

Abstract
Background & Aims:
The Nuclear Factor of Activated T-cells (NFAT) plays an important role in immune response by regulating the expression of inflammatory genes. However, it is not known whether it takes part in bile acid (BA)-stimulated expression of proinflammatory cytokines in hepatocytes in cholestatic livers.
Methods:
Gene and protein expression and cellular localization were assessed in primary hepatocyte cultures (mouse and human) and cholestatic liver tissues (murine models and patients with PBC and PSC) by Q-PCR, Western blot and immunohistochemistry. Specific NFAT inhibitors were used in vivo and in vitro. Gene reporter assay and ChIP-PCR were used to determine promoter activity.
Results:
NFAT isoform c1 and c3 were expressed in human and mouse hepatocytes. When treated with cholestatic levels of BA, both human and mouse hepatocytes increased NFATc3 nuclear translocation which was associated with elevated mRNA levels of IL-8, Cxcl2, and Cxcl10 in these cells. Blocking NFAT activation with pathway-specific inhibitors or knocking down Nfatc3 expression significantly decreased BA-induction of these cytokines in mouse hepatocytes. Nuclear expression of NFATc3/Nfatc3 protein was increased in cholestatic livers, both in mouse models (bile duct ligation or Abcb4−/− mice) and in patients with PBC and PSC in association with tissue elevations of Cxcl2 or IL-8, respectively. Gene reporter assays and ChIP-PCR demonstrated that NFAT response element in its promoter played a key role in BA-induced human IL-8 expression. Finally, blocking NFAT activation in vivo in Abcb4−/− mice reduced cholestatic liver injury.
Conclusions:
NFAT plays an important role in BA-stimulation of hepatic cytokines in cholestasis. Blocking hepatic NFAT activation may reduce cholestatic liver injury in humans.
Keywords: Bile acids, Nuclear Factor of Activated T-cells, Inflammatory cytokines, Cholestatic liver injury
Graphical Abstract
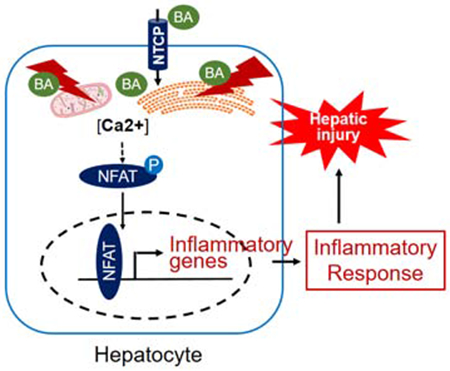
Lay summary:
Bile acid induces liver injury by stimulating the expression of inflammatory genes in hepatocytes through activation of transcription factor NFAT. Blocking this activation in vitro in hepatocyte cultures and in vivo in cholestatic mice, decreased the expression of inflammatory genes and reduced liver injury.
Introduction
Cholestasis is a syndrome where bile acids (BA) accumulate in the liver, resulting in liver injury. Cholestasis can be caused by genetic or developmental defects, as well as result from acquired diseases (1–3). In many cases, including primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and biliary atresia, the etiology is not known. Yet regardless of the initial cause, elevated hepatic BA concentrations are common to all. However, the pathogenetic role of BA in cholestasis remains unclear, hindering the development of effective therapies for these disorders. Recent studies indicate that the inflammatory response plays an important role in the pathogenesis of cholestasis and that BA induction of proinflammatory cytokines in hepatocytes is the initial pathophysiologic event (4–6). In the process of this event, BA cause endoplasmic reticulum (ER) stress and mitochondrial damage in hepatocytes. These injured mitochondria release DNA that then activates the Toll-like receptor (TLR/Tlr) 9 signaling pathway that results in the up-regulation of inflammatory cytokine expression(4). However, hepatic specific deficiency of Tlr9 diminishes, but does not eliminate, cholestatic liver injury in mice, indicating that signaling pathways independent of Tlr9 must also play a role in its pathogenesis (4). During these investigations we found that cyclosporine A (CsA)(4), a potent inhibitor of the calcium (Ca2+) / calcineurin / Nuclear Factor of Activated T-cells (NFAT) signaling pathway, significantly repressed taurocholic acid (TCA) induction of inflammatory cytokines in mouse hepatocytes. This observation prompted us to speculate that that the transcription factor NFAT is activated in cholestatic hepatocytes. NFAT is a highly phosphorylated family of proteins, described initially in T-cells (7–10). There are four isoforms of NFAT that are activated by intracellular Ca2+ signaling, i.e. NFATc1, c2, c3 and c4. These isoforms are differentially expressed in tissues and cells, and are activated in a stimulus-dependent and isoform-specific manner(11–13). At resting state, NFAT is localized in the cytosol. However, when there is a sustained elevation of intracellular [Ca2+], NFATc is dephosphorylated and translocates into the nucleus. In the nucleus NFATc regulates the expression of its targets either by binding directly to its specific response elements or by associating with other transcription factors on the promoter of the target. Many of NFAT targets in T-cells are involved in the immune response(14). However, it is not known whether NFAT plays any role in the BA-induced hepatic inflammatory response.
In this report, we examined the functional role of NFATc3 in cholestatic liver injury in mice and humans. We found that Nfatc3 mediated BA induction of inflammatory cytokines in mouse hepatocyte cultures. Knockdown of Nfatc3 reduced BA induction of these cytokines. Increased nuclear expression of NFATc3/Nfatc3 protein was also seen in the livers of patients with PBC and PSC as well as Abcb4−/− mice, associated with elevated tissue levels of IL-8 and Cxcl2 in humans and mice, respectively. BA-stimulated IL-8 expression is mediated through an NFAT response element in the human IL-8 promoter as determined by gene reporter assays. Blocking NFAT activation in Abcb4−/− mice reduced cholestatic liver injury. Together, these findings describe a previously unrecognized signaling pathway in hepatocytes that contributes to BA initiated hepatic inflammation, providing novel strategies for treating cholestatic liver diseases.
Materials and Methods
Materials.
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO), except where otherwise specified. Cell culture media (DMEM and Williams’ E), fetal bovine serum (FBS), penicillin/streptomycin, trypsin, phosphate buffered saline (PBS), Lipofectamine 2000 and Lipofectamine RNAiMax were from Life Technologies (Carlsbad, CA). Collagen coated plates and collagen were purchased from BD Sciences (Bedford, MA). Mouse Nfatc3 siRNA oligoes (siGENOME SMARTpool) and control siRNAs were purchase from Dharmacon (Lafayette, CO). FK506 was purchased from Cayman Chemical (Ann Arbor, MI). KN-62 and Inca-6 were from Tocris Bioscience (Minneapolis, MN). ECL reagents were from Thermo Scientific.
Animal experiments.
All animal experiment protocols were approved by the board of the Institutional Animal Care and Use Committee of Yale University (150 Munson St., New Haven, CT). Six-week-old Abcb4−/− female mice (FVB.129P2-Abcb4tm1Bor/J) were randomly divided into two groups (n=10-11) and administrated with either PBS (0.1 ml, solvent of the drug, as untreated disease control) or the NFAT blocker A285222 (15)(also called ACT-1061, 0.29 mg/kg body weight, provided by AbbVie Inc., North Chicago, IL) through daily i.p. injection for four weeks. The animals were sacrificed in random order after an overnight fast. Samples of plasma, urine (from the bladder), and liver were collected for further analyses.
Preparation and maintenance of mouse hepatocytes, cholangiocytes and human hepatocytes.
Mouse hepatocytes were isolated from 10-20 weeks old C57Bl/6 mice using collagenase perfusion. Primary cultures of mouse cholangiocyte were provided by Dr. Strazzabosco’s lab here in our Liver Center(16). Human hepatocytes were obtained through the Liver Tissue Cell Distribution System (Pittsburgh, Pennsylvania), which was funded by NIH Contract #N01-DK-7-0004 / HHSN267200700004C. Both human and mouse hepatocytes were maintained as previously described(17). All cell cultures were treated with indicated chemicals and collected within 96 hr after isolation. Protein and mRNA expression were detected as described(18). Of note, all Q-PCRs were assayed using TaqMan primer/probes (listed in supplementary CTAT Table). Antibodies against NFATc3 and HNF1α were purchased from Santa Cruz Biotechnology (Cat# sc-8405 and sc-8986, respectively), GAPDH antibody was from Sigma-Aldrich, Lamin B1 antibody (Cat#12586S) was from Cell Signaling Technology (Danvers, MA), and Nucleoporin p62 antibody (Cat#610497) was from BD Biosciences . To transfect mouse hepatocytes, siRNAs were mixed with Lipofectamine RNAiMax in Opti-MEM (Life Technologies) following manufacturer’s instruction and added to the culture medium 3 hr after the cells were plated on collagen coated plates (4x105 cell/well in a 12-well plate). Forty hours after transfection, the cells were treated with BA for 24 h, and collected for gene expression analyses.
Preparation of nuclear and cytoplasmic proteins from cells and tissues.
Frozen mouse liver was available from our previous studies(4, 18). These include tissues from 6-week old Abcb4+/+ and Abcb4−/− mice, 7-day bile duct ligated (BDL) mice, 7-day 1% cholic acid (CA) fed mice. De-identified human liver tissue specimens came from the Liver Tissue Cell Distribution System in University of Minnesota, Minneapolis, Minnesota, which was funded by NIH Contract # HSN276201200017C. These cholestatic liver specimens were explanted from patients with end-stage PBS or PSC, while normal human liver tissues were from deceased donors who did not have liver diseases. The clinical characteristics of these donors are listed in supplementary Table S1. To isolate cytoplasmic and nuclear protein from cells and tissue, a kit from Pierce Biotechnology (Cat# 78833, Rockford, IL) was used following manufacturer’s instructions. A protease inhibitor cocktail (Cat# 11873580001, Roche Diagnostic GmbH) was added to the solutions of the preparation. A dounce homogenizer was used to gently break up the liver tissue aggregates in PBS. The homogenate was centrifugated at 500xg for 3 min, and the pellet was used as the starting material.
Plasma biochemistry, liver histology, immunohistochemistry (IHC).
Plasma levels of liver enzyme alanine aminotransferase (ALT) and alkaline phosphatase (ALP) were determined by the clinical lab of Yale-New Haven Hospital (55 Part St., New Haven, CT). Bile acid concentration was measured as previously described(4). Formalin-fixed liver tissue was paraffin embedded and sections were stained with hematoxylin and eosin (H&E). Liver histology was blindly assessed for necrosis, bile duct proliferation, fibrosis and inflammation on a 1 to 4+ scale. For liver IHC, sections were labeled with antibodies against Nfatc1, Nfatc3, HNF1α and CD3. Specifically, the NovoLink Polymer Detection System (Cat# RE7140-K) from Leica Biosystems (Newcastle, UK) were used to detect Nfatc1 (mouse monoclonal antibody from Sigma-Aldrich, Cat# MABS409, 1:500 dilution) and Nfatc3 (mouse monoclonal antibody from Santa Cruz Biotechnology, Cat# sc-8405, 1:300 dilution) according to the manufacturer’s instructions. Rabbit polyclonal antibody against HNF1α were purchased from Santa Cruz Biotechnology (Cat# sc-8986, 1:200 dilution). Rabbit polyclonal antibody against CD3 was used to assess T-cells (Dako, Cat#A0452, 1:100 dilution) as previously described(19). Of note, the liver sections of five animals in each group were randomly selected for CD3 staining. The positive area was calculated using Image J software. Antigen retrieval was achieved with citrate buffer at pH 6.0 in a steamer for 20 min and primary antibody incubated on the tissue section overnight at 4°C. Sections were counter-stained with hematoxylin in CD3 detection.
Human IL-8 promoter reporter assay and Chromatin Immunoprecipitation (ChIP)-PCR assay.
The proximal promoter region of human IL-8 gene was amplified from a BAC clone (CH17-113M16, purchased from Children’s Hospital Oakland Research Institute, CA) using PCR with Forward primer: GCAT GGT ACCAG ATCTTCACCATCATGATAGCATCTGTA and Reverse primer: GCATGGATCCTGGCTCTTGTCCTAGAAGC. The amplified fragment (490 bp) was cloned into pGL3-basic vector, and verified by DNA-sequencing. The NFAT response element in human IL-8 promoter was mutated from GGAATTTCCTCT to GGAATTCTTTCT. For gene reporter assay, the reporter constructs were transfected into Huh7-BAT cells(20) (a cell line stably transfected with human NTCP, provided by Dr. Gregory Gores, Mayo Clinic) using Lipofectamine 2000 by following the manufacturer’s instruction. Twenty-four hours after transfection, the cells were treated with indicated concentration of BA for 16 hr. Dual-luciferase assay (purchased from Promega) were performed to determine promoter activity. Data are normalized to Renilla luciferase activity. For ChIP-PCR, Huh7-BAT cells were treated with indicated BA for 4 hr. After the cells were cross-linked, the assay was performed using a kit from Pierce Biotechnology (Pierce Agarose ChIP Kit, Cat#26156) by following the manufacturer’s instruction. Specifically, for each immunoprecipitation reaction, the nuclear fraction from 2x106 cells and 2 μg of antibody or control IgG were used. The primers for the detection PCR are TGGGCCATCAGTTGCAAATCG as the forward primer and the above-mentioned Reverse primer as the reverse primer. It generates a 170bp band.
Statistical Analysis.
One-way ANOVA followed by student t test were used to perform the statistical analysis for liver tissues using GraphPad Prism 8.4. Paired t-test was used for data from cell culture work. Data are presented as the means ± S.D. P<0.05 was considered statistically significant.
Results
Inhibitors involved in NFAT signaling repressed BA induction of chemokines in mouse hepatocytes.
We first verified that CsA could block the induction of pro-inflammatory cytokines Cxcl2 and Cxcl10 in a dose responsive manner when mouse hepatocytes were exposed to 50 μM glycocholic acid (GCA) (Fig 1A). We found an optimal concentration of 3 μM CsA. We next examined the effect of other inhibitors involved in the Ca2+-calmodulin-calcmeurm-NFAT signaling pathway. Mouse hepatocytes were exposed to 50 μM GCA in the presence of KN-62 (a specific Ca2+/calmodulin-dependent protein kinase inhibitor), FK506 (which binds to FKBP12 and blocks calcineurin activation) and Inca-6 (an inhibitor that prevents calcineurin and NFAT binding). Each of these inhibitors significantly reduced GCA and TCA induction of Cxcl2 (Fig 1C and data not shown). Together, these findings indicate that Ca2+/NFAT signaling pathway is involved in BA induction of chemokines in mouse hepatocytes.
Figure 1. Inhibitors involved in Ca2+/NFAT signaling pathway repressed bile acid (BA) induction of inflammatory chemokines in mouse hepatocyte cultures.
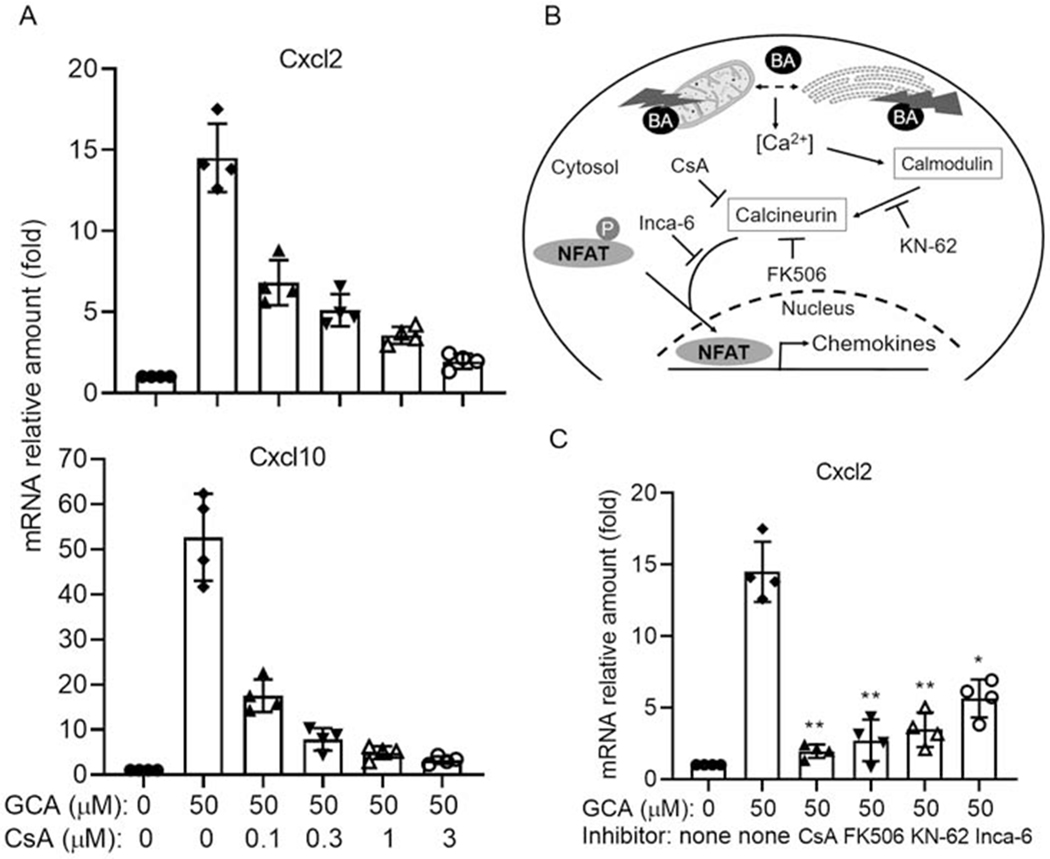
A, cyclosporin A (CsA) repressed GCA induction of Cxcl2 and Cxcl10 mRNA expression in mouse hepatocytes. B, A diagram of Ca2+/NFAT signaling pathway and inhibitors involved in NFAT activation. C, GCA induction of Cxcl2 expression were repressed by inhibitors involved in Ca2+/NFAT signaling in mouse hepatocytes. CsA (3 μM), FK506 (5 μM), KN-62 (3 μM), Inca-6 (40 μM). Data normalized to Gapdh, mean±SD, *p<0.05, **p<0.01 (t-test), n=4.
BA increased NFATc3 nuclear expression in mouse and human hepatocytes but not cholangiocytes
Nfatc1 and c3 are expressed in mouse cholangiocytes(21), but it is not known which isoforms are expressed in hepatocytes, nor whether BA alter their expression. To address these questions, we first analyzed the expression levels (mRNA) of the four different isoforms of NFATc/Nfatc in cultured primary hepatocytes from humans and mice. As shown in Figure 2A, NFATc1, c2 and c3 are expressed in human hepatocytes, whereas only Nfatc1 and c3 were detected in mouse hepatocytes. Neither cells expressed significant amount of NFATc4. We also confirmed that Nfatc1 and c3 are expressed in mouse cholangiocytes as previously described (21) (Data not shown). We next determined whether cholestatic levels of BA altered the mRNA expression of NFATc/Nfatc isoforms in human and mouse hepatocytes. As shown in Figure 2B, glycochenodeoxycholic acid (GCDCA, the major endogenous BA in humans) did not alter the mRNA expression of any NFATc isoform in human hepatocytes, whereas TCA (the major endogenous BA in rodents) slightly but significantly increased Nfatc1, c2 and c3 in mouse hepatocytes, where isoform c4 remained undetectable. Because NFATc3 is the most abundantly expressed isoform in both human and mouse hepatocytes, we next examined whether BA treatment could lead to nuclear translocation, indicating activation. We found that indeed nuclear expression of NFATc3/Nfatc3 was increased in both mouse (Fig 3A) and human (Fig 3B) hepatocytes when treated with pathophysiological concentrations of BA, indicating that human and mouse share a common mechanism in hepatic NFAT activation. Despite the high expression of Nfatc1 in mouse hepatocytes, TCA treatment did not result in nuclear translocation (Supplementary Fig S1). Additional testing demonstrated that GCA and TDCA also increased nuclear expression of Nfatc3 in mouse hepatocytes, and CsA, KN-62 and FK506 could inhibit the GCA-induced translocation (Fig 3C). This nuclear translocation did not occur when mouse cholangiocytes were treated with even at higher concentration of TCA (400 μM) (supplementary Fig S2) and there was no substantial induction of chemokines Ccl2, Cxcl2 and Cxcl10 when exposed to 1 mM TCA. Together, these findings establish the role of hepatic NFAT in BA induction of chemokines in cholestatic hepatocytes.
Figure 2. NFAT isoforms are expressed in hepatocytes.
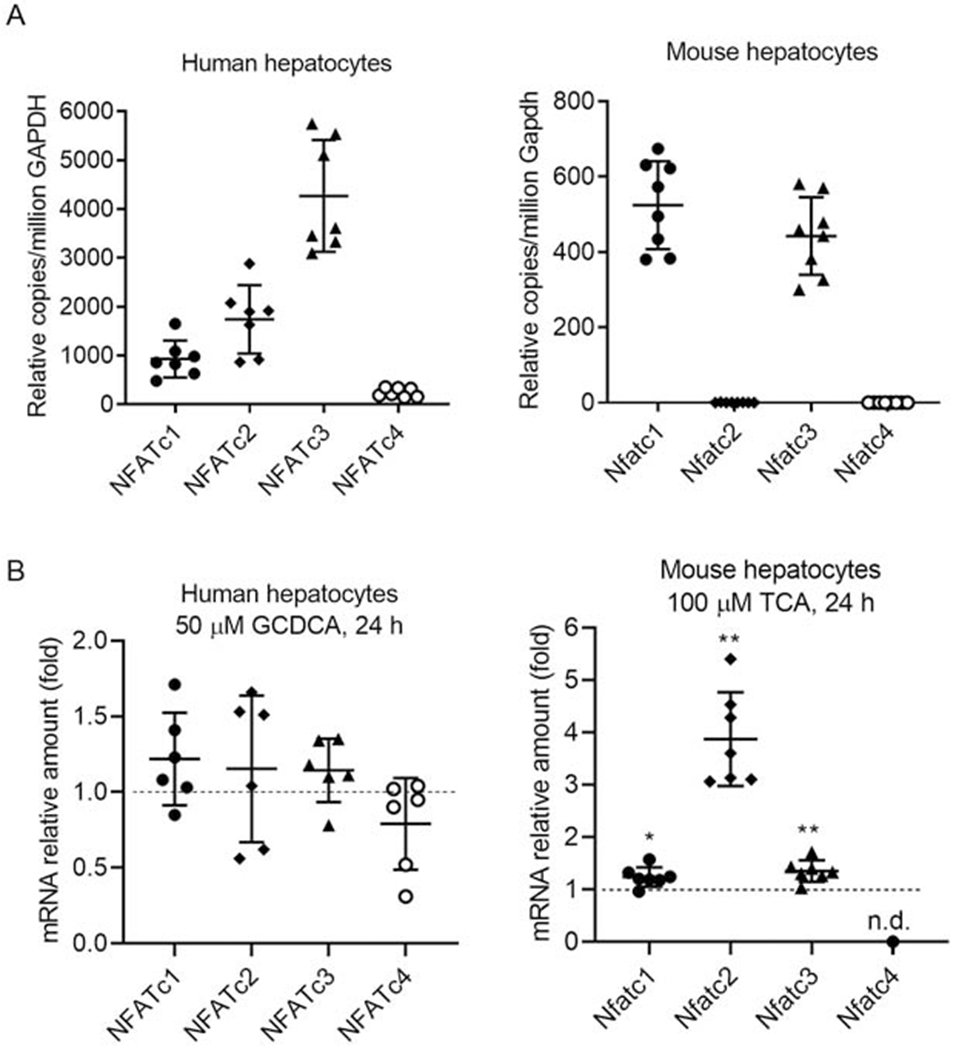
A, mRNA expression of NFATc isoforms in human and mouse hepatocytes. B, the speices-specific major endogenous bile acid altered NFAT isoform mRNA expression in mouse hepatocytes but not human hepatocytes. mean±SD, n=6-8, *P<0.05, **P<0.01, (t-test), n.d., non-detectable.
Figure 3. Bile acids stimulated NFATc3 nuclear translocation in hepatocytes.
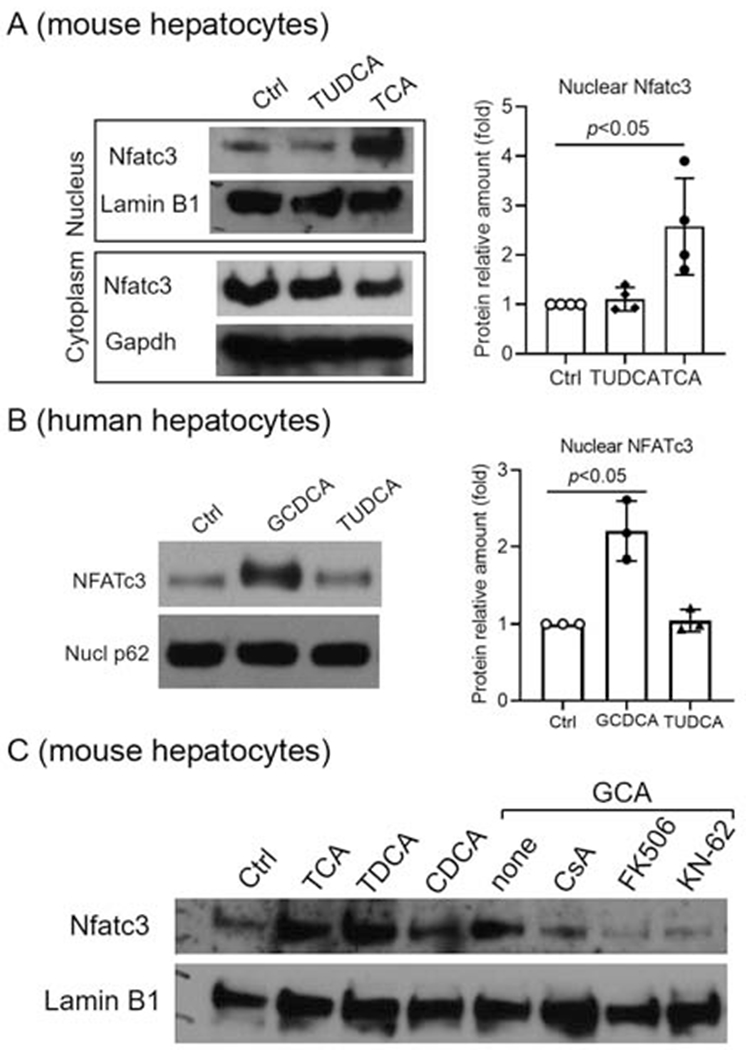
A. Western blots demonstrate that TCA significantly increased Nfatc3 protein nuclear expression in mouse hepatocytes. Lamin B1 and Gapdh as nuclear and cytoplasmic markers, respectively. B, Western blots of nuclear protein from human hepatocytes exposed to indicated bile acids. Nucleoporin p62 (Nucl p62) as loading control. C, Western blots of nuclear protein from mouse hepatocytes exposed to bile acids and inhibitors of Ca2+/NFAT signaling. Ctrl = treatment control; Except GCA (50μM), all other bile acids were at 100μM; CsA, 3μM; FK506, 5μM; KN-62, 3μM. P-value (t-test).
Increased NFAT nuclear expression in cholestatic mouse livers was associated with elevated hepatic levels of inflammatory cytokines
We next examined NFAT gene expression in normal mouse liver and liver from three cholestatic mouse models, which we have previously characterized (4,17). These three models represent a model for PSC (the Abcb4−/− mouse), an obstructive cholestatic model (BDL), and a BA overload model (1% cholic acid diet feeding). Q-PCR analysis revealed that both Nfatcl and c3 were moderately expressed in the livers of these cholestatic animals at essentially the same level but neither were substantially altered under cholestatic conditions. Similar to the isolated hepatocytes, Nfatc4 was undetectable in all livers and, the expression of Nfatc2 was very low in normal liver and significantly increased in the liver of all three cholestatic models (up to 6-fold in Abcb4−/− mice) (Fig 4A). Although Nfatc3 mRNA was unchanged, Western blot indicated higher nuclear expression of Nfatc3 in both Abcb4−/− livers and BDL livers than in their corresponding controls (Fig 4B and supplementary Fig S3). This was confirmed by immunohistochemical staining of liver sections from Abcb4−/− mice which showed increased staining throughout the liver lobules when compared to Abcb4+/+ livers (Fig 4C). In contrast, there was no difference in the nuclear expression of the transcription factor Hnf1α between Abcb4+/+ and Abcb4−/− livers (Fig 4B & 4C). Similar results were also found in BDL livers (Supplementary Figure S4). Immunohistochemistry for Nfatc1 revealed prominent staining of Kupffer cells as well as liver sinusoidal cells (Supplementary Fig S5). Staining of Nfatc1 protein in parenchymal cells was negligible when compared with these non-parenchymal cells. Finally, we analyzed the mRNA expression level of a few chemokines (e.g. Cxcl1, Cxcl2, Cxcl10) in these cholestatic mouse livers and found that their expression was significantly increased (Fig 4D).
Figure 4. Elevated hepatic chemokine expression was associated with increased nuclear expression of Nfatc3 protein in cholestatic mouse livers.
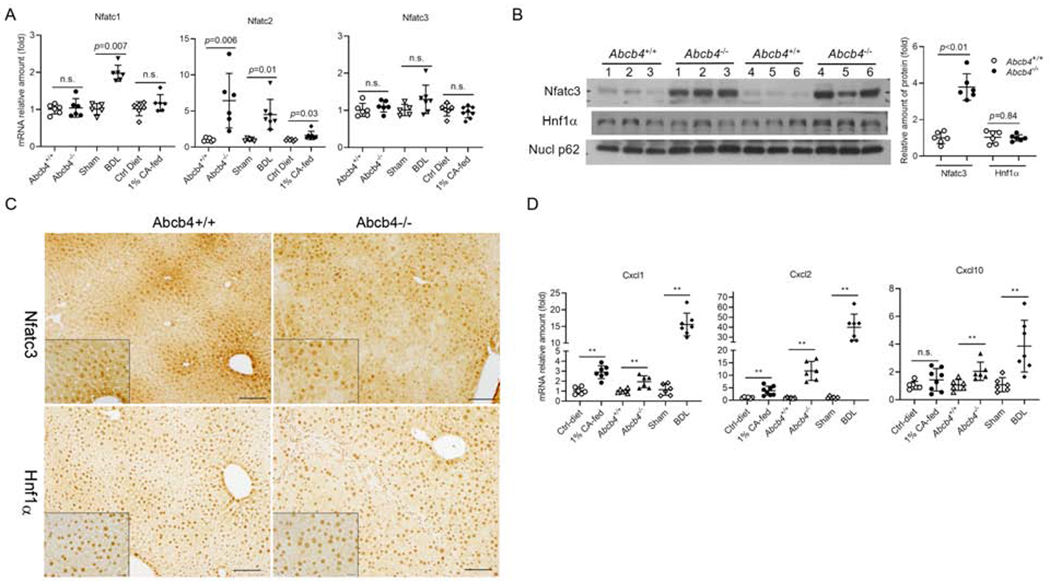
A, mRNA expression of Nfatc isoforms in cholestatic mouse livers. B, increased nuclear expression of Nfatc3 but not Hnf1α in Abcb4−/− mouse livers. C, immunohistochemical staining of Nfatc3 and Hnf1α in the liver of Abcb4+/+ and cholestatic Abcb4−/− mice. Bar=100μm. Inserts, Bar=20μm. D, hepatic mRNA expression of Cxcl1, Cxcl2 and Cxcl10 in Abcb4−/− BDL, and 1% cholic acid (CA)-fed mice. Nucl p62, Nucleoporin p62 as nuclear protein loading control. mean±SD, n=6-8, *P<0.05, **P<0.01 (t-test). n.s., not significant.
Knockdown of Nfatc3 reduced mRNA expression of inflammatory genes in mouse hepatocytes.
To determine whether Nfatc3 plays a direct role in BA induction of inflammatory genes in the liver, we knocked down Nfatc3 expression using siRNA transfection in mouse hepatocytes. As demonstrated in Figure 5, diminished Nfatc3 expression resulted in significant reduction of inflammatory genes at both basal levels and after TCA stimulation, including Ccl2, Cxcl1, Cxcl2, and Cxcl10, whereas the bile acid nuclear receptor Fxr (Nr1h4) mRNA was increased in these cells. Because a previous study (14) revealed a series of Nfatc2 targets in mouse T-cells, including Ccl3, Cxcr4, Egr1, Egr3, Icam1 and Vcam1, we also assayed the expression of these genes in Nfatc3 knockdown mouse hepatocytes. We found reduced expression of Egr1 and Icam1 (Fig 5). However, Vcam1 expression was not significantly changed, while Ccl3 and Egr3 were essentially undetectable (data not shown) in mouse hepatocytes. Together, these results indicate that NFAT plays an important role in BA induction of inflammatory genes in mouse hepatocytes, whereas there are also significant differences in NFAT targets between T-cells and hepatocytes.
Figure 5. Knockdown of Nfatc3 significantly reduced the mRNA expression of inflammatory genes in mouse hepatocyte cultures.
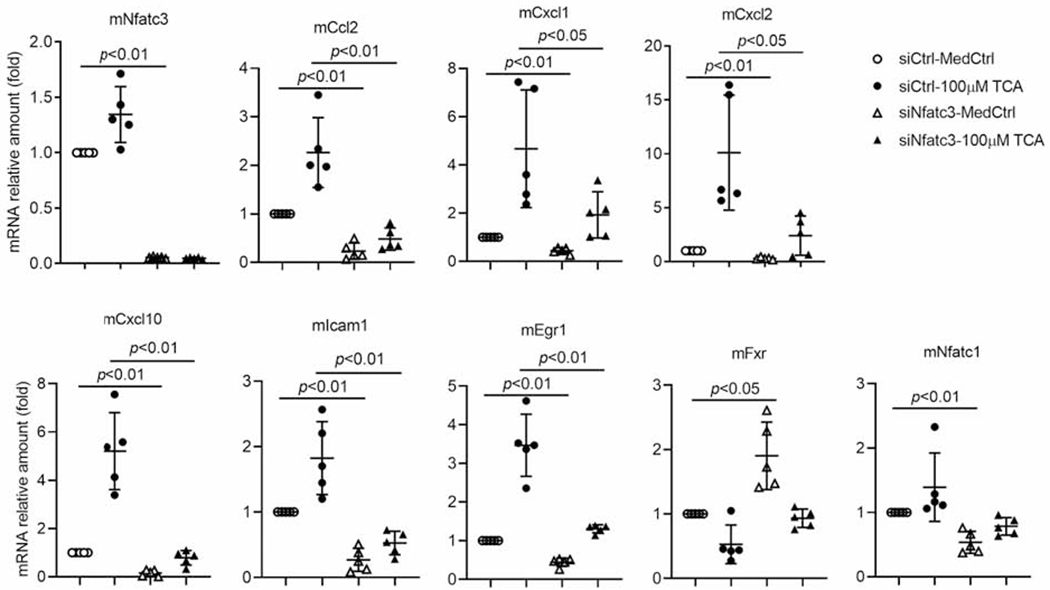
MedCtrl, control medium; TCA, taurocholic acid; siCtrl, scrambled siRNA control; siNfatc3, a smart pool of siRNAs targeting mouse Nfatc3. p-value (t-test).
Increased NFAT nuclear expression in the livers of patients with PBC and PSC is associated with elevated levels of IL-8
We next sought to verify the mouse findings in human cholestatic diseases. Q-PCR analysis revealed the relative mRNA abundance of the four NFAT isoforms in healthy control livers was NFATc1 = NFATc3 > NFATc2 >> NFATc4 (data not shown), similar to what was seen in mouse livers. Furthermore, mRNA levels for the four isoforms did not differ significantly between disease groups and the healthy controls (supplementary Fig S6). However, as demonstrated in Figure 6, the nuclear expression of NFATc3 protein was significantly increased in the livers of both PBC (3.8-fold) and PSC (3.4-fold) patients when compared to the non-diseased control livers. We also detected significant increases in mRNA of IL-8 in PBC and PSC livers (29 and 43-fold, respectively) when compared to normal liver tissue controls (Fig 6C). Further data analysis of IL-8 expression and the amount of NFATc3 nuclear protein in PBC livers indicated that the nuclear but not cytosolic expression of NFATc3 protein correlated directly with hepatic IL-8 mRNA levels (Fig 6D. r=0.663, p=0.005, n=16), suggesting that NFATc3 might up-regulate IL-8 expression in these cholestatic livers. This observation is consistent with elevated levels of Cxcll, Cxcl2 seen in cholestatic mouse livers (Fig 4D). Of note, the mouse does not have an IL-8 gene. Its homologs in mouse are Cxcll and Cxcl2 because they all activate the receptor CXCR2/Cxcr2.
Figure 6. Increased nuclear expression of NFATc3 protein in the liver of patients with PBC and PSC correlated with hepatic level of IL-8 mRNA.
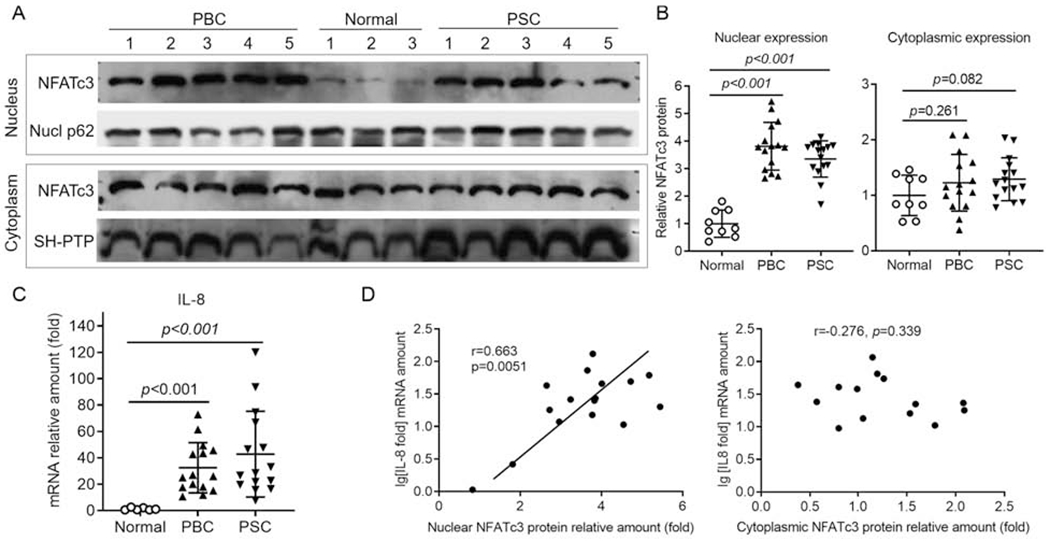
A, Western blots of NFATc3 in nuclear and cytoplasmic protein. Nucl p62 and SH-PTP as loading controls for nuclear and cytoplasmic protein, respectively. B, Quantification of NFATc3 protein by densitometry analysis. C, mRNA expression of hepatic IL-8 in patients with PBC and PSC. D, Correlation analysis of the hepatic levels of nuclear NFATc3 protein and IL-8 mRNA in PBC patients. p-value (t-test).
BA stimulated IL-8 expression through its NFAT response element.
Our previous studies indicated that cholestatic levels of BA-stimulated IL-8 mRNA expression in primary human hepatocyte cultures(4). To test whether NFAT mediates this stimulation, we generated a human IL-8 promoter reporter construct that contains an NFAT response element ((22) and Fig 7A). When this reporter construct was transfected into Huh7-BAT cells (a cell line that is stably transfected and expresses the bile acid uptake transporter NTCP/SLC10A1 (20)), it demonstrated strong basal activity, ~22-fold compared to the control vector pGL3-basic (Fig 7B). When the transfected cells were treated with BA (50 μM), GCDCA, GCA and TCA all significantly increased IL-8 promoter activity, whereas unconjugated cholic acid did not (Fig 7B). To examine whether the conjugated BA induction of the IL-8 promoter is NFAT dependent, we mutated its NFAT response element. As shown in Figure 7B, mutation of this NFAT response element greatly reduced both the basal activity and the BA inducibility of the IL-8 promoter. To further confirm NFAT’s role in BA-stimulated IL-8 expression, we performed ChIP-PCR in Huh7-BAT cells. Because GCA and TCA greatly stimulated IL-8 promoter activity in reporter assays (Fig 7B), we treated these cells with 50 μM GCA or TCA for 4 hr. Figure 7C demonstrated increased amounts of IL-8 promoter DNA in GCA and TCA treated cells than in the control cells. Together, these findings indicate that endogenous conjugated BA stimulate human IL-8 hepatic expression by activating NFAT.
Figure 7. Bile acids stimulate human IL-8 promoter activity through an NFAT response element (NFAT-RE).
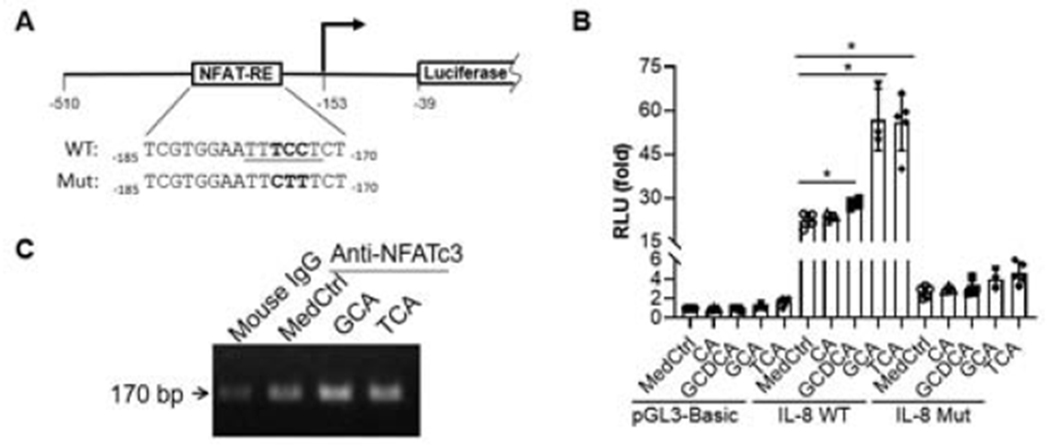
A, a diagram of human IL-8 promoter reporter constructs in pGL3-basic vector. B, Dual-luciferase reporter assay demonstrate that bile acids significantly stimulated human IL-8 promoter activities in transfected Huh7-BAT cells (an NTCP stably transfected cell line). C, Agarose gel electrophoresis from ChIP-PCR assay showed that bile acids increased NFATc3 binding to IL-8 promoter in Huh7-BAT cells. Reporter assay data was normalized to Renilla luciferase activity, and pGL3-basic activity in control medium (MedCtrl) was set as 1. mean±SD, n≥3, *p<0.05 (t-test).
Pharmacologically blocking NFAT activation reduced liver injury in Abcb4−/− mice
Finally, to assess whether blocking NFAT activation could reduce cholestatic liver injury in an animal model, we administrated the NFAT blocker A285222 (0.29 mg/kg body weight, a dose based on a previous report(23)) to 6-week old Abcb4−/− female mice for 4 weeks because these females develop more liver injury than their male counterparts(24). After this treatment, the levels of plasma ALT and ALP were significantly reduced by 36% and 27%, respectively when compared with the control group (Fig 8A). In addition, the bile acid levels in the plasma, liver tissue and urine were also significantly lower in these A285222 treated mice (Fig 8A and supplementary Fig S7B). Blinded assessment of liver histology indicated that both bile duct proliferation and liver fibrosis trended lower but did not reach statistical significance (supplementary Fig S7A). However, liver gene expression analysis revealed that mRNA expression of chemokines Ccl2, Cxcl1 and Cxcl10 were reduced in A285222 treated mice (Fig 8B), where Cyp7a1, the rate-limiting enzyme for bile acid synthesis, also trended lower (supplementary Fig S7C). Furthermore, IHC assay revealed that A285222 treatment significantly reduced the infiltration of T-cells by 50% in these livers (Fig 8C).
Figure 8. Blocking NFAT activation by A285222 administration reduced cholestatic liver injury in Abcb4−/− female mice.
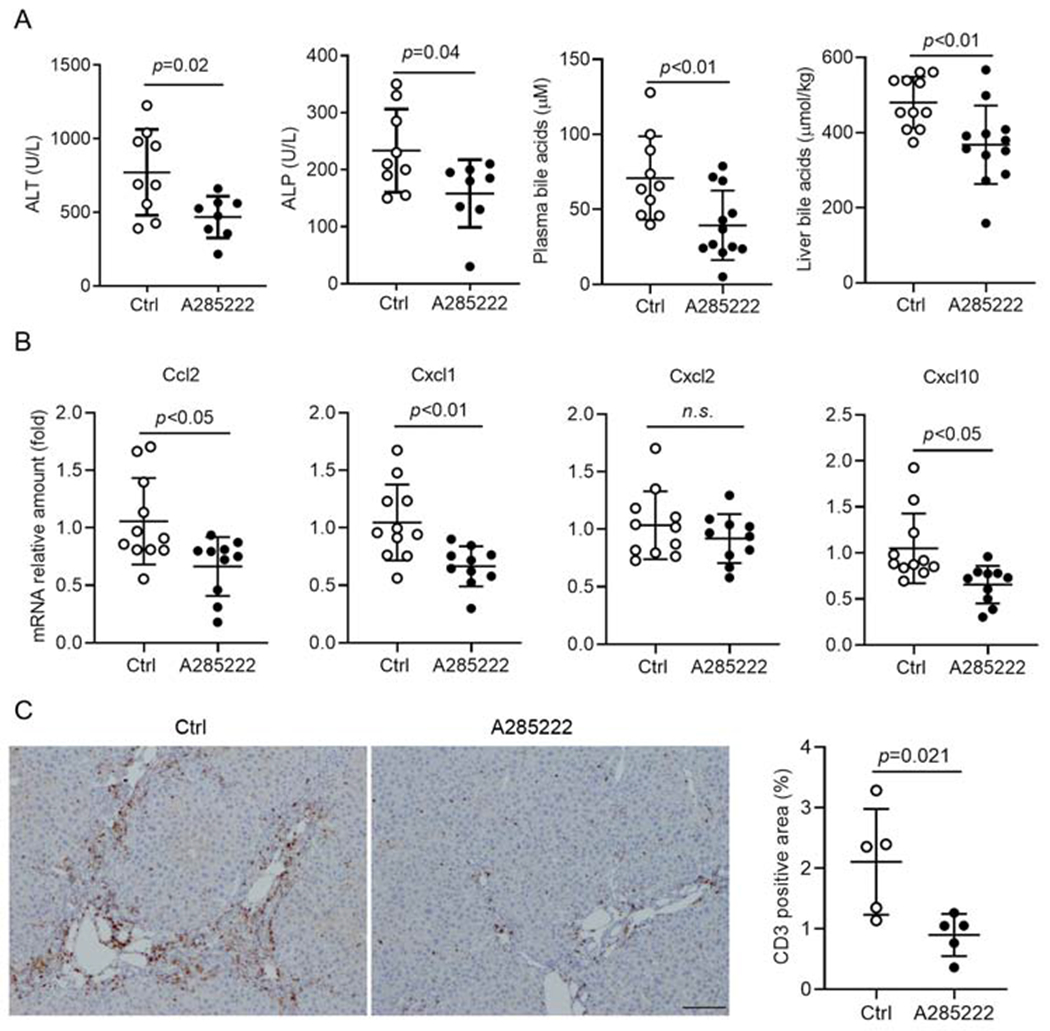
A, A285222 treatment significantly reduced plasma levels of ALT, ALP and bile acid and hepatic level of bile acids. B, A285222 treatment significantly decreased hepatic mRNA expression of Ccl2, Cxcll and Cxcl10. C, A285222 treatment significantly reduced liver infiltration of T-cells (CD3+ cells). Representative images of immunohistochemical staining of CD3 in liver sections (left) and their quantification (right). p-value (t-test).
Discussion
In this report, we examined the functional role of NFAT in cholestatic liver injury. We found that: 1) NFAT isoforms are expressed in hepatocytes from humans and mice (Fig 2); 2) Under cholestatic conditions, BA-stimulated expression of inflammatory cytokines in mouse and human hepatocytes was associated with the activation of the transcription factor NFAT (Figs 1 & 3). Blocking this activation by using pathway specific inhibitors or knocking down Nfatc3 expression reduced BA-induction of these proinflammatory genes in mouse hepatocyte cultures (Figs 1 & 5) and decreased cholestatic liver injury in Abcb4−/− mice (Fig 8); 3) Increased expression of NFATc3/Nfatc3 protein was found in the nucleus of cholestatic livers from both patients with PBC and PSC and in mice with Abcb4-deficiency or after BDL where elevated levels of inflammatory cytokines were also found (Figs 4 & 6 and Fig S3 & S4); Finally, 4) both gene reporter assays and ChIP-PCR demonstrated that BA-induced human IL-8 expression is mediated through NFAT response elements in its proximal promoter (Fig 7). Based on these observations, we conclude that NFAT plays an important role in the pathogenesis of cholestatic liver injury, a previously undescribed novel mechanism of BA initiated hepatic inflammatory response.
Studies in T-cells and other immune cells have established that NFATc isoforms are activated through the Ca2+/calmodulin/calcineurin signaling pathway. When there is a sustained elevation of intracellular Ca2+, the NFATc isoform translocates to the nucleus and acts as a transcription factor to modulate gene expression (7, 25, 26). There is a substantial literature indicating that BA increase cytosolic Ca2+ levels in hepatocytes (27–29). While those studies mainly demonstrate transient changes in cytosolic calcium, it seems likely that more chronic elevations could also occur. Both ER and mitochondria play a pivotal role in regulating Ca2+ signaling, and BA also cause ER-stress and mitochondrial damage when they reach cholestatic levels (30–32). Thus, it is likely that NFATc is activated in cholestatic hepatocytes since NFATc isoforms are expressed in these cells (Fig 2). In addition, Nfatc3 protein accumulates in the nucleus of BA treated mouse and human hepatocytes (Fig 3) but not in the livers of patients with other forms of liver injury, e.g. steatosis (Data not shown), further supporting the involvement of hepatic NFATc as a contributor to the cholestatic inflammatory response. Of note, NFATc3 is also present in the nuclei of hepatocytes under normal conditions as detected by both Western blot and IHC (Figs 3, 4C, & Fig S4), suggesting that hepatic NFATc3 may also play a role in maintaining other normal physiologic responses. Incomplete regeneration of the liver after partial hepatectomy seen in Nfatc3−/− mice would support this view (33). However, during cholestasis, nuclear NFATc3 expression increases in response to BA stress, resulting in the increased expression of inflammatory genes. These response also seems to be hepatocyte specific because BA did not increase the expression of nuclear Nfatc3 or chemokines in cholangiocyte cultures (Supplementary Fig S2 and (4)), nor did we observe an increase in Nfatc3 nuclear expression in the bile ducts ofAbcb4−/− livers (Fig 4B). Since there is little if any expression of NFATc4 in the liver (Fig 2), we conclude that this isoform does not play a role in cholestatic liver injury. Although Nfatc1 is also detected in mouse hepatocytes, BA did not increase its nuclear expression (Supplementary Fig S1). We propose that NFatc1 may not play a role in BA induction of inflammatory cytokines in mouse hepatocytes. This supports the view that NFATc isoforms are activated in a stimulus-dependent and a cell type-specific manner(11–13). However, we do not rule out Nfatcl play some roles in the pathogenesis of cholestatic liver injury as it is prominently expressed in Kupffer cells and sinusoidal cells (Fig S5) as well as cholangiocytes (21) . Because NFATc2 is detected in the liver (Figs 2, & S2 & (21)) although at much lower level than NFATc3, it remains to be determined whether it plays any role in BA-stressed cholestatic hepatocytes. Future studies may address these questions.
Activation of hepatic NFATc3 is at least partially responsible for BA induction of inflammatory genes because inhibitors blocking Nfatc3 nuclear translocation or knockdown of Nfatc3 significantly reduced BA stimulation of inflammatory gene expression in mouse hepatocytes (Figs 1, 3 & 5). The response elements for NFAT in many inflammatory genes have been identified by ChIP-Seq in mouse T-cells, including Icam1 and Egr1, supporting NFAT’s role in regulating the immune response (14). Most importantly, we demonstrated that BA-stimulated IL-8 expression was mediated through the NFAT response element in the human IL-8 promoter, as demonstrated by reporter and ChIP-PCR assays in Huh7-BAT cells (Fig.7), and consistent with elevated levels of IL-8 in the livers of patients with PBC and PSC (Fig.6). Because our findings indicate that NFAT plays an important role in BA-stimulation of inflammatory cytokine expression, and because the inflammatory response plays a critical role in the pathogenesis of cholestatic liver injury(34, 35), one may speculate that blocking hepatic NFAT activation would reduce liver injury in cholestasis. As expected, administration of an NFAT inhibitor reduced cholestatic liver injury in Abcb4−/− mice (Fig 8 and supplementary Fig. S7). Interestingly, a CsA analog, NIM811, has also been shown to reduce liver injury in the BDL cholestatic mouse (36). We found that NIM811 had similar effects as CsA in suppressing BA-induction of cytokines in mouse hepatocytes (Data not shown). Because NFAT inhibitor A285222 did not eliminate liver injury in Abcb4−/− mice, it is likely that Ca2+/NFAT signaling is not the only mechanism in BA induction of inflammatory cytokines as our previously report has indicated(4). We would also want to stress that knockout of Nfatc3 and the optimal dose of A285222 in these mice need further study.
Previous reports indicate that NFAT signaling cross talks with the innate immune response via TLR signaling(37, 38). Our prior study revealed that Tlr9 was involved in the BA-induced hepatic inflammatory response (4), while a recent report indicates that Ca2+/NFAT signaling can be down-stream of Tlr9 activation (39). In that report, activated Tlr9 stimulates Burton’s tyrosine kinase (BTK) activity, resulting in phosphorylation and activation of phospholipase Cγ. The activated phospholipase Cγ then initiates Ca2+/NFAT signaling. The involvement of Ca2+/NFAT signaling was also seen in TLR9-induced IL-10 secretion in human B-cells where Burton’s tyrosine kinase also played a role (40). Thus, it remains to be determined whether BA-activated Tlr9 and Ca2+/NFAT signaling are two independent signaling pathways or are part of the same cascade of events in cholestatic hepatocytes. If the latter, Ca2+/NFAT signaling would be downstream of TLR9. Blocking NFAT signaling would also stop TLR9 activation, suggesting that NFAT is the master controller in BA induction of proinflammatory genes in hepatocytes. Future studies may address this question.
In summary, our findings indicate that the transcription factor NFAT plays an important role in regulating the expression of inflammatory genes in cholestatic hepatocytes, a previously undescribed novel mechanism in the inflammatory response induced by BA. These findings suggest that blocking the activation of hepatic NFAT may provide a novel therapy for treating cholestatic disorders.
Supplementary Material
Highlights.
Bile acid activates transcription factor NFATc3 in mouse and human hepatocytes
This activation increases the expression of inflammatory genes in hepatocytes
Increased nuclear expression of NFATc3 expression is also seen cholestatic livers
Blocking this activation reduced cholestatic liver injury in mouse
Acknowledgements:
We are grateful to Kathy Harry and Maoxu Ge for their excellent technical support. We also thank Drs. Mario Strazzabosco and Romina Fiorotto in our section for providing primary mouse cholangiocytes, Dr. Paula Vidigal for help with liver Nfatc1 IHC characterization and Dr. Gregory Gores, Mayo Clinic, Rochester, Minnesota for sharing Huh7-BAT cells.
Grant Support: This study was supported by National Institutes of Health Grants DK34989 (Yale Liver Center), DK25636 (to J.L.B.) and a Yale Liver Center Pilot grant to SYC. Gilead Pharmaceuticals (Foster City, CA) also supported this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have declared that no conflict of interest exists.
Data availability statement: All data that support the findings of this report are available from the corresponding author, upon reasonable request.
Reference List
- (1).Hirschfield GM. Genetic determinants of cholestasis. Clin Liver Dis 2013. May;17(2):147–159. [DOI] [PubMed] [Google Scholar]
- (2).Keitel V, Droge C, Stepanow S, Fehm T, Mayatepek E, Kohrer K, et al. Intrahepatic cholestasis of pregnancy (ICP): case report and review of the literature. Z Gastroenterol 2016. December;54(12):1327–1333. [DOI] [PubMed] [Google Scholar]
- (3).Pollheimer MJ, Fickert P, Stieger B. Chronic cholestatic liver diseases: clues from histopathology for pathogenesis. Mol Aspects Med 2014. June;37:35–56. [DOI] [PubMed] [Google Scholar]
- (4).Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2017. March 9;2(5):e90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol 2011. January;178(1):175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang Y, Hong JY, Rockwell CE, Copple BL, Jaeschke H, Klaassen CD. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int 2012. January;32(1):58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003. September 15;17(18):2205–2232. [DOI] [PubMed] [Google Scholar]
- (8).Wu H, Peisley A, Graef IA, Crabtree GR. NFAT signaling and the invention of vertebrates. Trends Cell Biol 2007. June;17(6):251–260. [DOI] [PubMed] [Google Scholar]
- (9).Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell 2002. April;109 Suppl:S67–S79. [DOI] [PubMed] [Google Scholar]
- (10).Pan MG, Xiong Y, Chen F. NFAT gene family in inflammation and cancer. Curr Mol Med 2013. May;13(4):543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ulrich JD, Kim MS, Houlihan PR, Shutov LP, Mohapatra DP, Strack S, et al. Distinct activation properties of the nuclear factor of activated T-cells (NFAT) isoforms NFATc3 and NFATc4 in neurons. J Biol Chem 2012. November 2;287(45):37594–37609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kaminuma O, Kitamura F, Kitamura N, Hiroi T, Miyoshi H, Miyawaki A, et al. Differential contribution of NFATc2 and NFATc1 to TNF-alpha gene expression in T cells. J Immunol 2008. January 1;180(1):319–326. [DOI] [PubMed] [Google Scholar]
- (13).Kar P, Mirams GR, Christian HC, Parekh AB. Control of NFAT Isoform Activation and NFAT-Dependent Gene Expression through Two Coincident and Spatially Segregated Intracellular Ca(2+) Signals. Mol Cell 2016. November 17;64(4):746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 2015. February 17;42(2):265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Trevillyan JM, Chiou XG, Chen YW, Ballaron SJ, Sheets MP, Smith ML, et al. Potent inhibition of NFAT activation and T cell cytokine production by novel low molecular weight pyrazole compounds. J Biol Chem 2001. December 21;276(51):48118–48126. [DOI] [PubMed] [Google Scholar]
- (16).Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology 2011. October;141(4):1498–508, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cai SY, Gautam S, Nguyen T, Soroka CJ, Rahner C, Boyer JL. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology 2009. March;136(3):1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cai SY, Mennone A, Soroka CJ, Boyer JL. Altered expression and function of canalicular transporters during early development of cholestatic liver injury in Abcb4-deficient mice. Am J Physiol Gastrointest Liver Physiol 2014. April 15;306(8):G670–G676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yu D, Cai SY, Mennone A, Vig P, Boyer JL. Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int 2018. January 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Higuchi H, Bronk SF, Takikawa Y, Werneburg N, Takimoto R, El-Deiry W, et al. The bile acid glycochenodeoxycholate induces trail-receptor 2/DR5 expression and apoptosis. J Biol Chem 2001. October 19;276(42):38610–38618. [DOI] [PubMed] [Google Scholar]
- (21).Alpini G, Franchitto A, DeMorrow S, Onori P, Gaudio E, Wise C, et al. Activation of alpha(1) -adrenergic receptors stimulate the growth of small mouse cholangiocytes via calcium-dependent activation of nuclear factor of activated T cells 2 and specificity protein 1. Hepatology 2011. February;53(2):628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tsuge M, Hiraga N, Zhang Y, Yamashita M, Sato O, Oka N, et al. Endoplasmic reticulum-mediated induction of interleukin-8 occurs by hepatitis B virus infection and contributes to suppression of interferon responsiveness in human hepatocytes. Virology 2018. December;525:48–61. [DOI] [PubMed] [Google Scholar]
- (23).Zhang S, Zhang S, Garcia-Vaz E, Herwald H, Gomez MF, Thorlacius H. Streptococcal M1 protein triggers chemokine formation, neutrophil infiltration, and lung injury in an NFAT-dependent manner. J Leukoc Biol 2015. June;97(6):1003–1010. [DOI] [PubMed] [Google Scholar]
- (24).van Nieuwerk CM, Groen AK, Ottenhoff R, van WM, van den Bergh Weerman MA, Tytgat GN, et al. The role of bile salt composition in liver pathology of mdr2 (−/−) mice: differences between males and females. J Hepatol 1997. January;26(1):138–145. [DOI] [PubMed] [Google Scholar]
- (25).Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science 1988. July 8;241(4862):202–205. [DOI] [PubMed] [Google Scholar]
- (26).Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 1997;15:707–747. [DOI] [PubMed] [Google Scholar]
- (27).Thibault N, Ballet F. Effect of bile acids on intracellular calcium in isolated rat hepatocyte couplets. Biochem Pharmacol 1993. January 26;45(2):289–293. [DOI] [PubMed] [Google Scholar]
- (28).Beuers U, Nathanson MH, Boyer JL. Effects of tauroursodeoxycholic acid on cytosolic Ca2+ signals in isolated rat hepatocytes. Gastroenterology 1993. February;104(2):604–612. [DOI] [PubMed] [Google Scholar]
- (29).Marrero I, Sanchez-Bueno A, Cobbold PH, Dixon CJ. Taurolithocholate and taurolithocholate 3-sulphate exert different effects on cytosolic free Ca2+ concentration in rat hepatocytes. Biochem J 1994. June 1;300 (Pt 2):383–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate-induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J Clin Invest 1993. July;92(1):17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sokol RJ, Straka MS, Dahl R, Devereaux MW, Yerushalmi B, Gumpricht E, et al. Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr Res 2001. April;49(4):519–531. [DOI] [PubMed] [Google Scholar]
- (32).Adachi T, Kaminaga T, Yasuda H, Kamiya T, Hara H. The involvement of endoplasmic reticulum stress in bile acid-induced hepatocellular injury. J Clin Biochem Nutr 2014. March;54(2):129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pierre KB, Jones CM, Pierce JM, Nicoud IB, Earl TM, Chari RS. NFAT4 deficiency results in incomplete liver regeneration following partial hepatectomy. J Surg Res 2009. June 15;154(2):226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol 2012. September 28;18(36):4985–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li M, Cai SY, Boyer JL. Mechanisms of bile acid mediated inflammation in the liver. Mol Aspects Med 2017. August;56:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Rehman H, Ramshesh VK, Theruvath TP, Kim I, Currin RT, Giri S, et al. NIM811 (N-methyl-4-isoleucine cyclosporine), a mitochondrial permeability transition inhibitor, attenuates cholestatic liver injury but not fibrosis in mice. J Pharmacol Exp Ther 2008. December;327(3):699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Minematsu H, Shin MJ, Celil Aydemir AB, Kim KO, Nizami SA, Chung GJ, et al. Nuclear presence of nuclear factor of activated T cells (NFAT) c3 and c4 is required for Toll-like receptor-activated innate inflammatory response of monocytes/macrophages. Cell Signal 2011. November;23(11):1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ma B, Yu J, Xie C, Sun L, Lin S, Ding J, et al. Toll-Like Receptors Promote Mitochondrial Translocation of Nuclear Transcription Factor Nuclear Factor of Activated T-Cells in Prolonged Microglial Activation. J Neurosci 2015. July 29;35(30):10799–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Herbst S, Shah A, Mazon MM, Marzola V, Jensen B, Reed A, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med 2015. March;7(3):240–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ziegler S, Gartner K, Scheuermann U, Zoeller T, Hantzschmann J, Over B, et al. Ca(2+) - related signaling events influence TLR9-induced IL-10 secretion in human B cells. Eur J Immunol 2014. May;44(5):1285–1298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.