

Summary
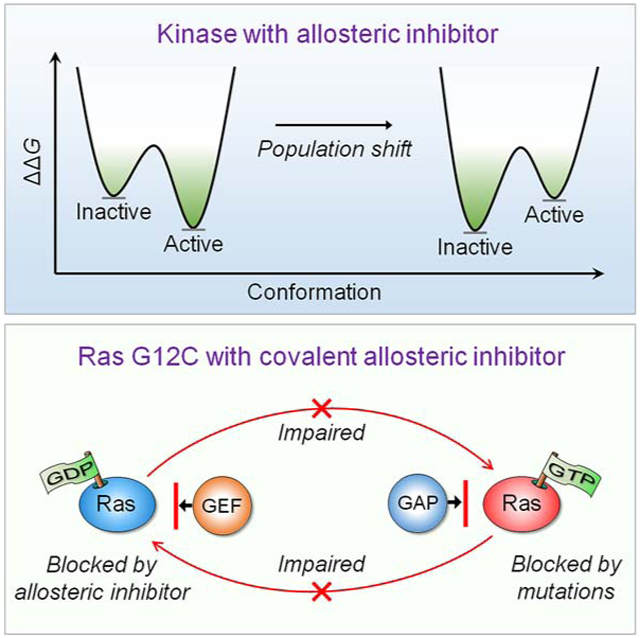
Intuitively, functional states should be targeted; not nonfunctional ones. So why could drugging the inactive K-Ras4BG12C work – but, drugging the inactive kinase will likely not? The reason is the distinct oncogenic mechanisms. Kinase driver mutations work by stabilizing the active state and/or destabilizing the inactive state. Either way, oncogenic kinases are mostly in the active state. Ras driver mutations work by quelling its deactivation mechanisms, GTP hydrolysis and nucleotide exchange. Covalent inhibitors that bind to the inactive GDP-bound K-Ras4BG12C conformation can thus work. By contrast, in kinases, allosteric inhibitors work by altering the active site conformation to favor orthosteric drugs. From the translational standpoint this distinction is vital: it expedites effective pharmaceutical development and extends the drug classification based on the mechanism of action. Collectively, here we postulate that drug action relates to blocking the mechanism of activation; not to whether the protein is in the active or inactive state.
Graphical Abstarct
eTOC Blurb
Nussinov et al. describe that the mechanism of mutational activation of kinases differs from Ras. Drug action should relate to the mechanism of activation, not to whether the protein is in active or inactive state. However, allosterically blocking the functional site will work for the inactive kinase and active Ras.
Background
Why targeting inactive Ras conformations can inhibit active ones, but targeting inactive kinase conformations likely will not? There are already examples of inhibition of inactive (nonfunctional) mutant K-Ras4BG12C (Goody et al., 2019; Janes et al., 2018; Kettle et al., 2020; Lou et al., 2019; McCarthy et al., 2019; Nagasaka et al., 2020; Nussinov and Tsai, 2015; Ostrem et al., 2013; Zeng et al., 2017), among them some with promising drugs undergoing clinical trials, and ongoing attempts to tame K-Ras dimers (Kessler et al., 2020). However, inhibition of nonfunctional kinases was not taken up. This is vastly important to understand as allosteric inhibitors are increasingly becoming a major drug discovery route (Gross et al., 2020; Khan et al., 2020b; Kostaras et al., 2020; Lu et al., 2018; Lu et al., 2020; Nussinov and Tsai, 2012, 2013, 2015; Ward et al., 2020). Below, we first briefly review Ras biology highlighting key attributes in Ras activation, some of which have been pursued in Ras pharmacology attempts. We follow with allosteric mutations and inhibitors aimed at nonfunctional Ras states, successes, failures and considerations. We distinguish these from oncogenic activation of kinases and consequent allosteric inhibitor strategies. In Ras, activating mutations work by interfering with deactivation; in kinases, they work by increasing the relative stability of the active versus the inactive state, thus, vastly increasing the number of molecules in the active state. It is exceedingly challenging to design a high affinity drug that would bind the inactive state and shift the ensemble back to this state, while still at non-toxic dosage. Allosteric drugs that alter the active site appear more tractable.
However, the location of the drug binding site matters (Guarnera and Berezovsky, 2019, 2020), and in kinases the inactive state may also be targeted. For example, Raf’s activation is through homo- or hetero-dimerization. If a drug binds at the dimer interface to prevent activation via dimerization, it is a successful inhibitor of a nonfunctional kinase conformation. On the other hand, an inhibitor of active GTP-bound Ras, binding at the interface of the effector binding site, still prevents the signal transduction of active Ras. That is, the drug action is related to blocking the protein mechanism of activation; not to whether the protein is in the active or inactive state. That is, we suggest drug classification based on its mechanism of action as it would help guide its design.
Ras: An Overview
Wild-type Ras regulates cell growth and division (Bryant et al., 2014; Cox and Der, 2010; Crespo and Leon, 2000; Drosten et al., 2010; Lu et al., 2016b; Pylayeva-Gupta et al., 2011; Wiesmuller and Wittinghofer, 1994). An activated receptor tyrosine kinase (RTK), such as epidermal growth factor receptor (EGFR) (Moran et al., 1991; Nussinov et al., 2020), recruits the Son of Sevenless (SOS), a Ras-specific guanine nucleotide exchange factor (GEF). Mediated by adaptor proteins SHC (Src homology 2 domain containing) and Grb2 (growth factor receptor-binding protein 2), RTK activates SOS which acts to substitute GDP by GTP at the membrane to activate Ras (Bandaru et al., 2019; Huang et al., 2019; Liao et al., 2018; Liao et al., 2020). Membrane-anchored Ras dimers or nanoclusters bind and activate Raf kinase (Nussinov et al., 2019b, c, 2020; Solman et al., 2015; Sutton et al., 2019; Zhou et al., 2018). In the absence of Ras, Raf is autoinhibited. Its Ras binding domain (RBD) and cysteine rich domain (CRD) loosely interact and occlude the kinase domain (KD) dimerization surface (Nussinov et al., 2018b). This conformation is stabilized by 14-3-3 (Kondo et al., 2019; Park et al., 2019; Rezaei Adariani et al., 2018). The high affinity Ras–Raf’s RBD interaction, enhanced by CRD membrane attachment (Fang et al., 2020; Li et al., 2018a; Li et al., 2018b; Okada et al., 1999; Sarkar and Garcia, 2020; Travers et al., 2018), shifts Raf’s equilibrium toward the open state and interaction with Ras (Nussinov et al., 2019a). RBDs’ interactions with spatially proximal Ras catalytic domains in Ras dimers or nanoclusters promote Raf’s KDs dimerization and activation (Holderfield et al., 2014; Lavoie et al., 2013; Lee et al., 2019; Muratcioglu et al., 2020; Muratcioglu et al., 2015). Active Raf phosphorylates MEK1/2 (MAPK kinase 1/2) (De Luca et al., 2012; Young et al., 2013), with signaling propagating down the mitogen-activated protein kinase (MAPK) pathway to activate extracellular signal-regulated kinase (ERK) and transcription factors, such as Elk-1 (Gao et al., 2019; Nussinov et al., 2014), with the resulting expressed proteins entering the G1 phase of the cell cycle, thereby linking the cellular environment to cell cycle progression. At the G1 phase, cyclin-dependent kinases (CDKs) promote passage to the S (Synthesis) phase.
The second major pathway activated by Ras at the membrane is Ras/PI3K/Akt/mTOR (Downward, 2008; Sheridan and Downward, 2013). RTK interacts with the nSH2 domain in the p85α regulatory domain of phosphatidylinositol 3-kinase (PI3Kα), recruiting the lipid kinase to the membrane and releasing its autoinhibition (Zhang et al., 2019a, b). Subsequent conformational changes expose the active site (Zhang et al., 2019a). Ras-GTP assists in the recruitment, acting to enhance the catalytically favored PI3K population at the membrane where signaling lipid substrate phosphatidylinositol-3,4-bisphosphate (PIP2) is located. PI3K phosphorylates PIP2 to produce phosphatidylinositol-3,4,5-bisphosphate (PIP3). PIP3 recruits Akt (also known as protein kinase B, PKB) and 3-phosphoinositide-dependent protein kinase 1 (PDK1) to the plasma membrane. PDK1 phosphorylates and activates Akt, with the signaling propagating downstream to activate transcription factors whose transcription products also enter the cell cycle at the G1 phase. Together with the MAPK products, they act in cell growth and division, accelerating proliferation (Gul et al., 2018; Lien et al., 2017; Nussinov et al., 2019a, 2020; Pokrass et al., 2020; Zhang et al., 2020). Countering PIP2 to PIP3 phosphorylation by PI3K is PTEN (phosphatase and tensin homologue), a phosphatase (Hosford et al., 2017; Ijuin, 2019; Yehia et al., 2020).
Ras anchors in the plasma membrane through its prenylated hypervariable region (HVR) at the C-terminal (Barklis et al., 2019; Nussinov et al., 2018a). Whereas the sequences and structures of the catalytic domains of H-Ras and N-Ras, and splice isoforms K-Ras4A and K-Ras4B, are highly similar, this is not the case for their HVRs (Lu et al., 2016b; Nussinov et al., 2017a; Pylayeva-Gupta et al., 2011), and the distinct combinations of their lipid posttranslational modifications – farnesyl (or geranylgeranyl) and palmitoyl. K-Ras4B is farnesylated, K-Ras4A can be farnesylated or farnesylated and palmitoylated; N-Ras is also farnesylated and palmitoylated, and H-Ras is farnesylated and double-palmitoylated. Their distinct chemistry makes them favor different membrane properties, thus segregation into homogeneous nanoclusters and dimers (Fujiwara et al., 2002; Murase et al., 2004; Simons and Vaz, 2004). It also influences Ras orientation at the membrane, accessibility and selective interaction with its effectors (Ahearn et al., 2011; Cao et al., 2019; Chavan et al., 2015; Jang et al., 2016a; Jang et al., 2020; Neale and Garcia, 2020; Nussinov et al., 2018a; Prakash and Gorfe, 2019; Prakash et al., 2020). Membrane attachment can also be regulated by ubiquitination (Baietti et al., 2016; Steklov et al., 2018; Thurman et al., 2020), phosphorylation of the C-terminal as well as other post-translational modifications, such as acetylation, and calmodulin (Jang et al., 2017; Jang et al., 2019; Nussinov et al., 2017b).
As members of the small GTPase family, Ras proteins function as molecular switches between their two states (Bourne et al., 1991; Cherfils and Zeghouf, 2013; Downward, 1990; Geyer and Wittinghofer, 1997; Grand and Owen, 1991; Lamontanara et al., 2014; Lowy et al., 1991; Sprang, 1997; Takai et al., 2001; Vetter and Wittinghofer, 2001; Wittinghofer and Pai, 1991; Wittinghofer and Vetter, 2011). Inactivation is mediated by GTPase-activating proteins (GAPs) that hydrolyze the GTP to GDP (Wittinghofer et al., 1997). Strong oncogenic mutations block GAP catalytic activity, retaining the active GTP-bound state, thus promoting downstream signaling and cell proliferation as in the case of G12D K-Ras4B mutant (Adjei, 2001; Schubbert et al., 2007; Smith and Ikura, 2014; Smith et al., 2013; Vigil et al., 2010). Weaker driver mutations can be associated with promoting a high rate of intrinsic and guanine nucleotide exchange factor-induced nucleotide exchange, as in the case of A146T K-Ras4B mutant (Poulin et al., 2019).
Below, we first distinguish between allosteric mutations in kinases and in Ras. In Ras, they work by quenching deactivation mechanisms, GTP hydrolysis and nucleotide exchange; inhibitors that bind to the inactive conformation block reactivation. By contrast, in kinases, they act by shifting the equilibrium toward the active state; accordingly, their allosteric drugs aim to modulate the active site conformation. The distinct mutation scenarios are the key to their inhibition strategy, and these considerations are general.
Allosteric Mutations in Ras and Kinases Work Differently; Thus, Their Inhibition Strategies Should Differ Too
Protein function hinges on the extent to which it populates its active conformation (Lu et al., 2014a; Lu et al., 2014b; Nussinov and Tsai, 2013). Molecular ensembles exist in a conformational equilibrium of active and inactive states (Henley et al., 2020; Nussinov, 2016; Nussinov and Wolynes, 2014). Aside from repressors, under physiological conditions, most enzymes and receptors are in their inactive states. When stimulated, either by an incoming signal or oncogenic mutations the ensemble shifts to populate the active state (Tsai and Nussinov, 2014). The relative populations of the states reflect their relative stabilities. Allosteric inhibitors whose shapes and chemistry fit a certain patch of surface of the enzyme or receptor in the inactive (or for repressors, active) state, are conformationally selected. The ensemble is equilibrated by a shift toward the stabilized inactive state.
Kinases fit into this description (Figure 1). They switch from the inactive αC-helix-out to the active αC-helix-in conformation through a rotation and shift movement, with a salt bridge between the β3-Lys and the αC-Glu, and R-spine formation (Nussinov and Tsai, 2013). The triggers for the conformational change vary among the kinase families. Oncogenic mutations stabilize the active αC-helix-in conformation or disrupt interactions that stabilize the αC-helix-out, as in the case of the L858R in EGFR. There, the mutation stabilizes the αC-helix-in conformation by heterodimerization. The mutation is in the middle of a hydrophobic core. Thus, the change to Arg also disrupts stabilizing hydrophobic interactions in the αC-helix-out conformation. The T790M mutation in EGFR, T315I in Bcr-Abl, T334I in c-Abl, T341I in Src, T670I in Kit, and T674I in PDGFRα (platelet-derived growth factor receptor A) work by stabilizing the hydrophobic R-spine, promoting enzymatically active conformation (Nussinov and Tsai, 2013). In another example, in PI3Kα lipid kinase (Zhang et al., 2019a) oncogenic mutations E542K and E545K in the helical domain, with charge reversal (Leontiadou et al., 2018) mimic the release of the autoinhibition. In the wild-type, it is triggered by nSH2 domain interacting with RTK’s pYXXM motif in the C-terminal. The outcome is exposing the active site at the membrane. Other mutations, e.g. Glu81, Gly106, Arg108, Lys111, and Gly118 in the adaptor binding domain (ABD), contribute to the exposure as well. These mutations reduce the transition state barrier (ka). Another common driver, H1047R, enhances the population time of the lipid substrate at the active site (km). In TRK tyrosine kinases, xDFG mutations confer resistance to type I inhibitors, but sensitize them to type II by stabilizing the inactive (DFG-out) conformations (Cocco et al., 2020).
Figure 1.

The free energy landscape ΔΔG of a kinase conformation (here EGFR), the effect of mutations and an allosteric inhibitor. Oncogenic driver mutations (L858R and T790M), destabilize the inactive state (left plot in the middle panel), stabilize the active state (center plot) or both (right plot). The outcome is a shift of the ensemble toward the constitutively active EGFR conformation. In the bottom panel, an allosteric drug binding EGFR’s mutant surface stabilizes this conformation, with a population shift toward this conformation. The drug modulates the active site shape.
Not all mutations can however be described by the free energy landscape. Ras’ major mutations, e.g. Gly12, Gly13, and Gln61, which block hydrolysis, cannot be described by the free energy landscape (Fernandez-Medarde and Santos, 2011; Nussinov et al., 2013; Tsai and Nussinov, 2014). Neither can mutations that block nucleotide exchange, such as K-Ras4BA146T. GTP hydrolysis involves proton transfer from an attacking water molecule to another, followed by transfer of a different proton from this water molecule to the GTP. Gln61 can stabilize the transient OH− and H3O+ molecules lowering the transition state barrier, which the Leu61 cannot (Martin-Garcia et al., 2012). As to the Gly12 mutants, their longer side chains interfere with GAP’s arginine finger insertion and hydrolysis (Krengel et al., 1990).
Despite the different mechanisms, both kinases and Ras GTPases are dynamic molecules, with multiple, not only two discrete, active and inactive states. Both types of proteins can be considered from the mechanistic free energy landscape standpoint, which epitomizes the ‘second molecular biological revolution’ (Nussinov and Wolynes, 2014). The paradigm fomented by this revolution postulated that the energy landscape (Frauenfelder et al., 1991) and the laws of quantum mechanics and structural chemistry apply to all biological systems and that biomolecules always interconvert between conformations with varying energies. This view inspired the dynamical statistical description of the molecules (Kumar et al., 2000; Ma et al., 2002; Tsai et al., 1999a; Tsai et al., 1999b), where the ensembles describe the relative conformational stabilities of the states, their transitions and their population shifts to retain their equilibrium (Arora and Brooks, 2007; Boehr et al., 2009; Huang et al., 2015; Kar et al., 2010; Mackereth et al., 2011; Motlagh et al., 2014; Nussinov and Ma, 2012; Okazaki and Takada, 2008). GDP-bound Ras can exist in inactive and active conformations. Similarly, Ras-GTP exists in active and inactive states, although the populations vary. Indeed, molecular dynamics (MD) simulations, and crystallography (Shima et al., 2010) suggest that the inactive state of Ras-GTP may exist in at least three substates. Notably, in reality, there are many inactive protein states and there is also an ensemble at the transition state. Classically, catalysis involves tight binding of the substrate to the transition-state structure to lower the activation energy. However, the transition state is a surface, not a single saddle point on the potential energy surface and the activated conformations in the transition-state ensemble can catalyze multiple reaction steps (Ma et al., 2000). NMR of GppNHp-bound wild-type H-Ras catalytic domain identified two conformational states that interconvert on a millisecond time scale, inactive state 1 and active state 2 (Geyer et al., 1996; Liao et al., 2008). The perturbations induced by the oncogenic mutations promote a redistribution of the ensemble. Crystal structures reveal allosteric effects on conformational states induced by mutations, and mutants display distinct outcomes in effector binding and in oncogenicity, as in the cases of G12V vs G12D. Further, K-Ras populates conformational states differently from its isoform H-Ras and oncogenic mutant K-Ras4BG12D (Parker and Mattos, 2018; Parker et al., 2018).
The free energy landscape describes the ensemble around the native state as populating the active and inactive states, with the states separated by kinetic barriers. The more stable the conformation the larger its population. Due to functional requirements, the barriers between states are not too high which allows conformations to switch between them. In kinases, oncogenic mutations work by creating new interactions and/or abolishing existing ones, resulting in higher stability (population) of the active state. In the case of Ras, these free energy considerations do not apply to the mutation actions. GTP binds the active state. With the high millimolar range concentration of the guanine nucleotide in the cell and its picomolar affinity, ~75% of K-RasG12C is GTP- bound in the steady state. Intrinsic hydrolysis, which is high for this mutant (Hunter et al., 2015) cycles K-Ras4BG12C into its GDP-bound state while allosteric drugs block the nucleotide exchange .
Thus, not all mutations can be described by the free energy landscape; only those working via a population shift can (Tsai and Nussinov, 2014). Accordingly, the nature of the drugs should be different. The nonfunctional states of Ras can be targeted; however, for kinases, shifting the ensemble away from the constitutively active state, by e.g. stabilizing the inactive autoinhibited state is challenging.
Imatinib (STI-571/Gleevec), the first synthetic kinase inhibitor to target an oncogene (Bcr-Abl), can serve as a classical example to illustrate our point (Nagar et al., 2003). The physiological state of Abl is autoinhibited via intradomain interactions between the SH3–linker–SH2 and the kinase domain. The fused Bcr abolishes the autoinhibition to constitutively activate it. The landmark Kuriyan and colleagues’ crystal structure of the kinase domain with imatinib bound at the ATP binding site reveals a collapsed activation loop blocking substrate binding, which is open and extended in all active kinase domains. This binding indicates that imatinib only can bind to the less populated, inactive kinase conformation of Bcr-Abl. The drug-resistant mutations observed by Daley and colleagues (Azam et al., 2003), either directly participate in imatinib binding or act allosterically to destabilize the autoinhibited Abl kinase conformation to which STI-571 preferentially binds. That is, they reduce drug binding affinity or increase the population of the active state by destabilizing the inactive conformation, counteracting the inhibiting action of imatinib.
However, as the inhibition of Raf (homo- or hetero-) dimerization shows, the location of the drug binding matters: if the drug is bound at the dimer interface to prevent activation via dimerization, it is a successful inhibitor bound to an inactive kinase conformation.
Approaches to Ras Inhibition
Driver mutations in K-Ras are common in cancers (Chen et al., 2015; Eser et al., 2014; Holderfield et al., 2014; Krens et al., 2010; Prior et al., 2012; Stephen et al., 2014); however, the massive community efforts to identify effective inhibitors have proven to be challenging (Cox et al., 2014; Ledford, 2015; Lu et al., 2016c; Milroy and Ottmann, 2014; Samatar and Poulikakos, 2014; Shao et al., 2014; Spiegel et al., 2014; Stites and Ravichandran, 2009; Wang et al., 2013; Zimmermann et al., 2013). There are many reasons for the difficulty, including Ras’ picomolar affinity for guanine nucleotide whose cellular concentration is in the millimolar range, making binding of nucleotide analogs disfavored (Khan et al., 2020a; Waters and Der, 2018). In addition, despite searches, deep pockets for small molecule binding have not been identified (Spiegel et al., 2014). The diverse strategies of potential Ras inhibitors have been reviewed extensively in the literature (Chatani and Yang, 2020; Cox et al., 2015; Cox et al., 2014; Dang et al., 2017; Gentile et al., 2017; Gorfe and Cho, 2019; Gupta et al., 2019; Khan et al., 2020a; Liu et al., 2010; Liu et al., 2019; Lu et al., 2016a; Lu et al., 2016c; Mattingly, 2013; McCormick, 2018; Mullard, 2019; O'Bryan, 2019; Papke and Der, 2017; Papke et al., 2016; Patricelli et al., 2016; Sakamoto et al., 2017; Sheridan, 2020; Singh et al., 2015; Spencer-Smith and O'Bryan, 2019; Spiegel et al., 2014; Zhang and Shokat, 2019; Zimmermann et al., 2013). Among those that have been explored are agents blocking Ras dimerization (e.g. NS1 and K13) (Spencer-Smith et al., 2017) and inhibition of farnesyl transferase (FTase), thus blocking cysteine farnesylation at the C-terminus, translocation from the endoplasmic reticulum (ER) to the plasma membrane and anchorage (e.g., tipifarnib, deltasonamide) (Baranyi et al., 2020; Cheng et al., 2020; Martin-Gago et al., 2017; Reid and Beese, 2004). Blocking Ras–GEF interaction (e.g., DCAI, HBS3) (Maurer et al., 2012; Patgiri et al., 2011), and Ras–Raf (e.g., Abd-7, 3344) (Bery et al., 2018; Quevedo et al., 2018) (Figure 2) have also been explored. In another strategy, an unbiased approach using synthetic single domain monobodies (Spencer-Smith et al., 2017) was employed. A NS1 monobody for Ras isoforms selectively inhibited oncogenic H-Ras and K-Ras, but not N-Ras proliferation (Khan et al., 2020a). Additional strategies for preventing Ras–plasma membrane anchorage include enhancing K-Ras phosphorylation, inhibiting K-Ras interaction with its chaperone protein and more (Cheng et al., 2020; Gorfe and Cho, 2019). With the aim of identifying functionally-productive states with the effector interacting surface exposed, Ras orientation at the membrane has also been considered (Cao et al., 2019; Chavan et al., 2015; Jang et al., 2016a; Jang et al., 2020; Neale and Garcia, 2020; Nussinov et al., 2018a; Prakash and Gorfe, 2019; Prakash et al., 2020). These approaches are hindered, however, by membrane dynamics and fluidity, toxicity, specificity, and thus altogether effectiveness. SML-8-73-1 (Hunter et al., 2014; Lim et al., 2014; Xiong et al., 2017) and SML-10-70-1 (Lim et al., 2014) are covalent small-molecule inhibitors binding at the active site of oncogenic G12C K-Ras mutant. Direct approaches of targeting Ras appear more promising, but still face hurdles. Early on, a small-molecule Kobe0065 and cyclen-metal complexes that bound to the inactive state were developed (Shima et al., 2013). A covalent allosteric inhibitor for oncogenic K-RasG12C obtained by a disulfide fragment-based screening approach (Ostrem et al., 2013) as well as inhibitors that target non-functional dimers (Kessler et al., 2019; Kessler et al., 2020; Tran et al., 2020) exploit the same rational.
Figure 2.

Examples of Ras inhibitors. The first row shows crystal structures of the GDP-bound H-Ras in complex with NS1 monobody (PDB: 5E95), farnesyltransferase (FTase) containing the α and β subunits in complex with farnesyl diphosphate (FPP) and the inhibitor Tipifarnib (R115777) (PDB 1SA4), and phosphodiesterase δ subunit (PDEδ) inhibited by Deltasonamide (PDB: 5ML3). Covalent drugs to the G12C mutation are shown in the second row; crystal structures of the GDP-bound K-Ras4BG12C with MRTX849 (PDB: 6UT0), the GDP-bound K-Ras4BG12C with ARS-1620 (PDB: 5V9U), and K-Ras4BG12C with the covalent GTP-competitive inhibitor SML-8-73-1 (PDB: 5KYK). The third row shows crystal structures of active Ras inhibited by non-covalent, small molecular drugs for the GCP-bound K-Ras4BG12D with DCAI (PDB: 4DST), the GNP-bound K-Ras4BQ61H with Abd-7 (PDB: 6FA4), and the GNP-bound K-Ras4BQ61H with 3344 (Abd-8) (PDB: 6F76).
Allosteric compounds that target inactive monomers and dimers may hold the key for future successes. Nonetheless, targeting dimers may also face additional hurdles: spatially proximal Ras nanoclusters can play a major role in MAPK activation; however, PI3K activation does not require Ras dimers, thus its activation can still go through (Nussinov et al., 2018a, 2019b).
Allosteric Drugs Targeting Nonfunctional Ras
Using a fragment-based screening approach, Shokat and co-workers pioneered the first compounds that covalently link to the cysteine in GDP-bound K-RasG12C, blocking SOS-catalyzed nucleotide exchange (Ostrem et al., 2013). Their initial leads had only millimolar affinity, however subsequent work by several groups led to other high-affinity drugs with some undergoing clinical trials (Sheridan, 2020). Among the more potent compounds are AMG510 (Sotorasib) by Amgen (phase I trial), MRTX849 (phase I/II) (Canon et al., 2019; Hallin et al., 2020; Mirati Therapeutics, 2020), JNJ-74699157 (formerly ARS-3248) (phase I, Johnson & Johnson, Wellspring Biosciences; earlier ARS-1620 (Janes et al., 2018)), and LY3499446 (phase I/II clinical trials, Eli Lilly). Illustrating the hardships in developing a drug against K-Ras, Eli Lilly exited the K-Ras race due to toxicity in its phase 1 effort and John & Johnson stopped recruiting (Adams, 2020). Initial results in patients with K-RasG12C mutation indicated that MRTX849 is a promising clinical candidate (Fell et al., 2020; Gabizon and London, 2020). Both AMG510 and MRTX seem to be moving at equal pace in the clinic. Phase 1 data for AMG510 in advanced colorectal cancer suggested that monotherapy provided prolonged disease control. Anti-tumor activity was also observed across multiple solid tumors (Hong et al., 2020). A Phase 2 monotherapy study in advanced colorectal cancer is getting under way. A targeted degradation strategy for oncogenic K-RasG12C by construction of a library of C12-directed covalent degrader molecules (PROTACs) was also explored; however initially the lead degrader was unable to effectively poly-ubiquitinate cellular K-RasG12C (Zeng et al., 2020). More recently, LC-2, a PROTAC capable of degrading endogenous K-RasG12C via E3 ligase VHL was developed. It too covalently binds K-RasG12C with a MRTX849 warhead, resulting in MAPK suppression (Bond et al., 2020). These allosteric compounds bind to nonfunctional K-RasG12C.
The covalent attachment first designed by Shokat and his colleagues involved disulfide tethering to the Cys, exploiting the Cys thiol reactivity (Ostrem et al., 2013). Exploiting this method, they obtained compounds that bound in an unknown pocket in the Switch II region near the nucleotide binding pocket (SW2 binding pocket). The pocket exists in the GDP-bound but not in the GTP-bound conformation; thus, these compounds bound only the GDP-bound state. Their binding resulted in conformational changes in the Switch I and Switch II regions inhibiting interactions with Ras effectors and activators. The impact of this work resulted from its significant and novel components: it was the first to exploit covalent allosteric drug on Ras and it exploited its inactive state. It provided hope that this concept can finally produce the highly coveted drug against a mutant Ras. Further, it showed that a drug that interacts with the inactive Ras state can work (Khan et al., 2020a). Continued efforts led to ARS-1620 (Janes et al., 2018) (Figure 2), with higher reaction kinetics and more potent drug action, inhibiting interaction with Raf, and consequently MAPK signaling. It resulted in several lead compounds by pharmaceutical companies with subsequent clinical trials as described above.
Covalent drugs, especially allosteric ones are powerful. However, to date their applicability has been limited to few cancers, particularly small cell lung cancer where the G12C mutation is abundant (Gorfe and Cho, 2019; Stephen et al., 2014). The account and promise shown by the impressive optimization of MRTX849 (Figure 2) that targets K-RasG12C emphasizes however the challenges in the development of covalent drugs in structure-based design to improve drug potency and pharmacological properties, reaching nM potency, and selectively in vivo (Fell et al., 2020; Gabizon and London, 2020).
Kessler et al. (Kessler et al., 2019) have targeted a pocket in K-Ras4B previously considered ‘undruggable’ due to its comparative shallowness and polarity, rather than hydrophobic, between Switch I and II (SI/II-pocket) (Cruz-Migoni et al., 2019; Maurer et al., 2012; Quevedo et al., 2018; Sun et al., 2012). They started from very weakly binding fragments and structure-based drug design. They discovered BI-2852 (compound 1) (Figure 3, upper left panel), a nanomolar inhibitor to this pocket distinct from covalent K-RasG12C inhibitors. The pocket it binds differs from that of covalent K-RasG12C inhibitors, which bind to the SII-P pocket in the Switch II region and is present in both the active (GTP-bound) and inactive (GDP-bound) forms of Ras proteins. It modulates MAPK signaling (pERK phosphorylation) and PI3K/Akt (pAkt) and inhibits KRAS mutant cell proliferation, suggesting that this pocket can be druggable.
Figure 3.

Ras inhibitor with nanomolar affinity. Crystal structure of the GCP-bound K-Ras4BG12D with BI-2852 (PDB: 6GJ8) (upper left panel). The compound 1 drug, BI-2852, exhibits nanomolar affinity to a pocket between Switch I and II. The BI-2852-induced Ras dimerization (right panel). Symmetric rotation of the K-Ras4B crystal structure reveals a β-sandwich dimer aligning its β1, β2, and γ3 strands by two BI-2852 molecules. The first BI-2852 (ligand-1) binds the Switch I and II pocket, the deep pocket formed by Lys5, Val7, Asp54, and Leu56 on the first Ras (Ras-1). The second BI-2852 (ligand-2), a copy of the first, binds the shallow pocket formed by Glu3, Leu52, and Asp54 on Ras-1. The complex is stabilized by several salt-bridges. The molecular dynamics (MD) simulations of Ras molecules at the anionic membrane (Jang et al., 2016b) previously reported the unfunctional β-sandwich dimer for K-Ras4B-GTP and H-Ras-GTP (lower left panel).
Tran et al. proposed an alternative interpretation to the Kessler et al. observation of BI-2852 inhibition at nanomolar affinity (Tran et al., 2020). Their analysis of the crystal structure of K-Ras4B with BI-2852, which included the neighboring unit cell, observed that BI-2852 induces a K-Ras4B dimer consisting of four molecules (two of BI-2852 and two of K-Ras4B) with rotational symmetry (Figure 3, right panel). This was confirmed experimentally by size exclusion column chromatography (SEC) at concentration 1,000 times higher (3 mM) than those in which the cellular effects of BI-2852 were observed (Kessler et al., 2020), mass spectrometry, measurement of binding affinity (ITC) and surface plasmon resonance (SPR). Analysis of the crystal structure (PDB: 6GJ8) indicated that the pocket is formed by two K-Ras4B molecules where one copy of BI-2852 binds, suggesting an asymmetry, with one K-Ras4B presenting a larger pocket (Switch I/II pocket) and the other a smaller one. The second BI-2852 indicates symmetric formation. The complex is stabilized by 4 salt-bridges, altogether, pointing to Kessler et al (Kessler et al., 2019) discovery of formation of a non-functional dimer, stabilized by two molecules of BI-2852.
To further explore stabilization of nonfunctional K-Ras4B dimers for KRAS-driven cancers Kessler et al. synthesized other possible dimeric Switch I/II pocket binders (Kessler et al., 2020). They discovered compound 2, which dimerizes K-Ras4B with a Kd of 3.8 μM. Soaking active K-Ras4BG12D (Bergner et al., 2019), they obtained K-Ras4B crystal structures of dimers with this compound at 1.9 Å resolution. Subsequent co-crystallization with compound 2 obtained a dimeric structure (at 1.57 Å resolution), a similar β-sandwich interface as with BI-2852, that resembles the one previously postulated by modelling and MD simulations of K-Ras4B molecules (Jang et al., 2016b). The modeled structure dimerizes through the effector lobe β-sandwich interface involving side-chain interactions of β1, β2, and β3 strands (Jang et al., 2016b) (Figure 3, lower left panel). The β-sandwich interface emerged from the exact β-sheet alignment due to hydrogen bonds (H-bonds) mismatch between the β2 strands. Especially noteworthy is that this dimeric interface was a minor dimeric form in the simulations, that is, with a smaller population, suggesting that compound 2 selects a minor species and stabilizes it.
Thus, whether monomers or dimers, allosteric interaction with a non-functional state will shift the ensemble away from the active state. Interestingly, the inactive K-Ras4B dimer that dimerizes through the effector lobe interface favors a shifted β-sheet extension through intermolecular H-bonds between the β2 strands formed by Glu37, Ser39, and Arg41 (Jang et al., 2016b). However, Raf easily competes with Ras molecules assembled through the effector lobe dimer interface due to the high-affinity interaction of RBD with the Ras effector binding site. The active K-Ras4B dimer, where the monomers interact through their allosteric lobes, favors an interaction through the α3-α4 helices, while H-Ras allosteric lobe dimerization favors the α4-α5 helical interface (Jang et al., 2016b; Jang et al., 2020).
Small molecules have also been developed to inhibit Ral GTPases, which are Ras GTPases identical in structure to K-Ras. The innovative strategy employed there similarly proposed to target GDP- rather than GTP-bound Ral, exploiting a new pocket (Yan et al., 2014). This pocket, which was not targeted before, harbors the K-RasG12C mutation. This work also led to a follow-up discovery in Meroueh’s lab of a covalent inhibitor that forms a bond with a tyrosine on Ral and Ras GTPases (Bum-Erdene et al., 2020), raising the possibility that covalent tyrosine linkage can open another venue in targeting of the Ras superfamily.
Drugging small GTPase Rheb is similarly challenging. Rheb activates the mechanistic target of rapamycin complex 1 (mTORC1), a master regulator of cellular growth and metabolism in the Ras/PI3K/Akt/mTOR pathway. Small molecule NR1 binds Rheb in the Switch II domain and selectively blocks mTORC1 signaling, but not mTORC2 (Mahoney et al., 2018).
Allosteric drugs are highly specific with fewer side effects and can tune the inhibition. Nonetheless, they still need to stably bind the protein, requiring some pockets. Covalent drugs also require a sufficiently reactive warhead though not too reactive as to avoid toxicity. The pocket should permit specific interactions that allow such warhead strength reduction.
Finally, K-RasG12C inhibitors target a somatic cysteine mutation. Since all cells are targeted, concerns about their likely toxicity persist (Sheridan, 2020). Selectivity against the wild type (therapeutic index) also depends upon the oncogene specificity in addition to the selectivity.
Significance
The mechanism of action of many drugs is known. Prior knowledge in the development stage of the mechanism of action – at the molecular level – of effective drugs for the targeted protein is enormously advantageous and reduces late stage failures. Here we interrogate distinct mechanisms embodied by allosteric drugs for two classes of major drug targets: Ras superfamily and kinases. Among the emerging allosteric strategies for Ras is targeting its nonfunctional state - which differs from the allosteric drug strategies for kinases. In kinases, allosteric drugs mostly target the active, functional state. We ask why drugging the inactive K-Ras4BG12C can work – but, drugging the inactive kinase will likely not. We propose that the reason is their distinct oncogenic mechanisms. In kinases, the mechanism of oncogenic kinase mutations involves increasing the population of the active conformations as compared to the inactive conformations. By contrast, Ras oncogenic mutations work by quenching the deactivation mechanisms, GTP hydrolysis and nucleotide exchange. Thus, covalent allosteric inhibitors that bind to the inactive GDP-bound K-Ras4BG12C conformation can work – which is not the case in kinases, whose allosteric inhibitors work by altering the active site conformation.
Drug discovery-wise the significance of our premise here is three-fold: (i) it postulates that the mechanism of drug action relates to the mechanism of the oncogenic driver mutations; not to whether the protein is in the active or inactive state. (ii) However, if drug binding allosterically blocks the target functional site, as in the case of Raf dimerization, it will work also for the inactive kinase states, and the GTP-bound Ras. That is, the location of the drug binding site matters. (iii) From the translational standpoint, this mechanism of action classification accelerates pharmaceutical development. It postulates that drug action is related to blocking the mechanism of activation – not to active or inactive state, extending drug classification charts.
Highlights.
Kinase mutations stabilize the active state or destabilize the inactive state
Ras mutations quell the deactivation mechanisms, GTP hydrolysis and GDP exchange
Drugging inactive K-Ras4BG12C can work, but likely not drugging inactive kinase
Inhibition relates to activation mechanism not to the active or inactive state
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the Intramural Research Program of the NIH Clinical Center.
Footnotes
Declaration of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams B, (2020). Eli Lilly crashes out of KRAS race as toxicity sees it ditch phase 1 effort. https://www.fiercebiotech.com/biotech/eli-lilly-crashes-out-kras-race-as-it-ditches-phase-1-effort [Google Scholar]
- Adjei AA (2001). Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 93, 1062–1074. [DOI] [PubMed] [Google Scholar]
- Ahearn IM, Haigis K, Bar-Sagi D, and Philips MR (2011). Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol 13, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora K, and Brooks CL 3rd. (2007). Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc Natl Acad Sci U S A 104, 18496–18501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M, Latek RR, and Daley GQ (2003). Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831–843. [DOI] [PubMed] [Google Scholar]
- Baietti MF, Simicek M, Abbasi Asbagh L, Radaelli E, Lievens S, Crowther J, Steklov M, Aushev VN, Martinez Garcia D, Tavernier J, et al. (2016). OTUB1 triggers lung cancer development by inhibiting RAS monoubiquitination. EMBO Mol Med 8, 288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru P, Kondo Y, and Kuriyan J (2019). The Interdependent Activation of Son-of-Sevenless and Ras. Cold Spring Harb Perspect Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranyi M, Buday L, and Hegedus B (2020). K-Ras prenylation as a potential anticancer target. Cancer Metastasis Rev 39, 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barklis E, Stephen AG, Staubus AO, Barklis RL, and Alfadhli A (2019). Organization of Farnesylated, Carboxymethylated KRAS4B on Membranes. J Mol Biol 431, 3706–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergner A, Cockcroft X, Fischer G, Gollner A, Hela W, Kousek R, Mantoulidis A, Martin LJ, Mayer M, Mullauer B, et al. (2019). KRAS Binders Hidden in Nature. Chemistry 25, 12037–12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bery N, Cruz-Migoni A, Bataille CJ, Quevedo CE, Tulmin H, Miller A, Russell A, Phillips SE, Carr SB, and Rabbitts TH (2018). BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions. Elife 7, e37122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, Nussinov R, and Wright PE (2009). The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 5, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MJ, Chu L, Nalawansha DA, Li K, and Crews CM (2020). Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent Sci 6, 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, and McCormick F (1991). The GTPase superfamily: conserved structure and molecular mechanism. Nature 349, 117–127. [DOI] [PubMed] [Google Scholar]
- Bryant KL, Mancias JD, Kimmelman AC, and Der CJ (2014). KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 39, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bum-Erdene K, Liu D, Gonzalez-Gutierrez G, Ghozayel MK, Xu D, and Meroueh SO (2020). Small-molecule covalent bond formation at tyrosine creates a binding site and inhibits activation of Ral GTPases. Proc Natl Acad Sci U S A 117, 7131–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAs(g12C) inhibitor AMG 510 drives antitumour immunity. Nature 575, 217–223. [DOI] [PubMed] [Google Scholar]
- Cao S, Chung S, Kim S, Li Z, Manor D, and Buck M (2019). K-Ras G-domain binding with signaling lipid phosphatidylinositol (4,5)-phosphate (PIP2): membrane association, protein orientation, and function. J Biol Chem 294, 7068–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatani PD, and Yang JC (2020). Mutated RAS: Targeting the "Untargetable" with T Cells. Clin Cancer Res 26, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan TS, Muratcioglu S, Marszalek R, Jang H, Keskin O, Gursoy A, Nussinov R, and Gaponenko V (2015). Plasma membrane regulates Ras signaling networks. Cell Logist 5, e1136374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Tao Y, and Jiang Y (2015). Apelin activates the expression of inflammatory cytokines in microglial BV2 cells via PI-3K/Akt and MEK/Erk pathways. Sci China Life Sci 58, 531–540. [DOI] [PubMed] [Google Scholar]
- Cheng J, Li Y, Wang X, Dong G, and Sheng C (2020). Discovery of Novel PDEδ Degraders for the Treatment of KRAS Mutant Colorectal Cancer. J Med Chem 63, 7892–7905. [DOI] [PubMed] [Google Scholar]
- Cherfils J, and Zeghouf M (2013). Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 93, 269–309. [DOI] [PubMed] [Google Scholar]
- Cocco E, Lee JE, Kannan S, Schram AM, Won HH, Shifman S, Kulick A, Baldino L, Toska E, Arruabarrena-Aristorena A, et al. (2020). TRK xDFG mutations trigger a sensitivity switch from type I to II kinase inhibitors. Cancer Discov, DOI: 10.1158/2159-8290.CD-1120-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, and Der CJ (2010). Ras history: The saga continues. Small GTPases 1, 2–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Der CJ, and Philips MR (2015). Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin Cancer Res 21, 1819–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, and Der CJ (2014). Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 13, 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo P, and Leon J (2000). Ras proteins in the control of the cell cycle and cell differentiation. Cell Mol Life Sci 57, 1613–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Migoni A, Canning P, Quevedo CE, Bataille CJR, Bery N, Miller A, Russell AJ, Phillips SEV, Carr SB, and Rabbitts TH (2019). Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc Natl Acad Sci U S A 116, 2545–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Reddy EP, Shokat KM, and Soucek L (2017). Drugging the 'undruggable' cancer targets. Nat Rev Cancer 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Maiello MR, D'Alessio A, Pergameno M, and Normanno N (2012). The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets 16 Suppl 2, S17–27. [DOI] [PubMed] [Google Scholar]
- Downward J (1990). The ras superfamily of small GTP-binding proteins. Trends Biochem Sci 15, 469–472. [DOI] [PubMed] [Google Scholar]
- Downward J (2008). Targeting RAS and PI3K in lung cancer. Nat Med 14, 1315–1316. [DOI] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, and Barbacid M (2010). Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 29, 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser S, Schnieke A, Schneider G, and Saur D (2014). Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 111, 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Lee KY, Huo KG, Gasmi-Seabrook G, Zheng L, Moghal N, Tsao MS, Ikura M, and Marshall CB (2020). Multivalent assembly of KRAS with the RAS-binding and cysteine-rich domains of CRAF on the membrane. Proc Natl Acad Sci U S A 117, 12101–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, Brown KD, Burgess LE, Burns AC, Burkard MR, et al. (2020). Identification of the Clinical Development Candidate MRTX849, a Covalent KRAs(g12C) Inhibitor for the Treatment of Cancer. J Med Chem 63, 6679–6693. [DOI] [PubMed] [Google Scholar]
- Fernandez-Medarde A, and Santos E (2011). Ras in cancer and developmental diseases. Genes Cancer 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H, Sligar SG, and Wolynes PG (1991). The energy landscapes and motions of proteins. Science 254, 1598–1603. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, and Kusumi A (2002). Phospholipids undergo hop diffusion in compartmentalized cell membrane. J Cell Biol 157, 1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon R, and London N (2020). Hitting KRAS When It's Down. J Med Chem 63, 6677–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhou J, Xie Z, Wang J, Ho CK, Zhang Y, and Li Q (2019). Mechanical strain promotes skin fibrosis through LRG-1 induction mediated by ELK1 and ERK signalling. Commun Biol 2, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile DR, Rathinaswamy MK, Jenkins ML, Moss SM, Siempelkamp BD, Renslo AR, Burke JE, and Shokat KM (2017). Ras Binder Induces a Modified Switch-II Pocket in GTP and GDP States. Cell Chem Biol 24, 1455–1466 e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer M, Schweins T, Herrmann C, Prisner T, Wittinghofer A, and Kalbitzer HR (1996). Conformational transitions in p21ras and in its complexes with the effector protein Raf-RBD and the GTPase activating protein GAP. Biochemistry 35, 10308–10320. [DOI] [PubMed] [Google Scholar]
- Geyer M, and Wittinghofer A (1997). GEFs, GAPs, GDIs and effectors: taking a closer (3D) look at the regulation of Ras-related GTP-binding proteins. Curr Opin Struct Biol 7, 786–792. [DOI] [PubMed] [Google Scholar]
- Goody RS, Muller MP, and Rauh D (2019). Mutant-Specific Targeting of Ras G12C Activity by Covalently Reacting Small Molecules. Cell Chem Biol 26, 1338–1348. [DOI] [PubMed] [Google Scholar]
- Gorfe AA, and Cho KJ (2019). Approaches to inhibiting oncogenic K-Ras. Small GTPases, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand RJ, and Owen D (1991). The biochemistry of ras p21. Biochem J 279 (Pt 3), 609–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross LZF, Sacerdoti M, Piiper A, Zeuzem S, Leroux AE, and Biondi RM (2020). ACE2, the Receptor that Enables Infection by SARS-CoV-2: Biochemistry, Structure, Allostery and Evaluation of the Potential Development of ACE2 Modulators. ChemMedChem 15, 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnera E, and Berezovsky IN (2019). Toward Comprehensive Allosteric Control over Protein Activity. Structure 27, 866–878 e861. [DOI] [PubMed] [Google Scholar]
- Guarnera E, and Berezovsky IN (2020). Allosteric drugs and mutations: chances, challenges, and necessity. Curr Opin Struct Biol 62, 149–157. [DOI] [PubMed] [Google Scholar]
- Gul A, Leyland-Jones B, Dey N, and De P (2018). A combination of the PI3K pathway inhibitor plus cell cycle pathway inhibitor to combat endocrine resistance in hormone receptor-positive breast cancer: a genomic algorithm-based treatment approach. Am J Cancer Res 8, 2359–2376. [PMC free article] [PubMed] [Google Scholar]
- Gupta AK, Wang X, Pagba CV, Prakash P, Sarkar-Banerjee S, Putkey J, and Gorfe AA (2019). Multi-target, ensemble-based virtual screening yields novel allosteric KRAS inhibitors at high success rate. Chem Biol Drug Des 94, 1441–1456. [DOI] [PubMed] [Google Scholar]
- Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. (2020). The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 10, 54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley MJ, Linhares BM, Morgan BS, Cierpicki T, Fierke CA, and Mapp AK (2020). Unexpected specificity within dynamic transcriptional protein-protein complexes. Proc Natl Acad Sci U S A 117, 27346–27353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderfield M, Deuker MM, McCormick F, and McMahon M (2014). Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. (2020). KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 383, 1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosford SR, Dillon LM, Bouley SJ, Rosati R, Yang W, Chen VS, Demidenko E, Morra RP Jr., and Miller TW (2017). Combined Inhibition of Both p110alpha and p110beta Isoforms of Phosphatidylinositol 3-Kinase Is Required for Sustained Therapeutic Effect in PTEN-Deficient, ER(+) Breast Cancer. Clin Cancer Res 23, 2795–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Wang G, Shen Q, Liu X, Lu S, Geng L, Huang Z, and Zhang J (2015). ASBench: benchmarking sets for allosteric discovery. Bioinformatics 31, 2598–2600. [DOI] [PubMed] [Google Scholar]
- Huang WYC, Alvarez S, Kondo Y, Lee YK, Chung JK, Lam HYM, Biswas KH, Kuriyan J, and Groves JT (2019). A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 363, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, et al. (2014). In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A 111, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Ijuin T (2019). Phosphoinositide phosphatases in cancer cell dynamics-Beyond PI3K and PTEN. Semin Cancer Biol 59, 50–65. [DOI] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 e517. [DOI] [PubMed] [Google Scholar]
- Jang H, Banerjee A, Chavan T, Gaponenko V, and Nussinov R (2017). Flexible-body motions of calmodulin and the farnesylated hypervariable region yield a high-affinity interaction enabling K-Ras4B membrane extraction. J Biol Chem 292, 12544–12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, Banerjee A, Chavan TS, Lu S, Zhang J, Gaponenko V, and Nussinov R (2016a). The higher level of complexity of K-Ras4B activation at the membrane. FASEB J 30, 1643–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, Banerjee A, Marcus K, Makowski L, Mattos C, Gaponenko V, and Nussinov R (2019). The Structural Basis of the Farnesylated and Methylated KRas4B Interaction with Calmodulin. Structure 27, 1647–1659 e1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, Muratcioglu S, Gursoy A, Keskin O, and Nussinov R (2016b). Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem J 473, 1719–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, Zhang M, and Nussinov R (2020). The quaternary assembly of KRas4B with Raf-1 at the membrane. Comput Struct Biotechnol J 18, 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar G, Keskin O, Gursoy A, and Nussinov R (2010). Allostery and population shift in drug discovery. Curr Opin Pharmacol 10, 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, Gollner A, Covini D, Fischer S, Gerstberger T, et al. (2019). Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A 116, 15823–15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D, Gollner A, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, Covini D, Fischer S, Gerstberger T, et al. (2020). Reply to Tran et al.: Dimeric KRAS protein-protein interaction stabilizers. Proc Natl Acad Sci U S A 117, 3365–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettle JG, Bagal SK, Bickerton S, Bodnarchuk MS, Breed J, Carbajo RJ, Cassar DJ, Chakraborty A, Cosulich S, Cumming I, et al. (2020). Structure-Based Design and Pharmacokinetic Optimization of Covalent Allosteric Inhibitors of the Mutant GTPase KRAS(G12C). J Med Chem 63, 4468–4483. [DOI] [PubMed] [Google Scholar]
- Khan I, Rhett JM, and O'Bryan JP (2020a). Therapeutic targeting of RAS: New hope for drugging the "undruggable". Biochim Biophys Acta Mol Cell Res 1867, 118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan ZM, Real AM, Marsiglia WM, Chow A, Duffy ME, Yerabolu JR, Scopton AP, and Dar AC (2020b). Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 588, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Ognjenovic J, Banerjee S, Karandur D, Merk A, Kulhanek K, Wong K, Roose JP, Subramaniam S, and Kuriyan J (2019). Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 366, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostaras E, Kaserer T, Lazaro G, Heuss SF, Hussain A, Casado P, Hayes A, Yandim C, Palaskas N, Yu Y, et al. (2020). A systematic molecular and pharmacologic evaluation of AKT inhibitors reveals new insight into their biological activity. Br J Cancer 123, 542–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krengel U, Schlichting I, Scherer A, Schumann R, Frech M, John J, Kabsch W, Pai EF, and Wittinghofer A (1990). Three-dimensional structures of H-ras p21 mutants: molecular basis for their inability to function as signal switch molecules. Cell 62, 539–548. [DOI] [PubMed] [Google Scholar]
- Krens LL, Baas JM, Gelderblom H, and Guchelaar HJ (2010). Therapeutic modulation of k-ras signaling in colorectal cancer. Drug Discov Today 15, 502–516. [DOI] [PubMed] [Google Scholar]
- Kumar S, Ma B, Tsai CJ, Sinha N, and Nussinov R (2000). Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci 9, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontanara AJ, Georgeon S, Tria G, Svergun DI, and Hantschel O (2014). The SH2 domain of Abl kinases regulates kinase autophosphorylation by controlling activation loop accessibility. Nat Commun 5, 5470. [DOI] [PubMed] [Google Scholar]
- Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, Duchaine J, Mao DY, Bouvier M, Sicheri F, et al. (2013). Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol 9, 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H (2015). Cancer: The Ras renaissance. Nature 520, 278–280. [DOI] [PubMed] [Google Scholar]
- Lee Y, Phelps C, Huang T, Mostofian B, Wu L, Zhang Y, Tao K, Chang YH, Stork PJ, Gray JW , et al. (2019). High-throughput, single-particle tracking reveals nested membrane domains that dictate KRas(G12D) diffusion and trafficking. Elife 8, e46393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontiadou H, Galdadas I, Athanasiou C, and Cournia Z (2018). Insights into the mechanism of the PIK3CA E545K activating mutation using MD simulations. Sci Rep 8, 15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jang H, Zhang J, and Nussinov R (2018a). Raf-1 Cysteine-Rich Domain Increases the Affinity of K-Ras/Raf at the Membrane, Promoting MAPK Signaling. Structure 26, 513–525 e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZL, Prakash P, and Buck M (2018b). A "Tug of War" Maintains a Dynamic Protein-Membrane Complex: Molecular Dynamics Simulations of C-Raf RBD-CRD Bound to K-Ras4B at an Anionic Membrane. ACS Cent Sci 4, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Shima F, Araki M, Ye M, Muraoka S, Sugimoto T, Kawamura M, Yamamoto N, Tamura A, and Kataoka T (2008). Two conformational states of Ras GTPase exhibit differential GTP-binding kinetics. Biochem Biophys Res Commun 369, 327–332. [DOI] [PubMed] [Google Scholar]
- Liao TJ, Jang H, Fushman D, and Nussinov R (2018). Allosteric KRas4B Can Modulate SOS1 Fast and Slow Ras Activation Cycles. Biophys J 115, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao TJ, Jang H, Nussinov R, and Fushman D (2020). High-Affinity Interactions of the nSH3/cSH3 Domains of Grb2 with the C-Terminal Proline-Rich Domain of SOS1. J Am Chem Soc 142, 3401–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien EC, Dibble CC, and Toker A (2017). PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol 45, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, et al. (2014). Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl 53, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Sjogren AK, Karlsson C, Ibrahim MX, Andersson KM, Olofsson FJ, Wahlstrom AM, Dalin M, Yu H, Chen Z, et al. (2010). Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc Natl Acad Sci U S A 107, 6471–6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Wang Y, and Li X (2019). Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B 9, 871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, Choi ALM, Mitchell DC, Swaney DL, Hann B, et al. (2019). KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy DR, Zhang K, DeClue JE, and Willumsen BM (1991). Regulation of p21ras activity. Trends Genet 7, 346–351. [DOI] [PubMed] [Google Scholar]
- Lu S, Huang W, and Zhang J (2014a). Recent computational advances in the identification of allosteric sites in proteins. Drug Discov Today 19, 1595–1600. [DOI] [PubMed] [Google Scholar]
- Lu S, Jang H, Gu S, Zhang J, and Nussinov R (2016a). Drugging Ras GTPase: a comprehensive mechanistic and signaling structural view. Chem Soc Rev 45, 4929–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, and Zhang J (2016b). Ras Conformational Ensembles, Allostery, and Signaling. Chem Rev 116, 6607–6665. [DOI] [PubMed] [Google Scholar]
- Lu S, Jang H, Zhang J, and Nussinov R (2016c). Inhibitors of Ras-SOS Interactions. ChemMedChem 11, 814–821. [DOI] [PubMed] [Google Scholar]
- Lu S, Ji M, Ni D, and Zhang J (2018). Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov Today 23, 359–365. [DOI] [PubMed] [Google Scholar]
- Lu S, Li S, and Zhang J (2014b). Harnessing allostery: a novel approach to drug discovery. Med Res Rev 34, 1242–1285. [DOI] [PubMed] [Google Scholar]
- Lu S, Qiu Y, Ni D, He X, Pu J, and Zhang J (2020). Emergence of allosteric drug-resistance mutations: new challenges for allosteric drug discovery. Drug Discov Today 25, 177–184. [DOI] [PubMed] [Google Scholar]
- Ma B, Kumar S, Tsai CJ, Hu Z, and Nussinov R (2000). Transition-state ensemble in enzyme catalysis: possibility, reality, or necessity? J Theor Biol 203, 383–397. [DOI] [PubMed] [Google Scholar]
- Ma B, Shatsky M, Wolfson HJ, and Nussinov R (2002). Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci 11, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackereth CD, Madl T, Bonnal S, Simon B, Zanier K, Gasch A, Rybin V, Valcarcel J, and Sattler M (2011). Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature 475, 408–411. [DOI] [PubMed] [Google Scholar]
- Mahoney SJ, Narayan S, Molz L, Berstler LA, Kang SA, Vlasuk GP, and Saiah E (2018). A small molecule inhibitor of Rheb selectively targets mTORC1 signaling. Nat Commun 9, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Gago P, Fansa EK, Klein CH, Murarka S, Janning P, Schurmann M, Metz M, Ismail S, Schultz-Fademrecht C, Baumann M, et al. (2017). A PDE6delta-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew Chem Int Ed Engl 56, 2423–2428. [DOI] [PubMed] [Google Scholar]
- Martin-Garcia F, Mendieta-Moreno JI, Lopez-Vinas E, Gomez-Puertas P, and Mendieta J (2012). The Role of Gln61 in HRas GTP hydrolysis: a quantum mechanics/molecular mechanics study. Biophys J 102, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattingly RR (2013). Activated Ras as a Therapeutic Target: Constraints on Directly Targeting Ras Isoforms and Wild-Type versus Mutated Proteins. ISRN Oncol 2013, 536529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, et al. (2012). Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A 109, 5299–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MJ, Pagba CV, Prakash P, Naji AK, van der Hoeven D, Liang H, Gupta AK, Zhou Y, Cho KJ, Hancock JF, et al. (2019). Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega 4, 2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick F (2018). Targeting KRAS Directly. In Annual Review of Cancer Biology, Jacks T, and Sawyers CL, eds. (ANNUAL REVIEWS; ), pp. 81–90. [Google Scholar]
- Milroy LG, and Ottmann C (2014). The renaissance of Ras. ACS Chem Biol 9, 2447–2458. [DOI] [PubMed] [Google Scholar]
- Mirati Therapeutics, (2020). KRAS G12C Inhibitor. https://www.mirati.com/pipeline/kras-g12c/
- Moran MF, Polakis P, McCormick F, Pawson T, and Ellis C (1991). Protein-tyrosine kinases regulate the phosphorylation, protein interactions, subcellular distribution, and activity of p21ras GTPase-activating protein. Mol Cell Biol 11, 1804–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh HN, Wrabl JO, Li J, and Hilser VJ (2014). The ensemble nature of allostery. Nature 508, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A (2019). Cracking KRAS. Nat Rev Drug Discov 18, 887–891. [DOI] [PubMed] [Google Scholar]
- Murase K, Fujiwara T, Umemura Y, Suzuki K, Iino R, Yamashita H, Saito M, Murakoshi H, Ritchie K, and Kusumi A (2004). Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys J 86, 4075–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratcioglu S, Aydin C, Odabasi E, Ozdemir ES, Firat-Karalar EN, Jang H, Tsai CJ, Nussinov R, Kavakli IH, Gursoy A, et al. (2020). Oncogenic K-Ras4B Dimerization Enhances Downstream Mitogen-activated Protein Kinase Signaling. J Mol Biol 432, 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratcioglu S, Chavan TS, Freed BC, Jang H, Khavrutskii L, Freed RN, Dyba MA, Stefanisko K, Tarasov SG, Gursoy A, et al. (2015). GTP-Dependent K-Ras Dimerization. Structure 23, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, and Kuriyan J (2003). Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–871. [DOI] [PubMed] [Google Scholar]
- Nagasaka M, Li Y, Sukari A, Ou SI, Al-Hallak MN, and Azmi AS (2020). KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat Rev 84, 101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale C, and Garcia AE (2020). The Plasma Membrane as a Competitive Inhibitor and Positive Allosteric Modulator of KRas4B Signaling. Biophys J 118, 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R (2016). Introduction to Protein Ensembles and Allostery. Chem Rev 116, 6263–6266. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Jang H, and Tsai CJ (2014). The structural basis for cancer treatment decisions. Oncotarget 5, 7285–7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Jang H, Tsai CJ, Liao TJ, Li S, Fushman D, and Zhang J (2017a). Intrinsic protein disorder in oncogenic KRAS signaling. Cell Mol Life Sci 74, 3245–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, and Ma B (2012). Protein dynamics and conformational selection in bidirectional signal transduction. BMC Biol 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, and Tsai CJ (2012). The different ways through which specificity works in orthosteric and allosteric drugs. Curr Pharm Des 18, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, and Tsai CJ (2013). Allostery in disease and in drug discovery. Cell 153, 293–305. [DOI] [PubMed] [Google Scholar]
- Nussinov R, and Tsai CJ (2015). The design of covalent allosteric drugs. Annu Rev Pharmacol Toxicol 55, 249–267. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2018a). Oncogenic Ras Isoforms Signaling Specificity at the Membrane. Cancer Res 78, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2019a). Does Ras Activate Raf and PI3K Allosterically? Front Oncol 9, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2019b). Is Nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant? Semin Cancer Biol 54, 114–120. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2019c). Oncogenic KRas mobility in the membrane and signaling response. Semin Cancer Biol 54, 109–113. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2020). Ras assemblies and signaling at the membrane. Curr Opin Struct Biol 62, 140–148. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Mattos C (2013). 'Pathway drug cocktail': targeting Ras signaling based on structural pathways. Trends Mol Med 19, 695–704. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Wang G, Tsai CJ, Jang H, Lu S, Banerjee A, Zhang J, and Gaponenko V (2017b). Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 3, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, and Wolynes PG (2014). A second molecular biology revolution? The energy landscapes of biomolecular function. Phys Chem Chem Phys 16, 6321–6322. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Zhang M, Tsai CJ, Liao TJ, Fushman D, and Jang H (2018b). Autoinhibition in Ras effectors Raf, PI3Kalpha, and RASSF5: a comprehensive review underscoring the challenges in pharmacological intervention. Biophys Rev 10, 1263–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Bryan JP (2019). Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol Res 139, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Hu CD, Jin TG, Kariya K, Yamawaki-Kataoka Y, and Kataoka T (1999). The strength of interaction at the Raf cysteine-rich domain is a critical determinant of response of Raf to Ras family small GTPases. Mol Cell Biol 19, 6057–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K, and Takada S (2008). Dynamic energy landscape view of coupled binding and protein conformational change: induced-fit versus population-shift mechanisms. Proc Natl Acad Sci U S A 105, 11182–11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke B, and Der CJ (2017). Drugging RAS: Know the enemy. Science 355, 1158–1163. [DOI] [PubMed] [Google Scholar]
- Papke B, Murarka S, Vogel HA, Martin-Gago P, Kovacevic M, Truxius DC, Fansa EK, Ismail S, Zimmermann G, Heinelt K, et al. (2016). Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat Commun 7, 11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG, Sharif H, Marto JA, Jeon H, and Eck MJ (2019). Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 575, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JA, and Mattos C (2018). The K-Ras, N-Ras, and H-Ras Isoforms: Unique Conformational Preferences and Implications for Targeting Oncogenic Mutants. Cold Spring Harb Perspect Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JA, Volmar AY, Pavlopoulos S, and Mattos C (2018). K-Ras Populates Conformational States Differently from Its Isoform H-Ras and Oncogenic Mutant K-RasG12D. Structure 26, 810–820 e814. [DOI] [PubMed] [Google Scholar]
- Patgiri A, Yadav KK, Arora PS, and Bar-Sagi D (2011). An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol 7, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al. (2016). Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov 6, 316–329. [DOI] [PubMed] [Google Scholar]
- Pokrass MJ, Ryan KA, Xin T, Pielstick B, Timp W, Greco V, and Regot S (2020). Cell-Cycle-Dependent ERK Signaling Dynamics Direct Fate Specification in the Mammalian Preimplantation Embryo. Dev Cell 55, 328–340 e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, et al. (2019). Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov 9, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, and Gorfe AA (2019). Probing the Conformational and Energy Landscapes of KRAS Membrane Orientation. J Phys Chem B 123, 8644–8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, Litwin D, Liang H, Sarkar-Banerjee S, Dolino D, Zhou Y, Hancock JF, Jayaraman V, and Gorfe AA (2020). Dynamics of Membrane-Bound G12V-KRAS from Simulations and Single-Molecule FRET in Native Nanodiscs. Biophys J 118, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, and Mattos C (2012). A comprehensive survey of Ras mutations in cancer. Cancer Res 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, and Bar-Sagi D (2011). RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevedo CE, Cruz-Migoni A, Bery N, Miller A, Tanaka T, Petch D, Bataille CJR, Lee LYW, Fallon PS, Tulmin H, et al. (2018). Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat Commun 9, 3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid TS, and Beese LS (2004). Crystal structures of the anticancer clinical candidates R115777 (Tipifarnib) and BMS-214662 complexed with protein farnesyltransferase suggest a mechanism of FTI selectivity. Biochemistry 43, 6877–6884. [DOI] [PubMed] [Google Scholar]
- Rezaei Adariani S, Buchholzer M, Akbarzadeh M, Nakhaei-Rad S, Dvorsky R, and Ahmadian MR (2018). Structural snapshots of RAF kinase interactions. Biochem Soc Trans 46, 1393–1406. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Kamada Y, Sameshima T, Yaguchi M, Niida A, Sasaki S, Miwa M, Ohkubo S, Sakamoto JI, Kamaura M, et al. (2017). K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem Biophys Res Commun 484, 605–611. [DOI] [PubMed] [Google Scholar]
- Samatar AA, and Poulikakos PI (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Sarkar S, and Garcia AE (2020). Presence or Absence of Ras Dimerization Shows Distinct Kinetic Signature in Ras-Raf Interaction. Biophys J 118, 1799–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, and Bollag G (2007). Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7, 295–308. [DOI] [PubMed] [Google Scholar]
- Shao T, Zheng Y, Zhao B, Li T, Cheng K, and Cai W (2014). Recombinant expression of different mutant K-ras gene in pancreatic cancer Bxpc-3 cells and its effects on chemotherapy sensitivity. Sci China Life Sci 57, 1011–1017. [DOI] [PubMed] [Google Scholar]
- Sheridan C (2020). Grail of RAS cancer drugs within reach. Nat Biotechnol 38, 6–8. [DOI] [PubMed] [Google Scholar]
- Sheridan C, and Downward J (2013). Inhibiting the RAS-PI3K pathway in cancer therapy. Enzymes 34 Pt. B, 107–136. [DOI] [PubMed] [Google Scholar]
- Shima F, Ijiri Y, Muraoka S, Liao J, Ye M, Araki M, Matsumoto K, Yamamoto N, Sugimoto T, Yoshikawa Y, et al. (2010). Structural basis for conformational dynamics of GTP-bound Ras protein. J Biol Chem 285, 22696–22705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, et al. (2013). In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci U S A 110, 8182–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, and Vaz WL (2004). Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct 33, 269–295. [DOI] [PubMed] [Google Scholar]
- Singh H, Longo DL, and Chabner BA (2015). Improving Prospects for Targeting RAS. J Clin Oncol 33, 3650–3659. [DOI] [PubMed] [Google Scholar]
- Smith MJ, and Ikura M (2014). Integrated RAS signaling defined by parallel NMR detection of effectors and regulators. Nat Chem Biol 10, 223–230. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Neel BG, and Ikura M (2013). NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A 110, 4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solman M, Ligabue A, Blazevits O, Jaiswal A, Zhou Y, Liang H, Lectez B, Kopra K, Guzman C, Harma H, et al. (2015). Specific cancer-associated mutations in the switch III region of Ras increase tumorigenicity by nanocluster augmentation. Elife 4, e08905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, et al. (2017). Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol 13, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R, and O'Bryan JP (2019). Direct inhibition of RAS: Quest for the Holy Grail? Semin Cancer Biol 54, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, and Waldmann H (2014). Small-molecule modulation of Ras signaling. Nat Chem Biol 10, 613–622. [DOI] [PubMed] [Google Scholar]
- Sprang SR (1997). G proteins, effectors and GAPs: structure and mechanism. Curr Opin Struct Biol 7, 849–856. [DOI] [PubMed] [Google Scholar]
- Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, Zhang M, Jang H, Renzi F, Cai Y, et al. (2018). Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 362, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, and McCormick F (2014). Dragging ras back in the ring. Cancer Cell 25, 272–281. [DOI] [PubMed] [Google Scholar]
- Stites EC, and Ravichandran KS (2009). A systems perspective of ras signaling in cancer. Clin Cancer Res 15, 1510–1513. [DOI] [PubMed] [Google Scholar]
- Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, and Fesik SW (2012). Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl 51, 6140–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MN, Lu Z, Li YC, Zhou Y, Huang T, Reger AS, Hurwitz AM, Palzkill T, Logsdon C, Liang X, et al. (2019). DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting RAS Clusters. Cell Rep 29, 3448–3459 e3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, and Matozaki T (2001). Small GTP-binding proteins. Physiol Rev 81, 153–208. [DOI] [PubMed] [Google Scholar]
- Thurman R, Siraliev-Perez E, and Campbell SL (2020). RAS ubiquitylation modulates effector interactions. Small GTPases 11, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TH, Alexander P, Dharmaiah S, Agamasu C, Nissley DV, McCormick F, Esposito D, Simanshu DK, Stephen AG, and Balius TE (2020). The small molecule BI-2852 induces a nonfunctional dimer of KRAS. Proc Natl Acad Sci U S A 117, 3363–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers T, Lopez CA, Van QN, Neale C, Tonelli M, Stephen AG, and Gnanakaran S (2018). Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci Rep 8, 8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, Kumar S, Ma B, and Nussinov R (1999a). Folding funnels, binding funnels, and protein function. Protein Sci 8, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, Ma B, and Nussinov R (1999b). Folding and binding cascades: shifts in energy landscapes. Proc Natl Acad Sci U S A 96, 9970–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, and Nussinov R (2014). The free energy landscape in translational science: how can somatic mutations result in constitutive oncogenic activation? Phys Chem Chem Phys 16, 6332–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter IR, and Wittinghofer A (2001). The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- Vigil D, Cherfils J, Rossman KL, and Der CJ (2010). Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 10, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kaiser CE, Frett B, and Li HY (2013). Targeting mutant KRAS for anticancer therapeutics: a review of novel small molecule modulators. J Med Chem 56, 5219–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RA, Fawell S, Floc'h N, Flemington V, McKerrecher D, and Smith PD (2020). Challenges and Opportunities in Cancer Drug Resistance. Chem Rev, DOI: 10.1021/acs.chemrev.1020c00383. [DOI] [PubMed] [Google Scholar]
- Waters AM, and Der CJ (2018). KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmuller L, and Wittinghofer F (1994). Signal transduction pathways involving Ras. Mini review. Cell Signal 6, 247–267. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, and Pai EF (1991). The structure of Ras protein: a model for a universal molecular switch. Trends Biochem Sci 16, 382–387. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, Scheffzek K, and Ahmadian MR (1997). The interaction of Ras with GTPase-activating proteins. FEBS Lett 410, 63–67. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, and Vetter IR (2011). Structure-function relationships of the G domain, a canonical switch motif. Annu Rev Biochem 80, 943–971. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Lu J, Hunter J, Li L, Scott D, Choi HG, Lim SM, Manandhar A, Gondi S, Sim T, et al. (2017). Covalent Guanosine Mimetic Inhibitors of G12C KRAS. ACS Med Chem Lett 8, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Liu D, Li L, Wempe MF, Guin S, Khanna M, Meier J, Hoffman B, Owens C, Wysoczynski CL, et al. (2014). Discovery and characterization of small molecules that target the GTPase Ral. Nature 515, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehia L, Keel E, and Eng C (2020). The Clinical Spectrum of PTEN Mutations. Annu Rev Med 71, 103–116. [DOI] [PubMed] [Google Scholar]
- Young A, Lou D, and McCormick F (2013). Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov 3, 112–123. [DOI] [PubMed] [Google Scholar]
- Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, Ficarro SB, Xiong Y, Ambrogio C, Paranal RM, et al. (2017). Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem Biol 24, 1005–1016 e1003. [DOI] [PubMed] [Google Scholar]
- Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, Nabet B, Gero TW, Feru F, Li L, et al. (2020). Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem Biol 27, 19–31 e16. [DOI] [PubMed] [Google Scholar]
- Zhang M, Jang H, and Nussinov R (2019a). The mechanism of PI3Kalpha activation at the atomic level. Chem Sci 10, 3671–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Jang H, and Nussinov R (2019b). The structural basis for Ras activation of PI3Kalpha lipid kinase. Phys Chem Chem Phys 21, 12021–12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Jang H, and Nussinov R (2020). PI3K inhibitors: review and new strategies. Chemical Science 11, 5855–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Shokat KM (2019). Bifunctional Small-Molecule Ligands of K-Ras Induce Its Association with Immunophilin Proteins. Angew Chem Int Ed Engl 58, 16314–16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Prakash P, Gorfe AA, and Hancock JF (2018). Ras and the Plasma Membrane: A Complicated Relationship. Cold Spring Harb Perspect Med 8, a031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, et al. (2013). Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 497, 638–642. [DOI] [PubMed] [Google Scholar]