

Abstract
Neuroblastoma (NB) is a pediatric cancer of the sympathetic nervous system and one of the most common solid tumors in infancy. Amplification of MYCN, copy number alterations, numerical and segmental chromosomal aberrations, mutations, and rearrangements on a handful of genes, such as ALK, ATRX, TP53, RAS/MAPK pathway genes, and TERT, are attributed as underlying causes that give rise to NB. However, the heterogeneous nature of the disease—along with the relative paucity of recurrent somatic mutations—reinforces the need to understand the interplay of genetic factors and epigenetic alterations in the context of NB. Epigenetic mechanisms tightly control gene expression, embryogenesis, imprinting, chromosomal stability, and tumorigenesis, thereby playing a pivotal role in physio- and pathological settings. The main epigenetic alterations include aberrant DNA methylation, disrupted patterns of posttranslational histone modifications, alterations in chromatin composition and/or architecture, and aberrant expression of non-coding RNAs. DNA methylation and demethylation are mediated by DNA methyltransferases (DNMTs) and ten-eleven translocation (TET) proteins, respectively, while histone modifications are coordinated by histone acetyltransferases and deacetylases (HATs, HDACs), and histone methyltransferases and demethylases (HMTs, HDMs). This article focuses predominately on the crosstalk between the epigenome and NB, and the implications it has on disease diagnosis and treatment.
Keywords: Epigenetics, DNA methylation, Histone modifications, MicroRNAs, Chromatin remodeling, Neuroblastoma
Introduction
The biological basis of neuroblastoma and the importance of the epigenome to NB
Neuroblastoma is a developmental neoplasm of the autonomic nervous system that primarily affects young children [1–3]. A hallmark of NB is its heterogeneous clinical presentation, ranging from tumors that spontaneously regress (~ 50% of infants) to widely disseminated tumors that are frequently resistant to multimodal treatments like chemoradiotherapy or a combination of stem cell transplantation and immunotherapy in the other half of the subjects [4–7]. In the latter patient cohort, namely high-risk NB, the survival rate is < 40% [6, 8], highlighting the elusive nature of the disease. Cancer is caused by heritable aberrations in genes that regulate cell division, proliferation, and death [9–12]. Such critical genetic factors underlying NB onset and progression include amplification of MYCN, deletions of TP53, mutations or amplifications of ALK, rearrangements of TERT, deletions or mutations of ATRX, and segmental chromosomal aberrations [1, 13–19]. However, recent whole-genome sequencing studies have identified a scarcity of recurrent somatic alterations [13, 14, 20], which has hampered the efforts to develop targeted therapeutics for all NB patients. Therefore, the primary challenge in the identification and validation of diagnostic tools and treatment agents is the accurate representation of NB biology and diversity.
Malignancies generally involve both the genetic and epigenetic components that work in a concerted and multilayered fashion. Epigenetic mechanisms, such as DNA methylation, histone modifications, and non-coding RNAs, coordinate the maintenance of the epigenome [21, 22]. Together, they determine whether chromatin is transcriptionally permissive or repressive. Changes in this balance can, in turn, alter transcriptional programs that promote cancer progression by increasing cancer cell plasticity or by directly silencing tumor suppressor genes [12, 21, 23]. Several lines of evidence in various cancer models have demonstrated that aberrations in the epigenome largely occur by disrupting the epigenetic machinery and are strongly correlated with cancer progression [24]. Large-scale genome sequencing projects have shown that roughly 50% of human cancers harbor mutations in chromatin proteins, causing malignant cells to exhibit genome-wide alterations in DNA methylation, chromatin structure, and regulatory element activities [25–27]. These findings not only imply a causative role for epigenetics in tumorigenesis, but also help to identify potential therapeutic targets.
Regulation of oncogenes and tumor suppressor genes by the epigenome
Epigenetic mechanisms are processes that have been associated with growth and development, and any dysregulations in this balance result in disease onset and progression. Aberrant gene expression as a result of the perturbed epigenome is a frequent event in cancer [21, 27]. DNA methylation primarily affects cytosines in the context of CpG dinucleotides, which is generally correlated with transcriptional repression [28, 29]. Other key chromatin remodeling activities include histone modifications, which undergo covalent posttranslational modifications (e.g., acetylation, methylation, and phosphorylation). While loss of histone acetylation has been established as a mark that is inversely correlated with gene expression, histone methylation can lead to either gene activation or silencing, depending on the degree of methylation (mono-, di-, or tri-methylation), type of amino acid residue impacted, and their location in the histone tails [30, 31]. Finally, microRNAs, which are short non-coding RNAs (17-22nt), negatively regulate gene expression, primarily by interacting with the 3′untranslated regions (UTR) of mRNAs [32]. Jointly, the abovementioned chromatin and gene regulatory mechanisms, and their role in oncogenesis in the context of NB will be the focus of this review. We will also discuss the current and emerging drug therapies that target these epigenetic regulators.
Role of DNA methylation in gene expression in neuroblastoma
DNA methylation at the fifth position of cytosine (5mC) in CpG dinucleotides is a major epigenetic mechanism involved in genome programming and reprogramming, and determination of cell fate during development, thus any disturbances in DNA methylation may give rise to disorders, such as cancer [21, 27]. Near gene promoters, transcription start sites, and/or first exons, CpG dinucleotides cluster in short DNA stretches, which are referred to as CpG islands (CGI), and most (80–95%) of them remain devoid of DNA methylation [33]. The majority of the CpG sites occur in intergenic regions and repetitive genomic sequences to maintain a transcriptionally inactive state [12]. Changes in the cancer epigenome are generally associated with loss of global DNA methylation and gain of DNA methylation at specific gene promoters. The consequences of loss of global methylation include chromosomal instability, loss of imprinting, and activation of transposable elements, thereby leading to disturbances in the genome [11, 22, 27] as exemplified in Fig. 1. Genome instability causes cancer genotypes to continuously change and evolve and can manifest itself genetically on several different levels, ranging from simple DNA sequence changes to structural and numerical abnormalities at the chromosomal level, all of which are observed in NB [34, 35].
Fig. 1.

Proposed structural changes of chromatin in neuroblastoma cells. In normal cells, the chromatin structure is tightly regulated by DNA methylation and histone modifications, ensuring proper gene activation/silencing as well as genome integrity. The epigenome of NB cells is characterized by abnormally global open nucleosome configuration (euchromatin), interspersed with silenced genes (heterochromatin). Abbreviations: amp, amplification; mut, mutation; rearrang, rearrangement; del, deletion. Chromosome drawing was adapted from https://smart.servier.com/
Understanding the impact of normal and aberrant DNA methylation is one key area of interest, considering that drugs that result in DNA hypomethylation have already been approved by the US Food and Drugs Administration (FDA) and/or the European Medicines Agency (EMA) for the treatment of certain malignancies. DNA methylation is carried out by the DNA methyltransferases (DNMTs) [36]. During cell division, DNA methylation is established by DNMT1 or the maintenance DNA methyltransferase, while DNMT3A&3B are responsible for the de novo methylation. Co-transfection of Dnmt3a&3b in murine NB cell lines was associated with increased cisplatin resistance, while inhibition of these enzymes with the DNMT inhibitor, 5′-azacytidine (5-aza), resulted in an increased cisplatin response [37]. Treatment of mouse and human NB cell lines with another DNMT inhibitor, 5-aza-2′-deoxycytidine (DAC), led to reduced DNA methylation levels and inhibited DNA synthesis, cell proliferation, and colony-forming activity [38, 39]. Furthermore, treatment of NB cell lines with DAC and chemotherapeutic drugs, including cisplatin, doxorubicin, and etoposide, resulted in a superior therapeutic outcome compared to the treatments without DAC [40]. DAC, also known as decitabine has been approved for the treatment of several hematological malignancies. A phase I clinical trial showed that low doses of this drug in combination with cyclophosphamide have tolerable toxicity levels, however, the doses of DAC needed to produce a clinically relevant response in NB tumors are far higher than the physiologically tolerable levels [41]. Use of SGI-1027, a DNMT pan-inhibitor (DNMT1/DNMT3A/DNMT3B), and nanomycin A, a DNMT3B inhibitor in NB cell lines, resulted in a higher cytotoxicity when given alone or in combinatorial treatment with doxorubicin, and this effect was independent of the MYCN amplification status [42]. In ganglioneuroblastoma, the expression of DNMT3B7, which is a truncated isoform of DNMT3B, is higher than in NB patients, and this results in reduced cell growth and induced differentiation. Analysis of the transcriptome of the DNMT3B7 overexpressing cells showed upregulation of genes involved in the retinoic acid pathway, and treatment with all-trans retinoic acid-induced NB cell differentiation [43]. Together, these studies suggest that inhibition of DNMTs promotes a phenotype that is characterized by increased cell differentiation and reduced proliferation, however, it remains to be seen how this would translate clinically in patients. Currently, three clinical trials have completed phase I studies for decitabine in NB, and a fourth study is ongoing (phase II) for genistein (Table 1).
Table 1.
Completed and ongoing clinical trials for DNMT and HDAC inhibitors in neuroblastoma (mentioned in this article)
| Category | Drug name | Clinical trial phase | Clinical trial ID |
|---|---|---|---|
| DNMT inhibitors | Decitabine | Phase I | NCT01241162 |
| Phase I | NCT00075634 | ||
| Phase I | NCT03236857 | ||
| Genistein | Phase II | NCT02624388 | |
| HDAC inhibitors | Vorinostat | Phase I | NCT00217412 |
| Phase I | NCT01132911 | ||
| Phase I | NCT01019850 | ||
| Phase I | NCT01208454 | ||
| Phase I | NCT04308330 | ||
| Phase II | NCT02035137 | ||
| Phase II | NCT02559778 |
DNMT, DNA methyltransferase; HDAC, histone deacetylase
Several studies in recent decades have used DNA methylation to distinguish healthy cells from diseased cells. Studies using targeted bisulfite sequencing for candidate genes and the various array platforms have suggested dysregulated methylation in NB also. Loss of apoptotic features by cancer cells is a commonly observed event in tumorigenesis. A study focused on investigating gene-specific hypermethylation as an alternative mechanism for loss of caspase 8 (CASP8) expression in NB cases that otherwise do not display deletions on this gene. Using methylation-sensitive PCR, the authors showed that in NB cell lines, loss of CASP8 expression was associated with increased levels of DNA methylation. Treatment with the DNA-demethylating drug, 5-aza, restored the expression of CASP8 in 2/3 of investigated cell lines. Caspase 8 is involved in apoptosis, and loss of its expression, exclusively in MYCN-amplified NB tumors, allows for unhinged cell proliferation. The cell line data were validated in a NB patient cohort as well [44]. Other genes involved in regulation of apoptosis include the anti-apoptotic decoy receptors (DcRs), DcR1 and DcR2. Methylation analyses in cell lines and NB patient samples showed dense methylation on these genes as a mechanism, which was linked to their loss of expression [45]. Interrogation of the methylation levels of 45 candidate genes in NB cell lines and tumors revealed, among others, another proapoptotic gene, TMS1, to be hypermethylated, and this was typically observed in stage 4 tumors [46]. Other studies analyzing the methylation levels of tumor suppressor genes showed loss of RASSF1A expression by promoter methylation [47, 48]. Additional studies focusing on nine specific tumor suppressor genes in cancer interrogated methylation levels at the CpG islands of these genes in NB patient samples. In addition to RASSF1A, the study identified BLU and DKFZp451I127, which were marked by aberrant methylation in NB. Methylation levels of PCDHB CGIs were used as a defining measure for the CpG island methylator phenotype (CIMP), a high-risk NB phenotype, and this was marked by poor overall survival (OS) in a Japanese cohort [49]. Similarly, another study showed that aberrant methylation of the PCDHB family members is associated with other clinical factors that indicate poor clinical outcome, such as MYCN amplification, metastatic tumor stage, and older age of the patient at the time of diagnosis [50]. The relevance of methylation in these CGIs was also tested in a German NB cohort, and the authors found even stronger correlation between CIMP-positive phenotype and poor OS as well as disease-free survival (DFS) compared to the CIMP-negative cases. Additionally, all cases in this cohort that displayed amplification of MYCN were marked as CIMP+ phenotype and affected OS and DFS independently of age and stage of disease [51].
Targeted DNA methylation analysis has identified several tumor suppressor genes, which are otherwise rarely mutated, to be regulated by methylation. Thus, study of DNA methylation status in NB has fueled the development of potential new screening strategies that could have a diagnostic and prognostic application. Earlier reports employing the Illumina 27k methylation array reported a set of 8 genes that were methylated in NB cell lines. Further investigations showed that 3 out of 8 genes (SCNN1A, PRKCDBP, and KRT19) were marked by differential methylation, which allowed to distinguish several subsets of individuals diagnosed with NB, where lower methylation levels were associated with favorable outcome in patients [52]. Another study using methyl-specific PCR along with methyl-CpG-binding domain (MBD) sequencing identified 43 genes that were marked by differences in DNA methylation in NB cell lines after treatment with DAC and primary NB samples. Validation studies in patient samples showed that the methylation levels of 2/43 genes (HIST1H3C and GNAS) were associated with OS and/or event-free survival (EFS) [53]. The same group proposed a panel of 58 gene signatures in a larger patient cohort (n = 396), suggesting that methylation levels of these genes allow for accurate OS and EFS prediction [54]. However, it is unclear why the genes identified in their first study were not present in the latter study, given that the same experimental approaches were utilized. Finally, a study performed in 105 NB patients determined DNA methylation by Illumina 450k methylation array showed distinct clusters that were separated based on methylation levels, age, and MYCN amplification status. Multivariate survival analysis showed distinct survival outcomes, where the groups that were marked by higher methylation levels were linked to poor outcome. However, this group was characterized by amplified MYCN or higher levels of MYCN [50], therefore, it is unclear to which extent the perturbed DNA methylation levels alone contribute to the outcome prediction.
DNA methylation is critical during development, and with NB being a neurodevelopmental tumor, it is pivotal to understand whether deranged methylation is linked to any of the specific developmental phases, thus providing more mechanistic insights into the disease etiology. A study of stage 4S NB, which generally undergoes spontaneous regression and is associated with excellent prognosis, aimed to understand the methylome of this specific subtype of NB. Using the MBD sequencing approach, the authors compared the methylation status between samples in stages 4S and 1, 2, and 4. None of the samples displayed MYCN amplification. They observed hypermethylation in genes associated with subtelomeric regions. Moreover, the study showed hypermethylation of genes that are involved with neural crest development and differentiation in 4S subset compared to the tumors in stages 1, 2, or 4 [55]. Another critical gene involved in telomere maintenance is telomerase reverse transcriptase (TERT). Expression of this gene is upregulated in high-risk NB, and this was associated with, among others, altered DNA methylation levels, which were also linked to poor clinical outcome [56]. ALK tyrosine kinase receptor (ALK) is another key gene in NB development, and several activating mutations have been reported in NB, and this was associated with unfavorable clinical outcome [57]. A study by Gómez et al. reported non-CpG methylation on the gene body of ALK, which was present in favorable NB tumors, but was otherwise absent in aggressive NB tumors. Furthermore, the authors suggest that post-chemotherapy, the non-CpG methylation in unfavorable NB, was restored along with the reduced expression levels of ALK [58]. However, non-CpG methylation was determined using the Illumina 450k methylation array, which covers relatively few non-CpG sites, and that bisulfite sequencing cannot discriminate between the 5mC and 5hmC modifications. Considering that non-CpG methylation and hydroxymethylation (5mC&5hmC) are prevalent in pluripotent stem cells as well as in neurodevelopment [59], it is pivotal to study the contribution of these modifications individually. In addition, for instance, the methylation status of ATRX and MYCN, key genes in NB pathology, is not known, albeit both contain a CpG island (Fig. 2).
Fig. 2.
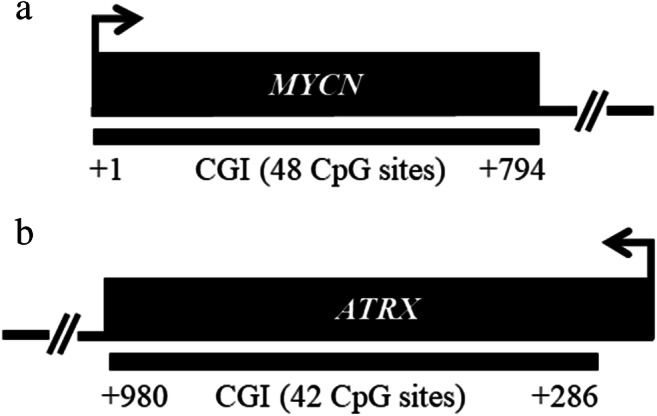
Identification of CpG islands (CGI) on MYCN and ATRX genes. Location of CGIs along with the number of CpG sites is indicated by black bars and was set relative to gene start (Ensemble Genome Browser, GRCh38). Only promoter and first exon regions were considered for the CGI identification. Arrows denote promoter orientation
DNA methylation is a reversible process, which is mediated by the TET proteins, where 5mC can be oxidized in an iterative manner to 5-hydroxymethyl (5hmC)-, 5-formyl (5fC), and 5-carboxyl-cytosine (5caC) [60, 61]. However, the roles of oxidized derivatives of 5mC have not been studied in NB. Moreover, most of the proposed genes for screening are dependent on MYCN amplification status, which on its own is sufficient to provide an accurate NB diagnosis. How the NB onset and progression is associated with predictable altered genome-wide levels of 5mC and its oxidized forms at base-pair resolution remains to be investigated.
Role of histone modifications and chromatin remodeling in gene expression in neuroblastoma
Chromatin structure and organization dictate gene activity throughout the genome. Posttranslational modifications of histone tails determine the chromatin configuration, enabling it to attain a more relaxed or condensed structure, which in turn allows for genes to become activated or silenced, respectively. The C- and N-terminal residues protruding from the nucleosomes undergo several posttranslational modifications, including acetylation, methylation, phosphorylation, and sumoylation, which primarily occur in lysine, arginine, serine, and/or threonine residues [31]. These covalent histone modifications are established and removed in a dynamic fashion by enzymes known as “writers” and “erasers,” allowing for chromatin plasticity.
Acetylation of lysine residues in the histones is mediated by histone acetyltransferases (HATs) and is generally associated with active gene transcription. Current evidence regarding the role of HATs in NB is limited, however, use of the synthetic compound BF1, a HAT inhibitor, in NB cell lines caused hypoacetylation of histone 3 (H3) and of lysine (K) 18 on H3 (H3K18), which was also marked by reduced cell growth [62]. Similarly, use of HAT inhibitors, PU139 and PU141, led to diminished lysine acetylation levels accompanied by reduced cell growth, both in vitro and in vivo. In addition, PU139 showed synergism with doxorubicin by reducing tumor growth [63, 64].
Histone acetylation is not a static entity, and as such the effects of HATs are antagonized by the catalytic activity of histone deacetylases (HDACs) [65]. In NB, the role of HDACs has been described by shedding light on the potential mechanisms involved in NB tumorigenesis, serving as potential markers aiding in patient stratification, as well as selection of novel therapeutic options in addition to the conventional treatment therapies. Studies showed that the following HDACs: HDAC2, SIRT1, and SIRT2 were upregulated by MYCN, which in turn promote stability and expression of MYCN protein, thus describing a positive feedback loop [66–68]. Inhibition of SIRT1 led to decreased tumorigenesis in mice [66]. Expression of several HDACs has been suggested as a potential approach for stratification of NB patients. Studies by Oehme et al. showed that the expression of HDAC8 and HDAC10 correlated with poor OS and EFS in NB [69, 70]. Pharmacological inhibition of HDAC8 in vitro and in vivo resulted in inhibition of proliferation and induction of differentiation [69, 71]. Additionally, levels of HDAC8 and HDAC10 anticorrelated with sensitivity to doxorubicin treatment in NB [70, 72]. Finally, kinome-wide RNAi screening revealed ALK as a candidate gene that gives rise to synthetic lethality in combination with HDAC8 inhibitors. Combined chemical inhibition of ALK and HDAC8 resulted in cell death or reduced cell viability in various cell lines with wild type ALK, or carrying amplified or activating mutations of ALK, respectively [73]. In recent years, several HDAC inhibitors have been included as a new category of anticancer treatment. Many ongoing clinical trials have included inhibitors of HDACs as a potential new line or as combinatorial treatment in NB. Treatment of NB cell lines with vorinostat (also known as suberanilohydroxamic acid, SAHA) resulted in growth arrest and promoted apoptotic pathways [74, 75], including in the cell lines that carry MYCN amplification [76]. In addition, vorinostat enhances the anti-tumor properties of various compounds, including flavopiridol and fenretinide [77, 78]. It also works in concert with conventional treatments, like chemotherapy (actinomycin D, paclitaxel) or radiotherapy [79–82]. Phase II clinical trials for the use of vorinostat in NB patients at the time of the submission of this review are still ongoing (Table 1). A report by Waldeck et al. showed that treatment of TH-MYCN transgenic mice for 9 continuous weeks with the pan-inhibitor of HDACs, panobinostat, increased survival rates compared to mice that were treated for 3 weeks only [83]. The combined treatment of high-risk NB cell lines with panobinostat and cisplatin, doxorubicin, or etoposide had synergistic effects in induction of apoptosis [84]. Finally, use of romidepsin in NB cell lines was also associated with reduced cell growth and increased cell apoptosis. This was evident in MYCN-amplified cell lines, p53 wild type or mutant cell lines, as well as those carrying ALK mutations [85, 86]. Growth inhibition by romidepsin was also evident in an immunocompromised mouse model [85, 86]. It remains to be seen how these drugs perform in clinical trials and whether they will be as efficient as in solid tumors or there will be a need to design more specific inhibitors.
Histone methylation can lead to either gene activation or repression, depending on the histone site that is methylated, the degree of methylation (e.g., mono-methylation, di-methylation, or tri-methylation), amino acid residues affected, and their position in the histone tail [31]. Histone methylation primarily occurs on the side chains of arginine (R), histidine (H), and lysine (K) residues and is a dynamic process mediated by histone methyltransferases (HMTs) and histone demethylases (HDMs). One of the most prominent HMTs, EZH2, a member of the polycomb repressive complex 2 (PRC2), which is responsible for establishing mono-, di-, and tri-methylation of H3K27, a marker of repressed chromatin (Table 2), is characterized by increased expression levels in NB [88]. Several studies reported that in MYCN-amplified NBs, the promoter of EZH2 is occupied by MYCN, both in vitro and in vivo, thereby driving its expression [89–91]. Another mechanism linked to elevated levels of EZH2 in NB was attributed to regional gains encompassing EZH2 gene in a NB patient [92]. Furthermore, studies in NB cell lines and patients showed that increased activity of PRC2 in MYCN-amplified tumors led to chromatin compaction, thus facilitating the silencing of gene networks that were marked by concomitant DNA hypermethylation [50]. Overexpression of EZH2 promoted an undifferentiated NB tumor phenotype and was associated with poor clinical outcome [93], while its pharmacological inhibition (tazemetostat) resulted in reduction of proliferation [94]. In addition, genetic silencing of EZH2 was marked by increased cell differentiation, which was mediated by NTRK1 [93]. Another histone methyltransferase that has been described in NB is EHMT2, which carries out mono- and di-methylation of H3K9, a mark associated with heterochromatin. Chemical inhibition of EHMT2 by BIX-01294 resulted in reduced cell proliferation and induced apoptosis [95]. Another study showed that inhibition of EHMT2 led to re-expression of several tumor suppressor genes, such as CLU, FLCN, AMHR2, and AKR1C1-3 [96]. Methylation on H3K79, a mark of permissive chromatin, is mediated by DOT1L. Inhibition of DOT1L using small molecule inhibitor SGC0946 led to lower methylation levels of H3K79, which was followed by reduced proliferation in cell lines carrying MYCN amplification, which also regulated DOT1L expression. Genetic ablation of DOT1L in vivo also reduced tumor growth and improved OS [97].
Table 2.
A simplified list of the members of histone demethylases and methyltransferases (mentioned in this article)
| Class of histone demethylases/methyltransferases | Histone demethylase /methyltransferase family |
Histone demethylase /methyltransferase |
Histone substrate | Gene expression |
|---|---|---|---|---|
| Erasers | ||||
| LSD demethylases | KDM1 | KDM1A* | H3K4me2/me1 | Repression |
| JMJC demethylases | KDM3 | KDM3A | H3K9me2/me1 | Activation |
| KDM4 | KDM4B | H3K9me3/me2 | Activation | |
| KDM5 | KDM5B | H3K4me3/me2/me1 | Repression | |
| KDM6 | KDM6A | H3K27me3/me2 | Activation | |
| KDM6B | H3K27me3/me2 | Activation | ||
| Writers | ||||
| EZH2 | EZH2 | EZH2 | H3K27me3/me2/me1 | Repression |
| EHMT2 | EHMT2 | EHMT2 | H3K9me2/me1 | Repression |
| DOT1L | DOT1L | DOT1L | H3K79me3/me2/me1 | Activation |
H histone, K lysine, me1 mono-methylation, me2 di-methylation, me3 tri-methylation, LSD lysine-specific demethylase, JMJC Jumonji C domain-containing protein demethylase, EZH2 enhancer of zeste 2 polycomb repressive complex 2 subunit, EHMT2 euchromatic histone lysine methyltransferase 2, DOT1L DOT1-like histone lysine methyltransferase
*In interaction with androgen receptor, KDM1A changes substrate preference to H3K9me2/me1, which is associated with gene expression activation [87]
Earliest research on the role of histone demethylation in NB showed overexpression of LSD1 (KDM1A), a histone demethylase that specifically demethylates H3K4me2/me1 to unmethylated H3K4, which is associated with gene silencing. This resulted in a phenotype, which was characterized by poorly differentiated cells and was associated with poor prognosis. Pharmacological inhibition of LSD1 resulted in inhibition of tumor growth in a mouse model [98]. In addition, LSD1 co-localizes with MYCN at the promoters of CDKN1A, CLU, and NDRG1 resulting in their repression of expression, where inhibition of LSD1 restored their expression, while combined inhibition of MYCN and LSD1 resulted in reduced cell proliferation and/or invasiveness in MYCN-amplified cell lines [99, 100]. Another critical process in tumorigenesis that is governed by LSD1 is autophagy by regulating the expression of mTORC1 through SESN2 [101]. What contributes to the high levels of LSD1 in NB tumors is not well known, however, studies reported miR-137 and miR-329 as regulators of LSD1 expression [102, 103]. Downregulation of miR-137 expression was linked to poor patient prognosis, and functional studies demonstrated that LSD1 is a direct target of miR-137 in NB cell lines [103]. The histone demethylase KDM3A, which removes me2/me1 from H3K9, was shown to be upregulated by MYCN in NB cell lines. This in turn led to increased proliferation by upregulating the expression of MALAT1 [104]. Expression profiling data showed that KDM4B, which catalyzes demethylation of H3K9me3/me2, a repressive chromatin mark, was upregulated in NB patients compared to ganglioneuroma or ganglioneuroblastoma. MYCN was shown to interact with and recruit KDM4B, and both genes could stratify a subgroup of patients that had poor clinical outcome [105]. The histone demethylase KDM5B, which targets H3K4me3/me2/me1, is upregulated in NB, and this confers the tumor cells with stem cell-like behavior and drug resistance. Paradoxically, the expression of KDM5B is negatively regulated by MYCN, and loss of expression of KDM5B resulted in reduced cell proliferation via Notch/Jagged signaling [106, 107]. Treatment of various NB cell lines with a small molecule inhibitor (GSK-J4) of KDM6A and KDM6B, demethylases of H3K27me3/me2 led to induced differentiation and apoptosis, as well as reduced proliferation in these cell lines. Moreover, GSK-J4 also effectively reduced the growth of tumors in patient-derived xenograft mouse models [108]. Similarly, another study showed that KDM6B promotes cell differentiation in NB, acting downstream of retinoic acid-HOXC9 axis. Expression of KDM6B is downregulated in high-risk NB, while high expression of KDM6B is a prognostic marker for better patient outcome [109]. The precise mechanisms by which the loss or gain of expression of these enzymes contributes to oncogenesis are yet to be fully investigated, especially since the inhibition of enzymes that promote permissive or repressive chromatin states both appear to contribute to reduced tumorigenic features of NB cells.
Role of non-coding RNAs in gene expression
MicroRNAs (miRNAs) are a family of small non-coding RNAs, which regulate an array of processes, and have been shown to be aberrantly regulated in cancers. They negatively regulate gene expression, either by perfectly matching the mRNA transcript, which results in degradation or by imperfect matching that results in prevention of translation [110]. The studies about the role of miRNAs and other non-coding RNAs in NB development and therapy resistance, as well as their expression regulation by MYCN, are too numerous to comprehensively describe in this review and have been extensively reviewed in these articles [111–115]. MiRNAs are relatively stable and can be secreted and circulate in the blood, which makes them excellent biomarker candidates [116], which will also be the prime focus of this review section. Currently, there are ~ 2300 human miRNAs identified [117]. A study by Bray et al. in a cohort of 145 NB patients with different genetic backgrounds studied the expression of 430 miRNAs. The study reported aberrant expression of 37 miRNAs in MYCN-amplified tumors compared to the ones carrying one copy of MYCN. The authors further showed that a set of 15 miRNAs was predictive of OS with a specificity of 86.5% and a sensitivity of 72.7% [118]. Another study using the same approach (analysis of 430 loci by stem-loop RT-qPCR) in 69 NB patients calculated EFS by means of support vector machines (SVM) and actual survival times with Cox regression-based models (CASPAR), which were highly predictable and accurate (SVM-EFS accuracy: 88.7% and CASPAR-EFS probability: 0.19%) and were validated in an independent cohort (SVM-EFS accuracy: 94.74% and CASPAR-EFS probability: 0.25%). Here, too, MYCN amplification correlated with the deregulated expression of miRNAs. Moreover, 37 miRNAs identified in this study correlated as well with TrkA expression, which is a marker of favorable clinical outcome. Expression of the most significant TrkA-correlated miRNA, miR-542-5p, also discriminated between local and metastatic disease, and was inversely correlated with MYCN amplification and EFS [119]. Again, using the same approach by screening for the expression of 430 mature miRNAs, a study in a large NB patient cohort (n = 534) showed that a set of 25 miRNAs was an independent predictor of patient survival, which was able to discriminate the test patients with regard to progression free and OS in both high-risk NB patients and general NB population [120]. In the three abovementioned studies, there was an overlap of 16 miRNAs in the first two studies, and an overlap of 2 miRNAs (miR-190, miR-488) between all the three studies, indicating a potential use for diagnosis and prognosis in NB. Using next generation sequencing, a study conducted in 128 NB patients reported a set of 23 miRNAs that were marked by deregulated expression in the tumors, regardless of MYCN amplification status compared to the controls. Target genes of these miRNAs were involved with various cancer pathways, such as DNA repair, apoptosis, and FGFR/EGFR signaling [121]. A study performed in 30 NB patients, out of which 11 were characterized as high-risk NB, while the rest as low to intermediate risk, screened for 754 miRNAs. The authors identified a set of 38 differentially expressed miRNAs between these two groups of patients [122]. A screening of 851 miRNAs in a discovery cohort of 13 patients identified a set of 17 miRNAs that were able to stratify low-risk from high-risk NB patients. In a validation cohort of 214 NB patients, 15/17 miRNAs were again able to discriminate the two different risk groups. Furthermore, miR-487b and miR-410 were marked by loss of expression in the high-risk group and were associated with DFS in non-MYCN-amplified tumors, whereas miR-487b expression was associated with OS and DFS, independent of the clinical covariates [123].
Use of serum to perform screening of the miRNome identified 36 upregulated and 46 downregulated miRNAs in murine models with high-risk NB [124]. In a cohort of 8 NB samples and 20 controls, using serum isolated RNA, Murray et al. identified a panel of 5 miRNAs (miR-124, miR-9, miR-218, miR-490, and miR-1538), which were overexpressed in MYCN-amplified NB subjects compared to controls [125]. A comprehensive study in 185 NB patients screened 743 miRNAs in serum, where levels of a set of 9 miRNAs (miR-375, miR-124-3p, miR-323a-3p, miR-129-5p, miR-218-5p, miR-490-5p, miR149-5p, miR-873-3p, and miR-10b-3p) were a feature of metastatic tumors compared to patients who only had localized primary tumors. The levels of these miRNAs were higher in serum of mouse NB xenografts, and these levels also correlated with the tumor volume [126]. Finally, a study tested a set of miRNAs associated with exosomes in the serum of 52 NB patients to potentially identify a signature that could discriminate between patients that have a favorable or poor response to chemotherapy. A set of 3 exosomal miRNAs (let-7b, miR-29c, and miR-342) could stratify patients that had a good response compared to the patients that were characterized by poor response to chemotherapy [127].
Although most of these studies have provided limited mechanistic insight and mainly present the connection between miRNAs and NB in a purely correlative manner, they have begun to shed light on the miRNome abnormities in NB tumors and elucidate how global sequencing platforms provide new means that can be leveraged in NB diagnosis and prognosis.
Regulation of the epigenome by core NB genes
Genetic lesions in chromatin-modifying enzymes and the consequent changes in the epigenetic landscapes have a direct impact on transcriptional programs that are fundamental to tumorigenesis [24–27]. Chromatin homeostasis is largely dependent on the interplay between the polycomb repressive complexes 1 and 2 (PRC), trithorax-group proteins (e.g., SWItch/Sucrose Non-Fermentable, SWI/SNF), and nucleosome remodelers [128]. Altered expression or mutations in constituents of these complexes disrupt this homeostasis. The PRC2 complex is responsible for trimethylated lysine 27 of histone 3, resulting in a shift towards a repressive chromatin state [27]. Overexpression of EZH2, which is the catalytic unit of PRC2, was reported in MYCN-amplified NB [50, 89, 91, 129], potentially silencing genes or networks of genes with a tumor suppressive role. The SWI/SNF complex opposes the effects of PRC2. Members of the SWI/SNF complex, most notably ARID1A/B, are among the most frequently mutated targets in all human cancers [130], including NB, resulting in a poor clinical outcome [20]. Alterations on ATRX, which codes for a SWI/SNF-like protein, have been reported in NB of children and adolescents, and are associated with overall poor survival and lack of appropriate treatments [131]. While the genetic role and to some extent also the epigenetic role of MYCN in NB are understood, the nature of epigenetic mechanisms involved in other types of NB, which encompass the majority of the NB cases, remains elusive.
Epigenetic regulation by MYCN in neuroblastoma
The pathological activation of MYCN is a typical feature of highly aggressive and relapsed NB tumors, which are characterized by a poor survival outcome and lack of treatments [132, 133]. Downregulation of MYCN expression induces apoptosis and differentiation, reverses tumor stem-like features, and accompanies senescence in MYCN-amplified NB cells. Pharmacological targeting of MYCN, albeit so far, only indirectly, is defined by reduced tumor growth in vitro and in vivo [134–143]. The role of MYCN as a master transcription regulator in controlling cell proliferation and survival is well-established [144–148]. In addition to the classical gene expression regulation, MYCN has been reported to regulate various epigenetic processes, such as histone modifying enzymes and miRNAs, which are described in Sections 2.2 and 2.3 of this review [149, 150]. MYCN along with chromatin-modifying proteins has a strong influence on disease progression and metastasis, which makes all of these molecules key targets for therapy. However, considering their expression in a broad range of healthy cells and their global contribution in gene regulation activity, it is pivotal to develop a better understating that would lead to generation of drugs that produce specific effects directly influencing oncogenesis.
Chromatin-modifying proteins are attractive as therapeutic targets for cancer since their aberrant expression has been reported in various tumor types [21, 31, 151]. Recent studies have started to emerge and have garnered attention about the role of MYCN and super-enhancers (SE). Gene regulatory circuitries consisting of master transcription factors (TFs) carefully control cell fate and identity by governing the expression of a well-defined set of genes. These types of TFs are generally recruited by a SE, which in turn is controlled by a master TF, thus regulating expression of cell type-specific developmental gene programs [152]. Such core regulatory circuitries (CRCs) have recently been described in NB, which were implicated in two phenotypically divergent cell states: committed ((nor)adrenergic)) and uncommitted (neural crest cell/mesenchymal cell-like type of cells), where each type of cell identity was well-defined by CRC models, including ASCL1, EYA1, PHOX2B, HAND1&2, GATA3, SIX3, and AP-1 and MEOX1, MEOX2, SIX1, SIX4, SOX9, SMAD3 and WWTR1, PRRX1, respectively [153, 154]. Additionally, in MYCN-amplified cell lines, another set of CRCs was identified, which included MYCN, HAND2, ISL1, PHOX2B, GATA3, and TBX2. Treatment with inhibitors of bromodomain and extra-terminal (BET) proteins, JQ1 and cyclin-dependent kinase 7 (CDK7)/SE, TZH1 resulted in a significant expression downregulation of genes that were associated with these CRCs [155, 156]. Other studies have shown that use of TZH1 resulted in downregulation of MYCN, followed by a significant suppression of MYCN-dependent gene transcriptional amplification. In vivo studies demonstrated tumor regression in a mouse model without significant systemic toxicity. The striking selective inhibitory effects of TZH1 in MYCN-amplified cells may be a result of downregulation of SE-associated genes or gene networks, including MYCN [157]. In addition, combinatorial treatment of TZH1 with ponatinib or lapatinib, which inhibits, among others, the protein kinase phosphatase 1 nuclear targeting subunit (PNUTS) that interacts with MYC protein and suppresses its degradation, resulted in a synergistically induced NB cell apoptosis, while having little effect on normal cells. Moreover, genetic ablation of PNUTS resulted in decreased MYCN protein, but not RNA expression. This was associated with reduced cell proliferation and cell survival, indicating another potential drug for use as an inhibitor of MYCN [158].
A new avenue of promising cancer therapeutics are the BET protein inhibitors, which have shown great efficacy in several malignancies, where a common target in all of these studies was downregulation of MYCN [159–161]. Treatment with the BET-bromodomain inhibitor, JQ1, resulted in reduced proliferation, induced apoptosis, and differentiation [141, 162–164]. This also was characterized by increased survival rates in vivo in three different NB models [141]. In NB, low expression of natural killer (NK) cell-activating receptors is inversely correlated with the expression of MYCN. However, treatment with JQ1 resulted in more resistant NB cell lines to NK cell-activating receptors, which—along with the inhibition of MYCN—impaired also the function of c-MYC and p53 that regulate the expression of specific ligands (ULBP1-3) for NKG2D-activating receptor [165]. Finally, as with other lines of treatment for malignancies, drug resistance is a major hurdle. A recent study pointed out that PI3K pathway promotes resistance to BET protein inhibitors, and as such, inhibitors of PI3K along with BET protein inhibitors were suggested as an upfront combination to circumvent this drug resistance [166]. Mechanisms of action for the BET protein inhibitors are currently being elucidated, and it appears that they target and regulate a selective group of genes, where expression downregulation seems to be mediated through inhibition of transcription elongation as a primary mode of action.
Chromatin remodeling in neuroblastoma by ATRX and ARID1A/1B
In the past decade, several major breakthroughs focusing on the identification of novel epigenetic enzymes that participate in epigenetic regulation of normal biological and disease processes have emerged. One such protein is ATRX, which when mutated leads to α-thalassemia, mental retardation, and X-linked (ATRX) syndrome. The ATRX gene encodes for a SWI/SNF-like protein, which plays diverse roles in chromatin remodeling [167, 168]. The SWI/SNF family of proteins restructures the nucleosome by regulating DNA-histone interactions, which change the conformation of the nucleosome, thus facilitating chromatin remodeling during transcription, DNA replication, and repair [169, 170]. Chromatin remodeling by ATRX is mediated by two of its highly conserved domains: (1) the N-terminal ATRX-Dnmt3-Dnmt3L (ADD) domain, which contains a GATA-like zinc finger, a PHD finger, and an alpha helix and (2) the ATPase/helicase domain that is located at the C-terminus. The ADD domain contains H3K9me3 binding pockets, and its binding is promoted by H3K9me3 and unmethylated H3K4, but is disrupted in the presence of H3K4me2/3 [171–173]. The ATP domain has DNA translocase and mononucleosome modulating pattern activities [174, 175]. Mutations in either domain are characterized by significant developmental delays and severe intellectual disabilities, among others [167, 176, 177]. The roles of ATRX mutations in pediatric and adult malignancies are emerging [178]. Our studies in a cohort of ~ 200 patients show that early disease progression and relapse are linked to, among others, intragenic deletions of the ATRX gene [179]. Similarly, others have reported alterations on the ATRX in NB of adolescents and young adults, which were associated with poor clinical outcome [131]. Additionally, ATRX mutation frequencies are prevalent in patients with INSS stage 4 disease and in the high-risk subgroup [180].
Mutations on ATRX and MYCN amplification are mutually exclusive [13, 180, 181]. ATRX and its interacting partner, DAXX, were shown to deposit histone variant H3.3 at heterochromatic regions (e.g., repetitive DNA, telomeres, etc.) [182–184]. NB specimens carrying mutations on ATRX or DAXX display an alternative lengthening of telomere phenotype [185]. However, recent evidence suggests that loss of ATRX-DAXX interaction is not critical in NB pathology [94]. Interestingly, DAXX mutations have not been reported in patients with ATRX syndrome [177], indicating potential tissue-specific interactions with other proteins. Moreover, mutations that generate in-frame fusion ATRX protein appear to change preference from repressive promoters to active promoters, thus regulating expression of neuronal genes through REST [94]. Another interacting partner of ATRX is MRN (Mre11, Rad50, and Nbs1), which was shown in HeLa and U2OS cell lines [186, 187], but whether this interaction persists in NB or not, and the role it plays, is not yet elucidated. Finally, ATRX contains a putative EZH2 interacting domain, which was identified through a yeast two-hybrid screen [188], but this has not yet been shown in mammalian cells. Recent studies show that NB cells are sensitive to EZH2 inhibitors, resulting in reduced cell growth [89, 94]. Whether this is mediated by interacting with ATRX or follows a different mechanism of action is not known. Thus, it is pivotal to identify and dissect the interacting partners of the ATRX complex, which will then allow proposing existing and new compounds for therapeutic use in NB.
Other members of the SWI/SNF complex, most notably ARID1A/B, are among the most frequently mutated targets in all human cancers [20, 130, 189, 190]. Mutations of these genes have also been reported in NB and are associated with treatment failure and poor survival rates [20, 191]. Similar to other SWI/SNF proteins, ARID1A&1B act to remodel chromatin and can have both activating and repressing effects on gene expression [192]. Loss of ARID1A expression in NB cell line, SK-N-SH, promoted cell invasion. In addition, this was associated with higher expression of matrix-metalloproteinases 2 and 9 (MMP2&9), along with increased expression of N-cadherin and nuclear translocation of β-catenin, whereas expression of E-cadherin was reduced [193]. Evidence from NB cell lines showed that treatment with 13-cis retinoic acid resulted in increased expression of ARID1A, which in turn reduced the expression of TERT. Promoter occupancy studies showed that ARID1A through SIN3A suppressed the expression of TERT, thus resulting in increased differentiation of NB cells. Patient data showed an inverse correlation between ARID1A and TERT expression, indicating a putative tumor suppressor role for ARID1A in NB [194].
These studies highlight that mutations in members of chromatin remodeling complexes are a feature of NB. The next step would be to elucidate the changes at any given nucleosome and associated gene expression, which will provide a strong base of knowledge in directing new avenues in epigenetic cancer therapies.
Conclusions
To date, intensive efforts have been made to identify biomarkers that are affected by the changes in the epigenome in NB. However, there is still no universal, effective epigenetic biomarker allowing for the diagnosis and prognosis of NB. Thus, lack of molecular biomarkers for early detection of the disease remains one of the major hurdles in NB diagnosis, treatment, and prognosis. Therefore, a clinically effective approach would be an unbiased take between control and NB, regardless of their genetic background based solely on epigenetic alterations. Additionally, it is pivotal to identify changes in the methylome at single base resolution as opposed to entire genic regions, which is the current approach. This would allow for quick and efficient targeted testing in clinical settings.
Funding
This work was supported by FWF Austrian Science Fund (# I 4162) and Vienna Science and Technology Fund (WTF LS18-111).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Irfete S. Fetahu, Email: irfete.fetahu@ccri.at
Sabine Taschner-Mandl, Email: sabine.taschner@ccri.at.
References
- 1.Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, Weiss WA. Neuroblastoma. Nature Reviews. Disease Primers. 2016;2:16078. doi: 10.1038/nrdp.2016.78. [DOI] [PubMed] [Google Scholar]
- 2.Irwin MS, Park JR. Neuroblastoma: Paradigm for precision medicine. Pediatric Clinics of North America. 2015;62:225–256. doi: 10.1016/j.pcl.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Maris JM. Recent advances in neuroblastoma. The New England Journal of Medicine. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nature Reviews. Clinical Oncology. 2014;11:704–713. doi: 10.1038/nrclinonc.2014.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KM, Hogarty M, on behalf of the COG Neuroblastoma Committee Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatric Blood & Cancer. 2013;60:985–993. doi: 10.1002/pbc.24433. [DOI] [PubMed] [Google Scholar]
- 6.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 7.Illhardt T, Toporski J, Feuchtinger T, Turkiewicz D, Teltschik HM, Ebinger M, Schwarze CP, Holzer U, Lode HN, Albert MH, Gruhn B, Urban C, Dykes JH, Teuffel O, Schumm M, Handgretinger R, Lang P. Haploidentical stem cell transplantation for refractory/relapsed neuroblastoma. Biology of Blood and Marrow Transplantation. 2018;24:1005–1012. doi: 10.1016/j.bbmt.2017.12.805. [DOI] [PubMed] [Google Scholar]
- 8.Ladenstein R, Pötschger U, Pearson ADJ, Brock P, Luksch R, Castel V, Yaniv I, Papadakis V, Laureys G, Malis J, Balwierz W, Ruud E, Kogner P, Schroeder H, de Lacerda AF, Beck-Popovic M, Bician P, Garami M, Trahair T, Canete A, Ambros PF, Holmes K, Gaze M, Schreier G, Garaventa A, Vassal G, Michon J, Valteau-Couanet D, SIOP Europe Neuroblastoma Group (SIOPEN) Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): An international, randomised, multi-arm, open-label, phase 3 trial. The Lancet Oncology. 2017;18:500–514. doi: 10.1016/S1470-2045(17)30070-0. [DOI] [PubMed] [Google Scholar]
- 9.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. The New England Journal of Medicine. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 12.Tsai HC, Baylin SB. Cancer epigenetics: Linking basic biology to clinical medicine. Cell Research. 2011;21:502–517. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, Schild L, Molenaar P, Stroeken P, van Noesel MM, Øra I, Santo EE, Caron HN, Westerhout EM, Versteeg R. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 14.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, McKenna A, Pedamallu CS, Ramos AH, Shefler E, Sivachenko A, Sougnez C, Stewart C, Ally A, Birol I, Chiu R, Corbett RD, Hirst M, Jackman SD, Kamoh B, Khodabakshi AH, Krzywinski M, Lo A, Moore RA, Mungall KL, Qian J, Tam A, Thiessen N, Zhao Y, Cole KA, Diamond M, Diskin SJ, Mosse YP, Wood AC, Ji L, Sposto R, Badgett T, London WB, Moyer Y, Gastier-Foster JM, Smith MA, Auvil JMG, Gerhard DS, Hogarty MD, Jones SJM, Lander ES, Gabriel SB, Getz G, Seeger RC, Khan J, Marra MA, Meyerson M, Maris JM. The genetic landscape of high-risk neuroblastoma. Nature Genetics. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attiyeh EF, London WB, Mossé YP, Wang Q, Winter C, Khazi D, McGrady P, Seeger RC, Look AT, Shimada H, Brodeur GM, Cohn SL, Matthay KK, Maris JM, Children's Oncology Group Chromosome 1p and 11q deletions and outcome in neuroblastoma. The New England Journal of Medicine. 2005;353:2243–2253. doi: 10.1056/NEJMoa052399. [DOI] [PubMed] [Google Scholar]
- 16.Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, Bartenhagen C, Walter E, Gecht J, Kerschke L, Volland R, Menon R, Heuckmann JM, Gartlgruber M, Hartlieb S, Henrich KO, Okonechnikov K, Altmüller J, Nürnberg P, Lefever S, de Wilde B, Sand F, Ikram F, Rosswog C, Fischer J, Theissen J, Hertwig F, Singhi AD, Simon T, Vogel W, Perner S, Krug B, Schmidt M, Rahmann S, Achter V, Lang U, Vokuhl C, Ortmann M, Büttner R, Eggert A, Speleman F, O’Sullivan RJ, Thomas RK, Berthold F, Vandesompele J, Schramm A, Westermann F, Schulte JH, Peifer M, Fischer M. A mechanistic classification of clinical phenotypes in neuroblastoma. Science. 2018;362:1165–1170. doi: 10.1126/science.aat6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schleiermacher G, Janoueix-Lerosey I, Delattre O. Recent insights into the biology of neuroblastoma. International Journal of Cancer. 2014;135:2249–2261. doi: 10.1002/ijc.29077. [DOI] [PubMed] [Google Scholar]
- 19.Schulte JH, Eggert A. Neuroblastoma. Critical Reviews in Oncogenesis. 2015;20:245–270. doi: 10.1615/critrevoncog.2015014033. [DOI] [PubMed] [Google Scholar]
- 20.Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA, Jr, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE, Hogarty MD. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nature Genetics. 2013;45:12–17. doi: 10.1038/ng.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature Reviews. Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- 24.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.You JS, Jones PA. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baylin, S. B., & Jones, P. A. (2016). Epigenetic determinants of Cancer. Cold Spring Harbor Perspectives in Biology, 8. [DOI] [PMC free article] [PubMed]
- 28.Bird A. DNA methylation patterns and epigenetic memory. Genes & Development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 29.Cedar H, Bergman Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nature Reviews. Genetics. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 30.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Research. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esteller M. Epigenetics in cancer. The New England Journal of Medicine. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 32.Rupaimoole R, Slack FJ. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nature Reviews. Drug Discovery. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 33.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes & Development. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fusco P, Esposito MR, Tonini GP. Chromosome instability in neuroblastoma. Oncology Letters. 2018;16:6887–6894. doi: 10.3892/ol.2018.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tonini GP, Capasso M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Reviews. 2020;39:275–285. doi: 10.1007/s10555-020-09843-4. [DOI] [PubMed] [Google Scholar]
- 36.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu YY, Mirkin BL, Dwivedi RS. Inhibition of DNA methyltransferase reverses cisplatin induced drug resistance in murine neuroblastoma cells. Cancer Detection and Prevention. 2005;29:456–463. doi: 10.1016/j.cdp.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Bartolucci S, Rossi M, Longo A, Rossi M, Estenoz M, Momparler RL, Santoro B, Augusti-Tocco G. 5-Aza-2′-deoxycytidine as inducer of differentiation and growth inhibition in mouse neuroblastoma cells. Cell Differentiation and Development. 1989;27:47–55. doi: 10.1016/0922-3371(89)90043-9. [DOI] [PubMed] [Google Scholar]
- 39.Carpinelli P, Granata F, Augusti-Tocco G, Rossi M, Bartolucci S. Antiproliferative effects and DNA hypomethylation by 5-aza-2′-deoxycytidine in human neuroblastoma cell lines. Anti-Cancer Drugs. 1993;4:629–635. doi: 10.1097/00001813-199312000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Charlet J, Schnekenburger M, Brown KW, Diederich M. DNA demethylation increases sensitivity of neuroblastoma cells to chemotherapeutic drugs. Biochemical Pharmacology. 2012;83:858–865. doi: 10.1016/j.bcp.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 41.George RE, Lahti JM, Adamson PC, Zhu K, Finkelstein D, Ingle AM, Reid JM, Krailo M, Neuberg D, Blaney SM, Diller L. Phase I study of decitabine with doxorubicin and cyclophosphamide in children with neuroblastoma and other solid tumors: A children’s oncology group study. Pediatric Blood & Cancer. 2010;55:629–638. doi: 10.1002/pbc.22607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Penter L, Maier B, Frede U, Hackner B, Carell T, Hagemeier C, Truss M. A rapid screening system evaluates novel inhibitors of DNA methylation and suggests F-box proteins as potential therapeutic targets for high-risk neuroblastoma. Targeted Oncology. 2015;10:523–533. doi: 10.1007/s11523-014-0354-5. [DOI] [PubMed] [Google Scholar]
- 43.Ostler KR, Yang Q, Looney TJ, Zhang L, Vasanthakumar A, Tian Y, Kocherginsky M, Raimondi SL, DeMaio JG, Salwen HR, Gu S, Chlenski A, Naranjo A, Gill A, Peddinti R, Lahn BT, Cohn SL, Godley LA. Truncated DNMT3B isoform DNMT3B7 suppresses growth, induces differentiation, and alters DNA methylation in human neuroblastoma. Cancer Research. 2012;72:4714–4723. doi: 10.1158/0008-5472.CAN-12-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nature Medicine. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- 45.van Noesel MM, et al. Tumor-specific down-regulation of the tumor necrosis factor-related apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is associated with dense promoter hypermethylation. Cancer Research. 2002;62:2157–2161. [PubMed] [Google Scholar]
- 46.Alaminos M, Davalos V, Cheung NK, Gerald WL, Esteller M. Clustering of gene hypermethylation associated with clinical risk groups in neuroblastoma. Journal of the National Cancer Institute. 2004;96:1208–1219. doi: 10.1093/jnci/djh224. [DOI] [PubMed] [Google Scholar]
- 47.Astuti D, Agathanggelou A, Honorio S, Dallol A, Martinsson T, Kogner P, Cummins C, Neumann HPH, Voutilainen R, Dahia P, Eng C, Maher ER, Latif F. RASSF1A promoter region CpG island hypermethylation in phaeochromocytomas and neuroblastoma tumours. Oncogene. 2001;20:7573–7577. doi: 10.1038/sj.onc.1204968. [DOI] [PubMed] [Google Scholar]
- 48.Yang Q, Zage P, Kagan D, Tian Y, Seshadri R, Salwen HR, Liu S, Chlenski A, Cohn SL. Association of epigenetic inactivation of RASSF1A with poor outcome in human neuroblastoma. Clinical Cancer Research. 2004;10:8493–8500. doi: 10.1158/1078-0432.CCR-04-1331. [DOI] [PubMed] [Google Scholar]
- 49.Abe M, et al. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Research. 2005;65:828–834. [PubMed] [Google Scholar]
- 50.Henrich KO, Bender S, Saadati M, Dreidax D, Gartlgruber M, Shao C, Herrmann C, Wiesenfarth M, Parzonka M, Wehrmann L, Fischer M, Duffy DJ, Bell E, Torkov A, Schmezer P, Plass C, Höfer T, Benner A, Pfister SM, Westermann F. Integrative genome-scale analysis identifies epigenetic mechanisms of transcriptional deregulation in unfavorable neuroblastomas. Cancer Research. 2016;76:5523–5537. doi: 10.1158/0008-5472.CAN-15-2507. [DOI] [PubMed] [Google Scholar]
- 51.Abe M, Westermann F, Nakagawara A, Takato T, Schwab M, Ushijima T. Marked and independent prognostic significance of the CpG island methylator phenotype in neuroblastomas. Cancer Letters. 2007;247:253–258. doi: 10.1016/j.canlet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 52.Carén H, Djos A, Nethander M, Sjöberg RM, Kogner P, Enström C, Nilsson S, Martinsson T. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer. 2011;11:66. doi: 10.1186/1471-2407-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Decock A, Ongenaert M, Hoebeeck J, de Preter K, van Peer G, van Criekinge W, Ladenstein R, Schulte JH, Noguera R, Stallings RL, van Damme A, Laureys G, Vermeulen J, van Maerken T, Speleman F, Vandesompele J. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biology. 2012;13:R95. doi: 10.1186/gb-2012-13-10-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Decock A, et al. Methyl-CpG-binding domain sequencing reveals a prognostic methylation signature in neuroblastoma. Oncotarget. 2016;7:1960–1972. doi: 10.18632/oncotarget.6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Decock A, Ongenaert M, de Wilde B, Brichard B, Noguera R, Speleman F, Vandesompele J. Stage 4S neuroblastoma tumors show a characteristic DNA methylation portrait. Epigenetics. 2016;11:761–771. doi: 10.1080/15592294.2016.1226739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Krämer A, Roncaioli JL, Sand F, Heuckmann JM, Ikram F, Schmidt R, Ackermann S, Engesser A, Kahlert Y, Vogel W, Altmüller J, Nürnberg P, Thierry-Mieg J, Thierry-Mieg D, Mariappan A, Heynck S, Mariotti E, Henrich KO, Gloeckner C, Bosco G, Leuschner I, Schweiger MR, Savelyeva L, Watkins SC, Shao C, Bell E, Höfer T, Achter V, Lang U, Theissen J, Volland R, Saadati M, Eggert A, de Wilde B, Berthold F, Peng Z, Zhao C, Shi L, Ortmann M, Büttner R, Perner S, Hero B, Schramm A, Schulte JH, Herrmann C, O’Sullivan RJ, Westermann F, Thomas RK, Fischer M. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015;526:700–704. doi: 10.1038/nature14980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trigg, R. M., & Turner, S. D. (2018). ALK in neuroblastoma: Biological and therapeutic implications. Cancers (Basel), 10. [DOI] [PMC free article] [PubMed]
- 58.Gómez S, Castellano G, Mayol G, Suñol M, Queiros A, Bibikova M, Nazor KL, Loring JF, Lemos I, Rodríguez E, de Torres C, Mora J, Martín-Subero JI, Lavarino C. DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics. 2015;7:1137–1153. doi: 10.2217/epi.15.49. [DOI] [PubMed] [Google Scholar]
- 59.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Secci D, Carradori S, Bizzarri B, Bolasco A, Ballario P, Patramani Z, Fragapane P, Vernarecci S, Canzonetta C, Filetici P. Synthesis of a novel series of thiazole-based histone acetyltransferase inhibitors. Bioorganic & Medicinal Chemistry. 2014;22:1680–1689. doi: 10.1016/j.bmc.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 63.Furdas SD, Shekfeh S, Bissinger EM, Wagner JM, Schlimme S, Valkov V, Hendzel M, Jung M, Sippl W. Synthesis and biological testing of novel pyridoisothiazolones as histone acetyltransferase inhibitors. Bioorganic & Medicinal Chemistry. 2011;19:3678–3689. doi: 10.1016/j.bmc.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 64.Gajer JM, Furdas SD, Gründer A, Gothwal M, Heinicke U, Keller K, Colland F, Fulda S, Pahl HL, Fichtner I, Sippl W, Jung M. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis. 2015;4:e137. doi: 10.1038/oncsis.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dawson MA, Kouzarides T. Cancer epigenetics: From mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 66.Marshall GM, Liu PY, Gherardi S, Scarlett CJ, Bedalov A, Xu N, Iraci N, Valli E, Ling D, Thomas W, van Bekkum M, Sekyere E, Jankowski K, Trahair T, MacKenzie KL, Haber M, Norris MD, Biankin AV, Perini G, Liu T. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genetics. 2011;7:e1002135. doi: 10.1371/journal.pgen.1002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim MK, Carroll WL. Autoregulation of the N-myc gene is operative in neuroblastoma and involves histone deacetylase 2. Cancer. 2004;101:2106–2115. doi: 10.1002/cncr.20626. [DOI] [PubMed] [Google Scholar]
- 68.Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, Koach J, Tee AE, Haber M, Norris MD, Toon C, Rooman I, Xue C, Cheung BB, Kumar S, Marshall GM, Biankin AV, Liu T. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death and Differentiation. 2013;20:503–514. doi: 10.1038/cdd.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clinical Cancer Research. 2009;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 70.Oehme I, Linke JP, Bock BC, Milde T, Lodrini M, Hartenstein B, Wiegand I, Eckert C, Roth W, Kool M, Kaden S, Grone HJ, Schulte JH, Lindner S, Hamacher-Brady A, Brady NR, Deubzer HE, Witt O. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2592–E2601. doi: 10.1073/pnas.1300113110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, Kopp-Schneider A, Fabian J, Schober A, Fernekorn U, von Deimling A, Deubzer HE, Milde T, Witt O, Oehme I. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death & Disease. 2015;6:e1657. doi: 10.1038/cddis.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao G, Wang G, Bai H, Li T, Gong F, Yang H, Wen J, Wang W. Targeted inhibition of HDAC8 increases the doxorubicin sensitivity of neuroblastoma cells via up regulation of miR-137. European Journal of Pharmacology. 2017;802:20–26. doi: 10.1016/j.ejphar.2017.02.035. [DOI] [PubMed] [Google Scholar]
- 73.Shen J, Najafi S, Stäble S, Fabian J, Koeneke E, Kolbinger FR, Wrobel JK, Meder B, Distel M, Heimburg T, Sippl W, Jung M, Peterziel H, Kranz D, Boutros M, Westermann F, Witt O, Oehme I. A kinome-wide RNAi screen identifies ALK as a target to sensitize neuroblastoma cells for HDAC8-inhibitor treatment. Cell Death and Differentiation. 2018;25:2053–2070. doi: 10.1038/s41418-018-0080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De los Santos M, Zambrano A, Aranda A. Combined effects of retinoic acid and histone deacetylase inhibitors on human neuroblastoma SH-SY5Y cells. Molecular Cancer Therapeutics. 2007;6:1425–1432. doi: 10.1158/1535-7163.MCT-06-0623. [DOI] [PubMed] [Google Scholar]
- 75.Mühlethaler-Mottet A, et al. Complex molecular mechanisms cooperate to mediate histone deacetylase inhibitors anti-tumour activity in neuroblastoma cells. Molecular Cancer. 2008;7:55. doi: 10.1186/1476-4598-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cortés C, Kozma SC, Tauler A, Ambrosio S. MYCN concurrence with SAHA-induced cell death in human neuroblastoma cells. Cellular Oncology (Dordrecht) 2015;38:341–352. doi: 10.1007/s13402-015-0233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang JM, Sheard MA, Ji L, Sposto R, Keshelava N. Combination of vorinostat and flavopiridol is selectively cytotoxic to multidrug-resistant neuroblastoma cell lines with mutant TP53. Molecular Cancer Therapeutics. 2010;9:3289–3301. doi: 10.1158/1535-7163.MCT-10-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheung BB, Tan O, Koach J, Liu B, Shum MSY, Carter DR, Sutton S, Po'uha ST, Chesler L, Haber M, Norris MD, Kavallaris M, Liu T, O'Neill GM, Marshall GM. Thymosin-β4 is a determinant of drug sensitivity for Fenretinide and Vorinostat combination therapy in neuroblastoma. Molecular Oncology. 2015;9:1484–1500. doi: 10.1016/j.molonc.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cortes CL, Veiga SR, Almacellas E, Hernández-Losa J, Ferreres JC, Kozma SC, Ambrosio S, Thomas G, Tauler A. Effect of low doses of actinomycin D on neuroblastoma cell lines. Molecular Cancer. 2016;15:1. doi: 10.1186/s12943-015-0489-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mueller S, Yang X, Sottero TL, Gragg A, Prasad G, Polley MY, Weiss WA, Matthay KK, Davidoff AM, DuBois SG, Haas-Kogan DA. Cooperation of the HDAC inhibitor vorinostat and radiation in metastatic neuroblastoma: Efficacy and underlying mechanisms. Cancer Letters. 2011;306:223–229. doi: 10.1016/j.canlet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhen Z, Yang K, Ye L, You Z, Chen R, Liu Y, He Y. Suberoylanilide hydroxamic acid sensitizes neuroblastoma to paclitaxel by inhibiting thioredoxin-related protein 14-mediated autophagy. Cancer Science. 2017;108:1485–1492. doi: 10.1111/cas.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DuBois SG, Groshen S, Park JR, Haas-Kogan DA, Yang X, Geier E, Chen E, Giacomini K, Weiss B, Cohn SL, Granger MM, Yanik GA, Hawkins R, Courtier J, Jackson H, Goodarzian F, Shimada H, Czarnecki S, Tsao-Wei D, Villablanca JG, Marachelian A, Matthay KK. Phase I study of vorinostat as a radiation sensitizer with 131I-metaiodobenzylguanidine (131I-MIBG) for patients with relapsed or refractory neuroblastoma. Clinical Cancer Research. 2015;21:2715–2721. doi: 10.1158/1078-0432.CCR-14-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Waldeck K, Cullinane C, Ardley K, Shortt J, Martin B, Tothill RW, Li J, Johnstone RW, McArthur GA, Hicks RJ, Wood PJ. Long term, continuous exposure to panobinostat induces terminal differentiation and long term survival in the TH-MYCN neuroblastoma mouse model. International Journal of Cancer. 2016;139:194–204. doi: 10.1002/ijc.30056. [DOI] [PubMed] [Google Scholar]
- 84.Wang G, Edwards H, Caldwell JT, Buck SA, Qing WY, Taub JW, Ge Y, Wang Z. Panobinostat synergistically enhances the cytotoxic effects of cisplatin, doxorubicin or etoposide on high-risk neuroblastoma cells. PLoS One. 2013;8:e76662. doi: 10.1371/journal.pone.0076662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Panicker J, Li Z, McMahon C, Sizer C, Steadman K, Piekarz R, Bates SE, Thiele CJ. Romidepsin (FK228/depsipeptide) controls growth and induces apoptosis in neuroblastoma tumor cells. Cell Cycle. 2010;9:1830–1838. doi: 10.4161/cc.9.9.11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hegarty SV, Togher KL, O’Leary E, Solger F, Sullivan AM, O’Keeffe GW. Romidepsin induces caspase-dependent cell death in human neuroblastoma cells. Neuroscience Letters. 2017;653:12–18. doi: 10.1016/j.neulet.2017.05.025. [DOI] [PubMed] [Google Scholar]
- 87.Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AHFM, Günther T, Buettner R, Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 88.Cohen AL, Piccolo SR, Cheng L, Soldi R, Han B, Johnson WE, Bild AH. Genomic pathway analysis reveals that EZH2 and HDAC4 represent mutually exclusive epigenetic pathways across human cancers. BMC Medical Genetics. 2013;6:35. doi: 10.1186/1755-8794-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, Wang EJ, Veschi V, Lam N, Qi J, Gustafson WC, Nasholm N, Vazquez F, Weir BA, Cowley GS, Ali LD, Pantel S, Jiang G, Harrington WF, Lee Y, Goodale A, Lubonja R, Krill-Burger JM, Meyers RM, Tsherniak A, Root DE, Bradner JE, Golub TR, Roberts CW, Hahn WC, Weiss WA, Thiele CJ, Stegmaier K. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. The Journal of Clinical Investigation. 2018;128:446–462. doi: 10.1172/JCI90793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Corvetta D, Chayka O, Gherardi S, D'Acunto CW, Cantilena S, Valli E, Piotrowska I, Perini G, Sala A. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: Functional and therapeutic implications. The Journal of Biological Chemistry. 2013;288:8332–8341. doi: 10.1074/jbc.M113.454280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsubota S, Kishida S, Shimamura T, Ohira M, Yamashita S, Cao D, Kiyonari S, Ushijima T, Kadomatsu K. PRC2-mediated transcriptomic alterations at the embryonic stage govern tumorigenesis and clinical outcome in MYCN-driven neuroblastoma. Cancer Research. 2017;77:5259–5271. doi: 10.1158/0008-5472.CAN-16-3144. [DOI] [PubMed] [Google Scholar]
- 92.Bate-Eya LT, Gierman HJ, Ebus ME, Koster J, Caron HN, Versteeg R, Dolman MEM, Molenaar JJ. Enhancer of zeste homologue 2 plays an important role in neuroblastoma cell survival independent of its histone methyltransferase activity. European Journal of Cancer. 2017;75:63–72. doi: 10.1016/j.ejca.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 93.Li Z, Takenobu H, Setyawati AN, Akita N, Haruta M, Satoh S, Shinno Y, Chikaraishi K, Mukae K, Akter J, Sugino RP, Nakazawa A, Nakagawara A, Aburatani H, Ohira M, Kamijo T. EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene. 2018;37:2714–2727. doi: 10.1038/s41388-018-0133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qadeer ZA, et al. ATRX in-frame fusion neuroblastoma is sensitive to EZH2 inhibition via modulation of neuronal gene signatures. Cancer Cell. 2019;36:512–527.e519. doi: 10.1016/j.ccell.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu Z, Tian Y, Salwen HR, Chlenski A, Godley LA, Raj JU, Yang Q. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anti-Cancer Drugs. 2013;24:484–493. doi: 10.1097/CAD.0b013e32835ffdbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bellamy J, Szemes M, Melegh Z, Dallosso A, Kollareddy M, Catchpoole D, Malik K. Increased efficacy of histone methyltransferase G9a inhibitors against. Frontiers in Oncology. 2020;10:818. doi: 10.3389/fonc.2020.00818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wong M, Tee AEL, Milazzo G, Bell JL, Poulos RC, Atmadibrata B, Sun Y, Jing D, Ho N, Ling D, Liu PY, Zhang XD, Hüttelmaier S, Wong JWH, Wang J, Polly P, Perini G, Scarlett CJ, Liu T. The histone methyltransferase DOT1L promotes neuroblastoma by regulating gene transcription. Cancer Research. 2017;77:2522–2533. doi: 10.1158/0008-5472.CAN-16-1663. [DOI] [PubMed] [Google Scholar]
- 98.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schüle R, Eggert A, Buettner R, Kirfel J. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: Implications for therapy. Cancer Research. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 99.Amente S, Milazzo G, Sorrentino MC, Ambrosio S, di Palo G, Lania L, Perini G, Majello B. Lysine-specific demethylase (LSD1/KDM1A) and MYCN cooperatively repress tumor suppressor genes in neuroblastoma. Oncotarget. 2015;6:14572–14583. doi: 10.18632/oncotarget.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ambrosio S, Amente S, Saccà CD, Capasso M, Calogero RA, Lania L, Majello B. LSD1 mediates MYCN control of epithelial-mesenchymal transition through silencing of metastatic suppressor NDRG1 gene. Oncotarget. 2017;8:3854–3869. doi: 10.18632/oncotarget.12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ambrosio S, Saccà CD, Amente S, Paladino S, Lania L, Majello B. Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene. 2017;36:6701–6711. doi: 10.1038/onc.2017.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang H, et al. miR-329 suppresses the growth and motility of neuroblastoma by targeting KDM1A. FEBS Letters. 2014;588:192–197. doi: 10.1016/j.febslet.2013.11.036. [DOI] [PubMed] [Google Scholar]
- 103.Althoff K, Beckers A, Odersky A, Mestdagh P, Köster J, Bray IM, Bryan K, Vandesompele J, Speleman F, Stallings RL, Schramm A, Eggert A, Sprüssel A, Schulte JH. MiR-137 functions as a tumor suppressor in neuroblastoma by downregulating KDM1A. International Journal of Cancer. 2013;133:1064–1073. doi: 10.1002/ijc.28091. [DOI] [PubMed] [Google Scholar]
- 104.Tee AE, Ling D, Nelson C, Atmadibrata B, Dinger ME, Xu N, Mizukami T, Liu PY, Liu B, Cheung B, Pasquier E, Haber M, Norris MD, Suzuki T, Marshall GM, Liu T. The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget. 2014;5:1793–1804. doi: 10.18632/oncotarget.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang J, et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. Journal of the National Cancer Institute. 2015;107:djv080. doi: 10.1093/jnci/djv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kuo YT, Liu YL, Adebayo BO, Shih PH, Lee WH, Wang LS, Liao YF, Hsu WM, Yeh CT, Lin CM. JARID1B expression plays a critical role in chemoresistance and stem cell-like phenotype of neuroblastoma cells. PLoS One. 2015;10:e0125343. doi: 10.1371/journal.pone.0125343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang L, Sokolowski N, Atmadibrata B, Liu T. Histone demethylase JARID1B promotes cell proliferation but is downregulated by N-Myc oncoprotein. Oncology Reports. 2014;31:1935–1939. doi: 10.3892/or.2014.3006. [DOI] [PubMed] [Google Scholar]
- 108.Lochmann TL, Powell KM, Ham J, Floros KV, Heisey DAR, Kurupi RIJ, Calbert ML, Ghotra MS, Greninger P, Dozmorov M, Gowda M, Souers AJ, Reynolds CP, Benes CH, Faber AC. Targeted inhibition of histone H3K27 demethylation is effective in high-risk neuroblastoma. Science Translational Medicine. 2018;10:eaao4680. doi: 10.1126/scitranslmed.aao4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang L, Zha Y, Ding J, Ye B, Liu M, Yan C, Dong Z, Cui H, Ding HF. Histone demethylase KDM6B has an anti-tumorigenic function in neuroblastoma by promoting differentiation. Oncogenesis. 2019;8:3. doi: 10.1038/s41389-018-0112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduction and Targeted Therapy. 2016;1:15004. doi: 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aravindan N, Subramanian K, Somasundaram DB, Herman TS, Aravindan S. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resistance. 2019;2:1086–1105. doi: 10.20517/cdr.2019.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Salomão KB, Pezuk JA, de Souza GR, Chagas P, Pereira TC, Valera ET, Brassesco MS. MicroRNA dysregulation interplay with childhood abdominal tumors. Cancer Metastasis Reviews. 2019;38:783–811. doi: 10.1007/s10555-019-09829-x. [DOI] [PubMed] [Google Scholar]
- 113.Pandey GK, Kanduri C. Long noncoding RNAs and neuroblastoma. Oncotarget. 2015;6:18265–18275. doi: 10.18632/oncotarget.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nallasamy P, Chava S, Verma SS, Mishra S, Gorantla S, Coulter DW, Byrareddy SN, Batra SK, Gupta SC, Challagundla KB. PD-L1, inflammation, non-coding RNAs, and neuroblastoma: Immuno-oncology perspective. Seminars in Cancer Biology. 2018;52:53–65. doi: 10.1016/j.semcancer.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chi, Y., Wang, D., Wang, J., Yu, W., & Yang, J. (2019). Long non-coding RNA in the pathogenesis of cancers. Cells, 8. [DOI] [PMC free article] [PubMed]
- 116.Cui M, Wang H, Yao X, Zhang D, Xie Y, Cui R, Zhang X. Circulating MicroRNAs in cancer: Potential and challenge. Frontiers in Genetics. 2019;10:626. doi: 10.3389/fgene.2019.00626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alles J, Fehlmann T, Fischer U, Backes C, Galata V, Minet M, Hart M, Abu-Halima M, Grässer FA, Lenhof HP, Keller A, Meese E. An estimate of the total number of true human miRNAs. Nucleic Acids Research. 2019;47:3353–3364. doi: 10.1093/nar/gkz097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bray I, Bryan K, Prenter S, Buckley PG, Foley NH, Murphy DM, Alcock L, Mestdagh P, Vandesompele J, Speleman F, London WB, McGrady PW, Higgins DG, O'Meara A, O'Sullivan M, Stallings RL. Widespread dysregulation of MiRNAs by MYCN amplification and chromosomal imbalances in neuroblastoma: Association of miRNA expression with survival. PLoS One. 2009;4:e7850. doi: 10.1371/journal.pone.0007850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schulte JH, Schowe B, Mestdagh P, Kaderali L, Kalaghatgi P, Schlierf S, Vermeulen J, Brockmeyer B, Pajtler K, Thor T, de Preter K, Speleman F, Morik K, Eggert A, Vandesompele J, Schramm A. Accurate prediction of neuroblastoma outcome based on miRNA expression profiles. International Journal of Cancer. 2010;127:2374–2385. doi: 10.1002/ijc.25436. [DOI] [PubMed] [Google Scholar]
- 120.De Preter K, et al. miRNA expression profiling enables risk stratification in archived and fresh neuroblastoma tumor samples. Clinical Cancer Research. 2011;17:7684–7692. doi: 10.1158/1078-0432.CCR-11-0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Megiorni F, Colaiacovo M, Cialfi S, McDowell HP, Guffanti A, Camero S, Felsani A, Losty PD, Pizer B, Shukla R, Cappelli C, Ferrara E, Pizzuti A, Moles A, Dominici C. A sketch of known and novel MYCN-associated miRNA networks in neuroblastoma. Oncology Reports. 2017;38:3–20. doi: 10.3892/or.2017.5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bienertova-Vasku J, Mazanek P, Hezova R, Curdova A, Nekvindova J, Kren L, Sterba J, Slaby O. Extension of microRNA expression pattern associated with high-risk neuroblastoma. Tumour Biology. 2013;34:2315–2319. doi: 10.1007/s13277-013-0777-0. [DOI] [PubMed] [Google Scholar]
- 123.Gattolliat CH, et al. Expression of miR-487b and miR-410 encoded by 14q32.31 locus is a prognostic marker in neuroblastoma. British Journal of Cancer. 2011;105:1352–1361. doi: 10.1038/bjc.2011.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ramraj SK, Aravindan S, Somasundaram DB, Herman TS, Natarajan M, Aravindan N. Serum-circulating miRNAs predict neuroblastoma progression in mouse model of high-risk metastatic disease. Oncotarget. 2016;7:18605–18619. doi: 10.18632/oncotarget.7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murray MJ, Raby KL, Saini HK, Bailey S, Wool SV, Tunnacliffe JM, Enright AJ, Nicholson JC, Coleman N. Solid tumors of childhood display specific serum microRNA profiles. Cancer Epidemiology, Biomarkers & Prevention. 2015;24:350–360. doi: 10.1158/1055-9965.EPI-14-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zeka, F., Decock, A., van Goethem, A., Vanderheyden, K., Demuynck, F., Lammens, T., Helsmoortel, H. H., Vermeulen, J., Noguera, R., Berbegall, A. P., Combaret, V., Schleiermacher, G., Laureys, G., Schramm, A., Schulte, J. H., Rahmann, S., Bienertová-Vašků, J., Mazánek, P., Jeison, M., Ash, S., Hogarty, M. D., Moreno-Smith, M., Barbieri, E., Shohet, J., Berthold, F., van Maerken, T., Speleman, F., Fischer, M., de Preter, K., Mestdagh, P., & Vandesompele, J. (2018). Circulating microRNA biomarkers for metastatic disease in neuroblastoma patients. JCI Insight, 3. [DOI] [PMC free article] [PubMed]
- 127.Morini, M., Cangelosi, D., Segalerba, D., Marimpietri, D., Raggi, F., Castellano, A., Fruci, D., de Mora, J. F., Cañete, A., Yáñez, Y., Viprey, V., Corrias, M. V., Carlini, B., Pezzolo, A., Schleiermacher, G., Mazzocco, K., Ladenstein, R., Sementa, A. R., Conte, M., Garaventa, A., Burchill, S., Luksch, R., Bosco, M. C., Eva, A., & Varesio, L. (2019). Exosomal microRNAs from longitudinal liquid biopsies for the prediction of response to induction chemotherapy in high-risk neuroblastoma patients: A proof of concept SIOPEN study. Cancers (Basel), 11. [DOI] [PMC free article] [PubMed]
- 128.Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. 2016;352:aad9780. doi: 10.1126/science.aad9780. [DOI] [PubMed] [Google Scholar]
- 129.Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nature Reviews. Cancer. 2016;16:803–810. doi: 10.1038/nrc.2016.83. [DOI] [PubMed] [Google Scholar]
- 130.Hodges, C., Kirkland, J. G., & Crabtree, G. R. (2016). The many roles of BAF (mSWI/SNF) and PBAF complexes in Cancer. Cold Spring Harbor Perspectives in Medicine, 6. [DOI] [PMC free article] [PubMed]
- 131.Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, Heguy A, Pappo AS, Federico S, Dalton J, Cheung IY, Ding L, Fulton R, Wang J, Chen X, Becksfort J, Wu J, Billups CA, Ellison D, Mardis ER, Wilson RK, Downing JR, Dyer MA, St Jude Children's Research Hospital–Washington University Pediatric Cancer Genome Project Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062–1071. doi: 10.1001/jama.2012.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clinical Cancer Research. 2012;18:2740–2753. doi: 10.1158/1078-0432.CCR-11-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. The New England Journal of Medicine. 1985;313:1111–1116. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- 134.Kang JH, Rychahou PG, Ishola TA, Qiao J, Evers BM, Chung DH. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochemical and Biophysical Research Communications. 2006;351:192–197. doi: 10.1016/j.bbrc.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bell E, Lunec J, Tweddle DA. Cell cycle regulation targets of MYCN identified by gene expression microarrays. Cell Cycle. 2007;6:1249–1256. doi: 10.4161/cc.6.10.4222. [DOI] [PubMed] [Google Scholar]
- 136.Yaari S, Jacob-Hirsch J, Amariglio N, Haklai R, Rechavi G, Kloog Y. Disruption of cooperation between Ras and MycN in human neuroblastoma cells promotes growth arrest. Clinical Cancer Research. 2005;11:4321–4330. doi: 10.1158/1078-0432.CCR-04-2071. [DOI] [PubMed] [Google Scholar]
- 137.Tonelli R, Purgato S, Camerin C, Fronza R, Bologna F, Alboresi S, Franzoni M, Corradini R, Sforza S, Faccini A, Shohet JM, Marchelli R, Pession A. Anti-gene peptide nucleic acid specifically inhibits MYCN expression in human neuroblastoma cells leading to cell growth inhibition and apoptosis. Molecular Cancer Therapeutics. 2005;4:779–786. doi: 10.1158/1535-7163.MCT-04-0213. [DOI] [PubMed] [Google Scholar]
- 138.Negroni A, Scarpa S, Romeo A, Ferrari S, Modesti A, Raschellà G. Decrease of proliferation rate and induction of differentiation by a MYCN antisense DNA oligomer in a human neuroblastoma cell line. Cell Growth & Differentiation. 1991;2:511–518. [PubMed] [Google Scholar]
- 139.Burkhart CA, Cheng AJ, Madafiglio J, Kavallaris M, Mili M, Marshall GM, Weiss WA, Khachigian LM, Norris MD, Haber M. Effects of MYCN antisense oligonucleotide administration on tumorigenesis in a murine model of neuroblastoma. Journal of the National Cancer Institute. 2003;95:1394–1403. doi: 10.1093/jnci/djg045. [DOI] [PubMed] [Google Scholar]
- 140.Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, Simonds EF, Seeger R, Matthay KK, Hertz NT, Eilers M, Shokat KM, Weiss WA. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell. 2014;26:414–427. doi: 10.1016/j.ccr.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, Stegmaier K. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discovery. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Taschner-Mandl S, Schwarz M, Blaha J, Kauer M, Kromp F, Frank N, Rifatbegovic F, Weiss T, Ladenstein R, Hohenegger M, Ambros IM, Ambros PF. Metronomic topotecan impedes tumor growth of MYCN-amplified neuroblastoma cells in vitro and in vivo by therapy induced senescence. Oncotarget. 2016;7:3571–3586. doi: 10.18632/oncotarget.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bogen D, Wei JS, Azorsa DO, Ormanoglu P, Buehler E, Guha R, Keller JM, Griner LAM, Ferrer M, Song YK, Liao H, Mendoza A, Gryder BE, Sindri S, He J, Wen X, Zhang S, Shern JF, Yohe ME, Taschner-Mandl S, Shohet JM, Thomas CJ, Martin SE, Ambros PF, Khan J. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget. 2015;6:35247–35262. doi: 10.18632/oncotarget.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 145.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zaytseva, O., & Quinn, L. M. (2017). Controlling the master: Chromatin dynamics at the MYC promoter integrate developmental signaling. Genes (Basel), 8. [DOI] [PMC free article] [PubMed]
- 148.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nature Reviews. Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 149.He S, Liu Z, Oh DY, Thiele CJ. MYCN and the epigenome. Frontiers in Oncology. 2013;3:1. doi: 10.3389/fonc.2013.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cotterman R, Jin VX, Krig SR, Lemen JM, Wey A, Farnham PJ, Knoepfler PS. N-Myc regulates a widespread euchromatic program in the human genome partially independent of its role as a classical transcription factor. Cancer Research. 2008;68:9654–9662. doi: 10.1158/0008-5472.CAN-08-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Feinberg AP, Tycko B. The history of cancer epigenetics. Nature Reviews. Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 152.Saint-André V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, Bradner JE, Young RA. Models of human core transcriptional regulatory circuitries. Genome Research. 2016;26:385–396. doi: 10.1101/gr.197590.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, Broekmans M, Haneveld F, Nowakowska NE, Bras J, van Noesel CJM, Jongejan A, van Kampen AH, Koster L, Baas F, van Dijk-Kerkhoven L, Huizer-Smit M, Lecca MC, Chan A, Lakeman A, Molenaar P, Volckmann R, Westerhout EM, Hamdi M, van Sluis PG, Ebus ME, Molenaar JJ, Tytgat GA, Westerman BA, van Nes J, Versteeg R. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nature Genetics. 2017;49:1261–1266. doi: 10.1038/ng.3899. [DOI] [PubMed] [Google Scholar]
- 154.Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugène C, Raynal V, Etchevers HC, Thomas S, Lermine A, Daudigeos-Dubus E, Geoerger B, Orth MF, Grünewald TGP, Diaz E, Ducos B, Surdez D, Carcaboso AM, Medvedeva I, Deller T, Combaret V, Lapouble E, Pierron G, Grossetête-Lalami S, Baulande S, Schleiermacher G, Barillot E, Rohrer H, Delattre O, Janoueix-Lerosey I. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nature Genetics. 2017;49:1408–1413. doi: 10.1038/ng.3921. [DOI] [PubMed] [Google Scholar]
- 155.Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, He S, Krill-Burger JM, Root DE, Vazquez F, Tsherniak A, Hahn WC, Golub TR, Young RA, Look AT, Stegmaier K. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nature Genetics. 2018;50:1240–1246. doi: 10.1038/s41588-018-0191-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Decaesteker B, Denecker G, van Neste C, Dolman EM, van Loocke W, Gartlgruber M, Nunes C, de Vloed F, Depuydt P, Verboom K, Rombaut D, Loontiens S, de Wyn J, Kholosy WM, Koopmans B, Essing AHW, Herrmann C, Dreidax D, Durinck K, Deforce D, van Nieuwerburgh F, Henssen A, Versteeg R, Boeva V, Schleiermacher G, van Nes J, Mestdagh P, Vanhauwaert S, Schulte JH, Westermann F, Molenaar JJ, de Preter K, Speleman F. TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nature Communications. 2018;9:4866. doi: 10.1038/s41467-018-06699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, Perez-Atayde A, Wong KK, Yuan GC, Gray NS, Young RA, George RE. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–1139. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tee AE, Ciampa OC, Wong M, Fletcher JI, Kamili A, Chen J, Ho N, Sun Y, Carter DR, Cheung BB, Marshall GM, Liu PY, Liu T. Combination therapy with the CDK7 inhibitor and the tyrosine kinase inhibitor exerts synergistic anticancer effects against MYCN-amplified neuroblastoma. International Journal of Cancer. 2020;147:1928–1938. doi: 10.1002/ijc.32936. [DOI] [PubMed] [Google Scholar]
- 159.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJP, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee S, Rellinger EJ, Kim KW, Craig BT, Romain CV, Qiao J, Chung DH. Bromodomain and extraterminal inhibition blocks tumor progression and promotes differentiation in neuroblastoma. Surgery. 2015;158:819–826. doi: 10.1016/j.surg.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mazar J, Gordon C, Naga V, Westmoreland TJ. The killing of human neuroblastoma cells by the small molecule JQ1 occurs in a p53-dependent manner. Anti-Cancer Agents in Medicinal Chemistry. 2020;20:1613–1625. doi: 10.2174/1871520620666200424123834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shahbazi J, Liu PY, Atmadibrata B, Bradner JE, Marshall GM, Lock RB, Liu T. The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N-Myc expression and induce anticancer effects. Clinical Cancer Research. 2016;22:2534–2544. doi: 10.1158/1078-0432.CCR-15-1666. [DOI] [PubMed] [Google Scholar]
- 165.Veneziani I, Fruci D, Compagnone M, Pistoia V, Rossi P, Cifaldi L. The BET-bromodomain inhibitor JQ1 renders neuroblastoma cells more resistant to NK cell-mediated recognition and killing by downregulating ligands for NKG2D and DNAM-1 receptors. Oncotarget. 2019;10:2151–2160. doi: 10.18632/oncotarget.26736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Iniguez AB, et al. Resistance to epigenetic-targeted therapy engenders tumor cell vulnerabilities associated with enhancer remodeling. Cancer Cell. 2018;34:922–938.e927. doi: 10.1016/j.ccell.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gibbons RJ, Pellagatti A, Garrick D, Wood WG, Malik N, Ayyub H, Langford C, Boultwood J, Wainscoat JS, Higgs DR. Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor ATRX in the alpha-thalassemia myelodysplasia syndrome (ATMDS) Nature Genetics. 2003;34:446–449. doi: 10.1038/ng1213. [DOI] [PubMed] [Google Scholar]
- 168.Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome) Cell. 1995;80:837–845. doi: 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 169.Kassabov SR, Zhang B, Persinger J, Bartholomew B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Molecular Cell. 2003;11:391–403. doi: 10.1016/s1097-2765(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 170.Tang L, Nogales E, Ciferri C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Progress in Biophysics and Molecular Biology. 2010;102:122–128. doi: 10.1016/j.pbiomolbio.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, Cochrane JC, Allis CD, Picketts DJ, Patel DJ, Li H, Shi Y. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nature Structural & Molecular Biology. 2011;18:769–776. doi: 10.1038/nsmb.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Eustermann S, Yang JC, Law MJ, Amos R, Chapman LM, Jelinska C, Garrick D, Clynes D, Gibbons RJ, Rhodes D, Higgs DR, Neuhaus D. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nature Structural & Molecular Biology. 2011;18:777–782. doi: 10.1038/nsmb.2070. [DOI] [PubMed] [Google Scholar]
- 173.Dhayalan A, Tamas R, Bock I, Tattermusch A, Dimitrova E, Kudithipudi S, Ragozin S, Jeltsch A. The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Human Molecular Genetics. 2011;20:2195–2203. doi: 10.1093/hmg/ddr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10635–10640. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mitson M, Kelley LA, Sternberg MJ, Higgs DR, Gibbons RJ. Functional significance of mutations in the Snf2 domain of ATRX. Human Molecular Genetics. 2011;20:2603–2610. doi: 10.1093/hmg/ddr163. [DOI] [PubMed] [Google Scholar]
- 176.Gibbons, R. J. (2012). α-Thalassemia, mental retardation, and myelodysplastic syndrome. Cold Spring Harbor Perspectives in Medicine, 2. [DOI] [PMC free article] [PubMed]
- 177.Gibbons RJ, Wada T, Fisher CA, Malik N, Mitson MJ, Steensma DP, Fryer A, Goudie DR, Krantz ID, Traeger-Synodinos J. Mutations in the chromatin-associated protein ATRX. Human Mutation. 2008;29:796–802. doi: 10.1002/humu.20734. [DOI] [PubMed] [Google Scholar]
- 178.Dyer, M. A., Qadeer, Z. A., Valle-Garcia, D., & Bernstein, E. (2017). ATRX and DAXX: Mechanisms and mutations. Cold Spring Harbor Perspectives in Medicine, 7. [DOI] [PMC free article] [PubMed]
- 179.Abbasi MR, Rifatbegovic F, Brunner C, Mann G, Ziegler A, Pötschger U, Crazzolara R, Ussowicz M, Benesch M, Ebetsberger-Dachs G, Chan GCF, Jones N, Ladenstein R, Ambros IM, Ambros PF. Impact of disseminated neuroblastoma cells on the identification of the relapse-seeding clone. Clinical Cancer Research. 2017;23:4224–4232. doi: 10.1158/1078-0432.CCR-16-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zeineldin M, Federico S, Chen X, Fan Y, Xu B, Stewart E, Zhou X, Jeon J, Griffiths L, Nguyen R, Norrie J, Easton J, Mulder H, Yergeau D, Liu Y, Wu J, van Ryn C, Naranjo A, Hogarty MD, Kamiński MM, Valentine M, Pruett-Miller SM, Pappo A, Zhang J, Clay MR, Bahrami A, Vogel P, Lee S, Shelat A, Sarthy JF, Meers MP, George RE, Mardis ER, Wilson RK, Henikoff S, Downing JR, Dyer MA. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nature Communications. 2020;11:913. doi: 10.1038/s41467-020-14682-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Farooqi AS, Dagg RA, Choi LMR, Shay JW, Reynolds CP, Lau LMS. Alternative lengthening of telomeres in neuroblastoma cell lines is associated with a lack of MYCN genomic amplification and with p53 pathway aberrations. Journal of Neuro-Oncology. 2014;119:17–26. doi: 10.1007/s11060-014-1456-8. [DOI] [PubMed] [Google Scholar]
- 182.Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Drané P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes & Development. 2010;24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kurihara S, Hiyama E, Onitake Y, Yamaoka E, Hiyama K. Clinical features of ATRX or DAXX mutated neuroblastoma. Journal of Pediatric Surgery. 2014;49:1835–1838. doi: 10.1016/j.jpedsurg.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 186.Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, Gibbons RJ. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nature Communications. 2015;6:7538. doi: 10.1038/ncomms8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Clynes D, Jelinska C, Xella B, Ayyub H, Taylor S, Mitson M, Bachrati CZ, Higgs DR, Gibbons RJ. ATRX dysfunction induces replication defects in primary mouse cells. PLoS One. 2014;9:e92915. doi: 10.1371/journal.pone.0092915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cardoso C, Timsit S, Villard L, Khrestchatisky M, Fontès M, Colleaux L. Specific interaction between the XNP/ATR-X gene product and the SET domain of the human EZH2 protein. Human Molecular Genetics. 1998;7:679–684. doi: 10.1093/hmg/7.4.679. [DOI] [PubMed] [Google Scholar]
- 189.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature Genetics. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, Ariizumi SI, Yamamoto M, Yamada T, Chayama K, Kosuge T, Yamaue H, Kamatani N, Miyano S, Nakagama H, Nakamura Y, Tsunoda T, Shibata T, Nakagawa H. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature Genetics. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 191.Lee SH, Kim JS, Zheng S, Huse JT, Bae JS, Lee JW, Yoo KH, Koo HH, Kyung S, Park WY, Sung KW. ARID1B alterations identify aggressive tumors in neuroblastoma. Oncotarget. 2017;8:45943–45950. doi: 10.18632/oncotarget.17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Raab JR, Resnick S, Magnuson T. Genome-wide transcriptional regulation mediated by biochemically distinct SWI/SNF complexes. PLoS Genetics. 2015;11:e1005748. doi: 10.1371/journal.pgen.1005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Li C, Xu ZL, Zhao Z, An Q, Wang L, Yu Y, Piao DX. ARID1A gene knockdown promotes neuroblastoma migration and invasion. Neoplasma. 2017;64:367–376. doi: 10.4149/neo_2017_307. [DOI] [PubMed] [Google Scholar]
- 194.Bui CB, le HK, Vu DM, Truong KDD, Nguyen NM, Ho MAN, Truong DQ. ARID1A-SIN3A drives retinoic acid-induced neuroblastoma differentiation by transcriptional repression of TERT. Molecular Carcinogenesis. 2019;58:1998–2007. doi: 10.1002/mc.23091. [DOI] [PubMed] [Google Scholar]