of species, ecotypes, and populations, and (iii) characterize the demographic history of speciation.
2. METHODS
2.1. Sampling and molecular techniques
From 2001 to 2015, we collected a total of 860 Sebastes spp. individuals, captured from 28 sampling sites in the Northwest Atlantic Ocean (Table 1 and Figure 1). Geographic coordinates of sampling sites are average latitude and longitude of sampling locations of all individuals considered in each site (see Table S1 for sampling location of each individual). Each site was sampled in only one year. For most sampling sites, adults were sampled except in the GSL and adjacent areas where juveniles of the new cohort were sampled (Figure S1). Individuals used for genotyping were selected based on their geometric morphometrics or their population classification using the 13 microsatellites developed by Valentin et al. (2014). Only individuals that were accurately identified at the species level and S. mentella ecotype level (i.e., shallow and deep only) were included in this study (Table 1 for detailed information). This sampling strategy consequently reduced the proportion of admixed individuals between the species and the ecotype in this study. This sampling bias is considered in the interpretation of the results. Genomic DNA was extracted using the DNA tissue kit protocol from Omega Bio‐tek. DNA quality and quantity were checked on 1% agarose gels. Libraries were prepared based on a GBS protocol modified from Mascher et al. (2013) using PstI and MspI (details in Perreault‐Payette et al., 2017). Individuals were barcoded with unique sequences and pooled in multiplexes of 48 individuals per library. Libraries were then sequenced on Ion Torrent Proton P1v2 chips.
TABLE 1.
Sampling sites analyzed in this study with year sampled, latitude, and longitude information as well as sampling depth
| Code | Year | Longitude | Latitude | Ngen | Depth | Genetic group |
|---|---|---|---|---|---|---|
| fas01 | 2014 | −54.07 | 52.93 | 29 | 387 | Fasciatus |
| fas02 | 2015 | −56.17 | 49.58 | 34 | NA | Fasciatus |
| fas03 | 2015 | −53.72 | 48.16 | 35 | NA | Fasciatus |
| fas04 | 2014 | −48.86 | 48.6 | 28 | 663 | Fasciatus |
| fas05 | 2014 | −49.01 | 48.25 | 35 | 383 | Fasciatus |
| fas06 | 2014 | −47.47 | 46.2 | 35 | 292 | Fasciatus |
| fas07 | 2015 | −50 | 43.76 | 30 | 253 | Fasciatus |
| fas08 | 2015 | −50.05 | 42.76 | 19 | 629 | Fasciatus |
| fas09 | 2015 | −54.2 | 44.73 | 23 | 306 | Fasciatus |
| fas10 | 2002 | −56.03 | 44.87 | 32 | 448 | Fasciatus |
| fas11 | 2002 | −59.74 | 48.3 | 32 | 248 | Fasciatus |
| fas12 | 2002 | −57.95 | 49.57 | 30 | NA | Fasciatus |
| fas13 | 2013 | −65.41 | 49.84 | 25 | 228 | Fasciatus |
| fas14 | 2001 | −59.18 | 49.82 | 30 | 217 | Fasciatus |
| fas15 | 2001 | −67.07 | 42.32 | 27 | 297 | Fasciatus |
| men16 | 2008 | −61.16 | 67.14 | 48 | 458 | Shallow |
| men17 | 2008 | −57.78 | 66.58 | 30 | 586 | Deep |
| men18 | 2015 | 60.88 | 62.44 | 29 | 547 | Shallow |
| men19 | 2015 | −62.9 | 60.91 | 22 | 288 | Shallow |
| men20 | 2015 | 61.54 | 60.86 | 25 | 319 | Shallow |
| men21 | 2014 | −52.09 | 53.14 | 34 | 606 | Deep |
| men22 | 2014 | −51.96 | 52.93 | 35 | 455 | Shallow |
| men23 | 2014 | −49.78 | 49.51 | 34 | 311 | Shallow |
| men24 | 2014 | −47.09 | 47.17 | 36 | 431 | Deep |
| men25 | 2014 | −57.56 | 45.11 | 30 | 290 | Gulf |
| men26 | 2014 | −57.3 | 47.2 | 29 | 234 | Gulf |
| men27 | 2013 | −58.08 | 50.13 | 31 | 228 | Gulf |
| men28 | 2013 | −67.3 | 49.31 | 33 | 230 | Gulf |
We used a code associated with the species identified (fas or men) and selected at the sampling location (01 to 28) to simplify graphic representations. We indicated the number of individuals genotyped per sampling site (Ngen) and the genetic group (Genetic group) inferred by Valentin et al. (2014) using 13 microsatellites.
2.2. Bioinformatics and genotyping
We used the process radtags module available in STACKS v.1.46 to demultiplex libraries (Catchen et al., 2013). Reads were truncated to 80 bp, and adapter sequences were removed with CUTADAPT (Martin, 2014). Then, reads were aligned to the reference genome of Sebastes aleutianus (id GCA_001910805.1_ASM191080v1) using BWA‐MEM (Burrows–Wheeler Aligner‐Maximal Exact Match) algorithm with default parameters (Li, 2013). Loci were required to have a minimum stack depth of three (m = 3) among reads with potentially variable sequences and identified using pstacks. The catalogue of consensus loci was created allowing up to three mismatches per sample. Genotypes were obtained after running populations module of STACKS v.1.46. SNPs were kept according to the following consecutive filtering steps and guidelines indicated in Benestan, Ferchaud, et al. (2016). First, SNPs genotyped in at least 60% of the individuals were retained. Potential paralogs were excluded by removing markers showing heterozygosity > 0.60 and −0.50 < FIS < 0.50 within sites, in more than 60% of the sites. Only SNPs showing a global minor allele frequency (MAF) > 0.01 or at least a MAF > 0.05 in one location were retained for the analysis. Only one SNP was kept per locus in order to avoid bias due to linkage disequilibrium (Table S2). This dataset was used for the analyses of population structure, the index of genetic differentiation F ST, the assignment tests, and the investigation of multivariate analyses. Another dataset of SNPs without the use of MAF filtering was also generated, with a total of 112,926 SNPs, to use with demographic models (more details in the next section). The resulting filtered VCF files were converted into the file formats necessary for the following analyses using PGDSpider v.2.0.5.0 (Lischer and Excoffier, 2012).
2.3. Population structure and validation with assignment tests
Clustering among samples was inferred using the Bayesian clustering method implemented in the program ADMIXTURE v.1.23 (Alexander and Lange, 2011). Admixture analyses were run using 20,000 bootstraps, and the number of clusters was set from 1 to 29 (K) (n + 1 sampling sites). The support for different values of K was assessed according to the likelihood distribution (i.e., lowest cross‐validation error) as well as a visual inspection of the co‐ancestry values for each individual. Admixture analyses were also performed at the species level in order to investigate finer patterns of genomic structure. A PCA, implemented in the adegenet package (Jombart, 2008), was applied to the 834 individuals genotyped at both the 13 microsatellites (dataset from Valentin et al., 2014) and at the filtered 24,603 SNPs (see results), in order to compare the degree of resolution obtained using these two types of genetic markers. The first two principal components were visualized using ggplot2 package available in R (Wickham, 2011). To determine the extent of genetic differentiation among the 28 sampling sites, we estimated pairwise Weir and Cokhram F ST using the fct_84 function available in the assigner package (Gosselin et al., 2020), using all 24,603 SNPs.
Two runs of assignments tests were performed to test for (1) the validity of population structure and (2) the number of SNPs necessary for accurate assignment. The first assignment test aimed to assess the validity of microsatellite classification by using genetic clusters inferred for each individual using ADMIXTURE. This first run was performed in GENODIVE (Meirmans and Van Tienderen, 2004) with the frequentist method of Paetkau et al. (2004) using 1,000 permutations. For the second run, our objective was to identify the number of SNP markers required to perform an efficient and accurate assignment test at the species and ecotype levels. For this purpose, we considered a subset of “pure” individuals for ecotypes and populations that had a q‐value higher than 0.9 to a genetic cluster according to species‐specific ADMIXTURE results. Then, we tested the assignment test success relatively to 1, 2, 10, 100, 200, 300, 400, 500, 600, 700, 800, 900, and 1000, and all the polymorphic markers were genotyped (for this subset of individuals, 24,219 polymorphic markers). The second run was achieved using the function assignment_ngs available in the package assigner (Gosselin et al., 2020) with the following parameters: sampling method = “ranked,” thl = 0.3, iteration_subsample = 10, and assignment analysis = “gsi_sim.” This function was designed to simultaneously test how many genetic markers were needed to accurately assign individuals at a species and population level while correcting for the problem of high‐grading bias (Anderson, 2010).
2.4. Drivers explaining genomic distances
We conducted distance‐based redundancy analyses (db‐RDA; Legendre and Andersson, 1999) to investigate the variables explaining redfish genomic variation among and within species. RDA is a direct extension of multiple linear regressions to model multivariate response data (Legendre and Gallagher, 2001). First, we calculated individual Euclidean genomic distance among individuals using the dist function available in package adegenet in R as described on the https://popgen.nescent.org webpage (Kamvar et al., 2017). The individual Euclidean genomic distance matrix was then used as response variable, while the explanatory variables included spatial patterns through Moran’s eigenvector maps (MEMs, Borcard and Legendre, 2002), fishing depth, and sampling years. We performed a principal coordinate analysis (PCoA) on the individual Euclidean distances, and we kept the resulting principal component (PC) axis. These values represent the position of individuals in the genomic multivariate space, and they can be used as response variable in the db‐RDA. MEMs are derived from spectral graph theory and characterize a wide range of autocorrelation structures based on the survey design (i.e., distances between the n sampling locations; Dray et al., 2006). Therefore, it is a spectral decomposition of the spatial relationships among the 59 GPS points of our entire dataset. This decomposition generates (n ‐ 1) eigenfunctions, which are orthogonal variables that can be used in statistical models as explanatory variables representing spatial structure at different scales among the studied locations. Given the large spatial extent of the study area, distances among locations accounted for the earth curvature using the gcd.hf function in the codep package. To compute the MEMs, the truncation threshold was set to the length of the longest edge of the minimum spanning tree and only positive MEMs were retained using the dbmem function in the adespatial package. At three sampling sites (i.e., fas02, fas03, and fas12), depth was not recorded and therefore the median of depth distribution was used instead. Years were coded as dummy variables and treated as factors, therefore not assuming a linear relationship among years, but testing for annual differences in redfish genomic variation. To overcome multicollinearity among variables, we used the variance inflation factor with a threshold of 5, meaning that if a strong correlation was found between two variables, we retained only one of the two variables.
A global db‐RDA model was run with all explanatory variables, and model probability and adjusted coefficient of determination (adjusted R2) were calculated. The adjusted R2 was quantified using the RsquareAdj function in vegan and accounts for the number of observations and number of degrees of freedom in the fitted model (Legendre et al., 2011; Peres‐Neto et al., 2006). A selection of variables contributing to the explained variation was achieved by using both forward and backward selection, and a stopping criterion (ordiR2step function in the vegan package, Blanchet et al., 2008). This criterion limits overfitting by preventing selected variables to explain more variation than the full model developed with all explanatory variables. The same approach was performed within species, using a genomic distance matrix computed for S. mentella and S. fasciatus separately, and subsets of explanatory variables associated with these individuals. Biplots were produced with a scaling of type 1 to illustrate distances among objects (i.e., individuals) and relationships with selected environmental variables. Species, ecotypes, and populations were identified by different colors in biplots to illustrate how genetic clusters were associated with selected variables. Maps of MEMs associated with redfish genomic variation were presented to visualize the most significant spatial scales.
2.5. Demographic history of genomic divergence
We used the diffusion approximation framework implemented in dadi and modified by Tine et al. (2014) to compare the joint site frequency spectrum (JSFS) of our data to those simulated under different demographic models (Gutenkunst et al., 2009). These models incorporate the effects of selection at linked sites locally affecting population size (Ne) and the effect of differential introgression affecting the rate of migration (m) along the genome (Rougeux et al., 2017; Tine et al., 2014). During the divergence process, accumulation of barriers to gene flow tends to reduce the effective migration rate locally in the genome (Barton and Bengtsson, 1986), while selection at linked sites increases the effect of genetic drift through local reduction in effective population size (Charlesworth et al., 1993). Neglecting these effects can bias demographic inferences (Ewing and Jensen, 2016; Schrider et al., 2016), but modeling different local migration rates m and Ne along the genome can help overcome this bias.
We considered four alternative models of historical divergence namely (i) a model of strict isolation (SI), (ii) a model of divergence with continuous gene flow or isolation with migration (IM), (iii) a model of divergence with migration during the first generation of divergence or ancient migration (AM), and (iv) a model of secondary contact (SC). Under IM, the two diverging populations exchange migrants at a constant rate at each generation and this gene flow can be asymmetric, so that two independent migration rates m12 (from population 2 to population 1) and m21 (from population 1 to population 2) were modeled. Under the SC model, the population evolved in strict isolation between Tsplit (i.e., time of the split of the ancestral population) and until Tsc (i.e., time of secondary contact) where a secondary contact (gene flow) occurs continuously up to present time. Gene flow was modeled as M = 2Nref.m. Heterogeneous introgression was modeled using two categories of loci occurring in proportions P (i.e., loci with a migration rate M12 and M21) and 1‐P (i.e., loci with a reduced effective migration rate Me12 and Me21) across the genome. The same procedure was used to account for linked selection by defining two categories of loci with varying effective size. Then, we quantified linked selection using a Hill–Robertson scaling factor (Hrf, Rougeux et al., 2017) in order to relate the effective population size of loci influenced by linked selection (Nr = Hrf * Ne) to that of neutral loci (Ne). Three JSFS were constructed using 112,926 SNPs available in the raw dataset: (i) S. fasciatus and S. mentella shallow, (ii) S. fasciatus and S. mentella deep, and (iii) S. mentella shallow and S. mentella deep. It is important to note that S. mentella GSL and S. fasciatus populations were not included since the complexity of the introgression pattern observed for those populations would make difficult the interpretation of demographic models. Details of the filtering procedure are provided in supplementary materials. Model choice was performed using Akaike’s information criterion (AIC) and ΔAIC values under each given model. We used a generation time of 9.05 years for species comparisons and of 9.8 years for ecotype comparisons (Gascon 2003) and a mutation rate of 1e‐8 mutation/bp/generation (e.g., Rougeux et al., 2017; Tine et al., 2014) for parameter estimation. Standard deviations around parameter estimates were obtained using the Godambe information matrix (Coffman et al., 2016) with 100 bootstraps. Finally, in order to place the population along the divergence continuum proposed by Roux et al. (2016), we computed the net divergence (Da) using haplotypes vcf file available from STACKS. The mscalc pipeline (Roux et al., 2013) was used to perform these computations after filtering the dataset by removing missing data and nonpolymorphic site as above.
3. RESULTS
3.1. Two species with intraspecific geographic structure
Following the filtering steps shown in Table S2, we genotyped 24,603 SNPs (mean genotyping rate = 95%) in 860 individuals: 416 and 444 individuals in S. mentella and S. fasciatus, respectively. ADMIXTURE analysis revealed two highly differentiated genetic clusters (Figure 2a), with the cross‐validation (CV) error that dropped from 0.382 for K = 1 to 0.225 for K = 2. Examination of K = 2 confirmed the existence of the two species with individuals showing always more than 90% of ancestry to their own genetic cluster, except for the GSL where admixture between S. mentella and S. fasciatus was observed. Indeed, all S. mentella individuals of the sampling sites from the GSL to the Laurentian Fan (i.e., men25, men26, men27, and men28) consistently showed an average of 18% of S. fasciatus ancestry (Figure 2a). Some S. mentella individuals located from 3K to 2G NAFO divisions also showed ancestry with S. fasciatus (between 2.1 and 8.6%, Figure 2a). Inversely, we observed an average of 6% S. mentella ancestry within S. fasciatus samples from offshore East Newfoundland and North GSL (i.e., fas01, fas04, fas13, and fas14, Figure 2a). From this K = 2, the CV progressively decreased until being the smallest at K = 8 (CV = 0.209; Figure S2) where a putative population structure within each species tended to appear and was similar to the one highlighted when each species dataset was run separately (Figure 2b,c).
FIGURE 2.
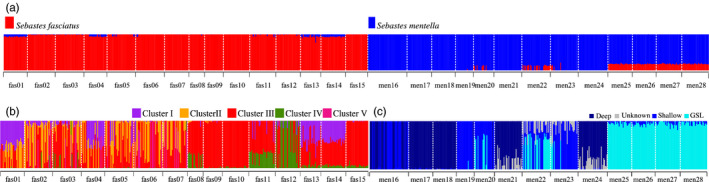
Population genomics analyses for inter‐ and intraspecific structure. Each individual is represented by a thin vertical line, which is partitioned into colored segments that represent the individual’s estimated ancestry into one of the genetic clusters observed using 24,603 SNPs. ADMIXTURE plots for (a) K = 2 for both species (n = 860), (b) K = 5 for S. fasciatus (n = 444), and (c) K = 4 for Sebastes mentella (n = 416). There are three ecotypes described in S. mentella: GSL (cyan), shallow (blue), and deep (dark blue). The unknown S. mentella genetic cluster is identified in light gray. There are five populations observed in S. fasciatus: I (purple), II (orange), III (red), IV (green), and IV (pink)
For S. mentella, the lowest cross‐validation value was for K = 4 (CV = 0.222). The four genetic clusters corresponded to two ecotypes already documented, shallow and deep, a new ecotype, GSL, and a fourth cluster designated as “Unknown” that had an unclear spatial distribution, with no individual having more than 50% ancestry of that cluster (Figure 2c). The Unknown cluster is not considered as an ecotype since its spatial distribution does not make the link with a particular habitat. As expected, S. mentella deep and shallow ecotypes were observed along the continental edge. The GSL ecotype was the unique ecotype observed among the juveniles of the 2011 cohort in the GSL (Figure 1). S. mentella ecotypes had distinct distributions in the Northwest Atlantic, albeit partially overlapping (Figure 1). For instance, S. mentella deep and shallow ecotypes were both sampled in men16. Also, 33 individuals sampled outside the GSL, in S. mentella shallow sampling sites (i.e., 7 individuals in men20, 22 individuals in men22, and 4 individuals in men23), were inferred to belong to GSL ecotype, suggesting the presence of a mixed ecotype composition in the NAFO divisions from 2G to 3K (Figures 1 and S3). Indeed, these individuals inferred to belong to GSL ecotype on the Labrador Shelf were adults (while only immature individuals were sampled in the GSL; Figure S1) and had a maximum estimated ancestry of 65% with GSL ecotype (Figure 2c). The presence of this mixed ecotypes composition on the Labrador Shelf was detected with SNP markers, whereas overlap between ecotypes was observed with the microsatellites (Figure S4).
For S. fasciatus, the genetic clusters detected were less obvious than for S. mentella since CV values dropped around K = 5 (CV = 0.196) and K = 8 (CV = 0.195). Yet, the clustering observed at K = 8 was less clear that the one documented at K = 5 since three genetic clusters out of 8 chaotically gather a few numbers of individuals (< 6 individuals) per sampling locations totalizing less than 18 individuals per cluster. In K = 5, 60% of all the S. fasciatus individuals (i.e., 266 out of 444 individuals) showed ancestry> 50.0% with population III (red; Figure 2b). The population III was present in all sampling sites, except fas01 (Figure 1), and was more frequent (>90.0% of individuals) in southern distribution of S. fasciatus, namely fas09, fas10, and fas15. The other four populations had different geographic distributions (Figure 1): population I (purple; fas01, fas04, fas05, fas13, and fas14; n = 50), population II (orange; fas02 to fas07; n = 81), population IV (green; fas12; n = 17), and population V (pink; fas01, fas04, fas05, fas06, fas11, fas13, and fas14; n = 30).
We compared F ST values and the results of ADMIXTURE analyses at the global and genetic cluster scales in order to examine the extent of inter‐ and intraspecific genetic differentiation. The heatmap based on F ST values gave similar results to the ADMIXTURE analyses. Both analyses identified the two species, as well as the three ecotypes and five populations within S. mentella and S. fasciatus, respectively (Figure S5). The smallest F ST value was between men27 and men28 (F ST = 0.0008, CIlow = 0.0004, CIhigh = 0.0013), whereas the largest F ST value was between fas12 and men18 (F ST = 0.6086; CIlow = 0.6025, CIhigh = 0.6147; Figure S5). We observed a pronounced genetic differentiation between samples from the two species (F ST = 0.5050, P‐value < 0.001), which was in line with ADMIXTURE results showing that the delimitation of the two species captured the majority of the signal of genetic differentiation. Within S. mentella, F ST values were twice larger—on average 0.0545 (ranging from 0.0004 to 0.1124)—than within S. fasciatus—on average 0.0257 (ranging from 0.0019 to 0.0715), which again was similar to ADMIXTURE finding a clearer population genomic structure in S. mentella than in S. fasciatus. Furthermore, the F ST heatmap showed that the sampling location men22 is genetically more similar to GSL than to shallow ecotype (Figure S5), which is likely due to a mixed ecotype composition of GSL and shallow individuals at the men22 sampling site as indicated by the ADMIXTURE analysis (Figure 2c).
3.2. Accuracy of assignment test at the species and ecotype level
We tested the validity of the clustering found by ADMIXTURE between and within species by performing individual assignment tests using all 860 individuals from both species. When considering the species and ecotype identity (shallow or deep) inferred from microsatellites and using the new 24,603 SNP markers genotyped, we obtained 100% assignment success at the species level.
At the ecotype level for S. mentella, we assigned 100% of the individuals coming from GSL and deep ecotypes, while for shallow ecotype assignment, success varies from 68% to 97% (except for men16 where deep and shallow ecotypes were both sampled). The sampling locations men22 and men20 showed the highest percent of misassigned individuals—on average 32%—and these individuals were inferred to belong to S. mentella GSL, which corroborate the pattern seen in ADMIXTURE (Figure 2c) and F ST analyses (Figure S5). Only a few percent of misassigned individuals sampled in shallow ecotype locations (i.e., men19, men20, men22, and men23) were assigned to deep ecotype (Figure 3a).
FIGURE 3.
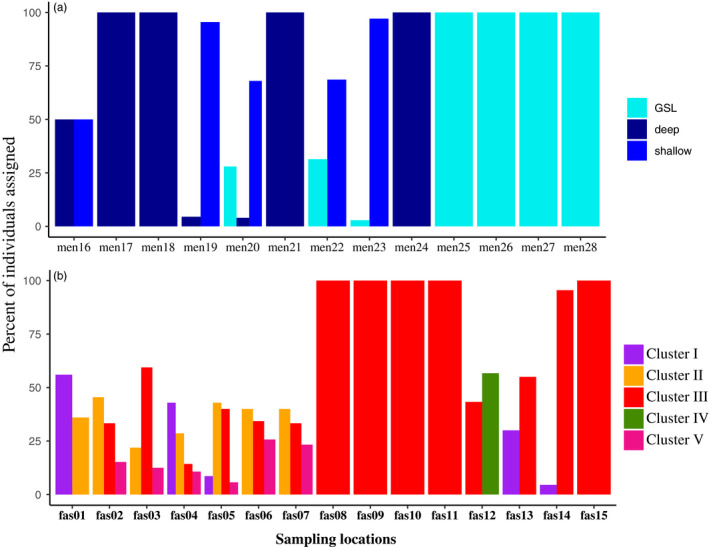
Assignment tests. Bar graph showing the percent of individuals correctly assigned for each sampling site and considering the three ecotypes and the five populations inferred from ADMIXTURE analyses in (a) Sebastes mentella and (b) S. fasciatus. Note that assignment tests were run by considering the genetic group inferred for each individual using the species‐specific ADMIXTURE analysis
The assignment success within S. fasciatus considering ADMIXTURE results revealed a very high assignment success always superior to 96.5% for every population. The populations II and V exhibited the lowest, albeit high, assignment success (96.5% and 97.5%, respectively), while populations I, III, and IV reached 100% of assignment success. Despite this high assignment success at the population level, we observed that sampling locations were actually composed of different populations except for fas08, fas09, fas09, fas11, and fas15 where all individuals belong to population III (Figure 3b). The only population that matched a unique sampling site (fas12) was population IV (Figure 3b).
By selecting a subset of 258 “pure” individuals assigned with up to 90% of ancestry to their genetic cluster, using ADMIXTURE results, we then assess the number of SNP markers required to reach a high assignment success at the species and ecotype levels (i.e., > 90%). This analysis revealed that only two SNPs (Figure S6) were needed to reach an assignment success of 100% at the species level while the use of a minimum of 2,000 highly divergent markers was required at the ecotype level for S. mentella (Figures 4 and S6).
FIGURE 4.
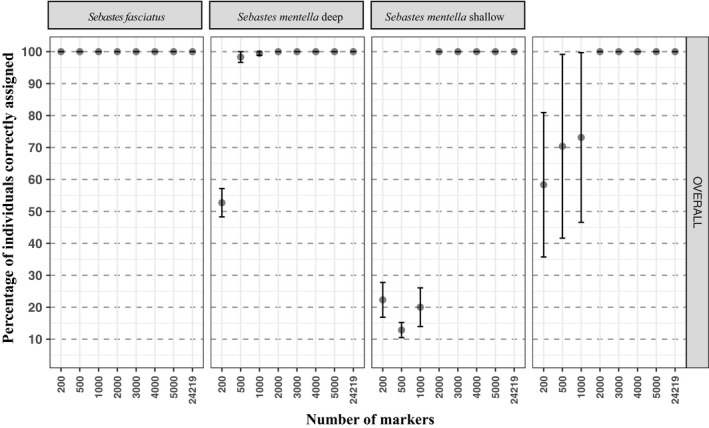
Assignment success related to the number of markers used (from 1 to 24,219 SNPs) based on ranked FST values of SNPs and using THL (Training, Holdout, Leave‐one‐out) method in S. mentella GSL, S. mentella shallow, S. mentella deep, and Sebastes fasciatus
3.3. Inter‐ and intraspecific genomic distances explained by spatial distribution, fishing depth, and sampling years
As the geographic distributions of Sebastes species, ecotypes, and populations surveyed in this study seemed distinct but overlapping (Figure 1), we investigated how environmental variables could explain the genomic distance using a db‐RDA (see Methods for more details). We used the individual sampling characteristics (i.e., geographic coordinates, depth, and year) for these analyses. For all samples, depth ranged from 134 to 708 m, with samples from S. mentella and S. fasciatus ranging between 228 to 708 m and 134 to 678 m, respectively. There are a total of 6 sampling years for both species (i.e., 2008, 2013, 2014, and 2015 for S. mentella; 2001, 2002, 2013, 2014, and 2015 for S. fasciatus).
First, explanatory variables were selected for the complete dataset (i.e., including both species). The elaboration of the MEMs based on geographic coordinates created 19 spatial explanatory variables (MEM1 to MEM19). The first explanatory variables (e.g., MEM1) corresponded to large spatial scales, while the last ones (e.g., MEM19) corresponded to fine spatial scales. A total of 21 explanatory variables were retained, namely 16 spatial variables, fishing depth, and sampling years (Table 2 and Figure 5a). The response variable was derived from a PCoA on the genetic distance matrix including 860 individuals from both species resulting in 859 PCs (i.e., n‐1) representing individuals’ genomic variation. All 859 PCs were used as response variables in the db‐RDA.
TABLE 2.
List of selected explanatory variables contributing to the genomic variation present among 860 individuals from both species
| Variable | Cum Adjusted R 2 | AIC | F | p‐value |
|---|---|---|---|---|
| MEM3 | 0.07597 | 7897.0 | 71.6277 | .002 |
| MEM1 | 0.14805 | 7828.2 | 73.5881 | .002 |
| MEM12 | 0.20215 | 7772.7 | 59.1131 | .002 |
| MEM2 | 0.24848 | 7722.3 | 53.7701 | .002 |
| Y2014 | 0.29454 | 7668.9 | 56.8162 | .002 |
| MEM7 | 0.33751 | 7615.8 | 56.3997 | .002 |
| MEM10 | 0.34889 | 7601.9 | 15.9108 | .002 |
| Y2008 | 0.35457 | 7595.4 | 8.4877 | .002 |
| MEM8 | 0.36108 | 7587.7 | 9.6702 | .002 |
| MEM15 | 0.36622 | 7581.7 | 7.9016 | .002 |
| Y2013 | 0.37161 | 7575.3 | 8.2740 | .002 |
| MEM4 | 0.37678 | 7569.2 | 8.0411 | .002 |
| MEM9 | 0.38072 | 7564.8 | 6.3821 | .002 |
| MEM14 | 0.38467 | 7560.2 | 6.4335 | .002 |
| MEM5 | 0.38793 | 7556.6 | 5.5016 | .002 |
| Depth | 0.39026 | 7554.3 | 4.2297 | .002 |
| MEM11 | 0.39317 | 7551.2 | 5.0350 | .002 |
| Y2002 | 0.39526 | 7549.2 | 3.9209 | .002 |
| MEM6 | 0.39706 | 7547.6 | 3.5122 | .002 |
| Y2001 | 0.39919 | 7545.6 | 3.9744 | .002 |
| MEM13 | 0.40057 | 7544.6 | 2.9218 | .002 |
Variable name, cumulative adjusted R 2, Akaike’s information criterion (AIC), F statistic, and probabilities (p‐value) are indicated.
FIGURE 5.
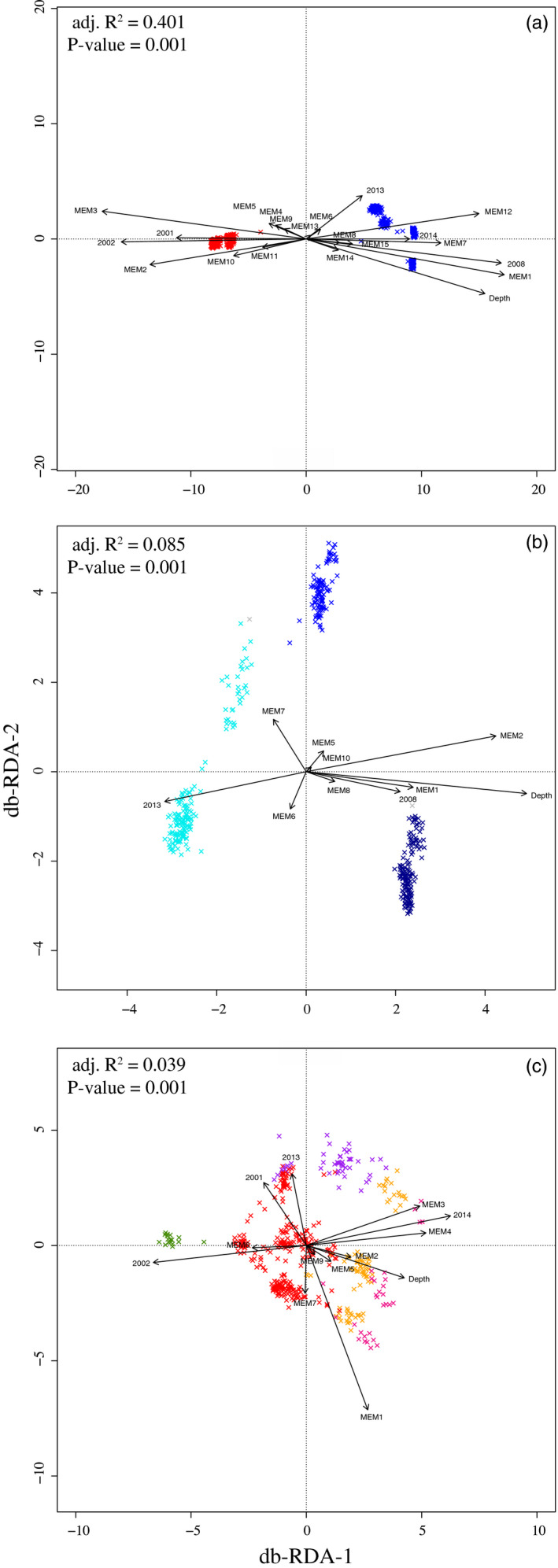
Db‐RDA ordination biplot (scaling 1), which preserves distances among objects and environmental variables for (a) 860 individuals, (b) 416 S. mentella individuals, and (c) 444 S. fasciatus individuals. (a) S. mentella is shown in blue and S. fasciatus in red. (b) There are three ecotypes described in S. mentella: GSL (cyan), shallow (blue), and deep (dark blue). The unknown S. mentella genetic cluster is identified in light gray. (c) There are five populations observed in S. fasciatus: I (purple), II (orange), III (red), IV (green), and IV (pink)
The global db‐RDA using all explanatory variables was significant (p‐value = .001) and accounted for 40.1% of the genomic variation (adjusted R 2 = 0.401). Using a forward and a backward selection approach, we identified the variables explaining most of the genomic variation. The db‐RDA biplot shows distances among objects and relationships with selected variables (Figure 5a). The first db‐RDA axis accounted for 92.3% of the explained variation, while the second accounted for 2.5%. On the left side of the biplot, S. fasciatus, in red, was mostly explained by the years 2002 and 2001, when most samples of that species were collected. On the right side of the figure, S. mentella, in blue, was mainly driven by 2008 and by greater fishing depths. MEM3 and MEM1 were the spatial variables best explaining species distribution (Figure S7).
With a subset of locations where S. mentella was present, 10 MEMs were obtained. The global db‐RDA for S. mentella using all explanatory variables (10 MEMs, fishing depth, and sampling years) was significant (p‐value = .001) and accounted for 8.5% of the genomic variation (adjusted R 2 = 0. 085). Variable selection identified seven MEMs, depth, and two sampling years (2008, 2013) as significant drivers of genomic variations within S. mentella (Table S3 and Figure 5B). The first db‐RDA axis accounted for 59.3% of the explained variation, while the second accounted for 13.7%. The first axis illustrated a gradient from the GSL ecotype in cyan on the left side, the shallow ecotype in intermediate blue in the middle, and the deep ecotype in dark blue on the right side. The second axis separated the deep ecotype in dark blue at the bottom, the GSL in cyan in the middle, and the shallow ecotype in intermediate blue at the top. Fishing depth, MEM2, and MEM1 were the variables most associated with the first axis. MEM2 corresponded to a large spatial gradient, where northern individuals shared more genetic similarities with southern individuals. MEM7 corresponded to a small spatial scale, where some individuals in the GSL or close to GSL had genomic differences compared to their surrounding counterparts (Figure S8).
Eight MEMs were obtained with the subset of locations where 444 S. fasciatus individuals were subsampled. The global db‐RDA (8 MEMs, fishing depth, and sampling years) was significant (P‐value = .001) and accounted for 3.9% of S. fasciatus genomic variation (adjusted R 2 = 0.039). Variable selection identified 8 MEMs, depth, and sampling years (2002, 2014, 2001, and 2013) as significant (Table S4 and Figure 5C). The first db‐RDA axis accounted for 24.2% of the explained variation, while the second accounted for 19.8%. The population I, shown in purple, was mostly associated with 2013 and MEM1, while the population IV, in green, was positively related to 2002. Populations II, III, and V in orange, red, and pink, respectively, had a large distribution in the db‐RDA space, and therefore, no clear relationships with explanatory variables could be identified (Figure 5c). MEM1 and MEM4 were the spatial variables best explaining S. fasciatus population distribution. MEM1 outlined a large‐scale east–west gradient. MEM4 also showed a large‐scale gradient where northeast individuals were different from their southwest counterparts (Figure S9).
3.4. Evidence for secondary contact at both species and ecotype level
We tested four major different demographic models, and the model choice indicated that the SC model was the best model for all three comparisons: S. mentella deep versus S. fasciatus, S. mentella shallow versus S. fasciatus, and S. mentella shallow versus deep (Table S5). In interspecific comparisons, the AIC and ΔAIC indicated that the SC model incorporating variation in drift (i.e., SC2N) received the highest support, while in the intraspecific comparisons, the model incorporating heterogeneous introgression (SC2m) was the best (Figure 6 and Table S5). Under SC2N, we found a simultaneous divergence time for the two comparisons between S. fasciatus and the ecotypes (i.e., S. mentella deep and shallow) with approximately 1 MYA (million years ago) of divergence for each comparison (see Table 3 for uncertainties around parameter estimates). The secondary contact represented on average 10.5% of the total time of divergence for these SC2N models, indicating that the species remained isolated most of their divergence time. The effective population sizes (Table 3) and the ratio of effective population sizes under all SC models revealed that both species showed large and relatively similar Ne (i.e., ratio Ne Fas/Ne Men ~ 0.92). Estimates of migration rate were low (m1 and m2 < 8e‐6; Table 3), suggesting that only a few migrants have been exchanged between species since secondary contact. We further found that approximately 50% of the genome displayed a reduced population size (Q = 0.50; Table 3) due to the action of linked selection. In these regions affected by linked selection, the Hrf indicated that the population size was 17% of the total population size.
FIGURE 6.
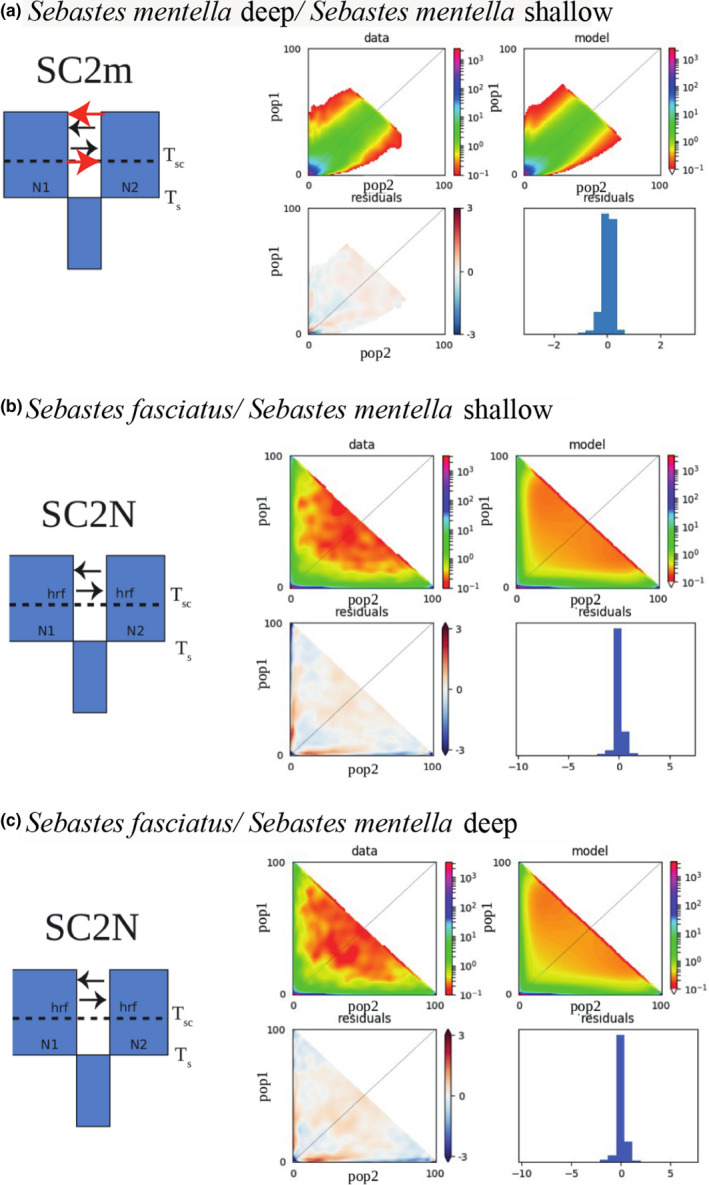
Observed best SC models (top panel) fitted to the folded site frequency spectrum (bottom panel) in the comparison between ecotypes (a) and between species (b and c). The suffix “2m” denotes heterogeneous migration and “2N” heterogeneous effective population size. AM = ancient migration. IM = isolation with migration, SC = secondary contacts, SI = strict isolation. All fittest models incorporate heterogeneous parameters, such as a heterogeneous rate of gene flow (2 m) and linked selection (2N). Each block corresponds to a population with effective population size of N1, N2, and θ. Horizontal dashed lines indicate the duration (Tsc) of the secondary contact period after the duration (Ts) of the allopatric period, while vertical dashed lines indicate change in effective size. Black arrows symbolize neutral gene flow, whereas red arrows symbolize heterogeneous introgression
TABLE 3.
Demographic parameter estimates obtained between the three major groups compared
| pop1 | Sebastes fasciatus | Sebastes fasciatus | Sebastes mentella deep |
|---|---|---|---|
| pop2 | Sebastes mentella deep | Sebastes mentella shallow | Sebastes mentella shallow |
| Best model | SC2N | SC2N | SC2m |
| AIC | 9588 | 9652 | 7426 |
| Log likelihood | −4786 | −4818 | −3704 |
| Theta | 21466 | 2077 | 1908 |
| Nref | 53,000 [44,000–62,000] | 51,000 [43,000–58,000] | 58,000 [57,000–59,000] |
| Ne1 | 119,000 [81,000–158,000] | 116,000 [88,000–143,000] | 63,000 [62,000–64,000] |
| Ne2 | 127,000 [89,000–165,000] | 126,000 [98,000–155,000] | 89,000 [88,000–90,000] |
| m1 ← 2 | 5,3E‐06 [3,8E‐06–6,7E‐06] | 6E‐06 [5,6E‐07–1,14E‐05] | 4E‐04 [0–0,001] |
| m2 ← 1 | 1,7E‐06 [1,3E‐06–2,2E‐06] | 3E‐06 [2,86E‐06–3,11E‐06] | 2E‐04 [1e‐04–2,4E‐04] |
| 2me1 ← 2 | NA | NA | 2,4E‐04 [2,2E‐04–2,6E‐04] |
| me2 ← 1 | NA | NA | 3,8E‐05 [3,3E‐05–4,4E‐05] |
| Tsplit | 1,027,000 [912,000–1,142,000] | 1,040,000 [1,024,000–1,054,000] | 417,900 [416,800–419,000] |
| Tsc | 124,700 [120,000–1290,00] | 89,000 [86,000–93,000] | 23,000 [22,000–24,000] |
| hrf | 0.17 | 0.16 | NA |
| P/Q | 0.50 | 0.50 | 0.70 |
AIC, Akaike’s information criterion; log likelihood, maximum likelihood; theta: effective mutation rate of the reference population, which here corresponds to the ancestral population; Nref, size of the ancestral; Ne1 and Ne2, effective population size of the compared pair; m1 ← 2 and m2 ← 1, migration from population 2 to population 1 and migration from population 1 to population 2; me12 and me21, effective migration rate estimated in the most differentiated regions of the genome; Ts, time of split of the ancestral population in the two species; Tsc, duration of the secondary contact; P, proportion of the genome freely exchanged (1‐P provides the proportion of the genome non‐neutrally exchanged); Q, proportion of the genome with a neutral Ne (1‐Q provides the proportion of the genome non‐neutrally exchanged); hrf = Hill–Robertson factor representing the reduction of Ne in the region (1‐Q) with reduced Ne.
For ecotype comparison, the median estimate of divergence time between S. mentella deep and shallow population was ~ 0.46 MYA under the SC2m models. The migration rate since secondary contact was higher than that observed between species and strongly asymmetric, with higher migration from the deep (m 12 = 0.0003) to the shallow population (m 21 = 9e‐5 Table 3). Overall, we found that ~28% (ranging from 26 to 38%) of the genome was undergoing reduced introgression since secondary contact (P parameter in Table 3). Finally, using haplotype data derived from the GBS loci, we found that the level of net divergence (Da) over all combined loci was an order of magnitude lower in intraspecific comparisons (Da ~ 0.0001) compared to interspecific comparisons (Da ~ 0.0015).
4. DISCUSSION
The goals of this study were to (i) assess population genomic structure with a dense marker set, (ii) investigate how environmental or temporal drivers may influence the population structure, and (iii) reconstruct the speciation history of two redfish species along the Northwest Atlantic Ocean. We confirmed the pronounced genetic distinction between S. mentella and S. fasciatus, and moderate genetic differentiation between two S. mentella ecotypes, deep and shallow. We identified a new ecotype of S. mentella GSL and at least five populations of S. fasciatus in the Northwest Atlantic. High individual assignment success was achieved at species, ecotype, and population levels for the first time, confirming that GBS data provide a powerful tool for traceability of the redfish fishery. Inter‐ and intraspecific genomic variations were best explained by large‐scale spatial distribution and fishing depth. We also uncovered that postglacial secondary contact was the most likely scenario to explain the speciation history of species and S. mentella ecotypes.
4.1. Highly structured Sebastes species in the northwest Atlantic
We observed a level of genetic differentiation spanning from weak to pronounced between the genetic clusters within both S. mentella and S. fasciatus, and all have distinct but overlapping distributions. The largest estimate of genetic differentiation was between S. mentella and S. fasciatus species (F ST = 0.505; P‐value < 0.001), supporting that these genetic clusters form independent demographic units.
Within S. mentella, we confirmed the presence of deep and shallow ecotypes and identified a new ecotype, GSL in the Northwest Atlantic with more genetic similarities to shallow than deep ecotypes. It is then possible that GSL ecotype may have diverged from the shallow ecotype. The shallow and deep ecotypes were previously identified and described as distinct genetic clusters inhabiting different depths along the continental slope in past studies across the North Atlantic (Cadrin et al., 2010; Saha et al., 2017; Valentin et al., 2014). Here, we suggest to use the term ecotype to describe these ecogeographically isolated groups. Ecotype was first defined and summarized as a genetic group specific to a particular habitat (Turesson, 1922). We considered that the three genetic groups of S. mentella described in this study correspond to that first definition and that results obtained from the db‐RDAs have shown that the genetic distance between these ecotypes is explained by differences in habitats through space and depth. Furthermore, the genetic composition of the S. mentella GSL ecotype shows consistent admixture with S. fasciatus (i.e., 18%) and it is the unique ecotype present in the GSL, a partially isolated area.
Within S. fasciatus, we identified five populations occurring from the Gulf of Maine to the Labrador Shelf with highly overlapping distribution. This contrasts with the more pronounced differentiation we observed between S. mentella ecotypes and consequently justifies the use of the term population (instead of ecotype) to describe these genetic clusters. Nevertheless, the five populations identified are mostly associated with distinct geographic areas along the Canadian coast (Figure 1), suggesting further investigation on the population structure of this species. The population I was the only genetic cluster that had up to 6% of S. mentella ancestry, which underlined that introgression may be the main cause of the distinctiveness observed in this particular population. Moreover, a higher genetic differentiation—in line with previous microsatellite observations (Valentin et al., 2014)—was detected for the semiclosed population located in Bonne Bay Fjord (fas12; population IV), for which we hypothesize that limited water exchange or limited physical connectivity may be responsible for this pattern, as previously proposed in the red algae Palmaria palmate (Li et al., 2015). Still, fas12 does not appear completely isolated and larvae may potentially migrate in the fjord since a mixture of two populations was observed at this location. Our results confirm that there is a unique population in Bonne Bay Fjord, which was in the past designated as “of special concern” because of its demographic isolation (COSEWIC, 2010); however, our results also suggest that the Bonne Bay Fjord could naturally be reseeded from other populations.
The spatially distinct distribution of species, ecotypes, and populations, as well as levels of genetic differentiation among them, suggests limited dispersal and migration of S. mentella and S. fasciatus in the Northwest Atlantic (ca. 0.505). For migratory fish species, estimates of genetic differentiation using thousands of neutral SNPs between trans‐Atlantic populations are of ca. 0.016 for Atlantic mackerel (Scomber scombrus; Rodríguez‐Ezpeleta et al., 2016) and ca. 0.081 for Atlantic cod (Gadus morhua; Berg et al., 2017). We observed on a much smaller spatial scale higher levels of genetic differentiation between species (ca. 0.5050) and within S. mentella (ca. 0.0545) suggesting limited dispersal during the larval stage and short migration distance. Their ovoviviparity, involving internal fertilization and larval extrusion, likely reduces the extent of dispersal and may reduce interspecific mating and produce reproductive isolation, thus favoring genomic differentiation on small geographic scales (Johns and Avise, 1998). Consequently, these genetically and ecologically distinct groups appear mostly closed (i.e., self‐recruiting) and thus need to be individually protected for proper conservation (Hellberg et al., 2002). Although hybrids have been documented between these two species in the past, no F1 was documented in a nonbiased sampling design (Roques et al., 2001). Along with our result, this tends to indicate that introgression events may be ancient between the two species, and thus, introgression is not limiting the establishment of genome‐wide barriers to gene flow between these species. Nevertheless, our current sampling design, which avoids potential hybrids by sampling only individuals clearly assignable to a priori groups, limits our interpretation of our results to properly answer to this question.
Although we identified pronounced intraspecific structure in S. mentella, some structure is still unexplained and more genotyping will be necessary to identify all the demographic units present in the Northwest Atlantic. Thus, an “unknown” genetic cluster was identified in S. mentella sampling zones (i.e., men21, men22, men23, and men24) that correspond to an area of contact with another Sebastes species, Sebastes norvegicus, which is present in the North Atlantic (Stransky, 2005). We also identified some admixture between the GSL and shallow ecotypes of the NAFO divisions from 3K to 2G. These samples were from adults and suggest that past larval migration through the Strait of Belle Isle was successful that these GSL ecotypes could reproduce with shallow ecotypes and have descendants who survived to adulthood. It would be of great interest in future studies to investigate whether the new GSL cohort has also dispersed to the Labrador Shelf and evaluate whether reproduction between GSL and shallow ecotypes can occur more than once.
It is also likely that our sampling under‐represented the S. fasciatus populations that may be present from the Gulf of Maine. Microsatellites indicated a clear genetic differentiation from Gulf of Maine sample (Roques et al., 2002; Valentin et al., 2014) which could not be tested with our sampling design due to the low number of individuals south of the 45th parallel that were genotyped. Clearly, the total number of S. fasciatus individuals that were available for this study most likely reduced the power to identify populations while structure is present (Puechmaille, 2016). Indeed, the initial design of the study was the investigation of the S. mentella and S. fasciatus differentiation, with a focus on the characterization of S. mentella individuals in the GSL, hence the lack of S. fasciatus samples. Future studies should sample more S. fasciatus over the whole species distribution to identify an accurate number of populations.
4.2. Spatial distribution, depth, and other factors explaining inter‐ and intraspecific barriers to gene flow
Defining population structure in marine species remains challenging to investigate considering the absence of obvious barriers to gene flow (Palumbi, 1994). Relying on tools from spatial ecology along with population genomics, we found that spatial distribution explained most of the genetic differentiation in the redfish system while fishing depth and sampling years significantly explained from a moderate to a small proportion of the genomic distances between individuals of Sebastes spp. Spatial patterns were strong predictive variables for species, ecotypes, and populations, which is in concordance with the emerging results of recent seascape genomic studies (reviewed in Riginos et al., 2016) on the American lobster (Homarus americanus; Benestan, Quinn, et al., 2016), the sea cucumber (Parastichopus californicus; Xuereb et al., 2018), the red mullet (Mullus surmuletus; Dalongeville et al., 2018), and the Eastern oyster (Crassostrea virginica; Bernatchez et al., 2019). In particular, this north/south genomic structure may reflect an historical signature of a glacial refugium during the Pleistocene followed by population expansion throughout the northwest. Pleistocene glacial cycles associated with geological and climatic changes have indeed clearly played a role in shaping the contemporary distribution and abundance of marine organisms in the North Atlantic (Wares and Cunningham, 2001).
Depth may play a role in the isolation of these species and other Sebastes species (Ingram, 2011; Shum et al., 2015). Despite the potential influence of bathymetry on marine organisms, no recent seascape genomics studies have already investigated and evidenced how this environmental feature can drive marine population genomics. Here, we uncovered that fishing depth was a significant explanatory variable for genomic distances between species, ecotypes, and populations of Sebastes spp. This outcome brings new evidence that habitat depth is a strong barrier to dispersal of Sebastes spp and contributes to the wide literature covering the topic specifically for S. mentella (Cadrin et al., 2010; Stefánsson et al., 2009) and other marine species such as the deep‐sea coral (Desmophyllum dianthus; Miller et al., 2011), the common protobranch bivalve (Nucula attacellana; Jennings et al., 2013), and the small‐spotted catshark (Scyliorhinus canicular; Kousteni et al., 2015). We also need to highlight that while depth was the most important significant predictor of diversification within S. mentella, it does not explain solely the barrier to gene flow between S. mentella and S. fasciatus since both species inhabit a similar depth range in our study area. Other pre‐ or postzygotic barriers to gene flow most likely played a role in their reproductive isolation.
Another significant predictor of the overall genomic variation was the year of sampling. This result is not surprising as we sampled a specific area each year due to sampling constraints (i.e., vessel availability), and thus, this is likely due to our nonrandom sampling design. If logistically possible, future population genomic studies on Sebastes spp. should sample all areas at different years to accurately test whether species and populations distribution remain constant interannually.
4.3. Physical separation followed by secondary contacts explains the speciation history for both species and S. mentella shallow and deep ecotypes
Our study is the first to extensively investigate the history of speciation for the Sebastes species inhabiting the North Atlantic using explicit demographic simulations. Here, we discovered that scenarios of secondary contact (SC) provided the best fit to the observed data for both species and ecotypes. Furthermore, modeling the process of speciation between S. mentella and S. fasciatus has never been done before, while for S. mentella deep vs. shallow comparison, Saha et al. (2017) previously identified the isolation with migration (IM) model as the best model to explain the divergence observed. However, the authors did not test for secondary contact and used a limited number of microsatellite markers, which are known to quickly converge to demographic equilibrium and therefore offer limited resolution to distinguish among models of divergence with gene flow (IM) and secondary contact (SC) (Bierne et al., 2013). In contrast, SNP markers are more suited to perform demographic inferences given their lower mutation rate (Ellegren, 2000). Here, we used a much higher number of markers, thus reaching sufficient power to accurately distinguish these two models. Our parameter estimates suggested that the two species diverged first (~1 MYA) followed by a more recent split of the ecotype (~0.5 MYA). These divergence times correspond to a pronounced drop of sea level, suggesting that the redfish species may tend to become genetically isolated during periods of expansion/contraction associated with the Pleistocene glacial oscillations (2.6 million to 11,700 years ago), as previously suggested by Hyde and Vetter (2007). Under these conditions, the genetic clusters had enough time to accumulate genomic incompatibilities that could act as a postzygotic mechanism of reproductive isolation upon secondary contact. Nevertheless, these estimates should be interpreted cautiously given that deviation from the mutation rate that we used here may substantially alter demographic parameter estimates (although the ratio of estimated time of secondary contact over divergence time still applies). Overall, our results are consistent with an increasing recent report of secondary contact in a wide range of marine species such as the European anchovy (Le Moan et al., 2016), the Atlantic cod (Gadus morhua; Fairweather et al., 2018) and European sea bass (Dicentrarchus labrax; Tine et al., 2014).
Our power to infer secondary contacts between ecotypes may have been favored by strong gene flow erasing previous genetic divergence outside of barrier loci and generating an excess of variants at intermediate frequencies (Alcala and Vuilleumier, 2014). Indeed, we pointed out that ~ 30% of the genome is undergoing reduced introgression between ecotypes, in line with previous estimates for marine species—ranging from ~ 15 to 45%—suggesting that most of the genome is still largely semipermeable to gene flow (Gagnaire et al., 2018; Le Moan et al., 2016). In these conditions, it is possible that upon secondary contact with S. fasciatus, genetic variants under positive selection in S. fasciatus and under negative selection in S. mentella may have allowed the adaptation of the shallow ecotype to an intermediate habitat depth. The observation of GSL ecotype that displayed relatively high historical admixture with S. fasciatus further suggests interspecific introgression events. Testing whether introgression is adaptive (Fraïsse et al., 2014; Hedrick, 2013) and resolving its role in the subsequent colonization of different depths would require an integrative study including whole‐genome sequencing of the four Sebastes species (i.e., Sebastes fasciatus, Sebastes mentella, Sebastes norvegicus, and Sebastes viviparous) across their whole distribution range. Nevertheless, this signal of introgression between S. mentella and S. fasciatus may also be due to the fact that both species and ecotypes showed large Ne and recent divergence time, which may result in incomplete lineage sorting indicating that some ancestral variants are still shared. Indeed, delineating species along a continuum of genomic divergence is a major challenge in evolutionary biology but can be tackled through appropriate demographic modeling by investigating the relation between net synonymous divergence (Da) and the probability of ongoing gene flow (Roux et al., 2016). Here, we found the Da estimate was ten times higher between species than ecotypes (Da = 0.0001 versus Da = 0.0015), which supports the idea that (i) ecotypes are more recently diverged than species, as well as the fact that (ii) species and ecotypes fall within the “grey‐zone of speciation” (Roux et al., 2016). Overall, we highlighted that models integrating linked selection and gene flow provided the best fit to our data for both species and ecotypes. These results supported a role for linked selection in shaping differentiation landscape, as is increasingly reported in the speciation literature (Burri et al., 2015; Cruickshank and Hahn, 2014; Rougemont and Bernatchez, 2018; Roux et al., 2016; Stankowski et al., 2019).
4.4. Improving management of Sebastes spp. in the Northwest Atlantic Ocean
Despite the plethora of studies in ecology and genetics that have shed light on their limited movement and their large genetic differentiation (Roques et al., 2002; Roques et al., 2001; Valentin et al., 2014; Valentin et al., 2002), the two redfish species, S. mentella and S. fasciatus, are still often reported as “redfish”—due to their morphological similarities—without any differentiation in stock assessment, except for Unit 1 and 2 stocks where species‐specific estimates are provided (DFO, 2020), and current Canadian management plans at some management units. Yet, reproductively isolated redfish aggregations can then be at risk of locally eliminating biological diversity and overfishing with the current large‐scale management plan that does not reflect the biology of the species (Benestan, 2020). Since the collapse of the vast majority of the stocks in the Northwest Atlantic, the Canadian government has undertaken significant efforts to improve the recovery of redfish populations but more efforts may be required.
We identified unexpected asymmetric migration through the Strait of Belle Isle. The ADMIXTURE and the assignment analyses pointed out that some individuals sampled in the habitat of the shallow ecotype from the Eastern Newfoundland coast to the Hudson Strait shared some ancestry with GSL ecotype (up to 65%). Yet, on the western Newfoundland coast, in the GSL, individuals shared ancestry only with the GSL ecotype and form the large new cohorts rebuilding the stock within the GSL (Senay et al., 2019). Similarly, S. fasciatus individuals belonging to the cluster V were also identified on the Labrador Shelf and Northern Grand Banks, uncovering possible asymmetric gene flow from North GSL to Labrador Shelf. These results imply that S. mentella and S. fasciatus may disperse from the North GSL through the Strait of Belle Isle to the Labrador Shelf, as it has been reported for the snow crab (Chionoecetes opilio; Puebla et al., 2008) and the capelin (Mallotus villosus; Kenchington et al., 2015, Cayuela et al., 2019). Since adult redfish were rarely observed in the shallow Strait of Belle Isle, we propose that larval drift may play a key role in linking subpopulations. Management Unit 1 and NAFO divisions 3K and 2G tend to then exchange more than 10% of individuals, which corresponds to a maximum threshold that can be used to infer demographic independence between two areas (Hastings and Harrison, 1994; Lowe and Allendorf, 2010; Waples and Gaggiotti, 2006). As the planning for possible re‐opening of the redfish fishery in the GSL following its signs of recovery (Senay et al., 2019), the recovery of this stock may strongly influence adjacent areas to the GSL, such as the south of Labrador Shelf. We then urge for a conjoint discussion across Atlantic provinces and the south of Labrador Sea for the purpose of establishing a sustainable plan for this marine fish.
Genetic resources such as microsatellite markers were used for species identification of Sebastes species over the last two decades (Roques et al., 1999, 2001, 2002; Valentin et al., 2014). Here, we found that only two SNPs were needed to accurately identify the two species. We also found that at least a subset of 2,000 SNPs could discriminate redfish S. mentella deep and shallow ecotype. This subset of 2,000 SNPs outperformed microsatellite markers for delineating ecotypes and improved assignment test success (i.e., ecotypes of S. mentella) as shown in a wide range of species with management or conservation concerns such as harbor porpoise (Phocoena phocoena; Lah et al., 2016), the brown trout (Salmo trutta; Lemopoulos et al., 2019), the Atlantic salmon (Salmo salar; Moore et al., 2014), and the great scallop (Pecten maximus; Vendrami et al., 2017). Therefore, a next step could be to develop a genotype assay (e.g., using Kaspar, Fluidigm, AmpliSeq, or Sequenom technologies)—based on the selection of these highly divergent SNP markers combined with an individual assignment test approach—in order to provide an efficient tool for traceability as previously validated for several marine resources in Europe (Nielsen et al., 2012). The DNA assay developed for Sebastes spp. would serve to distinguish the two species and three ecotypes within the fish catches in order to assess the true impacts of the redfish fishery in Canada. Furthermore, a surprising high assignment success was achieved for S. fasciatus populations, with up to 96.5% of success while the vast majority of the sampling locations always exhibit a mixing proportion of different genetic clusters. This surprising result indicates that more samples might be required for enhancing the resolution of population structure and assignment test and eventually develop similar DNA chip for S. fasciatus populations.
Supporting information
Fig S1
Fig S3
Fig S5
Supplementary Material
Acknowledgement
The authors would like to thank Jean‐Marie Sevigny for his scientific devotion to the research of redfish genetics since 1990 and his implication in the beginning of the project. We also want to acknowledge the work of Alexandra Valentin in the early steps of the project who cared for the sampling design and genotyping of the individuals. We would like to thanks Margaret Treble, Don Powers, Atlantic Groundfish Council (AGC), and the scientific teams of the Teleost and the Needler vessels for sampling efforts. This study was funded by grants from the Genomics Research and Development Initiative of Fisheries and Oceans Canada. Fisheries and Oceans Canada also financed this project through the “Fonds des partenariats” program (postdoctoral funding of L.B.). We are grateful to Ressources Aquatique Québec (RAQ) that provided financial support for genotyping and bioinformatics and Calcul Canada for computation resources. We also want to thank Audrey Bourret, Jean‐Martin Chamberland, and Hugo Bourdages for relevant comments and inputs that improved the overall quality of the present manuscript.
Benestan LM, Rougemont Q, Senay C, et al. Population genomics and history of speciation reveal fishery management gaps in two related redfish species (Sebastes mentella and Sebastes fasciatus). Evol Appl. 2021;14:588–606. 10.1111/eva.13143
[Correction added on 12 January 2021, after first online publication: the affiliation for Laura M. Benestan has been updated.]
Data Availability Statement
Data for this study are available in DRYAD (https://datadryad.org) after the manuscript is accepted for publication. Scripts on population genomics and spatial ecology are available on the GitHub page of Laura M. Benestan and Quentin Rougemont.
References
- Alcala, N. , & Vuilleumier, S. (2014). Turnover and accumulation of genetic diversity across large time‐scale cycles of isolation and connection of populations. Proceedings of the Royal Society B: Biological Sciences, 281(1794), 20141369 10.1098/rspb.2014.1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, D. H. , & Lange, K. (2011). Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics, 12, 246 10.1186/1471-2105-12-246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, E. C. (2010). Assessing the power of informative subsets of loci for population assignment: Standard methods are upwardly biased. Molecular Ecology Resources, 10(4), 701–710. 10.1111/j.1755-0998.2010.02846.x [DOI] [PubMed] [Google Scholar]
- Atkinson, D. B. (1987). The redfish resources off Canada’s east coast, pp. 15–33. In Proceedings of the Lowel Wakefield Fisheries Symposium: International Rockfish Symposium, Anchorage, AK, Alaska Sea grant College program report 97‐2. [Google Scholar]
- Barton, N. , & Bengtsson, B. O. (1986). The barrier to genetic exchange between hybridizing populations. Heredity, 57(3), 357–376. 10.1038/hdy.1986.135 [DOI] [PubMed] [Google Scholar]
- Benestan, L. M. , Ferchaud, A. L. , Hohenlohe, P. A. , Garner, B. A. , Naylor, G. J. P. , Baums, I. B. , Schwartz, M. K. , Kelley, J. L. , & Luikart, G. (2016). Conservation genomics of natural and managed populations: Building a conceptual and practical framework. Molecular Ecology, 25(13), 2967–2977. 10.1111/mec.13647 [DOI] [PubMed] [Google Scholar]
- Benestan, L. , Quinn, B. K. , Maaroufi, H. , Laporte, M. , Clark, F. K. , Greenwood, S. J. , Rochette, R. , & Bernatchez, L. (2016). Seascape genomics provides evidence for thermal adaptation and current‐mediated population structure in American lobster (Homarus americanus). Molecular Ecology, 25(20), 5073–5092. 10.1111/mec.13811 [DOI] [PubMed] [Google Scholar]
- Benestan, L. (2020). Population genomics applied to fishery management and conservation In Population genomics: Marine organisms (pp. 399‒421). Springer. [Google Scholar]
- Berg, P. R. , Star, B. , Pampoulie, C. , Bradbury, I. R. , Bentzen, P. , Hutchings, J. A. , Hutchings, J. A. , & Jentoft, S. (2017). Trans‐oceanic genomic divergence of Atlantic cod ecotypes is associated with large inversions. Heredity, 119(6), 418–428. 10.1038/hdy.2017.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatchez, S. , Xuereb, A. , Laporte, M. , Benestan, L. , Steeves, R. , Laflamme, M. , Bernatchez, L. , & Mallet, M. A. (2019). Seascape genomics of Eastern oyster (Crassostrea virginica) along the Atlantic coast of Canada. Evolutionary Applications, 12(3), 587–609. 10.1111/eva.12741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, W. C. IV , Baer, C. F. , Antolin, M. F. , & DuTeau, N. M. (2001). Population genomics: Genome‐wide sampling of insect populations. Annual Review of Entomology, 46(1), 441–469. 10.1146/annurev.ento.46.1.441 [DOI] [PubMed] [Google Scholar]
- Blanchet, F. G. , Legendre, P. , & Borcard, D. (2008). Modelling directional spatial processes in ecological data. Ecological Modelling, 215(4), 325–336. [Google Scholar]
- Bierne, N. , Gagnaire, P. A. , & David, P. (2013). The geography of introgression in a patchy environment and the thorn in the side of ecological speciation. Current Zoology, 59(1), 72–86. 10.1093/czoolo/59.1.72 [DOI] [Google Scholar]
- Borcard, D. , & Legendre, P. (2002). All‐scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecological Modelling, 153(1‐2), 51–68. 10.1016/S0304-3800(01)00501-4 [DOI] [Google Scholar]
- Burri, R. , Nater, A. , Kawakami, T. , Mugal, C. F. , Olason, P. I. , Smeds, L. , Suh, A. , Dutoit, L. , Bureš, S. , Garamszegi, L. Z. , Hogner, S. , Moreno, J. , Qvarnström, A. , Ružić, M. , Sæther, S.‐A. , Sætre, G.‐P. , Török, J. , & Ellegren, H. (2015). Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Research, 25(11), 1656–1665. 10.1101/gr.196485.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadrin, S. X. , Bernreuther, M. , Daníelsdóttir, A. K. , Hjörleifsson, E. , Johansen, T. , Kerr, L. , … Stransky, C. (2010). Population structure of beaked redfish, Sebastes mentella: Evidence of divergence associated with different habitats. ICES Journal of Marine Science, 67, 1617–1630. 10.1093/icesjms/fsq046 [DOI] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: An analysis tool set for population genomics. Molecular Ecology, 22, 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayuela, H. , Rougemont, Q. , Laporte, M. , Mérot, C. , Normandeau, E. , Dorant, Y. , Tørresen, O. K. , Hoff, S. N. K. , Jentoft, S. , Sirois, P. , Castonguay, M. , Jansen, T. , Praebel, K. , Clément, M. , & Bernatchez, L. (2020). Shared ancestral polymorphism and chromosomal rearrangements as potential drivers of local adaptation in a marine fish. Molecular Ecology, 29(13), 2379–2398. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B. , Morgan, M. T. , & Charlesworth, D. (1993). The effect of deleterious mutations on neutral molecular variation. Genetics, 134(4), 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman, A. J. , Hsieh, P. H. , Gravel, S. , & Gutenkunst, R. N. (2016). Computationally efficient composite likelihood statistics for demographic inference. Molecular Biology and Evolution, 33(2), 591–593. 10.1093/molbev/msv255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSEWIC (2010). COSEWIC assessment and status report on the Deepwater Redfish/Acadian Redfish complex Sebastes mentella and Sebastes fasciatus, in Canada [online]. Committee on the Status of Endangered Wildlife in Canada, Ottawa. Available from www.sararegistry.gc.ca/status/status_e.cfm
- Cruickshank, T. E. , & Hahn, M. W. (2014). Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Molecular Ecology, 23(13), 3133–3157. 10.1111/mec.12796 [DOI] [PubMed] [Google Scholar]
- Dalongeville, A. , Andrello, M. , Mouillot, D. , Lobreaux, S. , Fortin, M. J. , Lasram, F. , Belmaker, J. , Rocklin, D. , & Manel, S. (2018). Geographic isolation and larval dispersal shape seascape genetic patterns differently according to spatial scale. Evolutionary Applications, 11(8), 1437–1447. 10.1111/eva.12638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DFO (2020). Redfish (Sebastes mentella and S. fasciatus) stocks assessment in Unit 1 in 2019. DFO Can. Sci. Advis. Sec. Sci. Advis. Rep. 2020/000.
- Dray, S. , Legendre, P. , & Peres‐Neto, P. R. (2006). Spatial modelling: A comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM). Ecological Modelling, 196(3‐4), 483–493. 10.1016/j.ecolmodel.2006.02.015 [DOI] [Google Scholar]
- Ellegren, H. (2000). Microsatellite mutations in the germline: Implications for evolutionary inference. Trends in Genetics, 16(12), 551–558. 10.1016/S0168-9525(00)02139-9 [DOI] [PubMed] [Google Scholar]
- Endler, J. A. (1977). Geographic variation, speciation, and clines. Princeton University Press. [PubMed] [Google Scholar]
- Ewing, G. B. , & Jensen, J. D. (2016). The consequences of not accounting for background selection in demographic inference. Molecular Ecology, 25(1), 135–141. 10.1111/mec.13390 [DOI] [PubMed] [Google Scholar]
- Fairweather, R. , Bradbury, I. R. , Helyar, S. J. , deBruyn, M. , Therkildsen, N. O. , Bentzen, P. , Hemmer‐Hansen, J. , & Carvalho, G. R. (2018). Range‐wide genomic data synthesis reveals transatlantic vicariance and secondary contact in Atlantic cod. Ecology and Evolution, 8(23), 12140–12152. 10.1002/ece3.4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraïsse, C. , Roux, C. , Welch, J. J. , & Bierne, N. (2014). Gene‐flow in a mosaic hybrid zone: Is local introgression adaptive? Genetics, 197(3), 939–951. 10.1534/genetics.114.161380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnaire, P.‐A. , Lamy, J.‐B. , Cornette, F. , Heurtebise, S. , Dégremont, L. , Flahauw, E. , Boudry, P. , Bierne, N. , & Lapègue, S. (2018). Analysis of genome‐wide differentiation between native and introduced populations of the cupped oysters Crassostrea gigas and Crassostrea angulata . Genome Biology and Evolution, 10(9), 2518–2534. 10.1093/gbe/evy194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascon, D. (2003). Redfish multidisciplinary research zonal program (1995‐1998). Mont‐Joli: Fisheries and Oceans. [Google Scholar]
- Gosselin, T. , Anderson, E. C. , & Bradbury, I. (2020). Assigner: Assignment Analysis with GBS/RAD Data using R. R package version 0.5.8. http://thierrygosselin.github.io/assigner/. http://doi.org/10.5281/zenodo.592677
- Gutenkunst, R. N. , Hernandez, R. D. , Williamson, S. H. , & Bustamante, C. D. (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics, 5(10), e1000695 10.1371/journal.pgen.1000695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings, A. , & Harrison, S. (1994). Metapopulation dynamics and genetics. Annual Review of Ecology and Systematics, 25(1), 167–188. 10.1146/annurev.es.25.110194.001123 [DOI] [Google Scholar]
- Hedrick, P. W. (2013). Adaptive introgression in animals: Examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology, 22(18), 4606–4618. 10.1111/mec.12415 [DOI] [PubMed] [Google Scholar]
- Hellberg, M. E. , Burton, R. S. , Neigel, J. E. , & Palumbi, S. R. (2002). Genetic assessment of connectivity among marine populations. Bulletin of Marine Science, 70(1), 273–290. [Google Scholar]
- Hewitt, G. (2000). The genetic legacy of the quaternary ice ages. Nature, 405(6789), 907–913. 10.1038/35016000 [DOI] [PubMed] [Google Scholar]
- Hyde, J. R. , & Vetter, R. D. (2007). The origin, evolution, and diversification of rockfishes of the genus Sebastes (Cuvier). Molecular Phylogenetics and Evolution, 44(2), 790–811. 10.1016/j.ympev.2006.12.026 [DOI] [PubMed] [Google Scholar]
- Ingram, T. (2011). Speciation along a depth gradient in a marine adaptive radiation. Proceedings of the Royal Society B: Biological Sciences, 278(1705), 613–618. 10.1098/rspb.2010.1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, R. M. , Etter, R. J. , & Ficarra, L. (2013). Population differentiation and species formation in the deep sea: The potential role of environmental gradients and depth. PLoS ONE, 8(10), e77594 10.1371/journal.pone.0077594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns, G. C. , & Avise, J. C. (1998). Tests for ancient species flocks based on molecular phylogenetic appraisals of sebastes rockfishes and other marine fishes. Evolution, 52(4), 1135 10.2307/2411243 [DOI] [PubMed] [Google Scholar]
- Jombart, T. (2008). Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics, 24(11), 1403–1405. 10.1093/bioinformatics/btn129 [DOI] [PubMed] [Google Scholar]
- Kamvar, Z. N. , López‐Uribe, M. M. , Coughlan, S. , Grünwald, N. J. , Lapp, H. , & Manel, S. (2017). Developing educational resources for population genetics in R: An open and collaborative approach. Molecular Ecology Resources, 17(1), 120–128. 10.1111/1755-0998.12558 [DOI] [PubMed] [Google Scholar]
- Kenchington, E. L. , Nakashima, B. S. , Taggart, C. T. , & Hamilton, L. C. (2015). Genetic structure of capelin (Mallotus villosus) in the northwest Atlantic Ocean. PLoS ONE, 10(3), e0122315 10.1371/journal.pone.0122315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousteni, V. , Kasapidis, P. , Kotoulas, G. , & Megalofonou, P. (2015). Strong population genetic structure and contrasting demographic histories for the small‐spotted catshark (Scyliorhinus canicula) in the Mediterranean Sea. Heredity, 114(3), 333–343. 10.1038/hdy.2014.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lah, L. , Trense, D. , Benke, H. , Berggren, P. , Gunnlaugsson, P. , Lockyer, C. , Öztürk, A. , Öztürk, B. , Pawliczka, I. , Roos, A. , Siebert, U. , Skóra, K. , Víkingsson, G. , & Tiedemann, R. (2016). Spatially explicit analysis of genome‐wide SNPs detects subtle population structure in a mobile marine mammal, the harbor porpoise. PLoS ONE, 11(10), e0162792 10.1371/journal.pone.0162792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laikre, L. , Nilsson, T. , Primmer, C. R. , Ryman, N. , & Allendorf, F. W. (2009). Importance of genetics in the interpretation of favourable conservation status. Conservation Biology, 23(6), 1378–1381. 10.1111/j.1523-1739.2009.01360.x [DOI] [PubMed] [Google Scholar]
- Le Moan, A. , Gagnaire, P. A. , & Bonhomme, F. (2016). Parallel genetic divergence among coastal‐marine ecotype pairs of European anchovy explained by differential introgression after secondary contact. Molecular Ecology, 25(13), 3187–3202. 10.1111/mec.13627 [DOI] [PubMed] [Google Scholar]
- Legendre, P. , & Anderson, M. J. (1999). Distance‐based redundancy analysis: Testing multispecies responses in multifactorial ecological experiments. Ecological Monographs, 69(1), 1–24. 10.1890/0012-9615(1999)069[0001:DBRATM]2.0.CO;2 [DOI] [Google Scholar]
- Legendre, P. , & Gallagher, E. D. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia, 129(2), 271–280. 10.1007/s004420100716 [DOI] [PubMed] [Google Scholar]
- Legendre, P. , Oksanen, J. , & ter Braak, C. J. F. (2011). Testing the significance of canonical axes in redundancy analysis. Methods in Ecology and Evolution, 2(3), 269–277. 10.1111/j.2041-210X.2010.00078.x [DOI] [Google Scholar]
- Lemopoulos, A. , Prokkola, J. M. , Uusi‐Heikkilä, S. , Vasemägi, A. , Huusko, A. , Hyvärinen, P. , Koljonen, M.‐ L. , Koskiniemi, J. & Vainikka, A. (2019). Comparing RADseq and microsatellites for estimating genetic diversity and relatedness — Implications for brown trout conservation. Ecology and Evolution, 9(4), 2106–2120. 10.1002/ece3.4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). [Heng Li ‐ Compares BWA to other long read aligners like CUSHAW2] Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv Preprint arXiv:1303.3997 [q‐bio.GN]. [Google Scholar]
- Li, J. J. , Hu, Z. M. , & Duan, D. L. (2015). Genetic data from the red alga Palmaria palmata reveal a mid‐Pleistocene deep genetic split in the North Atlantic. Journal of Biogeography, 45(2), 902–913. 10.1111/jbi.12464 [DOI] [Google Scholar]
- Lischer, H. E. L. , & Excoffier, L. (2012). PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics, 28(2), 298–299. 10.1093/bioinformatics/btr642 [DOI] [PubMed] [Google Scholar]
- Lowe, W. H. , & Allendorf, F. W. (2010). What can genetics tell us about population connectivity? Molecular Ecology, 19(15), 3038–3051. 10.1111/j.1365-294X.2010.04688.x [DOI] [PubMed] [Google Scholar]
- Maggs, C. A. , Castilho, R. , Foltz, D. , Henzler, C. , Jolly, M. T. , Kelly, J. , Olsen, J. , Perez, K. E. , Stam, W. , Väinölä, R. , Viard, F. , & Wares, J. (2008). Evaluating signatures of glacial refugia for north Atlantic benthic marine taxa. Ecology, 89(sp11), S108–S122. 10.1890/08-0257.1 [DOI] [PubMed] [Google Scholar]
- Martin, M. (2014). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal, 17(1), 10 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Mascher, M. , Wu, S. , Amand, P. St , Stein, N. , & Poland, J. (2013). Application of genotyping‐by‐sequencing on semiconductor sequencing platforms: A comparison of genetic and reference‐based marker ordering in barley. PLoS ONE, 8(10), e76925 10.1371/journal.pone.0076925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirmans, P. G. , & Van Tienderen, P. H. (2004). GENOTYPE and GENODIVE: Two programs for the analysis of genetic diversity of asexual organisms. Molecular Ecology Notes, 4(4), 792–794. 10.1111/j.1471-8286.2004.00770.x [DOI] [Google Scholar]
- Miller, K. J. , Rowden, A. A. , Williams, A. , & Häussermann, V. (2011). Out of their depth? Isolated deep populations of the cosmopolitan coral Desmophyllum dianthus may be highly vulnerable to environmental change. PLoS ONE, 6(5), e19004 10.1371/journal.pone.0019004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J. S. , Bourret, V. , Dionne, M. , Bradbury, I. , O’Reilly, P. , Kent, M. , Chaput, G. , & Bernatchez, L. (2014). Conservation genomics of anadromous Atlantic salmon across its North American range: Outlier loci identify the same patterns of population structure as neutral loci. Molecular Ecology, 23(23), 5680–5697. 10.1111/mec.12972 [DOI] [PubMed] [Google Scholar]
- Nielsen, E. E. , Cariani, A. , Mac Aoidh, E. , Maes, G. E. , Milano, I. , Ogden, R. , Taylor, M. , Hemmer‐Hansen, J. , Babbucci, M. , Bargelloni, L. , Bekkevold, D. , Diopere, E. , Grenfell, L. , Helyar, S. , Limborg, M. T. , Martinsohn, J. T. , McEwing, R. , Panitz, F. , Patarnello, T. , … Carvalho, G. R. (2012). Gene‐associated markers provide tools for tackling illegal fishing and false eco‐certification. Nature Communications, 3(1), 1–7. 10.1038/ncomms1845 [DOI] [PubMed] [Google Scholar]
- Ovenden, J. R. , Berry, O. , Welch, D. J. , Buckworth, R. C. , & Dichmont, C. M. (2015). Ocean’s eleven: A critical evaluation of the role of population, evolutionary and molecular genetics in the management of wild fisheries. Fish and Fisheries, 16(1), 125–159. 10.1111/faf.12052 [DOI] [Google Scholar]
- Paetkau, D. , Slade, R. , Burden, M. , & Estoup, A. (2004). Genetic assignment methods for the direct, real‐time estimation of migration rate: A simulation‐based exploration of accuracy and power. Molecular Ecology, 13(1), 55–65. 10.1046/j.1365-294X.2004.02008.x [DOI] [PubMed] [Google Scholar]
- Palumbi, S. R. (1994). Genetic divergence, reproductive isolation, and marine speciation. Annual Review of Ecology and Systematics, 25(1), 547–572. 10.1146/annurev.es.25.110194.002555 [DOI] [Google Scholar]
- Peres‐Neto, P. R. , Legendre, P. , Dray, S. , & Borcard, D. (2006). Variation partitioning of species data matrices: Estimation and comparison of fractions. Ecology, 87(10), 2614–2625. 10.1890/0012-9658(2006)87[2614:VPOSDM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Perreault‐Payette, A. , Muir, A. M. , Goetz, F. , Perrier, C. , Normandeau, E. , Sirois, P. , & Bernatchez, L. (2017). Investigating the extent of parallelism in morphological and genomic divergence among lake trout ecotypes in Lake Superior. Molecular Ecology, 26(6), 1477–1497. 10.1111/mec.14018 [DOI] [PubMed] [Google Scholar]
- Puebla, O. , Sévigny, J. M. , Sainte‐Marie, B. , Brêthes, J. C. , Burmeister, A. D. , Dawe, E. G. , & Moriyasu, M. (2008). Population genetic structure of the snow crab (Chionoecetes opilio) at the Northwest Atlantic scale. Canadian Journal of Fisheries and Aquatic Sciences, 65(3), 425–436. 10.1139/F07-163 [DOI] [Google Scholar]
- Puechmaille, S. J. (2016). The program structure does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Molecular Ecology Resources, 16(3), 608–627. 10.1111/1755-0998.12512 [DOI] [PubMed] [Google Scholar]
- Reiss, H. , Hoarau, G. , Dickey‐Collas, M. , & Wolff, W. J. (2009). Genetic population structure of marine fish: Mismatch between biological and fisheries management units. Fish and Fisheries, 10(4), 361–395. 10.1111/j.1467-2979.2008.00324.x [DOI] [Google Scholar]
- Riginos, C. , Crandall, E. D. , Liggins, L. , Bongaerts, P. , & Treml, E. A. (2016). Navigating the currents of seascape genomics: How spatial analyses can augment population genomic studies. Current Zoology, 62(6), 581–601. 10.1093/cz/zow067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Ezpeleta, N. , Bradbury, I. R. , Mendibil, I. , Álvarez, P. , Cotano, U. , & Irigoien, X. (2016). Population structure of Atlantic mackerel inferred from RAD‐seq‐derived SNP markers: Effects of sequence clustering parameters and hierarchical SNP selection. Molecular Ecology Resources, 16(4), 991–1001. 10.1111/1755-0998.12518 [DOI] [PubMed] [Google Scholar]
- Roques, S. , Duchesne, P. , & Bernatchez, L. (1999). Potential of microsatellites for individual assignment: The North Atlantic redfish (genus Sebastes) species complex as a case study. Molecular Ecology, 8(10), 1703–1717. [DOI] [PubMed] [Google Scholar]
- Roques, S. , Sévigny, J. M. , & Bernatchez, L. (2002). Genetic structure of deep‐water redfish, Sebastes mentella, populations across the North Atlantic. Marine Biology, 140(2), 297–307. 10.1007/s002270100705 [DOI] [Google Scholar]
- Roques, S. , Sévigny, J. M. , & Bernatchez, L. (2001). Evidence for broadscale introgressive hybridization between two redfish (genus sebastes) in the north‐west Atlantic: A rare marine example. Molecular Ecology, 10(1), 149–165. 10.1046/j.1365-294X.2001.01195.x [DOI] [PubMed] [Google Scholar]
- Rougemont, Q. , & Bernatchez, L. (2018). The demographic history of Atlantic salmon (Salmo salar) across its distribution range reconstructed from approximate Bayesian computations. Evolution, 72(6), 1261–1277. 10.1111/evo.13486 [DOI] [PubMed] [Google Scholar]
- Rougeux, C. , Bernatchez, L. , & Gagnaire, P. A. (2017). Modeling the multiple facets of speciation‐with‐gene‐flow toward inferring the divergence history of lake whitefish species pairs (Coregonus clupeaformis). Genome Biology and Evolution, 9(8), 2057–2074. 10.1093/gbe/evx150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, C. , Fraïsse, C. , Castric, V. , Vekemans, X. , Pogson, G. H. , & Bierne, N. (2014). Can we continue to neglect genomic variation in introgression rates when inferring the history of speciation? A case study in a Mytilus hybrid zone. Journal of Evolutionary Biology, 27(8), 1662–1675. 10.1111/jeb.12425 [DOI] [PubMed] [Google Scholar]
- Roux, C. , Fraïsse, C. , Romiguier, J. , Anciaux, Y. , Galtier, N. , & Bierne, N. (2016). Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biology, 14(12), e2000234 10.1371/journal.pbio.2000234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, C. , Tsagkogeorga, G. , Bierne, N. , & Galtier, N. (2013). Crossing the species barrier: Genomic hotspots of introgression between two highly divergent Ciona intestinalis species. Molecular Biology and Evolution, 30(7), 1574–1587. 10.1093/molbev/mst066 [DOI] [PubMed] [Google Scholar]
- Rundle, H. D. , & Nosil, P. (2005). Ecological speciation: Ecological speciation. Ecology Letters, 8(3), 336–352. 10.1111/j.1461-0248.2004.00715.x [DOI] [Google Scholar]
- Saha, A. , Johansen, T. , Hedeholm, R. , Nielsen, E. E. , Westgaard, J. I. , Hauser, L ., Planque, B. , Cadrin, S. X. , & Boje, J. (2017). Geographic extent of introgression in Sebastes mentella and its effect on genetic population structure. Evolutionary Applications, 10(1), 77–90. 10.1111/eva.12429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senay, C. , Gauthier, J. , Bourdages, H. , Brassard, C. , Duplisea, D. , & Ouellette‐Plante, J. (2019). Redfish (Sebastes mentella and S. fasciatus) Stocks Status in Unit 1 in 2017. Ottawa, ON: Canadian Science Advisory Secretariat. [Google Scholar]
- Sévigny, J.‐M. , Gagné, P. , de Lafontaine, Y. , & Dodson, J. (2000). Identification and distribution of the larvae of redfish species (Sebastes fasciatus and S. mentella: Scorpaenidae) in the Gulf of St. Lawrence. Fishery Bulletin, 98, 375–388. [Google Scholar]
- Schrider, D. R. , Shanku, A. G. , & Kern, A. D. (2016). Effects of linked selective sweeps on demographic inference and model selection. Genetics, 204(3), 1207–1223. 10.1534/genetics.116.190223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum, P. , Pampoulie, C. , Sacchi, C. , & Mariani, S. (2014). Divergence by depth in an oceanic fish. PeerJ, 2, e525 10.7717/peerj.525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum, P. , Pampoulie, C. , Kristinsson, K. , & Mariani, S. (2015). Three‐dimensional post‐glacial expansion and diversification of an exploited oceanic fish. Molecular Ecology, 24(14), 3652–3667. 10.1111/mec.13262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankowski, S. , Chase, M. A. , Fuiten, A. M. , Rodrigues, M. F. , Ralph, P. L. , & Streisfeld, M. A. (2019). Widespread selection and gene flow shape the genomic landscape during a radiation of monkeyflowers. PLOS Biology, 17(7), e3000391 10.1371/journal.pbio.3000391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefánsson, M. Ö. , Sigurdsson, T. , Pampoulie, C. , Daníelsdóttir, A. K. , Thorgilsson, B. , Ragnarsdóttir, A. , Gíslason, D. , Coughlan, J. , Cross, T. F. , & Bernatchez, L. (2009). Pleistocene genetic legacy suggests incipient species of Sebastes mentella in the Irminger Sea. Heredity, 102(5), 514–524. 10.1038/hdy.2009.10 [DOI] [PubMed] [Google Scholar]
- Stransky, C. (2005). Geographic variation of golden redfish (Sebastes marinus) and deep‐sea redfish (S. mentella) in the North Atlantic based on otolith shape analysis. ICES Journal of Marine Science, 62(8), 1691–1698. 10.1016/j.icesjms.2005.05.012 [DOI] [Google Scholar]
- Tine, M. , Kuhl, H. , Gagnaire, P. A. , Louro, B. , Desmarais, E. , Martins, R. S. T. , Hecht, J. , Knaust, F. , Belkhir, K. , Klages, S. , Dieterich, R. , Stueber, K. , Piferrer, F. , Guinand, B. , Bierne, N. , Volckaert, F. A. M. , Bargelloni, L. , Power, D. M. , Bonhomme, F. , … Reinhardt, R. (2014). European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nature Communications, 5(1), 1–10. 10.1038/ncomms6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesson, G. (1922). The species and the variety as ecological units. Hereditas, 3(1), 100–113. 10.1111/j.1601-5223.1922.tb02727.x [DOI] [Google Scholar]
- Valentin, A. E. , Penin, X. , Chanut, J. P. , Power, D. , & Sévigny, J. M. (2014). Combining microsatellites and geometric morphometrics for the study of redfish (Sebastes spp.) population structure in the Northwest Atlantic. Fisheries Research, 154, 102–119. 10.1016/j.fishres.2014.02.008 [DOI] [Google Scholar]
- Valentin, A. , Sévigny, J. M. , & Chanut, J. P. (2002). Geometric morphometrics reveals body shape differences between sympatric redfish Sebastes mentella, Sebastes fasciatus and their hybrids in the Gulf of St Lawrence. Journal of Fish Biology, 60(4), 857–875. 10.1006/jfbi.2002.1889 [DOI] [Google Scholar]
- Vendrami, D. L. J. , Telesca, L. , Weigand, H. , Weiss, M. , Fawcett, K. , Lehman, K. , Clark, M. S. , Leese, F. , McMinn, C. , Moore, H. , & Hoffman, J. I. (2017). RAD sequencing resolves fine‐scale population structure in a benthic invertebrate: Implications for understanding phenotypic plasticity. Royal Society Open Science, 4(2), 160548 10.1098/rsos.160548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples, R. S. , & Gaggiotti, O. (2006). What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Molecular Ecology, 15(6), 1419–1439. 10.1111/j.1365-294X.2006.02890.x [DOI] [PubMed] [Google Scholar]
- Wares, J. P. , & Cunningham, C. W. (2001). Phylogeography and historical ecology of the North Atlantic intertidal. Evolution, 55(12), 2455–2469. 10.1111/j.0014-3820.2001.tb00760.x [DOI] [PubMed] [Google Scholar]
- Wickham, H. (2011). ggplot2. Wiley Interdisciplinary Reviews: Computational Statistics, 3(2), 180–185. 10.1002/wics.147 [DOI] [Google Scholar]
- Xuereb, A. , Benestan, L. , Normandeau, É. , Daigle, R. M. , Curtis, J. M. R. , Bernatchez, L. , & Fortin, M.‐J. (2018). Asymmetric oceanographic processes mediate connectivity and population genetic structure, as revealed by RADseq, in a highly dispersive marine invertebrate (Parastichopus californicus). Molecular Ecology, 27(10), 2347–2364. 10.1111/mec.14589 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S3
Fig S5
Supplementary Material
Data Availability Statement
Data for this study are available in DRYAD (https://datadryad.org) after the manuscript is accepted for publication. Scripts on population genomics and spatial ecology are available on the GitHub page of Laura M. Benestan and Quentin Rougemont.