

Summary
DNA-dependent protein kinase (DNA-PK), like all phosphatidylinositol 3-kinase-related kinases (PIKKs), is composed of conserved FAT and kinase domains (FATKIN) along with solenoid structures made of HEAT repeats. These kinases are activated in response to cellular stress signals, but the mechanisms governing activation and regulation remain unresolved. For DNA-PK, all existing structures represent inactive states with resolution limited to 4.3 Å at best. Here we report the cryoEM structures of DNA-PKcs (catalytic subunit) bound to a DNA end, or complexed with Ku70/80 and DNA, in both inactive and activated forms at resolutions of 3.7 Å overall, and 3.2 Å for FATKIN. These structures reveal the sequential transition of DNA-PK from inactive to activated forms. Most notably, activation of the kinase involves previously unknown stretching and twisting within individual solenoid segments and loosens DNA-end binding. This unprecedented structural plasticity of helical repeats may be a general regulatory mechanism of HEAT-repeat proteins.
Keywords: DNA-PKcs, Ku70, Ku80, PIKK, DNA-end binding
Graphical Abstract
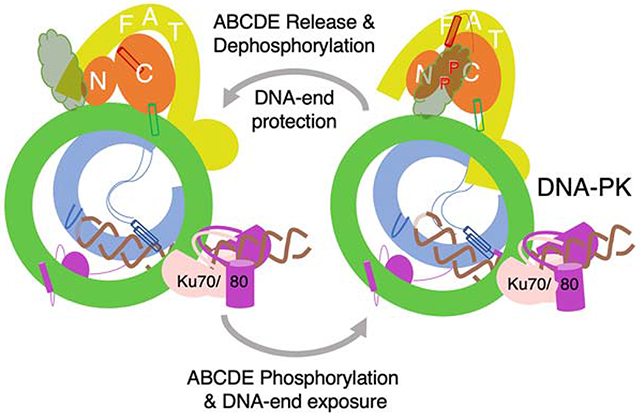
In Brief (eTOC blurb)
DNA-PK protects DNA ends when its ABCDE cluster is free and dephosphorylated, and coordinates DNA repair by non-homologous end joining (NHEJ). DNA-PK-DNA complex periodically toggles between kinase inactive and activated states. Linked by flexible HEAT repeats, autophosphorylation of DNA-PK allosterically exposes the DNA end to NHEJ repair factors.
Introduction
The DNA-dependent protein kinase (DNA-PK) is central to the process of non-homologous end joining of DNA (NHEJ) in both programmed gene arrangement and after unwanted DNA breakage (Davis et al., 2014). DNA-PK consists of a large catalytic subunit, known as DNA-PKcs, and the Ku70/80 heterodimer (Ku70/80) (Gottlieb and Jackson, 1993; Lees-Miller et al., 1990). DNA-PKcs is a Ser/Thr kinase of over 4000 residues and a member of the PI3K-related kinase (PIKK) family, which also includes mTOR, ATM, ATR and SMG1 (Baretic and Williams, 2014; Hartley et al., 1995). PIKK kinases play key roles in regulation of responses to nutrient stress (mTOR), misfolded and non-functional RNAs (SMG1), DNA double-strand breaks (DNA-PKcs and ATM) or single-stranded DNA (ATR) (Blackford and Jackson, 2017; Langer et al., 2020; Yang et al., 2017). The kinase activity of DNA-PKcs is modestly stimulated by DNA but becomes fully activated only in the presence of both DNA and Ku70/80, which is known to bind DNA ends (Chan and Lees-Miller, 1996; Hammarsten and Chu, 1998; Mimori et al., 1986; West et al., 1998). When activated, DNA-PK can phosphorylate both itself (auto-phosphorylation) and other repair factors (Meek et al., 2008). DNA-PKcs and Ku70/80 have also been implicated in telomere maintenance, RNA and ribosome biogenesis and the innate immune response to foreign DNAs (Hande, 2004; Meek, 2020; Shao et al., 2020).
Structures of the DNA-bound DNA-PK holoenzyme as well as DNA-PKcs associated with the C-terminal region (CTR) of Ku80 have been reported at 6.6 Å and 4.3-4.4 Å resolution, respectively (Baretic et al., 2019; Sharif et al., 2017; Sibanda et al., 2017; Yin et al., 2017). The first 3700 residues of DNA-PKcs are folded into ~65 α-helical HEAT repeats with a single β hairpin and arranged as an open N-HEAT (N for N-terminus) and a closed M-HEAT (M for middle) solenoid ring (Yin et al., 2017), which are nearly concentric, followed by the C-shaped FAT domain (Fig. 1A). The kinase domain at the C terminus occupies the hole in the FAT domain, and the two together, referred to as FATKIN (Baretic and Williams, 2014), form the “head” atop the doublering “body”. The helical repeats are folded sequentially, going in opposite directions in the consecutive N- and M-HEAT rings. These two rings are linked back-to-back at the neck between the head and body but are splayed apart on the opposite side (the bottom), where DNA and the Ku subunits bind (Yin et al., 2017). Upon DNA and Ku binding, the first half of N-HEAT undergoes a rotational movement and converts the half-circle N-HEAT to a nearly closed ring (Yin et al., 2017) (Fig. 1, Video 1). The shape of DNA-PK and locations of domains were clearly defined, but at the resolution of 4.3 – 6.6 Å, functional interfaces between protein domains and between protein and DNA were less certain.
Figure 1.
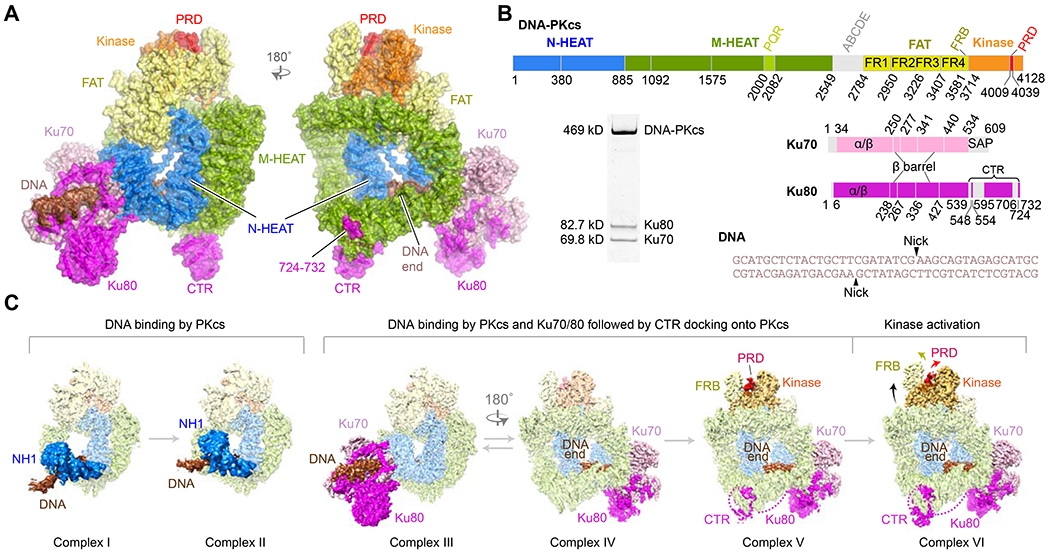
Structure characterization of DNA-PK complexed with DNA.
(A) Front and back views of the holo DNA-PK complex VI. Major parts are labeled and color coded.
(B) Diagrams of DNA-PKcs, Ku, and DNA sequences in the DNA-PK holo complexes. Quality of the protein sample is shown in the Coomassie-blue stained SDS gel.
(C) The cryoEM density maps of DNA-PK complexes I – VI and how they are related. Regions that differ among these complexes are highlighted in colors shown in 1a, while regions unchanged are shown in muted colors. Complexes I-III are shown in back view as the DNA changes the most among them, and complexes IV-VI are shown in the front view to illustrate the changes in Ku80-CTR binding and the activation of the kinase.
Multiple structures of other PIKK kinases complexed with accessory subunits, activation cofactors, ATP analogs, and even a substrate peptide have been reported, representing either active mTOR and SMG1 (Langer et al., 2020; Yang et al., 2017) or inhibited ATM and ATR (Jansma et al., 2020; Williams et al., 2020). In all cases the role of the HEAT-repeat structures, which are 5 to 9-fold larger than the kinase domain, remains unclear. Among PIKKs, DNA-PK is unique in its autonomous full activation by a short DNA duplex and auto-phosphorylation of its three subunits.
Here we report eight cryoEM structures of DNA-PKcs complexed with DNA or with DNA and Ku70/80 at 3.2 - 4.3 Å resolutions. These structures reveal the previously unknown function of DNA-end binding by DNA-PKcs, and in addition, an activated state of DNA-PK in association with the DNA end. Binding of Ku and a DNA end drive the intrinsic plasticity of DNA-PKcs toward kinase activation with coordinated stretching and twisting of helical repeats. The Slinky-like movement observed with DNA-PKcs may be general among HEAT-repeat proteins.
Results and Discussion
cryoEM structures of DNA-PK
DNA-PKcs purified from HeLa cell nuclear extract was mixed with Ku70/80 and a 40 bp DNA that contained identical blunt ends with two internal nicks (Fig. 1B). Formation of the complete DNA-PK complex was confirmed by size-exclusion chromatography. CryoEM analysis of DNA-PK (see Materials and Methods) revealed three major species of protein-DNA complexes: DNA-PKcs-DNA (two variants: complexes I and II), and inactive (complexes III, IV, and V) and activated holo-DNA-PK complex VI (Figs. 1C, S1). The FATKIN domains of the inactive and activated DNA-PK complexes were locally refined to 3.2 - 3.3 Å resolution (Table S1).
The two DNA-PKcs-DNA complex structures (I and II) at 4.3 Å resolution showed that DNA-PKcs alone can bind a DNA broken end and contact 15 bp. Complex I represented an initial DNA-binding state with DNA at the periphery of DNA-PKcs, whose protein structure was similar to a DNA-free DNA-PKcs (Sharif et al., 2017) (Fig. S2A), which was isolated from a sample containing DNA and Ku by cryoEM (Sharif et al., 2017). The structure of complex II showed that DNA and the associated N-HEAT region moved into the DNA-binding groove between N- and M-HEAT. The next three complexes (III to V) had DNA bound to DNA-PK in the inactive state at 4.1 to 3.9 Å resolutions. Ku80 was increasingly resolved: only its core domain was clear in complex III and IV, while the CTR became resolved in complex V (Fig. 1C). Structural differences between complexes III and IV probably reflected the dynamic flexibility of DNA-bound DNA-PK. With Ku80-CTR firmly docked, the structure of complex V became more stable. Interactions between DNA-PKcs and DNA were similar to those in complexes I and II, but specific contacts to the DNA end were formed in complexes III-V. As compared to the 6.6 Å DNA-PK structure (PDB: 5Y3R) (Yin et al., 2017), the protein and DNA compositions and overall arrangement are similar, but in complexes III-V DNA-PKcs is expanded by over 12 Å lengthwise, and the DNA end extends 2 bp beyond that in the 6.6 Å structure and directly contacts DNA-PKcs (Fig. S2B–C). Furthermore, we have corrected misassignments of the DNA-PKcs structure and located Ku80-CTR (see details below).
The activated DNA-PK complex (VI, at 3.7 Å resolution) exhibited extensive conformational changes. Compared to inactive complexes, the FATKIN head was raised, and the N- and C-lobes of kinase domain became open (Fig. 1C). Moreover, the PIKK regulatory domain (PRD, aa 4009-4039), which is partially disordered in mTOR and SMG1 (Langer et al., 2020; Yang et al., 2017), and closed in ATM, ATR (Jansma et al., 2020; Williams et al., 2020), and in all other DNA-PKcs and DNA-PK complexes, rotated 115° to expose the ATP- and substrate-binding groove. Concomitant to the protein movement, the DNA end is pulled out from DNA-PKcs by ~2 Å.
Structural comparison with apo DNA-PKcs
As our cryoEM density maps were of moderate resolution, to improve the structures and also for comparison, we took advantage of methionine locations well defined by selenium replacement of sulfur in the crystal structure of DNA-PKcs (PDB: 5LUQ, 4.3 Å) (Sibanda et al., 2017). In the process of building cryoEM models, we were able to re-refine the DNA-PKcs crystal structure (Table S2) (Fig. S2D–F), resulting in reduced R and Rfree (each by ~10%), reduced clash score and B values, and increased favorable Ramachandran distributions (Table S2). Re-assignment of secondary structures in DNA-PKcs was necessary to optimize this refinement (Fig. S3). The largest change was a 130-aa shift in the middle of the M-HEAT ring and involved over 200 residues in the crystal structure (PDB: 5LUQ) (Sibanda et al., 2017). Because the mis-assigned crystal structure was included in the 6.6 Å cryoEM structure (PDB: 5Y3R) (Yin et al., 2017), the corrections are also applicable to that work.
Domain and subdomain boundaries were determined by comparison of apo, inactive, and activated DNA-PKcs structures using Difference Distance Matrix Plot (DDMP) (Richards and Kundrot, 1988) (Fig. S4). According to established conventions (Sibanda et al., 2017; Yin et al., 2017), the three regions of DNA-PKcs are: N-HEAT (1-872), M-HEAT (890-2580), and FATKIN (2800-4128) (Fig. 2A–C). N-HEAT is broken into two segments (NH1 and NH2), each containing 8 helical repeats. NH1 (aa 1-380) binds 14 bp of DNA and interacts with Ku, while NH2 (aa 381-872) caps the DNA end and directly contacts M-HEAT and FATKIN (Fig. 2A). M-HEAT contains 32 helical repeats and assumes a heart shape in three segments (Fig. 2B). MH1 (aa 890-1092) forms domain connections at the neck; MH2 (aa 1093-1575) contacts NH2 and the N lobe of kinase; the large MH3 (aa 1576-2580) binds Ku and DNA and closes the M-HEAT ring (Fig. 2B). Based on DDMP analyses, M-HEAT, particularly MH3, is least changed through the series of DNA-PKcs structures (Fig. S4).
Figure 2.
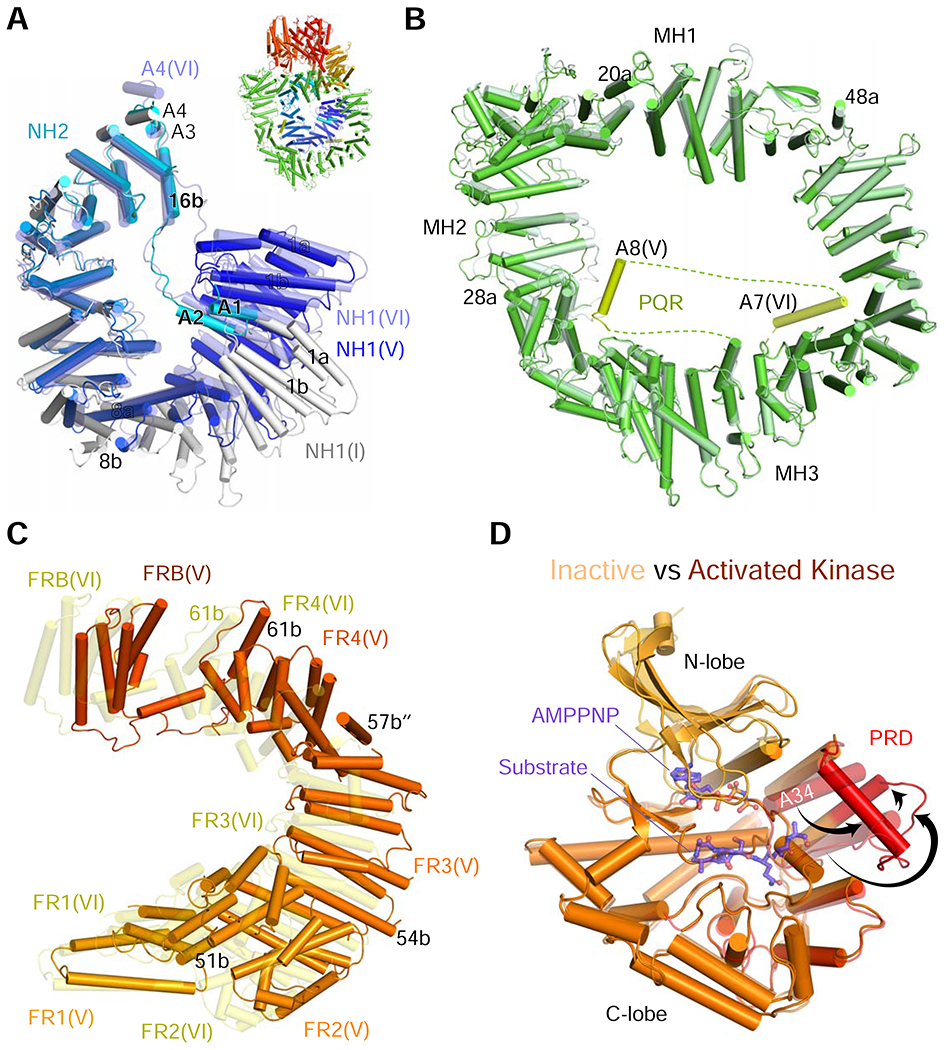
The DNA-PKcs structure.
(A) The N-HEAT structures of Complex I (light grey), V (gradient colors of dark blue to cyan for residues 1 to 872), and VI (semi-transparent slate blue). The overall structure of DNA-PKcs is inserted as a reference.
(B) M-HEAT structures in complexes V (solid green) and VI (semi-transparent pale green) are superimposed. MH2 (at the 10 o’clock position) is most changed between the two. Helices A7 (V) and A8 (VI) of PQR are highlighted in lime color.
(C) FAT in complex V (solid orange to red) and VI (semi-transparent yellow) are superimposed at FR1. FAT is stretched between FR2 and FRB in complex VI (activated).
(D) Kinase domain in complex V (semi-transparent) and VI (solid orange) are superimposed by the C lobe. AMPNPP and the substrate peptide (shown in semi-transparent purple sticks) are borrowed from the SMG1 structure (PDB: 6Z3R) after superimposing the conserved kinase domain. PRD in complex VI (highlighted in red) is open (as indicated by black arrows). The closed PRD in complex V would clash with the modeled substrate peptide.
The FAT domain starts at residue 2800, but the first segment FR1 (aa 2800-2944) actually moves with MH3 (Fig. 2C). The next segment FR2 (aa 2950-3199) contains non-HEAT-repeat helices and interacts extensively with N- and M-HEAT at the neck, where the C lobe of kinase is also docked. The last section FR4 (3407-3564) interacts with the FRB (FKBP12-rapamycin-binding) domain and the N lobe of kinase, but FR3 (aa 3226 – 3394) between FR2 and FR4 has few interactions with the rest of DNA-PKcs. The conserved kinase domain (aa 3714 - 4128) (Fig. 2D) includes FATC (aa 4000-4128) as an intrinsic part of the C-lobe, as noted previously (Yang et al., 2013).
DNA binding by DNA-PKcs
Previously, Ku80 was implicated in direct DNA-end binding by DNA-PK. However, the helices near the DNA end, initially assigned to Ku80 (Sibanda et al., 2017; Yin et al., 2017), actually belong to MH3 (as A7 in PQR, see explanation below) in the re-refined crystal structure and complexes I to VI (Fig. 2B). Only DNA-PKcs has direct contacts with the DNA end and the proximal 15 bp. This observation is supported by reports that DNA-PKcs by itself has weak DNA-end binding activity (Cary et al., 1997; West et al., 1998) and that its kinase activity is slightly activated by DNA in the absence of Ku (Chan and Lees-Miller, 1996; Hammarsten and Chu, 1998).
In the two DNA-PKcs-DNA structures (I and II), only 24 bp of DNA are ordered (Fig. 3A). The 5th to 15th bp of DNA are bound by five helical repeats (3rd to 7th, aa 161-331) of NH1 around the minor groove (Fig. 3B). Meanwhile the DNA end is contacted by the 9th to 11th helical repeats (aa 401-524) of NH2 in both complexes (Fig. 3C). In converting from complex I to II, the DNA and NH1 move together by a rotation of 20°, which leads to a 10-Å shift from their being at the periphery of the M-HEAT ring to its center (Video 2). As the DNA moves to the center, a pair of α helices (A1 and A2) (Fig. S3) extends from the last repeat of N-HEAT (16th) toward it, with one side contacting DNA across the major groove and the other side contacting the 1st and 2nd repeats of N-HEAT (Fig. 2A, 3B).
Figure 3.
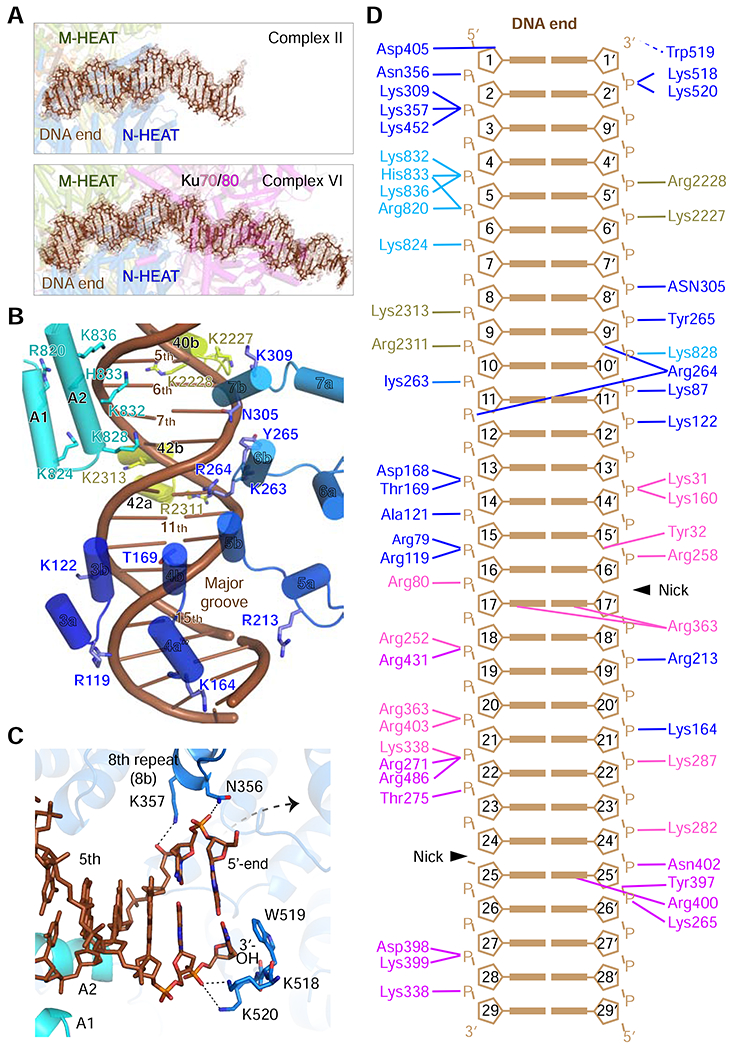
DNA binding by DNA-PK.
(A) Representative density maps of the DNA in complexes II and VI were contoured color at 5.5 and 5.0 σ, respectively, in brown. Ku stabilizes the DNA in complexes III-VI.
(B) Interactions between the DNA and NH1, the extension of the 16th helical repeat (A1, A2) and MH3 in complexes II-VI. Helices A1 and A2 are not docked onto DNA in complex I. Residues of DNA-PKcs at the interface with DNA are shown as sticks and labeled.
(C) In complexes III-VI, DNA-PKcs recognizes both DNA strands and caps the 3′ end specifically. Accommodation of a 5’-overhang is indicated by a dashed arrow.
(D) Diagram of the complete DNA interactions made by DNA-PKcs and Ku in complex VI.
Upon Ku binding and thus holo-DNA-PK formation (complexes III to VI), 37 of the 40 bp of DNA became traceable in these four structures, as Ku bound DNA next to DNA-PKcs. In the transition from without (I-II) to with Ku (III-V), the entire DNA and the associated N-HEAT moved toward M-HEAT and also outward from the center of the concentric ring by a 12° rotation and 8 Å translation, as if an arrow (DNA) were pulled (by Ku) from the bow (M-HEAT) (Video 3). In complexes III-V, the last helical repeat of NH1 (8th) finally makes contact with the DNA end, and in it N356, K357, and the positive helical dipole coordinate the 2nd and 3rd phosphates from the DNA 5′ end (Fig. 3C). The 5′ end, however, appears to be free. In contrast, the DNA 3′-OH is covered by W519, and the adjacent phosphates are bound by K518 and K520 (Fig. 3C). For the strand to continue as in a hairpin end, it has to bend away from the protein cap made of K518-K520. The asymmetric binding of 5′ and 3′ ends suggests that DNA-PKcs can easily accommodate a 5′ overhang of considerable length (Fig. 3C).
The interactions of internal DNA with DNA-PKcs become more extended when the holo-DNA-PK complex shifts toward the activated state. As the groove between the N- and M-HEAT rings that sandwich the DNA becomes narrower, the interface between DNA-PKcs and DNA increases from 918 Å2 in complex V to 1036 Å2 in complex VI. Previously disordered loops (aa 801-817 and 839-846 of the 16th helical repeat in N-HEAT) become ordered and contact M-HEAT at aa 2430-2511 (Fig. 3B). In addition to the protein-DNA interactions observed in complexes I-V, MH3 directly contacts the 4th - 5th bp via K2227, and the 9th - 10th bp via R2311 in complex VI (Fig. 3D). As the DNA is pushed against MH3 inside the HEAT rings, it is bent by ~20° away from M-HEAT and toward N-HEAT at the interface between DNA-PKcs and Ku. The nick between the 16th and 17th bp in our DNA is adjacent to the bend. Analysis of auto-phosphorylation and phosphorylation of Ku subunits by DNA-PKcs revealed that the nicked DNA stimulated the kinase activity at least 3-fold more than intact DNA of the same sequence (Fig. S5). Therefore, the nicked 40 bp DNA likely favored the activated state of complex VI. Interestingly, the internal bending of the DNA in the activated state (VI) pulled the DNA end away from its cap, NH2, by ~2 Å (Video 4, see the functional implication below).
Ku stabilizes DNA-PKcs-DNA interaction
Ku is situated outside of DNA-PKcs and contacts the NH1 of N-HEAT and the MH3 of M-HEAT, thus bridging two sides of the DNA binding groove in DNA-PKcs (Fig. 4A). Ku in the DNA-PK holoenzyme has essentially the same structure as in Ku-DNA complexes (Nemoz et al., 2018; Walker et al., 2001). Superimposition of the Ku80 structures with and without DNA-PKcs resulted in an RMSD of 1.5 Å over 500 pairs of Cα atoms. The N-terminal α/β domain of Ku70, which barely contacts DNA in the Ku-DNA complexes, rotates 28° relative to the rest of Ku protein and binds MH3 (aa 2350 – 2420) and DNA (14th – 16th bp) tightly in complexes III-VI (Fig. 4B). The broad Ku70/80 base (composed of α/β and β barrel domains) becomes an extension of the MH3 of DNA-PKcs in binding DNA, protecting 15 bp (14th to 28th) (Fig. 3D, 4A–B). While maintaining similar DNA interactions as Ku alone, the bridges of Ku70/80 contact the 3rd to 5th helical repeats of NH1 on the side opposite to the DNA (Fig. 4C). Therefore, Ku stabilizes the DNA-PKcs-DNA association not only by extending DNA binding, but also by fastening N-HEAT of DNA-PKcs onto DNA.
Figure 4.
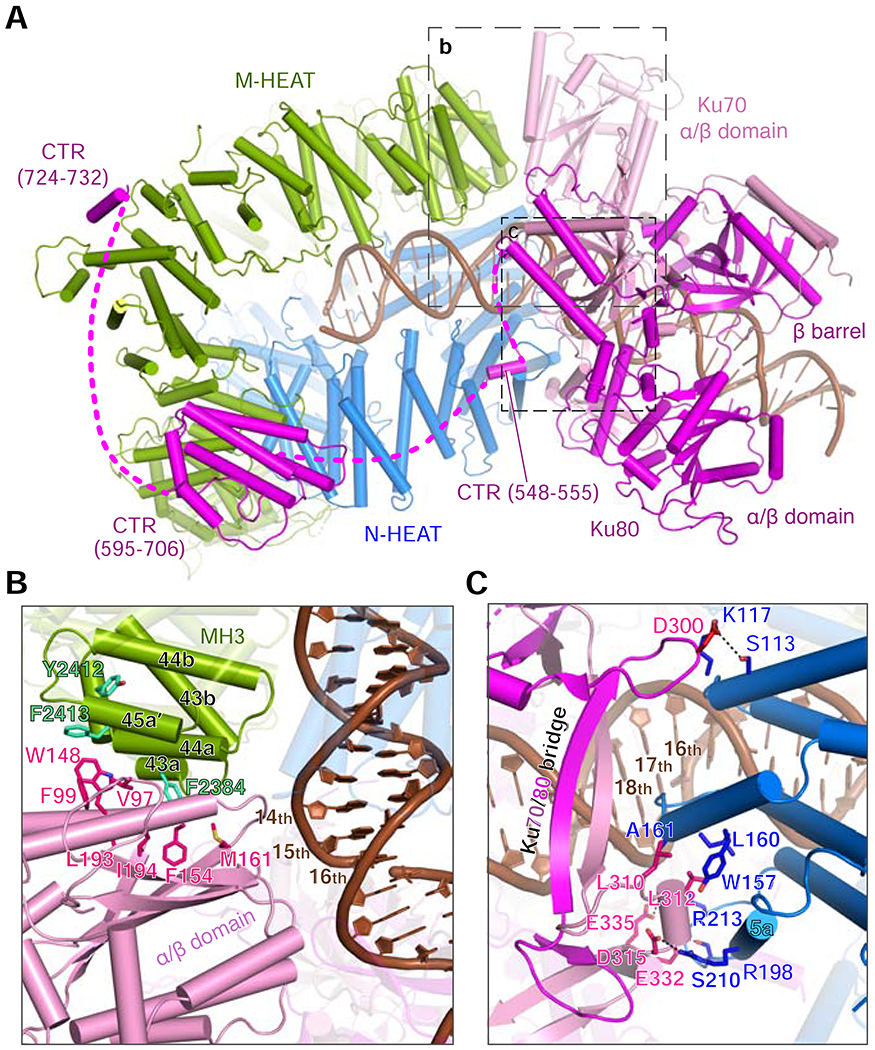
Ku association with DNA and DNA-PKcs.
(A) A bottom view of the DNA-PK holo-complex (VI is used as an example).
(B) The α/β domain of Ku70 interacts with DNA and MH3 of DNA-PKcs. Hydrophobic residues forming the interface are shown as sticks and labeled.
(C) The bridges of Ku70/80, which binds the DNA major groove, interact with NH1 (3rd to 5th helical repeat), which interacts with the adjacent minor groove. The interacting residues are shown as sticks and labeled.
While the first 30 residues and the C-terminal SAP domain of Ku70 (aa 535-609) remain undetectable as in other Ku structures, Ku80-CTR (aa 542-732) becomes traceable in complex V and VI structures (Fig. 1C). The folded domain in CTR (aa 595-706) is docked onto MH3 (aa 1700-1830), 65 Å away from the bulk of Ku, by a small interface of both hydrophobic and polar nature. In addition, a short helix of Ku80 (aa 548 - 555) is tucked between the 5th and 6th helical repeats of NH1 adjacent to the bridge and pillar of Ku70 (Fig. 4C). Selenium labeling of M731 (Sibanda et al., 2017) led to unequivocal identification of the last helix of Ku80 (aa 718-732), which is the only part of Ku80 present in the crystal structure of DNA-PKcs (Sibanda et al., 2017) (Fig. S2D–E). The last Ku80 helix is docked at aa 1910-1970 of MH3 60Å away from the folded CTR, resulting in Ku contacting all four corners of the DNA-PKcs DNA-end binding groove in complexes V and VI (Fig. 4A).
Although Ku is well known for its DNA end-binding activity, the cryoEM structures of complexes III-VI reveal that Ku does not specifically bind DNA ends. The apparent DNA-end recognition is probably due to Ku’s closed topology, which requires it to bind DNA only via an open end by threading. Once on DNA, Ku has no direct contact with either the 3′ or 5′ end and can slide along following the DNA contour. The stable and hydrophobic interaction between Ku80’s last helix and DNA-PKcs indicates that DNA-PK holoenzyme may form prior to DNA-end binding (Sibanda et al., 2017). As a holoenzyme, DNA-PK would find DNA ends more efficiently than either component alone. The closed topology of DNA-PK, however, requires first loading of Ku onto a broken DNA end.
Phosphorylation sites in DNA-PKcs
Residues 2581-2783 include the well-known auto-phosphorylation sites of DNA-PKcs, the ABCDE cluster (aa 2609-2647) (Ding et al., 2003; Neal et al., 2016). This 220-aa stretch was partially built in the apo crystal structure (PDB: 5LUQ and 7K17) inside the M-HEAT ring extending toward the center, where DNA would bind. In cryoEM maps of complexes I-VI, there is no density to support ABCDE inside the M-HEAT ring. Instead, a relatively featureless density approximating the volume of the missing 200 residues was found outside of the M-HEAT ring and adjacent to the substrate binding groove of the kinase domain. We suspect that this density represents the mobile ABCDE region (Fig. S6) and speculate that the displacement of ABCDE from inside to outside the M-HEAT ring is correlated with DNA-end binding. Being near the kinase active site in the complexes with DNA, ABCDE may be phosphorylated in cis and facilitate downstream events in NHEJ (Crowe et al., 2020).
The second phosphorylation cluster (aa 2023-2056), known as PQR (Cui et al., 2005; Neal et al., 2016), is most peculiar. Among HEAT repeats of DNA-PKcs, PQR is the only part that does not follow a sequential folding pattern (Fig. 2B). As compounded by disordered linkers between helices, PQR caused the largest mis-tracing in the crystal structure (Sibanda et al., 2017). Instead of being near helix A7 (aa 2000- 2018) and the 37th helical repeat (Fig. S3), helix A8 (aa 2037-2045) of PQR is located 55 Å away and adjacent to the 32nd and 33rd helical repeats (aa 1575-1640) (Fig. 2B). The linker between A7 and A8 contains only 20 residues, rather short for the 55-60 Å separating the two. Interestingly, helices A7 and A8 co-exist in the crystal structure with A7 shifted 5 Å towards A8, but in each of our six cryoEM structures, either A7 or A8 is found, but not both. Helix A8 is dominant in four out of the five inactive complexes, whereas in the activated DNA-PK complex (VI) helix A7 is found in 75% of the population. Although disordered, the linkers inside of the M-HEAT ring have to cross paths with the DNA end and parts of N-HEAT. Although phosphorylation sites in the PQR cluster remain disordered, their locations suggest trans phosphorylation (as it is 90 Å away from the cis kinase domain) and potential influence on binding of DNA ends and repair partners inside the M-HEAT ring.
Other phosphorylation sites of DNA-PKcs (except for S3205) are in ordered regions, and their functions can be rationalized. S72 is adjacent to the bridges of Ku; its phosphorylation would interfere with interactions of DNA-PKcs with Ku and consequently destabilize DNA binding by DNA-PK. Indeed, phosphorylation of S72 has been reported to inactivate the kinase (Neal et al., 2011). T946 and S1003, whose phosphorylation has no effect on the kinase activity but inhibits the NHEJ pathway, may have additive effects with the neighboring ABODE cluster in repair partner choices (Neal et al., 2011; Neal et al., 2016). T3950, which belongs to the activation loop of the kinase (Douglas et al., 2007), is buried in the available structures, and has to undergo conformational changes to be phosphorylated and modulate DNA-PK function.
Activation of the kinase
In the activated state (complex VI), the position of FATKIN relative to N- and M-HEAT and its internal structure have changed dramatically (Video 4) (Fig. 5A). In contrast, among complexes I to V, FATKIN moves little and only as a rigid body (Fig. S4). The telltale sign of the activated state was the rotation of the PRD loop outward by 115° and opening of the binding groove for ATP and kinase substrate (Fig. 2D). Unlike mTOR and SMG1, when ATP or non-hydrolyzable ATP analogs were added to our sample, the DNA-PK complexes became highly heterogeneous and structural characterization was not possible. In the absence of ATP or ATP analogs, however, both the inactive and activated forms of DNA-PK were present in our samples. Complex VI also revealed a potential binding site for inositol hexaphosphate (IP6), which has been shown to activate NHEJ (Hanakahi et al., 2000). An IP6-binding site was first found in SMG1 and was predicted to be general among PIKKs, but had been absent in the existing DNA-PKcs structures (Gat et al., 2019), all of which represent inactive states, as do complexes I to V. In the activated complex VI, a positively charged putative IP6-binding site emerges between FAT and kinase domains (Fig. 5B).
Figure 5.
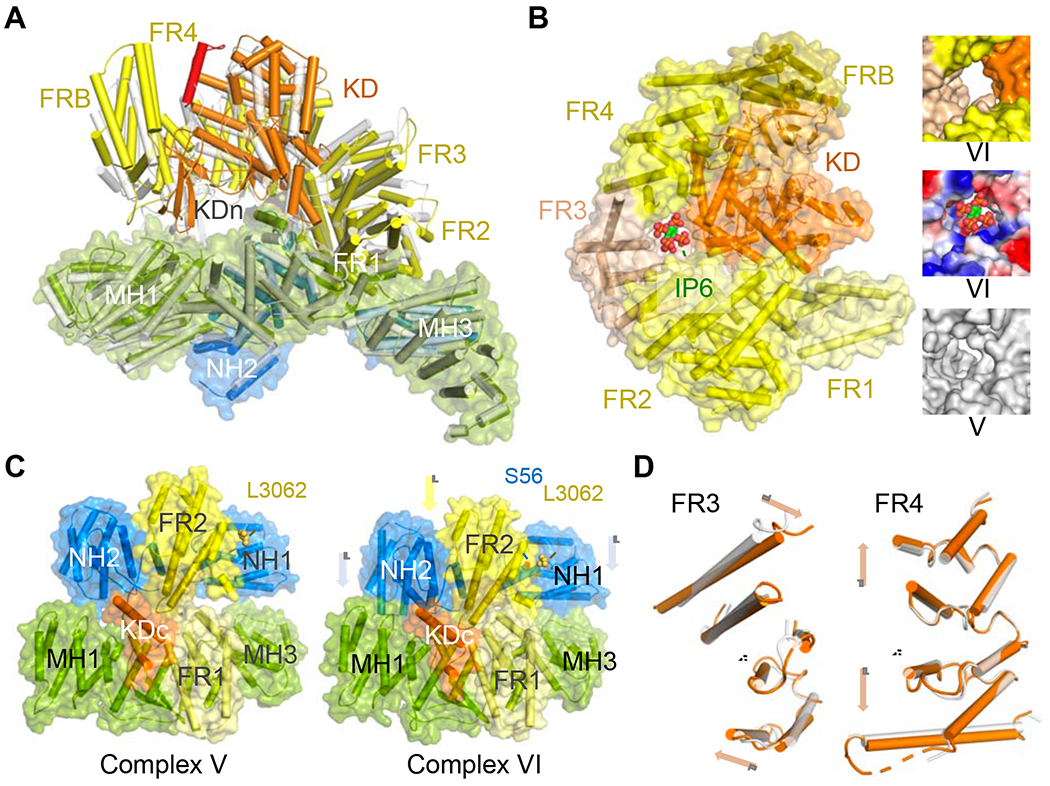
Activation of DNA-PK.
(A) The FATKIN “head” in complex VI (multicolor) is raised relative to the body (covered molecular surface) compared with complex V (semi-transparent grey cartoon).
(B) In complex VI, a positively charged pocket potentially binding IP6 is formed between the kinase domain (KD) and FR3. The pocket doesn’t exist in complex V as shown in the insert.
(C) During transition from complex V and VI, the N-HEAT ring with FR2 moves toward the M-HEAT ring (MH1, MH3 and FR1), where the kinase domain (aa 3882-3916) is docked. S56 contacts FR2 (near L3062) only in the activated state (complex VI).
(D) Comparison of FAT solenoid between complex V and VI. When one helix in FR3 or FR4 (in dashed circle) is superimposed, the surrounding helices twist (in FR3) or stretch (in FR4) in opposite directions as marked by arrowheads.
We asked how DNA and Ku activate the kinase activity of DNA-PKcs and what kinds of structural changes in the HEAT repeat rings are associated with this activation. Conformational changes from complex V to VI appear to emanate in the neck, where N- and M-HEAT, FAT and the C-lobe of the kinase converge. As shown in the animation (Video 4), the NH1 and FR2 segments move toward each other to close the N-HEAT ring, and the entire ring including FR2 moves downward and enters the M-HEAT ring (Fig. 5C). Meanwhile, helix A4, linking N-HEAT to M-HEAT (Fig. S3), is squeezed out of the neck and rises up 4Å toward the kinase. Adjacent to helix A4, the C-lobe of kinase rises as well, together with FAT except for the FR2 segment, giving the impression of a raised FATKIN head (Fig. 5A). As the C-lobe is at the C-terminus of DNA-PKcs, but physically adjacent to FR2, the upward movement of kinase and downward movement of FR2 lead to unusual changes of every portion between them (Video 4). As it is pulled in opposite directions, FR3 changes the twists between its helical repeats, and FR4 expands by increasing the distance between helical repeats (Fig. 5D). The solenoid made of FR3 and FR4 becomes more extended and more twisted in the activated state (Video 5). As these changes happen, the PRD loop pops open.
Like the effect of a DNA end on the conformational changes of DNA-PK, binding of the GTPase RHEB to mTORC1 leads to a similar squeezing motion of the extension of the kinase C-lobe (the neck) as shown in Fig. 5C (Video 6) (Yang et al., 2017). The “neck” connecting the activator-binding regions made of HEAT-repeats and FATKIN is also found in ATM, ATR and SMG1 (Jansma et al., 2020; Langer et al., 2020; Williams et al., 2020). We suspect the role of the “neck” in PIKK kinase activation to be conserved. The importance of FR2 in kinase activation is further supported by the observation that phosphorylation of S56 in NH1, which interfaces with FR2 in the activated state (Fig. 5C), causes kinase inactivation (Neal et al., 2011). Most interestingly, a point mutation isolated from a radiosensitive SCID patient, L3062R, is located in the interface of FR2 with NH1 in the activated state (Fig. 5C, S7), which explains the defects of this mutant in DNA-PK activation, NHEJ and DNA-PKcs-dependent DNA hairpin opening by Artemis (van der Burg et al., 2009).
The dramatic changes in FATKIN are supported by changes in the two HEAT rings. As the N-HEAT ring enters, the M-HEAT ring expands (Video 5), again by changing the distances and twisting angles between helical repeats. In the activated state, the N and C lobes of kinase also rotate away from each other, making room for substrate binding. This bidirectional movement appears to be correlated with the expansion of M-HEAT, which maintains interactions with the N-lobe of kinase by changing contacting residues.
DNA-PKcs alone is rather flexible, as evidenced in the differences between non-crystallographic symmetry mates (Sibanda et al., 2017) (Fig. S2E). But the intrinsic dynamics of DNA-PKcs are insufficient to re-arrange and activate the kinase. Binding of a DNA end leads to the re-organization of N-HEAT and brings NH1 and FR2 much closer than in apo structures, as shown in complex II. The closeness of NH1 and FR2 may explain the low kinase activity of DNA-PKcs in the presence of DNA (Chan and Lees-Miller, 1996). Binding of Ku70 and the entirety of Ku80 stabilizes the DNA-PKcs-DNA complex and shifts the ensemble of DNA-PKcs structures towards kinase activation.
A model of DNA-PK in NHEJ
Whether Ku alone or together with DNA-PKcs first finds a DNA end, DNA mediates and strengthens interactions between Ku70 and DNA-PKcs, which in turn leads to interactions between the folded domain of Ku80 CTR with DNA-PKcs (Fig. 4A). The full DNA-mediated contact between Ku70/80 and DNA-PKcs as observed in complex V leads to DNA-PKcs activation. Although DNA-PK primarily protects the DNA end, for NHEJ to take place it has to yield the DNA end for processing by nucleases and DNA polymerases and ligation by Ligase IV in complex with XRCC4, XLF and possibly PAXX (Zhao et al., 2020). Single-molecule microscopic analyses indicate that both end processing and ligation take place in the presence of DNA-PK (Graham et al., 2016; Wang et al., 2018). In the process, DNA-PKcs and Ku may be pushed inward along DNA, and XLF appears to stabilize the weakened DNA-PK interaction with DNA.
Autophosphorylation of ABCDE by DNA-PKcs is essential for NHEJ (Block et al., 2004; Crowe et al., 2020; Jiang et al., 2015; Reddy et al., 2004). Interestingly, phosphorylation sites in ABCDE are interchangeable and no one particular site is recognized for recruitment of NHEJ repair factors. When the DNA end is fully inserted into DNA-PKcs as in complex I-VI (Fig. S2C), the ABCDE cluster is positioned adjacent to the kinase substrate-binding groove (Fig. S6). Inhibition of ABCDE autophosphorylation blocks the DNA-end processing and repair by NHEJ (Block et al., 2004; Crowe et al., 2020; Cui et al., 2005; Reddy et al., 2004; van der Burg et al., 2009). During transition from the inactive (V) to the activated state (VI), the DNA end is pulled slightly away from DNA-PKcs (Video 4). We therefore propose that (1) the proximity of ABCDE to the kinase active site makes it a favorable target of auto-phosphorylation; (2) during active phosphorylation the DNA end becomes more exposed than in the inactive states; and (3) repeated autophosphorylation of the ABCDE cluster periodically exposes the DNA end and allows NHEJ factors to access the end for processing and ligation (Fig. 6). This model predicts that for the ABCDE to remain a substrate of autophosphorylation, it has to be regenerated by a phosphatase. Indeed, phosphatase PP6 has been shown to target DNA-PKcs (Douglas et al., 2014; Hosing et al., 2012; Mi et al., 2009), and knocking out PP6 leads to clear defects in NHEJ and to radiosensitivity (Dziegielewski et al., 2020).
Figure 6.
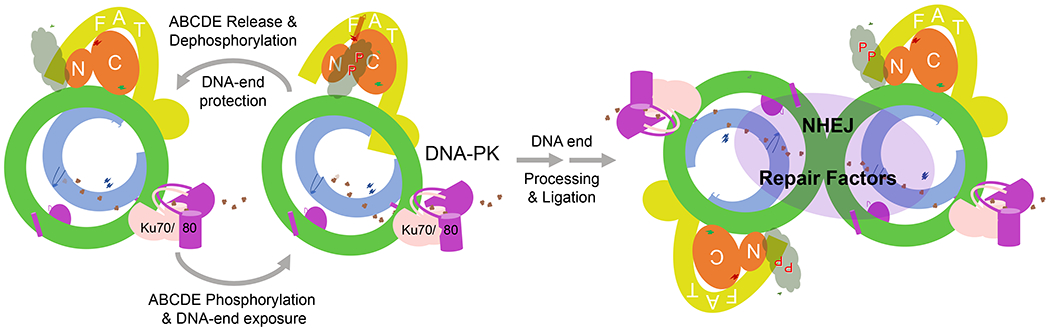
A model of DNA-PK in NHEJ
DNA-PK protects the DNA end (open or hairpin) and toggles between inactive and activated states. Autophosphorylation of the ABCDE cluster by DNA-PKcs allosterically loosens its grip on the DNA end. In the inactive state with ABCDE released from FATKIN, DNA-PK tightens its DNA-end binding again. Dephosphorylation regenerates the ABCDE cluster for repeated autophosphorylation. The result is periodic exposure of the DNA end for processing by NHEJ factors, which can take place in a single DNA-PK-DNA complex. When two DNA ends are complementary, ligation may take place between two linked DNA-PK-DNA complexes as depicted in the right panel.
Comparison with the latest DNA-PK structures
While this manuscript was under review, cryoEM structures of apo DNA-PKcs at 3.24 Å resolution and DNA-PK complexed with DNA at 3.9 Å resolution or below were published (Chaplin et al., 2020). The latest apo DNA-PKcs structure has corrected misassignments in the 4.3 Å crystal structure (PDB: 5LUQ) and is superimposable with our DNA-PKcs structures except for small local differences. In the Chaplin et al DNA-PK-DNA complex structures, the end of DNA is 4-5 bp away from the center of DNA-PKcs and appears “shorter” than ours as well as that in the 6.6 Å structure (PDB: 5Y3R) (Fig. S2C). The not fully inserted DNA end may explain why the ABCDE cluster remains inside of the M-HEAT ring as in the apo DNA-PKcs structures. Most interestingly, Chaplin et al. reported a dimeric DNA-PK structure, in which the last helix of Ku80 is domain swapped between two DNA-PKcs subunits. Domain swapped Ku80 C-terminal helices likely are the key for juxtaposing two DNA-PK-associated DNA ends for ligation.
Conclusion
The extensive global changes of DNA-PKcs that culminate in its activated state are results of local stretch and twist of HEAT repeats. Such movements within individual solenoids may be a common theme among HEAT-repeat proteins. For example, the fantastic shape changes of nuclear transport karyopherins exhibit local stretch and twist of helical repeats among large rigid-body conformational changes (Conti et al., 2006; Cook and Conti, 2010). We speculate that Slinky-like structural plasticity may be general among the vast number of HEAT proteins, PIKKs, karyopherins, condensins and cohesins alike.
Limitations
Although the resolution of our structures is sufficient to allow chain tracing of almost the whole DNA-PK-DNA, some parts are not as well resolved. In particular, the ABCDE cluster (aa 2609-2647), whose phosphorylation is important to DNA-PK’s role in progression of DNA repair by NHEJ, is poorly resolved and only its general location was identified.
STAR METHODS
RESOURCES AVAILABILITY
Lead Contact
Email contact for further information and reagent and resource sharing: weiy@niddk.nih.gov.
Materials Availability
All reagents generated in this study are available upon request from the Lead Contact without restriction.
Data and Code Availability
The structures and cryoEM maps have been deposited with EMDB and PDB with accession code of 7K19, 7K1B, 7K1J, 7K1K, 7K1N, 7K0Y, 7K11, 7K10 and EMD-22622, EMD-22623, EMD-22624, EMD-22625, EMD-22626, EMD-22618, EMD-22620, EMD-22619. The re-refined crystal structure has been deposited in PDB with accession code of 7K17. These data will be released immediately upon publication.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Growth conditions
The HEK293T cells were grown in Freestyle™ 293 Expression Medium (Thermo Fishier, Gibco) supplemented with 1% Fetal Bovine Serum (Thermo Fishier, Gibco) at 37°C in a shaker supplied with 5% CO2. 1 mg expression plasmids were mixed with 4 mg of polyethylenimine (Polysciences) in 35 ml of Hybridoma medium (Thermo Fishier, Gibco) to transfect 1 L of HEK293T cells grown in the suspension culture when the cell density reached 1.5 million/L.
METHOD DETAILS
Protein and DNA purification
DNA-PKcs was purified from HeLa cells (purchased from National Cell Culture Center, Minneapolis, MN) using a protocol we developed in 2009 based on a published method (Chan and Lees-Miller, 1996; Williams et al., 2008). The nuclear extracts were prepared according to the standard protocol (Abmayr et al., 2006) and then fractionated with 60% saturated ammonium sulfate. The precipitate was dissolved in DEAE loading buffer (50 mM HEPES pH 7.9, 75 mM KCI, 5% glycerol, 1 mM EDTA and 1 mM DTT) and clarified before loading onto a 200mL DEAE Sepharose FF column (GE Healthcare) pre-equilibrated with the loading buffer. DNA-PKcs fractions eluted from the DEAE column (in a gradient of 75-300 mM KCI) were pooled and diluted to a salt concentration of 150 mM KCI before loading onto a HiTrap Heparin HP column (GE Healthcare). A linear gradient of 150-500 mM KCI was applied to elute the protein, and the DNA-PKcs fractions were further purified using a Mono Q 10/100 GL anion exchange column (GE Healthcare) and eluted with a linear gradient of 150 to 350 mM KCI. The final purification step was on a Superose 6 10/300 GL size exclusion column (GE Healthcare) pre-equilibrated with 50 mM HEPES pH 7.9, 300 mM KCI, and 1 mM DTT. The purified DNA-PKcs protein was buffer exchanged to 50 mM HEPES pH 7.9, 300 mM KCl, 50% glycerol and 1 mM DTT, flash frozen in liquid nitrogen and stored at −80°C. All protein purification steps (including Ku purification described below) were carried out at 4°C, and protease inhibitors (100 mM PMSF, 1 mM pepstatin, 10 mg/mL aprotinin, 5 mg/mL leupeptin) were added to the buffer before each step of chromatography.
Ku70/80 protein was over-expressed and purified from HEK293T cells. Genes encoding full length Ku70 and Ku80 were cloned into pLEXm vector separately, and a His6-MBP tag and PreScission cleavage site were added to the N-terminus of Ku70 (Kim et al., 2015). HEK293T cells were pelleted 3 days after transfection and resuspended in 1/10 culture volume of lysis buffer (20 mM HEPES pH 7.9, 0.5M KCl, 5% glycerol, 0.5mM EDTA, 1 mM DTT and 1 tablet of Roche cocktail protease inhibitors). After sonication and centrifugation at 35,000 g for 1h, the clear supernatant was applied to an amylose affinity column. After thorough wash, Ku protein was eluted in 20 mM HEPES pH 7.9, 0.5 M KCl, 5% glycerol, 40 mM maltose, 0.5 mM EDTA and 1 mM DTT. After removal of the N-terminal His6-MBP tag by PreScission Protease, the protein was loaded onto a Mono Q 10/100 GL anion exchange column (GE Healthcare) in 20 mM HEPES pH 7.9, 100 mM KCl, 1 mM DTT, 0.5 mM EDTA. Ku protein was eluted in a linear gradient of 100-500 mM KCl. The purified Ku70/80 fraction was buffer-exchanged into 20 mM HEPES pH 7.9, 150 mM KCl, 50% glycerol, 1 mM EDTA, 1 mM DTT, flash frozen in liquid nitrogen and stored at −80°C.
DNA oligos of 24 nt (5′-GCATGCTCTACTGCTTCGATATCG-3′), 16 nt (5′-AAGCAGTAGAGCATGC-3′) and 40 nt (5′-GCATGCTCTACTGCTTCGATATCGAAGCAGTAGAGCATGC-3′) were purchased from IDT (Integrated DNA Technologies, Coralville, IA) and purified using an 8–15% TBE-urea PAGE gel in small gel cassettes (Life Technologies). The oligonucleotides extracted from gel were loaded onto a Glen Gel-Pak column (Glen Research) and eluted with deionized H2O. The purified 24 nt and 16 nt oligos were annealed in a buffer containing 20 mM Tris-HCl (pH 8.0), 50 mM NaCl and 0.5 mM EDTA in a Thermocycler to form a 40 bp nicked DNA (40N) with identical blunt ends and two internal nicks (Fig. 1B). 40N DNA was initially designed as a cheap and easy way to make symmetric DNA of various length for co-crystallization with DNA-PK. The self-complementary 40 nt oligo was annealed as described above to form the intact 40 bp DNA (40I).
Sample preparation and cryo-EM data collection
To assemble DNA-PK holoenzyme, purified DNA-PKcs, Ku70/80 and 40 bp DNA were mixed at the molar ratio of 1:1.2:1.2 in 50 mM HEPES pH 7.9, 100 mM KCl and 1mM DTT, and incubated at 4°C for 15 min. The mixture was purified over a Superose 6 10/300 GL column (GE Healthcare) pre-equilibrated with 50 mM HEPES 7.9, 100 mM KCl, 1mM DTT. Protein and DNA components of the holoenzyme were confirmed by SDS and TBE-urea PAGE gels. Fractions containing DNA-PK holoenzyme were pooled and concentrated to 0.5mg/ml for cryo-EM grid preparation.
The DNA-PK sample was loaded on either QUANTIFOIL R1.2/1.3 (Cu, 300 mesh) or Lacey grids (UC-A on holey 400 mesh Cu), 3 ul per grid at 100% humidity and 4°C in a Vitrobot, blotted for 4 s, and flash-frozen in liquid ethane. A total of 14742 micrographs from QUANTIFOIL grids were collected on the Titan Krios electron microscope operated at 300 kV at the Multi-Institute Cryo-EM Facility (MICEF) of NIH in the super-resolution mode of 130k nominal magnification (calibrated pixel size of 0.54 A, corresponding to 1.08 Å at the sample level) (Mastronarde, 2005). An additional 7730 micrographs (4298 from QUANTIFOIL and 3432 from Lacey grids) were collected on a Titan Krios electron microscope operated at 300 kV at the Frederick National Laboratory (Frederick, MD) in the counting mode with a nominal magnification of 175K (calibrated pixel size of 0.86 Å). Lacey grids alleviated the preferred orientation problems manifested with QUANTIFOIL grids.
Structure determination and model refinement
The software MotionCor2 (Zheng, 2017) was used for drift correction, during which dose-weighting was applied and the pixel size was binned to 1.16 Å/pixel to merge all micrographs from the two microscopes. CTF (contrast transfer function) estimation was measured with the dose-unweighted micrographs using Gctf (Zhang, 2016). 2,945,665 particles were picked on dose-weighted micrographs using Gautomatch (developed by K. Zhang; https://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch) and extracted with RELION-3.0.8 (Fernandez-Leiro, 2017; Scheres, 2012) using a box size of 352 × 352 pixels. An initial map was obtained with cryoSPARC (Punjani, 2017), and two-dimensional (2D) projections were generated for template-based particle picking. The re-picked 5,592,709 particles were subjected to 2D classifications in RELION-3.0.8. After excluding 3,258,196 bad particles, three-dimension (3D) classifications in RELION and cryoSPARC with and without alignment were applied to classify different conformations. The resulting good maps and the associated particles from 3D classifications were selected for further classifications and refinements according to the standard procedure (Fig. S1). All reported resolutions were determined based on the “gold standard” of the 0.143 Fourier shell correlation (FSC) criterion (Swint-Kruse, 2005). Local resolution was estimated using ResMap (Kucukelbir et al., 2014). For model building, we used the published 4.3-Å resolution apo DNA-PKcs structure (PDB: 5LUQ) (Sibanda et al., 2017) as the initial model to build cryo-EM structures of the 3.2 Å inactive FATKIN and the 3.7 Å DNA-PK complex VI, which then were used as the template for building Complexes I-V. We first fit the coordinates into the cryo-EM maps using Chimera (Pettersen et al., 2004), and then manually adjusted and rebuilt the models according to the cryo-EM density in COOT (Emsley, 2010). Real-space refinement in Phenix (Adams et al., 2010) was used to refine the models, and MolProbity (Chen et al., 2010) was used to validate the final model. The refinement statistics are summarized in Table S1. The detailed classifications and map qualities of the 8 structures reported in this manuscript are shown in a supplemental figure (Fig. S1).
For comparison and validation of our cryoEM structures of DNA-PK, we also rebuilt and refined the DNA-PKcs crystal structure (Sibanda et al., 2017) using the diffraction data deposited with the Protein Data Bank. Using our 3.7 Å cryoEM DNA-PK complex VI structure as the initial model, we got a solution by Molecular Replacement and rebuilt the DNA-PKcs structure against the 2 Fo-Fc as well as Se anomalous maps in COOT. The model was iteratively refined using strategies of rigid body, group B factor, TLS parameters, XYZ (reciprocal-space and real-space), NCS application and secondary structure restraint in Phenix. MolProbity was used to validate the final model. A total of 7293 residues were built per asymmetric unit, which included 3629 and 3646 residues of DNA-PKcs in chain A and chain B, respectively, and the C-terminal residues 724-732 of Ku80 in chain C and chain D. Rwork and Rfree of the re-refined structure of DNA-PKcs complexed with the C-terminal helix of Ku80 were 0.29 and 0.34 at 4.3 Å, respectively. The detailed refinement statistics are summarized in Table S2.
Kinase activity assay
To compare DNA-PK auto-phosphorylation activity in the presence of either intact or nicked 40 bp DNA, 50 nM DNA-PKcs, 60 nM Ku70/80, 60 nM DNA and 0.1 mM ATP (containing radioactive ATP, [γ-32P]) were mixed in 25 mM Tris-HCl pH 7.4, 100 mM KCl, 2 mM DTT, 0.1 mM EDTA and 10 mM MgCl2 at 37°C. Reaction products were collected at different time points and mixed with equal volume of 2X SDS-PAGE loading buffer (100 mM Tris-Cl pH 6.8, 4% (w/v) SDS, 0.2% (w/v) bromophenol blue, 20% (v/v) glycerol, 200 mM DTT) to stop the reaction. Radioactively phosphorylated DNA-PKcs and Ku70/80 were separated on 3%-8% SDS-PAGE and visualized on a Typhoon Phosphorlmager. The phosphorylated DNA-PKcs bands were quantified using ImageQuant NL (GE Healthcare). Average of triplicate measurements are shown in Fig. S5.
QUANTIFICATION AND STATISTICAL ANALYSIS
Kinase activity assay
In Figure S5, the experiments were performed with biological samples. In Figure S5B, the band intensity for each time point was shown as means and standard deviation (S.D.) of triplicates.
ADDITIONAL RESOURCES
Titan Krios electron microscopes
The microscopes belong to Multi-Institute Cryo-EM Facility (MICEF) of NIH (Bethesda, MD) and the Frederick National Laboratory (Frederick, MD).
Supplementary Material
Video 1. Morphing of DNA-PKcs from apo (crystal structure, chain A, PDB: 7K17) to its complex with Ku and DNA (complex V), related to Figure 1. The DNA-PKcs structure is shown in rainbow-colored (from blue N to red C terminus) cartoon diagram. The NH1 segment of N-HEAT lifts up to bind DNA. Both the upper “torso” of the N- and M-HEAT rings and the FATKIN head lift a little too.
Video 2. Movement of NH1 and DNA when complex I becomes complex II, related to Figure 3. DNA-PKcs is shown in cartoon diagram with domains in the same colors as in Fig. 1, blue N-HEAT, green M-HEAT, yellow FAT and FRB and orange and red kinase domain. The DNA is shown as brown tube-and-ladder. Only NH1 and DNA move in this movie.
Video 3. Movement of DNA and DNA-PKcs upon association with Ku, related to Figure 4. The protein and DNA are shown in the same scheme as in Movie 2.
Video 4. Activation of holo-DNA-PK complex, related to Figure 5. DNA-PKcs and DNA are shown as in Movies 2 and 3. Ku70 and Ku80 subunits are shown in pink and magenta cartoon diagrams. FR2 of FAT domain (yellow) moves downward and closes the N-HEAT ring, and together they enter the M-HEAT ring. Helical repeats in N-HEAT move counterclockwise, while M-HEAT expands in the opposite direction between the 9 and 10 o’clock position. The movement of N- and M-HEAT along with the DNA is stabilized by Ku. These movements support the changes in the FATKIN head. Along with the rise of helix A4 between the N- and M-HEAT ring, the head is raised and PRD (colored red) rotates 115° and opens the substrate-binding groove in the kinase domain.
Video 5. Movement of the M-HEAT ring and FAT during DNA-PK activation, related to Figure 5. Because there are many moving parts in Movie 4, M-HEAT and FAT (aa 885-3713) are shown here (without N-HEAT and kinase domain) in a cyan to orange color gradient. Different segments in these two domains move in different directions. The M-HEAT ring moves as if it were pinched at ~ 10 o’clock. FR1 (immediately after M-HEAT) moves upward, FR2 next to it (and behind) moves downward, and FR3, FR4 and FBR extend in a spiral.
Video 6. Activation of mTORC1 by RHEB GTPase, related to Figure 5. RHEB (magenta) binds between the N-HEAT (blue) and M-HEAT (green). The large movement of N-HEAT induced by RHEB binding pulls the FAT (yellow/orange) domain. The movement of FAT and shift of FR2 equivalent (yellow, with a Glu sidechain) toward the “neck” of kinase C lobe is analogous to the activation of DNA-PKcs.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human DNA-PKcs (full-length) | This paper | Residues 1-4128 |
| Human Ku70/80 | This paper | Residues 1-609 and 1-732 |
| Deposited Data | ||
| Re-refined apo-DNA-PKcs, X-ray (4.3 Å) | (Sibanda et al., 2017) | PDB: 7K17 |
| DNA-PKcs-DNA complex I, cryoEM (4.3 Å) | This paper | PDB: 7K19 EMDB: 22622 |
| DNA-PKcs-DNA complex II, cryoEM (4.3 Å) | This paper | PDB: 7K1B EMDB: 22623 |
| DNA-PK-DNA complex III, cryoEM (3.9 Å) | This paper | PDB: 7K1J EMDB: 22624 |
| DNA-PK-DNA complex IV, cryoEM (4.1 Å) | This paper | PDB: 7K1K EMDB: 22625 |
| DNA-PK-DNA complex V, cryoEM (3.9 Å) | This paper | PDB: 7K1N EMDB: 22626 |
| DNA-PK-DNA complex VI, cryoEM (3.7 Å) | This paper | PDB: 7K0Y EMDB: 22618 |
| Inactive FATKIN, cryoEM (3.2 Å) | This paper | PDB: 7K11 EMDB: 22620 |
| Active FATKIN, cryoEM (3.3 Å) | This paper | PDB: 7K10 EMDB: 22619 |
| Experimental Models: Cell Lines | ||
| HeLa-S3 cells | National Cell Culture Center | https://cellculturecompany.com/national-cell-culture-center/ |
| HEK293T cells | ATCC | CRL-3216 |
| Oligonucleotides | ||
| See DNA preparation in STAR Methods | This paper | N/A |
| Recombinant DNA | ||
| Ku70 in pLEXm plasmid | This paper | pWY2799 |
| Ku80 in pLEXm plasmid | This paper | pWY2802 |
| Software and Algorithms | ||
| PyMol | Schrodinger | https://pymol.org/2/ |
| Prism-8 | GraphPad Software | https://www.graphpad.com/ |
| CCP4 | CCP4 | http://www.ccp4.ac.uk |
| PHENIX | (Adams et al., 2010) | https://www.phenix-online.org |
| COOT | (Emsley, 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| cryoSPARC | (Punjani et al., 2017) | https://cryosparc.com/ |
| RELION | (Scheres, 2012); (Fernandez-Leiro and Scheres, 2017) | https://www2.mrc-lmb.cam.ac.uk/relion |
| SerialEM | (Mastronarde, 2005) | https://bio3d.colorado.edu/SerialEM/ |
Highlights.
Structure of activated DNA-PK differs significantly from inactive forms.
DNA-PKcs, not Ku, is responsible for recognition and binding of a DNA end.
Ku stabilizes the DNA-binding groove of DNA-PKcs and covers additional DNA.
Stretch and twist of HEAT repeats links DNA-end binding to the activation of kinase.
Acknowledgements
We are grateful to Drs. K. Meek and D. Leahy for critical reading of the manuscript. The research is supported by the NIH intramural research grants to M.G. (DK036167), W.Y. (DK036147).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing interest.
References
- Abmayr SM, Yao T, Parmely T, and Workman JL (2006). Preparation of nuclear and cytoplasmic extracts from mammalian cells. Curr Protoc Mol Biol Chapter 12, Unit 12 11. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretic D, Maia de Oliveira T, Niess M, Wan P, Pollard H, Johnson CM, Truman C, McCall E, Fisher D, Williams R, et al. (2019). Structural insights into the critical DNA damage sensors DNA-PKcs, ATM and ATR. Prog Biophys Mol Biol 147, 4–16. [DOI] [PubMed] [Google Scholar]
- Baretic D, and Williams RL (2014). PIKKs--the solenoid nest where partners and kinases meet. Curr Opin Struct Biol 29, 134–142. [DOI] [PubMed] [Google Scholar]
- Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Block WD, Yu Y, Merkle D, Gifford JL, Ding Q, Meek K, and Lees-Miller SP (2004). Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Res 32, 4351–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary RB, Peterson SR, Wang J, Bear DG, Bradbury EM, and Chen DJ (1997). DNA looping by Ku and the DNA-dependent protein kinase. Proc Natl Acad Sci U S A 94, 4267–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DW, and Lees-Miller SP (1996). The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem 271, 8936–8941. [DOI] [PubMed] [Google Scholar]
- Chaplin AK, Hardwick SW, Liang S, Kefala Stavridi A, Hnizda A, Cooper LR, De Oliveira TM, Chirgadze DY, and Blundell TL (2020). Dimers of DNA-PK create a stage for DNA double-strand break repair. Nat Struct Mol Biol. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti E, Muller CW, and Stewart M (2006). Karyopherin flexibility in nucleocytoplasmic transport. Curr Opin Struct Biol 16, 237–244. [DOI] [PubMed] [Google Scholar]
- Cook AG, and Conti E (2010). Nuclear export complexes in the frame. Curr Opin Struct Biol 20, 247–252. [DOI] [PubMed] [Google Scholar]
- Crowe JL, Wang XS, Shao Z, Lee BJ, Estes VM, and Zha S (2020). DNA-PKcs phosphorylation at the T2609 cluster alters the repair pathway choice during immunoglobulin class switch recombination. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, and Meek K (2005). Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol 25, 10842–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AJ, Chen BP, and Chen DJ (2014). DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 17, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, and Meek K (2003). Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol 23, 5836–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Cui X, Block WD, Yu Y, Gupta S, Ding Q, Ye R, Morrice N, Lees-Miller SP, and Meek K (2007). The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol Cell Biol 27, 1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Ye R, Trinkle-Mulcahy L, Neal JA, De Wever V, Morrice NA, Meek K, and Lees-Miller SP (2014). Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci Rep 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewski J, Bonkowska MA, Poniecka EA, Heo J, Du K, Crittenden RB, Bender TP, Brautigan DL, and Larner JM (2020). Deletion of the SAPS1 subunit of protein phosphatase 6 in mice increases radiosensitivity and impairs the cellular DNA damage response. DNA Repair (Amst) 85, 102737. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Leiro R, and Scheres SHW (2017). A pipeline approach to single-particle processing in RELION. Acta Crystallogr D Struct Biol 73, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gat Y, Schuller JM, Lingaraju M, Weyher E, Bonneau F, Strauss M, Murray PJ, and Conti E (2019). InsP6 binding to PIKK kinases revealed by the cryo-EM structure of an SMG1-SMG8-SMG9 complex. Nat Struct Mol Biol 26, 1089–1093. [DOI] [PubMed] [Google Scholar]
- Gottlieb TM, and Jackson SP (1993). The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72, 131–142. [DOI] [PubMed] [Google Scholar]
- Graham TG, Walter JC, and Loparo JJ (2016). Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Mol Cell 61, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarsten O, and Chu G (1998). DNA-dependent protein kinase: DNA binding and activation in the absence of Ku. Proc Natl Acad Sci U S A 95, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanakahi LA, Bartlet-Jones M, Chappell C, Pappin D, and West SC (2000). Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell 102, 721–729. [DOI] [PubMed] [Google Scholar]
- Hande MP (2004). DNA repair factors and telomere-chromosome integrity in mammalian cells. Cytogenet Genome Res 104, 116–122. [DOI] [PubMed] [Google Scholar]
- Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, Admon A, Lees-Miller SP, Anderson CW, and Jackson SP (1995). DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 82, 849–856. [DOI] [PubMed] [Google Scholar]
- Hosing AS, Valerie NC, Dziegielewski J, Brautigan DL, and Larner JM (2012). PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. J Biol Chem 287, 9230–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansma M, Linke-Winnebeck C, Eustermann S, Lammens K, Kostrewa D, Stakyte K, Litz C, Kessler B, and Hopfner KP (2020). Near-Complete Structure and Model of Tel1ATM from Chaetomium thermophilum Reveals a Robust Autoinhibited ATP State. Structure 28, 83–95 e85. [DOI] [PubMed] [Google Scholar]
- Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, Li C, Lee BJ, Dubois RL, Liu C, Yu X, et al. (2015). Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol Cell 58, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Lapkouski M, Yang W, and Gellert M (2015). Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 518, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, and Tagare HD (2014). Quantifying the local resolution of cryo-EM density maps. Nat Methods 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer LM, Gat Y, Bonneau F, and Conti E (2020). Structure of substrate-bound SMG1-8-9 kinase complex reveals molecular basis for phosphorylation specificity. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Chen YR, and Anderson CW (1990). Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol 10, 6472–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Meek K (2020). An Antiviral DNA Response without the STING? Trends Immunol 41, 362–364. [DOI] [PubMed] [Google Scholar]
- Meek K, Dang V, and Lees-Miller SP (2008). DNA-PK: the means to justify the ends? Adv Immunol 99, 33–58. [DOI] [PubMed] [Google Scholar]
- Mi J, Dziegielewski J, Bolesta E, Brautigan DL, and Larner JM (2009). Activation of DNA-PK by ionizing radiation is mediated by protein phosphatase 6. PLoS One 4, e4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori T, Hardin JA, and Steitz JA (1986). Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders. J Biol Chem 261, 2274–2278. [PubMed] [Google Scholar]
- Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, and Meek K (2011). Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol 31, 1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Xu Y, Abe M, Hendrickson E, and Meek K (2016). Restoration of ATM Expression in DNA-PKcs-Deficient Cells Inhibits Signal End Joining. J Immunol 196, 3032–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoz C, Ropars V, Frit P, Gontier A, Drevet P, Yu J, Guerois R, Pitois A, Comte A, Delteil C, et al. (2018). XLF and APLF bind Ku80 at two remote sites to ensure DNA repair by non-homologous end joining. Nat Struct Mol Biol 25, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14. [DOI] [PubMed] [Google Scholar]
- Reddy YV, Ding Q, Lees-Miller SP, Meek K, and Ramsden DA (2004). Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J Biol Chem 279, 39408–39413. [DOI] [PubMed] [Google Scholar]
- Richards FM, and Kundrot CE (1988). Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins 3, 71–84. [DOI] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Flynn RA, Crowe JL, Zhu Y, Liang J, Jiang W, Aryan F, Aoude P, Bertozzi CR, Estes VM, et al. (2020). DNA-PKcs has KU-dependent function in rRNA processing and haematopoiesis. Nature 579, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H, Li Y, Dong Y, Dong L, Wang WL, Mao Y, and Wu H (2017). Cryo-EM structure of the DNA-PK holoenzyme. Proc Natl Acad Sci U S A 114, 7367–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibanda BL, Chirgadze DY, Ascher DB, and Blundell TL (2017). DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 355, 520–524. [DOI] [PubMed] [Google Scholar]
- Swint-Kruse L, and Brown CS (2005). Resmap: automated representation of macromolecular interfaces as two-dimensional networks. Bioinformatics 21, 3327–3328. [DOI] [PubMed] [Google Scholar]
- van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ, et al. (2009). A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest 119, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, and Goldberg J (2001). Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412, 607–614. [DOI] [PubMed] [Google Scholar]
- Wang JL, Duboc C, Wu Q, Ochi T, Liang S, Tsutakawa SE, Lees-Miller SP, Nadal M, Tainer JA, Blundell TL, et al. (2018). Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nat Struct Mol Biol 25, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RB, Yaneva M, and Lieber MR (1998). Productive and nonproductive complexes of Ku and DNA-dependent protein kinase at DNA termini. Mol Cell Biol 18, 5908–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Lee KJ, Shi J, Chen DJ, and Stewart PL (2008). Cryo-EM structure of the DNA-dependent protein kinase catalytic subunit at subnanometer resolution reveals alpha helices and insight into DNA binding. Structure 16, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RM, Yates LA, and Zhang X (2020). Structures and regulations of ATM and ATR, master kinases in genome integrity. Curr Opin Struct Biol 61, 98–105. [DOI] [PubMed] [Google Scholar]
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, and Pavletich NP (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, and Pavletich NP (2013). mTOR kinase structure, mechanism and regulation. Nature 497, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Liu M, Tian Y, Wang J, and Xu Y (2017). Cryo-EM structure of human DNA-PK holoenzyme. Cell Res 27, 1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Rothenberg E, Ramsden DA, and Lieber MR (2020). The molecular basis and disease relevance of non-homologous DNA end joining. Nat Rev Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Morphing of DNA-PKcs from apo (crystal structure, chain A, PDB: 7K17) to its complex with Ku and DNA (complex V), related to Figure 1. The DNA-PKcs structure is shown in rainbow-colored (from blue N to red C terminus) cartoon diagram. The NH1 segment of N-HEAT lifts up to bind DNA. Both the upper “torso” of the N- and M-HEAT rings and the FATKIN head lift a little too.
Video 2. Movement of NH1 and DNA when complex I becomes complex II, related to Figure 3. DNA-PKcs is shown in cartoon diagram with domains in the same colors as in Fig. 1, blue N-HEAT, green M-HEAT, yellow FAT and FRB and orange and red kinase domain. The DNA is shown as brown tube-and-ladder. Only NH1 and DNA move in this movie.
Video 3. Movement of DNA and DNA-PKcs upon association with Ku, related to Figure 4. The protein and DNA are shown in the same scheme as in Movie 2.
Video 4. Activation of holo-DNA-PK complex, related to Figure 5. DNA-PKcs and DNA are shown as in Movies 2 and 3. Ku70 and Ku80 subunits are shown in pink and magenta cartoon diagrams. FR2 of FAT domain (yellow) moves downward and closes the N-HEAT ring, and together they enter the M-HEAT ring. Helical repeats in N-HEAT move counterclockwise, while M-HEAT expands in the opposite direction between the 9 and 10 o’clock position. The movement of N- and M-HEAT along with the DNA is stabilized by Ku. These movements support the changes in the FATKIN head. Along with the rise of helix A4 between the N- and M-HEAT ring, the head is raised and PRD (colored red) rotates 115° and opens the substrate-binding groove in the kinase domain.
Video 5. Movement of the M-HEAT ring and FAT during DNA-PK activation, related to Figure 5. Because there are many moving parts in Movie 4, M-HEAT and FAT (aa 885-3713) are shown here (without N-HEAT and kinase domain) in a cyan to orange color gradient. Different segments in these two domains move in different directions. The M-HEAT ring moves as if it were pinched at ~ 10 o’clock. FR1 (immediately after M-HEAT) moves upward, FR2 next to it (and behind) moves downward, and FR3, FR4 and FBR extend in a spiral.
Video 6. Activation of mTORC1 by RHEB GTPase, related to Figure 5. RHEB (magenta) binds between the N-HEAT (blue) and M-HEAT (green). The large movement of N-HEAT induced by RHEB binding pulls the FAT (yellow/orange) domain. The movement of FAT and shift of FR2 equivalent (yellow, with a Glu sidechain) toward the “neck” of kinase C lobe is analogous to the activation of DNA-PKcs.
Data Availability Statement
The structures and cryoEM maps have been deposited with EMDB and PDB with accession code of 7K19, 7K1B, 7K1J, 7K1K, 7K1N, 7K0Y, 7K11, 7K10 and EMD-22622, EMD-22623, EMD-22624, EMD-22625, EMD-22626, EMD-22618, EMD-22620, EMD-22619. The re-refined crystal structure has been deposited in PDB with accession code of 7K17. These data will be released immediately upon publication.