

Abstract
DNA replication forks use multiple mechanisms to deal with replication stress, but how the choice of mechanisms is made is still poorly understood. Here, we show that CARM1 associates with replication forks and reduces fork speed independently of its methyltransferase activity. The speeding of replication forks in CARM1-deficient cells requires RECQ1, which resolves reversed forks, and RAD18, which promotes translesion synthesis. Loss of CARM1 reduces fork reversal, increases single-stranded DNA gaps, but allows cells to tolerate higher replication stress. Mechanistically, CARM1 interacts with PARP1 and promotes PARylation at replication forks. In vitro, CARM1 stimulates PARP1 activity by enhancing its DNA binding and acts jointly with HPF1 to activate PARP1. Thus, by stimulating PARP1, CARM1 slows replication forks and promotes the use of fork reversal in stress response, revealing that CARM1 and PARP1 function as a regulatory module at forks to control fork speed and the choice of stress response mechanisms.
Graphical Abstract
eTOC Blurb
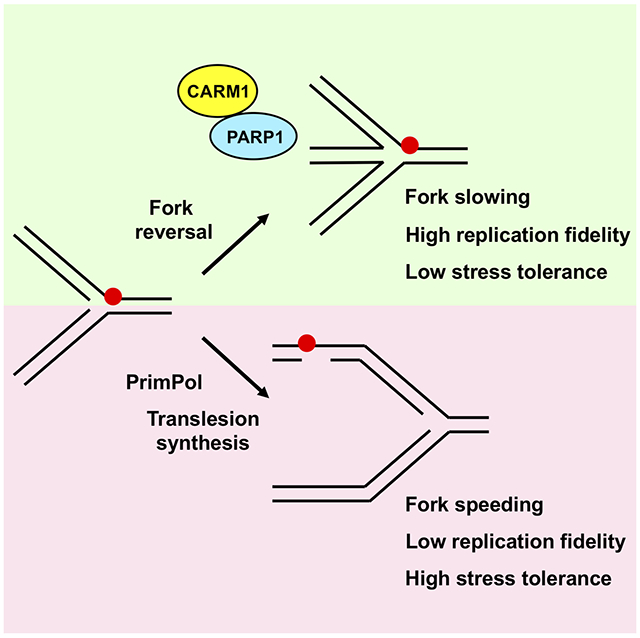
Genois et al. (2020) find that CARM1 has a methyltransferase-independent function at DNA replication forks to control fork speed and the choice of stress response mechanisms. By promoting PARP1 activation, CARM1 favors fork reversal over the use of PrimPol and translesion synthesis, slowing replication forks and maintaining replication fidelity.
Introduction
DNA replication, a complex cellular process essential for cell proliferation, is vulnerable to a variety of DNA damage, impediments in DNA, and cellular stresses (Zeman and Cimprich, 2014). When replication forks are stalled or stressed, they use multiple mechanisms to remove, bypass, or tolerate barriers, promoting fork recovery. For example, stressed replication fork can undergo a process called fork reversal, giving rise to four-way junctions (Berti et al., 2020; Quinet et al., 2017b). A number of factors promoting fork reversal, such as RAD51, SMARCAL1, ZRANB3, and HLTF, are implicated in the restart and/or progression of replication forks under stress (Kile et al., 2015; Kolinjivadi et al., 2017; Taglialatela et al., 2017; Vujanovic et al., 2017; Zellweger et al., 2015). Stressed replication forks can also use translesion synthesis (TLS) polymerases to bypass DNA lesions or tolerate replication errors, allowing them to continue DNA synthesis under stress (Yang and Gao, 2018). Furthermore, the DNA-directed primase/polymerase PrimPol can reprime for DNA synthesis ahead of stalled DNA polymerases, helping replication forks to bypass DNA lesions but leaving single-stranded DNA (ssDNA) gaps behind (Garcia-Gomez et al., 2013; Mourón et al., 2013; Quinet et al., 2020). Although it is clear that multiple mechanisms contribute to the stress response at replication forks, what controls the choice of mechanisms is still poorly understood. In BRCA-deficient cancer cells, multiple doses of cisplatin increase PrimPol levels, promoting PrimPol-mediated repriming while inhibiting fork reversal (Quinet et al., 2020). Loss of HLTF functions also promotes PrimPol-mediated repriming or TLS (Bai et al., 2020). However, it is still unknown how replication forks choose the mechanism of stress response when all pathways are functionally intact. Interestingly, several factors involved in fork reversal, including RAD51, ZRANB3, and HLTF, are required for preventing aberrant fork speeding, suggesting that fork reversal is a mechanism to slowdown fork progression (Bai et al., 2020; Vujanovic et al., 2017; Zellweger et al., 2015). In contrast, when TLS is hyperactive, forks progress faster under stress (Nayak et al., 2020). These findings raise a question as to whether replication fork speed is controlled by the choice of stress response mechanisms at forks.
PARP1/2 are activated by both DNA double-strand breaks (DSBs) and single-strand breaks (SSBs) (Luo and Kraus, 2012; Ray Chaudhuri and Nussenzweig, 2017), and they have been implicated in the regulation of replication fork through several mechanisms. First, because PARP1/2 are required for efficient SSB repair, loss of PARP1/2 activity leads to accumulation of SSBs in DNA, interfering with progressing replication forks (Bryant et al., 2005; Farmer et al., 2005). Second, endogenous PARP activity is detected at replication forks during normal S phase, and PARP1 is required for the repair of unligated Okazaki fragments in the lagging strand of replication forks (Hanzlikova et al., 2018). Third, in response to camptothecin (CPT)-induced DNA damage, PARP1 is required for replication fork reversal (Ray Chaudhuri et al., 2012). PARP1 carries out this function by promoting poly(ADP-ribosyl)ation (PARylation), which inhibits RECQ1, a helicase that resolves reversed forks (Berti et al., 2013). Finally, inhibition of PARP1/2 accelerates replication forks, which was attributed to the increased expression of p21 (Maya-Mendoza et al., 2018). While PARP1/2 impact replication forks in multiple ways, how PARP1/2 are regulated during DNA replication remains largely unknown.
To understand how replication forks recover from stress, we isolated proteins associated with restarting forks after cells were released from a replication block. From these proteins, we identified Coactivator-Associated Arginine Methyltransferase 1 (CARM1; also known as PRMT4), a member of the protein arginine methyltransferase (PRMT) family (Bedford and Richard, 2005). CARM1 is recruited to promoters during gene activation, where it catalyzes the asymmetric di-methylation of histone H3R17 (H3R17me2a) (Chen et al., 1999). CARM1 also has a number of non-histone substrates, such as the histone acetyltransferase p300/CBP, the p160 coactivator NCOA3 and the SWI/SNF complex component BAF155 (Shishkova et al., 2017). The methylation of p300 by CARM1 promotes the binding of p300 to BRCA1 and p53, contributing to the G1 arrest after DNA damage (Lee et al., 2011). However, CARM1 is not known to regulate DNA replication or the replication stress response.
Unexpectedly, we find that CARM1 plays a critical role in regulating fork speed and the choice of stress response mechanisms. In the absence of CARM1, replication forks are accelerated and stalled forks restart prematurely. CARM1 slows replication forks independently of its methyltransferase activity. The speeding of replication forks in CARM1-deficient cells requires RECQ1, which resolves reversed forks (Berti et al., 2013), as well as RAD18 and REV1, which promote TLS (Tissier et al., 2004; Watanabe et al., 2004). Upon replication stress, the lack of CARM1 reduces fork reversal, induces PrimPol-generated ssDNA gaps and mitotic aberrations, but increases stress tolerance. These results suggest that loss of CARM1 suppresses the use of fork reversal but promotes the use of PrimPol and TLS to deal with stress. Importantly, the function of CARM1 in regulating replication forks is dependent on PARP1. CARM1 interacts with PARP1, allows PARP1 to associate with replication forks, and enables PARylation at replication forks, suggesting that CARM1 promotes fork reversal through PARP1-mediated RECQ1 inhibition. In vitro, CARM1 directly stimulates PARP1 activity by enhancing its DNA binding, and it functions jointly with the histone PARylation factor 1 (HPF1) to activate PARP1 (Bonfiglio et al., 2017; Palazzo et al., 2018; Suskiewicz et al., 2020). Thus, CARM1 and PARP1 function as a regulatory module at replication forks to promote fork reversal and suppress the use of PrimPol and TLS in response to stress, enabling cells to constrain fork speed and maintain replication fidelity at the cost of reduced stress tolerance.
Results
CARM1 associates with progressing and restarting replication forks
To understand the recovery of stalled and stressed replication forks, we sought to identify the proteins associated with restarting replication forks after transient stalling. We treated HEK293T cells with 3 mM hydroxyurea (HU) for 2 hours to stall replication forks, and then released cells from HU in the presence of EdU and CDC7 inhibitor (CDC7i) (Fig. 1A). Since CDC7i prevents the firing of new replication origins, EdU was specifically incorporated into the nascent DNA synthesized by restarting replication forks. By monitoring EdU incorporation during the restart, we found that DNA synthesis resumed significantly 25-30 minutes after HU release (Fig. S1A). Furthermore, using isolation of proteins on nascent DNA (iPOND) (Sirbu et al., 2012), we found that PCNA, a core component of replication forks that is released in HU, reassociated with forks 30 minutes after HU removal (Fig. S1B). These results suggest that stalled replication forks undergo restart ~30 minutes after HU is removed.
Figure 1. CARM1 associates with progressing and restarting replication forks.

(A) A schematic of the iPOND-MS approach to identify proteins associating with restarting replication forks. The CDC7i PHA-767491 was used at 10 μM. To identify proteins at progressing forks and stalled forks, cells were labeled with EdU for 10 min, or were labeled with EdU for 10 min followed by incubation in 3 mM HU for 120 min. (B) A pipeline to identify the proteins that are enriched at restarting replication forks. Proteins were ranked according to the log2 ratio of LFQ intensities at restarting forks and on post-replicative DNA, and then selected by the cutoff of p-value ≤ 0.001 (LogProb=3). The top 15 proteins with known DNA or chromatin-related functions are shown. Red dots represent the proteins confirmed to be enriched at restarting forks in an independent iPOND-MS analysis. (C) Relative protein abundance based on LFQ intensities of CARM1, PCNA, RPA32 and PRMT1 protein at restarting forks and on post-replicative DNA after thymidine chase. The relative abundance of each protein at restarting forks was defined as 1. (D) Relative protein abundance based on LFQ intensities of CARM1, PCNA, RPA32 and PRMT1 at progressing forks and stalled forks. The intensity of each protein at progressing forks was defined as 1. (E) Proteins associating with restarting forks and post-replicative DNA were captured by iPOND and analyzed by Western blot. (F) Proteins associating with progressing forks and stalled forks were captured by iPOND and analyzed by Western blot.
Next, we used mass spectrometry (MS) to identify the proteins captured by iPOND 30 minutes after HU release (iPOND-MS) (Fig. 1A). To distinguish the proteins associated with the nascent DNA at or closely behind replication forks from those bound to DNA or chromatin independently of replication, we performed a 60-minute thymidine chase after EdU labeling (Fig. 1A). The proteins specifically associated with replication forks are expected to dissociate from EdU-labeled DNA after the thymidine chase (Sirbu et al., 2012). The relative abundance of the proteins identified by iPOND-MS was estimated by label-free quantification (LFQ) (Bern et al., 2012; Cox et al., 2014). From the proteins identified at restarting forks, we generated a short list of the proteins displaying the highest enrichment at restarting forks compared to post-replicative DNA and known to have DNA or chromatin-related functions (Fig. 1B, Table S1). The majority of these proteins was confirmed to be enriched at restarting forks in an independent iPOND-MS experiment (Fig. 1B, S1C, Table S2). Among the proteins confirmed to associate with restarting forks are several known replication fork components, including PCNA, RFC1, RFC2, and RFC4. Surprisingly, the arginine methyltransferase CARM1 was also enriched at restarting forks, raising the possibility that CARM1 may be involved in replication fork recovery.
To understand the specificity and behavior of the association of CARM1 with replication forks, we compared CARM1 with other replication proteins and chromatin modulators detected at restarting forks using the iPOND-MS data. Similar to PCNA and RPA, CARM1 dissociated from EdU-labeled DNA after thymidine chase, suggesting that CARM1 associates with restarting forks specifically (Fig. 1C, S1D). In contrast to CARM1, another protein arginine methyltransferase, PRMT1, associated with EdU-labeled DNA similarly before and after thymidine chase, suggesting that it binds to chromatin independently of replication (Fig. 1C, S1D). Similar to PCNA and RPA, CARM1 was also detected at progressing replication forks by iPOND-MS in the absence of HU (Fig. 1D). As reported, RPA was increased at HU-stalled forks (Dungrawala et al., 2015), whereas PCNA was reduced (Sirbu et al., 2011) (Fig. 1D). CARM1 was reduced at HU-stalled forks as PCNA (Fig. 1D). Using iPOND followed by Western blot, we confirmed that (1) CARM1 is associated with progressing forks, (2) CARM1 is reduced at stalled forks, and (3) CARM1 gradually reassociates with forks during the restart (Fig. 1E-F, S1E). Together, these results demonstrate that CARM1 is a dynamic component of replication forks, prompting us to further investigate whether CARM1 plays a role in regulating DNA replication.
CARM1 promotes slow fork progression and regulates fork restart
To test whether CARM1 is involved in fork restart, we knocked down CARM1 in MCF10A cells with siRNA and used DNA fiber assay to monitor fork restart after HU release (Fig. 2A, S2A). Knockdown of CARM1 in MCF10A cells did not significantly alter the cell cycle 2 days after CARM1 siRNA transfection (Fig. S2B). We transiently labeled nascent DNA with CldU, treated cells with a high concentration (2 mM) of HU for 2 hours, and then released cells in the presence of IdU (Fig. 2A). Depletion of CARM1 reduced the fraction of stalled forks (CldU+, IdU− replication tracts) 1 hour after HU release (Fig. 2A), showing that stalled forks restart more efficiently in the absence of CARM1. Furthermore, the DNA synthesized by restarted forks (IdU+ regions in CldU+, IdU+ tracts) were longer in CARM1 knockdown cells than in control cells (Fig. 2A), suggesting that either stalled forks restart faster or progress faster after restart upon CARM1 loss.
Figure 2. CARM1 promotes slowing of replication forks.

(A) MCF10A were transfected with control or CARM1 siRNAs for 48h and analyzed by DNA fiber assay as indicated. The percentage of stalled forks (CldU+ IdU−) was determined (n = 200, left panel), and the length of IdU+ region of CldU+ IdU+ replication tracts (referred to as IdU tracts below) was measured (n = 200, middle panel). Black bars indicate the mean length. Mean IdU tract length was determined in three independent experiments (n=3, right panel). Statistical significance was determined by Mann-Whitney test: (****) P<0.0001. (B) MCF10A (left panel) or U2OS cells (right panel) were transfected with control or CARM1 siRNAs for 48h. Carm1fl/fl KO MEFs (middle panel) were treated with 4-hydroxytamoxifen (4OHT, 2μM) for 3 days to deplete CARM1. Cells were analyzed by DNA fiber assay as indicated. The length of replication tracts (n ≥177 for MCF10A, n ≥151 for MEF, n = 200 for U2OS) was measured. Black bars indicate the mean length. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001. (C) MCF10A cells were transfected with control or CARM1 siRNA. Two days after transfection, cells were analyzed by DNA fiber assay as indicated. Cells were treated with increasing concentrations (25, 50, 100 μM) of HU during IdU labeling. The length of IdU tracts (n ≥158) was measured. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001. (D) U2OS cells were transfected with control or CARM1 siRNA. Two days after transfection, cells were treated with 0.5 mM HU for 30 min and released for 30 to 60 min. Levels of the indicated proteins or phosphorylated proteins were analyzed by Western blot. (E) MCF10A cells that inducibly express CARM1WT were transfected with CARM1 siRNA for 48h. One day after transfection, cells were treated with 1000 ng/mL doxycycline for 24h to induce expression of CARM1WT. Cells were analyzed by DNA fiber assay as indicated (right panel) and levels of the indicated proteins were analyzed by Western blot (left panel). The length of replication tracts (n=200) was measured. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001.
Next, we performed DNA fiber assay on CARM1 knockdown and control cells in the absence of HU to investigate the effects of CARM1 on progressing forks (Fig. 2B). Nascent DNA was sequentially labeled with CldU and IdU, and the lengths of CldU+, IdU+ replication tracts were measured. Knockdown of CARM1 in MCF10A cells with three independent siRNAs resulted in a significant increase of replication fork speed (Fig. 2B, S2A-C). Speeding of replication forks was also observed in conditional Carm1fl/fl knockout (KO) mouse embryonic fibroblasts (MEFs) (Cheng et al., 2007; Yadav et al., 2003) and CARM1 knockdown U2OS cells (Fig. 2B, S2A-C). A similar observation was made in U2OS cells using DNA combing assay (Fig. S2D). When cells were exposed to increasing concentrations of HU, replication forks progressed significantly faster in CARM1 knockdown cells than in control cells (Fig. 2C). The slowing and stalling of replication forks by HU elicit the ATR pathway (Marechal and Zou, 2013; Saldivar et al., 2017; Yazinski and Zou, 2016). After HU treatment, CARM1 knockdown cells displayed reduced levels of phosphorylated Chk1 (p-Chk1 S345) and RPA32 (p-RPA32 S33) compared with control cells (Fig. 2D), indicating a reduction in ATR activation. Thus, in the absence of CARM1, replication forks progress faster and fail to slow down adequately to activate ATR in response to stress.
To verify whether the effects of CARM1 knockdown on replication forks are specifically attributed to CARM1 loss, we knocked down endogenous CARM1 with an siRNA targeting the 3’ UTR and expressed exogenous wild-type CARM1 at the level of endogenous CARM1 (Fig. 2E). Wild-type CARM1 reduced the replication fork speed in CARM1 knockdown cells to the level in control cells (Fig. 2E, S2E), confirming that loss of CARM1 is indeed responsible for the speeding of replication forks.
CARM1 slows replication forks independently of its methyltransferase activity
CARM1 functions as an arginine methyltransferase in transcription regulation (Covic et al., 2005). To test whether the transcription function of CARM1 is required for slowing replication forks, we analyzed the DNA fibers from cells that were briefly treated with 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB), an inhibitor of RNA polymerase II (Fig. 3A). Consistent with the idea that replication forks are slowed by transcription (Hamperl and Cimprich, 2016), DRB increased fork speed (Fig. 3A). However, CARM1 knockdown increased fork speed similarly in control and DRB-treated cells (Fig. 3A, S3A), suggesting that CARM1 affects fork speed independently of transcription. To test whether the methyltransferase activity of CARM1 is involved in slowing replication forks, we treated cells with the CARM1 inhibitor (CARM1i) TP-064 (Nakayama et al., 2018). CARM1i efficiently decreased the CARM1-mediated PABP1 methylation at R455/R460 (Lee and Bedford, 2002). However, CARM1i did not speed up replication forks as CARM1 knockdown did (Fig. S3B), suggesting that CARM1 slows forks independently of its methyltransferase activity. Consistently, expression of the methyltransferase inactive CARM1R168A mutant in CARM1 knockdown cells significantly reduced fork speed (Fig. 3B, S3C) (Kim et al., 2010), lending further support to the notion that CARM1 regulates replication forks independently of protein methylation.
Figure 3. CARM1 functions with PARP1 to slow replication forks.

(A) MCF10A cells were transfected with control or CARM1 siRNA. Two days after transfection, cells were treated with DMSO or 100 μM DRB for 30 min, and then analyzed by DNA fiber assay as indicated. The length of replication tracts (n ≥135) was measured. Black bars indicate the mean length. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001. (B) MCF10A cells that inducibly express the methyltransferase inactive CARM1 mutant (CARM1R168A) were transfected with control or CARM1 siRNA for 48h. One day after transfection, cells were treated with 10 ng/mL doxycycline to induce CARM1R168A expression for 24h. Cells were analyzed by DNA fiber assay as indicated. The length of replication tracts (n=200) was measured. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001. (C) MCF10A cells were transfected with control or CARM1 siRNA for 48h. One day after transfection, cells were treated with 10 μM Veliparib (Velip.), Olaparib (Olap.), or Talazoparib (Talaz.) for 24h. Cells were analyzed by DNA fiber assay as indicated. The length of replication tracts (n ≥95) was measured. Statistical significance was determined by Mann-Whitney test: (ns) not significant. (D) U2OS cells were transfected with control or CARM1 siRNA. Two days after transfection, cells were labeled with EdU for 30 min then treated with 1 μM PARGi (PDD00017273) for 20 min. Levels of PAR immunofluorescence intensity in individual EdU-positive cells (n≥703) were quantified. Black bars indicate the mean PAR intensity; (a.u.) Arbitrary unit. A threshold is set at 446 a.u. above 97% of the control siRNA-transfected and DMSO-treated cells, and the fraction of cells above this threshold in each cell population was quantified. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001. (E) MCF10A cells were transfected with control, CARM1, and RECQ1 siRNAs for 48h as indicated. One day after transfection, the indicated cells were treated with 10 μM PARPi (Olaparib) for 24h. Cells were analyzed by DNA fiber assay as indicated. HU (200 μM) was used during IdU labeling. The length of replication tracts (n ≥126) was measured. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001; (***) P<0.001. (F) U2OS cells were transfected with control or CARM1 siRNA. Two days after transfection, cells were treated with 4 mM HU for 120 min and analyzed by electron microscopy (EM). The percentage of reversed forks is shown above each sample. The total number of forks analyzed is shown below each sample in brackets. (G) MCF10A cells were transfected with control, CARM1, and RAD18 siRNAs for 48h and analyzed by DNA fiber assay as indicated. HU (200 μM) was used during IdU labeling. The length of replication tracts (n ≥127) was measured. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001.
CARM1 and PARP1 share similar functions in slowing replication forks
The speeding of replication forks in CARM1-depleted cells is reminiscent to that induced by PARP1/2 inhibition (Maya-Mendoza et al., 2018), raising the possibility that CARM1 and PARP1/2 are functionally linked. To test this possibility, we first compared the potencies of three different PARPis (veliparib, olaparib, and talazoparib) in vitro (Fig. S3D). Consistent with previous reports (Murai et al., 2012; Murai et al., 2014), talazoparib was the most potent for inhibiting PARP1 auto-PARylation in vitro, which was followed by olaparib and then veliparib (Fig. S3D). In cells treated with the same concentrations of these PARPis, the extents of fork speeding correlated with the potencies of the inhibitors (Fig. 3C), with talazoparib as the most potent in speeding forks. Importantly, talazoparib did not increase fork speed in CARM1 knockdown cells (Fig. 3C), suggesting that CARM1 and PARP1/2 function in the same pathway or that forks have reached the maximal speed. Since talazoparib not only inhibits PARP1/2 but also traps them on DNA, we asked whether loss of PARP1/2 activity or trapping of PARP1/2 is responsible for fork speeding. Knockdown of PARP1 also increased fork speed (Fig. S3E), suggesting that trapping of PARP1 is unlikely the cause of accelerated fork progression. Similar to CARM1 knockdown, PARPi reduced HU-induced Chk1 phosphorylation and RPA32 phosphorylation at forks (Fig. S3F-G). Thus, CARM1 and PARP1/2 share similar functions in slowing replication forks.
Spontaneous PARylation was detected at replication forks during normal S phase when de-PARylation was blocked by PARG inhibitor (PARGi) (Hanzlikova et al., 2018), which is likely a response to intrinsic replication stress. Indeed, even in the absence of HU, PARGi increased poly(ADP-ribose) (PAR) levels above the background in cells undergoing DNA replication (Fig. 3D). Knockdown of CARM1 reduced the PARGi-induced PAR (Fig. 3D), suggesting that PARP1/2 are less active during replication in the absence of CARM1. These results suggest that CARM1 may promote PARP1/2 functions during DNA replication.
CARM1 inhibits RECQ1 function and increases fork reversal
PARP1/2 promote CPT-induced replication fork reversal by inhibiting RECQ1, a helicase that resolves reversed forks and a substrate of PARP1 (Berti et al., 2013; Larsen et al., 2018). In MCF10A and U2OS cells treated with a low concentration of HU, RECQ1 knockdown slightly reduced fork speed (Fig. 3E, S4A-B), indicating that reversed forks are not restarted efficiently. RECQ1 knockdown also reduced fork speed in PARPi-treated cells (Fig. 3E), supporting the idea that PARP1/2 slow forks by inhibiting RECQ1. Importantly, depletion of RECQ1 partially suppressed the speeding of forks in CARM1 knockdown MCF10A and U2OS cells (Fig. 3E, S4A-B, see S4F), suggesting that the fork speeding in the absence of CARM1 is mediated by RECQ1. If RECQ1 promotes fork speeding by restarting reversed forks in the absence of CARM1, CARM1 loss should reduce fork reversal. To directly analyze the effects of CARM1 on fork reversal, we used electron microscope (EM) to analyze the replication forks in CARM1 knockdown and control cells (Fig. 3F, S4C). As reported (Zellweger et al., 2015), HU significantly increased fork reversal in control cells (Fig. 3F). CARM1 knockdown reduced the frequency of reversed forks from 26% to 10% after HU treatment (Fig. 3F), providing direct evidence that CARM1 promotes the reversal of replication forks under stress.
Loss of CARM1 leads to altered stress response and increased stress tolerance
Defects of the fork reversal factor HLTF increase the use of TLS and PrimPol at stressed replication forks (Bai et al., 2020). Furthermore, hyperactive TLS increases fork speed under stress (Nayak et al., 2020). To test if TLS is responsible for the speeding of forks in CARM1 knockdown cells, we depleted RAD18, which is required for PCNA mono-ubiquitination, TLS, and possibly additional repair events dependent on PCNA poly-ubiquitination (Fig. 3G, S4D). Knockdown of RAD18 with three independent siRNAs reduced fork speed in CARM1-depleted cells (Fig. 3G), supporting the idea that TLS contributes to fork speeding in the absence of CARM1. Knockdown of REV1, a key factor for TLS, also reduced fork speed in CARM1-depleted cells (Fig. S4E). In contrast to RAD18 and REV1, knockdown of PrimPol in CARM1-depleted cells did not alter fork speed significantly (Fig. S4F-G). Furthermore, PrimPol depletion did not alter fork speed in PARPi-treated cells either (Fig. S4G). These results suggest that PrimPol is not a major contributor to fork speeding when fork reversal is compromised. However, in the EM data, we noticed an increase of ssDNA gaps in daughter strands of replication forks in HU-treated CARM1 knockdown cells (Fig. S5A), indicating PrimPol-mediated repriming. To confirm this finding, we treated cells with the S1 nuclease, which cleaves ssDNA gaps or DNA structures exposing ssDNA, right after DNA labeling (Quinet et al., 2017a). The replication tracts in CARM1 knockdown cells but not control cells were significantly shortened by the S1 nuclease (Fig. S5B), and this shortening was PrimPol-dependent (Fig. S5C), confirming the increase of PrimPol-generated ssDNA gaps in the absence of CARM1. Together, these results suggest that loss of CARM1 increases the use of PrimPol and TLS at stressed forks.
While CARM1 knockdown cells displayed an increase of ssDNA gaps, ATR is not efficiently activated in these cells (Fig. 2D), suggesting that the ssDNA gaps do not trigger the ATR-mediated G2/M checkpoint. To test whether the replication problems caused by CARM1 loss affect the subsequent mitosis, we treated CARM1fl/fl MEFs with 4-OHT to deplete CARM1 and then synchronize them in mitosis with nocodazole (Fig. S5D). Loss of CARM1 increased chromosomal breaks, chromosomal fusions, and premature separation of sister chromatids in mitotic cells (Fig. S5D), supporting the idea that reduced replication fidelity in CARM1-deficient cells increases mitotic aberrations. However, depletion of CARM1 decreased the levels of DSBs in neutral comet assay (Fig. S5E), indicating a reduction in lethal DNA damage. Furthermore, depletion of CARM1 reduced the cellular sensitivity to HU, CPT, and PARP inhibitor (PARPi) (Fig. S5F-G), showing a general increase of stress tolerance in CARM1-deficient cells. These results suggest that loss of CARM1 allows cells to use low-fidelity mechanisms to deal with replication stress and increase stress tolerance.
CARM1 interacts with PARP1 and promotes its association with replication forks
The functional link between CARM1 and PARP1/2 prompted us to investigate whether CARM1 physically interacts with PARP1. To test this possibility, we transiently expressed S, Flag, and Biotin-binding peptide (SFB)-tagged CARM1 or just the SFB tag in HEK293T cells and then immunoprecipitated PARP1 after DMSO, PARPi, or PARGi treatment (Fig. 4A). CARM1-SFB co-precipitated with endogenous PARP1 in DMSO-treated cells, and this interaction was drastically enhanced by PARPi (Fig. 4A). Consistently, immunoprecipitation of CARM1-SFB co-precipitated PARP1, and the interaction was also enhanced by PARPi (Fig. 4B). In the presence of PARGi, PAR levels were increased, but the levels of unmodified PARP1 and CARM-SFB in PARP1 immunoprecipitates were reduced (Fig. 4A). These results suggest that the CARM1-PARP1 interaction is inhibited by PARylation, possibly through a feedback mechanism. Nonetheless, even in the absence of PARPi, endogenous CARM1 and PARP1 co-precipitated with each other in reciprocal immunoprecipitations (Fig. 4C), showing that CARM1 interacts with functional PARP1.
Figure 4. CARM1 interacts with PARP1 and promotes its association with replication forks.

(A) CARM1-SFB or vector-SFB was transfected into HEK293T cells. Forty-four hours after transfection, cells were treated with DMSO or 10 μM PARPi (Olaparib) or 1 μM PARGi (PDD00017273) for 4 h. Endogenous PARP1 was immunoprecipitated using PARP1 antibody. Levels of the indicated proteins were analyzed by Western blot. (B) CARM1-SFB or vector-SFB was transfected into HEK293T cells. Forty-five hours and 30 min after transfection, cells were treated with DMSO or 10 μM PARPi (Olaparib) for 2.5 h. CARM1-SFB was immunoprecipitated using streptavidin magnetic beads (M280). Levels of the indicated proteins were analyzed by Western blot. (C) Endogenous PARP1 and CARM1 were immunoprecipitated from untreated HEK293T cells using PARP1 and CARM1 antibodies, respectively. Levels of the indicated proteins were analyzed by Western blot. (D) HEK293T cells were labeled with EdU for 15 min and collected immediately or after a 2 h treatment with 3 mM HU. Proteins at progressing forks and stalled forks were captured by iPOND and levels of the indicated proteins were analyzed by Western blot. (E) HEK293T were treated with DMSO or 10 μM PARPi (Olaparib) for 6 h 45 min. During the last 2 h 45 min in PARPi (Olaparib), cells were treated with 3 mM HU for 2 h, and then released in EdU for 45 min. Proteins at restarting forks were captured by iPOND, and levels of the indicated proteins were analyzed by Western blot. (F) HEK293T were transfected with control or CARM1 siRNA for 48 h. Levels of the indicated proteins in whole-cell extracts were analyzed by Western blot before iPOND (left panel). Cells were labeled with EdU for 15 min and analyzed by iPOND (right panel). The input of iPOND was prepared from cells permeabilized with detergent and washed two times.
Next, we used iPOND to compare the associations of CARM1 and PARP1 with replication forks. As shown in Fig. 1, CARM1 associates with progressing forks but is reduced at stalled forks after HU treatment. Similarly, PARP1 was also detected at progressing forks, and the association of PARP1 with forks was also reduced by HU (Fig. 4D). In the presence of PARPi, both CARM1 and PARP1 increasingly associated with restarting forks after HU release (Fig. 4E), showing that a robust CARM1-PARP1 interaction coincides with their efficient binding to forks. Furthermore, knockdown of CARM1 significantly reduced the PARP1 detected by iPOND (Fig. 4F), suggesting that CARM1 promotes the association of PARP1 with replication forks.
The interaction of CARM1 and PARP1 is important for slowing replication forks
To functionally evaluate the interaction between CARM1 and PARP1, we generated a set of truncated CARM1 variants (Fig. 5A). A CARM1 fragment lacking the C terminus (CARM1B) was significantly compromised for PARP1 binding, whereas another CARM1 fragment containing only the C terminus (CARM1D) still interacted with PARP1 quite efficiently (Fig. 5B). These results suggest that CARM1 binds PARP1 through its C terminus independently of the central catalytic domain. Interestingly, a CARM1 fragment lacking only the N terminus (CARM1C) was more defective for PARP1 binding than CARM1D (Fig. 5B), suggesting that the N terminus is important for PARP1 binding in the presence of the catalytic domain. In iPOND, neither CARM1B nor CARM1C associated with restarting forks as efficiently as wild-type CARM1 (CARM1WT) did (Fig. 5C). To test whether the interaction between CARM1 to PARP1 is functionally important, we expressed CARM1WT, CARM1B, and CARM1C at endogenous CARM1 levels in CARM1 knockdown cells (Fig. 5D). While CARM1WT reduced fork speed in CARM1 knockdown cells, CARM1B and CARM1C failed to do so (Fig. 5E). These results suggest that both the N and C termini of CARM1 contribute to the association of CARM1 with replication forks and its function in slowing forks.
Figure 5. CARM1 slows replication forks through its interaction with PARP1.

(A) A schematic representation of the CARM1 variants tested in this study. (N-term) N-terminal region; (C-term) C-terminal region; (WT) CARM1WT; (R168A) CARM1R168A. (B) CARM1-SFB or vector-SFB was transfected into HEK293T cells. Two days after transfection, cells were treated with 10 μM PARPi (Olaparib) for 2.5 h. SFB-tagged CARM1 variants were immunoprecipitated using streptavidin magnetic beads (M280). Levels of the indicated protein were analyzed by Western blot. (C) HEK293T cells were transfected with plasmids expressing SFB-tagged CARM1WT (WT) or CARM1B/C for 48 h. Cells were treated with 3 mM HU for 120 min and then released in EdU for 30 min. Proteins at restarting forks were captured by iPOND, and levels of indicated proteins were analyzed by Western blot. (D) MCF10A cells that inducibly express CARM1WT-HA, CARM1B-HA, or CARM1C-HA were transfected with CARM1 siRNA for 48 h. One day after transfection, cells were treated with 1000 ng/mL, 25 ng/mL or 40 ng/mL doxycycline for 24 h to induce expression of CARM1WT-HA, CARM1B-HA, or CARM1C-HA. Levels of the indicated proteins were analyzed by Western blot. (E) Cells described in (D) were analyzed by DNA fiber assay as indicated. The length of replication tracts (n=200) was measured (left panel). Black bars indicate the mean tract length. Mean tract length was determined from three independent experiments (n=3, right panel). Statistical significance was determined by Mann-Whitney test: (****) P<0.0001; (ns) not significant.
CARM1 stimulates PARP1 in vitro and promotes PARylation at replication forks
After establishing that CARM1 promotes PARP1 function at replication forks in cells, we turned to in vitro biochemistry to understand how CARM1 regulates PARP1. We purified recombinant CARM1WT, the methyltransferase inactive CARM1R168A mutant, and the PARP1 binding-defective CARM1B mutant from E. coli (Fig. 6A). As reported, the full-length CARM1 purified from E. coli migrated as three bands in SDS-PAGE (Chumanov et al., 2011), possibly because a fraction of the protein was truncated. Next, we tested the effects of purified CARM1 proteins on PARP1 activity in vitro. In the presence of nicked double-stranded DNA (dsDNA) and NAD+, purified PARP1 underwent efficient auto-PARylation (Shah et al., 1995) (Fig. 6B, S6A). PARP1 auto-PARylation was enhanced by CARM1WT in a concentration-dependent manner only in the presence of DNA (Fig. 6B, S6B), showing that CARM1 stimulates the activation of PARP1 by DNA directly. CARM1R168A also stimulated PARP1 (Fig. 6B), and CARM1i did not inhibit the stimulation of PARP1 by CARM1WT (Fig. S6B), showing that the methyltransferase activity of CARM1 is not required. Compared with CARM1WT, CARM1B stimulated PARP1 less efficiently even when used at higher concentrations than CARM1WT (Fig. 6C), suggesting that the interaction between CARM1 and PARP1 is important for the stimulation. Consistent with the roles of CARM1 in promoting PARP1 association with forks and stimulating PARP1 activity, knockdown of CARM1 drastically reduced the levels of PAR at progressing replication forks in PARGi-treated cells (Fig. 6D), highlighting the critical function of CARM1 in PARP1 activation at replication forks.
Figure 6. CARM1 stimulates PARP1 in vitro and promotes PARylation at replication forks.

(A) Purified proteins (300 ng) of full-length, wild-type CARM1WT (WT), CARM1R168A (R168A), and truncated CARM1B (B) were resolved by SDS-PAGE and stained with Coomassie blue. (B) In vitro PARP1 activation assay with purified PARP1 (0.5 nM), NAD+ (250 μM), and calf thymus dsDNA (25 μg/mL). As indicated, increasing concentrations (17-34 nM) of CARM1WT (WT) or CARM1R168A (R168A) were added to reactions. Levels of PAR and CARM1 variants were analyzed by Western blot with the indicated antibodies. (C) In vitro PARP1 activation assay was performed and analyzed as in (B). CARM1WT and CARM1B were added to reactions at the indicated concentrations. (D) HEK293T were transfected with control or CARM1 siRNA for 48 h. Cells were treated with 1 μM PARGi (PDD00017273) for 2 h 15 min, and were labeled with EdU during the last 15 min. Proteins at progressing forks were captured by iPOND, and levels of indicated proteins were analyzed by Western blot. (E) A model depicting how CARM1 and PARP1 control replication fork speed and the choice of stress response mechanisms.
Together, the results above suggest that CARM1 and PARP1 act as a functional module at replication forks to promote fork reversal and ensure efficient slowing/stalling of forks in response to stress (Fig. 6E). In the absence of CARM1, replication forks increasingly use PrimPol and TLS to deal with stress, reducing replication fidelity but enhancing stress tolerance.
CARM1 acts in concert with HPF1 to stimulate PARP1
Next, we investigated how CARM1 stimulates PARP1. As reported, treatment of cells with methyl methanesulfonate (MMS) and PARPi led to a significant increase of PARP1 on chromatin (Murai et al., 2012) (Fig. 7A), reflecting the binding of PARP1 to damaged DNA. Knockdown of CARM1 reduced the damage-induced chromatin binding of PARP1 (Fig. 7A), suggesting that CARM1 is important for the DNA binding of PARP1. To test this possibility more rigorously in vitro, we used biotinylated dsDNA to capture purified PARP1 in the presence and absence of CARM1 (Fig. 7B-C). In the presence of CARM1, an increased amount of PARP1 was captured by dsDNA (Fig. 7C), showing that CARM1 indeed enhances the binding of PARP1 to dsDNA. Interestingly, CARM1 itself was not efficiently captured by dsDNA (Fig. 7C). Similar observations were made when biotinylated ssDNA was used in the reactions (Fig. S7). These results suggest that CARM1 generally stimulates the DNA binding of PARP1, but its interaction with PARP1 is more transient/unstable than the PARP1-DNA interaction.
Figure 7. CARM1 acts in concert with HPF1 to activate PARP1.

(A) U2OS cells were transfected with control or CARM1 siRNA. Two days after transfection, cells were labeled with EdU for 30 min and then treated with 10 μM PARPi (Olaparib) and 0.01% MMS for 4 h. Cells were extracted with detergent, and the immunofluorescence intensity of chromatin-bound PARP1 in individual EdU-positive cells (n=527) were quantified. Black bars indicate the mean intensity; (a.u) Arbitrary unit. A threshold is set at 480 a.u. above 97% of the control siRNA-transfected and DMSO-treated cells, and the fraction of cells above this threshold in each cell population was quantified. Statistical significance was determined by Mann-Whitney test: (****) P<0.0001 (B) Purified proteins (10 ng) of full-length PARP1 (FL), the PARP1-ΔHD mutant, and HPF1 were run on an SDS-PAGE and stained with Sypro Ruby. (C) DNA-binding of PARP1 was analyzed using biotinylated dsDNA (60 nt) conjugated to streptavidin magnetic beads (M280). PARP1 (10 nM) (lane 1) and CARM1 (30 nM) (lane 2) were incubated with beads alone or beads carrying dsDNA as indicated. The PARP1 and CARM1 captured by beads were analyzed by Sypro Ruby staining. (D) In vitro PARP1 activation assay with purified PARP1-ΔHD (5 nM), NAD+ (250 μM), and dsDNA (25 μg/mL). Increasing concentrations (12-25 nM) of CARM1 were added to reactions. Levels of PAR and CARM1 were analyzed by Western blot with the indicated antibodies. (E) In vitro PARP1 activation assay with purified PARP1 (1 nM), NAD+ (250 μM), and dsDNA (25 μg/mL). CARM1 (10-20 nM) and HPF1 (10 nM) were added to reactions as indicated. Levels of PAR were analyzed by Western blot. The HPF1 and CARM1 used in the reactions were analyzed by Sypro Ruby staining. (F) In vitro PARP1 activation assay was performed and analyzed as in (E) except that HPF1 (10 −20 nM) and CARM1 (20 nM) were added to reactions as indicated. Levels of PAR, CARM1 and HPF1 were analyzed by Western blot (G) A model for concerted functions of CARM1 and HPF1 in activating PARP1.
Upon binding to DNA, the helical subdomain (HD) of PARP1 becomes unfolded, allowing PARP1 to bind NAD+ (Dawicki-McKenna et al., 2015; Langelier et al., 2018) and the activator protein HPF1 (Gibbs-Seymour et al., 2016; Suskiewicz et al., 2020). In addition, type I PARPi enhances the retention of PARP1 on DNA by unfolding HD (Zandarashvili et al., 2020). The PARP1 mutant lacking the HD was still stimulated by CARM1 (Fig. 7B, 7D), suggesting that CARM1 enhances the binding of PARP1 to DNA independently of HD. The enhancement of the DNA binding of PARP1 by CARM1 may allow HPF1 to stimulate PARP1 on DNA more efficiently. To test this possibility, we added both CARM1 and HPF1 to the in vitro PARP1 reactions (Fig. 7B, 7E-F). At a low concentration of HPF1, CARM1 further stimulated PARP1 in a dose-dependent manner (Fig. 7E). Similarly, at a low concentration of CARM1, HPF1 further stimulated PARP1 in a dose-dependent manner (Fig. 7F). These results suggest that CARM1 and HPF1 may function in concert to activate PARP1, supporting a model in which CARM1 and HPF1 act sequentially to stimulate the DNA binding and catalysis of PARP1 in response to DNA damage and replication stress (Fig. 7G).
Discussion
In this study, we find that CARM1 plays a critical role in controlling replication fork speed and the choice of stress response mechanisms at replication forks. In the absence of CARM1, replication forks are accelerated and unable to slow or stall properly in response to stress (Fig. 2B-C). Associated with the defect in slowing replication forks, CARM1-deficient cells fail to activate ATR efficiently, stalled forks restart prematurely, ssDNA gaps arise during replication, and chromosomal aberrations accumulate in mitosis (Fig. 2A, 2D, S5A-D). However, despite the reduction in replication fidelity, CARM1-deficient cells are more tolerant to replication stress (Fig. S5F-G). These findings are consistent with the recent observations in HLTF-deficient cells (Bai et al., 2020), suggesting that the regulatory role of CARM1 in replication favors replication fidelity over stress tolerance (Fig. 6E). It is likely that this function of CARM1 is beneficial to normal cells under physiological stress, and its loss in cancer cells promotes resistance to pathological stress and the DNA damage induced by therapy.
The effects of CARM1 on replication forks can be attributed to several factors involved in stress responses. The speeding of forks in CARM1-deficient cells requires RAD18 and REV1, two key factors driving TLS (Fig. 3G, S4E) (Tissier et al., 2004; Watanabe et al., 2004). The increase of ssDNA gaps in CARM1-deficient cells is dependent on PrimPol (Fig. S5A-C) (Quinet et al., 2020). These results suggest that both PrimPol and TLS are increasingly used at stressed forks in the absence of CARM1. In contrast to the increased use of PrimPol and TLS, fork reversal is suppressed in CARM1-deficient cells. RECQ1, which resolves reversed forks (Berti et al., 2013), promotes fork speeding upon CARM1 loss (Fig. 3E). Furthermore, EM data provide direct evidence that stress-induced fork reversal is reduced in CARM1-deficient cells (Fig. 3F). Thus, loss of CARM1 favors the use of PrimPol and TLS over fork reversal, exerting a clear impact on the choice of stress response mechanisms at forks. Interestingly, CARM1 reduces fork speed in the absence of HU (Fig. 2B), suggesting that the choice of stress response mechanisms influences replication even in unperturbed S phase. It is possible that replication forks frequently undergo stall-and-restart cycles in response to intrinsic stress, making the choice of stress response mechanisms an important determinant of fork speed and replication fidelity.
Our results reveal that CARM1 slows replication forks by promoting PARP1 activation at forks. To our surprise, this function of CARM1 is independent of its well-characterized methyltransferase activity, but dependent on its interaction with PARP1 (Fig. 3B, 5E, S3B). CARM1 promotes the association of PARP1 with replication forks, and is required for the PARylation at replication forks (Fig. 4F, 6D). In vitro, CARM1 directly stimulates the activity of PARP1 by enhancing its binding to DNA (Fig. 6C, 7C, S7). Importantly, the abilities of CARM1 to slow replication forks in cells and to stimulate PARP1 in vitro are dependent on the interaction between CARM1 and PARP1 (Fig. 5E, 6C), suggesting that these two proteins act as a functional module at replication forks. Interestingly, CARM1 and HPF1 stimulate PARP1 through distinct mechanisms. While CARM1 enhances the DNA binding of PARP1, HPF1 binds to the PARP1-DNA complex and modulates the active site of PARP1 (Suskiewicz et al., 2020). CARM1 and HPF1 activated PARP1 jointly in vitro (Fig. 7E-F), supporting the idea that they may act in concert to fully activate PARP1 in cells. Collectively, these results demonstrate that CARM1 is an important regulator of PARP1.
We found that both CARM1 and PARP1 associate with replication fork dynamically. Consistent with the effects of CARM1 and PARP1 on fork speed, both of these proteins associate with progressing replication forks (Fig. 1F, 4D). CARM1 and PARP1 are also associated with restarting forks, where CARM1 exerts its impact on fork restart (Fig. 1E, 2A, 4E). CARM1 interacts with PARP1 through both N and C terminal regions (Fig. 5A-B), and these interactions may be transient and regulated by other proteins in vivo. Interestingly, the association of both CARM1 and PARP1 with replication forks is reduced by HU (Fig. 1F, 4D). It is possible that CARM1 and PARP1 dissociate from forks together with other fork proteins, such as PCNA. Alternatively, the auto-PARylation of PARP1 at stalled forks may promote the release of both PARP1 and CARM1, providing a feedback loop to prevent persistent PARylation at forks (Mortusewicz et al., 2007). Consistent with the idea of a feedback loop, the interaction between CARM1 and PARP1 is inhibited by PARylation (Fig. 4A-B). The labile nature of the CARM1-PARP1 complex and its dynamic association with replication forks may enable the complex to control the choice of stress response mechanisms in a transient and regulated manner.
How is the CARM1-PARP1 complex activated at replication forks? PARP1 binds a variety of DNA structures in vitro, including DSBs, SSBs, and other ssDNA-containing structures (Langelier et al., 2014). At replication forks, PARP1 may bind ssDNA gaps with the help form CARM1, allowing PARP1 to sense replication stress. Indeed, we found that CARM1 stimulates the binding of PARP1 to ssDNA in vitro (Fig. S7). The binding of PARP1 to ssDNA gaps at forks is also consistent with the role of PARP1 in lagging strand maturation during unperturbed replication (Hanzlikova et al., 2018). PARP1 may also recognize the ssDNA and/or DNA ends in reversed forks, which are also induced by replication stress (Zellweger et al., 2015). The ability of CARM1-PARP1 to sense reversed forks may allow it to stabilize these forks by inhibiting RECQ1 (Berti et al., 2013). It should be noted that RECQ1 may not be the only protein regulated by CARM1-PARP1 at forks, and CARM1 may have PARP1-independent functions. The complete functions of CARM1 and PARylation at forks still need further investigations.
While both CARM1-PARP1 and ATR sense replication stress at forks (Zou and Elledge, 2003), the functions of CARM1 and ATR are clearly distinct. In contrast to CARM1, ATR generally promotes the progression of replication forks by suppressing origin firing and protecting stressed forks (Buisson et al., 2015; Couch et al., 2013; Eykelenboom et al., 2013; Iyer and Rhind, 2017; Petermann et al., 2010). Nevertheless, in response to DNA inter-strand crosslinks, ATR inhibition resulted in a reduction in detectable reversed forks (Mutreja et al., 2018). Interestingly, CARM1 facilitates ATR activation by promoting fork slowing (Fig. 2D). It is possible that the defective ATR activation in CARM1-deficient cells contributes to the genomic instability in these cells. However, the functions of CARM1 in controlling fork speed and the choice of stress response mechanisms are likely independent of ATR.
Clinical PARPis have different abilities to trap PARP1/2 on DNA (Murai et al., 2012; Murai et al., 2014; Zandarashvili et al., 2020). The abilities of different PARPis to induce fork speeding correlate with not only their potencies for PARP1/2 inhibition, but also their capabilities to trap PARP1/2 (Fig. 3C, S3D). Nonetheless, replication forks are similarly accelerated by PARPi treatments and PARP1 knockdown (Fig. 3C, S3E). Furthermore, replication forks are still accelerated when PARP1 is not recruited in the absence of CARM1 (Fig. 2B, 4F). These results suggest that the loss of PARP1 activity at replication forks, rather than the trapping of PARP1/2, is likely the direct cause of fork acceleration. It is interesting to note that loss of CARM1 renders cells resistant to PARPi (Fig. S5G), which could be a result of compromised DNA binding of PARP1 and reduced PARP1 trapping. In future studies, it is important to investigate whether alterations of CARM1 in tumors affect the efficacy of PARPi therapy.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Please direct any requests for further information or reagents to the lead contact, Dr. Lee Zou (zou.lee@mgh.harvard.edu), Massachusetts General Hospital Cancer Center, Harvard medical School, Boston, MA 02129, USA.
Materials Availability
All unique reagents generated in this study will be made available upon request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
Western blot raw data can be accessed in Mendeley: https://data.mendeley.com/datasets/yy797jhyd8/draft?a=36435c07-3368-4f2c-bbb8-948c030b72ed
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The human embryonic kidney HEK293T and human U2OS osteosarcoma cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 1 % penicillin/streptomycin (PS). The non-tumorigenic human mammary epithelial MCF10A cells (a gift from Drs. Valentine Comaills and Shyamala Maheswaran) were cultured as described (Debnath et al., 2003). MCF10 derivative lines expressing CARM1 were generated by infecting the cells with lentivirus expressing CARM1 under a doxycycline-inducible promoter (pINDUCER20) and selected with G418 (600 ug/mL). Carm1fl/fl KO mouse embryonic fibroblast (MEF) (a gift from Dr. Mark Bedford) were maintained in DMEM high glucose supplemented with 10% FBS, 1% PS, 1X non-essential amino acids solution (NEAA) and 3 μg/mL Blasticidin. To delete Carm1, MEFs were treated with 4-hydroxytamoxifen (4OHT, 2 μM) for 3 days (Yadav et al., 2003).
METHOD DETAILS
Cloning
The plasmid pENTR223-CARM1 was purchased at Harvard PlasmID Repository. From this plasmid, CARM1 variants were generated, fused with an NLS (CCCAAGAAGA-AGAGGAAAGTC) at the N-termini, and cloned into the pENTR-TOPO Gateway vector. These following primers were used: CARM1A (MMG8-9), CARM1B (MMG8-10), CARM1C (MMG11-12) and CARM1D (MMG12-13). Primers MMG23-24 were used to generated the catalytically inactive mutant CARM1R168A. For plasmid transfection, pENTR223-CARM1 and pENTR-TOPO-CARM1 variants were shuttled into pDEST-SFB vector (S, Flag, Biotin-binding peptide tags)(Marechal et al., 2014). For recombinant protein expression, pENTR-TOPO-CARM1B, pENTR223-CARM1WT and CARM1R168A were transferred into pDEST17 carrying 6xHis tag at the N-terminus. For lentiviral expression, pENTR-TOPO-CARM1B and CARM1C, pENTR223-CARM1WT and CARM1R168A were inserted into pINDUCER20 carrying an HA tag at the C-terminus. A start codon was added to pENTR-TOPO-CARM1B and CARM1C before the N-terminal NLS (MMG43-44) and the endogenous stop codon of CARM1 was removed (CARM1WT MMG33-34, CARM1R168A MMG33-34, CARM1B MMG37-38, CARM1C MMG39-40) to allow HA tag expression fused to the C-terminus.
Plasmid transfection
For immunoprecipitation and iPOND experiments using HEK293T cells, pDEST-SFB constructs were transfected using Transporter™5 (Polyscience). For viral production, HEK293T cells were transfected using X-tremeGENE 9 DNA transfection (Roche).
RNA interference
Reverse siRNA transfections were done using 5 nM Silencer Select pre-designed siRNAs (Thermo Fisher Scientific) with Lipofectamine RNAiMAX (Thermo Fisher Scientific) for 48 hours. We used BLOCK-iT™ RNAi Designer to design siRNAs (siCARM1 #4 and #5) targeting the 3’UTR region of CARM1 mRNA (Thermo Fisher Scientific).
| siRNAs used in this study | Sequence (5’-3’) or ID |
|---|---|
| siCTL | 4390843 |
| siCARM1 #3 | S20579 |
| siCARM1#4 (3’UTR) | UCACAGCUCUCUUUGCUAUTT |
| siCARM1#5 (3’UTR) | GGCUUGUCAUCUGCUGGAATT |
| siRECQ1 | S11903 |
| siPrimPol | S47416 |
| siPARP1 | S1099 |
| siRAD18-1 | S32296 |
| siRAD18-2 | GCAGUUUGCUUUAGAGUCATT |
| siRAD18-3 | GCUCUCUGAUCGUGAUUUATT |
| siREV1 | S28167 |
iPOND
iPOND was essentially performed as described previously with minor modifications (Sirbu et al., 2012). One hundred million of exponentially growing HEK293T cells were treated as described in figure legends. For capturing proteins associating with progressing forks, cells were pulse-labeled with 10 μM EdU for 10 or 15 minutes. For capturing proteins at stalled forks, 3 mM HU was added for 2 hours following EdU-labelling. For capturing proteins at restarting forks, cells were treated with 3 mM HU for 2 hours, washed with pre-equilibrated cell culture medium and rapidly released in 10 μM EdU for 30 minutes. For thymidine chase, cells were first released from HU in EdU for 30 minutes, and then washed and incubated with 10 μM thymidine for 60 minutes. Cells were crosslinked with 1 % formaldehyde for 20 minutes, quenched with 0.125M glycine and washed three times with PBS. Cells were then permeabilized with 0.25% Triton X-100 in PBS for 30 minutes at room temperature and washed twice with PBS. For the conjugation of EdU with biotin azide, cells were incubated in click reaction buffer (20 μM biotin azide, 10 mM sodium ascorbate and 2 mM CuSO4 in PBS) for 2 hours at room temperature. Cells were washed twice with PBS, and resuspended in lysis buffer (50 mM Tris-HCl, pH 8, and 1% SDS) supplemented with protease inhibitors. Chromatin was solubilized by sonication using a microtip sonicator 550 Sonic Dismembrator (ThermoFisher Scientific) for 1 minute 30 second at 4°C followed by centrifugation at 13,000 rpm for 10 minutes. Supernatants were diluted with 1:1 PBS (vol/vol) containing protease inhibitors and incubated overnight with streptavidin agarose beads. Beads were washed once with lysis buffer, once with low salt buffer (1% Triton, 20 mM Tris pH8, 2 mM EDTA, 150 mM NaCl), once with high salt buffer (1% Triton, 20 mM Tris pH8, 2 mM EDTA, 500 mM NaCl), and once with lysis buffer. Captured proteins were eluted by boiling beads for 30 minutes at 95°C in 2X Laemmli buffer. The beads were vortexed and spun, then the supernatants were loaded onto SDS-PAGE gel for Western blot analysis. The images were captured with a ChemiDoc XRS+ and analyzed with the Image Lab software.
For mass spectrometry analysis, after overnight incubation beads were washed twice with lysis buffer, once with low salt buffer, once with high salt buffer, twice with 50 mM Tris pH8 and twice with 75 mM ammonium bicarbonate pH 8.0. The beads were resuspended in 100 μL ammonium bicarbonate pH 8.0 and sent for proteomic analysis.
Mass spectrometry analysis
On-beads proteolysis
Isolated proteins were resuspended in one volume of 75 mM ammonium bicarbonate pH 8.0 solution for on-beads proteolysis. Proteins were first reduced with 10 mM DTT for 20 min at room temperature and alkylated with 50 mM iodoacetamide under the same conditions but protected from light. Two micrograms of a trypsin/Lys-C mix (Promega) were added to beads resuspensions. Proteins were digested overnight at 37°C on a rotating mixer. The digested products were acidified with trifluoroacetic acid (TFA) and the peptides were isolated and washed on C18 tips (Thermo Fisher Scientific, USA) according to the manufacturer's instructions. Finally, the purified peptides were dried by vacuum centrifugation and stored at −80 °C.
LC-MS/MS analysis
Peptide samples corresponding to two independent iPOND assays were analyzed using LC-MS/MS on two instruments. For the first iPOND analysis, samples were analyzed by nanoLC/MSMS using an Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) connected to a Dionex UltiMate 3000 nanoRSLC chromatography system (Thermo Fisher Scientific). Peptides were trapped at 20 μl/min in loading solvent (2% acetonitrile, 0.05% TFA) on a 5 mm x 300 μm C18 pepmap cartridge pre-column (Thermo Fisher Scientific) for 5 min. Then, the pre-column was switched online with a Pepmap Acclaim column (Thermo Fisher Scientific) 50 cm x 75 μm separation column and the peptides were eluted with a linear gradient from 5-40% solvent B (A: 0,1% formic acid, B: 80% acetonitrile, 0.1% formic acid) in 30minutes, at 300 nL/min. Mass spectra were acquired using a data dependent acquisition mode using Thermo XCalibur software version 4.1.50. Full scan mass spectra (350 to 1800 m/z) were acquired in the orbitrap using an AGC target of 4e5, a maximum injection time of 50 ms and a resolution of 120 000. Internal calibration using lock mass on the m/z 445.12003 siloxane ion was used. Each MS scan was followed by acquisition of fragmentation MSMS spectra of the most intense ions for a total cycle time of 3 seconds (top speed mode). The selected ions were isolated using the quadrupole analyzer in a window of 1.6 m/z and fragmented by Higher energy Collision-induced Dissociation (HCD) with 35% of collision energy. The resulting fragments were detected by the linear ion trap in rapid scan rate with an AGC target of 1e4 and a maximum injection time of 50 ms. Dynamic exclusion of previously fragmented peptides was set for a period of 30 sec and a tolerance of 10 ppm.
For the second iPOND analysis, trypsin-digested peptides were separated on a Dionex Ultimate 3000 nanoHPLC system coupled to a Q Exactive™ OrbiTrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). Ten μl of sample (a total of 1.5 μg) resuspended in 1% (vol/vol) formic acid were loaded with a constant flow of 4 μl/min onto an Acclaim PepMap100 C18 column (0.3 mm id x 5 mm, Dionex Corporation). After trap enrichment, peptides were eluted onto an EasySpray PepMap C18 nano column (75 μm x 50 cm, Dionex Corporation) with a linear gradient of 8-40% solvent B (80% acetonitrile with 0.1% formic acid) over 240 minutes with a constant flow of 200 nl/min. The HPLC system was coupled to the mass spectrometer via an EasySpray source. The spray voltage was set to 2.0 kV and the temperature of the column set to 40°C. Full scan MS survey spectra (m/z 350-1600) in profile mode were acquired in the Orbitrap with a resolution of 70,000 after accumulation of 1,000,000 ions. The ten most intense peptide ions from the preview scan in the Orbitrap were fragmented by collision-induced dissociation (normalized collision energy 25% and resolution of 17,500) after the accumulation of 50,000 ions. Maximal filling times were 250 ms for the full scans and 60 ms for the MS/MS scans. Precursor ion charge state screening was enabled and all unassigned charge states as well as singly, 7 and 8 charged species were rejected. The dynamic exclusion list was restricted to a maximum of 500 entries with a maximum retention period of 40 seconds and a relative mass window of 10 ppm. The lock mass option was enabled for survey scans to improve mass accuracy. Data were acquired using the XCalibur software.
Mass spectrometry data analysis
Mass spectra data generated by both instruments were searched using Byonic™ version 3.3.9 (Protein Metrics, USA) against the Homo sapiens reference proteome (canonical & isoforms, 74 811 entries, Uniprot) with a static modification of carbamidomethylation on Cys (+57.0215) and the following variable modifications: carbamidomethylation artifacts on His, Lys and peptide N-terminal (+57.0215), oxidation of Met (+15.9949 Da), formation of pyro-Glu from N-terminal glutamate and glutamine residues (−18.0105 Da for N-Term Glu and −17.0265 Da for N-term Gln) and N-terminal protein acetylation (+42.0105 Da). Semi-specific trypsin cleavage was specified and a maximum of two missed cleavage was allowed. For the Orbitrap Fusion™ Tribrid™, the mass tolerance was set to 5 ppm for the precursor ions and 0.6 Da for the fragment ions while the tolerance was set to 7 ppm for the precursor ions and 20 ppm for the fragment ions for data generated with the Q Exactive™ Orbitrap. A false discovery rate (FDR) of 1% was estimated using concatenated forward–reverse database search. A minimum Log Prob (i.e. -Log base 10 of the protein p-value) of 1.3 (p-value ≤ 0.05) was selected as protein identification cut off value. Decoys protein IDs and common contaminants were filtered out of the final dataset. Intensity-based label-free quantification (LFQ) values were used to estimate the relative abundance of proteins in each iPOND steps, which correspond to the sum of all peak intensities over all tandem mass spectra assigned to a particular protein. Missing LFQ data imputation was estimated by using a noise value corresponding to the lowest 1% percentile of the LFQ distribution. This noise value was imputed for each sample when the intensity value is missing (i.e. undetected proteins) and selected as the minimum LFQ intensity for low abundance protein identifications for which LFQ values fall below the 1% percentile background.
DNA fiber analysis
Exponentially growing MCF10A or U2OS cells were first pulse labeled with 50 μM CldU, washed twice with equilibrated PBS and then labeled with 250 μM IdU under the conditions specified in the figure legends. Collected cells were resuspended in cold PBS (1x106 cell per milliliter), and 2.5 μL was stretched on a glass slide after mixing with spreading buffer (7.5 μL) (0.5% SDS, 200 mM Tris-HCl pH 7.4, 50 mM EDTA). DNA fibers were fixed in methanol:acetic acid (3:1), denatured in 2.5 N HCl for 30 min, and blocked in 3% BSA/0.05% Tween-20 for 60 min at room temperature. CldU and IdU detection were done using rat anti-BrdU (1:100; abcam) and mouse anti-BrdU (1:20; BD Biosciences) for 2 hours at 37°C followed by Alexa-488 anti-mouse (1:100; Jackson ImmunoResearch) and Cy3 anti-rat (1:100; Jackson ImmunoResearch) for 45 minutes at room temperature. Slides were mounted with Prolong Gold and dried overnight. Fibers were imaged with a 60X objective on a Nikon 90i or 40X Echo Revolve microscope and quantified using FIJI.
S1 nuclease experiments were essentially performed as described previously (Quinet et al., 2017a). After IdU labeling, cells were permeabilized with CSK100 buffer (100 mM NaCl, 10 mM MOPS pH7, 3 mM MgCl2, 300 mM sucrose, 0.5 % Triton X-100) for 10 minutes at room temperature then washed with 1xPBS carefully. The cells were washed once with S1 nuclease buffer pH4.6 (30 mM NaAc, 10 mM ZnAc, 5% glycerol, 50 mM NaCl) and subsequently incubated with S1 buffer containing S1 nuclease (20 U/ mL) for 30 minutes at 37°C. The cells were washed once with 1xPBS containing 0.1% BSA to help precipitate nuclei, then collected and processed for staining as described previously.
DNA combing
Cells were labeled as described in DNA fiber analysis method and DNA solution was prepared according to the instructions provided by the FiberPrep®DNA Extraction Kit. Vinylsilane coated coverslips were incubated into the DNA solution for 15 minutes at room temperature and removed at a constant speed using the DNA combing system. Slides were dried for 2 hours at 60°C. DNA fibers were dehydrated 5 minutes in ethanol 70%, 90% and 100% then denatured in 1M NaOH for 20 minutes. Slides were washed five times one minute with 1xPBS and blocked in 3% BSA/0.05% Tween-20 for 60 min at room temperature. Primary and secondary antibody incubations were subsequently performed as described in DNA fiber analysis method following by ssDNA detection using (1:100, anti-DNA then 1:100, Alexa-647 anti-mouse) for 1 hour at 37°C for each incubation. Slides were mounted with Prolong Gold and dried overnight. Fibers were imaged with a 40X objective Echo Revolve microscope and quantified using FIJI.
Recombinant protein purification
The recombinant PARP1 WT and PARP1 ΔHD proteins from John Pascal lab and HPF1 protein from Ivan Ahel lab were prepared as described previously (Gibbs-Seymour et al., 2016; Langelier et al., 2018). CARM1WT, CARM1R168A and CARM1B were cloned in pDEST-17 carrying 6x His tag and expressed in Rosetta (DE3) cells (Novagen). Single clone was grown in LB medium and expression was induced at OD600 0.48 with 0.5 mM IPTG for 4 hours 30 minutes at 37°C. Cell pellets were lysed in P5 buffer (50 mM NaHPO4 pH 7.0, 500 mM NaCl, 10% glycerol, 0.05% Triton X-100, 5 mM imidazole supplemented with 1x protease inhibitors and 1 mM PMSF) then sonicated using a microtip sonicator 550 Sonic Dismembrator. Benzonase (20 U/mL) and 1 mM MgCl2 were added to the lysed sample and incubated for 1 hour 30 minutes at 4°C. Cell debris was removed by centrifugation (16,000 rpm, 30 minutes) and the cleared supernatant was loaded onto 150 μL TALON Metal Affinity Resin for 30 minutes at 4°C. The resin was washed two times with P5 buffer containing 30 mM imidazole and one time with 80 mM imidazole. His-tagged proteins were eluted in P5 buffer containing 500 mM imidazole and dialyzed against dialysis buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 10% glycerol,1 mM DTT) for 1 hour then stored at −80°C.
Immunoprecipitation
For PARP1 immunoprecipitation, HEK293T cells were transfected with the pDEST-CARM1-SFB construct for 48 hours and treated as described in the figure legends. Cell pellets were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, 1% NP40, 1% Triton X-100,1 mM DTT, 10 mM NaF, 1 mM Na2VO3, 1x protease inhibitors, 10 μM olaparib and 1 μM PARGi) and sonicated for 45 seconds using a microtip sonicator 550 Sonic Dismembrator. Cell extracts were incubated for 1 hour 30 minutes with Benzonase (5U/μL) at 4°C followed by centrifugation at 3,000 rpm for 10 min. The supernatant was diluted 1:1 in dilution buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM NaF, 1 mM Na2VO3, 1x protease inhibitors, 10 μM olaparib and 1 μM PARGi). Equal amount of proteins (1 mg) for each condition were incubated with Dynabeads™ Protein G conjugated with PARP1 antibody (2 μg/IP) for 2 hours at 4°C. Subsequently, beads were washed three times with wash buffer (50 mM Tris pH8, 150 mM NaCl, 1 mM DTT, 0.1% Tween20, 10 μM olaparib and 1 μM PARGi) and proteins were eluted by boiling beads for 5 min at 95°C in 2X Laemmli buffer. The beads were vortexed and spun, then the supernatants were loaded onto SDS-PAGE gels for Western blot analysis. Images were captured with a ChemiDoc XRS+ and analyzed with the Image Lab software.
For mapping the PARP1-interacting regions of CARM1, nuclear extracts were prepared from HEK293T and essentially performed as described previously with minor modifications (Mendez and Stillman, 2000). HEK293T cells were transfected with the pDEST-CARM1-SFB plasmid for 48 hours and treated as described in the figure legends. Cells were washed with 1xPBS and resuspended in 800 μl of buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.04 % NP40, 10 mM NaF, 1 mM Na2VO3, 1x protease inhibitors, 10 μM olaparib and 1 μM PARGi) then incubated on ice for 5 min. Cytoplasmic proteins were separated from nuclei by low-speed centrifugation (1,300 g, 4 minutes) and the pellets (nuclei) were lysed in 800 μl of buffer B (20 mM HEPES pH 7.9, 400 mM KCl, 1.5 mM MgCl2, 0.5 % NP40, 10 mM NaF, 1 mM Na2VO3, 1x protease inhibitors, 10 μM olaparib and 1 μM PARGi). After 20 minutes incubation at 4°C with buffer B, the nuclear soluble fraction was separated by centrifugation (1,700 g, 4 minutes) and incubated for 1 hour with Benzonase (5U/μL) at 4°C. Then, nuclear extracts were diluted 1:1 in dilution buffer (20 mM HEPES pH 7.9, 0.5 % NP40, 10 mM NaF, 1 mM Na2VO3, 1x protease inhibitors, 10 μM olaparib and 1 μM PARGi). Equal amount of proteins (1 mg) for each condition were incubated with Dynabeads™ M-280 Streptavidin beads overnight at 4°C. Subsequently, beads were washed three times with wash buffer (50 mM Tris pH8, 150 mM NaCl, 1 mM DTT, 0.5% NP40), and proteins were eluted by boiling beads for 5 min at 95°C in 2X Laemmli buffer. The beads were vortexed and spun, then the supernatants were loaded onto SDS-PAGE gels for Western blot analysis. Images were captured with a ChemiDoc XRS+ and analyzed with the Image Lab software.
For endogenous PARP1/CARM1 immunoprecipitation, nuclear extracts were prepared from HEK293T cells using similar protocol as described for mapping PARP1-interacting regions. Equal amount of proteins (200 μg for PARP1 IP and 100 μg for CARM1 IP) for each condition were incubated with PARP1 (2 μg/IP) or CARM1 (1 μg/IP) antibody for 2 hours at 4°C. Subsequently, lysates were incubated with Dynabeads™ Protein G conjugated for 30 minutes at 4°C. Then, beads were washed three times with wash buffer (50 mM Tris pH8, 150 mM NaCl, 1 mM DTT, 0.5% NP40, 10 μM olaparib and 1 μM PARGi) and proteins were eluted by boiling beads for 5 min at 95°C in 2X Laemmli buffer. The beads were vortexed and spun, then the supernatants were loaded onto SDS-PAGE gels for Western blot analysis. Images were captured with a ChemiDoc XRS+ and analyzed with the Image Lab software.
Immunofluorescence
To monitor DNA synthesis, U2OS cells were first pulse-labeled with 10 μM EdU for 30 minutes and then treated as described in the figure legends. Cells were pre-extracted with 1xPBS containing 0.2 % Triton X-100 for 2 minutes prior to fixation with 3% paraformaldehyde/2% sucrose for 15 minutes. Subsequently, cells were treated with blocking buffer (3% BSA in 1xPBS) for 30 minutes at room temperature. Primary antibodies, diluted to 1:100 in blocking buffer, were added to the cells for 2 hours at 37°C. After three washes with PBST (1xPBS with 0.05% Tween), cells were incubated in the dark with the secondary antibody conjugated to Cy3, diluted to 1:500 in blocking buffer, for 1 hour at room temperature. To visualize EdU incorporation, cells were washed three times with PBST and processed with Click-iT EdU Alexa Fluor 488 imaging Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Finally, after three washes with PBST, cells were stained with DAPI for 5 minutes and mounted on slides with Prolong Gold. Images were captured with Echo Revolve microscope and quantified using FIJI.
Flow cytometry analysis
Cells were labeled with 10 μM EdU for 30 minutes (U2OS) or 60 minutes (MCF10A) then fixed overnight at −20°C in 75% ethanol. Samples were processed with the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Thermo Fisher Scientific) following manufacturer’s instructions. Cell pellets were resuspended in 10 μg/mL Propidium Iodide (PI) and 50 μg/mL RNase A for 20 minutes at 37°C. Flow cytometry was done using a BD LSRII apparatus equipped with the FACS Diva software (BD Biosciences).
Electron microscopy
For EM analysis of replication intermediates, 5-10 x106 U2OS cells were collected immediately after treatment with 4 mM hydroxyurea for 2 hours. Untreated cells were also included. DNA was crosslinked by incubating with 10 μg/mL 4,5’,8-trimethylpsoralen followed by a 3-minute exposure to 366 nm UV light on a precooled metal block, for a total of three rounds. Cells were lysed and genomic DNA was isolated from the nuclei by proteinase K digestion and chloroform-isoamyl alcohol extraction. Genomic DNA was purified by isopropanol precipitation and digested with PvuII HF with the appropriate buffer for 4 hours at 37°C. Replication intermediates were enriched on a benzoylated naphthoylated DEAE-cellulose column. Samples were prepared for visualization by EM by spreading the purified, concentrated DNA on a carbon-coated grid in the presence of benzyl-dimethyl-alkylammonium chloride, followed by platinum rotary shadowing. Images were obtained on a JEOL JEM-1400 electron microscope using a bottom mounted AMT XR401 camera. Analysis was performed using ImageJ software (National Institute of Health). EM analysis allows distinguishing duplex DNA—which is expected to appear as a 10 nm thick fiber after the platinum/carbon coating step necessary for EM visualization—from ssDNA, which has a reduced thickness of 5-7 nm. Criteria used for the assignment of a three-way junction, indicative of a replication fork, include the joining of three DNA fibers into a single junction, with two symmetrical daughter strands and single parental strand. Reversed replication forks consist of four DNA fibers joined at a single junction, consisting of two symmetrical daughter strands, one parental strand and the addition of a typically shorter fourth strand, representative of the reversed arm. The length of the two daughter strands corresponding to the newly replicated duplex should be equal (b=c), whereas the length of the parental arm and the regressed arm can vary (a ≠ b = c ≠ d). Conversely, canonical Holliday junction structures will be characterized by arms of equal length (a = b, c = d). Particular attention is paid to the junction of the reversed replication fork in order to observe the presence of a bubble structure, indicating that the junction is opened up and that it is simply not the result of the occasional crossover of two DNA molecules. These four-way junctions of reversed replication forks may also be collapsed and other indicators such as daughter strand symmetry, presence of single-stranded DNA at the junction or the entire structure itself, all are considered during analysis (Neelsen et al., 2014). The frequency of reversed forks in a sample is computed using the Prism software.
In vitro PARP1 activation assay
Recombinant proteins were incubated in 25 μL PAR reaction buffer (100 mM Tris HCl pH8, 10 mM MgCl2, 10% glycerol, 10 % ethanol and 10 mM DTT) with 25 ug/mL calf thymus dsDNA. The PARylation reactions were started by adding 250 μM NAD+ to the reaction mixtures. Incubation time was 3 minutes for Figs. 6C, 7E-F, 10 minutes for Fig. S3D, 15 minutes for Figs. 6B, 7D, and S6A-B. All reactions were done at room temperature. To immediately terminate the reaction, 6X Laemmli buffer supplemented with 5% fresh β-mercaptoethanol was added followed by SDS-PAGE and Western blot analysis. Images were captured with a ChemiDoc XRS+ and analyzed with the Image Lab software.
Biotin DNA pull-down
Dynabeads M-280 Streptavidin (Thermo Fisher Scientific) containing 5’-biotinylated dsDNA or ssDNA 60-mer were pre-incubated with blocking buffer (25 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM DTT, 5 % glycerol and 10 mg/ml BSA) for 20 min at 37°C. The beads were captured with the magnetic particle separator, washed once with reaction buffer (25 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM DTT and 5 % glycerol) and resuspended in the same buffer. Purified PARP1 and CARM1 proteins were added to the beads and the reaction mixtures were incubated for the indicated time described in the figure legends. The beads were captured and were washed twice with 50 μl of reaction buffer. The beads were resuspended in 15 μl of 2X Laemmli buffer, vortexed and heated at 95°C for 5 minutes. Finally, the beads were spun down and the supernatants were loaded onto SDS-PAGE gels. Gels were stained with Sypro Ruby (Invitrogen) and proteins visualized with UV.
| Oligonucleotides for biotin DNA pull-down |
Sequence (5’-3’) |
|---|---|
| 5’ Biotin TYO111 | /5BiotinTEG/AAGCCCTCCCGTATCG TAGTTATCTACACGACGGGGAGTCAGGCA ACTATGGATGAACGA |
| TYO128 | TCGTTCATCCATAGTTGCCTGACTCC CCGTCGTGTAGATAACTACGATAC GGGAGGGCTT |
Chromosome spreads
Asynchronous Carm1fl/fl KO MEF cells were treated with 150 ng/mL nocodazole for 12 hours. Subsequently, mitotic cells were collected through mitotic shake-off. After one wash with 1xPBS, cells were resuspended in 75 mM KCl and immediately spun to avoid over swelling. Cell pellets were resuspended with 20 drops of fixative solution (Methanol: Acetic acid, 3:1) and spun again. Then, cells were washed twice with 5 mL fixative solution. Finally, cell pellets were resuspended with 10 drops of fixative solution and dropped onto wet slides tilted at a 45° angle. When the slides were dried, chromosomes were stained with DAPI for 10 minutes (1: 10 000) and mounted with Prolong Gold. Images were captured with a Nikon 90i microscope and quantified using FIJI.
Neutral comet Assay
Neutral comet assay (also known as single-cell gel electrophoresis assay) were carried out as per the instructions provided by the Comet Assay kit. At least 178 comet images from each condition were scored using OpenComet software.
Cell viability assay
Cell viability assays were performed in 96 well format and cells were treated as described in the figure legends. To determine the number of viable cells, CellTiter-Glo was used according to manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
FIJI software was used to quantify PAR/PARP1 intensities and measured CldU/IdU length. ImageJ software was used for electron microscopy. Statistical analyses were performed using the GraphPad Prism software. Statistical parameters are shown in figures and described in figure legends. For DNA fiber analysis, p-value significance was defined as follows: (ns) not significant; (****) P<0.0001;(***) P<0.001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CHK1 | Santa Cruz | Cat#: sc-8408; RRID: AB_627257 |
| CHK1 pS345 | Cell Signaling | Cat#: 2348; RRID: AB_331212 |
| CARM1 (for WB) | Bethyl | Cat#: A300-421A; RRID: AB_2068452 |
| CARM1 (for WB) | Bethyl | Cat#: A300-420A; RRID: AB_420963 |
| CARM1 (for IP) | Cell Signaling | Cat#: 12495; RRID: AB_2797935 |
| FLAG | Sigma | Cat#: F1804; RRID: AB_262044 |
| GAPDH | Millipore | Cat#: ABS16; RRID: AB_10806772 |
| HA | BioLegend | Cat#: 901501; RRID: AB_2565006 |
| His | Cell Signaling | Cat#: 2365; RRID: AB_2115720 |
| Histone H3 | Abcam | Cat#: ab1791; RRID: AB_302613 |
| KU70 | Genetex | Cat#: GTX70271; RRID: AB_372048 |
| PABP1 | Novus | Cat#: NB120-6125; RRID: AB_791470 |
| PABP1-meR455/R460 | Cell Signaling | Cat#:3505 ; RRID: AB_2298971 |
| PAR 96 :10 (PAR assay) | Home made | (Affar et al., 1999) |
| PAR (for IF and WB) | Trevigen | Cat#: 4336-BPC-100; RRID: AB_2721257 |
| PARP1 (for WB) | Cell Signaling | Cat#: 9542; RRID: AB_2160739 |
| PARP1 (for IP) | Santa Cruz | Cat#: sc-8007; RRID: AB_628105 |
| PARP1 (for IF) | Cell Signaling | Cat#: 9532; RRID: AB_659884 |
| PARG | Cell Signaling | Cat#: 66564; RRID: AB_2750890 |
| PCNA | Santa Cruz | Cat#: sc-56; RRID: AB_628110 |
| PrimPol | Home made | (Mourón et al., 2013) |
| RAD18 | Bethyl | Cat#:A301-340A; RRID: AB_937974 |
| RECQ1 | Bethyl | Cat#: A300-450A; RRID: AB_420916 |
| REV1 | Santa Cruz | Cat#: sc-393022; RRID:N/A |
| RPA32 | Thermo Fisher Scientific | Cat#: MA1-26418; RRID: AB_795362 |
| RPA32 pS33 | Bethyl | Cat#: A300-246A; RRID: AB_2180847 |
| RPA70 | Bethyl | Cat#: A300-241A; RRID: AB_2180681 |
| Anti-BrdU rat (for DNA fiber) | Abcam | Cat#: ab6326; RRID: AB_305426 |
| Anti-BrdU mouse (for DNA fiber) | Becton Dickinson | Cat#: 347580; RRID: AB_400326 |
| Anti-DNA, single stranded (for combing) | Millipore | Cat#:MAB3034; RRID: AB_94645 |
| Anti-mouse goat 647 IgG2a (for combing) | Thermo Fisher Scientific | Cat#:A-21241; RRID: AB_2535810 |
| Anti-mouse donkey 488 (for DNA fiber) | Jackson ImmunoResearch | Cat#: 715-545-151; RRID: AB_2341099 |
| Anti-rat donkey Cy3 (for DNA fiber) | Jackson ImmunoResearch | Cat#: 712-165-153; RRID: AB_2340667 |
| Anti-rabbit goat AlexaFluor 594 (for IF) | Jackson ImmunoResearch | Cat#: 115-585-144; RRID: N/A |
| Anti-HRP rabbit (for WB) | Jackson ImmunoResearch | Cat#: 111-035-003; RRID: AB_2313567 |
| Anti-HRP mouse (for WB) | Jackson ImmunoResearch | Cat#: 115-035-003; RRID: AB_10015289 |
| Chemicals and Recombinant Proteins | ||
| Doxycycline hyclate | Sigma-Aldrich | D9891 |
| Hydroxyurea | Sigma-Aldrich | H8627 |
| Camptothecin | Sigma-Aldrich | C9911 |
| Nocodazole | Sigma-Aldrich | M1404 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | 13778-150 |
| PureLink™ RNase A | Thermo Fisher Scientific | 12091-021 |
| Proteinase K | Thermo Fisher Scientific | 25530049 |
| 4,5’,8-trimethylpsoralen | Sigma-Aldrich | T6137 |
| Thymidine | Sigma-Aldrich | T1895 |
| EdU (5-ethynyl-2’-deoxyuridine) | Thermo Fisher Scientific | A10044 |
| CldU | Sigma-Aldrich | C6891 |
| IdU | Sigma-Aldrich | I7125 |
| DRB (5,6-dichloro-1-β-D-ribofuranosylbenzimidazole) | Cayman Chemical company | 10010302 |
| Propidium Iodide | Sigma-Aldrich | P4864 |
| Transporter™5 Transfection Reagent | Polysciences | 26008 |
| X-tremeGENE 9 DNA transfection | Roche | 06365779001 |
| Biotin azide | Thermo Fisher Scientific | B10184 |
| Dynabeads™ Protein G (for IP) | Thermo Fisher Scientific | 10004D |
| Dynabeads™ M-280 Streptavidin beads (for IP) | Thermo Fisher Scientific | 11205D |
| Streptavidin Agarose beads (for iPOND) | Novagen | 69203 |
| TALON Metal Affinity Resin | Takara | 635501 |
| Benzoylated naphthoylated DEAE-cellulose column | Sigma-Aldrich | B6385 |
| Benzonase nuclease | Millipore | E1014 |
| S1 Nuclease | Thermo Fisher Scientific | 18001-016 |
| Sodium L-ascorbate | Sigma-Aldrich | A4034 |
| IPTG | Millipore | 420322 |
| Methyl methanesulfonate (MMS) | Sigma-Aldrich | 129925 |
| CDC7i (PHA-767491) | Selleckchem | S2742 |
| CARM1i (TP064) | Tocris | 6008 |
| PARGi (PDD 00017273) | Tocris | 5952 |
| PARPi – Olaparib (AZD2281) | Selleckchem | S1060 |
| PARPi- Veliparib (ABT-888) | Selleckchem | S1004 |
| PARPi- Talazoparib (BMN 673) | Selleckchem | S7048 |
| PARP1 WT purified protein | (Langelier et al., 2018) | From John Pascal lab |
| PARP1 ΔHD purified protein | (Langelier et al., 2018) | From John Pascal lab |
| HPF1 purified protein | (Gibbs-Seymour et al., 2016) | From Ivan Ahel lab |
| Critical Commercial Assays | ||
| pENTR™/D-TOPO Cloning Kit | Thermo Fisher Scientific | 45-0218 |
| Gateway LR Clonase™ II Enzyme mix | Thermo Fisher Scientific | 11791-020 |
| Click-iT EdU Alexa Fluor 488 Imaging Kit | Thermo Fisher Scientific | C10337 |
| Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | C10635 |
| CometAssay®kit | Trevigen | 4250-050-K |
| CellTiter-Glo® Luminescent | Promega | G7570 |
| FiberPrep®DNA Extraction Kit | Genomic Vision | EXT-001 |
| CombiCoverslips™ | Genomic Vision | COV-002-10-RUO |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-11268 |
| U2OS | ATCC | HTB-96 |
| MCF10A | (Debnath et al., 2003) | From Shyamala Maheswaran lab |
| Carm1fl/fl KO MEF | (Yadav et al., 2003) | From Mark Bedford lab |
| Oligonucleotides | ||
| Primers for cloning | This study | See Table S3 |
| siRNAs sequence | This study | see Method below |
| Oligonucleotides for biotin DNA pull-down | This study | see Method below |
| Calf thymus dsDNA (for PAR assay) | Sigma-Aldrich | D4522 |
| Recombinant DNA | ||
| pENTR223-CARM1 | Harvard PlasmID Repository | N/A |
| pINDUCER20 | (Meerbrey et al., 2011) | Addgene plasmid#44012 |
| pINDUCER20-CARM1 WT-HA | This study | N/A |
| pINDUCER20-CARM1 mutant R168A - HA | This study | N/A |
| pINDUCER20-CARM1 mutant B-HA | This study | N/A |
| pINDUCER20-CARM1 mutant C-HA | This study | N/A |
| pDEST-SFB | (Marechal et al., 2014) | N/A |
| pDEST- CARM1 WT - SFB | This study | N/A |
| pDEST- NLS- CARM1 mutant A - SFB | This study | N/A |
| pDEST- NLS-CARM1 mutant B - SFB | This study | N/A |
| pDEST- NLS-CARM1 mutant C - SFB | This study | N/A |
| pDEST-NLS- CARM1 mutant D - SFB | This study | N/A |
| pDEST-17- CARM1 WT-HIS | This study | N/A |
| pDEST-17- CARM1 mutant B - HIS | This study | N/A |
| pDEST-17- CARM1 mutant R168A - HIS | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism 6 | GraphPad Software, Inc | https://www.graphpad.com/scientific-software/prism/ |
| NIS elements viewer | Nikon | https://www.microscope.healthcare.nikon.com/ |
| FIJI | (Schindelin et al., 2012) | https://fiji.sc/ |
| Image Lab | BioRad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
Highlights.
CARM1 associates with replication forks and reduces fork speed.
Loss of CARM1 reduces fork reversal but increases ssDNA gaps and stress tolerance.
CARM1 promotes PARP1 association and PARylation at replication forks.
CARM1 stimulates PARP1 activity in vitro by enhancing its DNA binding.
Acknowledgements
We thank Dr. Juan Mendez for the PrimPol antibody, Dr. Shyamala Maheswaran for MCF10A cells, members of the Zou, Dyson, Lan and Elia labs for discussions. M.-M.G. was partially supported by a postdoctoral fellowship from Fonds de recherche Santé Québec (FRQS). L.Z. is the James & Patricia Poitras Endowed Chair in Cancer Research, and was supported by a Jim & Ann Orr Massachusetts General Hospital Research Scholar Award. This work is supported by grants from the NIH grants GM126421 to M.T.B, CA237263 to A.V., CA248526 to A.V. and L.Z., and CA218856 and CA197779 to L.Z.
Footnotes
Declaration of interests
All authors declare no competing interests. L.Z. is a member of the Advisory Board of Molecular Cell.
References
- Affar EB, Duriez PJ, Shah RG, Winstall E, Germain M, Boucher C, Bourassa S, Kirkland JB, and Poirier GG (1999). Immunological determination and size characterization of poly(ADP-ribose) synthesized in vitro and in vivo. Biochim Biophys Acta 1428, 137–146. [DOI] [PubMed] [Google Scholar]
- Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, and Cimprich KA (2020). HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol Cell 78, 1237–1251 e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, and Richard S (2005). Arginine methylation an emerging regulator of protein function. Mol Cell 18, 263–272. [DOI] [PubMed] [Google Scholar]
- Bern M, Kil YJ, and Becker C (2012). Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics Chapter 13, Unit13 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Cortez D, and Lopes M (2020). The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 21, 633–651. [DOI] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfiglio JJ, Fontana P, Zhang Q, Colby T, Gibbs-Seymour I, Atanassov I, Bartlett E, Zaja R, Ahel I, and Matic I (2017). Serine ADP-Ribosylation Depends on HPF1. Mol Cell 65, 932–940 e936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Buisson R, Boisvert JL, Benes CH, and Zou L (2015). Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol Cell 59, 1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, and Stallcup MR (1999). Regulation of transcription by a protein methyltransferase. Science 284, 2174–2177. [DOI] [PubMed] [Google Scholar]
- Cheng D, Cote J, Shaaban S, and Bedford MT (2007). The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell 25, 71–83. [DOI] [PubMed] [Google Scholar]
- Chumanov RS, Kuhn PA, Xu W, and Burgess RR (2011). Expression and purification of full-length mouse CARM1 from transiently transfected HEK293T cells using HaloTag technology. Protein Expr Purif 76, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G, and Hottiger MO (2005). Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J 24, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawicki-McKenna JM, Langelier MF, DeNizio JE, Riccio AA, Cao CD, Karch KR, McCauley M, Steffen JD, Black BE, and Pascal JM (2015). PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol Cell 60, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eykelenboom JK, Harte EC, Canavan L, Pastor-Peidro A, Calvo-Asensio I, Llorens-Agost M, and Lowndes NF (2013). ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep 5, 1095–1107. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ, et al. (2013). PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs-Seymour I, Fontana P, Rack JGM, and Ahel I (2016). HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity. Mol Cell 62, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, and Cimprich KA (2016). Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 167, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, and Caldecott KW (2018). The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol Cell 71, 319–331 e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer DR, and Rhind N (2017). Replication fork slowing and stalling are distinct, checkpoint-independent consequences of replicating damaged DNA. PLoS Genet 13, e1006958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, and Cimprich KA (2015). HLTF's Ancient HIRAN Domain Binds 3' DNA Ends to Drive Replication Fork Reversal. Mol Cell 58, 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Lee J, Cheng D, Li J, Carter C, Richie E, and Bedford MT (2010). Enzymatic activity is required for the in vivo functions of CARM1. J Biol Chem 285, 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier MF, Riccio AA, and Pascal JM (2014). PARP-2 and PARP-3 are selectively activated by 5' phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 42, 7762–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier MF, Zandarashvili L, Aguiar PM, Black BE, and Pascal JM (2018). NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat Commun 9, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen SC, Hendriks IA, Lyon D, Jensen LJ, and Nielsen ML (2018). Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep 24, 2493–2505 e2494. [DOI] [PubMed] [Google Scholar]
- Lee J, and Bedford MT (2002). PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep 3, 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Bedford MT, and Stallcup MR (2011). Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev 25, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, and Kraus WL (2012). On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 26, 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jimenez AE, Jin J, and Zou L (2014). PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol Cell 53, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, and Bartek J (2018). High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284. [DOI] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, and Stillman B (2000). Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortusewicz O, Ame JC, Schreiber V, and Leonhardt H (2007). Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 35, 7665–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourón S, Rodriguez-Acebes S, Martínez-Jiménez MI, García-Gómez S, Chocrón S, Blanco L, and Méndez J (2013). Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20, 1383–1389. [DOI] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, and Pommier Y (2014). Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 13, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, and Lopes M (2018). ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep 24, 2629–2642 e2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Szewczyk MM, Dela Sena C, Wu H, Dong A, Zeng H, Li F, de Freitas RF, Eram MS, Schapira M, et al. (2018). TP-064, a potent and selective small molecule inhibitor of PRMT4 for multiple myeloma. Oncotarget 9, 18480–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK, and Cantor SB (2020). Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci Adv 6, eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, and Lopes M (2014). Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol 1094, 177–208. [DOI] [PubMed] [Google Scholar]
- Palazzo L, Leidecker O, Prokhorova E, Dauben H, Matic I, and Ahel I (2018). Serine is the major residue for ADP-ribosylation upon DNA damage. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, and Helleday T (2010). Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A 107, 16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017a). DNA Fiber Analysis: Mind the Gap! Methods Enzymol 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Quinet A, Lemacon D, and Vindigni A (2017b). Replication Fork Reversal: Players and Guardians. Mol Cell 68, 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Svikovic S, Lemacon D, Carvajal-Maldonado D, Gonzalez-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell 77, 461–474 e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, and Lopes M (2012). Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19, 417–423. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, and Nussenzweig A (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah GM, Poirier D, Duchaine C, Brochu G, Desnoyers S, Lagueux J, Verreault A, Hoflack JC, Kirkland JB, and Poirier GG (1995). Methods for biochemical study of poly(ADP-ribose) metabolism in vitro and in vivo. Anal Biochem 227, 1–13. [DOI] [PubMed] [Google Scholar]
- Shishkova E, Zeng H, Liu F, Kwiecien NW, Hebert AS, Coon JJ, and Xu W (2017). Global mapping of CARM1 substrates defines enzyme specificity and substrate recognition. Nat Commun 8, 15571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, and Cortez D (2012). Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc 7, 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, and Cortez D (2011). Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suskiewicz MJ, Zobel F, Ogden TEH, Fontana P, Ariza A, Yang JC, Zhu K, Bracken L, Hawthorne WJ, Ahel D, et al. (2020). HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 579, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissier A, Kannouche P, Reck MP, Lehmann AR, Fuchs RP, and Cordonnier A (2004). Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA Repair (Amst) 3, 1503–1514. [DOI] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell 67, 882–890 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, and Yamaizumi M (2004). Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 23, 3886–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav N, Lee J, Kim J, Shen J, Hu MC, Aldaz CM, and Bedford MT (2003). Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci U S A 100, 6464–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, and Gao Y (2018). Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu Rev Biochem 87, 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, and Zou L (2016). Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu Rev Genet 50, 155–173. [DOI] [PubMed] [Google Scholar]
- Zandarashvili L, Langelier MF, Velagapudi UK, Hancock MA, Steffen JD, Billur R, Hannan ZM, Wicks AJ, Krastev DB, Pettitt SJ, et al. (2020). Structural basis for allosteric PARP-1 retention on DNA breaks. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Western blot raw data can be accessed in Mendeley: https://data.mendeley.com/datasets/yy797jhyd8/draft?a=36435c07-3368-4f2c-bbb8-948c030b72ed