

Abstract
Background
Small RNAs (sRNAs) extensively mediate gene‐specific chromatin regulation in lower organisms. As a dominant type of functional sRNAs in mature mammals, microRNAs mainly regulate gene expression at post‐transcription level in the cytoplasm. Currently, whether there exists a type of nuclear‐localized sRNAs mediating gene‐specific epigenetic regulation in mature mammalian cells remains largely unclear. Here, we profiled sRNAs enriched in the nucleus and investigated their function in mediating gene‐specific epigenetic regulation in anti‐tumor immunity.
Methods
We established cytoplasmic and nuclear transcriptomes of sRNAs of dendritic cells (DCs) using high‐throughput sequencing. Transcription abundances of sRNAs and mRNAs were analyzed by reverse transcription‐quantitative polymerase chain reaction (RT‐qPCR) assay. The associations between sRNAs and Argonaute (AGO) proteins were detected by RNA immunoprecipitation analysis. Synthesized sRNAs and locked nucleic acid (LNA) ‐modified sRNA inhibitors were used to screen the function of sRNAs in innate immune cells. The effect of sRNA on the enrichment of either chromatin remodeler or histone modification at the gene promoter was analyzed by chromatin immunoprecipitation (ChIP)‐qPCR assay. Chromatin accessibility qPCR assay was used to detect the accessibility of gene promoters. A B16 melanoma‐bearing mouse model was established to determine the function of sRNAs in tumor‐associated macrophages (TAMs) and their effect on tumor growth.
Results
We identified a new class of nucleus‐localized sRNAs, named snRNA/snoRNA‐derived nuclear RNAs (sdnRNAs). Some sdnRNAs were Dicer‐independent and had no association with Argonaute proteins. sdnRNA‐3, the most abundant Dicer‐independent sdnRNAs identified in our analysis, was selected as a representative to examine the biological function of sdnRNAs. sdnRNA‐3 selectively inhibited the transcription of Nos2 in macrophages during innate immune response by repressing the chromatin accessibility at Nos2 gene promoter. sdnRNA‐3 promoted the enrichments of repressive chromatin‐remodeling regulator Mi‐2β and the repressive histone modification H3K27me3 at Nos2 gene promoter. In the B16 melanoma mouse model, we found higher expression of sdnRNA‐3 in M2 TAMs than M1 TAMs and DCs. Transfer of sdnRNA‐3‐silenced macrophages inhibited tumor growth with increased expression of inducible nitric oxide synthase (iNOS) in TAMs.
Conclusions
Our results demonstrated that the sdnRNA‐3 repressed the transcription of Nos2 by repressing chromatin accessibility at the promoter, providing new insights into the regulation of macrophage function in tumor immunity.
Keywords: dendritic cells, iNOS, Mi‐2β, Nos2, sdnRNA, small RNAs, tumor‐associated macrophages
Short abstract
Nuclear small RNAs conducting gene‐specific epigenetic regulation have not been clearly identified in adult mammals. Through establishing cytoplasmic and nuclear small RNomes in innate immune cells, we revealed that sdnRNA‐3, one of the new class of nucleus‐localized and dicer‐independent small RNAs derived from snRNAs/snoRNAs, inhibited transcription of Nos2 gene (encode iNOS) by decreasing chromatin accessibility of gene promoter in macrophages, increasing the tumor promoting function of tumor‐associated macrophages.
Abbreviations
- AGO
argonaute
- BGI
Beijing Genomics Institute
- CHD4
chromodomain‐helicase‐DNA‐binding protein 4
- ChIP
chromatin immunoprecipitation
- DCs
dendritic cells
- H3K27me3
trimethylated histone 3 lysine 27
- iNOS
inducible nitric oxide synthase
- IP
immunoprecipitation
- LINE
long interspersed nuclear elements
- LNA
locked nucleic acid
- lncRNAs
long non‐coding RNAs
- LPS
Lipopolysaccharides
- M1
classically activated macrophage
- M2
alternatively activated macrophage
- mDC
mature dendritic cells
- miRNAs
microRNAs
- MФ
macrophages
- NO
Nitric oxide
- piRNAs
piwi‐interacting RNAs
- rasiRNAs
repeat‐associated small interfering RNAs
- RISC
RNA induced silencing complex
- rRNA
ribosomal RNAs
- RT‐qPCR
reverse transcription‐quantitative polymerase chain reaction
- sdnRNAs
snRNA/snoRNA‐derived nuclear RNAs
- SINE
short interspersed nuclear elements
- siRNAs
small interfering RNAs
- SOAP
oligonucleotide alignment program
- sRNAs
small RNAs
- TAMs
tumor‐associated macrophages
- TLRs
toll‐like receptors
1. BACKGROUND
Epigenetic regulations, such as DNA methylation, histone modification, and chromatin remodeling play important roles in establishing cell lineage‐specific phenotypes and functions during mammalian development [1]. The mechanisms underlying the gene‐specific regulation of chromatin accessibility remain to be fully understood. [2]. One important component mediating gene‐specific epigenetic regulation is non‐coding regulatory RNAs, including long non‐coding RNAs (lncRNAs) and small RNAs (sRNAs) [3]. Among these small RNAs, microRNAs (miRNAs), small interfering RNAs (siRNAs), and Piwi‐interacting RNAs (piRNAs) are extensively studied as functional sRNAs [4]. In mammalian cells, exogenous siRNA and nuclear microRNA may be involved in epigenetic regulation by targeting gene promoters in a sequence‐dependent manner [5, 6, 7]. However, siRNAs and promoter‐associated sRNAs are not widely expressed in mammalian cells whereas miRNAs are mainly localized in the cytoplasm. Thus, whether there is a class of nuclear‐localized small RNAs that especially mediate gene‐specific epigenetic regulation deserves further investigation.
Epigenetic regulation is crucial for defining cell‐specific identity and function during lineage commitment. Chromatin status, especially in gene loci, is tightly regulated by epigenetic regulators, such as DNA methylation, histone modification, and ATP‐dependent chromatin remodeling [8]. During an innate immune response, innate mediators such as pro‐inflammatory cytokines are induced by Toll‐like receptors (TLRs) and other innate signals with dynamic transcription patterns due to different chromatin context in specific gene loci established by epigenetic regulators [9]. There are three classes of TLRs signal‐induced genes, namely, primary response genes, late primary response genes, and secondary response genes. Gene‐specific chromatin remodeling which tailors nucleosome composition for DNA accessing is required for transcription induction of secondary response genes [10]. However, the molecular mechanisms underlying the gene‐specific targeting of the chromatin remodelers have not been revealed.
Dendritic cells (DCs) and macrophages, two kinds of important innate immune cells, are not only capable of recognizing pathogens and initiating inflammatory responses during infection but can also regulate tumor immunity. Alternatively activated macrophage (M2), relative to classically activated macrophage (M1), promotes tumor growth, invasion, and immune evasion by low expression of M1‐associated immune effectors and high expression of immune‐repression molecules [11]. Like M2, tumor‐associated macrophages display suppressive capacity for tumor immunity, especially for T cell expansion and function in the tumor microenvironment. Thus, macrophages are potential targets for tumor immunotherapy [12, 13]. To explore new classes of nuclear‐localized sRNAs that could be involved in mediating gene‐specific epigenetic regulation in innate and tumor immunity, we established transcriptomes of small RNAs, termed small RNomes, from the nuclear and cytoplasmic fractions of mature DCs (mDC). By systematically analyzing the nuclear and cytoplasmic RNomes, we identified different kinds of nucleus‐localized sRNAs derived from different DC precursors. Functional studies further revealed that a Dicer‐independent snRNA/snoRNA derived nuclear sRNA (sdnRNA) was implicated in mediating gene‐specific epigenetic regulation of immune effector in the induction of anti‐tumor immunity.
2. MATERIALS AND METHODS
2.1. Mice and reagents
Male and female C57BL/6 mice (6‐8 weeks) were obtained from Joint Ventures Sipper BK Experimental Animal (Shanghai, China). No randomization of weight and sex or blinding was used for animal studies. Dicer1d/d mice (6‐8 weeks) were kindly gifted by Dr. Jiahuai Han (Xiamen University, Xiamen, Fujian, China) [14]. All animal experiments were performed following the National Institute of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of Second Military Medical University (SMMU, Shanghai, China). Lipopolysaccharides (LPS) (0111:B4) (Sigma‐Aldrich, St. Louis, Missouri USA) stimulation was performed following previously described methods [15]. Recombinant mouse GM‐CSF and IL‐4 were obtained from PeproTech (London, UK); restriction enzyme Pst1 from NEB (Ipswich, Massachusetts, USA); H3K27me3 antibody from Millipore (Burlington, Massachusetts, USA); Mi‐2β antibody from Santa Cruz (Santa Cruz, California, USA); iNOS antibody from Cell Signaling Technology (Beverly, Massachusetts, USA).
2.2. Cell culture and transfection
Murine bone marrow‐derived DCs and thioglycolate‐elicited peritoneal macrophages were prepared and cultured as previously reported [16, 17]. The macrophages were seeded into plates and maintained at 37°C and 5% CO2 overnight, then transfected with sRNAs using the INTERFERin reagent (Polyplus, Strasbourg, France) according to the standard protocol. The sequences of 2'‐O‐methyl modified sdnRNA‐3 mimics and the mutation form for sdnRNA‐3 were 5'‐ACCACGAGGACGAGACGTAGCG‐3', and 5'‐ACTGCTAGGACGAGACGTAGCG‐3'. LNA‐modified inhibitor of sdnRNA‐3 (reverse complementary sequence) was customized from Exiqon (Dusseldorf, Germany).
2.3. Nuclear and cytoplasmic RNA isolation
The nuclear and cytoplasmic RNAs of mature DCs were extracted as described previously with some modifications [18]. Briefly, cytoplasmic and nuclear fractions of mature DCs were isolated as described, however, the small RNAs of the two fractions were extracted using the mirVana™ miRNA Isolation Kit (Ambion, Waltham, Massachusetts, USA).
2.4. Next‐Generation sequencing and data analysis
Cytoplasmic and nuclear sRNAs were extracted and subjected to high‐throughput sequencing using Illumina Solexa Sequencing Technology (San Diego, California, USA). Data were mainly analyzed using the Short Oligonucleotide Alignment Program (SOAP) [19]. Four steps of data analysis were performed. First, we collected short RNAs at 18‐30 nt length, called clean reads, and mapped them to the mouse genome to obtain specific and common sequences of sRNAs. Second, we blasted these sRNAs against known non‐coding RNAs deposited in the Rfam database [20], fRNAbd [21], miRbase database [22], and NCBI GenBank sequence database [23], and classified sRNAs into different categories according to their annotations. Third, sRNAs were calculated as log2 ratio using the normalized TPM (transcripts per million reads) value that was taken as the abundance of each sRNA for a rigorous significance test to determine the expression difference of each sRNA from nuclear and cytoplasmic fractions [24]. We then picked up the superior nucleus‐localized sRNAs according to the criteria that the abundance of each sRNA was over 20 and x>10y (x: abundance of sRNA from nuclear fraction; y: abundance of sRNAs from cytoplasm fraction). Next, we analyzed the length distribution, 5' and 3' end nucleotide bias of these RNAs by using the value of abundances. Lastly, according to proximal genomic location, highly similar fragments only with variations at their 5' and 3' terminus were classified into one cluster and were named as cluster1, cluster2, etc.
2.5. Reverse transcription‐quantitative PCR (RT‐qPCR)
Total RNAs were extracted using the TRIzol reagent (Invitrogen, Waltham, Massachusetts, USA) following the manufacturer's instructions and sRNAs were extracted by using mirVana™ miRNA Isolation Kit (Ambion, Waltham, Massachusetts, USA). RT‐qPCR analysis was performed using PrimeScript™ RT reagent Kit (Takara, Kyoto, Japan), SYBR real‐time PCR kit (Takara, Kyoto, Japan), and LightCycler (Roche, Basel, Switzerland). Nuclear and cytoplasmic RNAs isolated from equal numbers of macrophages were used to test sdnRNA‐3 abundance. The stem‐loop primers used for small RNAs were described previously with some specific modifications for the analyzed small RNAs [25]. Data for transcripts expression were normalized with U6 or GAPDH levels in comparison with control groups. When analyzing the expression difference of each sRNA from nuclear and cytoplasmic fractions, synthesized sRNA (5'‐TTCTCCGAACGTGTCACGT‐3') that was used as spike‐in, were added equally to the nuclear and cytoplasmic RNAs, and parallel reversed using specific primers together with detected sRNAs. The signals from the spike‐in were used to normalize the two fractions. Primer sequences for all the tested sRNAs, cytokines, and others were listed in Table S1.
2.6. RNA immunoprecipitation
RNA binding protein immunoprecipitation was performed using EZ‐Magna RIPTM RNA‐Binding Protein IP Kit (Millipore, Burlington, Massachusetts, USA). The immunoprecipitated RNAs were extracted from RNA‐protein/beads complex using the mirVana™ miRNA Isolation Kit (Ambion, Waltham, Massachusetts, USA).
2.7. Western blot
Cells were lysed using the M‐PERTM Protein Extraction Reagent (Pierce, Rockford, Illinois, USA), supplemented with a protease inhibitor cocktail (Roche, Basel, Switzerland). Protein concentrations of the extracts were measured using the BCA assay (Pierce, Rockford, Illinois, USA) and equalized with the extraction reagent. Equal amounts of the protein were subjected to SDS‐PAGE and transferred onto nitrocellulose membranes (GE Healthcare, Boston, Massachusetts, USA) for blotting with antibodies as described previously [26].
2.8. ChIP‐qPCR assay
The cells were cross‐linked and processed according to the Chromatin Immunoprecipitation (ChIP) Assay Kit (Millipore, Burlington, Massachusetts, USA) protocol. ChIP results were analyzed by qPCR using 2% of each input as standards. The primers for Nos2 promoter amplification were listed in Table S1.
2.9. Chromatin accessibility qPCR assay
Experiments were performed as previously described with some modifications [27, 28]. Isolated cell nuclei added with limiting amounts of restriction enzyme (Pst1 for Nos2 promoter) were incubated at 37°C for 20 min, followed by genomic DNA isolation. Primers amplifying the cut region spanning the Pst1 recognition site for qPCR were the same as those in the ChIP assay. Primers for the uncut region (without Pst1 recognition site) as internal control were listed in Table S1. Accessibility index was normalized and calculated as uncut%.
2.10. Promoter pull‐down
The 1.5Kb upstream region at Nos2 or Il6 (negative control) promoter was amplified with 5’‐biotinylated forward primer and 5’‐unmodified reverse primer (Table S1). Nuclear fraction of macrophages were lysed by 90 min incubation in two volumes of nuclear lysis buffer (420 mM NaCl, 20 mM HEPES pH 7.9, 20% v/v glycerol, 2 mM MgCl2, 0.2 mM EDTA, 0.1% NP40, protease, and RNase inhibitor and 0.5 mM DTT), and sonicated to improve lysis. 5‐biotinylated promoters were immobilized on Dynabeads MyOne C1 (Invitrogen, Waltham, Massachusetts, USA) by incubating for 30 min at room temperature in binding buffer (1 M NaCl, 10 mM Tris‐HCl pH 8, 1 mM EDTA pH 8, and 0.05% NP‐40). Beads containing immobilized probes were then incubated with nuclear extracts of macrophages in protein binding buffer (50 mM Tris‐HCl pH8, 150 mM NaCl, 1 mM DTT, 0.25% NP‐40 and complete protease and RNase inhibitors) for 30 min at 37°C and another 2 h at 4°C in the presence of poly‐dAdT (Sigma‐Aldrich, St. Louis, Missouri, USA). Complex‐containing beads were washed extensively (using incubation buffer without poly‐dAdT), and RNA fractions were extracted using the Trizol reagent.
2.11. Tumor allografts and macrophage isolation
For the B16 melanoma model, B16‐F10 cells were injected subcutaneously into the right flank of each mouse (5 × 105 cells per injection in 100 μL Phosphate Buffered Saline. Male and female C57BL/6 mice (6‐8 weeks) were used for tumor inoculation. Tumor volume was determined by measuring the length (D, the larger diameter of tumor) and width (d, the smaller diameter of tumor) of the tumor with a caliper, and calculated by D*d2*0.5. The mice were killed mercifully before the longest diameter of the tumor reaching 15 mm.
All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of Second Military Medical University (Shanghai, China). For single‐cell suspensions, the tumors were surgically removed followed by dissociation with surgical scissors and digestion with Collagenase IV (Worthington, Worthington, California, USA) and Deoxyribonuclease I (Roche, Basel, Switzerland) in Roswell Park Memorial Institute 1640 medium for 30 min in a 37°C shaking incubator (150 rpm). After red blood cell lysis, live/dead cell discrimination was performed using 7‐Aminoactinomycin D (ThermoFisher, Waltham, Massachusetts, USA). Fragment of crystallization block was performed by staining with anti‐CD16/32 to avoid non‐specific binding. Surface staining was performed at 4°C for 30 minutes in Fluorescence‐activated cell sorting (FACS) staining buffer (PBS containing 5% Fetal Bovine Serum and 0.5% sodium azide) containing designated antibody cocktails. Antibodies for FACS analysis were purchased from BioLegend (San Diego, California, USA).
2.12. Statistical analysis
Error bars displayed throughout the manuscript represent Standard Deviation (sd) and were calculated from triplicate technical replicates of indicated biological sample described in the figure legends. Sample sizes were chosen by standard methods to ensure adequate power, and no randomization of weight and sex or blinding was used for animal studies. No statistical method was used to predetermine sample size. Statistical significance was determined using two‐tailed and unpaired Student's t‐tests between the two groups, or using one‐way ANOVA when more than two groups by Excel's descriptive statistics (Microsoft, Redmond, Washington, USA); * P < 0.05; ** P < 0.01.
3. RESULTS
3.1. Different components of nuclear and cytoplasmic transcriptomes of small RNAs in innate immune cells
To systematically analyze the differences of sRNAs enriched in the nucleus and cytoplasm, we sequenced sRNAs isolated from the cytoplasm and nucleus of mouse bone marrow‐derived and LPS‐matured DCs [18]. Sequenced tags were then mapped to the mouse genome and classified into categories according to different matching genomic elements. Sequence analysis demonstrated a marked difference in the unique reads between the nuclear and cytoplasmic sRNA fractions. All sequenced unique reads contained 25.65% specific sRNAs in the cytoplasm, 68.55% in the nucleus, and 5.80% were cross‐possessive, indicating that sRNAs in the nuclear fraction were much different from those in the cytoplasmic fraction (Figure 1A). Through analysis of normalized abundances of sRNAs, miRNAs were observed to represent a much larger fraction in the cytoplasm than in the nucleus, which is supported by the prevailing view that most miRNAs localize and perform their functions in the cytoplasm, while other precursor‐derived sRNAs were more enriched in the nuclear fraction (Figure 1B). Further origin analysis revealed that, except sRNAs derived from tRNAs, sRNAs derived from other analyzed precursors were more abundant in the nuclear fraction (Figure 1C). Among these, sRNAs originating from repeat elements are described as repeat‐associated small interfering RNAs (rasiRNAs). Because piRNAs belong to rasiRNAs and perform important genome defensive and epigenetic regulatory functions in the nucleus, rasiRNAs were therefore selected as a representative class for sub‐origin analysis [29]. We found that rasiRNAs originating from long interspersed nuclear elements (LINE) and short interspersed nuclear elements (SINE) were more enriched in the nuclear fraction (Figure 1D).
FIGURE 1.
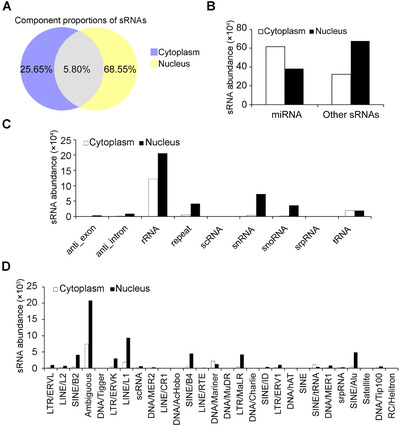
Analysis and comparison of sRNAs from the cytoplasm and nucleus of dendritic cells. (A) Component proportions of sRNAs with unique sequences in the cytoplasm and nucleus. (B) Abundances of miRNAs and the other sRNAs in the cytoplasm or nucleus. (C) Abundances of sRNAs among indicated sRNA origins in the cytoplasm and nucleus. (D) Abundances of repeat‐associated sRNAs classified by sub‐origin in the cytoplasm and nucleus. All the above analyses were from comparisons between cytoplasmic and nuclear fractions of sRNAs in dendritic cells.
miRNAs, microRNAs; sRNAs, small RNAs
3.2. snRNAs/snoRNAs‐derived nuclear RNAs (sdnRNAs) are most abundant among nucleus‐localized small RNAs in innate immune cells
In further analyses, hundreds of sRNAs were selected as nuclear‐localized sRNAs according to the criterion that x >10y (x: abundances of sRNAs in the nucleus, y: abundances of sRNAs in the cytoplasm) with the abundance of either sRNAs exceeding 20 (Table S2). Different from the length distribution of miRNAs with a significant peak at 22 nt [30], these sRNAs were predominantly 18‐24 nt in length (Figure 2A). Analysis of the 5' and 3' terminal nucleotide bias of these 18‐24 nt sRNAs revealed that a low C frequency, among all the size ranges, and a T bias appeared in the 5' termini of 18‐23 nt sRNAs, while the 3' termini of 19‐24 nt exhibited a low T frequency among all the size ranges and a high G frequency among the 19‐24 nt sRNAs (Figure 2B and C).
FIGURE 2.
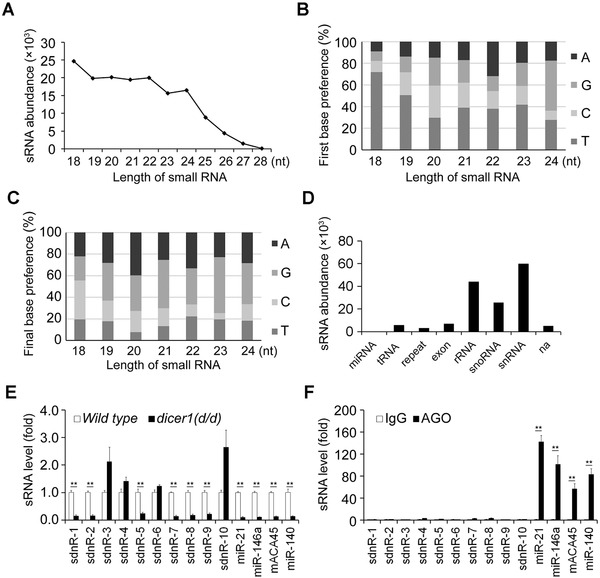
Features of nucleus‐localized sRNAs in dendritic cells. (A) Length distribution analysis of the sRNAs. (B) The proportions of first nucleotide bias in 18∼24 nt sRNAs. (C) The proportions of the last nucleotide bias in 18∼24 nt sRNAs. (D) The abundances of sRNAs in the indicated sRNA origins. (E) RT‐qPCR detection of relative abundances of indicated sRNAs in wild type and dicer1(d/d) mDCs. Data were normalized by U6 and compared with wild type group. (F) RT‐qPCR analysis of comparable abundances of indicated sRNAs in RNA immunoprecipitation (IP) fractions of AGO proteins compared to those in RNA IP fractions of IgG for mDCs. Enrichments were normalized with 1% RNA IP input from the same sample. Murine miRNA‐21 and miRNA‐146a were used as positive controls. Data were representative of three independent experiments. Error bars denoted SD. * P <0.05; ** P <0.01.
AGO, argonaute; IP, immunoprecipitation; mDCs, mature dendritic cells; na, not available;RT‐qPCR, reverse transcription‐quantitative polymerase chain reaction; sRNAs, small RNAs
Precursor analysis of these nucleus‐localized sRNAs indicated relatively high proportions of nucleus‐localized sRNAs derived from ribosomal RNAs (rRNA), snRNAs, and snoRNAs (Figure 2D). snRNAs and snoRNAs play a fundamental role in RNA splicing and modification mainly in the nucleus [31]. snRNAs and snoRNAs mainly localize in the nucleus and have hairpin containing secondary structures, so they are ideal precursors that may be directly processed into sRNAs in the nucleus. We analyzed sRNAs from snRNAs or snoRNAs together here. These snRNAs and/or snoRNAs‐derived sRNAs were most abundant among the nucleus‐localized sRNAs derived from all origins (Figure 2D). We designated these sRNAs as snRNA/snoRNA derived nuclear RNAs (sdnRNAs), regardless of whether they had been identified before as microRNAs. Some of these sRNAs exhibited a high degree of similarity with only a few variations at the 5' or 3' ends. Therefore, we clustered these similar sequences according to their overlapping genomic locations and selected the top 10 highly expressed sequences in different clusters (Table S3 and S4).
3.3. sdnRNAs are partially Dicer‐dependent and have no association with AGO proteins
Primary miRNAs are generally cleaved to form pre‐miRNAs by Drosha in the nucleus and exported to the cytoplasm where they are processed to mature miRNAs by Dicer [29]. Since the sdnRNAs were predominately localized in the nucleus, we investigated whether they were exclusively processed in a Dicer‐independent manner. Transcription abundances of sdnRNA1‐10 were analyzed by RT‐qPCR using specific stem‐loop primers [32]. Mature DCs were obtained from the variant Dicer1 allele (dicer1d/d) or wild type mice. The results demonstrated that the maturation of sdnRNA‐3, sdnRNA‐4, sdnRNA‐6, and sdnRNA‐10 were Dicer1‐independent, while the remainder were Dicer1‐dependent (Figure 2E). Dicer‐dependent sRNAs have always been considered to associate with Argonaute (AGO) family proteins. Further RNA IP analysis indicated that unlike microRNAs tested as the positive controls, all 10 sdnRNAs had no significant association with AGO proteins (Figure 2F).
AGO2‐associated sRNAs derived from snoRNAs were also reported: One sRNA, called ACA45, was Dicer‐dependent and was associated with AGO proteins in human cells [33]. The other one was identified as a miRNA, named miR‐140 [34]. We further investigated the difference between the AGO2‐associated sRNAs derived from snoRNAs and sdnRNAs. We found that although ACA45 and miR‐140 were also Dicer1‐dependent and associated with AGO proteins in murine DCs (Figure 2E and F), ACA45 and miR‐140 did not have much higher abundances in the nucleus than in the cytoplasm (Table S2). These results indicate that sRNAs derived from the same class of precursors may have different functions due to their different subcellular locations.
3.4. sdnRNA‐3 inhibits LPS‐induced iNOS expression in macrophages
As described above, miRNAs may perform their target‐specific transcription regulation in the nucleus through seed sequence‐dependent complementation with DNA sequences in some gene promoters. piRNAs, which are Dicer‐independent and do not associate with AGO proteins, perform epigenetic regulation in the nucleus. According to the functional features of such two classes of sRNAs, sdnRNA‐3 that has the highest abundance among the Dicer‐independent sdnRNAs was selected as the representative of sdnRNAs to perform the functional study.
According to our sequencing data, sdnRNA‐3 had two major isoforms with a 5' variation and position 3‐9 from the 5' end (Table S3). We further analyzed the expression of sdnRNA‐3 by measuring the most abundant form in sdnRNA‐3‐containing cluster. RT‐qPCR analysis revealed that there was no significant difference in sdnRNA‐3 expression between immature and mature DCs, sdnRNA‐3 was expressed even higher in mouse primary peritoneal macrophages (Figure 3A), indicating a more important function of sdnRNA‐3 in macrophages. sdnRNA‐3 was also nucleus‐enriched in macrophages, just as it was in mature DCs (Figure 3B and C). Thus, we investigated the regulatory role of sdnRNA‐3 in macrophages in the subsequent functional analysis. We screened the regulatory role of sdnRNA‐3 in the expression of M1‐associated innate immune mediators during LPS response using LNA‐modified sdnRNA‐3 inhibitor and found that inhibition of sdnRNA‐3 specifically increased the mRNA level of iNOS (encoded by Nos2 gene) (Figure 3D).
FIGURE 3.
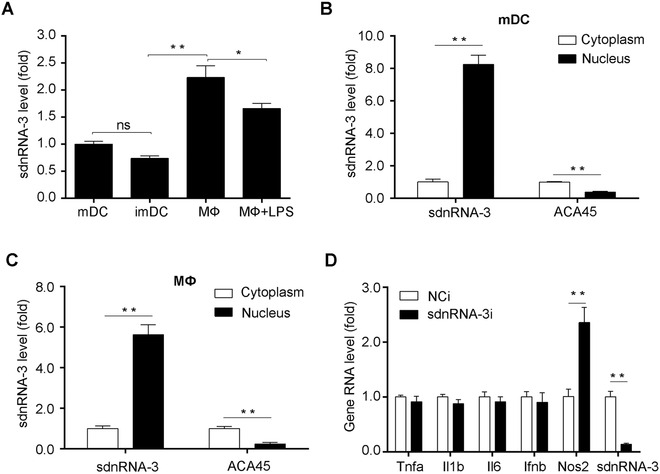
Expression of sdnRNA‐3 in DCs and macrophages. (A) RT‐qPCR detection of sdnRNA‐3 transcripts in imDCs and mDCs, mouse primary peritoneal macrophages (MФ) activated by 100 ng/mL LPS for 0h or 12h. (B, C) RT‐qPCR detection of nuclear and cytoplasmic sdnRNA‐3 and ACA45 transcripts in mDCs and MФ. ACA45 was the control for cytoplasm‐localized sRNAs. Data were normalized with synthesized sRNA (spike‐in) and were compared to cytoplasm groups. (D) MФ were transfected with LNA‐modified sdnRNA‐3 inhibitor (sdnRNA‐3i) and NCi for 36h. After the cells were stimulated by 100ng/mL LPS for 4 h, RNA levels of indicated genes were detected by RT‐qPCR. Data of RT‐qPCR analysis were normalized with GAPDH or U6 and compared to control groups. Data were representative of three independent experiments. Error bars denoted SD. * P <0.05; ** P <0.01.
imDCs, immature dendritic cells; LNA, locked nucleic acid; LPS, Lipopolysaccharides; mDCs, mature dendritic cells; MФ, mouse primary peritoneal macrophages; NCi, negative control inhibitor; ns, not significant; RT‐qPCR, reverse transcription‐quantitative polymerase chain reaction; sdnRNAs, snRNA/snoRNA‐derived nuclear RNAs
We further found that inhibiting sdnRNA‐3 increased the induction of iNOS mRNA expression by LPS stimulation at different time points, and transcription of Nos2 was also increased at the basal level within 1 hour after LPS stimulation in the presence of the sdnRNA‐3 inhibitor (Figure 4A). The same effects were observed for iNOS protein expression (Figure 4B). Transfection of 2'‐O‐methyl modified sdnRNA‐3 mimics into macrophages increased its endogenous level (Figure 4C). Overexpression of sdnRNA‐3 inhibited iNOS expression at both the mRNA and protein levels in LPS‐stimulated peritoneal macrophages, and partial mutation of the sdnRNA‐3 sequence significantly relieved this inhibitory effect (Figure 4D and E), indicating that sdnRNA‐3 regulates iNOS expression in a sequence‐dependent manner. These data indicate that sdnRNA‐3 can specifically inhibit Nos2 transcription in macrophages in response to innate signals such as LPS stimulation.
FIGURE 4.
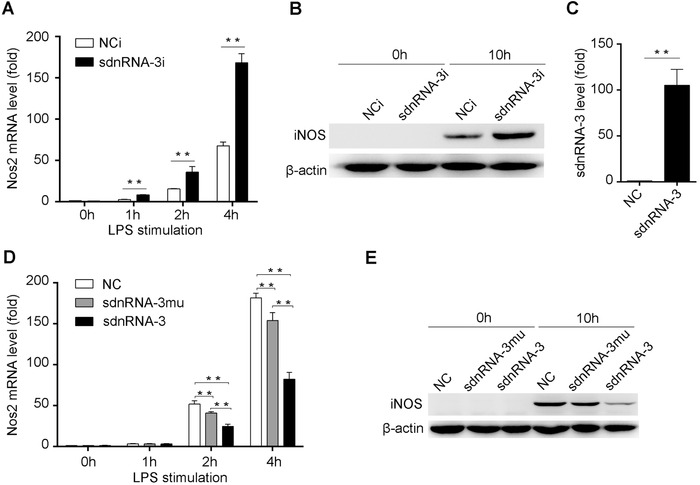
sdnRNA‐3 inhibits LPS‐induced iNOS expression in macrophages. (A, B) Peritoneal macrophages were transfected with LNA‐modified sdnRNA‐3 inhibitor (sdnRNA‐3i) and NCi for 36h. (C) Synthesized sdnRNA‐3 or negative control sRNAs were transfected into peritoneal macrophages for 24h, endogenous levels of sdnRNA‐3 were analyzed by RT‐qPCR. (D, E) Synthesized sdnRNA‐3, sequence partially mutant form of sdnRNA‐3 (sdnRNA‐3mu), and negative control sRNAs (NC) were transfected into peritoneal macrophages for 24h. After the cells were stimulated by 100ng/mL LPS at the indicated time points, iNOS (Nos2 gene) mRNA and protein levels were detected by RT‐qPCR and western blot, respectively. Data of RT‐qPCR analysis were normalized with GAPDH or U6 and compared to control groups. Data were representative of three independent experiments. Error bars denoted SD. * P <0.05; ** P <0.01.
LNA, locked nucleic acid; LPS, Lipopolysaccharides; NCi, negative control inhibitor; RT‐qPCR, reverse transcription‐quantitative polymerase chain reaction; sdnRNAs, snRNA/snoRNA‐derived nuclear RNAs
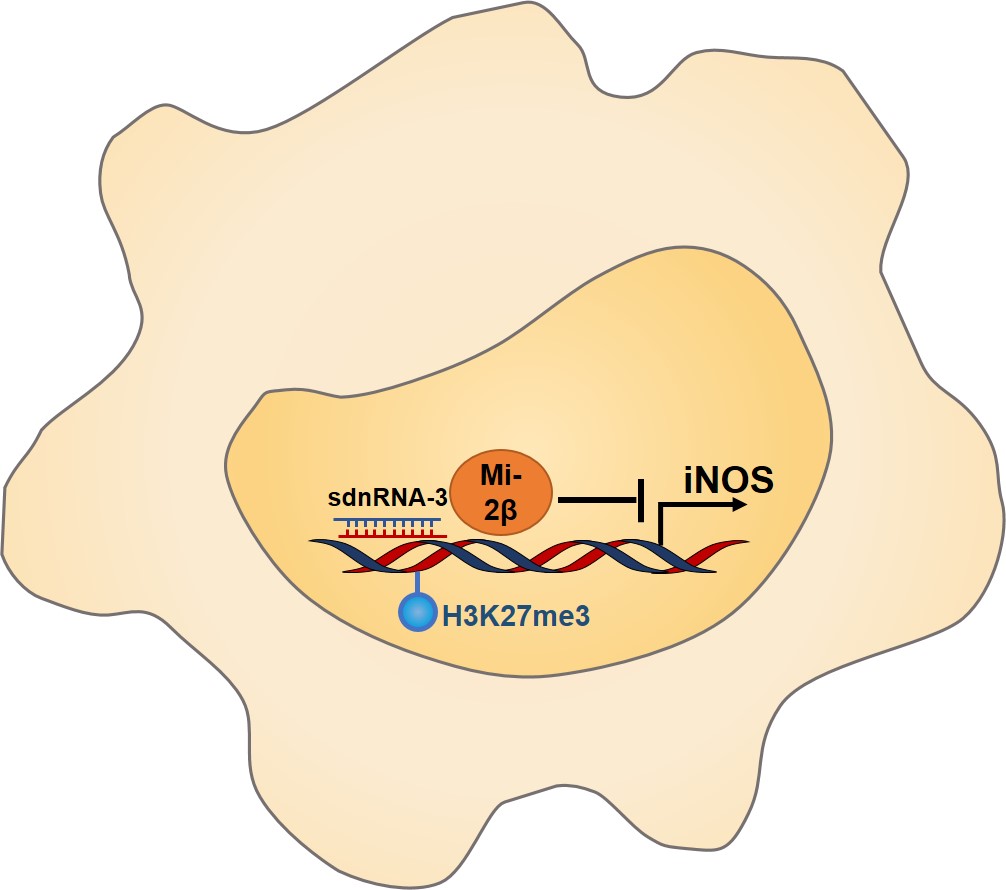
3.5. sdnRNA‐3 represses Nos2 transcription via recruitment of Mi‐2β and increase of H3K27me3
Nos2 is a secondary response gene and its promoter region has a closed chromatin structure in resting macrophages. Infection signal can increase the accessibility of Nos2 promoter for binding of transcription activators through epigenetic regulators. Among these regulators, chromodomain‐helicase‐DNA‐binding protein 4 (CHD4), also known as Mi‐2β, can specifically antagonize the opening of chromatin structure and limit the over‐activation of inflammatory mediators during pathogen infection. We speculated that sdnRNA‐3 might participate in these processes to mediate specific inhibition of iNOS expression by epigenetic regulators. Therefore, we studied the effect of sdnRNA‐3 on the enrichment of Mi‐2β to the Nos2 promoter. LPS stimulation increased the association of Mi‐2β with the promoter of Nos2, while overexpression of sdnRNA‐3 further increased the association of Mi‐2β with the Nos2 promoter during LPS response (Figure 5A). The sdnRNA‐3 inhibitor led to a decreased association of Mi‐2β with the Nos2 promoter before and after LPS stimulation compared to the control group (Figure 5B).
FIGURE 5.
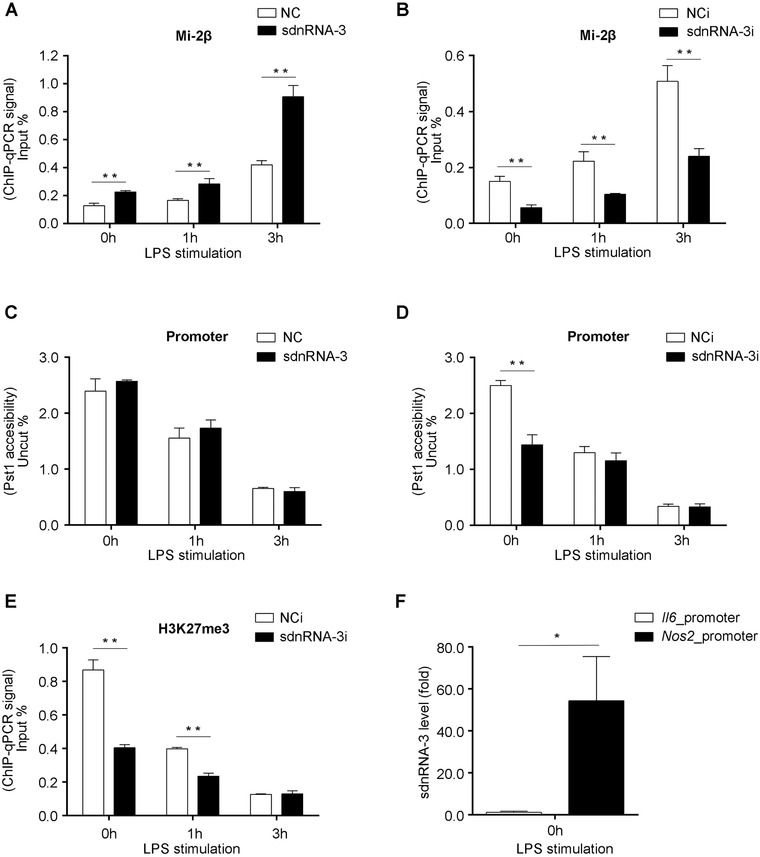
sdnRNA‐3 mediates epigenetic regulation of Nos2 promoter in macrophages. (A‐E) Peritoneal macrophages were transfected with synthesized sdnRNA‐3 and negative control sRNAs (NC) for 24h (A, C), or peritoneal macrophages were transfected with LNA‐modified sdnRNA‐3 inhibitor (sdnRNA‐3i) and NCi for 36h (B, D, E), and then, the cells were stimulated with 100ng/mL LPS at the indicated time points. ChIP‐qPCR was used to analyze Mi‐2β association (A, B), and H3K27me3 levels (E) in Nos2 promoter. Chromatin accessibility qPCR assay was performed using restriction enzyme Pst1 (C, D). ChIP‐qPCR results were quantified by input DNA (input %) from the same sample. Data of Chromatin accessibility qPCR were normalized to amplicons for the uncut region (uncut %). (F) Biotin‐labeled gene promoters were used for pulling down their‐associated small RNAs. The levels of sdnRNA‐3 were detected by RT‐qPCR, and the data were normalized by 1% input of nuclear fraction, compared with the normalized sdnRNA‐3 signal of the control group (Il6 promoter). Data were representative of three independent experiments. Error bars denoted SD. * P <0.05; ** P <0.01.
ChIP, chromatin immunoprecipitation; H3K27me3, trimethylated histone 3 lysine 27; LNA, locked nucleic acid; LPS, Lipopolysaccharides; NCi, negative control inhibitor; RT‐qPCR, reverse transcription‐quantitative polymerase chain reaction; sdnRNAs, snRNA/snoRNA‐derived nuclear RNAs
It was observed that inhibition of sdnRNA‐3 initiated Nos2 transcription at an earlier stage during LPS stimulation. Thus, we speculated that sdnRNA‐3 inhibition rendered the Nos2 promoter more sensitive to LPS stimulation and proposed that loss of sdnRNA‐3 could affect the closed chromatin structure of the Nos2 promoter in resting macrophages. Accordingly, we found that LPS stimulation increased accessibility of the Nos2 promoter, and sdnRNA‐3 inhibition could increase accessibility of the Nos2 promoter prior to LPS stimulation, while, overexpression of sdnRNA‐3 did not affect the Nos2 promoter structure (Figure 5C and D).
Although Nos2 promoter accessibility was not analyzed in a previous study, loss of Mi‐2β was reported to be insufficient to change the closed promoter structure of the secondary response genes [10]. Therefore, there may be other factors involved in repressing the expression of iNOS at the chromatin level. Trimethylated histone 3 lysine 27 (H3K27me3) is another chromatin silencing mediator. As H3K27me3‐mediated locus‐specific heterochromatin formation was reported to depend on siRNA‐mediated targeting in Tetrahymena [35], we investigated the role of H3K27me3 in chromatin regulation in resting and activated macrophages. LPS stimulation caused a prominent decrease in the H3K27me3 level at the Nos2 promoter. Before LPS stimulation, endogenous inhibition of sdnRNA‐3 reduced the H3K27me3 level at the Nos2 promoter compared to that in the control group. However, with the extension of stimulation time, the inhibitory effects of sdnRNA‐3 inhibitor on H3K27me3 levels declined (Figure 5E). Our results suggest that sdnRNA‐3 participates in the formation of a closed chromatin structure of the Nos2 promoter in resting macrophages, not only through recruiting Mi‐2β, but also by increasing the H3K27me3 level. Mi‐2β was previously reported to facilitate H3K27me3‐mediated gene silencing [36]. However, it is not known how these chromatin repression factors interact with each other to regulate the chromatin status of Nos2 promoter. To further investigate the gene‐specific regulation of iNOS by sdnRNA‐3, we used biotin‐labeled Nos2 promoter (0∼‐1.5Kb upstream transcription start site) as bait for pulling down its‐associated molecules in the nuclear fraction of macrophages and detected the existence of sdnRNA‐3 in pulled‐down fractions (Figure 5F).
3.6. Tumor‐promoting effect of M2 tumor‐associated macrophages with high expression of sdnRNA‐3
High expression of iNOS is a marker of M1 relative to M2, especially in the tumor microenvironment [37]. Expression of iNOS in tumor‐associated macrophages can suppress tumor growth and invasion by producing nitric oxide (NO) [38, 39]. To investigate the potential role of sdnRNA‐3 in tumor immunity, we established a B16 melanoma‐bearing mouse model. We isolated DCs and macrophage subsets from tumor tissues based on cell markers of DC (CD45+ (CD3/NK1.1/CD19/F4/80)– IA/E+ CD11c+), M1 (CD45+ (CD4/CD8/CD49/Siglec‐F)– Ly6G– Ly6Clow F4/80+ IAEhi) or M2 (CD45+ (CD4/CD8/CD49/Siglec‐F)– Ly6G– Ly6Clow F4/80+ IAElow), and found highest expression of sdnRNA‐3 in M2 TAMs, and lowest expression in DCs among the three tumor‐associated immune cell types (Figure 6A). Inhibition of sdnRNA‐3 in tumor‐associated M2 also increased the mRNA level of iNOS (Figure 6B). Furthermore, we tested the effect of sdnRNA‐3 on tumor growth using macrophage transfer analysis. Peritoneal macrophages transfected with control or sdnRNA‐3 inhibitor were transferred into tumor‐bearing mice every 4 days from day 0. We observed that adoptive transfer of macrophages with sdnRNA‐3 inhibition could inhibit tumor growth significantly, as compared to the control group (Figure 6C). Increased mRNA levels of iNOS were also observed in M2 TAMs isolated from the B16 allografts transferred with sdnRNA‐3‐inhibited macrophages, compared with the controls (Figure 6D). Taken together, these results primarily indicate that high expression of sdnRNA‐3 in M2 TAMs may contribute to the tumor‐promoting effect of M2 TAMs.
FIGURE 6.
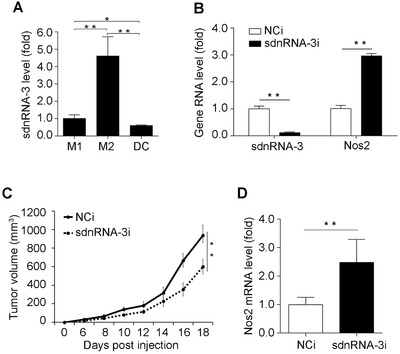
sdnRNA‐3 in macrophages promotes tumor growth by repressing Nos2 transcription. (A) RT‐qPCR analysis of sdnRNA‐3 in M1 and M2 TAMs, and DCs isolated from B16 allograft (n = 3). Data were normalized by U6 and compared with the M1 group. (B) M2 TAMs isolated from B16 allograft were transfected with LNA‐modified sdnRNA‐3 inhibitor (sdnRNA‐3i) and NCi for 36h, RNA levels of indicated genes were detected by RT‐qPCR. (C, D) Peritoneal macrophages were transfected with LNA‐modified sdnRNA‐3 inhibitor (sdnRNA‐3i) and NCi for 24h. 5 × 106 cells were injected intravenously into each recipient mouse (n = 6) every 4 days from day 0 when B16‐F10 cells were transplanted. Animals were monitored regularly for tumor growth (C). RNAs of M2 TAMs isolated from B16 allografts at day 18 were subjected for RT‐qPCR analysis of indicated gene RNAs (D). Data of RT‐qPCR analysis were normalized with GAPDH or U6 and compared to control groups. Data were representative of three independent experiments. Error bars denoted SD. * P <0.05; ** P <0.01.
DCs, dendritic cells; LNA, locked nucleic acid; M1, classically activated macrophage; M2, alternatively activated macrophage; NCi, negative control inhibitor; RT‐qPCR, reverse transcription‐quantitative polymerase chain reaction; sdnRNAs, snRNA/snoRNA‐derived nuclear RNAs; TAMs, tumor‐associated macrophages
4. DISCUSSION
In addition to miRNA, siRNA, and piRNA with limited functions, some other types of sRNAs derived from different precursors and their potential function have been reported [40, 41]. Although the functions of such sRNAs remain largely elusive, such sRNAs play regulatory roles in different biological processes, compared with miRNA and siRNA which are Dicer‐dependent. Different subcellular localization may determine their special biological functions. Although previous studies analyzed subcellular sRNAs in metazoans, the functions of these differently localized sRNA have not been fully uncovered [42, 43]. In this study, we established cytoplasmic and nuclear small RNomes in innate immune cells and identified nuclear‐localized small RNAs that originated differently from miRNAs in cytoplasmic fractions. Through systematic analysis, we found a new class of nuclear sRNAs derived from snRNAs/snoRNAs and named them as sdnRNAs. In addition to the discovery of such sdnRNAs, we chose sdnRNA‐3 as representative and investigated its potential function in innate immunity. sdnRNAs‐3‐mediated gene‐specific epigenetic repression of iNOS, an innate immune mediator, is also implicated in the function of the M2 TAMs in tumor immunity. Our nuclear small RNomes and the functional study on sdnRNAs indicate a novel class of small RNAs implicated in gene‐specific epigenetic regulation.
When considering the processing and regulating mechanisms of sdnRNAs, we classified newly‐discovered sdnRNAs into two subclasses: Dicer‐independent and Dicer‐dependent. For Dicer‐independent sdnRNAs, we found one of them could mediate gene‐specific epigenetic regulation in a sequence‐dependent manner. Our discovered sdnRNAs, which do not associate with AGO proteins, may have new functions through different intermediate mediators. However, these sdnRNAs‐associated functional proteins and the underlying mechanism for gene‐specific targeting have not been identified in this current study. Furthermore, whether these sRNAs are processed directly in the nucleus is waiting to be elucidated. For the Dicer‐dependent sdnRNAs, similar to previously reported snoRNA derived RNAs (sdRNAs) in lower organisms [44], they also do not associate with AGO proteins, different from microRNAs which associates with RNA Induced Silencing Complex (RISC) in the cytoplasm [45]. Thus, we thought they are a new class of sRNAs different from miRNA‐like sdRNAs, and AGO proteins and their associated sdRNAs‐mediated regulation may not be the main mechanism for gene‐specific epigenetic regulation in the nucleus of innate immune cells. Moreover, the underlying mechanisms for importing such Dicer‐dependent sdnRNAs back to the nucleus after they are processed by Dicer1 in the cytoplasm and their potential functions in the nucleus need to be further elucidated.
As a secondary response gene, chromatin remodeling by BRG1‐containing SWI/SNF complex is required for transcription induction of Nos2 gene. On the other hand, the Mi‐2β complex acts antagonistically to limit the induction of the Nos2 gene [10]. The underlying mechanisms for either maintaining a closed chromatin structure at promoters of secondary response genes, or targeting of chromatin remodeling complex are still elusive. In this study we investigated a negative role of sdnRNA‐3 in chromatin remodeling at Nos2 promoter, indicating that sdnRNAs in innate immune cells may be involved in decreasing the chromatin accessibilities at promoters of secondary response genes. sdnRNAs may directly or indirectly associate with and recruit repressive chromatin modifiers to these gene promoters in a sequence‐dependent manner, promoting the deposition of negative histone modifications there.
As an essential functional system in mammals, the immune system may have its own special sRNAs in determining immunological functions. In this study, we found a new class sdnRNAs, which were most abundant among our identified nuclear‐localized sRNAs in DCs. Although the abundances of sdnRNAs in other living systems have not been determined, we speculate that sdnRNAs may be a major class of special nuclear sRNAs in the innate immune system. DCs and macrophages play important roles not only in initiating innate immunity but also in regulating anti‐tumor immunity. Especially, macrophage subsets play different regulatory roles in tumor immunity depending on the expression of different immune‐modulating effectors under different polarization states. The epigenetic mechanisms underlying the expression of effectors, promoting or suppressing tumor growth and invasion in different macrophage subsets remain elusive. In this study, using a B16 melanoma‐bearing mouse model, we found a higher level of sdnRNA‐3 in M2 TAMs than in M1 cells, indicating that the repression of iNOS expression by sdnRNA‐3 was also involved in establishing M2‐specific gene expression pattern for tumor growth. High expression of iNOS is also a marker of M1 phenotype, although iNOS plays dual roles in tumor immunity [46]. High expression of iNOS in TAMs was reported to raise CD4+ Th1 chemokines and inhibit immune suppressive cytokines [38], in accordance with our investigation of decreased tumor growth when transferring sdnRNA‐3‐inhibited macrophages with increased expression of iNOS.
Our study revealed a small RNA‐mediated novel mechanism of genes‐specific epigenetic regulation of immune effectors in macrophages via the recruitment of Mi‐2β and increase of H3K27me3. Although we only used sdnRNA‐3 as an sdnRNA model in the functional studies, we thought that other infection‐induced molecules such as some cytokines and innate mediators may be also the targets of sdnRNAs with different sequences. Whether there are “seed” sequences in sdnRNAs like those in miRNAs or other mechanisms that exist for the gene‐specific targeting also need further investigation.
5. CONCLUSIONS
A new class of dicer‐independent small RNAs that are derived from snRNAs or snoRNAs localize in the nucleus of innate immune cells. Choosing sdnRNA‐3 as the representative for functional study, we found that sdnRNA‐3 inhibited transcription of Nos2 gene (encode iNOS) by decreasing chromatin accessibility of gene promoter in macrophages. Furthermore, we found that the inhibition of sdnRNA‐3 in tumor‐associated macrophages could repress tumor growth.
DECLARATIONS
CONSENT FOR PUBLICATION
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal experiments were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and the approval of the Scientific Investigation Board of Second Military Medical University (SMMU, Shanghai, China).
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interest.
AUTHORSHIP CONTRIBUTIONS
XTC and QZ designed the research; YS, QZS, QCS, and QZ performed research; QZ performed the bioinformatic analyses; XTC, QZ, YS, and QZS analyzed data and wrote the paper.
AVAILABILITY OF DATA AND MATERIALS
Sequencing data have been deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE162861), with the GEO accession number of GSE162861. All the other data of this study are available within the article and the supplementary materials and from the corresponding author upon reasonable request.
Supporting information
Supplementary Information
Supplementary Information
{kind=link}
ACKNOWLEDGMENTS
The authors thank Dr. Jiahuai Han for kindly providing Dicer1 allele (dicer1d/d) mice, and thank Fei Teng and Ning Li from Beijing Genomics Institute (BGI) for helping bioinformatics analyses. This work was supported by the National Natural Science Foundation of China (81922032, 31900660, 81788101) and the Young Elite Scientist Sponsorship Program by CAST (2018QNRC001).
Shi Y, Shi Q, Shen Q, Zhang Q, Cao X. Dicer‐independent snRNA/snoRNA‐derived nuclear RNA 3 regulates tumor‐associated macrophage function by epigenetically repressing inducible nitric oxide synthase transcription. Cancer Commun. 2021;41:140–153. 10.1002/cac2.12131
Contributor Information
Qian Zhang, Email: qianzhang@immunol.org.
Xuetao Cao, Email: caoxt@immunol.org.
REFERENCES
- 1. Alvarez‐Errico D, Vento‐Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. 2015;15(1):7‐17. [DOI] [PubMed] [Google Scholar]
- 2. Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20(4):207‐20. [DOI] [PubMed] [Google Scholar]
- 3. Anastasiadou E, Jacob LS, Slack FJ. Non‐coding RNA networks in cancer. Nat Rev Cancer. 2018;18(1):5‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Holoch D, Moazed D. RNA‐mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16(2):71‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA‐373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A. 2008;105(5):1608‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. Senescence is an endogenous trigger for microRNA‐directed transcriptional gene silencing in human cells. Nat Cell Biol. 2012;14(3):266‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA‐induced transcriptional gene silencing in human cells. Science. 2004;305(5688):1289‐92. [DOI] [PubMed] [Google Scholar]
- 8. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487‐500. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol. 2019;19(7):417‐32. [DOI] [PubMed] [Google Scholar]
- 10. Ramirez‐Carrozzi VR, Nazarian AA, Li CC, Gore SL, Sridharan R, Imbalzano AN, et al. Selective and antagonistic functions of SWI/SNF and Mi‐2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20(3):282‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pathria P, Louis TL, Varner JA. Targeting Tumor‐Associated Macrophages in Cancer. Trends Immunol. 2019;40(4):310‐27. [DOI] [PubMed] [Google Scholar]
- 12. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer. 2019;19(4):215‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Otsuka M, Jing Q, Georgel P, New L, Chen J, Mols J, et al. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1‐deficient mice is due to impaired miR24 and miR93 expression. Immunity. 2007;27(1):123‐34. [DOI] [PubMed] [Google Scholar]
- 15. Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, et al. Induction of Siglec‐G by RNA Viruses Inhibits the Innate Immune Response by Promoting RIG‐I Degradation. Cell. 2013;152(3):467‐78. [DOI] [PubMed] [Google Scholar]
- 16. Xu S, Liu X, Bao Y, Zhu X, Han C, Zhang P, et al. Constitutive MHC class I molecules negatively regulate TLR‐triggered inflammatory responses via the Fps‐SHP‐2 pathway. Nat Immunol. 2012;13(6):551‐9. [DOI] [PubMed] [Google Scholar]
- 17. An H, Hou J, Zhou J, Zhao W, Xu H, Zheng Y, et al. Phosphatase SHP‐1 promotes TLR‐ and RIG‐I‐activated production of type I interferon by inhibiting the kinase IRAK1. Nat Immunol. 2008;9(5):542‐50. [DOI] [PubMed] [Google Scholar]
- 18. Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315(5808):97‐100. [DOI] [PubMed] [Google Scholar]
- 19. Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966‐7. [DOI] [PubMed] [Google Scholar]
- 20. Kalvari I, Argasinska J, Quinones‐Olvera N, Nawrocki EP, Rivas E, Eddy SR, et al. Rfam 13.0: shifting to a genome‐centric resource for non‐coding RNA families. Nucleic Acids Res. 2018;46(D1):D335‐D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kin T, Yamada K, Terai G, Okida H, Yoshinari Y, Ono Y, et al. fRNAdb: a platform for mining/annotating functional RNA candidates from non‐coding RNA sequences. Nucleic Acids Res. 2007;35(Database issue):D145‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kozomara A, Birgaoanu M, Griffiths‐Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155‐D62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sayers EW, Beck J, Bolton EE, Bourexis D, Brister JR, Canese K, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li X, Chen J, Hu X, Huang Y, Li Z, Zhou L, et al. Comparative mRNA and microRNA expression profiling of three genitourinary cancers reveals common hallmarks and cancer‐specific molecular events. PLoS One. 2011;6(7):e22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang P, Gu Y, Zhang Q, Han Y, Hou J, Lin L, et al. Identification of resting and type I IFN‐activated human NK cell miRNomes reveals microRNA‐378 and microRNA‐30e as negative regulators of NK cell cytotoxicity. J Immunol. 2012;189(1):211‐21. [DOI] [PubMed] [Google Scholar]
- 26. Liu X, Zhan Z, Li D, Xu L, Ma F, Zhang P, et al. Intracellular MHC class II molecules promote TLR‐triggered innate immune responses by maintaining activation of the kinase Btk. Nat Immunol. 2011;12(5):416‐24. [DOI] [PubMed] [Google Scholar]
- 27. Schor IE, Rascovan N, Pelisch F, Allo M, Kornblihtt AR. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci U S A. 2009;106(11):4325‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11(11):1068‐75. [DOI] [PubMed] [Google Scholar]
- 29. Farazi TA, Juranek SA, Tuschl T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development. 2008;135(7):1201‐14. [DOI] [PubMed] [Google Scholar]
- 30. Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18(4):610‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matera AG, Terns RM, Terns MP. Non‐coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol. 2007;8(3):209‐20. [DOI] [PubMed] [Google Scholar]
- 32. Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR‐199a/b‐3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19(2):232‐43. [DOI] [PubMed] [Google Scholar]
- 33. Ender C, Krek A, Friedlander MR, Beitzinger M, Weinmann L, Chen W, et al. A human snoRNA with microRNA‐like functions. Mol Cell. 2008;32(4):519‐28. [DOI] [PubMed] [Google Scholar]
- 34. Scott MS, Avolio F, Ono M, Lamond AI, Barton GJ. Human miRNA precursors with box H/ACA snoRNA features. PLoS Comput Biol. 2009;5(9):e1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, Allis CD. RNAi‐dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 2007;21(12):1530‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morey L, Brenner C, Fazi F, Villa R, Gutierrez A, Buschbeck M, et al. MBD3, a component of the NuRD complex, facilitates chromatin alteration and deposition of epigenetic marks. Mol Cell Biol. 2008;28(19):5912‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low‐dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589‐602. [DOI] [PubMed] [Google Scholar]
- 39. Sektioglu IM, Carretero R, Bender N, Bogdan C, Garbi N, Umansky V, et al. Macrophage‐derived nitric oxide initiates T‐cell diapedesis and tumor rejection. Oncoimmunology. 2016;5(10):e1204506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Z, Ender C, Meister G, Moore PS, Chang Y, John B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012;40(14):6787‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rosace D, Lopez J, Blanco S. Emerging roles of novel small non‐coding regulatory RNAs in immunity and cancer. RNA Biol. 2020;17(8):1196‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taft RJ, Simons C, Nahkuri S, Oey H, Korbie DJ, Mercer TR, et al. Nuclear‐localized tiny RNAs are associated with transcription initiation and splice sites in metazoans. Nat Struct Mol Biol. 2010;17(8):1030‐4. [DOI] [PubMed] [Google Scholar]
- 43. Liu H, Lei C, He Q, Pan Z, Xiao D, Tao Y. Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol Cancer. 2018;17(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS. Small RNAs derived from snoRNAs. RNA. 2009;15(7):1233‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol. 2016;16(5):279‐94. [DOI] [PubMed] [Google Scholar]
- 46. Vannini F, Kashfi K, Nath N. The dual role of iNOS in cancer. Redox Biol. 2015;6:334‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Information
Data Availability Statement
Sequencing data have been deposited in the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE162861), with the GEO accession number of GSE162861. All the other data of this study are available within the article and the supplementary materials and from the corresponding author upon reasonable request.