

Abstract
Microglia play an important role in the central sensitization and chronic pain. However, a direct connection between microglial function and pain development in vivo remains incompletely understood. To address this issue, we applied chemogenetic approach by using CX3CR1creER/+:R26LSL-hM4Di/+ transgenic mice to enable expression of inhibitory Designer Receptors Exclusively Activated by Designer Drugs (Gi DREADD) in microglia. We found that microglial Gi DREADD activation inhibited spinal nerve transection (SNT)-induced microglial reactivity as well as chronic pain in both male and female mice. Gi DREADD activation downregulated the transcription factor interferon regulatory factor 8 (IRF8) and its downstream target pro-inflammatory cytokine interleukin 1 beta (IL-1β). Using in vivo spinal cord recording, we found that activation of microglial Gi DREADD attenuated synaptic transmission following SNT. Our results demonstrate that microglial Gi DREADD reduces neuroinflammation, synaptic function and neuropathic pain after SNT. Thus, chemogenetic approaches provide a potential opportunity for interrogating microglial function and neuropathic pain treatment.
Keywords: Microglia, CX3CR1, Gi DREADD, Chemogenetics, IL-1β, IRF8, SNT, Neuropathic Pain
Introduction:
It has previously been demonstrated that neuroimmune interactions within the central nervous system (CNS) can mediate the pathogenesis of chronic pain (Ji et al., 2016; Ren and Dubner, 2010). In particular, microglia as CNS resident immune cells play a central role in the development of central sensitization and neuropathic pain (Inoue and Tsuda, 2018; Ji and Suter, 2007). Following peripheral nerve injury, microglia become activated and promote chronic pain by releasing a number of glial mediators that sensitize spinal neurons (Inoue et al., 2009; Zhuo et al., 2011). Indeed, specific ablation of microglia delayed the development of neuropathic pain (Peng et al., 2016). However, the direct connection between microglial function and neuropathic pain has not been clearly demonstrated in vivo.
G-protein-coupled receptors (GPCRs) and their downstream signaling regulate most physiological and pathological processes (Marinissen and Gutkind, 2001). To this end, Designer Receptors Exclusively Activated by Designer Drugs (DREADD) have been developed to precisely control various types of GPCR signaling (i.e., Gq, Gs, Gi) using biologically inert agonists (Armbruster et al., 2007; Urban and Roth, 2015). DREADDs have been commonly used to modulate neuronal activity and interrogate neural circuitry of behaviors (Ilg et al., 2018). Recently, DREADD approaches in peripheral or central neurons were used to control pain transmission. For instance, activation of Gi DREADD in TRPV1+ DRG sensory neurons was able to decrease neuronal excitability, producing analgesia (Saloman et al., 2016), while activation of Gi DREADD in spinal dorsal horn GABAergic interneurons induced robust spontaneous nocifensive behaviors (Koga et al., 2017). Consequently, DREADD approaches offer a powerful tool for studying pain transmission and circuits.
While multiple studies have utilized neuronal DREADD approaches to modulate behaviors, few have investigated the effects of DREADD in glial cells. Previously, it has been shown that astrocytic Gq DREADD activation can elicit a number of physiological alterations, including changes in cardiovascular function, body temperature, activity-related behaviors, motor coordination (Agulhon et al., 2013; Sciolino et al., 2016) and improved cognitive performance (Adamsky et al., 2018). As for microglia, it has recently been reported that AAV9-mediated DREADD expression in rat spinal microglia could affect morphine-induced nociceptive sensitivity (Grace et al., 2016) and peripheral nerve injury-induced neuropathic pain (Grace et al., 2018). More recently, there are two studies reported microglial DREADD approaches using genetic mouse models (Binning et al., 2020; Saika et al., 2020). Among these, Saika et al. found that activation of Gi DREADD in CX3CR1 cells was able to reduce neuropathic pain induced by peripheral nerve injury or chemotherapy (Saika et al., 2020). However, chemogenetic interrogations of mouse microglia specifically and their downstream signaling pathways in chronic pain have so far been largely undetermined.
Microglia express a number of GPCRs that are associated with Gi-signaling important for various microglia functions (Kettenmann et al., 2011). In particular, microglia signature P2Y12 receptor is a Gi-coupled GPCR. Our previous studies have found that P2Y12 receptors participate in microglia-neuron communication (Eyo and Wu, 2019) and in neuropathic pain (Gu et al., 2016a). However, it is still largely unknown how Gi signaling itself and its downstream pathways mediate microglial mechanism of pain. In this study, we used CX3CR1creER/+:R26LSL-hM4Di/+ transgenic mice to enable selective expression of Gi DREADD in microglia. Our results showed that chemogenetic inhibition of microglial function via Gi DREADD suppressed the development of neuropathic pain after spinal nerve transection (SNT). Moreover, we examined the molecular mechanisms by which microglial Gi DREADD reduced chronic pain. These results demonstrate that modulation of microglial Gi signaling directly impacts neuropathic pain pathogenesis and microglial chemogenetic approaches represent a potential opportunity for chronic pain treatment.
Results:
Cre-dependent expression of hM4Di Gi DREADD in CX3CR1+ cells.
The chemokine receptor CX3CR1 is highly expressed by microglia in the CNS and cells of mononuclear origin in the periphery (Jung et al., 2000). To study the role of CX3CR1+ cells in neuropathic pain, we first generated CX3CR1creER/+:R26LSL-hM4Di/+ mice to enable tamoxifen (TM) inducible Cre-mediated expression of Gi DREADD in CX3CR1+ cells (Figure 1A–B). The mCitrine and HA-tag in R26LSL-hM4Di/+ mice allows one to determine spatial localization of Gi DREADD after Cre-mediated removal of the STOP cassette (Zhu et al., 2016). Indeed, after TM injection, both mCitrine and HA were expressed and colocalized in the spinal and brain slices (Figure 1C and Figure S1). To validate that Gi DREADD expression was specific to CX3CR1+ microglia, we examined the co-localization of microglial marker Iba1 with HA tag in mice with or without TM treatment (Figure 1C and Figure S1). As expected, in mice receiving TM, there was near total co-localization of HA tag with Iba1 in spinal dorsal horn (SDH) but not in control group without TM treatment (Figure 1C). Similarly, Iba1 positive cell co-localized with HA in the cortex of with TM mice unlike the without TM mice. (Figure S1). These results indicate that we were able to induce Cre-dependent expression of Gi DREADD specifically in CX3CR1+ cells in the CNS.
Figure 1. Microglial Gi DREADD activation delays the development of chronic pain after SNT.
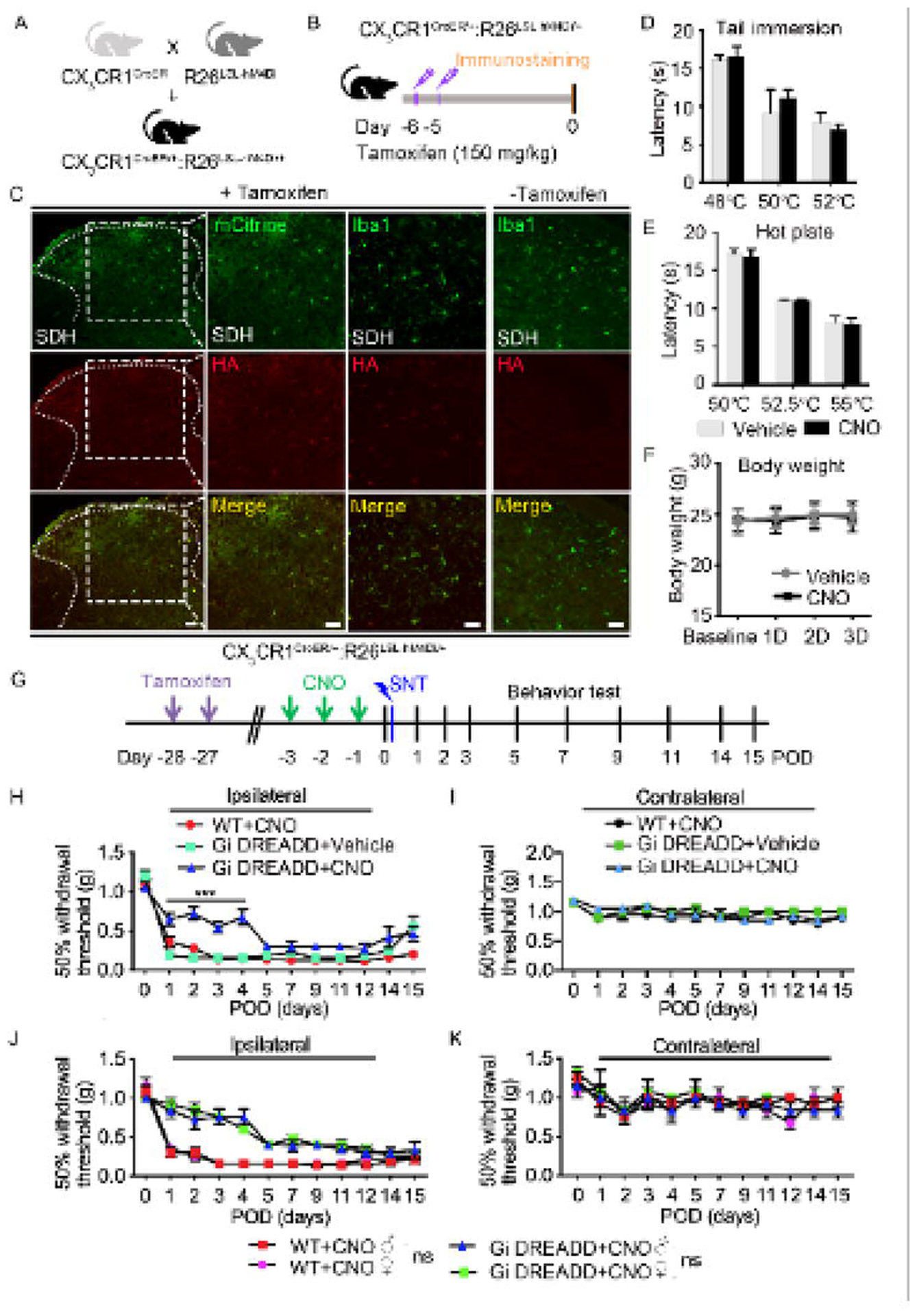
(A) Generation of CX3CR1creER/+:R26LSL-hM4Di/+ transgenic mice. (B) Timeline of experimental procedures for tamoxifen (TM) injection and immunostaining. (C) Representative images of mCitrine (green) and HA immunostaining (red) in spinal cord dorsal horn (SDH) of CX3CR1creER/+:R26LSL-hM4Di/+ mice after TM. Co-labeling Iba1 (green) and HA (red) showed that HA expression was co-localized with Iba1+ cells in SDH region of CX3CR1creER/+:R26LSL-hM4Di/+ mice with TM, but not in CX3CR1creER/+:R26LSL-hM4Di/+ mice without TM. Scale bar, 40 μm. n=5 mice/group. (D, E) Effect of Gi DREADD on acute pain was tested by analyzing tail withdrawal latencies in tail immersion test (D) and hot plate test (E). (F) Body weight was measured in CX3CR1creER/+:R26LSL-hM4Di/+ mice with vehicle (grey) or with CNO treatment (black). The results show no significant change between the vehicle and CNO treated groups. Data represented as mean ± SEM, n=5 mice/group. (G) Timeline of experimental procedures. (H, I) Pre-SNT activation of microglial Gi DREADD delayed mechanical hypersensitivity in Gi DREADD+CNO group when compare to control groups (WT+CNO or Gi DREADD+Vehicle) ipsilaterally (H), but no significant change was observed contralaterally (I). Data represented as mean ± SEM, n=8 mice/group, *** p < 0.001. (J) Pain behavioral tests in the ipsilateral side showed pre-SNT activation of microglial Gi DREADD reduced mechanical hypersensitivity in both male and female CX3CR1creER/+:R26LSL-hM4Di/+ mice but not WT mice. There is no significant difference in pain response between male and female mice. (K) No difference was observed between all groups contralaterally. Data represented as mean ± SEM, n=5 mice/group.
Activation of Gi DREADD in CX3CR1+ cells attenuates chronic pain after SNT.
To address the function of Gi DREADD in CX3CR1+ cells in chronic pain, we used SNT model to induce neuropathic pain in three experimental groups: WT mice with DREADD ligand CNO (WT+CNO); CX3CR1creER/+:R26LSL-hM4Di/+ mice with vehicle (Gi DREADD+Vehicle), and CX3CR1creER/+:R26LSL-hM4Di/+ mice with CNO (Gi DREADD+CNO). We injected TM into these mice starting 6d before SNT to induce cre-medicated Gi DREADD expression in CX3CR1+ cells, then either CNO or vehicle was administered starting 3d before SNT (pre-SNT) (Figure S2A). We compared SNT induced chronic pain behaviors in mice with or without Gi DREADD activation. Our results showed that pre-SNT Gi DREADD activation significantly attenuated mechanical allodynia for a 4 days period in the ipsilateral side of SNT in Gi DREADD+CNO group, when compared to Gi DREADD+Vehicle control mice. CNO itself has no effect on chronic pain induced by SNT in WT mice (Figure S2B). In addition, no difference was observed between any groups in contralateral response (Figure S2C). These results indicate that pre-SNT activation of Gi DREADD in CX3CR1+ cells can delay the development of neuropathic pain.
We also wanted to understand the effect of Gi DREADD activation in CX3CR1+ cells on the maintenance of neuropathic pain. Therefore, we administered CNO at postoperative days (POD) 3 to POD5 after SNT (post-SNT) and examined chronic pain behaviors (Figure S3A). We found that post-SNT Gi DREADD activation significantly attenuated mechanical allodynia starting POD5 in Gi DREADD+CNO group, but not in Gi DREADD+Vehicle control or WT+CNO groups (Figure S3B). Interestingly, the effect of post-SNT Gi DREADD activation only lasted for 3 days (Figure S3B). No significant difference between all three groups was observed in contralateral side of SNT (Figure S3C). Therefore, post-SNT activation of Gi DREADD in CX3CR1+ cells temporally reversed the maintenance of mechanical allodynia induced by peripheral nerve injury.
To ensure that these observations were specific to SNT-induced chronic pain but not acute pain, we examined the effects of Gi DREADD activation by CNO on tail immersion (Figure 1D) and hot plate test in Gi DREADD+CNO groups and Gi DREADD+Vehicle groups (Figure 1E). For both assays no difference between two groups was observed. Gi DREADD activation also had no apparent effect on body weight (Figure 1F). Taken together, our results indicate that both pre- and post-SNT activation of Gi DREADD in CX3CR1+ cells can suppress the development and the maintenance of neuropathic pain.
Activation of Gi DREADD specific to microglia inhibits neuropathic pain.
Previous studies have demonstrated that CX3CR1+ cells, including microglia, monocytes, NK cells are all critically involved in neuropathic pain (Ji et al., 2016). To exclusively evaluate the function of microglial Gi DREADD in chronic pain, we injected TM into CX3CR1creER/+:R26LSL-hM4Di/+ mice 28d before SNT (Figure 1G). This is to take advantage of the significant difference in turnover rates between microglia and peripheral mononuclear cells, allowing for exclusive expression of Gi DREADD within microglia (Parkhurst et al., 2013). We found pre-SNT activation of Gi DREADD by CNO significantly reduced mechanical allodynia following SNT in ipsilateral hindpaws of Gi DREADD+CNO group, but not in WT+CNO mice or in Gi DREADD+Vehicle group (Figure 1H). No significant difference between all three groups was observed in the contralateral side (Figure 1I). Thus, pre-SNT activation of microglial Gi DREADD delays the development of mechanical allodynia following SNT.
It has been reported that microglia play more important role in pathological pain in male than in female mice (Chen et al., 2018; Sorge et al., 2015). Therefore, we wanted to examine whether microglial Gi DREADD function in neuropathic pain was also sex dependent. To this end, we compared the CNO effect on pain behavior in male and female CX3CR1creER/+:R26LSL-hM4Di/+ mice (or WT mice as control) following SNT. Interestingly, we found no difference in the inhibitory effect of microglial Gi DREADD on pain response between male and female mice (Figure 1J), and also no significant difference in contralateral side (Figure 1K). Together, our results highlight that microglia specific Gi DREADD activation delays the development of neuropathic pain in both male and female mice following SNT.
Activation of Gi DREADD suppresses SNT-induced microglial activation.
There is significant proliferation and activation of spinal microglia after peripheral nerve injury (Inoue and Tsuda, 2018; Zhang et al., 2008). As such, we first examined microglial numbers at POD3 after SNT (Figure 2A), when it is the peak of spinal microglia proliferation in response to peripheral nerve injury (Gu et al., 2016b; Tashima et al., 2016). Consistently, the number of microglia labeled by Iba1 immunostaining in the ipsilateral dorsal horn was dramatically increased following SNT. Further, we found that pre-SNT Gi DREADD activation by CNO in CX3CR1creER/+:R26LSL-hM4Di/+ mice largely reduced SNT-induced microglial numbers compared with the vehicle group (Figure 2B, C). We examined L1 to S1 segments of spinal cord and found that the reduction of microglial number is mainly restricted to L4 after CNO treatment after SNT (Figure S4). Using CD11b immunostaining, we also observed the similar inhibition of microglial numbers by pre-SNT CNO treatment (data not shown). Microglial proliferation is the major source of spinal microgliosis after peripheral neve injury (Gu et al., 2016a; Tashima et al., 2016). Therefore, we tested the effect of Gi DREADD on microglial proliferation using thymidine analog 5-bromo-2’-deoxyuridine (BrdU, i.p. 100 mg/kg, 2 pulses/day) in Iba1+ microglia in SDH after SNT. Indeed, we found that the percentage of proliferated microglia (BrdU+Iba1+ cells) of the total Iba1+ cells was dramatically reduced by pre-SNT CNO treatment (Figure 2D, E). Thus, pre-SNT activation of microglial Gi DREADD signaling inhibits the increase of spinal microglial numbers and proliferation induced by peripheral nerve injury.
Figure 2. Microglial Gi DREADD activation reduces microglia activation.

(A) Timeline of experimental procedures. (B) Representative immunostaining images show that the number of Iba1+ microglia was reduced in the ipsilateral dorsal horn at POD3 by pre-SNT activation of microglial Gi DREADD in CX3CR1creER/+:R26LSL-hM4Di/+ mice. Scale bar, 40 μm. Data represented as mean ± SEM, n=8 mice/group. (C) Summarized data showing microglial numbers were reduced by CNO treatment but not in vehicle group. Scale bar, 40 μm. Data represented as mean ± SEM, n=5 mice/group, *** p < 0.001. (D) Immunofluorescence images showing co-localization of BrdU (red) and Iba1 (green) at POD3 following SNT. BrdU (i.p. 100 mg/kg was applied before SNT at 2 pulses/day for 5 days (n=4 mice). Scale bar, 40 μm. n=5 mice/group, **** p < 0.0001. (E) Quantitative summary showing the percentage of BrdU+Iba1+ cells of the total Iba1+ cells were significantly reduced by pre-SNT CNO treatment but not in Vehicle group. Data are presented as mean ± s.e.m. n=4 mice/group, **** p < 0.0001. (F) Representative single microglia images in the spinal cord following SNT by Iba1 immunostaining and after being skeletonized. (G) Sholl analysis showing CNO treatment increased the complexity of microglia compared with vehicle group. Data represented as mean ± SEM, n=5 mice/group, **** p < 0.0001.
It has been shown that microglial morphology has been directly related to their activation state. For instance, microglial processes are visibly shorter and less complex within a day of injury (Gu et al., 2016a; Inoue and Tsuda, 2009). Therefore, here we further examined the effects of Gi DREADD activation on microglial morphology in CX3CR1creER/+:R26LSL-hM4Di/+ mice following SNT. Using Sholl analysis, we compared the complexity of spinal microglia after CNO or vehicle treatment (Figure 2F). Consistent with the reduced microglial numbers, we found that pre-SNT Gi DREADD activation reversed SNT-induced morphological change (Figure 2F, G). Consequently, these results further indicate that Gi DREADD inhibits microglial activation in response to SNT-induced pain.
Activation of Gi DREADD reduces microglial IRF8 and IL-1β expression.
Under neuropathic pain conditions, the transcription factor interferon regulatory factor 8 (IRF8) plays a significant role in microglial response (Masuda et al., 2012). Therefore, we tested the possibility that microglial Gi DREADD impacts neuropathic pain by modulating IRF8. To this end, we examined IRF8 expression after SNT with Gi DREADD activation (Figure 3A). Our results showed that IRF8 is selectively expressed in Iba1+ microglia in the ipsilateral dorsal horn at POD3 after SNT (Figure 3B). Pre-SNT activation of Gi DREADD by CNO significantly reduced SNT-induced IRF8 upregulation at POD3 (Figure 3B, C). Western blot analysis revealed that SNT increased IRF8 expression for at least 5 days (Figure 3D), and Gi DREADD activation reversed SNT-induced increase from POD1–3 days (Figure 3E). IRF8 expression returned to comparable levels as control by POD5, which is also when mechanical allodynia became comparable to controls.
Figure 3. Microglial Gi DREADD activation inhibits IRF8 and IL1β expression.
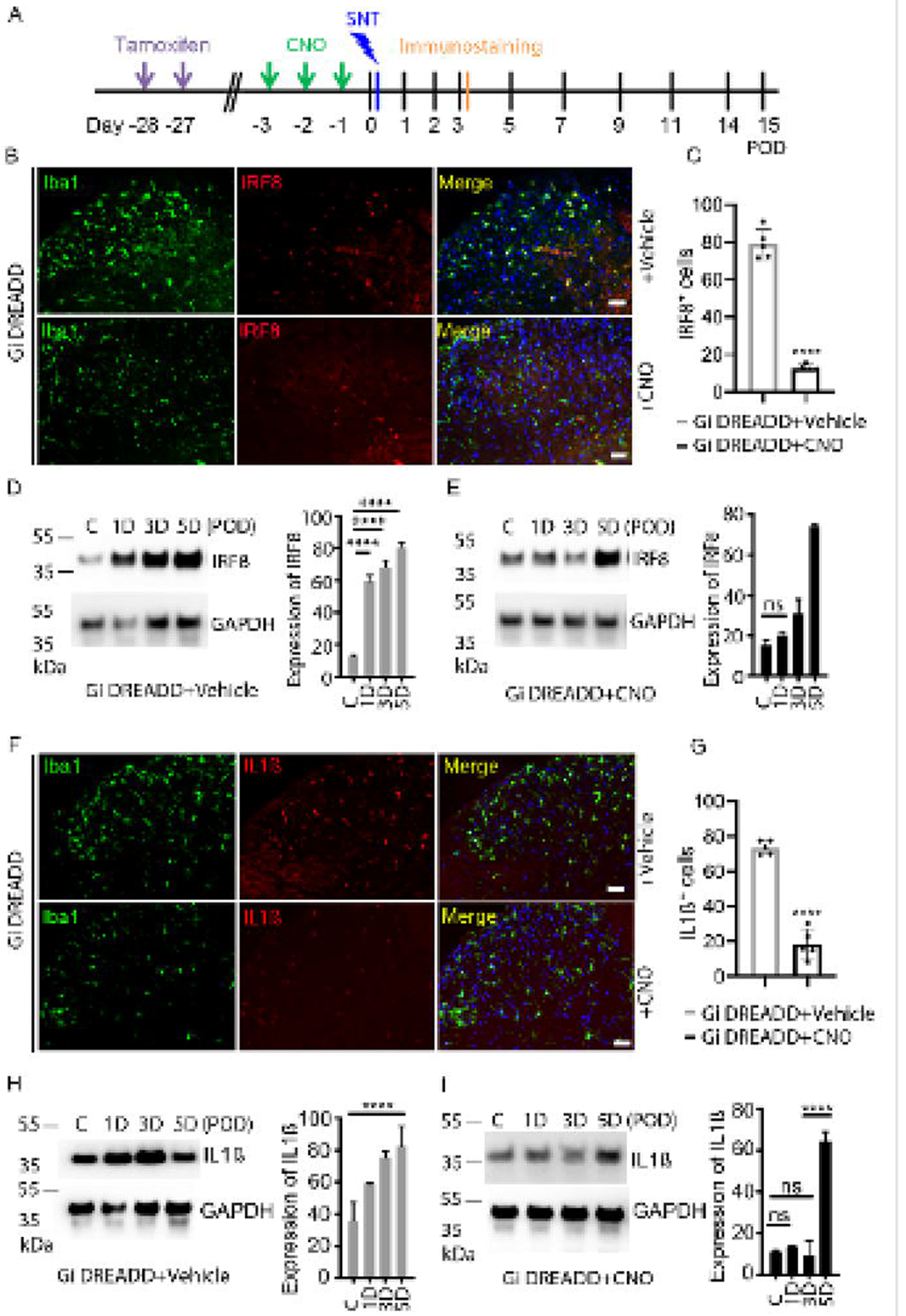
(A) Timeline of experimental procedures. (B) Immunofluorescence images showed that IRF8 (red) co-localized with Iba1+ cells (green) and Dapi (blue) in the ipsilateral dorsal horn at POD3. The pre-SNT CNO treatment suppressed IRF8 expression. Scale bar, 40 μm. (C) Pooled results show significantly reduced IRF8+ cells after CNO treatment as compared with that in vehicle group. n=5 mice/group, **** p < 0.0001. (D) Representative Western blot images and quantification data showing that IRF8 expression in the L4–5 level of the dorsal horn increased after SNT n=5–8 mice/group, GAPDH was used as internal control. **** p < 0.0001. (E) Pre-SNT activation of microglial Gi DREADD by CNO suppressed the increase of IRF8 expression following SNT at POD1–3. n=5–8 mice/group. (F) Immunofluorescence images showing that IL1β (red) is co-localized with Iba1+ microglial cells (green) in the ipsilateral dorsal horn at POD3 following SNT. CNO treatment reduced SNT-induced IL1β expression in microglia. Scale bar, 40 μm. (G) Pooled results show significantly reduced IL1β+ cells after CNO treatment compared with that in vehicle treated group. n=5 mice/group, **** p < 0.0001. (H, I) Representative Western blot images and quantification data showing that IL1β expression in the L4–5 level of the dorsal horn was increased after SNT (H) which was suppressed by pre-SNT activation of microglial Gi DREADD by CNO (I). n=5–8 mice/group, GAPDH was used as internal control. **** p < 0.0001.
IRF8 is a critical transcription factor for the regulation of pro-inflammatory interleukin 1 beta (IL-1β), which is important for the pathogenesis of chronic pain (Gui et al., 2016; Ren and Torres, 2009). Thus, we investigated the possibility that the effect of Gi DREADD upon IRF8 also extended to IL-1β production by microglia. Consistently, we found that pre-SNT Gi DREADD activation significantly reduced IL-1β expression largely expressed in Iba1+ microglia (Figure 3F, G). Moreover, Western blot analysis showed that up-regulation of IL-1β persists over POD1–5 after SNT (Figure 3H), while CNO suppressed IL-1β upregulation through POD3 (Figure 3I). In addition, since BDNF is highly implicated in microglia-neuron interactions in neuropathic pain (Coull et al., 2005), we examined the effect of Gi DREADD activation on BDNF expression using Western blot analysis. We observed that BDNF protein levels were significantly upregulated at POD3–5 in control group. After Gi DREADD activation by CNO treatment, BDNF expression was similar to that in control group (Figure S5). Taken together, these results suggest that microglial Gi DREADD may exert its influence on chronic pain via the modulation of neuroinflammation, including IRF8 and IL-1β expression.
Post-SNT Gi DREADD activation in microglia attenuates chronic pain and neuroinflammation.
Our previous study using cell ablation approaches report that microglia are transiently required for the maintenance of neuropathic pain (Peng et al., 2016). Here, to directly assess the role of the post-SNT microglial Gi DREADD activation in the maintenance of neuropathic pain, we applied CNO at POD3–5 after SNT in CX3CR1creER/+:R26LSL-hM4Di/+ mice (Figure 4A). WT+CNO group and Gi DREADD+Vehicle group were examined as controls. Our result indicated that post-SNT activation of Gi DREADD in microglia reduced mechanical hypersensitivity in ipsilateral side, which lasted for 4 days in CX3CR1creER/+:R26LSL-hM4Di/+ mice but not in control groups (Figure 4B). In addition, we found no sex difference in the inhibitory effect of post-SNT activation of Gi DREADD on chronic pain maintenance between male and female mice (Figure 4C).
Figure 4. Post-SNT activation of microglial Gi DREADD reverses mechanical hypersensitivity and neuroinflammation.

(A) Timeline of experimental procedures. (B) Behavioral test showing that post-SNT activation of microglial Gi DREADD attenuated mechanical hyperactivity in CX3CR1creER/+:R26LSL-hM4Di/+ mice (Gi DREADD+CNO) when compared to control groups (WT+CNO or Gi DREADD+Vehicle). Data represented as mean ± SEM, n=5 mice/group. ** p < 0.01, *** p < 0.001. (C) Post-SNT activation of microglial Gi DREADD attenuated mechanical hyperactivity in both male and female CX3CR1creER/+:R26LSL-hM4Di/+ mice (Gi DREADD+CNO). Data represented as mean ± SEM, n=5 mice/group. WT mice with CNO were considered to be controls. There was no difference between male and female groups. (D) Immunofluorescence images and summarized results show that IRF8 (red) co-localized with Iba1+ microglial cells (green) in the ipsilateral dorsal horn at POD5 following SNT. Post-SNT CNO treatment to activate microglial Gi DREADD reversed the increase of IRF8 expression by SNT. Scale bar, 40 μm. n=5 mice/group, **** p < 0.0001. (E) Immunofluorescence images and summarized results show increased expression of IL1β which is co-localized with Iba1+ microglia cells in the ipsilateral dorsal horn at POD5 following SNT. CNO treatment reversed the increase of IL1β expression by SNT. Scale bar, 40 μm. n=5 mice/group, **** p < 0.0001.
We also explored the effect of post SNT activation of microglial Gi DREADD on IRF8 and IL-1β expression using immunostaining. We found that microglial Gi DREADD activation at POD3–5 by CNO reduced the expression of IRF8 in Iba1+ microglia at POD5 after SNT compared to that of vehicle treatment (Figure 4D). In addition, post-SNT activation of microglial Gi DREADD similarly reduced IL-1β expression in Iba1+ microglia (Figure 4E). Thus, our results suggest that microglial Gi DREADD attenuates the maintenance of chronic pain likely through reduced neuroinflammation such as the downregulation of IRF8 and IL-1β in microglia.
Gi DREADD reduces C-fiber-evoked field potential after SNT.
Finally, we wanted to determine the effect of Gi DREADD upon C-fiber mediated nociceptive transmission which is intimately related to chronic pain induction and maintenance (Sandkuhler, 2007). To this end, we used in vivo recording in the spinal cord dorsal horn in anesthetized mice to study on C-fiber-evoked field potentials (Liu et al., 2017; Zhou et al., 2019). Three groups of mice were used: WT sham, WT SNT (POD3) and Gi DREADD SNT (POD3). For each experimental group, CNO was injected after 30 mins of baseline recording (Figure 5A). We found that basal C-fiber-evoked field potentials were strongly decreased by CNO in Gi DREADD mice after SNT, but not in WT sham control or WT SNT group (Figure 5B, C). After normalization to pre-drug values (baseline; −30–0 min before CNO administration), C-fiber-evoked field potential were maximally decreased by 44.8% ± 18.9% after CNO treatment (Figure 5C). Interestingly, the inhibitory effect of Gi DREADD on C-fiber response in SNT group lasted less than one hour (Figure 5C). These results indicate that nociceptive transmission after peripheral nerve injury is inhibited by the activation of microglial Gi DREADD.
Figure 5. Microglial Gi DREADD activation attenuates C-fiber responses in the spinal dorsal horn after SNT.
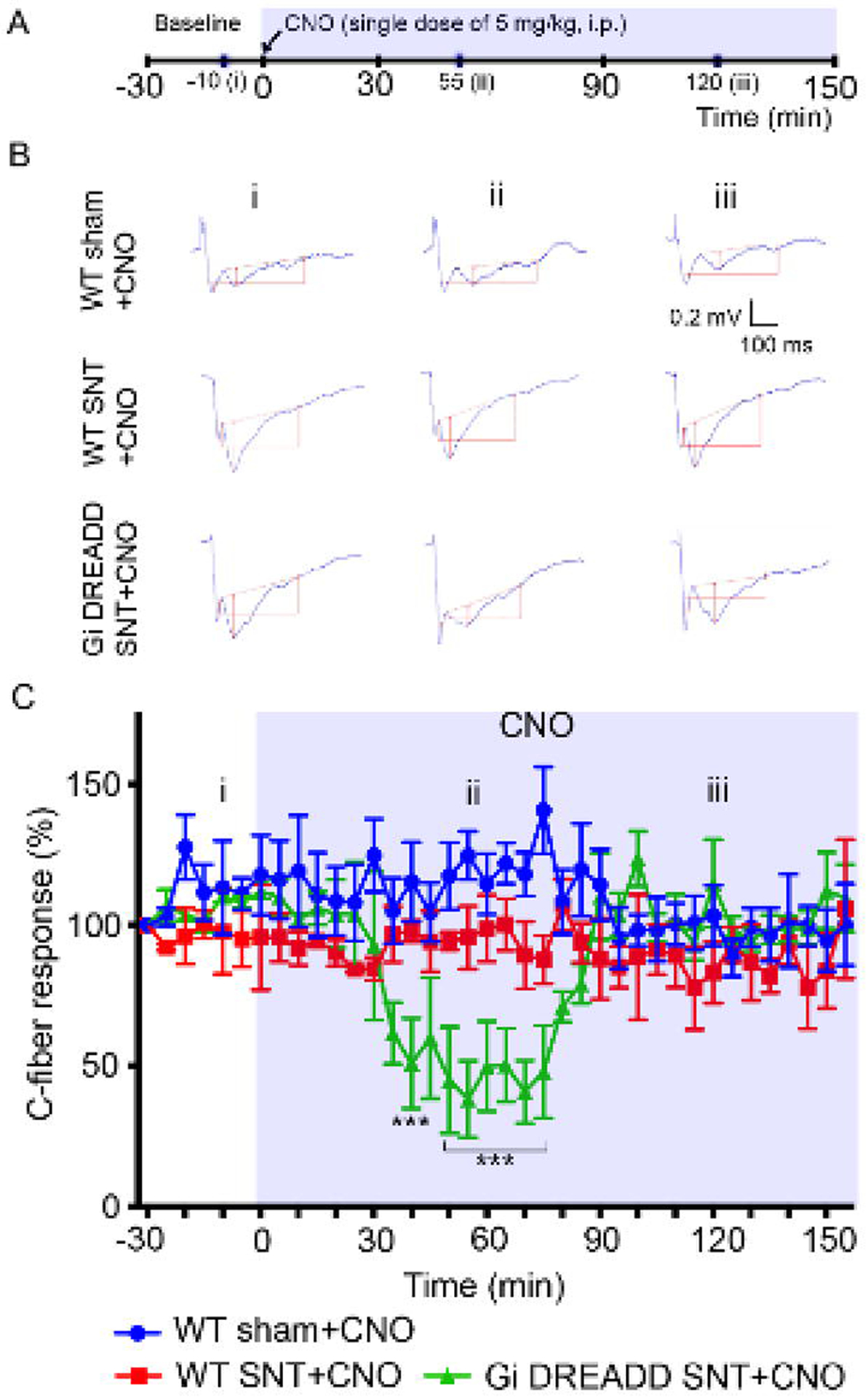
(A) Timeline of experimental procedures. (B) Representative traces of C-fiber field potentials from three groups (WT sham, WT SNT, and Gi DREADD SNT), recorded at baseline (i), after CNO treatment, 55 min (ii) and 120 min (iii). The amplitude of C-fiber-evoked response (red vertical line) was determined with parameter extraction software WinLTP. n=5 mice/group. Scale bars, 100 ms (X) and 0.2 mV (Y). (C) Pooled results showing the time course of C-fiber field potentials in response to the CNO application. CNO reduced C-fiber responses in Gi DREADD SNT group but not in control groups. C-fiber-evoked field potential was normalized to baseline. Data represented as mean ± SEM, n=5 mice/group. *** p < 0.001.
Discussion:
In this study, we used transgenic Gi DREADD approach as a novel tool to examine the role of microglia in neuropathic pain. Taking advantage of CX3CR1creER/+:R26LSL-hM4Di/+ transgenic mice, we were able to controllably activate Gi DREADD in microglia. Our results showed that microglial Gi DREADD attenuated SNT-induced microglial activation and chronic pain hypersensitivities. Mechanistically, we found that microglial Gi DREADD signaling reduced IRF8 upregulation, IL-1β production, and C-fiber evoked responses after peripheral injury. Together, these results demonstrate that microglial Gi signaling by chemogenetic manipulation attenuates chronic pain via inhibition of neuroinflammation.
Microglial DREADD approaches to study microglia function
Chemogenetic approaches like DREADD have only recently been employed to study glial function, in particular, astrocyte function in motor coordination and memory (Agulhon et al., 2013; Nam et al., 2019; Sciolino et al., 2016). Chemogenetic approaches have also been used to study oligodendrocyte control of neural activity (Ou et al., 2019). Finally, the Watkins’ group utilized DREADD approaches to study microglia in the spinal cord of rats via AAV9 mediated expression (Grace et al., 2016; Grace et al., 2018). By selectively expressing Gi DREADD under a CD68 promotor, the group elucidated microglial function in morphine-induced nociceptive sensitivity (Grace et al., 2016) and cytokine production in neuropathic pain (Grace et al., 2018). Interestingly, a very recent study reported that activation of Gi DREADD constitutively epxressed in CX3CR1 cells exerts sex-dependent alleviation of neuropathic pain in only male mice (Saika et al., 2020). However, our results showed that microglial Gi DREADD activation reduced neuropathic pain in both male and female mice. The discrepancy could be due to the following differences: 1) Different cre mouse lines. Saika et al. utilized constitutive CX3CR1-Cre promoter which results in the Gi DREADD expression all CX3CR1 cells including microglia and monocytes. In addition, there has been a debate about the potential leakage of this mouse line (Zhao et al., 2019). 2) Different CNO administration paradigms. Saika et al. used one time injection of CNO (10mg/kg) while we injected daily CNO (5mg/kg) for three days; 3) Different time windows for Gi DREADD activation. Saika et al. chose the late time point (POD7–29) after peripheral nerve injury to study the effect of Gi DREADD in pain maintenance while our study mainly focused POD0–5 after SNT. Our previous studies have demonstrated that microglia are more effective in the development of neuropathic pain (Gu et al., 2016b; Peng et al., 2016).
However, further study is needed to understand microglial DREADD downstream signaling in chronic pain behaviors. To this end, we employed a genetic mouse model to express Gi DREADD specifically in microglia. Our results are largely consistent with the study from Watkin’s group and Kiguchi’s group (Grace et al., 2018; Saika et al., 2020). However, our current study is exciting in several regards. First, we used genetic mouse model to manipulate microglia function specifically in vivo using chemogenetic approaches and found that microglia participate in the initiation of neuropathic pain in both male and female mice. Second, our results suggest that expression of IRF8, IL-1β, and synaptic transmission can be regulated by Gi signaling during the development of neuropathic pain. Third, we showed that microglia are transiently required for the maintenance of neuropathic pain. Therefore, this study provides proof-of-principle that Gi signaling in microglia can suppress their pro-inflammatory activation and thus is a powerful tool to manipulate microglia function in chronic pain. One caveat is that recent study demonstrated leaky issues from CX3CR1CreER mice depending on the length of the STOP cassette of loxp system (Van Hove et al., 2020). Although Our Gi DREADD system (Rosa-CAG-LSL-HA-DREADD-P2A-mCitirine-WPRE) includes short length (0.9 kb) of STOP cassette (Zhu et al., 2016), we confirmed the specific expression of Gi DREADD in Iba1+ microglia after TM injection in the spinal cord and brain. Gi DREADD is also expressed in supraspinal microglia and thus may contribute to the analgesic effects of systemic CNO on SNT-induced pain. To circumvent this issue, local activation of microglial Gi DREADD is required in the future studies to delineate the specific function of spinal and supraspinal microglia in chronic pain.
Microglia mechanism of neuropathic pain
In the present study we found that IL-1β, which is a critical mediator of neuropathic pain as well as a promising therapeutic target for pain control (Ji et al., 2014; Masuda et al., 2012), was decreased by microglial Gi DREADD activation. These results demonstrate that pro-inflammatory cytokines produced by microglia can be regulated via Gi signaling. In addition, recent studies have concluded that IRF8 is a major regulator of IL-1β (Masuda et al., 2012). In the periphery, IRF8 plays a pivotal role in the immune system (Honda and Taniguchi, 2006; Taniguchi et al., 2001). We further demonstrated a link between microglial Gi DREADD signaling and IRF8 expression after SNT. Interestingly, ectopic expression of IRF8 causes marked upregulation of P2Y12 which is Gi-protein-coupled receptor in cultured microglia in vitro (Honda et al., 2001; Masuda et al., 2012). Therefore, this may provide the self-regulation mechanism for neuroinflammation in chronic pain. Collectively, our results suggest that expression of IRF8 and IL-1β are regulated by Gi signaling pathways which can modulate microglial activation and subsequent pain hypersensitivity. However, to confirm the causality of IRF8 and IL1β downregulation after GI DREADD activation for pain control, additional studies will be needed to address whether Gi DREADD inhibits pain directly via IL1β and further dissect how Gi signaling directly impacts IRF8 expression.
We also demonstrated with in vivo electrophysiology experiments that Gi DREADD significantly inhibited C-fiber-evoked field potentials following SNT. We observed the single dose of CNO reversed the nociceptive transmission from around 40 min post injection until at least 70 min post injection. These findings broadly match the known pharmacokinetic profile of CNO in the activation of DREADD (Jendryka et al., 2019). It is unlikely that CNO has off-target effects for two reasons because it did not alter C-fiber responses in the sham control mice or WT mice after SNT. The mechanism is still unknown regarding how Gi DREADD activation acutely inhibits synaptic transmission. However, multiple lines of evidence have suggested that microglia can regulate neuronal activity. For example, microglia were able to dampen neuronal hyperactivity under seizure context (Eyo et al., 2014; Kato et al., 2016) or even under physiological conditions (Cserep et al., 2020; Peng et al., 2019). Particularly, selective microglial activation in the spinal cord was sufficient to facilitate synaptic strength between primary afferent C-fibers and spinal neurons (Clark et al., 2015; Zhou et al., 2019). Thus, considering the increased microglia reactivity under neuropathic pain condition, our results are plausible that Gi DREADD activation in microglia inhibited C-fiber synaptic transmission. Interestingly, we found that three doses of CNO were able to reduce chronic pain and the inhibitory effects lasted for several days. In addition, microglia Gi DREADD activation was accompanied by reduced neuroinflammation such as IRF8 and IL-1β. Thus, the longer term activation of Gi signal transduction via DREADD may lead to a biochemical cascade over multiple days. However, it still remains unknown how transient inhibition of synaptic transmission results in the long lasting effect on SNT-induced chronic pain after multiple times of microglial Gi DREADD activation.
Microglia DREADD as potential therapeutic avenues
Recent reports indicate that Gi DREADD can be successfully expressed and functional in non-human primates (Nagai et al., 2016). Therefore, it is plausible to explore the therapeutic potential of Gi DREADD in neuropathic pain. For example, AAV-CD68-hM4Di was used to express Gi DREADD in rat microglia (Grace et al., 2016). Using similar AAV vectors to express Gi DREADD in microglia in humans is possible, as they are commonly used for gene therapy in clinical patients (Naso et al., 2017). Moreover, consistent with its pharmacological inertness, CNO has been administered to humans without apparent toxic effect (Jann et al., 1994). However, CNO is not a drug that has been approved for use in humans by the Food and Drug Administration (FDA) (Lieb et al., 2019). Alternatively, FDA approved olanzapine is a second-generation atypical antipsychotic which is able to activate Gi DREADD (Weston et al., 2019). Nevertheless, this study is a proof-of-concept that Gi DREADD manipulation of microglia could influence neuropathic pain, suggesting its potential application for pain treatment in the long run.
Materials and Methods:
Animals:
7–12 weeks old male mice, unless otherwise indicated, were used in accordance with institutional guidelines as approved by the animal care and use committee at Mayo Clinic. C57BL/6J (000664) and R26LSL-hM4Di (026219) (Zhu et al., 2016), and CX3CR1CreER/CreER (021160) mice (Parkhurst et al., 2013) were purchased from Jackson Laboratory (Bar Harbor, ME). CX3CR1CreER/CreER mice were crossed with R26LSL-hM4Di mice to obtain CX3CR1creER/+:R26LSL-hM4Di/+ mice. These mice were assigned to an experimental group randomly within a litter. Experimenters were blind to drug treatments.
Gi DREADD activation:
To induce Gi DREADD expression in CX3CR1+ cells, 150 mg/kg tamoxifen (TM; T5648, Millipore-Sigma, Burlington, MA) in corn oil (20 mg/mL) was administered via i.p. injection twice daily for 2 days. To activate Gi DREADD, 5 mg/kg Clozapine N-oxide (CNO; 16882, Cayman Chemical, Ann Arbor, MI) was administered i.p. daily for 3 days. WT (C57BL/6J) mice receiving TM and CNO and CX3CR1creER/+:R26LSL-hM4Di/+ receiving TM and vehicle (but not CNO) were used for controls.
Spinal nerve transection (SNT) induced pain induction:
SNT surgery was performed under 2% isoflurane anesthesia. Briefly, an incision was made along the mid-line of the lumbar spine. The left paraspinal muscles in front of the pelvic bone were separated to expose the L5 transverse process. The L5 transverse process was then removed to expose the L4 spinal nerve. The L4 spinal nerve was separated, transected and removed 1–1.5 mm from the end to DRG. The wound was then irrigated with sterile PBS and sutured.
Behavioral assessments:
Mechanical allodynia was assessed by measuring paw withdrawal threshold test via von Frey filaments (0.04–2 g). Briefly, mice were placed on an elevated metal grid and filaments of increasing weight were applied to the plantar surface at a vertical angle for up to 3 s. Fifty percent withdrawal threshold values were determined using the up-down method (Chaplan et al., 1994).
Thermal hyperalgesia was assessed by measuring paw withdrawal latency to radiant heat stimuli. Briefly, mice were placed in elevated chambers with a plexiglass floor and allowed to habituate for 20 min. A radiant heat source was then applied to the center of the plantar surface of the hind paw four times with at least 3-min intervals. The average withdrawal latency of the four trials was recorded as the response latency.
Tissue collection:
At various time points post SNT, mice were deeply anesthetized and perfused transcardially with 20 mL PBS followed by 20 mL of cold 4% paraformaldehyde (PFA) in PBS. Spinal cords were then removed and post-fixed with 4% PFA for 6 h at 4°C. The samples were then transferred to 30% sucrose in PBS overnight. 15 μm thick sections were prepared via cryosection and applied to charged glass slides. Slides were then stored at −20°C until use.
Fluorescent immunostaining:
Tissue slides were blocked with 5% goat serum in 0.3% triton X-100 (Sigma) in PBS buffer for 60 min, and then incubated overnight at 4°C with primary antibody for rat anti-HA (1:200; 12013819001, RRID:AB_390917, Roche, Basel, Switzerland), rabbit anti-GFP (1:200; ab290, RRID:AB_303395, Abcam, Cambridge, United Kingdom), rabbit anti-Iba1 (1:500; ab178847, RRID:AB_2832244, Abcam), mouse anti-IRF8 (1:400; sc-365042, RRID:AB_10850401, Santa Cruz Biotechnology, Dallas, Texas), mouse anti-IL-1β (1:400; 12242, RRID:AB_2715503, Cell Signaling Technology, Danvers, MA), and rat anti-BrdU (1:1000; ab6326, RRID:AB_305426, Abcam). The sections were then incubated for 60 min at room temperature, with goat anti-rat (1:500; A-11006, RRID:AB_2534074, Thermo Fisher Scientific, Waltham, MA), goat anti-rabbit (1:500; A-11035, RRID:AB_2534093, Thermo Fisher Scientific) or goat anti-mouse secondary antibodies (1:500; A-11029, RRID:AB_2534088, Thermo Fisher Scientific). The sections were then mounted with Fluoromount-G (Southern Biotech) and fluorescent images were obtained with an EVOS FL Imaging System (Thermo Fisher Scientific) and confocal microscope (LSM510, Zeiss).
BrdU labeling
The thymidine analog bromodeoxyuridine (BrdU) (B5002, Sigma) was used to label proliferating and recently postmitotic cells in the spinal cord after SNT surgery. The BrdU solution was diluted in 1M PBS just before use and intraperitoneally administered (i.p. 100 mg/kg). For BrdU staining, the spinal cord sections were hydrated in Tris-buffered saline (TBS) for 10 s and transferred to a 50% form-amide in 2× saline sodium citrate (SSC) solution at 65°C for 2 hr. Slides with spinal cord sections were then placed in a 2× SSC solution at room temperature for 15 min and were transferred to a 2 N HCl solution at 37°C for 20 min and then a 0.1 M borate for 10 min at RT.
Western blot analysis:
Lumbar 4–5 spinal dorsal horns were collected at various time points and protein was extracted. 50 μg of protein from each group was then loaded and separated by SDS-PADGE, transferred to a PVDF membrane, blocked with 5 % skim milk in TBST, and incubated overnight with primary antibodies at 4°C. Primary antibodies include, mouse anti-IRF8 (1:1000; sc-365042, RRID:AB_10850401, Santa Cruz Biotechnology, Dallas, Texas), mouse anti-IL-1β (1:1000; 12242, RRID:AB_2715503, Cell Signaling Technology, Danvers, MA), rabbit anti-BDNF (1:1000; ANT-010, RRID:AB_2039756, Alomone labs™), and GAPDH (1:1000; sc-32233, RRID:AB_627679, Santa Cruz Biotechnology). Membranes were incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (1:3,000; 115-035-003, RRID:AB_10015289, Jackson ImmunoResearch Labs, West Grove, Pennsylvania) and peroxidase-conjugated goat anti-rabbit IgG (1:3,000; 111-036-045, RRID:AB_2337943, Jackson ImmunoResearch Labs) for 1 hr at room temperature. Membranes were then treated with West Pico substrate (34078, Thermo Fisher Scientific) and chemiluminescence signal was detected with a G:BOX Chemi XRQ gel doc (Syngene, Frederick, MD). Optical density of each band was then determined using Fiji, (NIH).
Sholl analysis
Z-stack confocal live microglia images (30 μm) were acquired at 3-μm intervals using a 40× objective of confocal microscope (LSM510, Zeiss). Consecutive Z-stack Images were converted to a maximum intensity projection image using Fiji software. Using the Image5D plugin (Fiji, NIH), z-stack images were condensed into a maximum intensity projection image over which concentric circles were drawn (concentric circles plugin, fiji), centered on the soma, beginning at 0.5 μm radii and increasing 0.1 μm with every circle. Sholl analysis was manually performed for each cell by counting the number of intersections between microglia branches and each increasing circle to create a Sholl plot. Additional measures to characterize each cell included the number of branch number, length (manual count) and cell soma area (Fiji Analysis).
In vivo electrophysiology recording in the spinal cord dorsal horn:
Under anesthesia, a T12-L1 laminectomy was performed to expose the lumbar enlargement of the spinal cord and the dura was removed. The left sciatic nerve was then exposed for electrical stimulation with a bipolar platinum hook electrode. A test stimulus (0.5 ms duration, every 1 min, at C-fiber intensity) was then delivered to the sciatic nerve. C-fiber evoked fEPSP was recorded from the dorsal horn with a glass microelectrode filled with 0.5 M sodium acetate (impedance 0.5–1 MU). An A/D converter card (M-Series PCI-6221, National Instruments, Austin, TX) was used to digitize and store data at a sampling rate of 10 kHz. C-fiber evoked fEPSP was determined with WinLTP Standard Version (WinLTP Ltd., Bristol, United Kingdom). For each experiment the average amplitude of five consecutive fEPSP was recorded at 30 sec intervals. The mean amplitude of the responses before CNO administration served as baseline. To observe the effect of DREADD activation on C-fiber evoked field potentials, CNO was injected i.p. 30 mins after stable recording of C-fiber evoked field potentials.
Statistical analysis:
Pain behaviors were analyzed using two-way ANOVA to test for main effects between groups followed by post-hoc testing for significant differences by day. Two group analyses utilized Student’s t-test. Data were represented as mean ± SEM. All statistical analyses were performed using Prism6 (GraphPad, San Diego, CA). Level of significance is indicated with *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001
Supplementary Material
Figure S1. CX3CR1creER/+:R26LSL-hM4Di/+ transgenic mice enable selective expression of Gi DREADD in microglia of the brain. Immunofluorescence images of the hippocampus and cortex labeled with mCitrine and HA in CX3CR1creER/+:R26LSL-hM4Di/+ mice after TM injection show expression of Gi DREADD. Co-staining of HA (red) with Iba1 (green) in brain cortex of CX3CR1creER/+:R26LSL-hM4Di/+ mice with TM indicates co-localization which is absent without TM injection. Scale bar, 40 μm. n=5 mice/group.
Figure S2. Activation of Gi DREADD in CX3CR1+ cells delays the development of chronic pain after SNT. (A) Timeline of experimental procedures for TM injection, pre-SNT CNO administration, SNT surgery, and pain behavioral test. (B, C) Measurement of mechanical hypersensitivity in WT mice with CNO (WT+CNO), CX3CR1creER/+:R26LSL-hM4Di/+ mice with vehicle (Gi DREADD+Vehicle), and CX3CR1creER/+:R26LSL-hM4Di/+ mice with CNO (Gi DREADD+CNO). Activation of Gi DREADD in CX3CR1+ cells following SNT showed that initiation of mechanical hypersensitivity was delayed when compared to control groups ipsilaterally (B), but no significant change was observed contralaterally (C). Data represented as mean ± SEM, n=5–8 mice/group, *** p < 0.001.
Figure S3. Activation of Gi DREADD in CX3CR1+ cells attenuates chronic pain after SNT. (A) Timeline of experimental procedures for TM injection, SNT surgery, post-SNT CNO administration, and pain behavioral test. (B) Mechanical hypersensitivity showing that post-SNT activation of Gi DREADD significantly reversed the pain behaviors ipsilaterally in Gi DREADD+CNO group when compared to control groups. (C) However, no difference was found between the three groups in the contralateral side. Data represented as mean ± SEM, n=8 mice/group, ** p < 0.01, *** p < 0.001.
Figure S4. Serial sections of Iba1 immunostaining in the SDH. Somatotopic distribution of Iba1+ cells in the ipsilateral spinal dorsal horn (L1–S1) from a Gi DREADD+Vehicle control group (left, n=3 mice) and Gi DREADD+CNO group (right, n=3 mice) at POD3 after SNT in CX3CR1creER/+:R26LSL-hM4Di/+ mice. Representative images are showing that the reduced number of microglia is mainly restricted to L4 after CNO treatment. Scale bar: 40 μm
Figure S5. SNT-induced BDNF upregulation not affected by microglial Gi DREADD activation. (A) Timeline of experimental procedures. (B) Representative Western blot images and quantification data showing that BDNF expression in the L4–5 level of the dorsal horn increased gradually at POD3–5 after SNT in Vehicle control group. n=4–5 mice/group, GAPDH was used as internal control. * p < 0.05, *** p < 0.001. (C) Pre-SNT activation of microglial Gi DREADD by CNO did not alter the BDNF upregulation seen in Gi DREADD+Vehicle control group. n=4–5 mice/group. GAPDH was used as internal control. * p < 0.05, *** p < 0.001.
Acknowledgements:
This work is supported by National Institutes of Health (R01NS088627, R01NS110825, R01NS110949, R01NS112144, R21AG064159) to L.J.W., and by a postdoctoral fellowship from the Mayo Clinic Center for Multiple Sclerosis and Autoimmune Neurology to T.C. We thank Dr. Anthony Umpierre and Dr. Aastha Dheer for critical reading of the paper and members of Wu lab at Mayo for insightful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare no competing financial interests.
References:
- Adamsky A, Kol A, Kreisel T, Doron A, Ozeri-Engelhard N, Melcer T, Refaeli R, Horn H, Regev L, Groysman M, London M, Goshen I, 2018. Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell 174, 59–71 e14. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, McCarthy KD, 2013. Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J Physiol 591, 5599–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL, 2007. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binning W, Hogan-Cann AE, Yae Sakae D, Maksoud M, Ostapchenko V, Al-Onaizi M, Matovic S, Lu WY, Prado MAM, Inoue W, Prado VF, 2020. Chronic hM3Dq signaling in microglia ameliorates neuroinflammation in male mice. Brain Behav Immun 88, 791–801. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL, 1994. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Chen G, Luo X, Qadri MY, Berta T, Ji RR, 2018. Sex-Dependent Glial Signaling in Pathological Pain: Distinct Roles of Spinal Microglia and Astrocytes. Neurosci Bull 34, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkuhler J, 2015. Selective activation of microglia facilitates synaptic strength. J Neurosci 35, 4552–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y, 2005. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. [DOI] [PubMed] [Google Scholar]
- Cserep C, Posfai B, Lenart N, Fekete R, Laszlo ZI, Lele Z, Orsolits B, Molnar G, Heindl S, Schwarcz AD, Ujvari K, Kornyei Z, Toth K, Szabadits E, Sperlagh B, Baranyi M, Csiba L, Hortobagyi T, Magloczky Z, Martinecz B, Szabo G, Erdelyi F, Szipocs R, Tamkun MM, Gesierich B, Duering M, Katona I, Liesz A, Tamas G, Denes A, 2020. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367, 528–537. [DOI] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ, 2014. Neuronal Hyperactivity Recruits Microglial Processes via Neuronal NMDA Receptors and Microglial P2Y12 Receptors after Status Epilepticus. J Neurosci 34, 10528–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Wu LJ, 2019. Microglia: Lifelong patrolling immune cells of the brain. Prog Neurobiol 179, 101614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, Fabisiak TJ, Anderson ND, Cheng K, Greene LI, Berkelhammer D, Zhang Y, Ellis AL, Yin HH, Campeau S, Rice KC, Roth BL, Maier SF, Watkins LR, 2016. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci U S A 113, E3441–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Wang X, Strand KA, Baratta MV, Zhang Y, Galer EL, Yin H, Maier SF, Watkins LR, 2018. DREADDed microglia in pain: Implications for spinal inflammatory signaling in male rats. Exp Neurol 304, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Eyo UB, Murugan M, Peng J, Matta S, Dong H, Wu LJ, 2016a. Microglial P2Y12 receptors regulate microglial activation and surveillance during neuropathic pain. Brain Behav Immun 55, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, Ren Y, DiCicco-Bloom E, Young W, Dong H, Wu LJ, 2016b. Spinal Microgliosis Due to Resident Microglial Proliferation Is Required for Pain Hypersensitivity after Peripheral Nerve Injury. Cell Rep 16, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui WS, Wei X, Mai CL, Murugan M, Wu LJ, Xin WJ, Zhou LJ, Liu XG, 2016. Interleukin-1beta overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol Pain 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Taniguchi T, 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6, 644–658. [DOI] [PubMed] [Google Scholar]
- Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S, 2001. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci 21, 1975–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilg AK, Enkel T, Bartsch D, Bahner F, 2018. Behavioral Effects of Acute Systemic Low-Dose Clozapine in Wild-Type Rats: Implications for the Use of DREADDs in Behavioral Neuroscience. Front Behav Neurosci 12, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Tsuda M, 2009. Microglia and neuropathic pain. Glia 57, 1469–1479. [DOI] [PubMed] [Google Scholar]
- Inoue K, Tsuda M, 2018. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci 19, 138–152. [DOI] [PubMed] [Google Scholar]
- Inoue N, Ito S, Tajima K, Nogawa M, Takahashi Y, Sasagawa T, Nakamura A, Kyoi T, 2009. Etodolac attenuates mechanical allodynia in a mouse model of neuropathic pain. J Pharmacol Sci 109, 600–605. [DOI] [PubMed] [Google Scholar]
- Jann MW, Lam YW, Chang WH, 1994. Rapid formation of clozapine in guinea-pigs and man following clozapine-N-oxide administration. Arch Int Pharmacodyn Ther 328, 243–250. [PubMed] [Google Scholar]
- Jendryka M, Palchaudhuri M, Ursu D, van der Veen B, Liss B, Kätzel D, Nissen W, Pekcec A, 2019. Pharmacokinetic and pharmacodynamic actions of clozapine-N-oxide, clozapine, and compound 21 in DREADD-based chemogenetics in mice. Sci Rep 9, 4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Chamessian A, Zhang YQ, 2016. Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Suter MR, 2007. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, Gao YJ, 2014. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov 13, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR, 2000. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20, 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato G, Inada H, Wake H, Akiyoshi R, Miyamoto A, Eto K, Ishikawa T, Moorhouse AJ, Strassman AM, Nabekura J, 2016. Microglial Contact Prevents Excess Depolarization and Rescues Neurons from Excitotoxicity. eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A, 2011. Physiology of microglia. Physiol Rev 91, 461–553. [DOI] [PubMed] [Google Scholar]
- Koga K, Kanehisa K, Kohro Y, Shiratori-Hayashi M, Tozaki-Saitoh H, Inoue K, Furue H, Tsuda M, 2017. Chemogenetic silencing of GABAergic dorsal horn interneurons induces morphine-resistant spontaneous nocifensive behaviours. Sci Rep 7, 4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb A, Weston M, Kullmann DM, 2019. Designer receptor technology for the treatment of epilepsy. EBioMedicine 43, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG, 2017. TNF-alpha Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J Neurosci 37, 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS, 2001. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22, 368–376. [DOI] [PubMed] [Google Scholar]
- Masuda T, Tsuda M, Yoshinaga R, Tozaki-Saitoh H, Ozato K, Tamura T, Inoue K, 2012. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep 1, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Kikuchi E, Lerchner W, Inoue KI, Ji B, Eldridge MA, Kaneko H, Kimura Y, Oh-Nishi A, Hori Y, Kato Y, Hirabayashi T, Fujimoto A, Kumata K, Zhang MR, Aoki I, Suhara T, Higuchi M, Takada M, Richmond BJ, Minamimoto T, 2016. PET imaging-guided chemogenetic silencing reveals a critical role of primate rostromedial caudate in reward evaluation. Nat Commun 7, 13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam MH, Han KS, Lee J, Won W, Koh W, Bae JY, Woo J, Kim J, Kwong E, Choi TY, Chun H, Lee SE, Kim SB, Park KD, Choi SY, Bae YC, Lee CJ, 2019. Activation of Astrocytic mu-Opioid Receptor Causes Conditioned Place Preference. Cell Rep 28, 1154–1166 e1155. [DOI] [PubMed] [Google Scholar]
- Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR, 2017. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 31, 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Z, Ma Y, Sun Y, Zheng G, Wang S, Xing R, Chen X, Han Y, Wang J, Lu QR, Zhao TJ, Chen Y, 2019. A GPR17-cAMP-Lactate Signaling Axis in Oligodendrocytes Regulates Whole-Body Metabolism. Cell Rep 26, 2984–2997 e2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB, 2013. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Gu N, Zhou L, Eyo UB, Murugan M, Gan WB, Wu LJ, 2016. Microglia and Monocytes Synergistically Promote the Transition from Acute to Chronic Pain after Nerve Injury Nat Commun 7, 12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Liu Y, Umpierre AD, Xie M, Tian DS, Richardson JR, Wu LJ, 2019. Microglial P2Y12 receptor regulates ventral hippocampal CA1 neuronal excitability and innate fear in mice. Mol Brain 12, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R, 2010. Interactions between the immune and nervous systems in pain. Nat Med 16, 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Torres R, 2009. Role of interleukin-1beta during pain and inflammation. Brain Res Rev 60, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saika F, Matsuzaki S, Kobayashi D, Ideguchi Y, Nakamura TY, Kishioka S, Kiguchi N, 2020. Chemogenetic Regulation of CX3CR1-Expressing Microglia Using Gi-DREADD Exerts Sex-Dependent Anti-Allodynic Effects in Mouse Models of Neuropathic Pain. Front Pharmacol 11, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloman JL, Scheff NN, Snyder LM, Ross SE, Davis BM, Gold MS, 2016. Gi-DREADD Expression in Peripheral Nerves Produces Ligand-Dependent Analgesia, as well as Ligand-Independent Functional Changes in Sensory Neurons. J Neurosci 36, 10769–10781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkuhler J, 2007. Understanding LTP in pain pathways. Mol Pain 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciolino NR, Plummer NW, Chen YW, Alexander GM, Robertson SD, Dudek SM, McElligott ZA, Jensen P, 2016. Recombinase-Dependent Mouse Lines for Chemogenetic Activation of Genetically Defined Cell Types. Cell Rep 15, 2563–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS, 2015. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 18, 1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, Takaoka A, Tanaka N, 2001. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 19, 623–655. [DOI] [PubMed] [Google Scholar]
- Tashima R, Mikuriya S, Tomiyama D, Shiratori-Hayashi M, Yamashita T, Kohro Y, Tozaki-Saitoh H, Inoue K, Tsuda M, 2016. Bone marrow-derived cells in the population of spinal microglia after peripheral nerve injury. Sci Rep 6, 23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban DJ, Roth BL, 2015. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol 55, 399–417. [DOI] [PubMed] [Google Scholar]
- Van Hove H, Antunes ARP, De Vlaminck K, Scheyltjens I, Van Ginderachter JA, Movahedi K, 2020. Identifying the variables that drive tamoxifen-independent CreERT2 recombination: Implications for microglial fate mapping and gene deletions. Eur J Immunol 50, 459–463. [DOI] [PubMed] [Google Scholar]
- Weston M, Kaserer T, Wu A, Mouravlev A, Carpenter JC, Snowball A, Knauss S, von Schimmelmann M, During MJ, Lignani G, Schorge S, Young D, Kullmann DM, Lieb A, 2019. Olanzapine: A potent agonist at the hM4D(Gi) DREADD amenable to clinical translation of chemogenetics. Sci Adv 5, eaaw1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Vadakkan KI, Kim SS, Wu LJ, Shang Y, Zhuo M, 2008. Selective activation of microglia in spinal cord but not higher cortical regions following nerve injury in adult mouse. Mol Pain 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XF, Alam MM, Liao Y, Huang T, Mathur R, Zhu X, Huang Y, 2019. Targeting Microglia Using Cx3cr1-Cre Lines: Revisiting the Specificity. eNeuro 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LJ, Peng J, Xu YN, Zeng WJ, Zhang J, Wei X, Mai CL, Lin ZJ, Liu Y, Murugan M, Eyo UB, Umpierre AD, Xin WJ, Chen T, Li M, Wang H, Richardson JR, Tan Z, Liu XG, Wu LJ, 2019. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep 27, 3844–3859 e3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Aryal DK, Olsen RH, Urban DJ, Swearingen A, Forbes S, Roth BL, Hochgeschwender U, 2016. Cre-dependent DREADD (Designer Receptors Exclusively Activated by Designer Drugs) mice. Genesis 54, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Wu G, Wu LJ, 2011. Neuronal and microglial mechanisms of neuropathic pain. Mol Brain 4, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CX3CR1creER/+:R26LSL-hM4Di/+ transgenic mice enable selective expression of Gi DREADD in microglia of the brain. Immunofluorescence images of the hippocampus and cortex labeled with mCitrine and HA in CX3CR1creER/+:R26LSL-hM4Di/+ mice after TM injection show expression of Gi DREADD. Co-staining of HA (red) with Iba1 (green) in brain cortex of CX3CR1creER/+:R26LSL-hM4Di/+ mice with TM indicates co-localization which is absent without TM injection. Scale bar, 40 μm. n=5 mice/group.
Figure S2. Activation of Gi DREADD in CX3CR1+ cells delays the development of chronic pain after SNT. (A) Timeline of experimental procedures for TM injection, pre-SNT CNO administration, SNT surgery, and pain behavioral test. (B, C) Measurement of mechanical hypersensitivity in WT mice with CNO (WT+CNO), CX3CR1creER/+:R26LSL-hM4Di/+ mice with vehicle (Gi DREADD+Vehicle), and CX3CR1creER/+:R26LSL-hM4Di/+ mice with CNO (Gi DREADD+CNO). Activation of Gi DREADD in CX3CR1+ cells following SNT showed that initiation of mechanical hypersensitivity was delayed when compared to control groups ipsilaterally (B), but no significant change was observed contralaterally (C). Data represented as mean ± SEM, n=5–8 mice/group, *** p < 0.001.
Figure S3. Activation of Gi DREADD in CX3CR1+ cells attenuates chronic pain after SNT. (A) Timeline of experimental procedures for TM injection, SNT surgery, post-SNT CNO administration, and pain behavioral test. (B) Mechanical hypersensitivity showing that post-SNT activation of Gi DREADD significantly reversed the pain behaviors ipsilaterally in Gi DREADD+CNO group when compared to control groups. (C) However, no difference was found between the three groups in the contralateral side. Data represented as mean ± SEM, n=8 mice/group, ** p < 0.01, *** p < 0.001.
Figure S4. Serial sections of Iba1 immunostaining in the SDH. Somatotopic distribution of Iba1+ cells in the ipsilateral spinal dorsal horn (L1–S1) from a Gi DREADD+Vehicle control group (left, n=3 mice) and Gi DREADD+CNO group (right, n=3 mice) at POD3 after SNT in CX3CR1creER/+:R26LSL-hM4Di/+ mice. Representative images are showing that the reduced number of microglia is mainly restricted to L4 after CNO treatment. Scale bar: 40 μm
Figure S5. SNT-induced BDNF upregulation not affected by microglial Gi DREADD activation. (A) Timeline of experimental procedures. (B) Representative Western blot images and quantification data showing that BDNF expression in the L4–5 level of the dorsal horn increased gradually at POD3–5 after SNT in Vehicle control group. n=4–5 mice/group, GAPDH was used as internal control. * p < 0.05, *** p < 0.001. (C) Pre-SNT activation of microglial Gi DREADD by CNO did not alter the BDNF upregulation seen in Gi DREADD+Vehicle control group. n=4–5 mice/group. GAPDH was used as internal control. * p < 0.05, *** p < 0.001.