

Abstract
Dilated cardiomyopathy (DCM) is a major cause of heart failure and cardiovascular mortality. In the past 20 years, there has been an overwhelming focus on developing therapeutics that target common downstream disease pathways thought to be involved in all forms of heart failure independent of the initial etiology. While this strategy is effective at the population level, individual responses vary tremendously and only approximately one third of patients receive benefit from modern heart failure treatments. In this perspective, we propose that DCM should be considered as a collection of diseases with a common phenotype of left ventricular dilation and systolic dysfunction rather than a single disease entity, and that mechanism-based classification of disease subtypes will revolutionize our understanding and clinical approach towards DCM. We discuss how these efforts are central to realizing the potential of precision medicine and how they are empowered by the development of new tools that allow investigators to strategically employ genomic and transcriptomic information. Finally, we outline an investigational strategy to 1) define DCM at the patient level, 2) develop new tools to model and mechanistically dissect subtypes of human heart failure, and 3) harness these insights for the development of precision therapeutics.
Keywords: engineered heart tissue, next generation sequencing, personalized medicine, functional genomics, stem cells, heart failure
Cardiovascular diseases including heart failure and coronary artery disease represent important causes of morbidity and mortality worldwide. Over the past several decades, the predominant focus of cardiovascular medicine has centered on identifying common factors that contribute to the progression of these prevalent diseases. Such initiatives have formed the basis of our current approach to patient care, which is generally agnostic to the underlying cause of a patient’s disease. For example, patients diagnosed with heart failure are offered essentially identical treatments regardless of whether their disease was caused by blockages in their coronary arteries or genetic mutations. Mechanistically, heart failure therapeutics target a process termed adverse remodeling, a common pathway by which the adult heart is thought to universally respond to injury. Pathologically, adverse remodeling is defined by cardiomyocyte hypertrophy and fibrosis [1,2]. Landmark clinical trials have established that therapeutics which target adverse remodeling including β-adrenergic, angiotensin receptor II, and aldosterone signaling inhibitors reduce mortality and improve left ventricular systolic function in adult heart failure patients [3].
While this “one-size-fits-all’ approach has led to improvements in clinical outcomes when large populations are examined, individual response rates vary tremendously, and it is difficult to distinguish patients who will achieve a favorable response from those who will experience disease progression and ultimately succumb to their illness. Consequently, many individuals are left inadequately treated and current 5-year transplant-free survival rates remain above 50% [4].
Intriguingly, therapies targeting adverse remodeling have substantially less efficacy in pediatric populations. The Pediatric Carvedilol Study failed to demonstrate improvements in clinical outcomes for children with symptomatic heart failure [5–7]. Registry data further revealed that adult heart failure therapeutics have provided no survival benefit in children over digoxin and diuretic based regimens that were established in the 1970s [8]. Our group has previously demonstrated that patients with pediatric heart failure displayed markedly reduced adverse remodeling at the histopathologic, electron microscopic, and gene expression levels compared to adult heart failure patients [9]. These observations indicate that pediatric and adult heart failure represent distinct entities, provide a mechanistic rationale for why children display substantially lower rates of ventricular arrhythmias and sudden cardiac death compared to adults [10,11], and highlight the clinically unmet need to identify novel approaches for pediatric cardiomyopathies.
In the cardiovascular field, we are now just beginning to appreciate that heart failure and coronary artery disease may actually represent a compilation of unique pathologies that are driven by complex interactions among a diverse array of genetic perturbations, environmental risk factors, and host responses to tissue injury or chronic disease. For example, due to the increased use of genetic sequencing in the clinic, inherited or spontaneously occurring mutations in genes important for cardiac contractility, structure, and metabolism are increasingly found in patients with idiopathic dilated cardiomyopathy (DCM), a common etiology of heart failure in adult and pediatric populations [12,13]. It is likely that different mutations give rise to distinct disease entities with differing phenotypes, environmental interactions, and responses to current medical regimens. For example, in the case of hypertrophic cardiomyopathy, the relative incidence of heart failure, ventricular arrhythmias, and atrial fibrillation varies depending on whether a patient has a mutation in a sarcomeric protein or not [14]. We believe that an individualized approach to cardiovascular medicine targeting the mechanisms that initiate disease and drive disease progression has the potential to be transformative and will ultimately lead to new treatments and hope for our patients.
Large gaps in knowledge must be overcome to realize the potential of cardiovascular precision medicine. Specifically, we must: reclassify cardiovascular diseases based on the molecular and cellular mechanisms that drive pathogenesis; develop diagnostic strategies to identify patients with shared disease subtypes; and, generate therapeutics that target the specific mechanisms that underlie disease subtypes. Realizing this potential will require functional studies, powered by new technologies, to study and model patient-specific phenotypes at the molecular and cellular levels, and then translate these insights into the development of precision therapies. Here, we will focus on the application of this approach to DCM, which represents a major cause of heart failure and mortality; however, these tools and approaches are broadly applicable to a host of cardiovascular diseases. We believe that this approach will reshape our clinical and research philosophy and realize the potential for precision medicine in the cardiovascular field.
Functional insights are necessary to translate from genomic insights to precision medicine
Recent innovations in sequencing technology has made it feasible and reasonably inexpensive to sequence whole human exomes and genomes. The large volume of available sequencing data has revealed surprising diversity in the human gene pool, and as such, has become a focal point for precision medicine. Large scale sequencing and genome wide association studies (GWAS) have changed how we think about cardiovascular disease, and we direct the reader to some of the excellent reviews written on this topic [15–17]. They have helped to uncover the genetic bases of monogenic diseases with common presentations, such as DCM, Marfan’s Syndrome, and Long Q-T syndrome, as well as polygenic risk factors for diseases including atherosclerosis, hypertension, and heart failure [18,19]. In the case of DCM, at least 25% [20] can be attributed to specific mutations in a subset of genes that encode for sarcomeric (Troponin C, Troponin T, Myosin Heavy Chain, Tropomyosin, Myosin Binding Protein C3), structural (Titin, Desmin), mechanotransduction (Lamin A/C), calcium handling (Phospholamban, SERCA), signaling (Integrin-Linked Kinase), and metabolic or mitochondrial proteins [12,13]. It should be noted that some of these proteins can serve multiple roles in the cell. While the majority of genotype-positive patients have heterozygous missense mutations that produce proteins with altered activity [21,22], abundance [23], cellular localization, and/or stability, some mutations produce splice site variants or truncations [13].
While the genetic basis for DCM has become better understood, this advancement has yet to impact patient care beyond family screening. One challenge arises from the fact that despite sharing the common feature of cardiac remodeling and having a prevalence of 1:250 in the population, DCM can be caused by hundreds of mutations [12]. For newly discovered mutations, it is difficult to determine whether a given variant is pathogenic, especially if examined families are not sufficiently large or the mutation occurred spontaneously. Genetic testing can be quite useful for individuals or families carrying a known or well-characterized variant that causes DCM. Individuals at risk for developing disease can easily be identified and subjected to either careful monitoring or aggressive treatment regimens focused on reducing cardiovascular risk factors and early initiation of anti-remodeling therapies. It is important to note that the genetic background and environment of a patient impact the presentation and prognosis of the disease. As a result, the timing of onset and heart failure phenotypes may differ between matched siblings and between parents and children. While some have suggested that genetic information alone could be used to decide whether to treat a patient with anti-remodeling therapies prophylactically, there is no consensus in the field as to the efficacy of such an approach with respect to the entire population of DCM or in regards to individual mutations [24].
Unfortunately, the direct translation of genomic data into precision medicine has been frustrated by several limitations. The most important limitation is that many genomic studies are correlative and do not necessarily provide actionable mechanistic insights into the disease pathogenesis. Surprisingly little is known regarding whether patients who harbor mutations in different genes display distinct heart failure phenotypes and/or clinical outcomes. It is possible that DCM could be better reclassified based on the identification of mutations that give rise to common disease phenotypes. Moreover, mutations within the same molecule can lead to different gross phenotypes. For example, mutations within troponin T can lead to hypertrophic [25], dilated [26], restricted cardiomyopathy, or no effect depending on the specific variant [27]. In fact, it has been shown that point mutations at the same residue with different amino acid substitutions can lead to different phenotypes [28].
Realizing the promise of precision medicine will require us to pair insights from genomic studies with mechanistic studies of the disease pathogenesis. Here, we discuss our approach and the fields progress towards precision medicine in 1) defining disease at the patient level, 2) developing new tools to mechanistically understand and model subtypes of human heart failure, and 3) harnessing these insights for the development of precision therapeutics (Figure 1).
Figure 1:
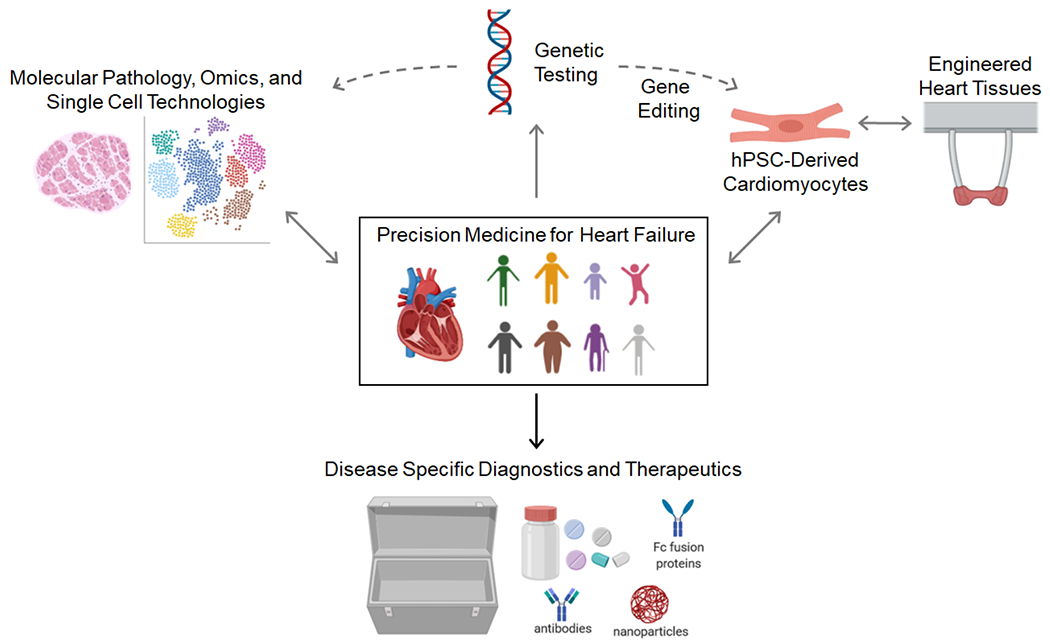
Schematic describing a precision medicine approach to heart failure and other cardiovascular diseases. Images were generated using Biorender.
Harnessing new technologies to delineate patient-specific phenotypes
The establishment of cardiac tissue biobanks have provided critical opportunities to explore cellular and molecular mechanisms that contribute to heart failure pathogenesis. Early histopathology and gene expression profiling studies have provided key evidence that heart failure is more heterogeneous and complex than previously appreciated [29]. In fact, standard classification schemes dividing cardiomyopathies based on ischemic or non-ischemic etiologies are not sufficient to account for variability between individual patients [30]. It is evident from rudimentary pathological analysis that dramatic differences exist between different forms of ischemic and non-ischemic cardiomyopathies (Figure 2). These observations highlight the need to develop new techniques and approaches to investigate, appropriately reclassify, and identify causative mechanisms that give rise to different forms of human heart failure.
Figure 2:
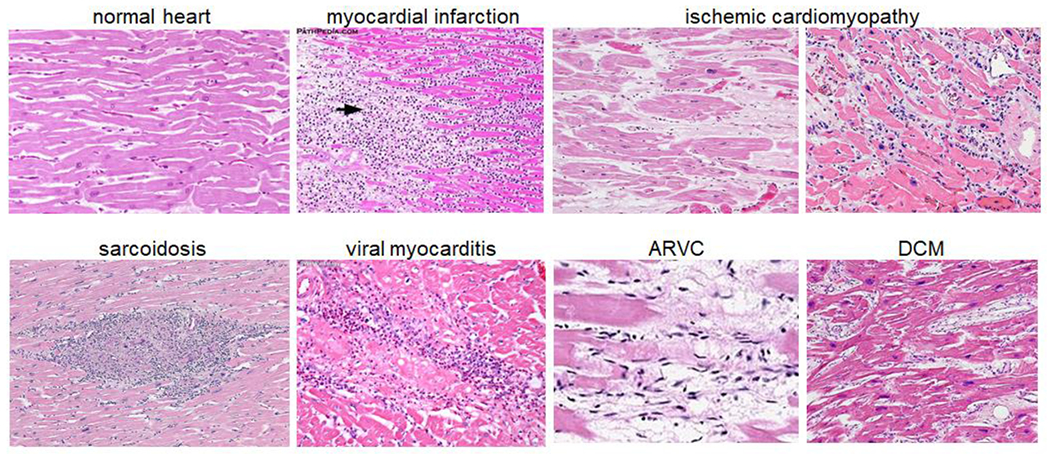
Individual heart failure etiologies display distinct histopathology features. Arrow denotes site of myocardial infarction. ARVC: arrhythmogenic right ventricular cardiomyopathy, DCM: dilated cardiomyopathy. Images from Wikipedia and Pathpedia.
Over the past several years, an explosion of molecular pathology techniques built on the shoulders of next generation nucleic acid sequencing technologies have revolutionized modern pathology. With the advent of single cell and single nuclei RNA sequencing approaches, it is now possible to measure gene expression at single cell resolution. Such technologies enable investigators to define the cellular composition of a given tissue, discover new cell types, and compare gene expression within a given cell type across experimental conditions or diseases within a single comprehensive and unbiased workflow. The introduction of single nuclei RNA sequencing has expanded these promising capabilities to encompass cell types that cannot be recovered from enzymatically digested tissues. Most importantly, single nuclei RNA sequencing is readily adaptable to cryopreserved specimens, fueling unprecedented exploration of biobanked tissues and reinvigorating enthusiasm for developing and expanding human tissue repositories. These technologies have already provided critical new insights into disease diversity and new cell types that contribute to the pathogenesis of diseases ranging from cancer to autism [31–34].
A limitation of single cell and single nuclei RNA sequencing approaches is the loss of spatial information, most notably the location of cell types within a tissue and the existence of unique tissue niches composed of various cell populations. Within current workflows, this information must be acquired retrospectively using either immunohistochemistry or in situ hybridization. Spatial transcriptomics and advanced small sample input tissue acquisition systems offer viable solutions. These technologies provide transcriptomic level information from fresh or fixed tissue sections, respectively. Spatial transcriptomics provides an unbiased platform for spatial resolved transcriptional profiling [35]. In this system, fresh frozen tissue sections are placed over a slide containing uniquely barcoded oligo-dT primers spaced every 40-100 microns printed over a 6mm × 6mm area. Co-registration of an H&E or antibody stained image with the position of the barcoded oligos allows the integration of spatial and transcriptomic data. Advanced laser capture and other small input tissue capture systems provide an alternative approach by allowing investigators to select particular regions of interest [36]. These platforms have the unique advantage of working on an array of tissue types ranging from fresh frozen to formalin fixed paraffin embedded tissues.
Today, there is incredible opportunity to apply these technologies to cardiovascular diseases. Understanding, the cellular composition of the healthy and diseased human heart is likely to provide unprecedented opportunities to identify disease potentiating cell types and delineate the functional diversity of human heart disease. For example, we know very little regarding the exact immune and fibroblast cell types that orchestrate myocardial fibrosis, whether mutations in distinct DCM genes produce unique tissue pathologies, or the underlying signaling pathways that contribute to inflammatory and infiltrative cardiomyopathies including cardiac sarcoidosis, giant cell myocarditis, and cardiac amyloidosis. Most importantly, next generation molecular pathology will undoubtedly allow investigators to glean critical information directly from the human disease itself rather than relying on oversimplified or potentially inaccurate animal or cellular models.
New tools to mechanistically understand and model subtypes of human heart failure in vitro
Recent technological advances have also opened the door to modeling human heart failure subtypes in vitro. These new tools can be leveraged for the development of precision therapeutics for heart failure. Here, we will focus on recent advances in stem cell technologies, gene editing, and tissue engineering, and their translational potential for cardiovascular precision medicine.
Gene editing and human pluripotent stem cells
There are several model systems that have been applied to model heart failure. Each of these systems comes with its own set of advantages and caveats. Tissue from patients provides useful information about the specific patient phenotype in human tissues [37]; however, it is often not possible to obtain genetically matched control tissues for functional experiments, and it is difficult to obtain sufficient volumes of tissue for most functional experiments. Moreover, human tissue is usually only available from patients in the end-stages of the disease, either after the implantation of a ventricular assist device, heart transplantation, or postmortem. Transgenic animals including drosophila, zebrafish, and mice are excellent systems for studying some genetic forms of cardiovascular disease [24]; however, they do not always recapitulate the disease phenotype seen in humans due to physiological differences between species [38–43]. In the case of mice, mouse hearts beat 500-600 times per minute, compared to approximately 60-80 times per minute for humans. To achieve this faster heart rate, murine hearts have different proteins isoforms for calcium handling, ion channels, and contractile proteins. For example, murine hearts express the alpha myosin isoform (MYH6), which has a speed (as measured by the ADP release rate from actomyosin) that is ~10-times greater than the beta cardiac isoform (MYH7) expressed in human ventricles [44]. These differences can make it so that mouse models of heart failure do not recapitulate the human disease phenotype, and efforts have been made to make more humanized mice [39,45,46]. Given known limitations of rodent and other model systems, there has been a great push to develop new experimental platforms that focus on human tissues or cell-based systems.
Recent advances in stem cell technologies and gene editing have made it easier to study mutations that cause human heart failure in experimentally tractable systems. The derivation of human induced pluripotent stem cells (hiPSCs) [47] from a patient blood sample, urine sample, or skin biopsy, has enabled the study of patient-specific disease-causing mutations. hiPSCs can be differentiated into cardiomyocytes (hiPSC-CMs) using small molecules that activate developmental pathways [48,49]. hiPSCs can undergo genome editing using the CRISPR/Cas9 system [50] to enable the modeling of human disease on a controlled genetic background. One caveat of this system is that hiPSC-CMs are developmentally immature compared to adult cardiomyocytes [51,52]. These cells differ from mature cardiomyocytes in several ways. For example, hiPSC-CMs have a lower mitochondrial content, minimal t-tubular structures, show sarcomeric disarray, have higher membrane resting potentials, generate less force in response to activation, and have altered action potentials [53,54]. That being said, the field is actively developing approaches to promote maturity, including providing hiPSC-CMs with mechanical [55–61], electrical [62–64], and chemical [51,65] cues that mimic the native environment of the heart.
Single hiPSC-CMs can be extensively characterized using deep phenotyping tools. Transcriptional profiles can be probed using RNA sequencing. Precise measurements of contractility can be obtained using tools such as traction force microscopy or atomic force microscopy [55,66,67]. Cellular metabolism can be measured using tools such as the Seahorse Analyzer. Cellular and sarcomeric structural organization can be examined in fixed cells using immunofluorescence or in live cells using fluorescently tagged proteins [68]. E-C coupling can be investigated using single cell patch clamping and voltage/calcium sensitive dyes [69]. Moreover, these profiling techniques can be used for drug screening of individual patient-specific cell lines.
Using genome edited hiPSC-CMs, it is now possible to engineer single cells carrying disease-causing mutations on a controlled genetic background. The CRISPR/Cas9 system [50] can be used both to introduce mutations into control lines or to correct mutations found in patient cells (i.e., generate a genetically matched healthy control lines). The use of gene editing on a controlled genetic background removes confounding factors that arise when comparing two non-matched patients or even siblings. This gene editing approach has been used to model several forms of cardiac diseases at the single cell level including hypertrophic cardiomyopathy, long QT syndrome, Duchenne’s Muscular Dystrophy, and dilated cardiomyopathy [70]. While hiPSC-CMs are developmentally immature [51] and do not currently capture the late stages of the disease, single cell assays have been able to recapitulate many features of the early disease phenotype including cellular hypertrophy, disrupted calcium transients, altered gene expression, and altered action potentials [71]. This can be seen as a key advantage to study disease initiation.
In the case of DCM, several patient-specific mutations have been modeled in hiPSC-CMs, including mutations in troponin T [21,67], lamin A/C [72], titin [23], dystrophin [73], and phospholamban [74]. Moreover, hiPSC-CMs have been used to model diabetic cardiomyopathy [75,76]. These studies have been used to identify new mechanisms involved in the disease pathogenesis, such as aberrant PDGFR signaling [72] and disrupted responses to mechanobiological cues [21]. hiPSC-CMs have also been used to test the effects of potential therapeutics for individual patient-specific mutations [67], revealing interesting similarities and differences between specific mutations, supporting the power of this experimental system for precision medicine approaches.
One place where this technology shows great promise for precision medicine is in determining whether a given variant is pathogenic. For many DCM-associated variants, there are not enough patients with a particular variant to definitively determine whether it is likely pathogenic or not using linkage analysis. Gene edited hiPSC-CMs were recently used to demonstrate the likely pathogenicity of a mutation that causes cardiomyopathy [77]. Cells from both healthy and diseased patients were obtained, reprogrammed, and differentiated to form hiPSC-CMS. The hiPSC-CMs from the diseased patients showed alterations in calcium transients, contractility, sarcomeric structure, and gene expression compared to healthy controls. Next, the healthy lines underwent gene editing to introduce the mutation, and the diseased patient line underwent editing to fix the mutation. The data clearly demonstrate that cellular dysfunction is dependent of the presence of the mutation, establishing that the mutation is likely pathogenic.
Human engineered heart tissues
Human engineered heart tissues (EHTs) provide a unique system for human disease modeling and the development of novel therapeutics. EHTs are in vitro systems that recapitulate aspects of the 3D environment of the heart [78,79]. As such, they provide a more physiologically relevant environment for studying cardiomyocyte function. The heart is a complex environment, consisting of many cell types including cardiomyocytes, fibroblasts, endothelial cells, and immune cells. These cells interact with each other, and these interactions can affect the contractile and electrophysiological properties of the myocardium [80]. In EHTs, cardiomyocytes are mixed with desired stromal cells in the presence of extracellular matrix proteins. These tissues will self-assemble in engineered devices where they can undergo extensive phenotyping. EHTs can be assembled using hiPSC-CMs, giving flexibility to examine patient-specific mutations that cause cardiovascular disease.
Multiple EHT platforms have been designed (Figure 3), each with its own set of strengths and weaknesses [81–84,63]. These platforms have varied geometries for tissue formation and are designed to examine different functional parameters [85]. These platforms can incorporate elements for measuring contractility, tissue organization, perfusion, electrophysiology, and conduction velocities. Moreover, systems have been designed to provide various cues to promote tissue maturity and to model environmental interactions including electrical stimulation [86,62], afterload [59], exogenous chemicals, and specific extracellular matrix scaffolds. hiPSC-CMs cultured in EHTs show improvements over single cells in mitochondrial content, sarcomeric organization, gene expression, T-tubule structure, electrophysiology, and contractility [62]. EHT systems have also been designed for higher throughput drug screening [87,88]. As such, EHTs show great promise for human disease modeling and drug discovery.
Figure 3:
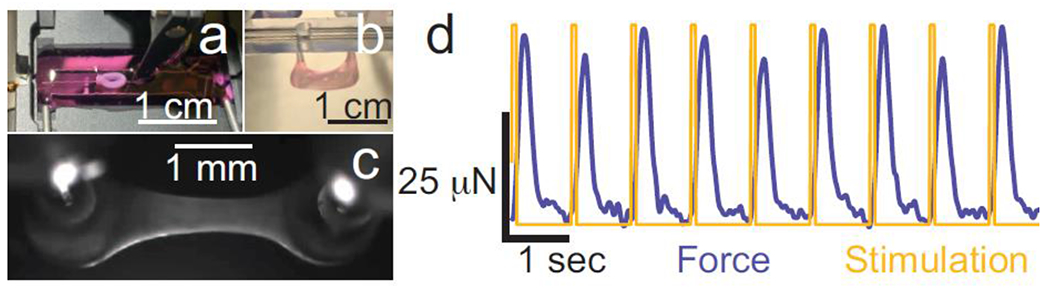
Examples of engineered heart tissues (EHTs) generated for disease modeling. (a) A circular EHT consisting of hiPSC-CMs and fibroblasts was mounted in between a force transducer and a length mover (Aurora Scientific) for active mechanical measurements. (b-c) An EHT consisting of hiPSC-CMs and fibroblasts was grown between two PDMS posts (EHT Technologies). The tissue self assembles and contracts spontaneously within 5 to 7 days after seeding. (d) An EHT grown between two PDMS posts was electrically stimulated (yellow). As the tissue contracts, it displaces the PDMS posts, enabling the calculation of the force of contraction (blue) for the tissue.
EHT systems have been used to model patient-specific mutations in several forms of heart disease. These studies have revealed diversity in the disease pathogenesis and highlight the potential role for EHTs in cardiovascular precision medicine. For example, an EHT model of DCM-causing truncations of the muscle protein titin revealed that these mutations likely exert their effect through haploinsufficiency [23]. EHTs were also used to model hypertrophic cardiomyopathy caused by mutations in myosin binding protein C (MyBPC), and it was used as a platform to demonstrate that expression of a phosphomimetic MyBPC could restore cardiac function in a particular mutant [89]. EHTs have also been used to study patient specific sensitivity to the chemotherapeutic agent, sunitinib, and to reveal that some heart specific effects of the drug become more pronounced with increased cardiac afterload [90].
Control of the genetic, chemical, cellular, and mechanical environment in EHTs gives them unique qualities that can be harnessed for precision medicine. For example, these systems could be used for diagnostic purposes, testing whether a given variant identified in a patient is pathogenic or for dissecting how multiple genetic polymorphisms contribute to the development of complex polygenic diseases. Moreover, one can completely define the EHT environment to mimic different physiological or pathological conditions. For example, it is possible to examine the effects of different cell types in the heart, and their relative roles in disease. One can examine how changes in mechanics, such as increased afterload in hypertension or stiffening of the myocardium in fibrosis affects the disease development [91,59,90]. Moreover, it is now possible to examine how changes in the extracellular matrix that accompany disease affect EHT function.
The road to precision therapies
The ultimate goal of precision medicine is to develop and appropriately match therapeutics to patients. This approach requires upfront knowledge of the patient’s diagnosis, disease classification, and therapeutic options. Thus, the generation of new diagnostic tools is an essential element that cannot be ignored. Diagnostic classifications based on genetic testing, imaging, and/or serum biomarkers represent feasible options. For example, genetic testing for DCM variants or molecular imaging for particular immune cell populations would provide physicians with the requisite information to prescribe tailored or individualized therapies that target specific genotypes of DCM or inflammatory mediators, respectively. As opposed to our current practice, this approach maximizes benefit for a given individual and minimizes risk associated with adverse effects. These tools can be applied to other diseases with cardiac involvement. For example, hiPSC-CMs from cancer patients were recently screened for cardiotoxicity to tyrosine kinase inhibitors [92]. Moreover, precision diagnostic tools are also likely to yield useful prognostic information regarding the anticipated natural history of an individual’s disease.
As highlighted above, EHTs provide a human model of heart disease for drug discovery, development, and cardiotoxicity studies. By using patient specific hiPSC-derived cardiomyocytes or engineering an individualized mutation into an established hiPSC cell line, EHTs can be used to predict an individual’s response to a particular therapy. The use of human EHTs has several advantages over animal systems for early drug discovery, including fewer ethical concerns, easier scaling for high throughput screening [88,87], and lower cost per experiment. Moreover, murine hearts do not express the hERG channel that is expressed in humans. Many drugs bind to this channel, and as such, undetected cardiotoxicity in mouse models is one of the leading causes for clinical trial failures [93,92].
There are several opportunities for the development of precision treatments for DCM, that expand beyond the current “one-size-fits-all” treatments that target remodeling. It is likely that therapeutics which correct the activity of individual DCM variants, prevent their incorporation into sarcomeres, or promote their removal through selective degradation would constitute a highly effective strategy to delay or reverse the natural history of DCM. For example, there are several new compounds in development that target the sarcomere to correct altered contractile protein function [94–96]. Moreover, gene therapy and exon skipping strategies to correct or mitigate the functional effects of mutant proteins have continued to evolve [97,98]; however, there are substantial challenges that still must be overcome before these technologies can be brought to the clinic. Alternatively, since the vast majority of DCM patients harbor heterozygous missense mutations that display reduced or absent activity, it might be possible to target mutant proteins for selective degradation. For many but not all of these mutants, the incorporation of these mutant proteins into sarcomeres (or other complexes) occurs in a stoichiometric fashion and results in a dominant effect on contractile function. For example, the TNNT2ΔK210 variant encodes a mutant protein with markedly reduced sensitivity to calcium [21]. Incorporation of this mutant protein into sarcomeres containing wild type TNNT2 leads to reduced force generation and abnormal relaxation [99,26]. Targeting of mutant protein for degradation or removal from the sarcomere has the possibility of rescuing the disease phenotype. Taken together, there are multiple exciting avenues for the development of precision therapeutics for cardiovascular disease.
We anticipate that the application of precision medicine to cardiovascular diseases will have profound translational impacts and result in paradigm shifts that will ultimately reshape our approach towards treating heart failure and other cardiovascular diseases. We envision that these initiatives will yield new diagnostics that define specific disease subtypes with differing etiologies, prognoses, and treatment responses. Ultimately, the deployment of precision therapies will finally arm physicians with the appropriate tools to treat disease on an individual rather than a population level. While these “forward-looking” initiatives may appear futuristic, technological advances are propelling the field at unprecedented speed and investigators are now only limited by their imagination and creativity rather than technical expertise.
Acknowledgments
Funding: The authors would like to acknowledge funding support from the National Institutes of Health (R01HL141086 to M.J.G, R01 HL138466 to K.J.L., R01 HL139714 to K.J.L.), Burroughs Welcome Fund (1014782 to K.J.L.), the March of Dimes Foundation (FY18-BOC-430198 to M.J.G.), Foundation of Barnes-Jewish Hospital (8038–88 to K.J.L.), and Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CH-ll-2017–628 to K.J.L., PM-LI-2019-829 to K.J.L. and M.J.G.).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflicts of interest/Competing interests: Kory Lavine - Medtronic: DT-PAS/APOGEE trial advisory board. Michael Greenberg - None.
References
- 1.Burchfield JS, Xie M, Hill JA (2013) Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation 128 (4):388–400. doi: 10.1161/CIRCULATIONAHA.113.001878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie M, Burchfield JS, Hill JA (2013) Pathological ventricular remodeling: therapies: part 2 of 2. Circulation 128 (9):1021–1030. doi: 10.1161/CIRCULATIONAHA.113.001879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg LR (2010) In the clinic. Heart failure. Ann Intern Med 152 (11):ITC61–15; quiz ITC616. doi:10.7326/0003-4819-152-6-201006010-0100610.7326/0003-4819-152-6-201006010-0100610.7326/0003-4819-152-11-201006010-0100610.7326/0003-4819-152-11-201006010-01006 [DOI] [PubMed] [Google Scholar]
- 4.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE (2006) Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 296 (15):1867–1876. doi: 10.1001/jama.296.15.1867 [DOI] [PubMed] [Google Scholar]
- 5.Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, Ross RD, Pahl E, Blume ED, Dodd DA, Rosenthal DN, Burr J, LaSalle B, Holubkov R, Lukas MA, Tani LY, Pediatric Carvedilol Study G (2007) Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA 298 (10):1171–1179. doi: 10.1001/jama.298.10.1171 [DOI] [PubMed] [Google Scholar]
- 6.Bristow MR, Gilbert EM, Abraham WT, Adams KF, Fowler MB, Hershberger RE, Kubo SH, Narahara KA, Ingersoll H, Krueger S, Young S, Shusterman N (1996) Carvedilol produces dose-related improvements in left ventricular function and survival in subjects with chronic heart failure. MOCHA Investigators. Circulation 94 (11):2807–2816 [DOI] [PubMed] [Google Scholar]
- 7.Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH (1996) The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med 334 (21):1349–1355. doi: 10.1056/NEJM199605233342101 [DOI] [PubMed] [Google Scholar]
- 8.Kantor PF, Abraham JR, Dipchand AI, Benson LN, Redington AN (2010) The impact of changing medical therapy on transplantation-free survival in pediatric dilated cardiomyopathy. J Am Coll Cardiol 55 (13):1377–1384. doi: 10.1016/j.jacc.2009.11.059 [DOI] [PubMed] [Google Scholar]
- 9.Patel MD, Mohan J, Schneider C, Bajpai G, Purevjav E, Canter CE, Towbin J, Bredemeyer A, Lavine KJ (2017) Pediatric and adult dilated cardiomyopathy represent distinct pathological entities. JCI Insight 2 (14). doi: 10.1172/jci.insight.94382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everitt MD, Sleeper LA, Lu M, Canter CE, Pahl E, Wilkinson JD, Addonizio LJ, Towbin JA, Rossano J, Singh RK, Lamour J, Webber SA, Colan SD, Margossian R, Kantor PF, Jefferies JL, Lipshultz SE, Pediatric Cardiomyopathy Registry I (2014) Recovery of echocardiographic function in children with idiopathic dilated cardiomyopathy: results from the pediatric cardiomyopathy registry. J Am Coll Cardiol 63 (14):1405–1413. doi: 10.1016/j.jacc.2013.11.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pahl E, Sleeper LA, Canter CE, Hsu DT, Lu M, Webber SA, Colan SD, Kantor PF, Everitt MD, Towbin JA, Jefferies JL, Kaufman BD, Wilkinson JD, Lipshultz SE, Pediatric Cardiomyopathy Registry I (2012) Incidence of and risk factors for sudden cardiac death in children with dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol 59 (6):607–615. doi: 10.1016/j.jacc.2011.10.878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNally EM, Mestroni L (2017) Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circulation research 121 (7):731–748. doi: 10.1161/CIRCRESAHA.116.309396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McNally EM, Golbus JR, Puckelwartz MJ (2013) Genetic mutations and mechanisms in dilated cardiomyopathy. The Journal of clinical investigation 123 (1):19–26. doi: 10.1172/JCI62862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM, Olivotto I (2018) Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 138 (14):1387–1398. doi: 10.1161/CIRCULATIONAHA.117.033200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Ende MY, Said MA, van Veldhuisen DJ, Verweij N, van der Harst P (2018) Genome-wide studies of heart failure and endophenotypes: lessons learned and future directions. Cardiovascular research 114 (9):1209–1225. doi: 10.1093/cvr/cvy083 [DOI] [PubMed] [Google Scholar]
- 16.Tayal U, Prasad S, Cook SA (2017) Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med 9 (1):20. doi: 10.1186/s13073-017-0410-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puckelwartz MJ, McNally EM (2014) Genetic profiling for risk reduction in human cardiovascular disease. Genes (Basel) 5 (1):214–234. doi: 10.3390/genes5010214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah S, Henry A, Roselli C, Lin H, Sveinbjornsson G, Fatemifar G, Hedman AK, Wilk JB, Morley MP, Chaffin MD, Helgadottir A, Verweij N, Dehghan A, Almgren P, Andersson C, Aragam KG, Arnlov J, Backman JD, Biggs ML, Bloom HL, Brandimarto J, Brown MR, Buckbinder L, Carey DJ, Chasman DI, Chen X, Chen X, Chung J, Chutkow W, Cook JP, Delgado GE, Denaxas S, Doney AS, Dorr M, Dudley SC, Dunn ME, Engstrom G, Esko T, Felix SB, Finan C, Ford I, Ghanbari M, Ghasemi S, Giedraitis V, Giulianini F, Gottdiener JS, Gross S, Guethbjartsson DF, Gutmann R, Haggerty CM, van der Harst P, Hyde CL, Ingelsson E, Jukema JW, Kavousi M, Khaw KT, Kleber ME, Kober L, Koekemoer A, Langenberg C, Lind L, Lindgren CM, London B, Lotta LA, Lovering RC, Luan J, Magnusson P, Mahajan A, Margulies KB, Marz W, Melander O, Mordi IR, Morgan T, Morris AD, Morris AP, Morrison AC, Nagle MW, Nelson CP, Niessner A, Niiranen T, O’Donoghue ML, Owens AT, Palmer CNA, Parry HM, Perola M, Portilla-Fernandez E, Psaty BM, Regeneron Genetics C, Rice KM, Ridker PM, Romaine SPR, Rotter JI, Salo P, Salomaa V, van Setten J, Shalaby AA, Smelser DT, Smith NL, Stender S, Stott DJ, Svensson P, Tammesoo ML, Taylor KD, Teder-Laving M, Teumer A, Thorgeirsson G, Thorsteinsdottir U, Torp-Pedersen C, Trompet S, Tyl B, Uitterlinden AG, Veluchamy A, Volker U, Voors AA, Wang X, Wareham NJ, Waterworth D, Weeke PE, Weiss R, Wiggins KL, Xing H, Yerges-Armstrong LM, Yu B, Zannad F, Zhao JH, Hemingway H, Samani NJ, McMurray JJV, Yang J, Visscher PM, Newton-Cheh C, Malarstig A, Holm H, Lubitz SA, Sattar N, Holmes MV, Cappola TP, Asselbergs FW, Hingorani AD, Kuchenbaecker K, Ellinor PT, Lang CC, Stefansson K, Smith JG, Vasan RS, Swerdlow DI, Lumbers RT (2020) Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun 11 (1):163. doi: 10.1038/s41467-019-13690-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young EP, Stitziel NO (2019) Capitalizing on Insights from Human Genetics to Identify Novel Therapeutic Targets for Coronary Artery Disease. Annu Rev Med 70:19–32. doi: 10.1146/annurev-med-041717-085853 [DOI] [PubMed] [Google Scholar]
- 20.Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D (2011) Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol 108 (8):1171–1176. doi: 10.1016/j.amjcard.2011.06.022 [DOI] [PubMed] [Google Scholar]
- 21.Clippinger SR, Cloonan PE, Greenberg L, Ernst M, Stump WT, Greenberg MJ (2019) Disrupted mechanobiology links the molecular and cellular phenotypes in familial dilated cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America 116 (36):17831–17840. doi: 10.1073/pnas.1910962116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ujfalusi Z, Vera CD, Mijailovich SM, Svicevic M, Yu EC, Kawana M, Ruppel KM, Spudich JA, Geeves MA, Leinwand LA (2018) Dilated cardiomyopathy myosin mutants have reduced force-generating capacity. The Journal of biological chemistry 293 (23):9017–9029. doi: 10.1074/jbc.RA118.001938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, Haghighi A, Homsy J, Hubner N, Church G, Cook SA, Linke WA, Chen CS, Seidman JG, Seidman CE (2015) HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349 (6251):982–986. doi: 10.1126/science.aaa5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fatkin D, Huttner IG, Kovacic JC, Seidman JG, Seidman CE (2019) Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. Journal of the American College of Cardiology 74 (23):2921–2938. doi: 10.1016/j.jacc.2019.10.011 [DOI] [PubMed] [Google Scholar]
- 25.Chandra M, Tschirgi ML, Tardiff JC (2005) Increase in tension-dependent ATP consumption induced by cardiac troponin T mutation. American journal of physiology Heart and circulatory physiology 289 (5):H2112–2119. doi: 10.1152/ajpheart.00571.2005 [DOI] [PubMed] [Google Scholar]
- 26.Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, Lu QW, Wang YY, Zhan DY, Mochizuki M, Kita S, Miwa Y, Takahashi-Yanaga F, Iwamoto T, Ohtsuki I, Sasaguri T (2007) Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circulation research 101 (2):185–194. doi: 10.1161/CIRCRESAHA.106.146670 [DOI] [PubMed] [Google Scholar]
- 27.Lynn ML, Lehman SJ, Tardiff JC (2018) Biophysical Derangements in Genetic Cardiomyopathies. Heart Fail Clin 14 (2):147–159. doi: 10.1016/j.hfc.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He H, Javadpour MM, Latif F, Tardiff JC, Ingwall JS (2007) R-92L and R-92W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophysical journal 93 (5):1834–1844. doi: 10.1529/biophysj.107.107557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurmani S, Squire I (2017) Acute Heart Failure: Definition, Classification and Epidemiology. Curr Heart Fail Rep 14 (5):385–392. doi: 10.1007/s11897-017-0351-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM (2014) Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 129 (9):1009–1021. doi: 10.1161/CIRCULATIONAHA.113.003863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, Kriegstein AR (2019) Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364 (6441):685–689. doi: 10.1126/science.aav8130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, Richoz N, Frazer GL, Staniforth JUL, Vieira Braga FA, Botting RA, Popescu DM, Vento-Tormo R, Stephenson E, Cagan A, Farndon SJ, Polanski K, Efremova M, Green K, Del Castillo Velasco-Herrera M, Guzzo C, Collord G, Mamanova L, Aho T, Armitage JN, Riddick ACP, Mushtaq I, Farrell S, Rampling D, Nicholson J, Filby A, Burge J, Lisgo S, Lindsay S, Bajenoff M, Warren AY, Stewart GD, Sebire N, Coleman N, Haniffa M, Teichmann SA, Behjati S, Clatworthy MR (2019) Spatiotemporal immune zonation of the human kidney. Science 365 (6460): 1461–1466. doi: 10.1126/science.aat5031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartoschek M, Oskolkov N, Bocci M, Lovrot J, Larsson C, Sommarin M, Madsen CD, Lindgren D, Pekar G, Karlsson G, Ringner M, Bergh J, Bjorklund A, Pietras K (2018) Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 9 (1 ):5150. doi: 10.1038/s41467-018-07582-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camp JG, Platt R, Treutlein B (2019) Mapping human cell phenotypes to genotypes with single-cell genomics. Science 365 (6460): 1401–1405. doi: 10.1126/science.aax6648 [DOI] [PubMed] [Google Scholar]
- 35.Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg A, Ponten F, Costea PI, Sahlen P, Mulder J, Bergmann O, Lundeberg J, Frisen J (2016) Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353 (6294):78–82. doi: 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- 36.Kamal M, Saremi S, Klotz R, Iriondo O, Amzaleg Y, Chairez Y, Tulpule V, Lang JE, Kang I, Yu M (2019) PIC&RUN: An integrated assay for the detection and retrieval of single viable circulating tumor cells. Sci Rep 9 (1):17470. doi: 10.1038/s41598-019-53899-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fananapazir L, Dalakas MC, Cyran F, Cohn G, Epstein ND (1993) Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America 90 (9):3993–3997. doi: 10.1073/pnas.90.9.3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowey S, Bretton V, Gulick J, Robbins J, Trybus KM (2013) Transgenic mouse alpha- and beta-cardiac myosins containing the R403Q mutation show isoform-dependent transient kinetic differences. The Journal of biological chemistry 288 (21): 14780–14787. doi: 10.1074/jbc.M113.450668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ford SJ, Mamidi R, Jimenez J, Tardiff JC, Chandra M (2012) Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. Journal of molecular and cellular cardiology 53 (4):542–551. doi: 10.1016/j.yjmcc.2012.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J, Robbins J (2008) Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. The Journal of biological chemistry 283 (29):20579–20589. doi: 10.1074/jbc.M800554200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roh J, Houstis N, Rosenzweig A (2017) Why Don’t We Have Proven Treatments for HFpEF? Circulation Research 120 (8): 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conceição G, Heinonen I, Lourenço AP, Duncker DJ, Falcão-Pires I (2016) Animal models of heart failure with preserved ejection fraction. Netherlands Heart Journal 24 (4):275–286. doi: 10.1007/s12471-016-0815-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, Shkreli M, Delp S, Pomerantz JH, Artandi SE, Blau HM (2010) Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143 (7):1059–1071. doi: 10.1016/j.cell.2010.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deacon JC, Bloemink MJ, Rezavandi H, Geeves MA, Leinwand LA (2012) Identification of functional differences between recombinant human alpha and beta cardiac myosin motors. Cellular and molecular life sciences : CMLS 69 (13):2261–2277. doi: 10.1007/s00018-012-0927-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyska MJ, Hayes E, Giewat M, Seidman CE, Seidman JG, Warshaw DM (2000) Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circulation research 86 (7):737–744 [DOI] [PubMed] [Google Scholar]
- 46.Nag S, Sommese RF, Ujfalusi Z, Combs A, Langer S, Sutton S, Leinwand LA, Geeves MA, Ruppel KM, Spudich JA (2015) Contractility parameters of human beta-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv 1 (9):e1500511. doi: 10.1126/sciadv.1500511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4):663–676. doi: 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 48.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP (2012) Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proceedings of the National Academy of Sciences of the United States of America 109 (27):E1848–1857. doi: 10.1073/pnas.1200250109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L (2001) Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. The Journal of clinical investigation 108 (3):407–414. doi: 10.1172/JCI12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337 (6096):816–821. doi: 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, Bruneau BG, Seidman JG, Seidman CE (2016) Single-Cell Resolution of Temporal Gene Expression during Heart Development. Developmental cell 39 (4):480–490. doi: 10.1016/j.devcel.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsa E, Ahrens JH, Wu JC (2016) Human Induced Pluripotent Stem Cells as a Platform for Personalized and Precision Cardiovascular Medicine. Physiol Rev 96 (3):1093–1126. doi: 10.1152/physrev.00036.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Addis RC, Epstein JA (2013) Induced regeneration--the progress and promise of direct reprogramming for heart repair. Nat Med 19 (7):829–836. doi: 10.1038/nm.3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iorga B, Schwanke K, Weber N, Wendland M, Greten S, Piep B, Dos Remedios CG, Martin U, Zweigerdt R, Kraft T, Brenner B (2017) Differences in Contractile Function of Myofibrils within Human Embryonic Stem Cell-Derived Cardiomyocytes vs. Adult Ventricular Myofibrils Are Related to Distinct Sarcomeric Protein Isoforms. Front Physiol 8:1111. doi: 10.3389/fphys.2017.01111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ribeiro AJ, Ang YS, Fu JD, Rivas RN, Mohamed TM, Higgs GC, Srivastava D, Pruitt BL (2015) Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proceedings of the National Academy of Sciences of the United States of America 112 (41):12705–12710. doi: 10.1073/pnas.1508073112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feaster TK, Cadar AG, Wang L, Williams CH, Chun YW, Hempel JE, Bloodworth N, Merryman WD, Lim CC, Wu JC, Knollmann BC, Hong CC (2015) Matrigel Mattress: A Method for the Generation of Single Contracting Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circulation research 117 (12):995–1000. doi: 10.1161/CIRCRESAHA.115.307580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, Sanger JW, Sanger JM, Discher DE (2008) Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. Journal of cell science 121 (Pt 22):3794–3802. doi: 10.1242/jcs.029678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fink C, Ergun S, Kralisch D, Remmers U, Weil J, Eschenhagen T (2000) Chronic stretch of engineered heart tissue induces hypertrophy and functional improvement. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 14 (5):669–679. doi: 10.1096/fasebj.14.5.669 [DOI] [PubMed] [Google Scholar]
- 59.Leonard A, Bertero A, Powers JD, Beussman KM, Bhandari S, Regnier M, Murry CE, Sniadecki NJ (2018) Afterload promotes maturation of human induced pluripotent stem cell derived cardiomyocytes in engineered heart tissues. Journal of molecular and cellular cardiology 118:147–158. doi: 10.1016/j.yjmcc.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimko VF, Claycomb WC (2008) Effect of mechanical loading on three-dimensional cultures of embryonic stem cell-derived cardiomyocytes. Tissue engineering Part A 14 (1):49–58. doi: 10.1089/ten.a.2007.0092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mihic A, Li J, Miyagi Y, Gagliardi M, Li SH, Zu J, Weisel RD, Keller G, Li RK (2014) The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials 35 (9):2798–2808. doi: 10.1016/j.biomaterials.2013.12.052 [DOI] [PubMed] [Google Scholar]
- 62.Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G (2018) Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556 (7700):239–243. doi: 10.1038/s41586-018-0016-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Masse S, Gagliardi M, Hsieh A, Thavandiran N, Laflamme MA, Nanthakumar K, Gross GJ, Backx PH, Keller G, Radisic M (2013) Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nature methods 10 (8):781–787. doi: 10.1038/nmeth.2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radisic M, Park H, Shing H, Consi T, Schoen FJ, Langer R, Freed LE, Vunjak-Novakovic G (2004) Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proceedings of the National Academy of Sciences of the United States of America 101 (52):18129–18134. doi: 10.1073/pnas.0407817101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Birket MJ, Casini S, Kosmidis G, Elliott DA, Gerencser AA, Baartscheer A, Schumacher C, Mastroberardino PG, Elefanty AG, Stanley EG, Mummery CL (2013) PGC-1alpha and reactive oxygen species regulate human embryonic stem cell-derived cardiomyocyte function. Stem Cell Reports 1 (6):560–574. doi: 10.1016/j.stemcr.2013.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ribeiro AJS, Schwab O, Mandegar MA, Ang YS, Conklin BR, Srivastava D, Pruitt BL (2017) Multi-Imaging Method to Assay the Contractile Mechanical Output of Micropatterned Human iPSC-Derived Cardiac Myocytes. Circulation research 120 (10):1572–1583. doi: 10.1161/CIRCRESAHA.116.310363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC, Wu JC (2012) Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Science translational medicine 4 (130):130ra147. doi: 10.1126/scitranslmed.3003552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toepfer CN, Sharma A, Cicconet M, Garfinkel AC, Mucke M, Neyazi M, Willcox JAL, Agarwal R, Schmid M, Rao J, Ewoldt J, Pourquie O, Chopra A, Chen CS, Seidman JG, Seidman CE (2019) SarcTrack. Circulation research 124 (8):1172–1183. doi: 10.1161/CIRCRESAHA.118.314505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Meer BJ, Krotenberg A, Sala L, Davis RP, Eschenhagen T, Denning C, Tertoolen LGJ, Mummery CL (2019) Simultaneous measurement of excitation-contraction coupling parameters identifies mechanisms underlying contractile responses of hiPSC-derived cardiomyocytes. Nat Commun 10 (1):4325. doi: 10.1038/s41467-019-12354-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, Terzic A, Wu JC, American Heart Association Council on Functional G, Translational B, Council on Cardiovascular Disease in the Y, Council on C, Stroke N (2018) Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 11 (1):e000043. doi: 10.1161/HCG.0000000000000043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sallam K, Kodo K, Wu JC (2014) Modeling inherited cardiac disorders. Circ J 78 (4):784–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee J, Termglinchan V, Diecke S, Itzhaki I, Lam CK, Garg P, Lau E, Greenhaw M, Seeger T, Wu H, Zhang JZ, Chen X, Gil IP, Ameen M, Sallam K, Rhee JW, Churko JM, Chaudhary R, Chour T, Wang PJ, Snyder MP, Chang HY, Karakikes I, Wu JC (2019) Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572 (7769):335–340. doi: 10.1038/s41586-019-1406-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsurumi F, Baba S, Yoshinaga D, Umeda K, Hirata T, Takita J, Heike T (2019) The intracellular Ca2+ concentration is elevated in cardiomyocytes differentiated from hiPSCs derived from a Duchenne muscular dystrophy patient. PloS one 14 (3):e0213768. doi: 10.1371/journal.pone.0213768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karakikes I, Stillitano F, Nonnenmacher M, Tzimas C, Sanoudou D, Termglinchan V, Kong CW, Rushing S, Hansen J, Ceholski D, Kolokathis F, Kremastinos D, Katoulis A, Ren L, Cohen N, Gho J, Tsiapras D, Vink A, Wu JC, Asselbergs FW, Li RA, Hulot JS, Kranias EG, Hajjar RJ (2015) Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat Commun 6:6955. doi: 10.1038/ncomms7955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burkart AM, Tan K, Warren L, Iovino S, Hughes KJ, Kahn CR, Patti ME (2016) Insulin Resistance in Human iPS Cells Reduces Mitochondrial Size and Function. Sci Rep 6:22788. doi: 10.1038/srep22788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, Gerard R, Badi L, Kam-Thong T, Bu L, Jiang X, Hoflack JC, Kiialainen A, Jeworutzki E, Aoyama N, Carlson C, Burcin M, Gromo G, Boehringer M, Stahlberg H, Hall BJ, Magnone MC, Kolaja K, Chien KR, Bailly J, Iacone R (2014) Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep 9 (3):810–821. doi: 10.1016/j.celrep.2014.09.055 [DOI] [PubMed] [Google Scholar]
- 77.Ma N, Zhang JZ, Itzhaki I, Zhang SL, Chen H, Haddad F, Kitani T, Wilson KD, Tian L, Shrestha R, Wu H, Lam CK, Sayed N, Wu JC (2018) Determining the Pathogenicity of a Genomic Variant of Uncertain Significance Using CRISPR/Cas9 and Human-Induced Pluripotent Stem Cells. Circulation 138 (23):2666–2681. doi: 10.1161/CIRCULATIONAHA.117.032273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eschenhagen T, Fink C, Remmers U, Scholz H, Wattchow J, Weil J, Zimmermann W, Dohmen HH, Schafer H, Bishopric N, Wakatsuki T, Elson EL (1997) Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: a new heart muscle model system. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 11 (8):683–694. doi: 10.1096/fasebj.11.8.9240969 [DOI] [PubMed] [Google Scholar]
- 79.Greenberg MJ, Daily NJ, Wang A, Conway MK, Wakatsuki T (2018) Genetic and Tissue Engineering Approaches to Modeling the Mechanics of Human Heart Failure for Drug Discovery. Front Cardiovasc Med 5:120. doi: 10.3389/fcvm.2018.00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M (2017) Macrophages Facilitate Electrical Conduction in the Heart. Cell 169 (3):510–522 e520. doi: 10.1016/j.cell.2017.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Legant WR, Pathak A, Yang MT, Deshpande VS, McMeeking RM, Chen CS (2009) Microfabricated tissue gauges to measure and manipulate forces from 3D microtissues. Proceedings of the National Academy of Sciences of the United States of America 106 (25):10097–10102. doi: 10.1073/pnas.0900174106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boudou T, Legant WR, Mu A, Borochin MA, Thavandiran N, Radisic M, Zandstra PW, Epstein JA, Margulies KB, Chen CS (2012) A microfabricated platform to measure and manipulate the mechanics of engineered cardiac microtissues. Tissue engineering Part A 18 (9–10):910–919. doi: 10.1089/ten.TEA.2011.0341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zimmermann WH, Schneiderbanger K, Schubert P, Didie M, Munzel F, Heubach JF, Kostin S, Neuhuber WL, Eschenhagen T (2002) Tissue engineering of a differentiated cardiac muscle construct. Circulation research 90 (2):223–230 [DOI] [PubMed] [Google Scholar]
- 84.Huebsch N, Loskill P, Deveshwar N, Spencer CI, Judge LM, Mandegar MA, Fox CB, Mohamed TM, Ma Z, Mathur A, Sheehan AM, Truong A, Saxton M, Yoo J, Srivastava D, Desai TA, So PL, Healy KE, Conklin BR (2016) Miniaturized iPS-Cell-Derived Cardiac Muscles for Physiologically Relevant Drug Response Analyses. Sci Rep 6:24726. doi: 10.1038/srep24726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loskill P, Huebsch N (2019) Engineering Tissues from Induced Pluripotent Stem Cells. Tissue engineering Part A 25 (9–10):707–710. doi: 10.1089/ten.TEA.2019.0118 [DOI] [PubMed] [Google Scholar]
- 86.Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Bornchen C, Muller C, Schulz H, Hubner N, Stenzig J, Stoehr A, Neuber C, Eder A, Luther PK, Hansen A, Eschenhagen T (2014) Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. Journal of molecular and cellular cardiology 74:151–161. doi: 10.1016/j.yjmcc.2014.05.009 [DOI] [PubMed] [Google Scholar]
- 87.Marquez JP, Legant W, Lam V, Cayemberg A, Elson E, Wakatsuki T (2009) High-throughput measurements of hydrogel tissue construct mechanics. Tissue Eng Part C Methods 15 (2):181–190. doi: 10.1089/ten.tec.2008.0347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lam V, Wakatsuki T (2011) Hydrogel tissue construct-based high-content compound screening. J Biomol Screen 16 (1):120–128. doi: 10.1177/1087057110388269 [DOI] [PubMed] [Google Scholar]
- 89.Dutsch A, Wijnker PJM, Schlossarek S, Friedrich FW, Kramer E, Braren I, Hirt MN, Breniere-Letuffe D, Rhoden A, Mannhardt I, Eschenhagen T, Carrier L, Mearini G (2019) Phosphomimetic cardiac myosin-binding protein C partially rescues a cardiomyopathy phenotype in murine engineered heart tissue. Sci Rep 9 (1 ):18152. doi: 10.1038/s41598-019-54665-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Truitt R, Mu A, Corbin EA, Vite A, Brandimarto J, Ky B, Margulies KB (2018) Increased Afterload Augments Sunitinib-Induced Cardiotoxicity in an Engineered Cardiac Microtissue Model. JACC Basic Transl Sci 3 (2):265–276. doi: 10.1016/j.jacbts.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rodriguez ML, Werner TR, Becker B, Eschenhagen T, Hirt MN (2019) A magnetics-based approach for fine-tuning afterload in engineered heart tissues. ACS Biomater Sci Eng 5 (7):3663–3675. doi: 10.1021/acsbiomaterials.8b01568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, Churko JM, Kitani T, Wu H, Holmstrom A, Matsa E, Zhang Y, Kumar A, Fan AC, Del Alamo JC, Wu SM, Moslehi JJ, Mercola M, Wu JC (2017) High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Science translational medicine 9 (377). doi: 10.1126/scitranslmed.aaf2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC, Wu JC (2013) Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 127 (16):1677–1691. doi: 10.1161/CIRCULATIONAHA.113.001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Woody MS, Greenberg MJ, Barua B, Winkelmann DA, Goldman YE, Ostap EM (2018) Positive cardiac inotrope omecamtiv mecarbil activates muscle despite suppressing the myosin working stroke. Nat Commun 9 (1):3838. doi: 10.1038/s41467-018-06193-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R, Baliga R, Cox DR, Garard M, Godinez G, Kawas R, Kraynack E, Lenzi D, Lu PP, Muci A, Niu C, Qian X, Pierce DW, Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen YT, Vatner SF, Morgans DJ (2011) Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331 (6023):1439–1443. doi: 10.1126/science.1200113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG, Seidman CE (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351 (6273):617–621. doi: 10.1126/science.aad3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, Darby H, Van Dyken C, Li Y, Kang E, Park AR, Kim D, Kim ST, Gong J, Gu Y, Xu X, Battaglia D, Krieg SA, Lee DM, Wu DH, Wolf DP, Heitner SB, Belmonte JCI, Amato P, Kim JS, Kaul S, Mitalipov S (2017) Correction of a pathogenic gene mutation in human embryos. Nature 548 (7668):413–419. doi: 10.1038/nature23305 [DOI] [PubMed] [Google Scholar]
- 98.Gramlich M, Pane LS, Zhou Q, Chen Z, Murgia M, Schotterl S, Goedel A, Metzger K, Brade T, Parrotta E, Schaller M, Gerull B, Thierfelder L, Aartsma-Rus A, Labeit S, Atherton JJ, McGaughran J, Harvey RP, Sinnecker D, Mann M, Laugwitz KL, Gawaz MP, Moretti A (2015) Antisense-mediated exon skipping: a therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol Med 7 (5):562–576. doi: 10.15252/emmm.201505047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, Ohta M, Sasaguri T, Ohtsuki I (2002) Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America 99 (2):913–918. doi: 10.1073/pnas.022628899 [DOI] [PMC free article] [PubMed] [Google Scholar]