

Abstract
Background & Aims:
Hepatocellular carcinoma (HCC) is an increasing cause of cancer-related deaths worldwide, due in part through prevalent obesity-related nonalcoholic steatohepatitis (NASH). Hepatocyte Notch pathway is a pathogenic factor to NASH-associated fibrosis, but its role in HCC is less defined. We characterized the molecular and clinical features of Notch-active human HCC, and investigated the mechanisms of how Notch affects NASH-driven HCC.
Methods:
Using a 14-gene Notch score, we stratified human HCCs in multiple datasets with comprehensive profiling. We performed gene set enrichment analyses comparing Notch-active HCCs to published HCC subtype signatures. Next, we sorted Notch-active hepatocytes from Notch reporter mice for RNA sequencing to study how Notch activation contributes to tumorigenesis in NASH, and characterized Notch-active tumors in a HCC model combining carcinogen and NASH-inducing diet. We used genetic mouse models to manipulate hepatocyte Notch to investigate the sufficiency and necessity of Notch in NASH-driven tumorigenesis.
Results:
Notch-active signatures were found in ~30% of human HCCs that transcriptionally resemble cholangiocarcinoma-like HCC with lack of activating CTNNB1 (β-catenin) mutations and overall poor prognosis. Endogenous Notch activation in hepatocytes is associated with repressed β-catenin signaling and hepatic metabolic functions, in lieu of increased interactions with extracellular matrix in NASH. Constitutive hepatocyte Notch activation is sufficient to induce β-catenin-inactive HCC in mice with NASH. Notch and β-catenin show a pattern of mutual exclusivity in carcinogen-induced HCC; in this mouse model, chronic blockade of Notch led to the tumor development reliant on β-catenin.
Conclusions:
Notch activity characterizes a distinct HCC molecular subtype with unique histology and prognosis. Sustained Notch signaling in chronic liver diseases is a driver of tumor formation without acquiring specific genomic mutations.
Lay Summary:
Notch pathway activity is increased in a subset of liver cancers with poor outcomes, and aberrant Notch activity in obese mice can drive HCC.
Keywords: Hepatocellular carcinoma, Nonalcoholic steatohepatitis, Fibrosis, Notch signaling, β-catenin, Mouse Model
Graphical Abstract
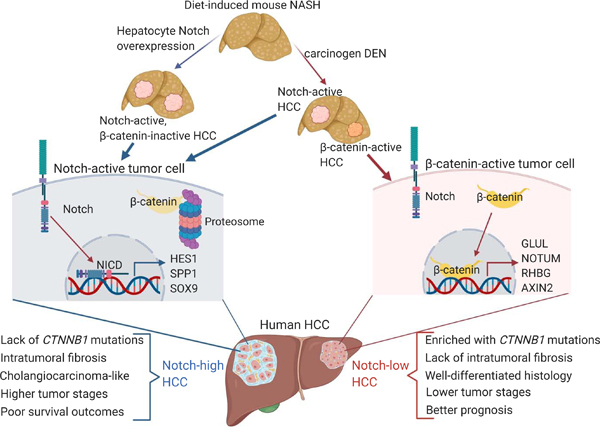
Introduction
Hepatocellular carcinoma (HCC) is a malignant primary liver cancer, and the fourth leading cause of cancer-related deaths worldwide,1 with a 5-year survival rate of less than 18%.2 HCC typically arises in the setting of chronic liver diseases of various etiologies, with approximately 90% of tumors occurring in fibrotic livers.3 Of chronic liver diseases, obesity-related nonalcoholic steatohepatitis (NASH) is the fastest-growing etiology for HCC in the United States.4 Comprehensive characterizations of large human HCC datasets, including The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC), revealed that HCCs are highly heterogenous with distinct histological and molecular subtypes.5 Only a few genes are frequently mutated (> 5% of HCCs), and aside from “hotspot” mutations in CTNNB1 (β-catenin),6 TP53,7 and TERT (Telomerase reverse transcriptase) promoter,8 most mutations are not recurrent.9 Adding to this complexity, recent sequencing efforts reported that heterogeneity also exists at inter- and intra-tumoral levels in HCC.10 Elucidation of additional molecular mechanisms underlying HCC diversity is necessary for personalized medicine to cope with the growing obese population.
Notch signaling is a conserved cell-to-cell communication pathway. Notch ligands (Jagged1/2, and Delta-like ligands 1/3/4) on a signal-sending cell bind Notch receptors (Notch1–4) on a physically proximal signal-receiving cell, resulting in proteolytic cleavage and release of the Notch intracellular domain (NICD). NICD is a transcriptional coactivator that binds to Rbpj and Mastermind (MAM) to activate expression of Notch target genes, including Hairy enhancer of split (Hes) and Hes-related Hey family genes.11 Notch is best known for its role in cell fate determination. During liver development, Notch activation in progenitor cells near the portal vein prompts commitment to the biliary lineage.12 Post-development, Notch signaling remains dormant in hepatocytes, but is aberrantly activated in the setting of obesity and NASH as a maladaptive repair signal that facilitates fibrosis development, which can be ameliorated by pharmacologic Notch inhibitors13.
As NOTCH1 gain-of-function mutations are frequently associated with human T-cell acute lymphoblastic leukemia (T-ALL),14 we hypothesized that increased hepatocyte Notch activity might push NASH to HCC. But Notch pathway mutations are rarely reported in HCC,15 although this has not been systematically evaluated. More intriguingly, Notch actions in solid tumors is highly context-dependent,16 and there are even reported Notch loss-of-function mutations in some lung cancers,17, 18 suggesting potential tumor suppressive functions of this pathway. Animal studies have been similarly inconclusive, with hepatoblast-specific NICD transgenic mice developing HCC,19, 20 but Notch activity also found to be tumor-suppressive in a HCC model of Retinoblastoma (Rb) pathway deficiency.21
To clarify these issues, we created a 14-gene transcriptional Notch score to stratify human HCCs. Using this scoring system, we found that Notch-active HCCs were not enriched in somatic mutations in Notch pathways, but were notable for lack of activating CTNNB1 mutations and repressed metabolic processes. Notch-active HCCs expressed a cholangiocarcinoma-like signature with enriched intratumoral fibrosis and had higher tumor stages and worse prognosis than Notch-inactive HCCs. We observed a similar transcriptional profile – decreased hepatocyte metabolic processes and β-catenin signaling, but increased ECM/stromal interactions and growth factor signaling – in Notch-active hepatocytes in reporter mice fed a NASH provoking-diet, as well as fully-penetrant, diet-dependent HCC in mice with constitutive hepatocyte Notch activity. Consistently, in HCC induced by carcinogen and NASH diet-feeding, Notch activity defined the majority of tumors. In this model, Notch inhibition did not impact overall tumor burden, but provoked compensatory β-catenin activity in residual tumors. Overall, these data suggest that NASH-induced persistent Notch activation prompts tumorigenesis even in the absence of genomic driver events such as activating mutations in CTNNB1 or Notch pathway components.
Materials and Methods
Human HCC datasets.
For analyses of human HCC datasets (TCGA, NCI, Korean, and Fudan cohorts), please see Supplementary Materials and Methods.
Mouse Studies.
Notch-Venus,22 homozygous RosaNICD,23 and RosaDNMAM24 male mice were crossed with female C57BL/6J (strain #000664, the Jackson Laboratory) mice to generate heterozygous transgenic mice for experiments. Mice were weaned to standard chow (PicoLab, #5053) in all experiments. For the DEN NASH model, 8-week-old male C57BL/6J mice were first treated with 100 mg/kg diethylnitrosamine (DEN, Sigma #73861), then started on NASH diet (Teklad, TD.190142) with fructose-containing drinking water (23.1 g fructose and 18.9 g glucose dissolved in 1 L water, then filter-sterilized) two weeks later. 8-week-old RosaNICD or RosaDNMAM mice were transduced by tail vein injections with 1.5×1011 genome copies/mouse of Adeno-associated virus subtype 8 (AAV8) expressing hepatocyte-specific Cre recombinase (AAV-TBG-Cre, 107787-AAV8) or control (AAV-TBG-LacZ, 105534-AAV8) from Addgene, to generate Cre− control, L-NICD, and L-DNMAM mice. After transduction, mice were maintained on chow diet or started on NASH or high-fat (Research Diets, D12492) diet for 16 weeks. Animals were housed in standard cages at 22°C in a 12-hour light and 12-hour dark cycle, and monitored for overall well-being and signs of distress with body weight measured weekly. Upon completion of each studies, all mice were weighed and euthanized. Numbers and diameters of macroscopically visible tumors were recorded, and livers were weighed, excised and split for histology and molecular characterization. Specifically, tumors (diameter ≥ 5 mm) and surrounding non-tumor tissues were carefully separated for comparison including qPCR and RNA sequencing. The Columbia University Institutional Animal Care and Use Committee approved all animal procedures.
Statistical analysis.
Data are shown as mean ± SEM unless indicated otherwise. Statistical analysis was performed using Prism software (version 8, GraphPad Software). Contingency comparison was done by Fisher exact tests (2 groups) or Chi-square tests (3 or more groups). Survival analyses were done by log-rank tests. Differences between 2 groups were calculated using 2-tailed Student’s t tests. Analysis involving multiple groups was performed using one-way ANOVA, followed by Dunnett’s multiple comparison tests for two specific groups. P < 0.05 was considered statistically significant.
For further details, please refer to CTAT table and Supplementary Materials and Methods.
Results
Notch signaling is transcriptionally active in a subset of human HCCs that lack activating CTNNB1 mutations.
To define the relationship between tumor Notch activity and HCC progression, we applied a 14-gene Notch transcriptional signature score (hence, Notch score) to the TCGA HCC dataset.9 Specifically, we calculated the z-score average of Notch receptors (NOTCH1, 2, 3, 4) and ligands (JAG1, JAG2, DLL1, DLL4), core transcription coactivator (MAML1), canonical targets (HES1, HEY1, HEYL) and liver-enriched downstream effectors (SOX9, SPP1) in 373 HCCs of mixed etiologies with available transcription data (Fig. S1A), and defined three groups: Notch-low (29.5%), -mid (41%), and -high (29.5%) (Fig. 1A). Of note, Notch-high HCCs were observed at a similar frequency to the percentage (31.8%) of human hepatitis C virus-related HCCs reported as having an active Notch signature.19
Fig. 1. Notch signaling is active in a subset of HCC characterized by a lack of activating CTNNB1 mutations.
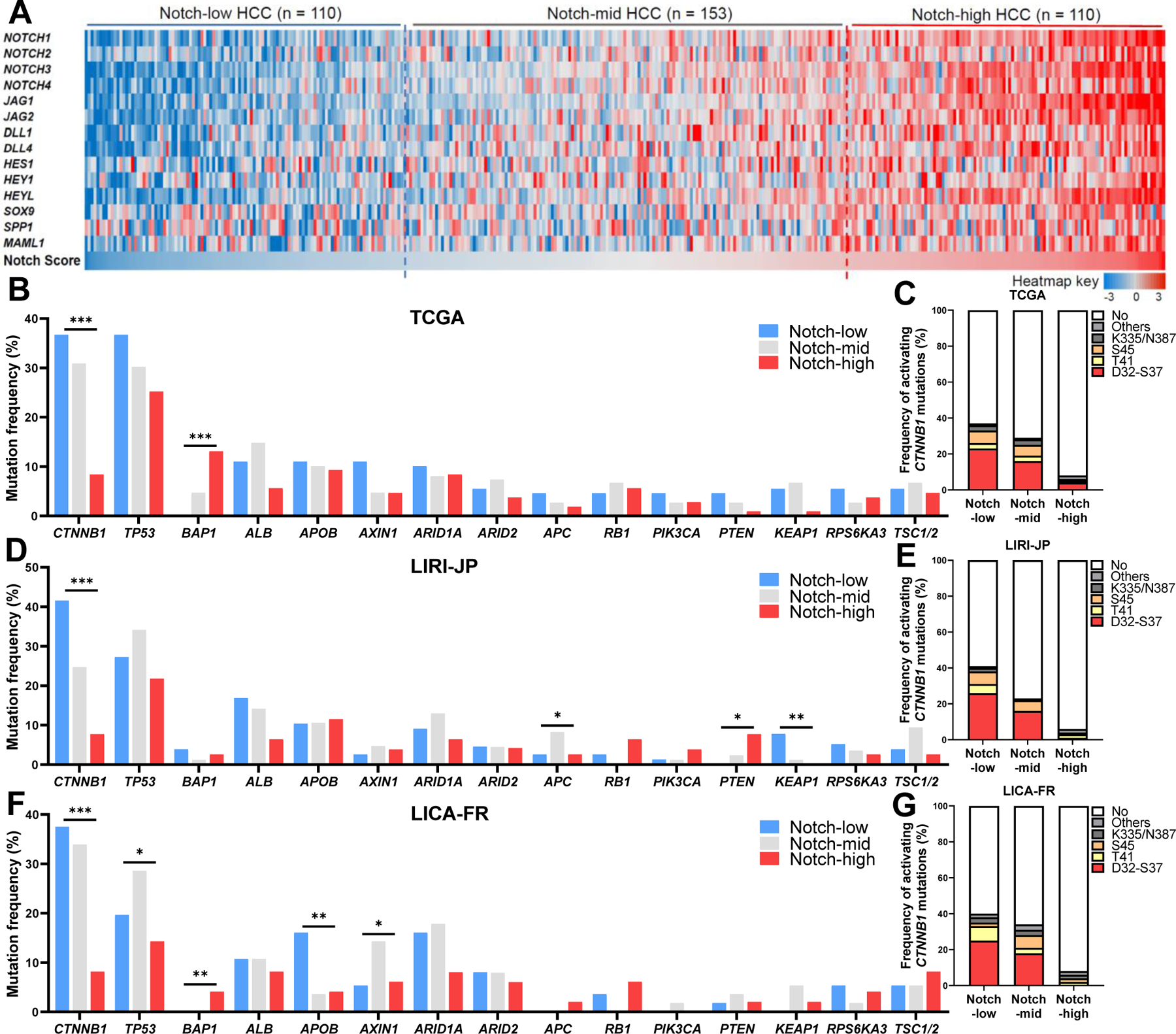
(A) Heatmap of mRNA expression z-scores of 14 Notch pathway-related genes that collectively define the Notch Score, based on which the 373 tumors in the TCGA HCC dataset were sub-divided into Notch-low, -mid and -high groups. (B to G) Frequencies of commonly found somatic mutations and recurrent activating CTNNB1 mutations in (B and C) TCGA, (D and E) LIRI-JP and (F and G) LICA-FR HCC datasets, based on Notch score. *, P < 0.05, **, P < 0.01 and ***, P < 0.001, calculated by Chi-square tests.
We next defined molecular characteristics of Notch-high HCCs. Compared to Notch-inactive groups, Notch-high HCCs are mostly (~90%) deprived of activating mutations of CTNNB16 (Fig. 1B, C), a frequently mutated oncogene in HCC that activates a unique set of downstream genes associated with periportal hepatocytes, including GLUL, AXIN2, RHBG, and NOTUM25 (Fig. S1B). To validate these data, we next applied our Notch score to an additional two cohorts with both genomic and transcriptomic data – LIRI-JP26 and LICA-FR27 from ICGC (Fig. S1C–F) – both of which showed a lower number of activating CTNNB1 mutations in Notch-high tumors (Fig. 1D–G). The inverse correlation between Notch activity and CTNNB1 mutations are reminiscent of opposing actions of Notch and Wnt/β-catenin in development28 and liver injury response,29 suggesting two distinct mechanistic routes of HCC development.
Notch genomic mutations are not causal to transcriptional Notch activity in HCC.
Although activating NOTCH1 mutations are found in over 50% of T-ALL patients,14 the role of Notch in solid tumors is highly context-dependent with both gain- and loss-of-function mutations reported,16 though none thus far in HCC.15, 19 We queried 1221 HCC samples from five different datasets on cBioPortal, and found that ~10% of tumors harbor Notch pathway somatic mutations, with the majority in Notch receptor genes (Fig. S2A). To investigate the correlation between these mutations and tumoral Notch activity, we grouped these mutations based on the Notch score in TCGA, LIRI-JP and LICA-FR datasets (Table S1–3). This analysis revealed primarily missense mutations without recurrent “hotspots” associated with known oncogenic or tumor suppressive functions, with inconsistent relationship between Notch score and mutations (Fig. S2B), suggesting likely passenger mutations in genes of relatively large size. We did find several mutations, especially in Notch receptor genes (Fig. S2B) that could lead to Notch gain or loss of function, and theoretically drive HCC. For instance, the frameshift insertion mutation E2143Lfs*5 in the last exon of NOTCH2 results in loss of the C-terminal proline-glutamic acid-serine-threonine-rich (PEST) domain responsible for ubiquitination and degradation of NICD, resulting in persistent NOTCH2-ICD activation.30 Other truncations (G1453Vfs*100 and A1917Efs*13) likely result in dominant-negative proteins that bind to ligands without transcriptional capabilities.31 But these data overall suggest that somatic mutations in the Notch pathway that may drive HCC are rare, whereas the 14-gene Notch score revealed far greater frequency of Notch activity in tumors.
Notch-active HCCs are cholangiocarcinoma-like and enriched with intratumoral fibrosis.
Next, we compared transcriptomics from Notch-high and -low HCCs. Gene set enrichment analysis (GSEA) further confirmed that Notch-high HCCs display a previously identified Notch-induced liver cancer signature19 (Fig. S3A), while Notch-low HCCs fit the CTNNB1 activation subclass32 (Fig. S3B). Canonical β-catenin target genes associated with the presence of activating CTNNB1 mutations, including GLUL, AXIN2, NOTUM, are downregulated in Notch-high HCCs; whereas genes related to extracellular matrix (ECM) and myofibroblast markers (PDGFRA/B and VIM) are highly induced in these tumors (Fig. 2A). Pathway analysis confirmed that the top pathways upregulated in Notch-high HCCs are related to ECM deposition, while downregulated pathways are primarily metabolic, suggesting that Notch-high HCCs are less differentiated and more fibrotic (Fig. 2B and Fig. S3C). Consistently, Notch-high tumors show marked intratumoral fibrosis (Fig. 2C), and enrichment of stroma transcription signatures33, 34 (Fig. S3D).
Fig. 2. Notch-high HCCs show unique molecular and histologic features, and lower survival.
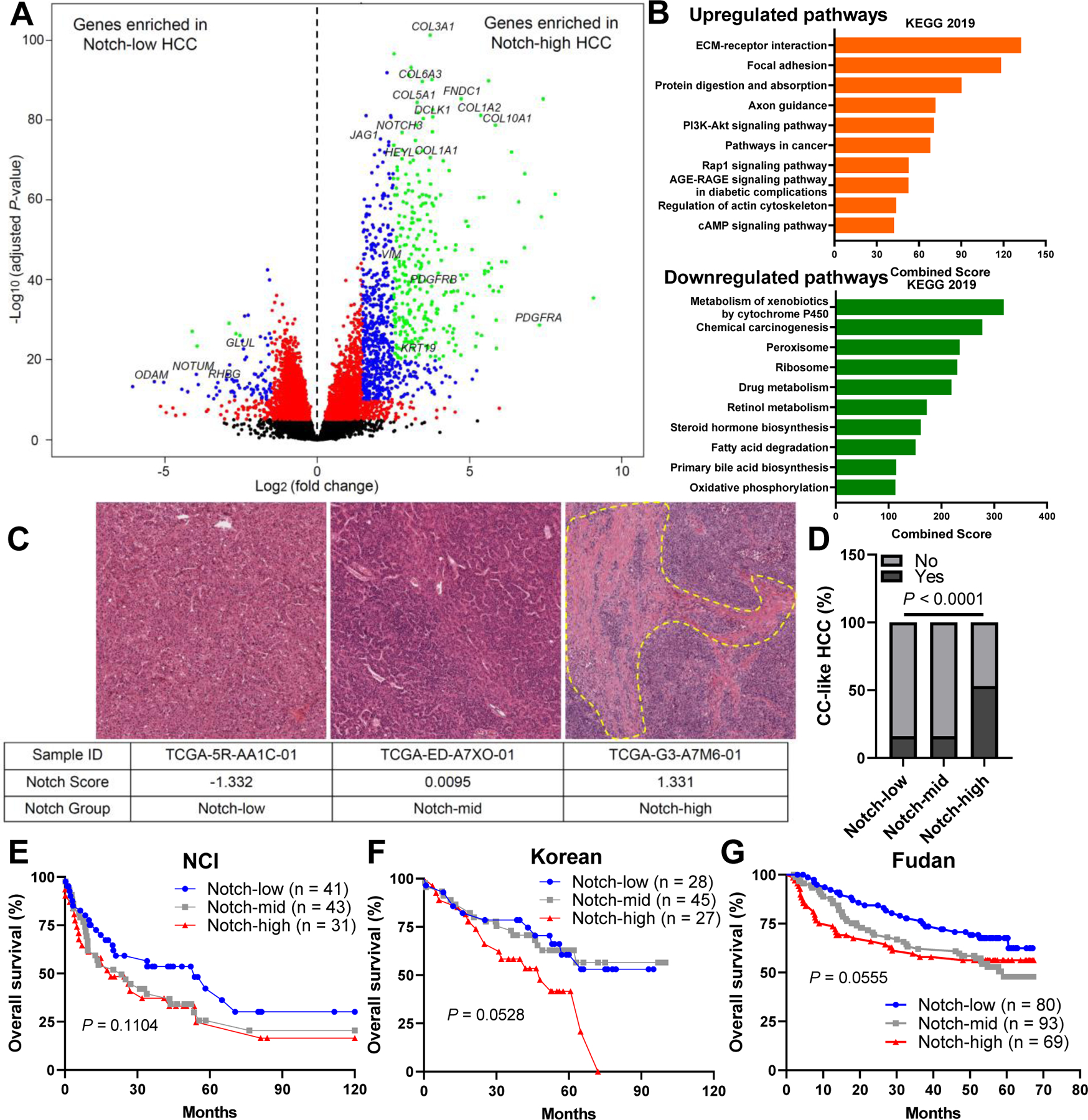
(A) Volcano plot showing differentially expressed genes (DEGs, adjusted P-value < 0.05) in Notch-low and -high HCCs (n = 110 per group) from the TCGA dataset. (B) Upregulated and downregulated pathways from KEGG 2019 in Notch-high HCCs, ranked by combined enrichment scores. (C) Representative H&E images of Notch-low, -mid and -high tumors. (D) Percentage of cholangiocarcinoma (CC)-like HCC in Notch-low, -mid and -high tumors in the TCGA dataset. (E) Kaplan-Meier survival curves of patients with Notch-low, -mid and -high tumors in NCI, (D) Korean, and (G) Fudan cohorts. P-values calculated by log-rank tests.
Notch-high HCCs also showed increased expression of cholangiocyte/progenitor markers (KRT19 and DCLK1) (Fig. 2A), suggestive of cholangiocarcinoma (CC)-type features. CCs also show a highly desmoplastic stroma,3 and these data are reminiscent of the role of Notch in promoting biliary lineage during liver development and mouse data suggesting potential of Notch to transform mouse hepatocytes into intrahepatic CC (ICC).35 Thus, we applied a CC-like HCC transcriptional signature, which marks a HCC subtype with similarly poor outcomes as combined HCC-CC and CC despite lacking the overall histopathological features of CC36, and found that over 50% of Notch-high tumors fit this signature in TCGA HCC datasets (Fig. 2D), while GSEA using another signature of Cytokeratin 19 (KRT19)-positive HCCs37 demonstrate similar results (Fig. S3E). Consistently, expression of Notch pathway genes, β-catenin targets, and hepatocyte markers of Notch-high HCC fall between Notch-low HCC and ICC (Fig. S4A–C), with some notable differences in expression of HCC markers (i.e. AFP and GPC3) which are far higher in Notch-high HCC compared to ICC (Fig. S4D). Consistent with the RNA seq results, Notch-high HCCs have higher expression of genes related to cholangiocyte/progenitor cell identity and ECM/fibrosis than Notch-low HCC (Fig. S4E–F), with a similar pattern observed in LICA-FR and LIRI-JP cohorts (Fig. S5A–F). These data suggest that Notch activity marks HCCs with CC features.
Notch activity is associated with HCC subtypes of higher cancer stage and poor prognosis.
We next compared the transcriptomic signature of Notch-high HCCs to previously defined molecular subtypes. Upregulated genes from Notch-high HCCs are highly enriched in Subclass 1 (S1), a subtype associated with TGF-β signaling, less differentiated tumors and poor survival; conversely, downregulated genes are enriched in S3, a subtype of well-differentiated tumors defined by activating CTNNB1 mutations38 (Fig. S6A). Notch-high HCCs similarly overlaps with the transcriptional signature of Subgroup 1 (G1), a subtype enriched in genes of developmental signaling and fetal liver39 (Fig. S6B). Notch-high HCCs also share molecular homology with a Proliferation subclass characterized by elevated serum a-fetoprotein (AFP) and chromosomal instability32 (Fig. S6C).
These data suggested that Notch-high HCCs would be associated with worse outcome. To test this, we applied TCGA iCluster to categorize each tumor. Consistent with our other analyses, Notch-low HCCs primarily fell in iClusters 2 and 3 characterized by CTNNB1 mutations, while > 65% of Notch-high tumors clustered with iCluster 1 (Fig. S6D), which have the worst survival outcomes.9 We performed analogous comparisons of TCGA samples to three different classification signatures, and found that Notch-high HCCs are predicted to show progenitor features40 and poorer survival41 with greater risks of metastasis42 (Fig. S6E–G). Next, we compared the clinical data based on Notch activity. Although subtype-matching analyses suggest that patients with Notch-high HCCs would have worse prognosis, and Notch-high group has a higher percentage of advanced stage tumors, with a trend towards higher levels of serum AFP, overall and recurrence-free survival curves were not different (Fig. S6H–K), which may be due to the relatively short follow-up times of TCGA cohort.9 We evaluated another three long-term follow-up HCC cohorts with clinical characterization and microarray-based expression data,40–42 where we observed that higher Notch activity in the tumors (Fig. S7A–C) is associated with worse survival (Fig. 2E–G) and more frequent relapse (Fig. S7D–F), and Notch-high group in general displayed higher tumor stages, larger tumor sizes, and higher AFP levels (Fig. S7G–N).
Overall, these data from human HCC datasets demonstrate that Notch-high HCCs adopt a CC-like histology with stromal fibrosis, and are associated with advanced tumor stages and generally poorer outcomes. On the contrary, Notch-inactive HCCs are well-differentiated neoplasms, and typically associated with CTNNB1 mutations that portend better prognosis.25
Notch signaling in hepatocytes promotes their loss of identity but enhances stromal activity.
We next asked whether hepatocyte Notch activation, known to activate hepatic stellate cells (HSC) to trigger liver fibrosis in NASH,13 can directly contribute to NASH-induced HCC development. First, using a Notch-Venus reporter mouse,22 we found that liver Notch signaling is generally confined to nonparenchymal cells (Fig. S8A), but activated in ~15% hepatocytes13 in diet-induced NASH (Fig. 3A). To understand the impact of this, we isolated Notch-active and -inactive hepatocyte populations from NASH diet-fed reporter mice (Fig. 3B) and performed RNA sequencing. Notch-active hepatocytes showed increased expression of pathways that mediate interactions with ECM, consistent with non-cell autonomous activation of stromal cells; conversely, metabolic pathways and β-catenin signaling were repressed, consistent with previous finding43 that Notch reprograms hepatocytes to progenitor/cholangiocyte-like cells (Fig. 3C, D and Fig. S8B). Intriguingly, the transcriptional signatures of Notch-active hepatocytes strongly overlapped with human Notch-high HCCs (Fig. 3E and Fig. S8C) and mouse data from Notch overexpression studies (Fig. S8D), suggesting that Notch-induced change of hepatocyte identity and remodeling of ECM may represent a mechanism that persists during the tumorigenic progression from NASH to HCC.
Fig. 3. Notch activation is associated with reduced hepatocyte metabolic function, but a signature of intercellular crosstalk with stromal cells.
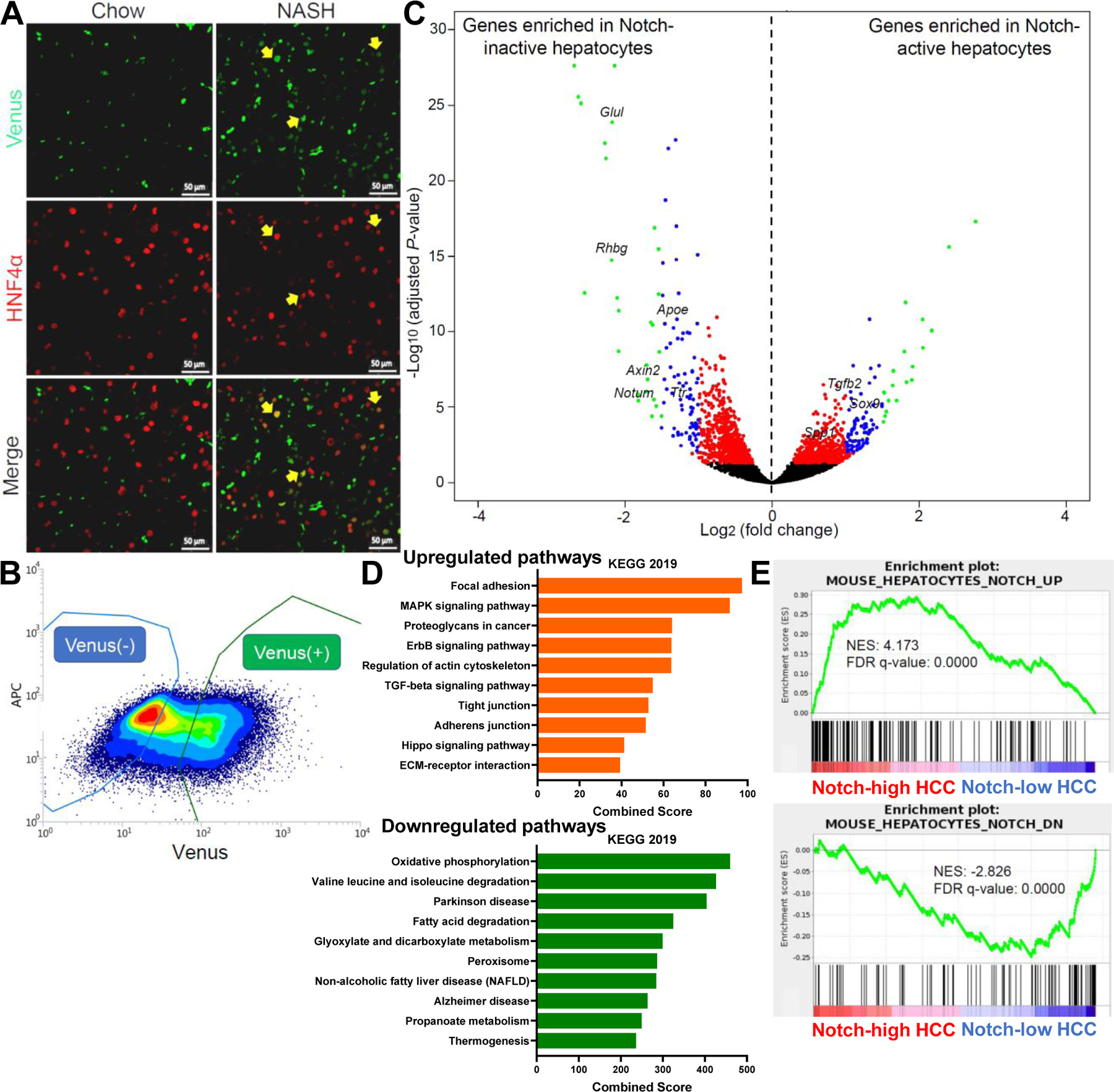
(A) Representative images of Venus (green) and HNF4α (red) co-staining in chow- and NASH diet-fed Notch-Venus reporter mice. (B) Flow cytometry plot and (C) Volcano plot showing DEGs in Notch-active and -inactive hepatocytes (n = 3 per group) isolated from NASH diet-fed Notch-Venus reporter mice. (D) Upregulated and downregulated pathways from KEGG 2019 in Notch-active hepatocytes, ranked by combined enrichment scores. (E) Gene set enrichment analyses (GSEA) of Notch-active hepatocyte signatures in human Notch-high or low HCCs from TCGA. UP, upregulated. DN, downregulated. NES, normalized enrichment scores. FDR, false discovery rate.
Notch signaling is active in a subset of tumors in a mouse model of HCC induced by carcinogen and NASH diet.
To test whether Notch is active in mouse HCC, we combined the carcinogen diethylnitrosamine (DEN) with NASH diet-feeding (henceforth, DEN-NASH) in wildtype (WT) mice, similar to prior studies,44 which led to progressive development of multifocal liver tumors (Fig. S9A, B). These tumors were histologically HCC, with varying morphologies including steatotic and steatohepatitic features which are more commonly described in patients with NASH45 (Fig. S9C). We applied this experimental paradigm to Notch-Venus reporter mice (Fig. 4A), which prompted robust tumorigenesis (Fig. 4B) with elevated HCC markers (Fig. 4C). By co-staining with A6 antiserum that marks some mouse HCC,46 we observe both Notch-inactive and -active HCCs (Fig. 4D). Given our data showing Notch and β-catenin mutual exclusivity observed in human HCC, we predicted this would hold true in this mouse model. We found that over half of the tumors are Notch-active, while immunoreactivity of tumors with β-catenin target glutamine synthetase (GS, encoded by Glul)46 was low in DEN-NASH HCC (~10%), and Notch/Venus and GS signals are mutually exclusive (Fig. 4E), even in non-tumor areas (Fig. S10A). We confirmed this result with Osteopontin (Opn, encoded by Spp1) staining, a sensitive readout of Notch activity in hepatocytes13 (Fig. S10B–D). These data suggest that similar to human HCC, Notch- and β-catenin-active HCCs are fundamentally different in mice.
Fig. 4. Notch and β-catenin activity mark distinct carcinogen-induced HCC subsets.
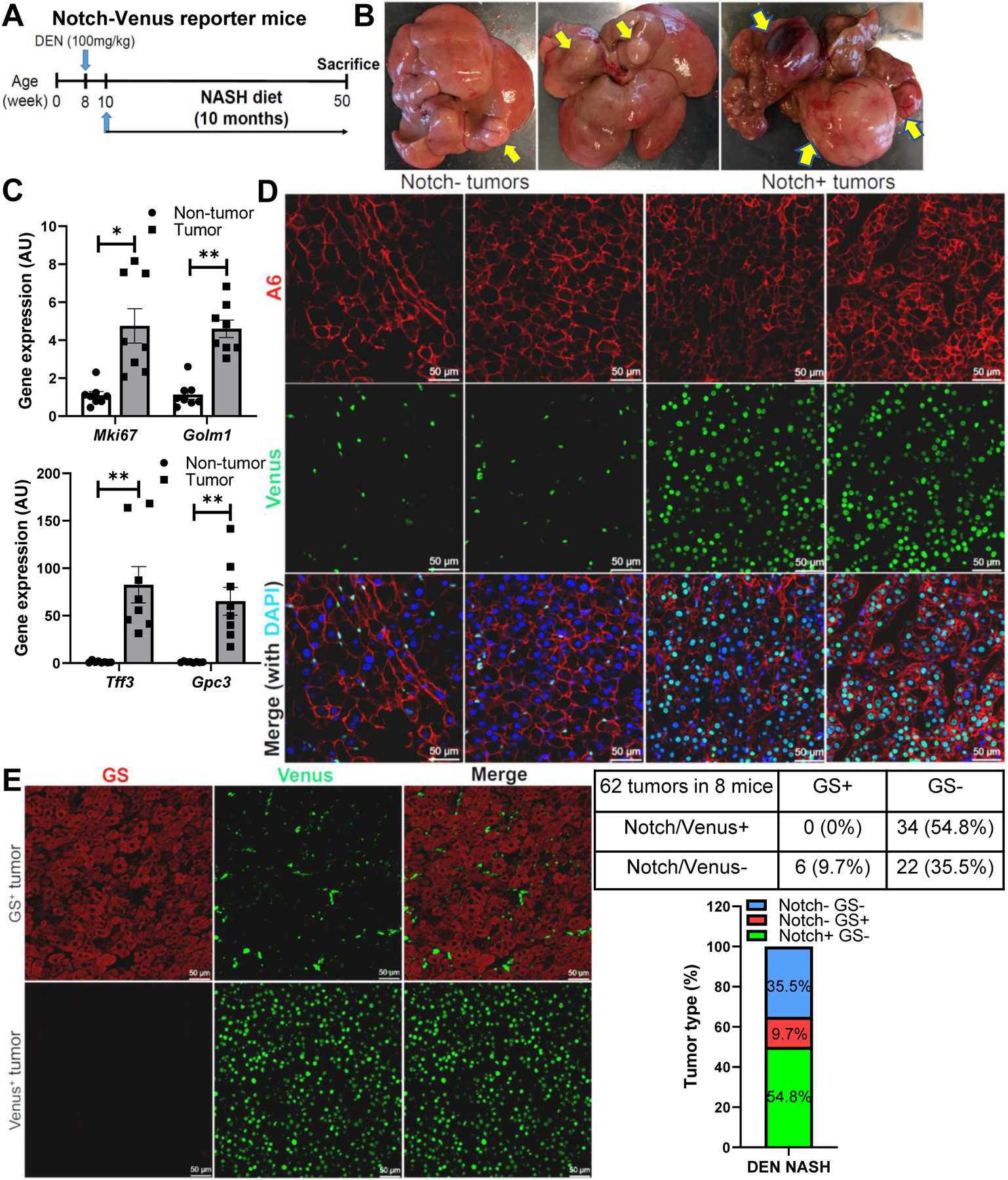
(A) Notch-Venus reporter mice were dosed with diethylnitrosamine (DEN, 100 mg/kg), then fed NASH diet for 10 months prior to sacrifice. (B) Representative images of liver tumors, and (C) Non-tumor and Tumor expression of HCC marker genes in Notch-Venus mice (n = 8 per group). (D) Representative images of A6 (red), Venus (green), and DAPI (blue) co-staining in Notch-negative and –positive tumors from Notch-Venus mice. (E) Representative images of β-catenin target glutamine synthetase (GS, red) and Venus (green) co-staining in GS+ and Venus+ tumors from Notch-Venus mice, with quantification of GS+ and/or Notch/Venus+ tumors. *, P < 0.05 and **, P < 0.01 as compared to the non-tumor group by two-tailed t tests. AU, arbitrary unit. Data are shown as means ± SEM.
Notch activity is sufficient to drive tumorigenesis in the context of NASH.
To determine if hepatocyte Notch activation is sufficient to drive HCC and model Notch-high HCCs in humans, we transduced young adult male RosaNICD mice23 with AAV8-Tbg-Cre (henceforth, L-NICD mice) to overexpress NICD specifically in hepatocytes,47 followed by 4-month exposure to the NASH diet (Fig. 5A). NASH diet-fed L-NICD mice developed fully penetrant, multifocal large liver tumors in both male (Fig. 5B–D) and female mice (Fig. S11A–C). In contrast, L-NICD mice fed regular chow or a similarly obesogenic high-fat diet did not develop HCC within the same timeframe (Fig. S11D). Histologically, most of the tumors were well-differentiated and steatotic HCCs (Figure 5E), with loss of reticulin fibers (Fig. 5F). Tumors stained positive for the hepatocyte marker HNF4α; consistently, taking advantage of Cre-inducible nuclear eGFP gene in the same locus as NICD for lineage tracing,23 we confirmed that tumor cells derived from hepatocytes (Fig. S11E). L-NICD tumors also stained positive for the liver progenitor antigen A6 (Fig. 5G) and proliferation marker Ki67 (Fig. 5H), but negative for AFP (Fig. S11F), consistent with well-differentiated and proliferative HCCs with some immature features. Finally, consistent with the mutual exclusivity between Notch and β-catenin signaling, Glul expression was downregulated in L-NICD livers and further decreased in the tumors (Fig. S11G–H), leading to the complete lack of GS staining in the tumors (Fig. 5I). Overall, these findings indicate that constitutive Notch activation in hepatocytes leads to Notch-active, β-catenin-deficient HCCs, but only in the fibrotic niche evoked by NASH diet-feeding, consistent with human HCC development in the context of chronic liver diseases.
Fig. 5. Hepatocyte Notch activation induces HCC in NASH diet-fed mice.
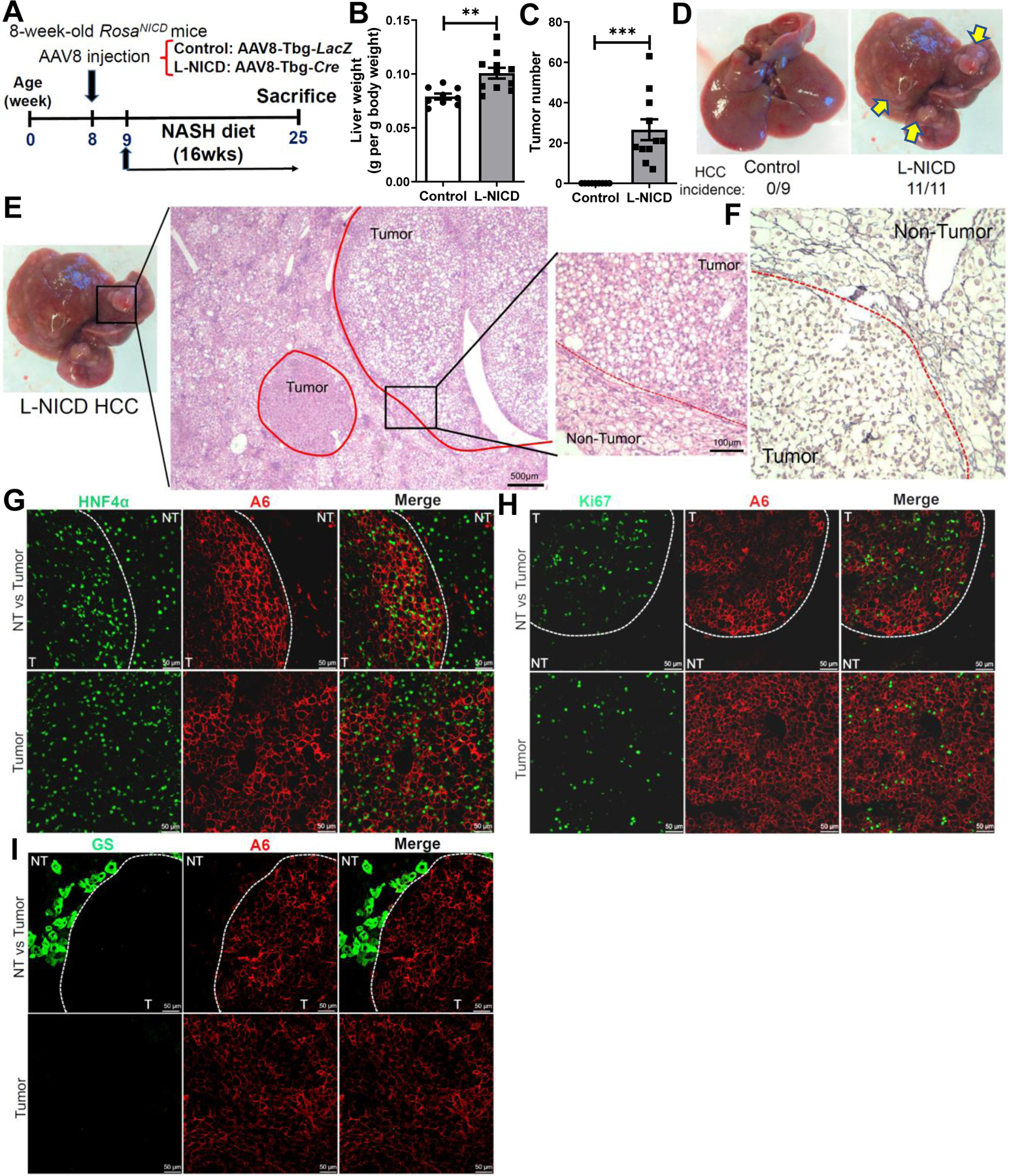
(A) Male RosaNICD mice were transduced with AAV8-Tbg-LacZ (control) or AAV8-Tbg-Cre (L-NICD) and then fed with NASH diet for 16 weeks. (B) Liver-to-body weight ratios and (C) tumor number in control and L-NICD mice (n = 9–11 per group). (D) Representative images of livers from control and L-NICD mice, with (E) H&E and (F) reticulin staining in a representative tumor from L-NICD mice. (G) Representative images of HCC antigen A6 (red) co-staining with HNF4α (green), (H) Ki67 (green), and (I) GS (green) in tumors from L-NICD mice. **, P < 0.01 and ***, P < 0.001 as compared to the control group by two-tailed Student’s t tests. Data are shown as means ± SEM.
NASH diet-fed L-NICD mice recapitulates major aspects of human Notch-active HCC.
To determine if HCCs in NASH diet-fed L-NICD mice model Notch-high human HCC, we performed RNA sequencing of the mouse tumors. Compared to surrounding non-tumor (NT) liver, HCCs from L-NICD mice showed upregulation of pathways related to cell cycle and proliferation, with increased expression of the minichromosome maintenance protein complex (MCM) family of genes that are critical for DNA replication and dysregulated in cancers,48 and downregulation of liver metabolic functions (Fig. 6A, B and Fig. S12A).
Fig. 6. NASH-diet fed L-NICD mice transcriptionally recapitulates human Notch-active HCC.
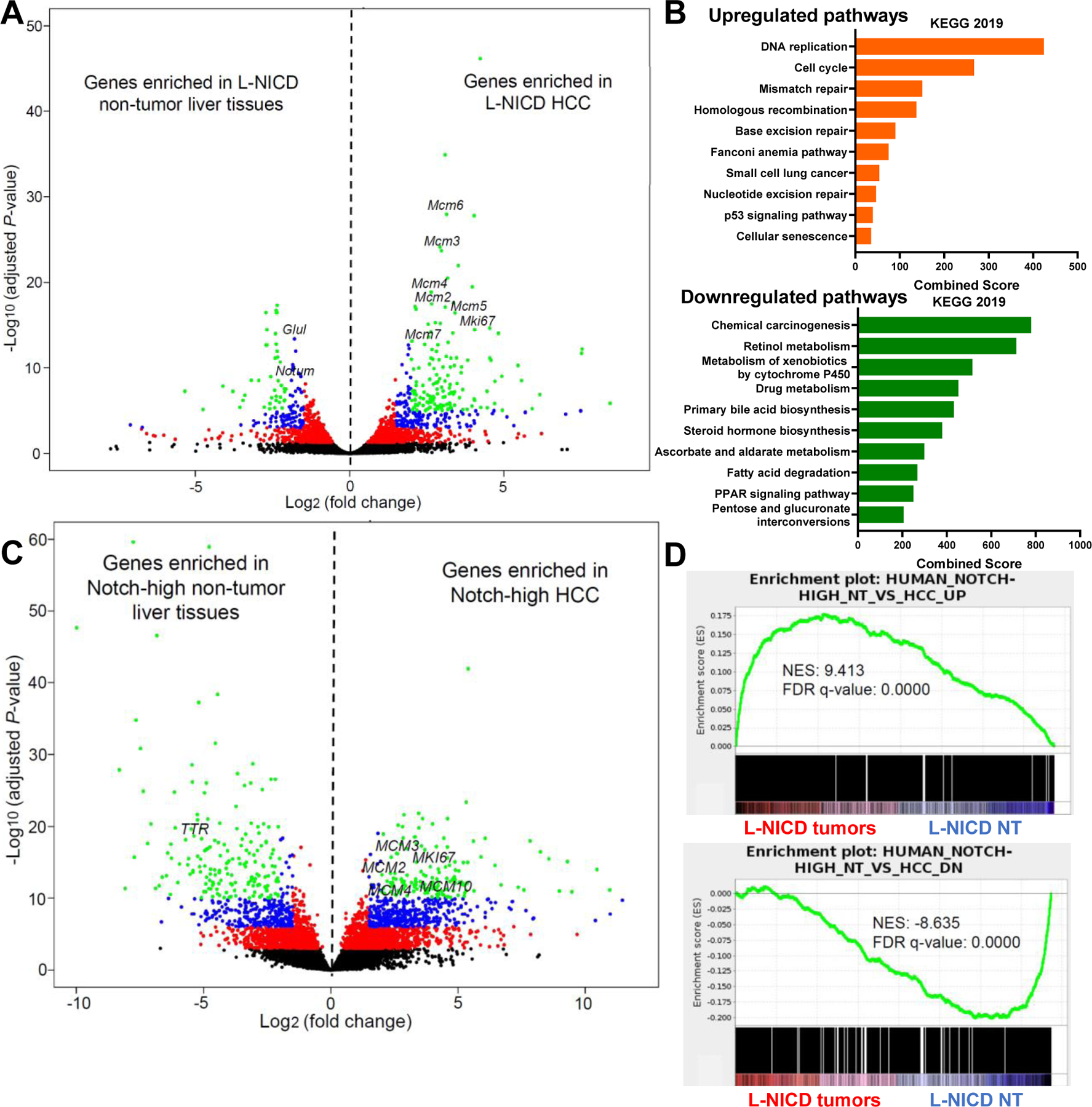
(A) Volcano plot showing DEGs in tumors as compared to non-tumor surrounding tissues (n = 3 per group) in NASH diet-fed L-NICD mice. (B) Upregulated and downregulated pathways from KEGG 2019 in L-NICD HCCs, ranked by combined enrichment scores. (C) Volcano plot showing DEGs and (D) GSEA in human Notch-high HCCs as compared to adjacent non-tumor liver tissues (n = 10 per group) from the TCGA dataset.
In order to select appropriate human Notch-high HCCs from TCGA for comparison, we first calculated the Notch scores in the surrounding normal liver tissues, and selected Notch-high NT samples (Fig. S12B, C) to parallel increased Notch activity in NT and tumor areas in L-NICD mice. Differential gene expression analysis of human Notch-high HCCs normalized to NT (Fig. 6C) showed enrichment of altered pathways similar to L-NICD HCCs (Fig. 12D, E). GSEA revealed that L-NICD tumors transcriptionally resemble the signatures from human Notch-high HCCs (Fig. 6D), as well as hepatocytes with endogenous Notch activity isolated from Notch reporter mice fed on NASH diet (Fig. S12F), ruling out the possible supraphysiologic effects due to NICD overexpression in this model.
GSEA of established molecular signatures of HCC subtypes found that mouse L-NICD HCCs and human Notch-high HCCs share the signatures of both KRT19-expressing HCC37 (Fig. S13A) and Proliferation subclass32 (Fig. S13B), especially the subgroups (G1-G3 groups) with chromosomal instability and overexpression of cell cycle-related genes39 (Fig. S13C). However, unlike human Notch-high HCCs, L-NICD HCCs resemble S2, a subclass of HCC related to high rate of proliferation, rather than S138 (Fig. S13D), possibly due to lower expression of stromal ECM-related genes (Fig. S13E). Indeed, in comparison to human Notch-high HCCs, tumors from NASH diet-fed L-NICD mice did not show increased intratumoral fibrosis or numbers of hepatic stellate cell-derived myofibroblasts (Fig. S14A–D) as compared to surrounding non-tumor tissues. Finally, we probed the mutational landscape of 9 HCC samples from NASH diet-fed L-NICD mice using the MSK mouse Integrated Mutation Profiling of Actionable Cancer Targets (M-IMPACT) assay.49 M-IMPACT sequencing found no activating mutations of Ctnnb1, but unique somatic mutations in 27 genes (Fig. S14E and Table S4), including recurrent mutations in epigenetic regulators Kmt2a and Kmt2d,50 as well as Amer1 which is a negative regulator of β-catenin stability.51
Chronic inhibition of Notch results in switch of molecular subtypes in mouse HCC.
These data establish a potential mouse model for Notch-active HCC, but also that hepatocyte Notch activation is sufficient to promote tumorigenesis in the context of NASH. To investigate if Notch signaling is necessary for HCC development, we transduced DEN-treated, NASH diet-fed RosaDNMAM mice carrying a dominant-negative Mastermind (DNMAM) allele24 with AAV8-Tbg-Cre to block all Notch receptors-mediated transcription in hepatocytes (L-DNMAM mice) (Fig. 7A). At the end of the experiment, we found no difference in tumor number or size between the two groups (Fig. S15A–C), despite a reduction in liver fibrosis (Fig. S15D, E), maintained expression of the Dnmam transgene (Fig. S15F) and lowered Notch targets (Fig. 7B and Fig. S15G), including lower tumor Opn staining (Fig. 7C). These data suggest that hepatocyte Notch signaling is not required for tumorigenesis in the DEN NASH model, even though half of the tumors in this model are normally Notch-active. We hypothesized that a shift in tumor origin would explain this seeming contradiction. Indeed, we observed that β-catenin target genes Glul and Axin2 are upregulated in the L-DNMAM tumors (Fig. 7D and Fig. S15H). Consistently, the majority of the tumors in control animals are Opn+, but GS reactivity is seen in more than half of the tumors in L-DNMAM mice (Fig. 7E, F). Additionally, there is a small increase in the number of tumors negative for both Opn and GS. In sum, these data suggest that chronic Notch inhibition does not prevent carcinogen-induced tumor development, but rather alters HCC molecular subtype likely by de-repression of β-catenin signaling.
Fig. 7. Chronic Notch inhibition in carcinogen-treated mice leads to β-catenin-dependent HCC.
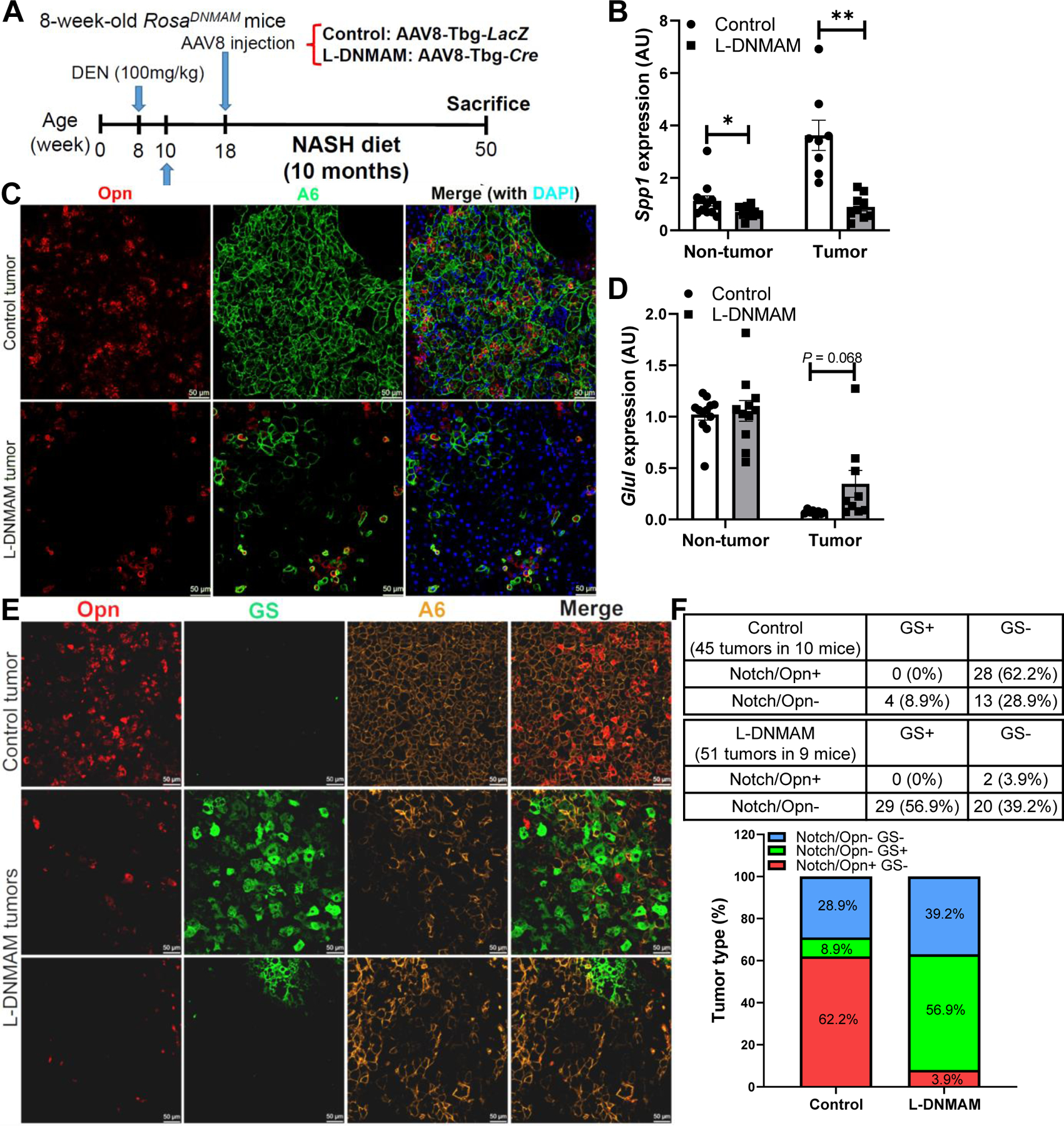
(A) RosaDNMAM mice were dosed with DEN (100 mg/kg), fed NASH diet, then transduced with AAV8-Tbg-LacZ (control) or AAV8-Tbg-Cre (L-DNMAM) (n = 9–10 per group). (B) L-DNMAM tumors show reduced expression of the Notch target Spp1, (C) with correspondingly lower tumor co-staining of Opn (red). (D) L-DNMAM tumors show increased tumoral expression of the β-catenin target Glul, (E) with correspondingly increased tumor co-staining with GS (green). (F) Quantification of Notch/Opn+ and GS+ tumors. *, P < 0.05 and **, P < 0.01 as compared to the indicated control by two-tailed t tests. AU, arbitrary unit. Data are shown as means ± SEM.
Discussion
HCC is a heterogenous cancer at molecular and histologic levels. This heterogeneity challenges broad-strokes treatment; hence, approved therapeutics are only effective in limited patient subsets.52 High-throughput profiling and phenotype-to-genotype efforts have identified HCCs with distinct mutational and transcriptomic signatures that are correlated with clinical outcomes.5 Deciphering molecular mechanisms underlying HCC heterogeneity is needed to understand tumor behavior, especially in the absence of CTNNB1 or other genomic driver mutations.
Our data reveal that Notch activation, not mutations, characterizes a subset of β-catenin-inactive HCCs, and suggest these two pathways represent different routes for tumor evolution. This dichotomy may be context-dependent. In NASH, Notch represents a maladaptive regenerative response to chronic hepatocellular injury; unopposed Notch signaling in NASH induces HCC without the need to acquire specific driver mutations. In contrast, activating CTNNB1 mutations have been demonstrated in benign dysplastic nodules and hepatocellular adenomas,53 which show variable progression to HCC. The difference between Notch- and β-catenin-active cancers may also lie in the cellular origin of tumor cells. For example, pericentral hepatocytes show intrinsically high Wnt/β-catenin activity.54 It is possible that these cells are polarized towards Notch-inactive CTNNB1-mutated tumors in the setting of a second hit, although this has not been explicitly studied.
Along these lines, several potential mechanisms underlie Notch-β-catenin mutual exclusivity in HCC. β-Catenin activity promotes nuclear localization of Dimerization partner 155 and activates expression of Numb,29 the sum total of these processes is to promote Notch degradation. Intriguingly, Notch reciprocally promotes β-catenin degradation,56 but further study is required to test whether these mechanisms are causal to the Notch vs. β-catenin dichotomy in mouse and human HCC. Interestingly, Notch and β-catenin cooperativity has been reported in other types of cancer,57, 58 suggesting highly context-dependent functions of the two pathways.
Notch controls a key HCC niche factor, liver fibrosis. Interestingly, Notch gain-of-function mice show similar fibrosis with both standard chow or NASH diet-feeding,13 but we find HCC development only with NASH diet-feeding. These data suggest that the obesogenic environment in NASH likely affects other aspects of the tissue environment and/or represents a niche-independent HCC risk factor. Somewhat surprisingly, mice with chronic Notch loss-of-function in hepatocytes showed less liver collagen accumulation, but similar tumor numbers as control mice. These data point to the possibility that β-catenin-active tumors may be less reliant on a fibrotic niche, while Notch-active tumors thrive in an ECM-rich environment. However, our study cannot answer whether existing or novel Notch inhibitors in development59 may be effective for treatment of Notch-active HCCs; this remains an area of active research in our lab.
Finally, the necessity of this gene-environment interaction supports use of L-NICD mice to model Notch-active HCC that arises from NASH and potentially other chronic liver diseases, given similar molecular phenotypes as human Notch-high HCCs and an experimentally practical and pathophysiologically appropriate timeline. In contrast, prior work with AFP- or Albumin-Cre–driven epithelial Notch activation19, 20 may interfere with liver development, and transposase-mediated tail vein delivery of oncogenic plasmids rapidly induces liver tumors60 which is difficult to reconcile with timespan of human HCC development in the setting of chronic liver diseases. There are some interesting differences between L-NICD mice and human Notch-high HCCs, in particular the desmoplastic nature of Notch-active human tumors. Additionally, although human Notch-high tumors display less differentiated CC-like features, HCCs from NASH diet-fed L-NICD mice are histologically steatotic and well-differentiated, consistent with the observations that HCCs in NASH patients are usually well-differentiated.61 Future work is needed to characterize Notch-active NASH-driven human HCCs, and determine how Notch and NASH interact to influence tumorigenesis and stromal microenvironment.
In summary, we found that Notch activity predicts CC-like HCC with enriched intratumoral fibrosis and worse clinical outcomes in patients, and is mutually exclusive with activating CTNNB1 mutations. In mice, Notch activation promotes loss of hepatocyte identity and simultaneously enhances stromal cell activity, and in the inflammatory and fibrotic tissue environment of NASH, is sufficient to induce HCC. However, Notch activity is not required per se for carcinogen-induced HCC, likely due to de-repression of β-catenin signaling normally suppressed by Notch. These data demonstrate the context-dependent role of Notch signaling in HCC development, and puts forth a mouse HCC model for preclinical testing of therapeutic combinations for precision medicine of Notch-active HCC.
Supplementary Material
Highlights.
Notch activation induces liver fibrosis, an important niche factor for tumorigenesis
Notch is active in 30% of human hepatocellular carcinomas (HCCs) with poor prognosis
Notch-active HCCs are fibrotic, and have low β-catenin activity in mice and humans.
Forced hepatocyte Notch activity in mice induces diet-dependent HCC.
Acknowledgments:
We thank A. Flete, T. Kolar, and J. Weber for excellent technical support, W. Wang, L. Lu, S. Shah and S. Ho for their assistance with cell sorting, and members of the Pajvani, Lowe, and Schwabe laboratories for insightful discussion. We also gratefully acknowledge H. Grajal and J. Kitajewski (UIC) for sharing mouse strains. The graphical abstract was created with BioRender.com.
Financial support: This work was supported by NIH DK103818 and NIH DK105303 (U.B.P.), NIH CA087497 (S.W.L.), and an AHA Predoctoral Fellowship 17PRE33120000 (C.Z.). S.W.L. is a Howard Hughes Medical Institute Investigator. Cell sorting experiments were supported by NIH S10OD020056 (the CCTI Flow Cytometry Core) and NIH 5P30DK063608 (the Diabetes and Endocrinology Research Center Flow Core Facility).
Abbreviations:
- AAV
adeno-associated virus
- AFP
α-fetoprotein
- CC
cholangiocarcinoma
- DEN
diethylnitrosamine
- DNMAM
dominant-negative Mastermind
- ECM
extracellular matrix
- GSEA
gene set enrichment analysis
- HCC
hepatocellular carcinoma
- HSC
hepatic stellate cell
- ICC
intrahepatic cholangiocarcinoma
- ICGC
International Cancer Genome Consortium
- M-IMPACT
mouse Integrated Mutation Profiling of Actionable Cancer Targets
- NASH
nonalcoholic steatohepatitis
- NICD
Notch intracellular domain
- NT
non-tumor tissue
- TCGA
The Cancer Genome Atlas
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: The authors declare that they have no competing interest in the work described.
Data availability statement: All data supporting the findings of this study are available from the first and corresponding authors upon request.
Data and materials availability. RNA sequencing data (Venus mouse hepatocytes and L-NICD mouse HCC) that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GSE154553. All other data supporting the findings of this study are available from the first and corresponding authors upon request.
References
- [1].Villanueva A Hepatocellular Carcinoma. The New England journal of medicine 2019;380:1450–1462. [DOI] [PubMed] [Google Scholar]
- [2].Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J Natl Cancer Inst 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Affo S, Yu LX, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annual review of pathology 2017;12:153–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, Duseja A, et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin Gastroenterol Hepatol 2019;17:748–755.e743. [DOI] [PubMed] [Google Scholar]
- [5].Calderaro J, Ziol M, Paradis V, Zucman-Rossi J. Molecular and histological correlations in liver cancer. J Hepatol 2019;71:616–630. [DOI] [PubMed] [Google Scholar]
- [6].Rebouissou S, Franconi A, Calderaro J, Letouzé E, Imbeaud S, Pilati C, et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 2016;64:2047–2061. [DOI] [PubMed] [Google Scholar]
- [7].Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991;350:427–428. [DOI] [PubMed] [Google Scholar]
- [8].Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 2013;4:2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341.e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour evolution in hepatocellular carcinoma. Nature reviews Gastroenterology & hepatology 2020;17:139–152. [DOI] [PubMed] [Google Scholar]
- [11].Bolos V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocrine reviews 2007;28:339–363. [DOI] [PubMed] [Google Scholar]
- [12].Zong Y, Stanger BZ. Molecular mechanisms of liver and bile duct development. Wiley Interdisciplinary Reviews: Developmental Biology 2012;1:643–655. [DOI] [PubMed] [Google Scholar]
- [13].Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Science translational medicine 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004;306:269–271. [DOI] [PubMed] [Google Scholar]
- [15].Luiken S, Fraas A, Bieg M, Sugiyanto R, Goeppert B, Singer S, et al. NOTCH target gene HES5 mediates oncogenic and tumor suppressive functions in hepatocarcinogenesis. Oncogene 2020;39:3128–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Aster JC, Pear WS, Blacklow SC. The Varied Roles of Notch in Cancer. Annual review of pathology 2017;12:245–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S, et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012;143:1660–1669.e1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dill MT, Tornillo L, Fritzius T, Terracciano L, Semela D, Bettler B, et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 2013;57:1607–1619. [DOI] [PubMed] [Google Scholar]
- [21].Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med 2011;208:1963–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nowotschin S, Xenopoulos P, Schrode N, Hadjantonakis AK. A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Dev Biol 2013;13:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A 2003;100:14920–14925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tu L, Fang TC, Artis D, Shestova O, Pross SE, Maillard I, et al. Notch signaling is an important regulator of type 2 immunity. J Exp Med 2005;202:1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Desert R, Rohart F, Canal F, Sicard M, Desille M, Renaud S, et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 2017;66:1502–1518. [DOI] [PubMed] [Google Scholar]
- [26].Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet 2016;48:500–509. [DOI] [PubMed] [Google Scholar]
- [27].Bayard Q, Meunier L, Peneau C, Renault V, Shinde J, Nault JC, et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat Commun 2018;9:5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hayward P, Kalmar T, Arias AM. Wnt/Notch signalling and information processing during development. Development 2008;135:411–424. [DOI] [PubMed] [Google Scholar]
- [29].Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med 2012;18:572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Isidor B, Lindenbaum P, Pichon O, Bezieau S, Dina C, Jacquemont S, et al. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat Genet 2011;43:306–308. [DOI] [PubMed] [Google Scholar]
- [31].Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A 2011;108:17761–17766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008;68:6779–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zender S, Nickeleit I, Wuestefeld T, Sorensen I, Dauch D, Bozko P, et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013;23:784–795. [DOI] [PubMed] [Google Scholar]
- [36].Woo HG, Lee JH, Yoon JH, Kim CY, Lee HS, Jang JJ, et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res 2010;70:3034–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Villanueva A, Hoshida Y, Battiston C, Tovar V, Sia D, Alsinet C, et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology 2011;140:1501–1512.e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 2009;69:7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007;45:42–52. [DOI] [PubMed] [Google Scholar]
- [40].Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006;12:410–416. [DOI] [PubMed] [Google Scholar]
- [41].Kim SM, Leem SH, Chu IS, Park YY, Kim SC, Kim SB, et al. Sixty-five gene-based risk score classifier predicts overall survival in hepatocellular carcinoma. Hepatology 2012;55:1443–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res 2010;70:10202–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev 2013;27:719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Salomao M, Remotti H, Vaughan R, Siegel AB, Lefkowitch JH, Moreira RK. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum Pathol 2012;43:737–746. [DOI] [PubMed] [Google Scholar]
- [46].Friemel J, Frick L, Unger K, Egger M, Parrotta R, Boge YT, et al. Characterization of HCC Mouse Models: Towards an Etiology-Oriented Subtyping Approach. Molecular cancer research : MCR 2019. [DOI] [PubMed]
- [47].Mu X, Espanol-Suner R, Mederacke I, Affo S, Manco R, Sempoux C, et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest 2015;125:3891–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bochman ML, Schwacha A. The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiology and molecular biology reviews : MMBR 2009;73:652–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sarris M, Nikolaou K, Talianidis I. Context-specific regulation of cancer epigenomes by histone and transcription factor methylation. Oncogene 2014;33:1207–1217. [DOI] [PubMed] [Google Scholar]
- [51].Tanneberger K, Pfister AS, Kriz V, Bryja V, Schambony A, Behrens J. Structural and functional characterization of the Wnt inhibitor APC membrane recruitment 1 (Amer1). J Biol Chem 2011;286:19204–19214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nature reviews Gastroenterology & hepatology 2019;16:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rebouissou S, Franconi A, Calderaro J, Letouze E, Imbeaud S, Pilati C, et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ss-catenin activity associated with liver tumor progression. Hepatology 2016;64:2047–2061. [DOI] [PubMed] [Google Scholar]
- [54].Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature 2015;524:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim W, Khan SK, Gvozdenovic-Jeremic J, Kim Y, Dahlman J, Kim H, et al. Hippo signaling interactions with Wnt/beta-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest 2017;127:137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kwon C, Cheng P, King IN, Andersen P, Shenje L, Nigam V, et al. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat Cell Biol 2011;13:1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rodilla V, Villanueva A, Obrador-Hevia A, Robert-Moreno A, Fernández-Majada V, Grilli A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A 2009;106:6315–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Peignon G, Durand A, Cacheux W, Ayrault O, Terris B, Laurent-Puig P, et al. Complex interplay between β-catenin signalling and Notch effectors in intestinal tumorigenesis. Gut 2011;60:166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Richter LR, Wan Q, Wen D, Zhang Y, Yu J, Kang JK, et al. Targeted Delivery of Notch Inhibitor Attenuates Obesity-Induced Glucose Intolerance and Liver Fibrosis. ACS nano 2020. [DOI] [PMC free article] [PubMed]
- [60].Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014;158:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol 2012;56:1384–1391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.