

Abstract
Rationale:
We recently discovered pivotal contributions of stress kinase JNK2 in increased risk of atrial fibrillation (AF) through enhanced diastolic sarcoplasmic reticulum (SR) Ca2+ leak via ryanodine receptors (RyR2). However, the role of JNK2 in the function of the SR Ca2+-ATPase (SERCA2), essential in maintaining [Ca2+]SR cycling during each heartbeat, is completely unknown.
Objective:
To test the hypothesis that JNK2 increases SERCA2 activity [Ca2+]SR and exacerbates an arrhythmic [Ca2+]SR leak-load relationship.
Methods and Results:
We used confocal Ca2+ imaging in myocytes and HEK cells, biochemistry, dual Ca2+/voltage optical mapping in intact hearts from alcohol-exposed or aged mice (where JNK2 is activated). We found that JNK2, but not JNK1, increased SERCA2 uptake and consequently elevated [Ca2+]SR load. JNK2 also associates with and phosphorylates SERCA2 proteins. JNK2 causally enhances SERCA2-ATPase activity via increased Vmax, without altering Ca2+ affinity (Km). Unlike the CaMKII-dependent JNK2 action in SR Ca2+ leak, JNK2-driven SERCA2 function was CaMKII-independent (not prevented by CaMKII inhibition). With CaMKII blocked, the JNK2-driven SR Ca2+ loading alone did not significantly raise leak. However, with JNK2-CaMKII-driven SR Ca2+ leak present, the JNK2-enhanced SR Ca2+ uptake limited leak-induced reduction in SR Ca2+, normalizing Ca2+ transient amplitude, but at a higher arrhythmogenic SR Ca2+ leak. JNK2-specific inhibition completely normalized SR Ca2+ handling, attenuated arrhythmic Ca2+ activities, and alleviated AF susceptibility in aged and alcohol-exposed myocytes and intact hearts.
Conclusions:
We have identified a novel JNK2-induced activation of SERCA2. The dual-action of JNK2 in CaMKII-dependent arrhythmic SR Ca2+ leak and a CaMKII-independent uptake exacerbates atrial arrhythmogenicity, while helping to maintain normal levels of Ca2+ transients and heart function. JNK2 modulation may be a novel therapeutic target for AF prevention and treatment.
Keywords: c-jun N-terminal kinase, sarcoplasmic reticulum, calcium uptake, diastolic calcium leak, SERCA2, atrial fibrillation, Subject Terms
INTRODUCTION
Atrial fibrillation (AF) is the most common cardiac arrhythmia and leads to a high risk of mortality and associated morbidities, including stroke and heart failure (HF) among a large patient population.1-5 To date, effective options for pharmacological AF treatment and prevention are limited and the incomplete understanding of the underlying mechanisms of AF greatly hinders the development of new therapeutic strategies.
Binge alcohol drinking is known as an independent risk factor for cardiac arrhythmias and often occurs during holidays and weekends, which has been termed as Holiday Heart Syndrome (HHS).5, 6 And AF is the most frequently diagnosed arrhythmias among patients suffering from HHS.5-8 Meanwhile, advanced age is an inevitable and predominant risk factor for AF.1-4 Our group recently found that both aging and excessive binge alcohol exposure drive activation of the stress kinase c-Jun N-terminal kinase2 (JNK2) in the atria, which consequently plays a critical role in atrial arrhythmogenesis in both humans and animal models.1, 2, 9, 10 While the contributions of JNK1 in cellular apoptosis and proliferation have been well-studied, the function of JNK2, one of the two major cardiac isoforms, has received less attention.10-12 We reported for the first time that JNK2 specifically enhanced atrial arrhythmic Ca2+ events and AF susceptibility mediated by a CaMKII-dependent diastolic SR Ca2+ leak through a kinase-to-kinase crosstalk (JNK2 activated CaMKII) in both aged and binge alcohol exposed hearts.1, 2 Remarkably, we found that this JNK2-driven SR Ca2+ leak was associated with elevated [Ca2+]SR load, opposite to the lower levels one would expect from the high level of diastolic leak. However, underlying mechanisms of this discrepant phenomenon of JNK2-drive [Ca2+]SR cycling remain incompletely understood.
Dynamic [Ca2+]SR cycling is essential to the function of cardiac muscles. During each heartbeat, Ca2+ is released from the SR via Ca2+-triggered Ca2+ release channel (RyR2), which increases intracellular Ca2+ ([Ca2+]in) and initiates myocyte contraction.13 In the myocyte relaxation phase, the SR Ca2+ release almost completely shuts off (~99%) and cytosolic Ca2+ is pumped back into the SR by the Ca2+ pump (SERCA2) and extruded from the cell via the Na+/Ca2+ exchanger (NCX).13 Under pathological conditions, aberrant diastolic SR Ca2+ release (diastolic SR Ca2+ leak) occurs slowing down the diastolic [Ca2+]in decline, which can lead to arrhythmogenic Ca2+ activities.14, 15 While SERCA2 is critical in maintaining the normal [Ca2+]SR dynamic cycling via its function of SR Ca2+ uptake, increased uptake could result in overloaded [Ca2+]SR and consequently augment diastolic leak. Here, we used multiple methods including biochemical assays and confocal Ca2+ imaging in atrial myocytes and HEK-RyR2 cells as well as dual channel (Ca2+ and voltage) optical mapping in intact hearts revealing a previously unrecognized JNK2-specific regulation of SERCA activity and the functional consequences in [Ca2+]SR dynamic cycling and enhanced atrial arrhythmogenicity.
METHODS
All data and supporting materials have been provided with the published article.
An expanded Methods Section is available in the Supplemental Material and all essential research materials are listed in the Major Resources Table in the Data Supplement.
Animal, Cell, Human Atrial Preparations.
All animal studies followed the Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition, 2011) and were approved by the Institutional Animal Care and Use Committees of Rush University Medical Center. New Zealand white male rabbits and wild-type (WT) C57B/6j male young and aged mice as well as phospholamban knockout (PLNKO) and WT littermate mice were studied as previously described.2, 16 Human atrial tissues were obtained from fifteen human donors provided by the Illinois Gift of Hope Organ & Tissue Donor Network (GOH). All studies were approved by the Human Study Committees of Rush University Medical Center, and Illinois GOH. Consent was obtained by GOH from the donors’ families for use of donor hearts for research.
Electrophysiological Studies and Biochemical Assays.
Frequencies of Ca2+ waves and the time constant (tau) of Ca2+ transient decay of the confocal images in rabbit atrial and ventricular myocytes, aged mouse myocytes, HL-1 atrial myocytes, and HEK-RyR2 cells transfected with tGFP-JNK1 or tGFP-JNK2 vectors were analyzed. And tetracaine-sensitive SR Ca2+ leak, caffeine-released [Ca2+]SR load, thapsigargin-sensitive SERCA2 uptake were measured using our well-established protocols as previously described.1, 2, 10, 15, 17-20 Dual channel (Ca2+/Vm) optical mapping and AF inducibility were performed in young and aged mouse intact hearts as previously described.1, 2, 9, 10, 21 JNK2 action on SERCA2 activity in human atrial SR microsomes was assessed using a well-established protocol as previously described.1 Immunoblotting assays and co-immunoprecipitation were used to assess protein abundance and the association between JNK2 and SERCA2 proteins as previously described.1, 9, 10, 15 The action of JNK2 on SERCA2 phosphorylation and activity was detected by Pro-Q Diamond gel staining and ADP-Glo™ Kinase assay and SERCA-ATPase activity assay as previously described.1, 22 Additional details are described in the Supplemental Methods.
RESULTS
JNK2 Increases Diastolic [Ca2+]SR Uptake and Load in Myocytes.
We imaged [Ca2+]in transients at 0.5 Hz pacing in isolated rabbit atrial myocytes with and without 0.23% ethanol (Alc; N.B) pretreatment for 24 hr followed by Alc washout 1 hr prior to [Ca2+]in measurements. Fig. 1A shows the kinetics of twitch [Ca2+]in decline before and after complete SERCA2 inhibition by thapsigargin (TG) to infer the TG-sensitive SERCA2 dependent [Ca2+]SR uptake in situ.18, 19 To avoid complication by our recently described JNK2-induced CaMKII-dependent diastolic SR Ca2+ leak during the uptake measurement,1, 2 we employed the widely used CaMKII inhibitor KN93.1, 15 We found that Alc-pretreatment accelerated [Ca2+]SR uptake rate in rabbit atrial myocytes compared to sham controls (Fig. 1B, middle vs. left bar), suggesting roughly a doubling of SERCA2-specific Ca2+ uptake. This SERCA2 activation was completely suppressed by our well-validated JNK2-specific inhibitor (JNK2I),1, 9, 10 which slowed myocyte [Ca2+]SR uptake to that in sham controls (Fig. 1B, right vs. left bars). To test whether this JNK2 action of enhanced [Ca2+]SR uptake is atrial-specific, we also tested this in isolated ventricular myocytes from the same rabbits as used above, which showed similar enhancement of SERCA2 uptake as that in atrial myocytes (Online Fig. IA). Consistent with the findings in Alc-exposed rabbit myocytes, aged mouse ventricular myocytes also showed markedly faster [Ca2+]SR uptake rate (Online Fig. IB).
Figure 1.

JNK2, but not JNK1 increases SR Ca2+ uptake and load. A) An example trace of the thapsigargin (TG)-sensitive SERCA uptake confocal measurement protocol. B) Summarized data showing shortened tau of TG-sensitive Ca2+ removal by SERCA2, reflecting an increased SR Ca2+ uptake, in alcohol (Alc)-exposed rabbit atrial myocytes (in the presence of 1 μM CaMKII inhibitor KN93 to inhibit JNK2-driven diastolic leak) compared to controls. JNK2 inhibition by 170 nM JNK2 inhibitor (JNK2I) reverses the uptake to normal levels seen in controls. C-D) Pooled data of increased [Ca2+]SR load (measured with 10 mM caffeine-induced Ca2+-released transients) in alcohol-exposed rabbit atrial myocytes and HL-1 myocytes; JNK2 specific inhibition by JNK2 inhibitor (JNK2I) completely prevented this alcohol-elevated [Ca2+]SR load. E) Pooled data of a higher [Ca2+]SR load (caffeine-induced Ca2+ transients) in tGFP-JNK2 positively expressed HEK-RyR2 cells (tGFP-JNK2) compared to tGFP-JNK1 positive cells (tGFP-JNK1) and sham controls; the JNK2 action in [Ca2+]SR load is completely eliminated by the JNK2 inhibitor (JNK2I) treatment. F) Summarized quantitative data of tGFP fluorescence intensity from those imaged HEK-RyR2 cells with positively expressed tGFP-JNK1 and tGFP-JNK2 cells and no tGFP in non-transfected controls (Sham). All quantified tGFP-JNK1 and tGFP-JNK2 fluorescence signals were subtracted to non-specific fluorescence background in non-transfected sham control cell images. G) Representative immunoblotting images (reflecting the mean values of quantitative data) showing a similar expression amount of endogenous SERCA2 and exogenous JNK2 and JNK1 proteins (detected by the tGFP antibody) in tGFP-JNK1 and tGFP-JNK2 transfected HEK-RyR cells vs non-transfected controls (Sham).
Since activated JNK2 increases SR Ca2+ uptake rate, the diastolic [Ca2+]SR load would be expected to increase. Indeed, we found significantly increased [Ca2+]SR load by measuring the amplitude of 10 mM caffeine-induced Ca2+ transients in Alc-exposed rabbit atrial myocytes compared to sham controls (Fig. 1C, left vs. middle bars). Again, JNK2-specific inhibition by JNK2I eliminated this Alc-evoked elevation of [Ca2+]SR load (Fig. 1C, far right bar). The contribution of JNK2 to elevated [Ca2+]SR load was further confirmed in our well-characterized HL-1 atrial myocytes.9, 10 Fig. 1D shows increased [Ca2+]SR load in Alc-exposed HL-1 atrial myocytes, while JNK2 inhibition completely prevents the elevation of [Ca2+]SR load. Similar to the finding in atrial myocytes, JNK2 also drives Alc-enhanced [Ca2+]SR load in rabbit ventricular myocytes (Online Fig. IC). Thus, JNK2 increases diastolic SR Ca2+ uptake and consequently elevates [Ca2+]SR load in both atrial and ventricular myocytes.
JNK2, but not JNK1, Elevates [Ca2+]SR Load in a Heterologous Cell Model.
JNK1 and JNK2 are the major isoforms in the heart, while JNK3 is expressed at a much lower level.23 To further determine the specific action of JNK2, but not JNK1 isoform, in the [Ca2+]SR load, we transfected HEK-RyR2 cells24 with tGFP-JNK2 or tGFP-JNK1 vectors, respectively, followed by the measurement of caffeine-released [Ca2+]SR load in tGFP positively expressing cells using confocal cellular Ca2+ imaging. We found that JNK2-expressing cells exhibited significantly increased [Ca2+]SR load compared to sham controls, while JNK1-expressing cells did not (Fig. 1E). Moreover, additional JNK2 inhibition by JNK2I (24 hr) in JNK2-expressing cells completely prevented the elevation of the [Ca2+]SR load observed in tGFP-JNK2 cells (Fig. 1E, far right vs middle bars). This indicates that active JNK2 is responsible for enhancing [Ca2+]SR load. Quantitative tGFP fluorescence intensities in those imaged cells showed comparable levels between tGFP-JNK2 and tGFP-JNK1 transfected cells, while no tGFP was detected in non-transfected sham control cells (Fig. 1F). Immunoblotting using tGFP antibody further confirmed the similar abundance of exogenous tGFP-JNK2 and tGFP-JNK1 proteins between tGFP-JNK2 and tGFP-JNK1 transfected cells compared to undetectable tGFP in sham control cells (Fig. 1F). Moreover, the expression level of endogenous SERCA2 proteins detected by SERCA2-specific antibody using immunoblotting was also comparable between tGFP-JNK2 and tGFP-JNK1 transfected cells as well as sham control cells (Fig. 1G), suggesting that the protein abundance of endogenous SERCA2 and exogenous tGFP-JNK1 and tGFP-JNK2 proteins in the cells were unlikely to influence the specific functional impact of JNK2 on the [Ca2+]SR load. Thus, active JNK2, but not JNK1, specifically enhances [Ca2+]SR load.
JNK2 is Colocalized with SERCA2 Proteins.
We next performed co-immunoprecipitation (co-IP) experiments to determine whether JNK2 and SERCA2 proteins are associated within a molecular complex. First, JNK2 proteins were immunoprecipitated (IPed) using tGFP antibody-conjugated agarose beads from tGFP-tagged JNK2-expressing cell lysates followed by incubation with solubilized SERCA2 vesicles isolated from an insect expression system as previously described.25 We found that SERCA2 was associated with JNK2 proteins, evidenced from co-IPed SERCA2 with IPed tGFP (Fig. 2A). Conversely, pure active human JNK2 protein (EMD) was incubated with solubilized SERCA2 vesicles and JNK2 was found to co-IP with the pulled-down SERCA2 protein (Online Fig. II). Moreover, active hJNK2 was associated with SERCA2 in tGFP-JNK2 and Flag-SERCA2 co-transfected HEK cells vs. Flag-SERCA2-only controls, evidenced by both tGFP-JNK2 and phosphorylated JNK2 pulled down by SERCA2-specific antibody (Fig. 2B). Furthermore, this JNK2-SERCA2 association was confirmed by co-IP JNK2 with pulled-down SERCA2 in human atria (Fig. 2C). Finally, double immunofluorescence staining of SERCA2 and JNK2 in rabbit myocytes revealed that JNK2 like SERCA2, shows similar sarcomeric striation patterns, with some regions of co-localization (yellow, Fig. 2D). Thus, JNK2 is associated with SERCA2, which could directly impact SERCA2 function.
Figure 2.

JNK2 is physically associated with SERCA2 and phosphorylates SERCA2. A) Immunoblotting images of co-immunoprecipitated SERCA2 proteins are associated with pull-down tGFP-JNK2 proteins with a tGFP-specific antibody. B) Immunoblotting images of co-immunoprecipitated JNK2 including phosphorylated JNK2 (JNK-P, detected by phospho-specific JNK antibody) and tGFP-tagged JNK2 (detected by tGFP antibody) with SERCA2-specific antibody pull-down Flag-tagged SERCA2 proteins (Flag-SERCA2, detected by Flag antibody) in HEK cells co-transfected with tGFP-JNK2 and Flag-SERCA2 vectors, while negative control (Flag-SERCA2 alone transfection) is in the left lane. C) Immunoblotting images of co-immunoprecipitated SERCA2 proteins with a SERCA2-specific antibody are associated with pull-down JNK2 proteins in human atrial homogenates, while negative control (no SERCA2 antibody) is in the left lane. D) Double immunofluorescence staining of JNK2 and SERCA2 antibodies in rabbit myocytes showing a typical striation pattern of SR for both JNK2 (green) and SERCA2 (red). Far right panel shows an enlarged image of well-colocalized JNK2 and SERCA2 signals (yellow arrows) distributing in a striation pattern. E) Summarized data of increased ADP production of pure active-hJNK2 (but not nonactive-hJNK2) phosphorylation of hSERCA2-PN proteins, with either 10 nM Ca2+ (left panel) or 10 μM Ca2+ (right panel), while the level of ADP production was minimal in the baseline controls of either hJNK2 alone and SERCA alone. F) A Pro-Q Diamond staining gel image of phosphorylated SERCA2-PN proteins (160 ng) by active-hJNK2 (far-right lane) and baseline phosphor-signals of SERCA2-PN alone (middle lane), and undetectable signal of active-hJNK2 alone (far-left lane).
JNK2 Phosphorylates SERCA2.
To investigate whether the physically associated JNK2 directly phosphorylates SERCA2, we incubated pure human active JNK2 (hJNK2, Millipore-Sigma) with pure recombinant human SERCA2 protein (hSERCA2-PN) that contains the nucleotide-binding (N) and phosphorylation (P) domains (Val314-Met756, Origene), but that lacks the transmembrane and actuator domains. Useful for us here is that this truncated hSERCA2-PN lacks appreciable ATPase activity at either low or high Ca2+ (10 nM or 10 μM), when compared to human atrial SR vesicles (Online Fig. IIIA). This allowed us to use pure hSERCA2-PN and hJNK2 alone in kinase assays using ADP-Glo™ (Fig. 2E). Non-active hJNK2, with or without hSERCA2-PN, exhibited no ATP consumption (right 2 bars). This confirms that the Nonactive hJNK2 is functionally inactive (also tested in Online Fig. IIIB) and that hSERCA2-PN does not consume ATP even at high Ca2+. The active purified hJNK2 alone only consumes a tiny amount of ATP (2 white bars), but is greatly increased by the presence of hSERCA2-PN as a kinase substrate (at both low and high Ca2+; red bars). The actual phosphorylation of the hSERCA2-PN was also confirmed and visualized by the Pro-Q™ Diamond phosphoprotein gel staining (Fig. 2F). Therefore, our results demonstrate that activated JNK2 directly phosphorylates the SERCA2 protein in a simple two-protein only system.
JNK2 Enhances SERCA2-ATPase Activity.
To further assess the JNK2 action in the SERCA2 function, a well-validated SERCA2-ATPase activity assay12, 13 was conducted using isolated human atrial SR vesicles. We found that 0.23% Alc-exposure (30 min, 37°C) significantly increased SERCA2-ATPase activity at 10 μM Ca2+, and JNK2 inhibition (JNK2I) eliminated this Alc-evoked SERCA2-ATPase activity but had no effect on sham controls (Fig. 3A). These results suggest that the JNK2 action on the SERCA2-ATPase activity was not due to any off-target effects of the JNK2 and the baseline levels of the SERCA2-ATPase activity were not affected by JNK2I. Next, we incubated purified hJNK2 proteins with human atrial SR vesicles for different times (at 37 °C). Within 20 min, there was a stable increase in the Vmax of SERCA2-ATPase activity, without a change in (Km) compared to sham controls (Fig. 3B). This ~20% increase in ATPase Vmax is similar to the Alcohol-induced increase in Fig. 3A that was prevented by JNK2 Inhibitor. The SERCA2-specific inhibitor thapsigargin completely suppressed Ca2+-dependent SERCA2 activity (Fig. 3B). In addition, the pure non-active-hJNK2 had no effect on the Ca2+-dependent ATPase compared to sham controls (Online Fig. IIID). In line with these results, pure active-hJNK2 phosphorylation of pure SERCA2-PN protein in the 2-protien system showed a time-dependent increase (Fig. 3D). These results demonstrate that JNK2 is a critical enhancer of SERCA2 activity via a Vmax effect, which differs from the primarily Km effect of PLB and PLB phosphorylation.
Figure 3.

JNK2 increases the maximal rate of SERCA2-ATPases activity. A) Summarized data showing alcohol exposure (0.23%, 24hr) increases thapsigargin (TG)-sensitive SERCA2-ATPase activity in isolated human atrial SR vesicles, but the treatment of JNK2-specific inhibitor (JNK2I) attenuates the alcohol-enhanced SERCA2-ATPase activity. JNK2I (170 nM) treatment shows no inhibitory effect on the baseline activity of SERCA2 in sham human atrial SR vesicles. B-C) pooled data of Ca2+-dependent curves of SERCA2-ATPase activity (B) and Ca2+ affinity (Km, C) in human atrial SR vesicles incubated with pure active hJNK2 (64ng) at reaction times of 10 (green), 20 (blue), 30 (pink), and 60 (red) min compared to sham controls (black). And summarized data showing suppressed Ca2+-dependent responses of SERCA2-ATPase activities by incubating with 0.5 μM thapsigargin (brown open squares) compared to sham controls (black dots). E) Pooled data of time-dependent phosphorylation of hSERCA2-PN by 10 ng pure active hJNK2 (10 μM Ca2+).
JNK2 Enhances SERCA2 Function in a CaMKII-Independent Manner.
CaMKII is a well-known pro-arrhythmic signaling kinase that can promote SERCA2 activity via phospholamban.1, 15 However, CaMKII inhibition did not suppress the JNK2-enhanced SERCA2-ATPase activity, indicating this SERCA2 activation is CaMKII-independent (Fig. 4A, far right bar). Consistent with those findings, CaMKII inhibition by KN93 also failed to attenuate Alc-evoked elevation of [Ca2+]SR load in Alc-exposed HL-1 myocytes (Fig. 4B), which contrasts with the profound rescue effect of CaMKII inhibition on the diastolic SR Ca2+ leak we have previously demonstrated.1, 2 To further test the contribution of JNK2, we treated HL-1 atrial myocytes with a previously characterized JNK activator, anisomycin, for 24 hr to activate JNK2.9, 10 As for Alc-pretreatment, the anisomycin-induced elevation of [Ca2+]SR load was also unaffected by CaMKII inhibition (Fig. 4C).
Figure 4.

JNK2 increases SERCA2-ATPase activity in a CaMKII-independent manner. A) Summarized data showing active purified human JNK2 (hJNK2) proteins increase thapsigargin-sensitive SERCA2-ATPase activity in isolated human SR vesicles, but additional treatment of CaMKII inhibitor KN93 (1 μM) has no inhibitory effect on this JNK2-enhanced SERCA2-ATPase activity. B) Pooled data of elevated [Ca2+]SR load in 0.23% alcohol (Alc)-exposed (24 hr) HL-1 myocytes with the treatment of CaMKII inhibitor KN93 compared to sham controls (sham) and positive controls treated with KN92 (an inactive analogue of KN93), suggesting lack of action of CaMKII on alcohol-elevated load. C) Summarized data showing CaMKII inhibition has no effect on 50 nM anisomycin (Aniso)-induced increase of [Ca2+]SR load in KN93+Aniso treated as well as KN92+Aniso treated HL-1 myocytes compared to sham controls (sham). D) Summarized data of [Ca2+]SR load in isolated myocytes from alcohol-exposed PLNKO mice showing an increase in [Ca2+]SR load compared to vehicle-treated PLNKO sham controls and JNK2 inhibitor JNK2I treatment precluded this alcohol-evoked increase of [Ca2+]SR load, while lack of PLB in PLNKO mice (vehicle-treated sham controls) markedly increases [Ca2+]SR load compared to WT littermate controls. E) Pooled data of Ca2+-dependent curves of SERCA2-ATPase activity in anti-PLB antibody 2D12 (6 ug) pre-treated human atrial SR vesicles incubated with (pink) and without (blue) pure active hJNK2 (64 ng) compared to sham controls (black).
PLB is known as an important regulator of SERCA2 function through phosphorylation of PLB at the threonine 17 site (PLB17) by CaMKII and the serine 16 site (PLB16) by PKA. Alcohol exposure could enhance PKA-dependent PLB16 phosphorylation, but we found unchanged PLB16 phosphorylation or PKA expression in Alc-exposed human atria (Online Figs. IVA-IVB). While SERCA2 expression was unchanged in alcohol-exposed human, rabbit, and mouse atria (Online Figs. IVC-IVE), we have previously shown that JNK2 leads to increased CaMKII-dependent PLB17 phosphorylation.1 However, the fact that CaMKII inhibition does not prevent the JNK2 induced increase in SR load suggests that PLB phosphorylation is not predominant in JNK2-induced [Ca2+]SR load. We then performed additional experiments to measure [Ca2+]SR load in binge alcohol-exposed PLB knockout (PLNKO) mice.16 We found that the higher SR Ca2+ content expected in PLNKO mice vs. WT littermates (Fig. 4D, left bars). But in binge alcohol-exposed PLNKO myocytes there was an apparent rise in SR Ca2+ content induced by alcohol pretreatment (gray arrow) that was not significant, perhaps because the SR Ca2+ content is very close to maximal in the PLNKO myocytes. However, there was a significant JNK2I-sensitive component of [Ca2+]SR load (right bars, black arrow). This indicates that PLB is not required for the JNK2-induced increase in SERCA2 function.
Next, the PLB independence of the JNK2 specific action on SERCA2-ATPase activity of human atrial SR vesicles was further tested by using an anti-PLB specific antibody (2D12) that displaces PLB from SERCA2.25 Fig. 4E shows that 2D12 shifted the Ca2+-dependent SERCA2-ATPase curve to the left, without altering Vmax, confirming the acute removal of PLB effect on SERCA2. In this setting, purified active hJNK2 proteins significantly increased Vmax. This additional evidence further supports that there is a PLB-independent mechanism by which JNK2 enhances the Vmax of SR Ca2+ uptake. And these complementary tests confirm that the JNK2-induced enhancement of SERCA2 function is PLB-independent. Thus, JNK2 is a critical enhancer of SERCA2 activity and this JNK2-specific action in SERCA2 function is both CaMKII-and PLB-independent.
JNK2 has No Effect on NCX-Mediated Ca2+ Extrusion in Myocytes.
During myocyte relaxation, Ca2+ is removed from the cytosol via pumping back to SR by SERCA2 and extrusion from the cell by NCX.13 Thus, NCX changes could also influence diastolic [Ca2+]in decline and indirectly influence [Ca2+]SR load. We thus measured NCX-mediated Ca2+ extrusion via assessing the tau of [Ca2+]in decay during a rapid application of 10 mM caffeine.1 Figs. 5A-5B show that NCX function was unchanged by Alc-pretreatment in both rabbit and HL-1 atrial myocytes compared to sham controls, nor did JNK2 inhibition by JNK2I alter NCX function.
Figure 5.
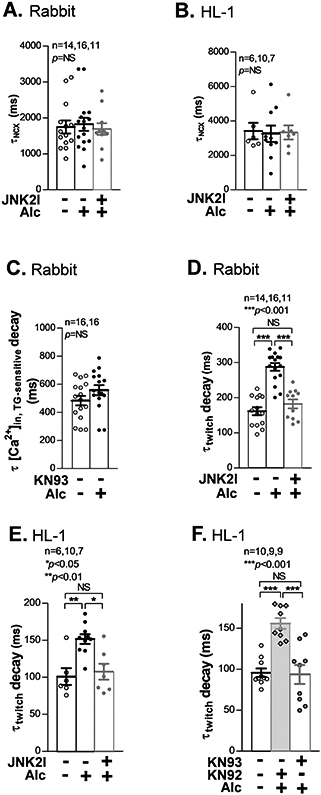
JNK2 prolongs twitch [Ca2+]in decline due to increased diastolic [Ca2+]SR leak (CaMKII-dependent). A-B) Summarized data showing unchanged tau of [Ca2+]in decay of caffeine-induced Ca2+ transients (τNCX), reflecting unchanged contribution of NCX to the diastolic [Ca2+]in removal, in alcohol-exposed rabbit (A) and HL-1 (B) atrial myocytes vs sham controls. C) Summarized data of increased tau of thapsigargin-sensitive [Ca2+]in (τ[Ca]in.TG-sensitive) decay in the absence of KN93, indicating that the presence of CaMKII-dependent [Ca2+]SR leak masks the contribution of [Ca2+]in removal by SERCA2 in 0.23% alcohol-exposed rabbit atrial myocytes compared to controls. D-E) Pooled data of a prolonged tau of [Ca2+]in (τtwitch) decay in alcohol-exposed rabbit myocytes (D) and HL-1 myocytes (E) compared to sham controls; and prevented by 170 nM JNK2I treatment (far right bars). F) Pooled data showing a completely suppressed tau of [Ca2+]in (τtwitch) decay, to the level seen in sham controls (sham), by CaMKII inhibition (1 μM KN93 treatment) in 0.23% alcohol-exposed HL-1 myocytes compared to a significantly prolonged [Ca2+]in decline in alcohol-exposed myocytes treated with 1 μM KN92 (inactive analogue of KN93).
JNK2-induced [Ca2+]SR Leak Slows Twitch [Ca2+]in Decline in Atrial Myocytes.
We have recently revealed that JNK2 drives diastolic SR Ca2+ leak via a CaMKII-dependent RyR2 channel dysfunction.1, 2 The JNK2-driven SERCA2 uptake and RyR2-mediated leak would individually cause opposing effects on both the rate of twitch [Ca2+]in decline and [Ca2+]SR load. So, which effect is dominant? The fact that diastolic [Ca2+]SR load was increased in the absence of CaMKII inhibitor (when both JNK2 effects are functional; Fig. 1C) demonstrates that the SERCA activation is dominant for diastolic [Ca2+]SR load. Conversely, the JNK2-induced acceleration of TG-sensitive twitch [Ca2+]in decline was not seen when leak was also enhanced in Alc-exposed rabbit atrial myocytes in the absence of CaMKII inhibition by KN93 (Fig. 5C). This JNK2 action was also consistently found in both Alc-exposed and aged ventricular myocytes (Online Figs. VA-VB). That is clearly different from Fig. 1B where CaMKII (and leak) were inhibited, and Alc-accelerated TG-sensitive [Ca2+]in decline become detectable. Furthermore, with the JNK2-induced SR Ca2+ leak present the overall rate of twitch [Ca2+]in decline was substantially slowed (τtwitch, Fig. 5D). Thus, during twitch relaxation, the enhanced SR Ca2+ leak is the more dominant effect. This indicates that despite the faster SERCA2 pumping rate, some Ca2+ that had been taken up by the SR then leaked out during the same beat, to be pumped in again. Exactly the same pattern of effects was observed in HL-1 myocytes (Figs. 5E-5F), where the Alc-induced slowing of overall twitch [Ca2+]in decline (τtwitch) occurred, but this effect was reversed by either JNK2 inhibition (JNK2I) or CaMKII inhibition (KN93). Thus, JNK2 activation enhances both diastolic [Ca2+]SR uptake and leak, but the JNK2-enhanced uptake is sufficient to overcome the leak during diastole to setup a new leak-uptake relationship, resulting in an elevated diastolic [Ca2+]SR load. In contrast, the robust leak during the twitch is sufficient to delay the overall rate of [Ca2+]in decline.
JNK2 Alters the SR Ca2+ Leak-Load Relationship to Enhance Arrhythmogenicity of Atrial Myocytes and Intact Atria.
We next assessed JNK2 actions in the SR Ca2+ leak-load relationship. We measured both [Ca2+]SR load (caffeine-induced Ca2+ transients) and tetracaine-sensitive diastolic leak using our well-established tetracaine-sensitive protocol (with NCX blocked)1, 2, 15, 20 in HL-1 myocytes with either Alc-pretreatment or anisomycin to activate JNK2 and JNK2I or KN93 to inhibit JNK2 or CaMKII, respectively. Alcohol and anisomycin raised SR Ca2+ leak and load, and both were fully reversed by selective JNK2 inhibition. In contrast, CaMKII inhibition with KN93 (vs. KN92 control), selectively suppressed the rise in SR Ca2+ leak, but not the rise in the load (Figs. 6A-6B). These results provide further support that JNK2-elevated [Ca2+]SR load is CaMKII-independent, while JNK2-driven diastolic leak is CaMKII-dependent. Notably, the JNK2-elevated [Ca2+]SR load alone (with KN93 present) did not cause a substantial increase in diastolic leak. This agrees with the finding in Fig. 5F, where CaMKII inhibition completely rescued tau of twitch [Ca2+]in decline induced by Alc-evoked JNK2 activation. In other words, CaMKII inhibition nearly completely stopped JNK2-enhanced diastolic leak and prevented JNK2-induced slowing of twitch [Ca2+]in decline, but without suppressing the higher load.
Figure 6.

JNK2 regulates a [Ca2+]SR load (CaMKII-independent) and leak (CaMKII-dependent) relationship, which driving arrhythmic activities in alcohol-exposed rabbit atrial myocytes. A-B) Plot of the amount of diastolic [Ca2+]SR leak and load in alcohol-exposed (A) and JNK activator anisomycin (Aniso or A)-treated (B) HL-1 myocytes in the presence of 1 μM KN93 or 1 μM KN92 compared to sham controls. Red arrows indicate completely attenuated higher SR Ca2+ leak and load by JNK2-specific inhibition (JNK2I) in both alcohol-exposed and anisomycin (Aniso or A)-treated (B) HL-1 myocytes and green arrows indicate abolished leak but unaffected load by CaMKII inhibition by KN93 but not KN92, an inactive analogue of KN93. C) Representative confocal images of normal Ca2+ transients (0.5Hz) from a sham-control myocyte (upper), multiple Ca2+ waves from an alcohol-exposed myocyte (middle), and normal Ca2+ transients from a JNK2I treated alcohol-exposed atrial myocyte (lower). D) Summarized data showing significantly increased frequency of pacing (0.5Hz)-induced Ca2+ waves in alcohol (Alc)-exposed atrial myocytes compared to sham controls, while these alcohol-evoked changes are eliminated by either JNK2 inhibitor (JNK2I) or CaMKII inhibitor KN93 treatment (far right bars). E) Representative action potential traces of alcohol-exposed (0.23%, 24 hr) and sham control rabbit myocytes showing delayed afterdepolarization (DAD) events at 1Hz electrical pacing, while no DAD events were induced in sham controls. DADs are indicated by the arrows in red.
Using confocal Ca2+ imaging, we found that Alc-exposed atrial myocytes exhibited increased Ca2+ waves (vs sham controls) when a bout of electric pacing (0.5 Hz) was applied (Figs. 6C-6D). However, if those myocytes were pretreated with either the JNK2-specific inhibitor JNK2I or CaMKII inhibitor KN93 during the 24 hr Alc-pretreatment, the Alc-evoked arrhythmic Ca2+ waves were prevented (Figs. 6C-6D). These arrhythmogenic events at the cellular level are consistent with the Alc-induced increase in diastolic leak and load as well as prolongation of diastolic [Ca2+]in decay, which are inherently larger in amplitude and are hence, more likely to trigger myocyte-wide Ca2+ sparks and waves and may consequently cause the larger DADs recorded using patch clamp (Fig. 6E, 1Hz, 37 °C). In addition, we performed dual channel optical mapping1, 2, 9, 21 in Langendorff-perfused aged mouse hearts where JNK2 activates leak.1 We found significantly increased spatiotemporal heterogeneity in the delay between the Ca2+ and voltage (Vm) signals (ΔtVm-Ca), evidenced by increased standard deviation of ΔtVm-Ca in aged intact atria subjected to 10 Hz electrical pacing (Figs. 7A-7B). Consistently, aged atria also showed increased pacing-induced AF compared to young controls (Fig. 7C). With regards to the cellular effects, the highly arrhythmogenic atrial spatial heterogeneity of ΔtVm-Ca and pacing-induced AF were completely prevented by the in vivo JNK2 inhibition (5 mg/kg JNK2I, I.P, for five treatments) in aged mice compared to vehicle-treated aged controls (Figs. 7B-7C). Thus, JNK2 drives diastolic [Ca2+]SR mishandling that promotes arrhythmic Ca2+ activities and enhances AF susceptibility.
Figure 7.

JNK2 drives the heterogeneous relationship between Vm and Ca2+ in aged intact atria. A) Representative isochronal optical maps of simultaneously recorded membrane voltage potentials (Vm; Rh237) and calcium transients (Ca2+; Rhod2) in aged and young atria subjected to a pacing cycle length (CL) of 100 ms (10 Hz). B-C) Summarized data showing a markedly enhanced heterogeneous relationship between Vm and Ca2+ signals. (B) along with increased AF inducibility (C) in aged mouse atria, evidenced by the significantly increased standard deviation of the ΔCa-Vm values from the mapping field vs young controls. And 10 mg/kg JNK2-specific inhibitor (JNK2I) in vivo I.P. treatment in aged WT mice attenuated heterogeneity of ΔCa-Vm and pacing induced AF events (far right bars) compared to untreated aged and young control WT mice.
DISCUSSION
In the current study, we revealed for the first time that the stress kinase JNK2 is a previously unrecognized enhancer of SERCA2 function. We found that JNK2 and SERCA2 proteins are physically associated with each other, and that JNK2 directly elevates the maximal rate of the SERCA2 activity by phosphorylating SERCA2. We also revealed that JNK2 (but not JNK1) plays a dual role in the SR Ca2+ dynamics - JNK2-ehnanced SR Ca2+ uptake partially compensates for the greater SR Ca2+ leak and maintains a normal level of Ca2+ transients and normal cardiac function, while the greater JNK2-driven leak acts as a key contributor to enhanced arrhythmogenicity (Fig. 8). Another novel finding is that this JNK2-specific action in the SERCA2 function is CaMKII-independent, which is unlike the JNK2 action in the diastolic SR Ca2+ leak (CaMKII-dependent) as we recently reported.1, 2 Our new findings are significant because these JNK2 specific actions on the [Ca2+]SR mishandling shed new light on modulating JNK2 as a promising anti-AF therapeutic strategy.
Figure 8.

Schematic diagram of demonstrated mechanism of JNK2-driven diastolic leak-uptake relationship, which enhances triggered arrhythmic activities and AF risk.
JNK2 is a Newly Identified Enhancer of SERCA2 Function.
A novel finding in the current studies is the identification of JNK2 as a previously unrecognized functional enhancer of SERCA2 function. SERCA2 pump activity is known to be regulated by PLB, sarcolipin, myoregulin, and a newly identified peptide Dworf.25-27 Our findings demonstrate that JNK2 and SERCA2 proteins are associated within a molecular complex suggesting a close spatial link between the two proteins. We also reveal, for the first time, that JNK2 increases SR Ca2+ uptake via its direct action on SERCA2 phosphorylation and consequently the SERCA2 pump activity. Our finding of JNK2-specific phosphorylation of SERCA2 within the SERCA2 P & N domains using purified human SERCA2-PN proteins was consistent with a recent report showing the direct phosphorylation of SERCA2-N domain by a striated muscle preferentially expressed protein kinase (SPEG).28 Whether SPEG is related to this JNK2-specific action in the SERCA2 activity may be worthy of further investigations.
We have previously shown that JNK2 drives diastolic SR Ca2+ leak via a CaMKII-mediated RyR2 channel dysfunction.1, 2 Here, we discovered that the JNK2-specific action on the SERCA2 function is CaMKII-independent. JNK2 promoted CaMKII-dependent PLB17 phosphorylation was actually found in aged atria as we recently reported.1 We originally speculated that this CaMKII-dependent PLB17 phosphorylation could contribute to the surprisingly elevated SR load. However, our current findings demonstrate a predominant contribution of JNK2 in enhanced SR Ca2+ uptake and elevated load via a direct regulation of SERCA2 pump activity, which is clearly independent of CaMKII. It is generally believed that PLB regulates SERCA2 Ca2+ affinity (Km) but does not change the maximal rate (Vmax). Unlike the mechanism of PLB regulation, our new findings here demonstrated that active JNK2 significantly enhanced the Vmax of SERCA2-ATPase activity but not the Km. Moreover, we found that JNK2 still mediates alcohol-enhanced [Ca2+]SR load and elevated Vmax of SERCA2 activity in the complete absence of PLB (in PLNKO mice) or after acute removal of PLB from SERCA2 by the 2D12 antibody. These results strongly support a direct regulation of JNK2 in enhanced SERCA2 function independent of PLB, but do not preclude an additional JNK2 effect on the PLB-SERCA2 interaction, in a manner that partially relieves PLB-dependent inhibition of the SERCA2 function.
The intriguing JNK2-SERCA2 functional link revealed in the current study was supported by evidence from several biological systems including atrial and ventricular myocytes, intact atria, HEK-RyR2 cells, and human atrial SR vesicles. Remarkably, this JNK2-SERCA functional link was consistently demonstrated in each of those biological systems from different species including rabbits, mice, and humans. Thus, this JNK2-regulated SERCA2 function likely represents a conserved mechanism of ‘adaptive response’ in the heart during the process of cardiac remodeling under stress conditions such as aging, binge alcohol abuse and other stressors. These new findings open a new door to further understand the molecular mechanism by which JNK2 regulates SERCA2 activity under physiological and pathological conditions.
Dual Role of JNK2 in SR Ca2+ Leak and Load, which Maintains Normal Ca2+ Transients but Exacerbates Atrial Arrhythmogenicity.
Another significant new finding of the current studies is that the dual JNK2 specific actions shift the relationship between SR Ca2+ leak and uptake. JNK, a member of the important MAPK family, is a well-characterized stress kinase that is activated in response to multiple stresses involved in the development of various cardiovascular diseases (HF, myocardial infarction, atherosclerosis etc.) and other diseases such as arthritis, and cancer.11, 29, 30 Our group has shown that activation of JNK2 causes enhanced atrial arrhythmic Ca2+ activities and AF susceptibility, mediated by a CaMKII-dependent diastolic SR Ca2+ leak in both aged and binge alcohol-exposed hearts.1, 2 However, our prior work left an unexplained dichotomy: JNK2 activation causes elevated SR leak and slowed twitch [Ca2+]i decline (suggestive of slowed SERCA2 function), but elevated higher [Ca2+]SR load. This opposes the expectation that higher SR Ca2+ leak and slowed Ca2+ reuptake would deplete SR load. This is what occurs in HF15, 31 and when HF is induced by transgenic CaMKIIδ overexpression,32 where increased diastolic SR leak is accompanied by reduced [Ca2+]SR load, which contributes to reduced Ca2+ transients and contractions.
Our discovery here, that JNK2 directly activates SERCA2, explains the aforementioned dichotomy as to how neither diastolic [Ca2+]SR nor systolic Ca2+ transients in atria are depressed via JNK2 activation induced by aging, binge-alcohol or anisomycin. Indeed, the joint JNK-induced increases in SR Ca2+ leak and uptake rate may be adaptive in maintaining cardiac output, but at the cost of increased arrhythmogenicity. These two JNK2-specific actions shift the diastolic SR Ca2+ leak -load relationship as illustrated in Fig. 6A-B. Thus, our conclusion here is that JNK2 activation indeed promotes both diastolic SR Ca2+ uptake and leak, but the JNK2-driven pump stimulation is sufficient to elevate the diastolic [Ca2+]SR load, despite a greater diastolic leak. We also unmasked the JNK2-mediated increase in SERCA2 uptake by analyzing the thapsigargin-sensitive rate of twitch [Ca]i decline with CaMKII inhibition. (Fig. 1B). This had been masked by the JNK2-driven and CaMKII-mediated increase in leak, that was large enough to cause slowing of [Ca]i decline, despite the accelerated SERCA2 function (Fig. 5C-F). That indicates that the CaMKII-induced SR leak (or continued release) is sufficient also to slow diastolic Ca2+ removal. Notably, cardiac function (normal ejection fraction and fraction shortening) was preserved with JNK2 activation in both aged and binge alcohol exposed hearts and in humans and animal models.1, 2, 9, 10 In another words, JNK2 may be a stress-induced regulator to maintain a high SR Ca2+ uptake and load in order to preserve cardiac function, even when SR Ca2+ leak is enhanced. Indeed, we found preserved normal amplitude of systolic Ca2+ transients (despite increased SR leak and a higher load) in both aged and binge alcohol exposed atrial myocytes.1, 2 Thus, this dual regulation of JNK2 in Ca2+ handling could be an adaptive response in the heart during the process of cardiac remodeling to heart failure under various of stressed conditions such as aging and binge alcohol abuse.
On the other hand, JNK2 critically enhances arrhythmogenesis through JNK2-specific actions in SR Ca2+ leak via sensitized RyR2 channels as we recently reported.1 This may in part be due to a delayed shut-off of Ca2+ release during the relaxation phase that occurs at JNK2-driven high [Ca2+]SR loads and diastolic leak (RyR2 channel sensitization mediated by the JNK2-induced CaMKII activation).1, 33 Thus, the robust leak during the twitch is sufficient to delay the overall rate of [Ca2+]in decline, which is likely to trigger arrhythmic activities. Although we surprisingly found that JNK2-enhanced [Ca2+]SR load by itself (in the absence of CaMKII-dependent RyR2 channel sensitization) is insufficient to cause significant diastolic leak, a combined higher load and CaMKII-sensitized RyR2 channel may promote the diastolic leak as we and others have previously demonstrated.1, 15, 31, 34 A highly analogous leak-dependent slowing of twitch [Ca2+]in decline was indeed found in CaMKIIδC transgenic mice (exhibiting CaMKII-dependent RyR2 leak) upon crossing with PLB-knockout mice (with boosted [Ca2+]SR uptake and load).19, 31 In that study, the PLNKO restored SR Ca2+ uptake, load, and normal contractility in individual myocytes, but the prominent Ca2+ sparks during twitch [Ca2+]in decline and diastole created increased arrhythmogenic activities and animal mortality. So, enhancing SERCA2 function to raise load in the face of a CaMKII-dependent SR Ca2+ leak may exacerbate the leak and have severe consequences (higher arrhythmogenesis). Interestingly, a concordant increase of SR Ca2+ leak and load was also reported in patients with paroxysmal AF.35 Thus, our findings emphasize the clinical significance of this particular mechanistic Ca2+ handling scenario in AF development, especially in aging and excessive binge alcohol exposure where we’ve demonstrated that the JNK2-specific actions on SERCA2 uptake, diastolic SR Ca2+ leak, and atrial arrhythmogenicity take place.1, 2
Implications.
It has been widely recognized that aging, alcohol abuse, and other cellular and environmental stressors increase atrial arrhythmogenicity, which significantly increases AF risk.1-8 AF is known to substantially increase the risk of morbidity and mortality, and these AF risk factors increase the vulnerability of developing HF.3, 4 With our findings revealing JNK2 as a previously unrecognized functional enhancer of SERCA2, it is especially intriguing to further investigate contributions of JNK2 in cardiac contractile function during the development of HF. Possible atria-ventricle chamber differences of the JNK2 actions during cardiac remodeling also warrant further studies. Moreover, our studies here reveal a novel underlying mechanism of stress kinase JNK2-driven diastolic SR Ca2+ uptake-leak relationship that is critical for cardiac arrhythmic remodeling and enhanced AF risk in the settings of aging and excessive binge alcohol exposure. Importantly, our studies provide new evidence showing the clinical potential of modulating JNK2 and SR Ca2+ leak as novel therapeutic strategies in anti-AF drug development.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Atrial fibrillation (AF), the most common sustained arrhythmia, is a major public health issue and lacks effective pharmacological therapies.
Stress kinase JNK2 contributes to enhanced AF risk in the settings of advanced age and alcohol abuse,.
Stress kinase JNK2 also drives diastolic SR Ca leak and promotes arrhythmic Ca activities that, in turn, enhances atrial arrhythmogenicity in the aged oralcohol exposed hearts.
What New Information Does This Article Contribute?
We have identified JNK2 as a previously unrecognized regulator of cardiac SERCA2 function through the phosphorylation of SERCA2 protein and consequently increased the maximal rate of the SERCA2-ATPase activity.
We found the JNK2-specific action in an arrhythmic [Ca2+]SR leak-uptake relationship, a likely mechanism coupling arrhythmia and biological stressors.
Our results identify JNK2 inhibition as a potential target for developing new therapeutic strategies to prevent and/or treat AF.
We recently identified the stress kinase JNK2 as playing a pivotal role in the development of atrial fibrillation, the most common arrhythmia associated with a high risk of mortality and morbidity. However, the role of JNK2 in the observed higher [Ca2+]SR load and greater diastolic [Ca2+]SR leak in the settings of aging, binge alcohol abuse, and paroxysmal AF was poorly defined. We demonstrate that JNK2 is a previously unidentified enhancer of SERCA2, an essential Ca pump in [Ca2+]SR dynamic cycling during each cardiac contraction. This JNK2-specific enhancement in the maximal rate of the SERCA2-ATPase activity occurs through phosphorylation of SERCA2 proteins. Further, we establish that JNK2 causes a [Ca2+]SR leak-uptake balance, which maintains normal Ca2+ transients but drives the arrhythmic Ca2+ activities and triggers atrial arrhythmias. Our findings show how SERCA2 activity is controlled by different regulators under physiological and various pathological conditions, and also demonstrate a potential therapeutic target (JNK2) that can be further studied for the treatment of AF..
Acknowledgments
SOURCES OF FUNDING
This work was supported by the Heart and Stroke Foundation Chair in Cardiovascular Research [to SRWC] and National Institutes of Health [R01-HL142282 and P01-HL141084 to DMB; P01-HL06426 to RJS & XA, R01-AA024769 to SRWC & XA; R01-HL113640, R01-HL146744, and AA024769S2 to XA].
Nonstandard Abbreviations and Acronyms:
- AF
atrial fibrillation
- Ag
aged
- Alc
alcohol-treated
- Ca2+
calcium
- [Ca2+]in
intracellular Ca2+ concentration
- [Ca2+]SR
sarcoplasmic reticulum Ca2+ content
- CaMKII
Ca2+/calmodulin-dependent kinase II
- co-IP
co-immunoprecipitation
- co-IPed
co-immunoprecipitated
- DAD
delayed after-depolarization
- ΔtVm-Ca
activation time difference between action potential and Ca2+ transient
- HF
heart failure
- HHS
Holiday Heart Syndrome
- hJNK
human JNK
- IP/IPed
immunoprecipitation/immunoprecipitated
- I.P.
intraperitoneal injection
- JNK
c-Jun N-terminal Kinase
- JNK1
c-Jun N-terminal Kinase isoform 1
- JNK2
c-Jun N-terminal Kinase isoform 2
- JNK2I
JNK2 inhibitor
- Km
SERCA2 Ca2+ affinity
- NCX
Na+/Ca2+-exchanger
- PLB
phospholamban
- PLNKO
PLB knockout mice
- RyR2
ryanodine receptor isoform 2
- SERCA2
sarcoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- tGFP
turbo-GFP
- tGFP-JNK1
turbo-GFP tagged JNK1
- tGFP-JNK2
turbo-GFP tagged JNK2
- tau/τ
time constant
- Vmax
maximal rate
Footnotes
DISCLOSURES
None.
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Yan J, Zhao W, Thomson JK, Gao X, DeMarco DM, Carrillo E, Chen B, Wu X, Ginsburg KS, Bakhos M, Bers DM, Anderson ME, Song LS, Fill M and Ai X. Stress Signaling JNK2 Crosstalk With CaMKII Underlies Enhanced Atrial Arrhythmogenesis. Circ Res. 2018;122:821–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan J, Thomson JK, Zhao W, Gao X, Huang F, Chen B, Liang Q, Song LS, Fill M and Ai X. Role of Stress Kinase JNK in Binge Alcohol-Evoked Atrial Arrhythmia. J Am Coll Cardiol. 2018;71:1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV and Singer DE. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–5. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ and Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. 1994;271:840–4. [PubMed] [Google Scholar]
- 5.Tonelo D, Providencia R and Goncalves L. Holiday heart syndrome revisited after 34 years. Arquivos brasileiros de cardiologia. 2013;101:183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ettinger PO, Wu CF, De La Cruz C Jr., Weisse AB, Ahmed SS and Regan TJ. Arrhythmias and the "Holiday Heart": alcohol-associated cardiac rhythm disorders. Am Heart J. 1978;95:555–62. [DOI] [PubMed] [Google Scholar]
- 7.Mandyam MC, Vedantham V, Scheinman MM, Tseng ZH, Badhwar N, Lee BK, Lee RJ, Gerstenfeld EP, Olgin JE and Marcus GM. Alcohol and vagal tone as triggers for paroxysmal atrial fibrillation. The American journal of cardiology. 2012;110:364–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voskoboinik A, Prabhu S, Ling LH, Kalman JM and Kistler PM. Alcohol and Atrial Fibrillation: A Sobering Review. J Am Coll Cardiol. 2016;68:2567–2576. [DOI] [PubMed] [Google Scholar]
- 9.Yan J, Thomson JK, Zhao W, Wu X, Gao X, DeMarco D, Kong W, Tong M, Sun J, Bakhos M, Fast VG, Liang Q, Prabhu SD and Ai X. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J Mol Cell Cardiol. 2017;114:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan J, Kong W, Zhang Q, Beyer EC, Walcott G, Fast VG and Ai X. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc Res. 2013;97:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose BA, Force T and Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogoyevitch MA. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): differences revealed by gene targeting. Bioessays. 2006;28:923–34. [DOI] [PubMed] [Google Scholar]
- 13.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–81. [DOI] [PubMed] [Google Scholar]
- 14.Kass RS and Tsien RW. Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys J. 1982;38:259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ai X, Curran JW, Shannon TR, Bers DM and Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22. [DOI] [PubMed] [Google Scholar]
- 16.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T and Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res. 1994;75:401–9. [DOI] [PubMed] [Google Scholar]
- 17.Yan J, Thomson JK, Wu X, Zhao W, Pollard AE and Ai X. Novel methods of automated quantification of gap junction distribution and interstitial collagen quantity from animal and human atrial tissue sections. Plos One. 2014;9:e104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bassani JW, Bassani RA and Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo T, Zhang T, Ginsburg KS, Mishra S, Brown JH and Bers DM. CaMKIIdeltaC slows [Ca]i decline in cardiac myocytes by promoting Ca sparks. Biophys J. 2012;102:2461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon TR, Ginsburg KS and Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. [DOI] [PubMed] [Google Scholar]
- 21.Fast VG. Simultaneous optical imaging of membrane potential and intracellular calcium. J Electrocardiol. 2005;38:107–12. [DOI] [PubMed] [Google Scholar]
- 22.Abrol N, Smolin N, Armanious G, Ceholski DK, Trieber CA, Young HS and Robia SL. Phospholamban C-terminal residues are critical determinants of the structure and function of the calcium ATPase regulatory complex. J Biol Chem. 2014;289:25855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P and Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–76. [DOI] [PubMed] [Google Scholar]
- 24.Kong H, Jones PP, Koop A, Zhang L, Duff HJ and Chen SR. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. The Biochemical journal. 2008;414:441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Autry JM and Jones LR. Functional Co-expression of the canine cardiac Ca2+ pump and phospholamban in Spodoptera frugiperda (Sf21) cells reveals new insights on ATPase regulation. J Biol Chem. 1997;272:15872–80. [DOI] [PubMed] [Google Scholar]
- 26.Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel-Duby R and Olson EN. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 2016;351:271–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bluhm WF, Kranias EG, Dillmann WH and Meyer M. Phospholamban: a major determinant of the cardiac force-frequency relationship. Am J Physiol Heart Circ Physiol. 2000;278:H249–55. [DOI] [PubMed] [Google Scholar]
- 28.Quan C, Li M, Du Q, Chen Q, Wang H, Campbell D, Fang L, Xue B, MacKintosh C, Gao X, Ouyang K, Wang HY and Chen S. SPEG Controls Calcium Reuptake Into the Sarcoplasmic Reticulum Through Regulating SERCA2a by Its Second Kinase-Domain. Circ Res. 2019;124:712–726. [DOI] [PubMed] [Google Scholar]
- 29.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. [DOI] [PubMed] [Google Scholar]
- 30.Karin M and Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–95. [DOI] [PubMed] [Google Scholar]
- 31.Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annual review of physiology. 2014;76:107–27. [DOI] [PubMed] [Google Scholar]
- 32.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH and Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circulation research. 2003;92:904–11. [DOI] [PubMed] [Google Scholar]
- 33.Zima AV, Picht E, Bers DM and Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D and Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circulation Arrhythmia and electrophysiology. 2008;1:93–102. [DOI] [PubMed] [Google Scholar]
- 35.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S and Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.