

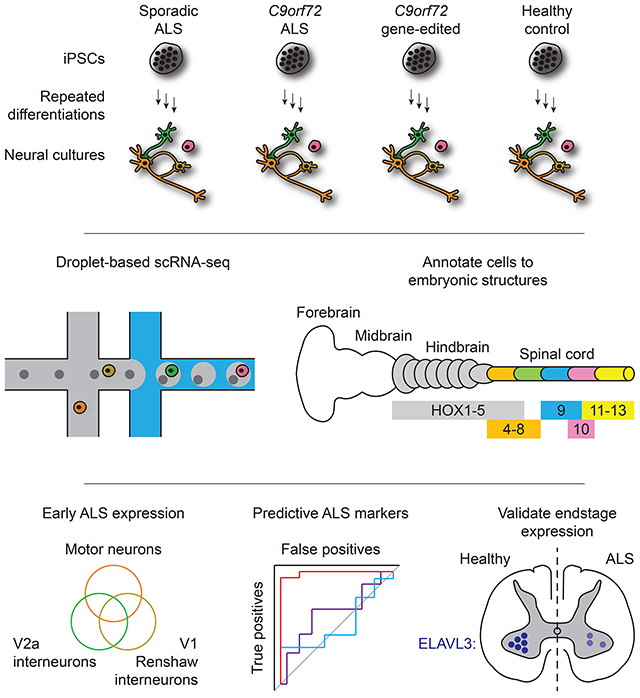
Summary
Induced pluripotent stem cell- (iPSC) derived neural cultures from amyotrophic lateral sclerosis (ALS) patients can model disease phenotypes. However, heterogeneity arising from genetic and experimental variability limits their utility, impacting reproducibility and the ability to track cellular origins of pathogenesis. Here, we present methodologies using single-cell RNA-sequencing (scRNA-seq) analysis to address these limitations. By repeatedly differentiating and applying scRNA-seq to motor neurons (MNs) from healthy, familial ALS, sporadic ALS, and genome-edited iPSC lines across multiple patients, batches, and platforms, we account for genetic and experimental variability towards identifying unified and reproducible ALS signatures. Combining HOX and developmental gene expression with global clustering, we anatomically classified cells into rostrocaudal, progenitor, and postmitotic identities. By relaxing statistical thresholds, we discovered genes in iPSC-MNs that were concordantly dysregulated in postmortem MNs and yielded predictive ALS markers in other human and mouse models. Our approach thus revealed early, convergent, and MN-resolved signatures of ALS.
Graphical Abstract
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by cortical and spinal motor neuron (MN) death resulting in weakness and paralysis of voluntary muscles (Ragagnin et al., 2019; Swinnen and Robberecht, 2014). While numerous molecular pathways and cell types associated with ALS have been described, definitive mechanisms responsible for MN degeneration remain elusive (Taylor et al., 2016). The vast majority of ALS cases are sporadic, with no known genetic link. In familial cases, ALS can be traced to a set of genetic mutations, for instance GGGGCC hexanucleotide repeat expansions (HREs) in the intronic sequence between alternate 5' exons in C9orf72, a gene that regulates endosomal trafficking and autophagy. Furthermore, symptom onset for both familial and sporadic ALS varies across body regions, thereby compounding difficulty in discerning disease etiology. Despite this variation, common clinical presentations are observed across familial and sporadic cases, suggesting that molecular features may converge across ALS patients.
Induced pluripotent stem cells (iPSCs) derived from ALS patients carry great potential to experimentally model molecular events underlying ALS pathogenesis. However, identifying early, MN-resolved, and reproducible gene expression changes has posed challenges. First, iPSC-differentiated tissues do not recapitulate mature and adult-like states (Ho et al., 2016; Stein et al., 2014). This suggests that signals representing dysfunctional physiologies experienced by in vivo tissues in late onset diseases may not be recapitulated with high fidelity in in vitro disease models. Second, recent studies investigating transcriptomic ALS signatures in human iPSC models have profiled cultures in bulk (Fujimori et al., 2018) or transgenically labeled MNs (Kiskinis et al., 2014; Shi et al., 2018), all of which were performed at time points where overt disease phenotypes such as neurite degeneration of cell death have emerged. Bulk transcriptomic profiles cannot distinguish whether disease signatures originate from MNs or other cell types in culture, and sampling after prolonged differentiation times cannot distinguish early transcriptomics events from secondary transcriptomic events responding to the overt disease phenotypes observed at these times. Thirdly, studies using iPSC models have rarely addressed whether experimental variations in expression data arising from repeated differentiations or transcriptomic profiling platforms could impact the reproducible biology of disease signatures (Volpato and Webber, 2020).
Here, we overcome these challenges and present an approach for generating iPSC-based experimental models of ALS that exhibit early, MN-specific, and reproducible transcriptomic disease signatures. We differentiated MNs from iPSCs derived from patients with familial ALS, sporadic ALS, healthy controls, and CRISPR-Cas9-edited C9orf72 HREs. These cultures were profiled using single-cell RNA-seq (scRNA-seq) at the earliest time when postmitotic neurons arise and exhibit no degeneration. We validated that iPSC-derived MNs (iPSC-MNs) express appropriate fetal hindbrain and spinal cord development markers. Analyzing this data uncovered early transcriptional signatures of ALS in MNs, which were distinctive from interneuron subtypes that are concomitantly differentiated in culture. These were verified in gene expression data sets from other human iPSC-MN models, mouse models, and postmortem patient samples, demonstrating that these signatures persist from early to endstage disease in both familial and sporadic ALS. In total, our results highlight the utility of iPSC-based experimental models to capture early, dysregulated gene expression in MNs common across a wide range of ALS patients.
Results
Production of control, sporadic, C9orf72 ALS and isogenic iPSC derived MNs
MNs were differentiated from iPSCs reprogrammed from either fibroblasts or peripheral blood monocytes from four healthy subjects: 0083, 0179, 0025, and 0465, two sporadic ALS subjects: 2XWC and 8BRM, and four familial ALS subjects with C9orf72 HRE: 0028, 0029, 0052, and 6ZLD (Table S1 and Figure 1A). To isolate C9orf72 HRE effects from inherent genetic variability, isogenic patient lines were established from two C9orf72 ALS lines (0029 and 0052) using CRISPR-Cas9-mediated gene editing to remove the HREs (Table S1 and Figure S1A). Edited iPSC clones were kayotypically normal (Figure S1B-E), and retained the ability to differentiate into MNs over a 30 day in vitro differentiation protocol (Yang et al., 2013) at a comparable efficiency to parental C9orf72 ALS cell lines (Figure S2A). Removal of the HREs resulted in two-fold increased expression of all C9orf72 transcript variants back to levels observed in normal controls (Figures S2B and S2C) and eliminated sense and antisense RNA foci (Figures S2D and S2E). Furthermore, polyGP dipeptide repeats, which accumulated in C9orf72 ALS MN cultures (Figure S2F), were reduced to control subject levels (Figure S2G). Isogenic MN cultures thus enabled direct attribution of molecular phenotypes to HREs in parental C9orf72 ALS lines.
Figure 1. iPSC-MN cultures recapitulate developmental gene expression patterns.

A. Immunostaining day 18 cultures from three individual subject lines for expression of MN markers ISL1 and SMI-32.
B. Experiment schematic for analyzing the 18 day time course of MN differentiation.
C - D. Violin plots indicate gene expression during MN differentiation. C depicts number of unique molecular identifiers (nUMI), and D depicts number of detectable genes per time point.
E. tSNE of MN differentiation time course samples.
F. Monocle analysis projects samples into a pseudo-time axis consistent with the order of time points.
G. Histogram displays meta data for all samples profiled in this study. This data is also presented in Figure S4.
H. UMI counts were summed for each gene across the expression matrix for each sample, simulating bulk RNA-seq data. Simulated bulk gene expression profiles for each sample were correlated to bulk RNA-seq gene expression profiles from fetal hindbrain and spinal cord at various Carnegie stages analyzed in de Kovel et al., 2017. For each row of pairwise correlations, the top three ranked correlations are outlined, indicating which sample from de Kovel et al., 2017 is most globally similar to each sample analyzed by single-cell RNA-seq. For each column of pairwise correlations, the median Spearman correlation was calculated and displayed along the bottom row. These data summarize for each sample from de Kovel et al., 2017 what is the frequency of global gene expression correlations from iPSC-MN cultures. The top three ranked correlations along the bottom row are outlined, indicating which sample from de Kovel et al., 2017 are most globally similar to iPSC-MNs.
See also Figures S1, S2, S3, and S4, and Table S1.
iPSC-derived MN cultures recapitulate developmental gene expression patterns
Next, MN differentiations from iPSCs (iPSC-MNs) were characterized at the single-cell level with the Illumina® Bio-Rad® SureCell™ WTA 3' Library Prep Kit for the ddSEQ™ System for one control line (0083) using a more rapid 18 day differentiation protocol that produces cranial and spinal MNs and interneurons (Maury et al., 2015) (Figure 1B). Consistent with previous observations, pluripotent cells undergo a reduction in overall transcriptional activity upon differentiation, suggesting a refinement of transcriptional programs from the pluripotent to progenitor state (Efroni et al., 2008) (Figures 1C and 1D). Unique molecular identifiers (nUMIs) per cell increased between days 12 and 18, suggesting a state of specialized physiology and functions. Global clustering resolved each time point into distinct clusters, where day 12 and day 18 populations further resolved into subpopulations (Figure 1E). Pseudotime analysis of cells from all time points through Monocle (Qiu et al., 2017) arranged each time point in the expected order of progressively differentiating cell states (Figure 1F). 20 marker genes for spinal MN development and maturation (Ho et al., 2016) were expressed along the pseudotime axis in a pattern consistent with fetal-like tissues derived in vitro from iPSCs (Figure S3A).
iPSC-MN cultures globally resemble fetal hindbrain and spinal cord
We next performed scRNA-seq on MN cultures from several ALS and control subject lines at 18 days of differentiation in order to establish a pool of single cells we could use to determine regional specificity along the rostrocaudal axis of the neural tube as well as the presence of ALS signatures (Table S1 and Figure 1G). Because only a finite amount of samples could be captured and processed within each experiment, we collected samples across six batches of differentiation (A-F). We also aimed to establish the robustness of any signal across two different scRNA-seq platforms: the Illumina Bio-Rad Single-Cell Sequencing Solution (DDSEQ) and the 10X Genomics Chromium (10X) (Table S1 and Figure 1G). Immunostaining and quantification of day 18 cultures indicated no significant differences in ISL1 and SMI-32 positive MNs between ALS and control. This suggests that an overt disease phenotype such as cell death, as shown in previous studies (Fujimori et al., 2018; Sareen et al., 2013), has not manifested at this relatively early differentiation time point (Figure S3B). In total, we analyzed 21,702 cells that passed quality control filters. These filters required that A) genes not expressed in any cell in any sample were excluded, and B) cells with 1) percent mitochondrial genes, 2) total number of expressed genes, and 3) total number of UMIs that were greater or less than three standard deviations of the sample population were excluded (see Methods for details). To gauge the developmental and maturation states of these cultures, we correlated their expression profiles to spinal MN maturation gene expression (Ho et al., 2016) (Figures S4A and S4B) and to fetal hindbrain and spinal cord tissue ranging from Carnegie stages 13 to 23 (de Kovel et al., 2017) (Figure 1H). By 18 days, iPSC-derived cells showed transcriptional states that most globally resemble fetal hindbrain and spinal cord tissue at Carnegie stage 17, or about 42 days of in vivo development.
In order to establish the rostrocaudal identity of individual cells, we focused on the family of homeobox (HOX) transcriptional regulators of morphological patterning. Based on previous genetic studies (Di Bonito et al., 2013; Lippmann et al., 2015; Philippidou and Dasen, 2013), we composed a model for relative HOX gene expression along rhombomeres two to eight of the developing hindbrain and cervical to caudal segments of the spinal cord (Figures 2A, 2B, and Table S2A). RNA expression levels of each HOX gene in the fetal hindbrain and spinal cord samples from de Kovel et al., 2017 were consistent with our model, and classification of segment identity based on the highest correlation for each sample resolves the sample types (Figures 2B and 2C). While correlation of bulk profiles from day 18 iPSC-MN cultures suggested that cultures globally resembled hindbrain more than spinal cord, we hypothesized that some rare cells may have differentiated into more caudal identities. We therefore applied this classification approach for each cell in the day 18 cultures. These results indicated that while a majority of cells (33.19%) were not assigned (NA) categories, either due to a lack of any detectable HOX gene expression or failure to meet the correlation cutoff, the second majority of cells (25.75%) were classified as rhombomere eight, and a third majority of cells (10.78%) were classified as the cervical segment (Figure 2C and Table S2C). Notably, there were some cells classified as brachial (1.32%) and thoracic (1.40%) segments, suggesting that the 18 day protocol can achieve differentiation into cell types within the spinal cord that reflect upper limb sites of disease onset for most subjects represented in this study (Table S1). However, no cells classified as lumbar segment, possibly due to the early differentiation time point of these cultures.
Figure 2. iPSC-MN cultures globally resemble rhombomere eight and cervical spinal cord.

A. Models of cell type organization along representative hindbrain and spinal cord segments. Left, HOX gene expression patterns determine rostrocaudal axis identities, adapted and simplified from Di Bonito et al., 2013; Lippmann et al., 2015; and Philippidou and Dasen, 2013. Center, a simplified, two-dimensional schematic of the dorsoventral and mediolateral axes of a representative hemisection of a spinal cord segment adapted from Alaynick et al., 2011 and Lu et al., 2015. Each subclass of progenitors in the ventricular zone (VZ) are labeled along the dorsoventral axis, represented by the y-axis, are colored distinctly. As each of these progenitors give rise to subclasses of postmitotic neurons, they migrate laterally into distinct dorsoventral and mediolateral locations in the mantle zone (MZ), represented by the x-axis. These progenies are labeled and colored similarly to their respective progenitors. Descriptions of each cell type are indicated in Table S2B. Locations of each cell type are approximately relative to each other and are not to scale. Right, three-dimensional model of the center panel, including both hemispheres of the spinal cord. All cell types present within the hindbrain and spinal cord are registered based on HOX and lineage-specific transcription factor expression. This model is not to scale.
B. Left: Unit expression matrix of HOX genes that developmentally determine hindbrain and spinal cord segment identity. R2-8: rhombomeres 2-8, Ce: cervical, Br: brachial, Th: thoracic, Lu: lumbar, Sa: sacral, Ca: caudal. Right: Expression heatmap of HOX genes in fetal hindbrain and spinal cord at various Carnegie stages analyzed in de Kovel et al., 2017. Labels below each sample column indicate the segment that each sample most resembles, based on the highest Spearman correlation to the expression patterns for each segment shown in the unit expression matrix and with Benjamini-Hochberg adjusted P-values < 0.05.
C. Histogram of segment identities assigned to fetal hindbrain samples, fetal spinal cord samples, and individual cells analyzed from day 18 cultures based on the highest Spearman correlation to the expression patterns for each segment shown in the unit expression matrix. NA: not assigned. For assignment of fetal hindbrain and fetal spinal cord samples from de Kovel et al., 2017, samples with Benjamini-Hochberg adjusted P-values less than 0.05 are assigned to each class along the y-axis; all other other samples are assigned as NA. For assignment of cells in day 18 cultures, cells with Benjamini-Hochberg adjusted P-values less than 0.1 are assigned to each class along the y-axis; all other cells are assigned as NA.
Developmental gene expression profiles and global clustering classify ventricular zone (VZ) progenitor and mantle zone (MZ) postmitotic neuronal identities
The induction of neural differentiation occurs after embryonic regionalization of the anterioposterior axis (Metzis et al., 2018). The programmed expression of genes encodes a two-dimensional coordinate system of morphogen gradients regulating dorsoventral and mediolateral axes and progression of neural progenitors to postmitotic neurons in a representative spinal cord segment (Alaynick et al., 2011; Lu et al., 2015) (Figures 2A, S5A, and Table S2B). We resolved individual neural identities using 105 of these genes. By correlating each cell type in our model with one another based on these genes, these profiles can systematically distinguish each identity (Figure S5B). Assignment of individual cells along the 18 day differentiation to either VZ progenitors or MZ postmitotic neurons illustrated a cell fate progression consistent with the functions of morphogenic components used during induction (Figures 1B, 3A, and Table S2C). Few astrocytes were seen (Figure 3A) indicating the rapid 18 day differentiation did not promote a glial program.
Figure 3. Developmental gene expression profiles and global clustering classify VZ progenitor and MZ postmitotic neuronal identities.

A. Based on the expression profile of 105 developmental genes (Table S2B), individual cells from the time course of differentiation over 18 days were classified as identities belonging to the VZ, MZ, astrocyte, or not assigned (NA).
B. tSNE showing global clustering of 17,531 cells from day 18 cultures across experimental batches, which by Seurat determined four clusters indicated by four colors. Significantly differentially expressed genes in each cluster relative to the other clusters were analyzed for enriched GO terms, and these are displayed below the tSNE plot.
C. The same tSNE as in B with each cell colored by classification based on 105 developmental genes.
D. The same tSNE as in B with each cell colored by relative expression level of each indicated gene, which were determined to be among the most significantly differentially expressed genes in clusters 1 (SOX2, TOP2A), 2 (COL3A1, TAGLN), 3 (STMN2, ONECUT2), and 4 (PHOX2A, PHOX2B). These data are also presented in Figure S3G.
E. tSNE showing global clustering of 10,866 cells classified into clusters 3 and 4 in B. Seurat analysis determined 18 clusters.
F. The same tSNE as in E with each cell colored red if they were classified as the indicated cell type based on 105 developmental genes.
G. The same tSNE as in E with each cell colored by their reclassification into clusters formed by merging the clusters identified in E based on overlap for identities shown in F. This data is also presented in Figure 4.
H. The same tSNE as in E with each cell colored by relative expression level of each indicated gene, which are regarded as distinct markers of each identity listed above each plot in F.
I. Boxplots of Spearman correlation values between gene expression profiles of LC MNs from Ho et al., 2016; summed gene expression profiles across all cells for each day 18 sample (Bulk); and summed gene expression profiles across cells classified as MN in G. P-value calculated from a paired, two-tailed t-test.
J. Bar graph of segment identities assigned to individual cells within each of the seven clusters based on the highest Spearman correlation to the HOX expression patterns for each segment shown in the unit expression matrix (Table S2A) and with Benjamini-Hochberg adjusted P-value < 0.1. R2-8: rhombomeres 2-8, Ce: cervical, Br: brachial, Th: thoracic, Lu: lumbar, Sa: sacral, Ca: caudal, NA: not assigned.
See also Figures S4, S5, and S6, and Table S2.
Additionally, we used unsupervised global gene expression profiles to unbiasedly cluster distinct identities present in day 18 cultures. However, dimensional reduction and projection using principal component analysis (PCA) and t-distributed stochastic neighbor embedding (tSNE) of raw expression data primarily separated cells based on single-cell technology platform (Figure S3C). This was despite comparable UMIs and genes per cell, albeit DDSEQ had a higher fraction of reads aligning to intergenic regions (Figure S3D). Experimental batch effects were also evident for samples processed within the same platform (Figure S3C). This highlighted the need to correct the scRNA-seq expression data prior to discovering common variations between ALS and control conditions. To this end, we compared several approaches to correct for experimental batch and platform effects, including multi-canonical correlation analysis (MultiCCA) in Seurat (Butler et al., 2018), Harmony (Korsunsky et al., 2019), Liger (Welch et al., 2019), and FastMNN (Haghverdi et al., 2018) (Figure S3C). Evaluation of batch integration using either a Chi-squared test (kBET) or average silhouette width (Büttner et al., 2019) (Figure S3E), revealed that most methods improved batch correction over uncorrected data. Seurat 2 (MultiCCA) ranked as the 2nd best method assessed by kBET and the best performing method assessed by average silhouette width (Figure S3E). We therefore continued subsequent analyses on data corrected by MultiCCA, which also effectively integrated samples, case and controls, genotypes, and cell lines (Figure S5C). By optimizing clustering parameters after batch correction to yield a maximum modularity value of all communities (Blondel et al., 2008; Waltman and van Eck, 2013), this analysis revealed four major populations of cells that distinctly expressed genes associated with a variety of gene ontology (GO) terms (Figures 3B-D, S3F, S5D, and Table S2C). Overlaying marker gene expression on tSNE plots further demonstrated that batch integration using MultiCCA generated tight clusters of neuronal cells (STMN2), neural progenitors (SOX2), and smooth muscle-like cells (TAGLN), suggesting that cluster generation is driven more by cell identity than by batch effects once data has been corrected (Figure S3G). Altogether, these analyses enable the resolution of major populations present in this rapid differentiation protocol, identifying not only postmitotic neurons generated from iPSCs, but also persistent progenitors and another population of non-neuronal cells.
In order to specifically detect neuronal subtype signatures in these cultures, we repeated subpopulation detection by removing non-neuronal cells and progenitors and then performing a new batch correction and global clustering. This analysis assigned 18 major populations of cells (Figures 3E, S5E, and Table S2C). Six of the VZ and MZ labels formed patterns in the tSNE that visually overlapped with globally defined clusters. We therefore renamed them based on these observations (Figure 3F-3H and Table S2C). For example, cells assigned as MNs of the lateral motor column (MN LMC) were enriched in clusters 0, 4, 7, 11, 15, and 17, and these cells expressed MN markers PHOX2B (Pla et al., 2008) and ISL1 (Liang et al., 2011). We therefore renamed this group MN hereafter (Figure 3G). Immunostaining cultures confirmed protein co-expression of PHOX2B with ISL1, and distinct expression of V2a and V2c interneuron markers VSX2 and SOX1, respectively (Figure S6A). Overall, based on overlapping classifications and expression of key marker genes, subsets of the 18 populations were merged to assign seven major populations (Figure 3G and Table S2C).
We then assessed whether cells classified as MN, when segregated from the rest of the culture, showed more of an adult MN expression profile than if all cells were analyzed in bulk. By correlating only pooled MN expression profiles to our previously characterized data set (Ho et al., 2016), MNs were significantly more correlated to in vivo adult MNs (Figures 3I, S4C-E). By subsetting cells into seven populations, reanalysis of rostrocaudal identity based on HOX gene expression demonstrated that distributions of hindbrain and spinal cord segments are largely consistent across all populations (Figure 3J). Cluster 1, V1 Renshaw, V2a, and V2c populations contained a modest number of cells resembling brachial and thoracic identities. These results highlight the value of scRNA-seq in resolving cell types to enable more accurate measures of similarity between in vitro iPSC-derived models and in vivo cell types.
Pooling sparse transcriptional changes detected by scRNA-seq defines cell type-specific ALS responses
Having defined seven populations, we performed differential gene expression between ALS and control conditions. After dividing each population into ALS and control groups, comparable numbers of cells remained for each condition (Figure 4A), supporting results determined by protein immunostaining for MN markers at this time point (Figure S3B). Tracking scRNA-seq platforms also demonstrated equal representation of ALS and control groups assayed within each platform (Figure 4A). There were sufficient numbers of MNs, V1 Renshaw, and V2a interneurons from each experimental batch to perform differential gene expression analysis, and we focused on analyzing gene expression changes in these populations. The Pearson correlation profiles for these cell types based on 105 marker genes are reasonably distinct (MN LMC vs. V1 Renshaw: 0.38, MN LMC vs. V2a: 0.36, V1 Renshaw vs. V2a: 0.40) (Figure S5B). Conducting comparisons between ALS and control conditions (which included isogenic C9orf72 HRE-corrected lines) yielded genes called significantly differentially expressed (data not shown). However, latent categorical variables such as experimental batch and scRNA-seq technology platform effects mainly drove these differences, illustrating the pitfalls of performing differential gene expression analysis without accounting for these properties. Thus, we next applied a meta-analysis approach by conducting comparisons between ALS and control or isogenic samples within each experimental batch and cataloged genes called significant (Figure 4B and Tables S3-7). For each ALS to control comparison (sporadic ALS samples presented in orange, C9orf72 ALS samples in magenta, control samples in black, and isogenically corrected HRE samples in green), the list of significantly upregulated genes (enumerated in red) were intersected with all other ALS to control comparisons, and the red heatmap indicates the Jaccard index, a measure of overlap between gene sets (Figure 4B). A similar analysis was performed on downregulated genes (enumerated in blue) and presented in the blue heatmap. The number and concordance of genes called significantly dysregulated were highly variable across several comparisons, including repeated comparisons performed between two subject lines across different experimental batches (Figure 4B, 4C, and Table S6). While there is a slight trend in increased Jaccard indices when replicate comparisons are analyzed (Figure 4C), this indicated that despite assaying the same genetic comparisons, batch effects are evident, which may have arisen either by distinct biological responses to repeated differentiation experiments or by distinct technical effects across sample processing, both within and across commercial scRNA-seq platforms. Furthermore, there was low concordance of dysregulated genes when C9orf72 ALS lines were compared directly to their isogenically corrected lines. This observation highlights a challenge in detecting a reproducible gene expression signature of the C9orf72 HRE using scRNA-seq analysis of iPSC models, even when genetic variation is controlled.
Figure 4. ALS enacts distinct gene expression changes in neuronal subtypes.

A. Left to right: same tSNE as in Figure 3G, with each cell colored by ALS or control (CTR) condition, colored by experimental platform, and bar graph quantifying the number of cells assigned to each class.
B. Jaccard indices of overlapping gene sets across ALS to CTR comparisons within the neuronal subtypes MN, V1 Renshaw, and V2a. Each pairwise ALS to CTR comparison is shown along left and top axes, and heatmap indicates Jaccard index for each intersection of gene sets. The number of upregulated genes in each ALS to CTR comparison is printed in red along the right axis, and the number of downregulated genes in each ALS to CTR comparison is printed in blue along the bottom axis. Upregulated gene sets were only compared to other upregulated gene sets, and downregulated gene sets were only compared to other downregulated gene sets. The red and blue heat maps represent the Jaccard indices for intersections of upregulated and downregulated gene sets, respectively. When an ALS cell line was compared to a CTR or isogenic HRE-corrected (ISO) cell line in more than one experimental batch, the Jaccard indices of these gene sets (replicate comparisons) can be compared to Jaccard indices of gene sets calculated for comparisons using the same ALS, CTR, or ISO cells lines within the same experimental batch that were not repeated in other batches (non-replicate comparisons) in C. Panels with solid squares indicate replicate comparisons (n = 7) and open squares indicate non-replicate comparisons (n = 41). These are only indicated for upregulated genes in MNs, but the same conditions were analyzed for downregulated genes and V1 Renshaw and V2a in C.
C. Comparison of upregulated and downregulated genes sets among replicate comparisons and non-replicate comparisons described in B. P-values were calculated using the Wilcox test.
D. Left: Differentially expressed genes between ALS and control conditions for neuronal subtypes MN, V1 Renshaw, V2a, and simulated bulk RNA-seq. Genes were categorized as upregulated or downregulated by ALS, and Chow-Ruskey diagrams present the intersection among each neuronal subtype as well as with bulk comparisons. Right: Euler diagrams present the intersection of GO terms enriched among genes that are reproducibly dysregulated by ALS uniquely within each neuronal subtype in the upregulated or downregulated categories. Representative GO terms are displayed from each overlapping or unique set. See Table S7H-M for full set of uniquely enriched GO terms. No GO terms were significantly enriched among genes upregulated or downregulated by ALS in bulk comparisons, and no GO terms were enriched among genes uniquely downregulated by ALS in V2a interneurons. See Tables S3-7 and Methods for details on how differentially expressed gene and GO lists were generated.
Given the sparseness of genes that were reproducibly dysregulated across experimental batches, we next cataloged and pooled upregulated and downregulated genes called significant in at least two ALS to control or isogenic sample comparisons. This was done for C9orf72 ALS lines (12 comparisons) and sporadic ALS lines (9 comparisons) (Tables S3, S4, and S5). Since our goal was to find early, convergent signatures across familial and sporadic forms of ALS, we respectively compared the extent of overlap between upregulated and downregulated gene sets for each category between C9orf72 ALS and sporadic ALS conditions. Through hypergeometric testing, all comparisons indicated that gene sets cataloged for both ALS conditions overlapped significantly (Figure S6B). We therefore combined the sparse set of differentially expressed genes from C9orf72 ALS lines together with sporadic ALS lines to amass gene sets large enough to pursue subsequent enrichment analyses. To this end, we cataloged and pooled genes called significant in at least two of the 21 ALS to control or isogenic sample comparisons drawn across all scRNA-seq experiments. With this approach, we generated a list of upregulated and downregulated genes for each of these three majority populations in our cultures (Tables S6 and S7A), and we compared these gene lists across all three populations (Figure 4D). Furthermore, we compared these gene lists to differentially expressed genes calculated by bulk analysis of all cells (Table S7A). This comparison demonstrated ALS can induce some overlapping but mostly distinct gene expression changes in each of the three iPSC-derived neuronal populations. Resolving cells into subpopulations was necessary to detect reproducibly disrupted genes, because analysis on the bulk expression profiles of the whole culture did not yield a high number of genes in either upregulated or downregulated categories (Figure 4D and Table S7A).
GO analysis on the entire list of upregulated or downregulated genes from each cell type determined overlapping and distinct GO terms enriched among each list (Figure S6C and Table S7B-G). Analysis on the upregulated and downregulated genes that were unique to each cell type further refined GO terms (Figure 4D and Table S7H-M). Components involved with translation and ribosomal subunits were commonly enriched among upregulated genes in all three neuronal cell types, but functions in cholesterol and isoprenoid synthesis were enriched among genes uniquely upregulated in V1 Renshaw interneurons. While translational components were also enriched among genes downregulated in all three neuronal cell types, components of neuronal processes including dendrite and growth cone were enriched among genes uniquely downregulated in MNs.
ALS iPSC-MN cultures exhibit transcriptional changes detectable in postmortem ALS spinal MNs
We next tested the pathological relevance of these iPSC-MN defined gene sets by examining postmortem, adult spinal MNs. In previous work, we defined 52 co-expression modules using Weighted Gene Co-expression Network Analysis (WGCNA) (Zhang and Horvath, 2005) from laser capture micro-dissected MNs (LC MNs) from postmortem sporadic ALS and control subjects (Ho et al., 2016; Rabin et al., 2010), herein referred to as data set A. Some of these modules significantly correlated or anti-correlated to a principal component that distinguished sporadic ALS from control conditions. We systematically tested whether each list of upregulated or downregulated genes from MNs, V1 Renshaw, and V2a interneurons were enriched in each of the 52 modules (Figure 5A). Markedly, genes upregulated and downregulated by ALS in MNs were significantly enriched among modules that were respectively upregulated and downregulated by sporadic ALS in postmortem spinal MNs. This concordant response to ALS was not observed for V1 Renshaw and V2a interneurons. A repeated analysis between our scRNA-seq data set and an independent but similar postmortem study (Krach et al., 2018), herein referred to as data set B, demonstrated reproducibly concordant gene expression changes (Figure 5B). The robustness of networks defined in each of the postmortem data sets were also examined using module preservation z-statistics (Langfelder et al., 2011), which indicates the likelihood that the network structures of each module occurred by random chance. The most significantly overlapping modules, namely the Magenta, Midnightblue, Blue (Figure 5A), and Darkgreen modules (Figure 5B) possessed some of the most preserved network structures across data sets A and B (Figures S6D and S6E), suggesting they support critical functions in MNs. Dysregulation of these network genes in iPSC-MNs suggests that their disruption by ALS conditions occurs as early as embryonic development.
Figure 5. ALS iPSC-MN cultures exhibit transcriptional changes detectable in postmortem ALS spinal MNs.

A. Hypergeometric test for each of 52 modules defined from WGCNA on LC MNs (data set A) from postmortem sporadic ALS (sALS) and non-ALS subjects (Ho et al., 2016) to detect enrichment for ALS dysregulated genes from each of the neuronal subtype categories identified in Figure 4D. Upper panel: WGCNA modules and the sample traits (sALS disease status and PC1 sALS component) with which they are significantly associated outlined in black rectangles (Pearson correlation between module eigengene and sample trait, Bonferroni-adjusted P-value < 0.01). Z-summary value for each module measures the extent of module preservation between data set A and B (Krach et al., 2018) (Figure 5B). Bar graphs above and to the right indicate the number of genes represented in data set A and scRNA-seq data set, respectively. A matrix of P-values from hypergeometric tests performed for each module to neuronal subtype category comparison were adjusted by the Benjamini–Hochberg method, and subsequent P-values < 0.05 are marked as black squares in the matrix.
B. As in A, except for 32 modules define from data set B (Krach et al., 2018).
C. Euler diagram of intersecting gene sets among the Magenta module from data set A, the Steelblue module from data set B, and the MN upregulated in ALS genes. P-values indicate the likelihood of multi-set intersections using SuperExactTest (Wang et al., 2015). Genes are listed for overlapping sets, bolded genes are associated with the representative GO terms listed below.
D. As in C, except among the Blue module from data set A (left), the Midnightblue module from data set A (right), the Darkgreen module from data set B, and the MN downregulated in ALS genes.
E. Split dot plots indicate percent of MNs within each sample that express a non-zero value of each gene and also indicate the average gene expression among MNs within each sample that express non-zero values of that gene.
F. Scatterplots depict gene expression in each LC MN sample from data set A (y-axis) against the coordinate for that sample along the first principal component (PC1 sALS component, x-axis). Prior to performing PCA on the expression data set, the ten genes shown in this panel were removed from the expression matrix to eliminate autocorrelation. sALS samples are colored red, and control samples are colored blue. The Spearman correlation between expression and PC1 coordinate is indicated next to the gene symbol, and the nominal P-value of the correlation is indicated below the gene symbol.
G. As in F, except applied to data set B.
See also Figure S6.
A closer examination of upregulated genes overlapping among the Magenta module in data set A, the Steelblue module in data set B, and MNs highlighted genes previously implicated in ALS and other motor neuropathies, and the overlapping genes and GO terms enriched among them are consistent with reports of disrupted mRNA and protein processing pathways (Deshaies et al., 2018; Kim et al., 2013, 2008; Montibeller and de Belleroche, 2018) (Figure 5C). Similarly, examination of downregulated genes overlapping among the Blue and Midnightblue modules in data set A, the Darkgreen module in data set B, and MNs highlighted genes previously implicated in ALS (Lederer et al., 2007; Saris et al., 2009; Umahara et al., 2016) (Figure 5D). The GO term regulation of neuronal projection development was significantly represented among the overlapping, downregulated genes (Figure 5D), consistent with recent models suggesting that deficiencies in maintaining axonal projections may underlie ALS (Klim et al., 2019; Melamed et al., 2019).
Auditing the average expression as well as percent expression of these overlapping genes in MNs demonstrated their dysregulation in ALS conditions (Figure 5E). Neuronatin (NNAT), which has been associated with neuronal development as well as degeneration (Joseph, 2014), was upregulated in ALS MNs in the greatest number of ALS to control comparisons (Table S6A) while not observed as belonging to any modules significantly associated with sporadic ALS in postmortem data sets. Auditing the expression of ten overlapping genes in LC MNs from data sets A and B demonstrated high correlation between their expression and the first principal component that distinguishes sporadic ALS from control conditions (Figures 5F and 5G), further supporting the efficacy and fidelity of our discovery approach. A deeper investigation into the module genes disrupted in sporadic ALS conditions revealed a significant enrichment of module genes previously associated with spinal MN maturation and aging (Ho et al., 2016) (Figure 6). Genes involved with neurite growth, axon guidance, and neurotransmitter release, which were classified into co-expression modules that significantly correlated with spinal MN maturation and aging were also found to be downregulated in postmortem, sporadic ALS conditions (Ho et al., 2016). Since these genes were observed as downregulated in iPSC-MNs from ALS subjects, this suggests that disruptions to homeostatic processes that occur after fetal developmental stages are already occurring in MNs and thereby priming them for disease during later stages of life. Altogether, these data indicate that scRNA-seq analysis of iPSC-MNs can detect early ALS-signatures affecting important maturation and age-related gene expression networks whose disruption can possibly lead to MN degeneration.
Figure 6. ALS iPSC-MNs exhibit early transcriptional changes counteracting homeostatic maturation and aging.

A. Euler diagram of intersecting gene sets among the Magenta, Blue, Midnightblue, or Yellow modules from data set A, the MN dysregulated genes in ALS identified in Figure 4D, and genes assigned to modules that significantly correlate with MN maturation and aging (Ho et al., 2016). The number of genes within each set is indicated. P-values indicate the likelihood of multi-set intersections using SuperExactTest (Wang et al., 2015). Genes with pathogenic variants in the ClinVar database are indicated on the right.
B. Heatmap of genes listed in A demonstrating expression kinetics as tissues progress from embryonic, fetal, and adult spinal cord tissues (Ho et al., 2016). Pluripotent stem cells (PSCs) include embryonic stem cells and iPSCs.
Predictive ALS markers are detectable in iPSC-MNs
While pooling of sparsely dysregulated genes in iPSC-MNs enriched for concordantly dysregulated genes in postmortem MNs, the average expression of these genes in each cell and the percent of cells expressing each gene varied considerably across subject lines (Figure 5E). This demonstrated a challenge in discovering consistently dysregulated genes by applying a significance threshold on a sample to sample basis across many scRNA-seq samples. We therefore took an alternative approach to discover genes that are consistently altered in iPSC-MNs from ALS subjects. We considered a combined expression score that reflected the average expression and percent expression for each gene in the MN populations at day 18 per subject (n = 22) in the scRNA-seq data set (see Methods). We then performed t-tests comparing combined expression scores between all ALS and control and isogenic samples and ranked them by increasing nominal p-values. Among the top 20 ranking genes, we found six genes were concordantly downregulated in ALS conditions in data sets A and B, and they exhibited more uniform downregulation in ALS iPSC-MNs compared to controls (Figure 7A). We found no genes concordantly upregulated in all three data sets. Observing expression kinetics of these genes over the course of embryonic, fetal, and adult spinal cord tissues (Ho et al., 2016) showed that some positively correlated with spinal MN maturation (ADCYAP1, ELAVL3, and NUAK1), DNMT3B anti-correlated with spinal MN maturation, and NDUFAF5 as well as NNAT were upregulated during fetal spinal cord stages (Figure 7B).
Figure 7. Single-cell analysis of iPSC-MNs reveals predictive ALS marker genes.

A. Split dot plots indicate percent of MNs within each sample that express a non-zero value of each of six predictive marker genes. Plots also indicate the average gene expression among MNs within each sample that express non-zero values of that gene.
B. Heatmap of five genes listed in A, along with NNAT demonstrating expression kinetics as tissues progress from embryonic, fetal, and adult spinal cord tissues (Ho et al., 2016). CARS2 was not annotated in this data set. Pluripotent stem cells (PSCs) include embryonic stem cells and iPSCs.
C - G. Receiver-operator characteristic (ROC) analysis performed on several data sets classifying samples in each data set as ALS or non-ALS, based on sample coordinates along the first, second, or both principal components using six predictive marker genes. See methods for calculations. P-values for each area under the curve (AUC) are calculated using the Mann-Whitney U test to determine whether the AUC differs significantly from 0.5 (diagonal grey line), which indicates an uninformative test. The number of samples for each ALS and non-ALS case used in the analysis, along with their genotypes are indicated. The number of genetically distinct subjects included in the analysis are indicated in parentheses. Familial ALS subjects with pathogenic variants in C9orf72 (C9O), CHMP2B (CHM), FUS, SOD1 (SOD), TARDBP (TDP). Sporadic ALS subjects with no known pathogenic variants (SPO). Non-ALS control subjects (CTR). C9orf72 or SOD1 subject lines in which the pathogenic variant has been genome edited (ISO). Spinal muscular atrophy subjects classified as non-ALS (SMA). C. ROC analysis performed on subpopulation data generated from scRNA-seq data set based on a combined metric of average and percent expression for the six predictive marker genes. D. ROC analysis performed on LC MN expression data sets (Cox et al., 2010; Highley et al., 2014; Kirby et al., 2011; Krach et al., 2018; Rabin et al., 2010). E. ROC analysis performed on mouse LCM MNs (Nardo et al., 2013). F. ROC analysis performed on bulk RNA-seq data sets generated by the NeuroLINCS Consortium for undifferentiated iPSCs (day 0), two MN differentiation protocols at days 18 and 90, and iPSC-MNs from Fujimori et al., 2018; Kiskinis et al., and 2014; Shi et al., 2018. G. ROC analysis performed on bulk proteomics data sets generated by the NeuroLINCS Consortium for two MN differentiation protocols at days 18 and 90. ELAVL3 protein expression is used as the prediction.
H - J. Representative images of lumbar spinal cord sections from control, sporadic, and C9orf72 ALS subjects immunostained for ELAVL3 and counterstained with hematoxylin. Scale bar, 2.5 mm for H, I, and J; 250 μm for inset images H’, I’, and J’.
K. Comparative distributions of ELAVL3 optical densities.
See also Figure S7.
The classification accuracy of ALS cases versus controls, as measured by the area under the curve (AUC), using PCA based on these six genes was significant in the MN population (Figures 7C and S7A). However, classification accuracy of ALS cases versus controls was not significant in V1 Renshaw, V2a, or by using bulk expression data (Figures 7C and S7A). Classification of sporadic ALS cases versus control postmortem adult spinal MNs in data set A (Rabin et al., 2010) and data set B (Krach et al., 2018) was also significant (Figure 7D and S7B). These results were expected, because the classifier genes were defined by these data sets. However, validating the accuracy of these six genes in classifying external test data sets would underscore their predictive power. In separate test data sets of postmortem adult spinal MNs from familial and sporadic ALS cases, which include variants in C9orf72, CHMP2B, and SOD1 (Cox et al., 2010; Highley et al., 2014; Kirby et al., 2011), classification using these genes significantly distinguished ALS from control subjects (Figures 7D and S7B). Additionally, in a disease progression study of SOD1G93A transgenic mouse spinal MNs (Nardo et al., 2013), classification of ALS versus control conditions based on these genes increased accuracy as mice progressed to disease endstage (Figures 7E and S7C). We also focused analysis on these genes from the NeuroLINCS Consortium bulk RNA-seq data set (Keenan et al., 2018), which analyzed undifferentiated human iPSCs and iPSCs differentiated into MN cultures over 18 days. This demonstrated that ALS could not accurately be distinguished from control conditions (Figure 7F and S7D). However, using a longer iPSC-MN differentiation protocol (Sareen et al., 2013) where cultures were extended up to 90 days and again profiled by bulk RNA-seq, analysis of these six genes demonstrated a significant accuracy in classifying ALS cases from control as well as from spinal muscular atrophy cases (Figures 7F and S7D). Additionally, these signature genes could distinguish SOD1A4V ALS patient samples from zinc-finger nuclease corrected isogenic samples in iPSC-derived, HB9-RFP positive MNs at 39 days of differentiation (Kiskinis et al., 2014) (Figures 7F and S7D). Similarly, these genes also distinguished C9orf72 ALS patient derived, HB9-RFP positive MNs from control samples, and further distinguished isogenic control samples in which one or two copies of the C9orf72 HRE were targeted into the genome with CRISPR-Cas9 (Shi et al., 2018) (Figure 7F). Finally, this panel of genes distinguished control subject iPSC-MN cultures from sporadic and familial ALS subjects, including those with variants in FUS, SOD1, and TARDBP (Fujimori et al., 2018) (Figures 7F and S7B). Among the six genes quantifiable by RNA in all expression data sets tested, ELAVL3 was the only gene quantifiable as a protein when analyzing the NeuroLINCS proteomics data sets at 18 and 90 days of differentiation. Classification of iPSC-MN cultures based solely on ELAVL3 protein expression demonstrated that it was an accurate classifier for ALS versus control only in extended cultures at 90 days, where it was also decreased in ALS conditions (Figure 7G). However, analysis of ELAVL3 RNA alone showed less overall accuracy when compared to joint RNA analysis of all six genes (Figures S7E-H). Lastly, decline in ELAVL3 protein per MN was also detectable in postmortem spinal cords in both sporadic and C9orf72 ALS cases versus control (Figures 7H-7K). Altogether, these data reveal that despite globally resembling in vivo fetal tissue, single-cell analysis of iPSC-MNs can model early, common signatures of familial and sporadic ALS that persist into the endstage of disease.
Discussion
Recent scRNA-seq studies have characterized diverse neuronal populations in in vivo mouse spinal cords (Delile et al., 2019; Sathyamurthy et al., 2018). However, scRNA-seq has not been used to rebuild the spinal cord from complex cell mixtures in cultures differentiated from iPSCs. Our approach described here is ideally suited to achieve this goal and demonstrates initial steps toward building a human iPSC-based cellular atlas of the developing hindbrain and spinal cord to provide an anatomical context for human embryonic development as well as disease modeling. This anatomical classification lays the foundation for future work with iPSC models to investigate instrinsically different physiologies across regions of the hindbrain and spinal cord.
As variable molecular readouts caused by genetic background is becoming increasingly acknowledged by experimentalist in human iPSC disease modeling, experimental design must account for the genetic backgrounds of several individual subjects as well as isogenic controls in order to isolate reproducible disease-related effects (Fujimori et al., 2018; Kiskinis et al., 2014; Shi et al., 2018). In line with this outlook, we incorporated iPSC lines from several ALS and control subjects and repeatedly assayed MN differentiations , aiming to detect reproducible transcriptional signatures in distinct cellular subpopulations. However, repeated experimental sampling presented the challenge of coping with batch effects, which in the process of scRNA-seq analysis, severely affected global clustering approaches towards cell type annotation such as Louvain community detection and tSNE dimensionality reduction (Hicks et al., 2018; Luecken and Theis, 2019). We alleviated these effects through MultiCCA (Butler et al., 2018) and other batch correction methods. This shows the feasibility of integrating scRNA-seq data generated from iPSC models across several experimental batches and platforms, demonstrating a suitable approach for consortia-driven projects.
Recent iPSC-based transcriptomic reports performed RNA profiling at time points during differentiation concomitant with various observed ALS phenotypes which include nuclear RNA foci (Sareen et al., 2013), decreased neurite length (Fujimori et al., 2018), reduced neurite repair after injury (Klim et al., 2019; Melamed et al., 2019), and MN death (Kiskinis et al., 2014; Shi et al., 2018). Several of these protocols differentiated iPSCs for over 30 days, and many required a relatively prolonged maturation phase, the presence of glia, and additional stressor conditions in order to provoke a disease phenotype. Thus, it is unclear whether the transcriptional events observed precede the disease phenotypes, are concomitant, or are immediate consequences of other prior events. We elected to profile transcription in postmitotic MNs at an earlier point, day 18 at which their identity was established and in the absence of glial cells. This event was demonstrated as early as day 14 of differentiation (Maury et al., 2015). Our approach satisfied two objectives. One was to capture a transcriptional signature as early as possible, prior to the manifestation of disease phenotypes ranging from neurite repair to overt cell death. The other was to reduce heterogeneity across subject lines and experimental batches that could be augmented by a longer time of differentiation in culture. Within this early developmental time point, we detected common signatures across familial and sporadic ALS conditions prior to disease phenotypes, suggesting that these transcriptomic events precede and are potentially causative of later phenotypes. In light of our findings altogether, there may nevertheless be other differentiation time points that can exhibit more prominent differentially expressed genes in ALS.
Future iPSC-based studies that distinguish bulbar from spinal onset ALS patients can build upon the data reported here to help correlate region of onset in the patient with the pathology in specific MNs associated with those regions. Our anatomical assessment of iPSC-MN models establishes a cellular and molecular framework to address how MN degeneration and paralysis spreads throughout the body of ALS patients, mechanisms which are of great interest to develop accurate prognostic assessments or interventional therapies (Turner et al., 2010). While the fidelity of our iPSC-MNs to in vivo MNs was based on pooled LC MNs, recent advances in single nuclei RNA-seq of human postmortem tissues of the central nervous system (Gaublomme et al., 2019; Mathys et al., 2019) will provide an expanded resolution of cellular and disease signatures with which our data can be reconciled. This comparison will enable better interpretation of molecular signatures and cellular compositions as they arise in early stages of ALS and progress into the endstages of ALS, thus enabling a better understanding of disease etiology. Finally, the analysis reported here provides a methodological resource for iPSC-based disease models of not only ALS, but also for several other late onset diseases standing to benefit from single-cell resolved investigations.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Clive N. Svendsen (clive.svendsen@cshs.org).
MATERIALS AVAILABILITY
iPSC lines generated in this study are listed in Table S1 and are available through the Cedars-Sinai Biomanufacturing Center.
DATA AND CODE AVAILABILITY
The scRNA-seq source data have been deposited at Gene Expression Omnibus and are publicly available under the accession number: GSE138121.
The original codes used for the analyses reported in this study are publicly available at https://github.com/ritchieho/2020_scRs_iPSC_ALS.
The scripts used to generate the figures reported in this paper are available at https://github.com/ritchieho/2020_scRs_iPSC_ALS.
Any additional information required to reproduce this work is available from the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All human iPSC lines are banked and available through the Cedars-Sinai Biomanufacturing Center. Cell lines were routinely characterized through mycoplasma testing, Alkaline Phosphatase staining, immunostaining for pluripotency markers, karyotyping by G-banding, PluriTest, Trilineage Differentiation Potential (assessed via TaqMan hPSC Scorecard Assay), and Cell Line Authentication (assessed via STR Analysis) to match primary donor tissue. Relevant clinical and experimental data about iPSC donor subjects (e.g. age, sex, tissue source) are presented in Table S1 and in the Key Resources Table. All protocols were performed in accordance with the Institutional Review Board guidelines at Cedars-Sinai Medical Center under the auspices of IRB-SCRO Protocol 21505.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal IgG anti-Human ISL1 | R&D Systems | AF1837; RRID: AB_2126324 |
| Mouse monoclonal IgG1 anti-NF-H (SMI-32) | BioLegend | 801701; RRID: AB_2564642 |
| Rabbit polyclonal IgG anti-PHOX2B | GeneTex | GTX109677; RRID: AB_1951223 |
| Rabbit polyclonal IgG anti-Human ELAVL3/HUC (for immunohistochemistry staining) | LSBio | LS-C408905-50 |
| Rabbit polyclonal IgG anti-CHX10 (VSX2) | Novus | NBP1-84476; RRID: AB_11022841 |
| Rabbit polyclonal IgG anti-SOX1 [EPR4766] | GeneTex | GTX62974 |
| Goat polyclonal anti-ChAT | Millipore | AB144P; RRID: AB_2079751 |
| Rabbit polyclonal Poly(GP) | N/A | Rb9259 |
| Biological Samples | ||
| Human lumbar spinal cord tissue sections | UC San Diego | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| mTeSR1 | StemCell Technologies | 85850 |
| DMEM | Thermo Fisher Scientific | 11995081 |
| IMDM | Thermo Fisher Scientific | 12440061 |
| F12 | Thermo Fisher Scientific | 11765062 |
| Neurobasal medium | Thermo Fisher Scientific | 21103049 |
| B27 (+vitamin A) | Thermo Fisher Scientific | 17504044 |
| N2 | Thermo Fisher Scientific | 1780240 |
| NEAA | Thermo Fisher Scientific | 1114050 |
| GlutaMax | Life Tech | 35050061 |
| PSA | Thermo Fisher Scientific | 15240062 |
| D-(+)-Glucose | Sigma | G7021 |
| Y-27632 dihydrochloride | Sigma | Y0503 |
| Y-27632 dihydrochloride | Tocris | 1254 |
| CHIR99021 | Xcess Biosciences | M60002 |
| LDN193189 | Selleck | S2618 |
| SB431542 | Stemgent | 04-0010-10 |
| SB431542 | Cayman Chemicals | 13031 |
| Dorsomorphin | Sigma | P5499 |
| SAG | Sigma | 566660 |
| SAG | Cayman Chemicals | 11914 |
| Retinoic acid | Sigma | R2625 |
| All-trans retinoic acid | Stemgent | 040021 |
| BDNF | R&D | 248-BDB-005 |
| BDNF | Peprotech | 45002 |
| EGF | Peprotech | AF-100-15 |
| FGF2 | Peprotech | 100-18B |
| GDNF | Peprotech | 45010 |
| Ascorbic acid | Millipore | A4403 |
| Compound E | Calbiochem | 565790 |
| DAPT | Cayman Chemicals | 13917 |
| Ara-C | Sigma | C1768 |
| db-cAMP | Millipore | 28745 |
| Purmorphamine | Millipore | 540220 |
| Accutase | Millipore | SCR005 |
| Versene | Life Technologies | 15040-066 |
| Trypsin-EDTA solution | Sigma | T4049 |
| Laminin (mouse) | Millipore | L2020 |
| Poly-ornithine | Sigma | P4638 |
| Matrigel (growth factor reduced) | Corning | 354230 |
| Triton X-100 | Sigma | X100 |
| Tween-20 | Sigma | P1379 |
| Betaine hydrochloride | Millipore | B3501-100G |
| Diethyl pyrocarbonate | Sigma | D5758 |
| ProLong™ Gold Antifade Mountant | Thermo Fisher Scientific | P36930 |
| Citrisolv | Thermo Fisher Scientific | 04-355-121 |
| Antigen Unmasking Solution, Tris-Based | Vector Laboratories | HH-3301 |
| FBS | Atlanta Biologicals | 511150 |
| Hematoxylin | Thermo Fisher Scientific | HHS128 |
| Critical Commercial Assays | ||
| SureCell WTA 3’ Library Prep Kit for the ddSEQ System | Illumina | 20014280 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10X Genomics | PN-120237 |
| Chromium Single Cell A Chip Kit | 10X Genomics | PN-120236 |
| Chromium i7 Multiplex Kit | 10X Genomics | PN-120262 |
| FastStart™ PCR Master | Sigma | 4710436001 |
| TOPO™ TA Cloning™ Kit for Sequencing | Sigma | K457501 |
| PrimeSTAR® HS DNA Polymerase (premix) | Takara | R040A |
| Papain Dissociation System | Worthington | LK003150 |
| PureLink RNA Mini Kit | Thermo | 12183018A |
| Promega Reverse Transcription System | Promega | A3500 |
| ImmPRESS® HRP Horse Anti-Rabbit IgG Polymer Detection Kit, Peroxidase | Vector Laboratories | MP-7401 |
| ImmPACT® DAB Substrate, Peroxidase (HRP) | Vector Laboratories | SK-4105 |
| Deposited Data | ||
| Single cell RNA sequencing data for iPSC-MN cultures | This study, Gene Expression Omnibus | GSE138121 |
| Experimental Models: Cell Lines | ||
| 0083_CTR_CTR | Cedars-Sinai Biomanufacturing Center | CS83iCTR-33nxx |
| 0179_CTR_CTR | Cedars-Sinai Biomanufacturing Center | CS0179iCTR-nxx |
| 0025_CTR_CTR | Cedars-Sinai Biomanufacturing Center | CS25iCTR-18nxx |
| 0465_CTR_CTR | Cedars-Sinai Biomanufacturing Center | EDi034-A |
| 0028_C9O_ALS | Cedars-Sinai Biomanufacturing Center | CS28iALS-C9nxx |
| 0029_C9O_ALS | Cedars-Sinai Biomanufacturing Center | CS29iALS-C9nxx |
| 0029_ISO_CTR | Cedars-Sinai Biomanufacturing Center | CS29iALS-C9n1.ISOxx |
| 0052_C9O_ALS | Cedars-Sinai Biomanufacturing Center | CS52iALS-C9nxx |
| 0052_ISO_CTR | Cedars-Sinai Biomanufacturing Center | CS52iALS-C9n6.ISOxx |
| 6ZLD_C9O_ALS | Cedars-Sinai Biomanufacturing Center | CS6ZLDiALS-nxx |
| 2XWC_SPO_ALS | Cedars-Sinai Biomanufacturing Center | CS2XWCiALS-nxx |
| 8BRM_SPO_ALS | Cedars-Sinai Biomanufacturing Center | CS8BRMiALS-nxx |
| Oligonucleotides | ||
| C9orf72 Sanger sequencing primer forward: AAAGAACAGGACAAGTTGCCCCGCC | Sigma | N/A |
| C9orf72 Sanger sequencing primer reverse: GCAGGCACCGCAACCGCAG | Sigma | N/A |
| C9orf72 repeat primed PCR anchor (forward): TACGCATCCCAGTTTGAGACG | Sigma | N/A |
| C9orf72 repeat primed PCR repeat-plus-anchor (forward) TACGCATCCCAGTTTGAGACGGGGGCCGGGGCCGGGGCCGGGG | Sigma | N/A |
| C9orf72 repeat primed PCR rev-plus-6FAM (reverse) 6-FAM-AGTCGCTAGAGGCGAAAGC | Sigma | N/A |
| C9orf72 total qPCR primer forward: CAGTGATGTCGACTCTTTG | Sigma | N/A |
| C9orf72 total qPCR primer reverse: AGTAGCTGCTAATAAAGGTGATTTG | Sigma | N/A |
| C9orf72 TV2 qPCR primer forward: CGGTGGCGAGTGGATATCTC | Sigma | N/A |
| C9orf72 TV2 qPCR primer reverse: TGGGCAAAGAGTCGACATCAC | Sigma | N/A |
| C9orf72 TV3 qPCR primer forward: GTGTGGGTTTAGGAGATATC | Sigma | N/A |
| C9orf72 TV3 qPCR primer reverse: TGGGCAAAGAGTCGACATCAC | Sigma | N/A |
| RPL13A qPCR primer forward: CCTGGAGGAGAAGAGGAAAGAGA | Sigma | N/A |
| RPL13A qPCR primer reverse: TTGAGGACCTCTGTGTATTTGTCAA | Sigma | N/A |
| C9orf72 sense FISH probe | Product #500150, Exiqon Inc. Woburn, MA, USA | 5TYE563/CCCCGGCCCCGGCCCC |
| C9orf72 antisense FISH probe | Product #500150, Exiqon Inc. Woburn, MA, USA | 5TYE563/GGGGCCGGGGCCGGGG |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) plasmids | Addgene | plasmid # 48138; RRID: Addgene_48138 |
| Software and Algorithms | ||
| RStudio | N/A | https://rstudio.com |
| Seurat version 2.3.0 | Butler, et al., 2018. PMID: 29608179 | https://github.com/satijalab/seurat/releases/tag/v2.3.0 |
| Monocle 2.12.0 | Qiu et al., 2017. PMID: 28114287 | http://cole-trapnell-lab.github.io/monocle-release/docs |
| Illumina bcl2fastq | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Illumina Single-Cell RNA Seq BaseSpace Workflow (v1.0.0) | Illumina | https://basespace.illumina.com |
| STAR Aligner v2.5.1 and v2.5.2b | Dobin et al., 2013. PMID: 23104886 | https://github.com/alexdobin/STAR |
| 10X Genomics Cell Ranger (v2.1.0) | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression |
| Fiji ImageJ | Schindelin, Arganda-Carreras, and Frise et al., 2012. PMID: 22743772 | https://fiji.sc |
| DESeq | Anders and Huber, 2010. PMID: 20979621 | http://bioconductor.org/packages/release/bioc/html/DESeq.html |
| Vennerable | N/A | https://github.com/js229/Vennerable |
| WGCNA | Langfelder and Horvath, 2008. PMID: 19114008 | https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA |
| SuperExactTest | Wang, Zhou, and Zhang, 2015, PMID: 26603754 | https://github.com/mw201608/SuperExactTest |
| ROCR | Sing et al., 2005. PMID: 16096348 | https://rocr.bioinf.mpi-sb.mpg.de |
| psych 1.9.12.31 | Revell, 2019. | https://www.rdocumentation.org/packages/psych/versions/1.9.12.31 |
| Seurat Wrappers version 0.2.0 | N/A | https://github.com/satijalab/seurat-wrappers |
| Harmony | Korsunsky et al., 2019. PMID: 31740819 | https://github.com/immunogenomics/harmony |
| kBET version 0.99.6 | Büttner et al., 2019. PMID: 30573817 | https://rdrr.io/github/theislab/kBET |
| Custom R scripts | This study | https://github.com/ritchieho/2020_scRs_iPSC_ALS |
METHODS DETAILS
Culture of human iPSCs
All iPSC lines were maintained in complete mTeSR1 growth medium on Growth Factor Reduced Matrigel and passaged every seven days using the StemPro EZ Passaging Tool or Versene and typically split between 1:4 and 1:9 ratios.
Genome editing of C9orf72 repeats in iPSCs
CRISPR guides were designed to target regions immediately 5’ and 3’ of the C9orf72 hexanucleotide repeat expansion using the Zhang lab CRISPR design tool (Shi et al., 2018). Guides were cloned into pSpCas9(BB)-2A-GFP (PX458) plasmids, (gifted from Feng Zhang, Addgene plasmid #48138). Each iPSC line was transfected with both 5’ and 3’ targeting plasmids using the Neon Electroporation System (Thermo Fisher). After 48 hours, iPSCs were dissociated and flow sorted by GFP fluorescence to isolate successfully transfected cells. These cells were plated, cultured for 1 week, passaged, and allowed to grow to confluency. Cells were then subcloned as follows: iPSCs were Accutase-dissociated into single cells and replated sparsely at 30,000 cells/10 cm dish. Rock inhibitor (Y-27632) was included for 24 hours after plating to promote iPSC survival. Once individual cells formed small colonies, pipette tips were used to manually transfer subclones from the 10 cm dish into individual wells of a 96 well plate. These subclones were passaged with Versene into two 96 well plates, one for further propagation and one for gDNA extraction and sequencing. The C9orf72 locus was PCR-amplified using PrimeStar Polymerase with 1M betaine. To determine the sequence of each allele, PCR products were cloned using the TOPO Cloning Kit for Sequencing (Invitrogen). Plasmids were used to transform TOP10 competent bacteria, which were plated on agar dishes containing ampicillin and incubated at 37°C overnight. Individual colonies were transferred to a new agar dish containing ampicillin and an indexed grid with a clean pipette tip and grown overnight, and these plates were sent to Genewiz to perform direct colony sequencing through rolling circle amplification. Subclones lacking the C9orf72 HRE sequence were expanded and characterized.
Repeat primed PCR assay for HRE
100 ng of genomic DNA template was amplified using FastStart Master Mix (Roche), 1M betaine (Sigma), 7% DMSO (Sigma), 0.18 mM 7-deaza-dGTP (New England Biolabs), 0.9 mM magnesium chloride (Sigma), 1.4 μM C9orf72 repeat primed PCR anchor (forward primer), 0.7 μM C9orf72 repeat primed PCR repeat-plus-anchor (forward primer), and 1.4 μM C9orf72 repeat primed PCR rev-plus-6FAM (reverse primer) using the following cycling conditions: 1x 95°C for 15 min, 2x 94°C for 1 min -> 70°C for 1 min -> 72°C for 3 min, 3x 94°C for 1 min -> 68°C for 1 min -> 72°C for 3 min, 4x 94°C for 1 min -> 66°C for 1 min -> 72°C for 3 min, 5x 94°C for 1 min -> 64°C for 1 min -> 72°C for 3 min, 6x 94°C for 1 min -> 62°C for 1 min -> 72°C for 3 min, 7x 94°C for 1 min -> 60°C for 1 min -> 72°C for 3 min, 8x 94°C for 1 min -> 58°C for 1 min -> 72°C for 3 min, 5x 94°C for 1 min -> 56°C for 1 min -> 72°C for 3 min, and 1x 72°C for 10 min. PCR products were then sent to Genewiz for fragment analysis.
Karyotype
All patient-derived and control iPSC lines are routinely authenticated for cytogenetic integrity by G-band karyotype chromosomal analysis. For every iPSC line, we store early passage seed banks, from which subsequent distribution banks of iPSC lines can be generated. Analysis is performed to confirm a normal karyotype before a distribution bank is used. Specifically, the 0029 and 0052 gene-edited lines were confirmed to have normal karyotype three separate times at different passages. G-band karyotyping is performed by the Cedars-Sinai Clinical Cytogenetics Core for Cytogenetic analysis using G-banding at the 425-475 band level of resolution on slides of cultured iPSCs. For each karyotype per cell line, metaphase spreads of typically 20 cells are counted with their chromosomal complement. The cytogeneticist reviews whether any consistent numerical or structural abnormality is observed. A consistent numerical or structural abnormality that is observed in greater than one cell is classified as an abnormal karyotype for the iPSC culture.
Differentiating iPSC-MN cultures
For Figures S2A-E, iPSC-MNs were differentiated as previously described (Yang et al., 2013). In brief, iPSCs were dissociated into single cells, cultured in Neural Induction Media (NIM) consisting of Neurobasal (Gibco), 1.1 μM ascorbic acid (Sigma), 1% non-essential amino acids (NEAA) (Gibco), 1% GlutaMax (Gibco), 2% B27 (Gibco), 0.16% D-glucose solution, and 1% Penicillin-Streptomycin-Amphotericin (PSA) solution. 10 μM Y-27632 ROCK inhibitor (Sigma) was included in the media or the first 48 hours to improve survival of iPSCs following dissociation. On days 1-4, NIM was supplemented with 10 μM SB431542 (StemGent), 1μM Dorsomorphin (Sigma), and 10 ng/mL bFGF (PeproTech). This media was changed every other day, and bFGF was replenished daily. On day 5, cells were cultured in NIM supplemented with SB431542, Dorsomorphin, 10 ng/mL BDNF (R&D), and 1 μM retinoic acid (Sigma). On days 7 and 9, the media was changed to NIM with BDNF, retinoic acid, and 1 μM smoothened agonist (Sigma). The cells were densely plated onto poly-ornithine/laminin coated dishes and cultured in the same media on day 11. This media was further supplemented with 2 μM DAPT (Cayman Chemicals) on days 13-20, with media changes every 2-3 days. On days 20-30, cells were fed every 2-3 days with NIM containing BDNF, smoothened agonist, 1 μM retinoic acid, and 2 μM Ara-C (Sigma). Cells were then gently dissociated using papain (Worthington), plated on poly-ornithine/laminin coated dishes, and cultured in NIM with the addition of 1% N2 supplement (Gibco), 4 μM Ara-C, and 40 ng/mL each of growth factors BDNF and GDNF (PeproTech). For Figures S2F and S2G, iPSC-MN cultures were differentiated using the 18 day protocol as described below for polyGP ELISA.
For the 18 day iPSC-MN differentiation, mTeSR1 was removed from iPSCs at 30-40% confluency and replaced with Stage 1 media (1:1 mixture of Iscove's Modified Dulbecco's Medium (IMDM):F12 basal media supplemented with 1% NEAA, 2% B27, 1% N2, 1% PSA, 0.2 μM LDN193189 (Selleck), 10 μM SB431542, and 3 μM CHIR99021 (Xcess Biosciences)) for six days with daily media changes. The cells were then Accutase-treated to single-cell suspension and centrifuged in 50 ml conical tubes, resuspended in Stage 2 media (Stage 1 media further supplemented with 0.1 μM all-trans retinoic acid (Stemgent) and 1 μM Sonic hedgehog agonist (SAG) (Cayman Chemicals)), and plated onto Matrigel-coated plates or coverslips. Stage 2 media was changed every two days until day 12, when Stage 3 media (1:1 mixture of IMDM:F12 basal media supplemented with 1% NEAA, 2% B27, 1% N2, 1% PSA, 0.1 μM Compound E (Calbiochem), 2.5 μM DAPT, 0.1 μM dibutyryl cyclic adenosine monophosphate (db-cAMP), 0.5 μM all-trans retinoic acid, 0.1 μM SAG, 200 ng/ml ascorbic acid, 10 ng/ml BDNF, and 10 ng/ml GDNF) was then used to feed cells every two days until day 18, when cultures were analyzed.
The 90 day iPSC-MN differentiation was performed as previously described (Ho et al., 2016). In brief, 80% confluent iPSC cultures were Accutase-treated into single cells suspension and centrifuged in 384-well Matrigel coated PCR plates. The cells were maintained in Neural Differentiation Media (NDM): IMDM/F12 supplemented with 2% B27-vitamin A , 1% N2, 1% NEAA, 0.2 μM LDN193189, and 10 μM SB431542. On day two, neural aggregates were collected and transferred into Poly-Hema coated T75 flasks and the aggregates were cultured for three more days in NDM. On day seven, aggregates were collected and transferred onto poly-ornithine/laminin coated wells with fresh NDM. After five days, cells were cultured in MN Specification Media: NDM supplemented with 0.25 μM all-trans retinoic acid, 1 μM purmorphamine, 1 μM db-cAMP, 200 ng/mL ascorbic acid, 20 ng/mL BDNF, and 20 ng/mL GDNF. Once rosettes were observed, they were collected with STEMdiff Neural Rosette Selection Reagent and cultured in MN Precursor Expansion Media: NDM supplemented with 0.1 μM all-trans retinoic acid, 1 μM purmorphamine (Millipore), 100 ng/mL EGF, and 100 ng/mL FGF2. After day 26, the iPSC-MN precursor spheres (iMPS) are expanded by using a chopping method every seven to ten days. The iMPS are matured into MNs for 21 days in MN Maturation Media: Neurobasal supplemented with 1% NEAA, 0.5% Glutamax, 1% N2, 10 ng/ml BDNF, 10 ng/ml GDNF, 200 ng/ml ascorbic acid, 1 μM db-cAMP and 0.1 μM all-trans retinoic acid.
Immunofluorescent staining, imaging, and quantification of iPSC-MN cultures
iPSC-MNs were fixed in 4% paraformaldehyde, rinsed with PBS, incubated in 0.5% Triton-X in PBS, rinsed with 0.2% Tween-20 in PBS, incubated in blocking solution (5% normal donkey serum and 0.2% Tween-20 in PBS). Primary antibody solution in blocking solution containing various combinations of goat polyclonal IgG anti-Human ISL1 (1:200) ( R&D Systems AF1837, RRID: AB_2126324), mouse monoclonal IgG1 anti-NF-H (SMI-32) (1:200) (BioLegend 801701, RRID: AB_2564642), goat polyclonal anti-ChAT (1:200) (Millipore AB144P, RRID: AB_2079751), rabbit polyclonal IgG anti-PHOX2B (1:200) (GeneTex GTX109677, RRID: AB_1951223), mouse monoclonal IgG2b, rabbit polyclonal IgG anti-CHX10 (VSX2) (1:200) (Novus NBP1-84476, RRID: AB_11022841), and rabbit polyclonal IgG anti-SOX1 [EPR4766] 1:200) (GeneTex GTX62974) were incubated, rinsed with 0.2% Tween-20 in PBS, and incubated in species-specific Alexa-fluor secondary antibodies (1:2,000), and rinsed with 0.2% Tween-20 in PBS with DAPI staining. Fluorescent images were acquired using ImageXpress Micro XLS system (Molecular Devices) at 10X magnification. For a complete analysis, total 9 sites per well were captured. The captured images were quantified for the cellular population using MetaXpress software (Molecular Devices).
Quantification of C9orf72 transcript variants
RNA was extracted from iPSCs using the PureLink RNA mini Kit (Invitrogen) and reverse-transcribed into cDNA using the Promega Reverse Transcription System. Quantitative PCRs were conducted in triplicate using SYBR Green and primers amplifying all C9orf72 transcripts as well as specific transcript variants. PCR cycles consisted of the following steps: [1x 95°C for 10 min, 40x 95°C for 30 seconds -> 58°C for 60s, and 1x 72°C for 5 min].
FISH of C9orf72 sense and antisense RNA foci and imaging
RNA FISH was performed as previously described in (Sareen et al., 2013). Briefly, cells were cultured on chamber slides (Lab-Tek II chamber slide system, Thermo Fischer Scientific, Cat #154917). Cells were then fixed in 4% paraformaldehyde, permeabilized with diethylpyrocarbonate (DEPC)-PBS/0.2% Triton X-100, and washed with (DEPC)-PBS. Cells were incubated with hybridization buffer containing 50% formamide, DEPC-2xSSC (300 mM sodium chloride, 30 mM sodium citrate, pH 7.0), 10% w/v dextran sulfate, and DEPC-50 mM sodium phosphate, pH 7.0 for 30 min at 66°C. This was followed by hybridization with 40 nM of a Locked Nucleic Acid probe for C9orf72 HREs in hybridization buffer for 3 hours at 66°C. Afterwards, the cells were rinsed once in DEPC-2xSSC/0.1% Tween-20 at room temperature and three times in DEPC-0.1xSSC at 65°C. The cells were then stained with DAPI, mounted using ProLong Gold antifade reagent, and analyzed with fluorescence microscopy.
PolyGP Response
PolyGP in iPSC-MNs were measured blinded to C9orf72 HRE and disease status using a previously described sandwich immunoassay that utilizes Meso Scale Discovery electrochemiluminescence detection technology, and an affinity purified rabbit polyclonal polyGP antibody (Rb9259) as both capture and detection antibody (Gendron et al., 2015; Su et al., 2014).
Single-cell RNA-seq of MN cultures
iPSC and iPSC-MN differentiation cultures were washed with PBS, incubated at 37°C with 0.25% Trypsin-EDTA between 5 and 20 minutes, and diluted with an equal volume of the complete culture media in which they were grown. After pelleting cells at 200 x g for five minutes at 4°C, cells were resuspended in PBS, observed for clumps, and further triturated with a fire polished glass pipet. The cell suspension was filtered through a Miltenyi 30 μm filter, counted on a hemocytometer, and the concentration was adjusted prior to loading onto the Illumina Bio-Rad ddSEQ System or 10X Genomics Chromium scRNA-seq platforms in accordance with the respective instructions for each kit for targeting approximately 1,000 cells per sample. Library preparation kits used were Illumina® Bio-Rad® SureCell™ WTA 3' Library Prep Kit for the ddSEQ™ System and 10X Chromium Single-cell 3’ Library & Gel Bead Kit v2. Libraries were sequenced on Illumina NextSeq500 targeting 100,000 reads per cell. Raw sequencing reads were demultiplexed and processed to FASTQ using Illumina bcl2fastq. Sample reads were aligned to the transcriptome and uniquely mapped reads were counted and assigned to cell specific barcodes. For ddSEQ libraries, reads were aligned and demultiplexed to cell barcodes using Illumina Single-Cell RNA Seq BaseSpace Workflow (v1.0.0) with STAR Aligner (v2.5.2b) (Dobin et al., 2013) and hg19 reference genome. For 10X libraries, reads were aligned and demultiplexed using 10X Genomics Cell Ranger (v2.1.0) with STAR Aligner (v2.5.1) and GRCh38 reference genome. Ensembl gene IDs were annotated to HGNC symbols. In instances of multiple ENSG IDs mapping to unique HGNC symbols, the sum of unique molecular identifiers (UMIs) across ENSG IDs was calculated and used as the UMI for the unique HGNC symbol. The summarized UMI count tables for each experimental batch are deposited in GEO under accession number GSE138121.
Immunohistochemistry and quantification of ELAVL3 in spinal cords
Human tissues were obtained using a short-postmortem interval acquisition protocol that followed HIPAA-compliant informed consent procedures and were approved by Institutional Review Board (Benaroya Research Institute, Seattle, WA IRB# 10058 and University of California San Diego, San Diego, CA IRB# 120056). For IHC, 8 sporadic ALS, 4 C9 ALS, and 5 control lumbar spinal cord sections were studied. Sections with 6 μm thickness were formalin-fixed and paraffin-embedded. On day one, sections were deparaffinized with Citrisolv (Fisher Scientific #04-355-121) and hydrated with different dilutions of alcohol. Endogenous peroxidase activity was quenched with 0.06% H2O2 for 15 min. Antigen retrieval was performed in an Antigen Unmasking Solution (Vector Laboratories #H-3301) in a pressure cooker for 20 min at a temperature of 120 °C. Following antigen retrieval, sections were permeabilized with 1% FBS (Atlanta Biologicals #511150) and 0.2% Triton X-100 in PBS for 15 min and then blocked with 2% FBS in PBS for 25 min. The sections were incubated overnight with the primary antibody, rabbit polyclonal ELAVL3, 1:1000, LSBio, Cat# LS-C408905. On the second day, after 60-min incubation with the secondary antibody (Immpress reagent kit, anti-Rabbit, Vector Laboratories #MP-7401) in room temperature, signals were detected using Immpact DAB (Vector Laboratories #SK-4105) for 5–10 min. Counterstaining was performed with hematoxylin (Fisher #HHS128). For IHC visualization, slides were scanned with Hamamatsu Nanozoomer 2.0HT Slide Scanner at 40X magnification. At least 6 motor neurons per spinal cord were evaluated, and across all samples totaled a combined number of 199 neurons from sporadic ALS subjects, 77 neurons from C9 ALS subjects, and 313 neurons from control subjects. Images were deconvoluted using Fiji ImageJ (Schindelin et al., 2012) and the optical density (OD) was measured for each neuron, where OD = log (max intensity/1/Mean intensity), where max intensity = 255 for 8-bit images.
QUANTIFICATION AND STATISTICAL ANALYSIS
Pseudotime analysis
Monocle version 2.12.0 (Qiu et al., 2017) was used to perform pseudotime analysis of the 18 day differentiation time course. Genes with minimum average expression of 0.1 and detectable in at least 10 cells were filtered. Cells were further filtered for those whose total UMI count was within three standard deviations of the average log10 UMI across all time points. Tests for differential expression of each gene as a function of the time course was calculated using the full generalized linear model, and genes with a q-value less than 0.1 from this test was filtered. These genes were used in dimensional reduction of the time course samples onto two components through Discriminative Dimensionality Reduction with Trees. All cells were ordered along this pseudotime trajectory, and expression of select genes were plotted against the cells ordered along this pseudotime.
Seurat Version 2.3.0 was used to process, normalize, cluster, and analyze scRNA-seq data for day 18 MN cultures. UMI count tables for each of the six experimental batches were each loaded as Seurat objects as well as cell barcodes and sample covariates for meta data. For downstream analysis, quality control filters for genes and cells were applied. Genes with at least one UMI in at least one cell were kept. The percent of mitochondrial genes was calculated for each cell and stored as meta data. Z-scores were calculated for three columns in the meta data for each cell: nGene, nUMI, and percent mitochondrial genes. Cells were then filtered based on these z-scores; any cell that had a z-score greater than 3 or less than −3 (greater than 3 standard deviations away from the mean of that meta data) in any of the three columns were excluded from further analysis. Next, the global scaling normalization method normalizes the gene expression measurement for each cell by the total expression, multiplies this by a scale factor, and log transforms the result. The maximum UMI detected in the experimental batch was used as the scaling factor. Next, highly variable genes (HVGs) in the experimental batch were calculated. The mean expression for all detected ( i.e. non-zero value) genes was calculated as well as the log transformed ratio of variance to mean expression (regarded as the dispersion). Genes were then binned into 20 intervals, and within each interval, the z-score for dispersion was calculated for each gene. This helps control for the relationship between variability and average expression. Genes with z-score for dispersion values greater than 2 were deemed to be HVGs. After all six experimental batches were processed as Seurat objects, samples were subsetted out of each Seurat object, totaling 22 samples. 279 HVGs were calculated in at least 11 of the 22 samples, and these were kept for subsequent dimensional reduction.
Data set normalization, identity assignment, and clustering
Multiple Canonical Correlation Analysis (MultiCCA) was performed on the 22 samples to correct for experimental batch and platform effects. Up to 20 dimensions were evaluated, and the first 18 dimensions were determined to be used for subspace alignment. Prior to subspace alignment, cells whose expression profiles cannot be well-explained by low-dimensional CCA compared to low-dimensional PCA (less than a two-fold ratio) were removed. 17,531 cells remained. Subsequently, samples were aligned using dynamic time warping along the first 18 dimensions, and the resulting batch integrated Seurat object holding all 22 samples was used for downstream analysis.
Alternative packages for batch correction were applied to the same Seurat object as MultiCCA through the Seurat Wrapper package version 0.2.0. For batch correction through Harmony, the RunHarmony command was applied to the Seurat object after running the raw counts matrix through the following pipe: NormalizeData() %>% FindVariableFeatures() %>% ScaleData() %>% RunPCA. For batch correction through Liger, the raw counts matrix was run through the following pipe: NormalizeData() %>% FindVariableFeatures(). Subsequently, the ScaleData command was applied with the following parameters: split.by = "EXP_BATCH" or “PLATFORM”, do.center = FALSE. The RunOptimizeALS command was applied with k = 20, lambda = 5, split.by = "EXP_BATCH" or “PLATFORM”. Finally, the RunQuantileNorm command was applied with split.by = "EXP_BATCH" or “PLATFORM”. For batch correction through FastMNN, the raw counts matrix was run through the following pipe: NormalizeData() %>% FindVariableFeatures(). The Seurat object was split by experimental batch or scRNA-seq platform, and the RunFastMNN command was applied to this split object list. kBET version 0.99.6 was used to calculate the acceptance rate and average silhouette width for all batch correction methods along with uncorrected data. The kBET command was applied to the cell embeddings within the dimensionally reduced projections calculated by each method and considering either experimental batch or scRNA-seq platform with k0 = 30 and do.PCA = TRUE; all other parameters were kept at default. The prcomp command was applied to the cell embeddings within the dimensionally reduced projections calculated by each method, and the batch_sil command was applied to the first three principal components to determine the average silhouette width.
In scRNA-seq analysis, increasing attention is directed towards understanding how various scRNA-seq analysis programs, particularly using pre-defined yet tunable parameters can influence the outcome of tasks such as cell clustering (Kiselev et al., 2019; Krzak et al., 2019). To determine the optimal number of communities to cluster, several resolution settings were tested using the FindClusters command in Seurat. The first 18 dimensions from the reduction through CCA were used, and 30 nearest neighbors were considered for each resolution setting. All other parameters were kept at default values. The original Louvain algorithm determined the modularity for each setting, and the maximum modularity observed after 100 iterations was recorded for each number of communities. A polynomial trendline was calculated, and the residuals for each setting greater than zero was considered to determine the optimal number of communities. Based on the independently optimized tSNE calculations and visualizations for 17,531 cells, a resolution setting of 0.125 yielding 4 communities was selected to proceed with downstream analysis. When projecting all cells on two two dimensional tSNE plots using the RunTSNE command, the same 18 dimensions were used as for the FindClusters command, and all other parameters were kept at default values . A perplexity setting of 100 was selected based on the visual concordance with the 4 communities determined.
To analyze only the postmitotic, neuronal subtypes from these 17,531 cells, we repeated the FindClusters command using a resolution parameter of 0.04, which detected 2 communities, and the postmitotic community containing 11,120 cells was subsetted into a new Seurat object. Once again, 22 samples were subsetted out of this Seurat object. HVGs were again calculated in each of the 22 samples using the same parameters stated above. 158 HVGs were calculated in at least 11 of the 22 samples, and these were kept for subsequent dimensional reduction. MultiCCA using 22 dimensions was applied to this batch integrated Seurat object, and the final data set comprised of 10,866 cells. The optimal parameters for resolution set to 1 and perplexity set to 0.75 were selected for FindClusters and RunTSNE, respectively, and this produced 18 communities, which were subsequently re-annotated based on key marker gene expression.
To assign rostrocaudal segment or cell type identity considering the expression pattern of HOX genes (Table S2A) or 105 developmental genes (Table S2B), respectively, all expression values were log transformed after adding a pseudocount of 1. Pearson correlations were performed using pairwise complete observations. Benjamini-Hochberg-corrected p-values for each Pearson correlation were calculated using the corr.test function in the psych 1.9.12.31 package, and the correlation with the lowest p-value, meeting the specified threshold was used to assign the segment or cell type identity. Multiple identities with the highest correlation were randomly selected for assignment.
Differential expression, gene set enrichment, and classifier accuracy analyses
To perform differential gene expression analysis between any two populations, the FindMarkers command in Seurat was applied with the bimodal expression likelihood test and the log fold change threshold was set to 0.1. Genes with Bonferroni adjusted P-values less than 0.05 were called significantly changed. Additionally, differentially expressed genes between ALS and control conditions were calculated in DESeq (Anders and Huber, 2010) by summing all scRNA-seq UMI counts for each gene across the expression matrix for each sample to simulate bulk RNA-seq expression.
Jaccard indices were calculated by tabulating genes called differentially upregulated or downregulated in each ALS to control of isogenic comparison within each experimental batch and intersecting each set of genes among all experimental batches. The Jaccard index is the ratio of the number of intersecting genes divided by the sum of the union of all genes across the two sets being compared. Gene Ontology (GO) analysis was performed on gene lists using official gene symbols for homo sapiens through the DAVID functional annotation chart. The following categories were tested: OMIM disease, GO Term BP direct, GO Term CC direct, GO Term MF direct, BIOCARTA, KEGG, and REACTOME. Thresholds used were minimum count of 2 and EASE score of 0.1, and GO and pathway sets with Benjamini-Hochberg-corrected P-values of less than 0.05 were called significant and reported. The Vennerable package was used to create Euler and Chow-Ruskey plots. WGCNA and module preservation was performed as previously described in Ho et al., 2016.
Multiset enrichment analysis was performed using SuperExactTest (Wang et al., 2015). Lists of gene sets to be intersected were input, the expected and observed number of overlaps were calculated, and the P-value indicates the likelihood of overlap among all possible comparisons.
To generate a combined expression score for each gene within a population of cells in each sample, we calculated the average UMI counts using all cells within a specified population with a non-zero UMI value. For each gene, the minimum average UMI count among all 22 samples was subtracted from the average UMI in each sample so that the minumum average UMI count among all samples was transformed to zero, and the average UMI counts for all other samples were linearly scaled. From this transformed set of values, the maximum among all 22 samples was subsequently used as a divisor for each transformed value to secondarily transform the maximum average UMI count to 1 and proportionally scale the values of all other samples. This effectively bounded the set of transformed, average UMI counts between zero and 1. For each gene in each population in each sample, the secondarily transformed average expression was summed with the percent UMI counts for all cells within the specific population, which includes zero UMI values, to generate a combined expression score that equally weights average UMI expression with percent UMI expression. This combined expression score was used to perform statistical test for changes in distribution between all ALS samples and all control and isogenic samples across all experimental batches.
To define MN-specific marker genes for ALS classification, a table of combined expression scores were generated from iPSC-MN scRNA-seq data, which contained 1,281 genes. A t-test was performed between all ALS and all control and isogenic samples; 39 genes obtained nominal p-values less than 0.05, and none of these retained this status after Benjamini-Hochberg correction. Therefore, the genes were ranked from lowest to highest nominal p-values, and the top 20 genes were selected to be intersected with the LCM MN transcriptomic data from Rabin et al., 2010 as analyzed in Ho et al., 2016 as well as Krach et al., 2018. Among these, six genes were concordantly changed between ALS and control conditions in all three data sets; the combined expression score was lower in ALS compared to control and isogenic iPSC-MNs, and the gene significance to the sALS component was negative in LCM MNs.
To incorporate the six ALS marker genes into a single prediction metric, principal component analysis (PCA) was applied to samples using the expression values of these six genes, and sample coordinates along the first, second, or a sum of both principal components was used as the prediction metric. For analyzing the scRNA-seq samples, the combined expression score for the six genes were used as input. For analyzing bulk RNA-seq data, the log transformed expression values for the six genes were used as input. In some data sets, five of the six genes were used; NDUFAF5 was not annotated in Highley et al., 2014; CARS2 was not annotated in Cox et al., 2010, Kirby et al., 2011, and Kiskinis et al., 2014. For Krach et al., 2018, Highley et al., 2014, and Shi et al., 2018, sample coordinates along PC2 were used as the prediction metric. For Fujimori et al., 2018, the signed values for PC2 coordinates of samples were reversed to place control samples concordant with their placement along PC1. Both PC1 and PC2 coordinates were floored to zero by subtracting the minimum of each PC coordinate, and the sum of the floored PC1 and PC2 coordinates were used as the prediction metric. Coordinates along PC1 were used as the prediction metric for all other data sets. The ROCR package (Sing et al., 2005) was used to plot the Receiver Operator Characteristics and calculate the Area Under the Curve (AUC). The P-value of the Wilcox Rank Sum test was used to determine whether the AUC significantly differs from 0.5, the AUC of an uninformative test.
Supplementary Material
Table S1. Subject cell line data, Related to Figure 1. Subject clinical and experimental attributes.
Table S2. HOX expression, VZ, MZ, expression, and cell meta data and annotations, Related to Figures 2 and 3.
A. Unit expression matrix of HOX genes as they are expressed along the rostrocaudal axis of the developing embryo.
B. Unit expression matrix of ventricular and mantle zone developmental marker genes as they are expressed along the dorsoventral and medial lateral axes of the developing embryonic spinal cord.
C. Cell meta data and annotations based on rostrocaudal, VZ, and MZ gene expression and global clustering.
Table S3. Differentially expressed genes in MN, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within MN for each experimental batch, separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison.
Table S4. Differentially expressed genes in V1 Renshaw, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within V1 Renshaw for each experimental batch, separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison.
Table S5. Differentially expressed genes in V2a, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within V2a for each experimental batch, separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison.
Table S6. Differentially expressed genes in MNs, V1 Renshaw, and V2a interneurons, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within each of three populations: MN (A and B), V1 Renshaw (C and D), and V2a (E and F) interneurons, respectively separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison. Genes called significant are highlighted blue. The number of instances each gene was called significant within each category is listed to the right: comparisons, ALS cell lines, CTR or ISO cell lines, and batches. The ratio of each number to total number of comparisons, ALS cell lines, CTR or ISO cell lines, or batches is also displayed. The sum of ratios is calculated to the far right of each table. Genes are displayed based on ranking their ratio sums from highest to lowest. Genes determined to be significant in at least two comparisons were grouped into each set used for subsequent analysis in Figures 4, 5, and 6.
Table S7. Enriched Gene Ontology terms among genes differentially expressed in MNs, V1 Renshaw, and V2a interneurons, Related to Figure 4.
A. Differentially expressed genes (DEG) table shows the number of comparisons in which genes were significantly, differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Genes observed to be differentially expressed in two or more comparisons were deemed to be reproducibly dysregulated and thereby kept for subsequent GO analysis.
B - G. Tables of Gene Ontology (GO) terms enriched among genes that are reproducibly dysregulated by ALS within each neuronal subtype in the upregulated or downregulated categories.
H - M. Tables of Gene Ontology (GO) terms enriched among genes that are reproducibly dysregulated by ALS uniquely within each neuronal subtype in the upregulated or downregulated categories.
GO terms are displayed based on ranking Benjamini-Hochberg-corrected P-values from lowest to highest. Benjamini-Hochberg-corrected P-values < 0.05 are highlighted blue. Bonferroni-corrected P-values < 0.01 are highlighted yellow.
Acknowledgements
The authors gratefully acknowledge the following: Tania F. Gendron and Leonard Petrucelli for the polyGP immunoassay data, Victoria Dardov and Jennifer Van Eyk for providing mass spectrometry data for ELAVL3; Kathleen Kurowski, Berhan Mandefro, and Dylan West for assistance with experiments and reagent organization; Soshana Svendsen for critical reading and comments on the manuscript. This work was supported by the following grants: ALS Association (J.R., C.N.S.), California Institute for Regenerative Medicine (RN3-06530, R.H.B.), NIA (K99AG056678, R.H.), NINDS (R01NS069669, R.H.B.), NINDS (U54NS091046, C.N.S.), Target ALS (J.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Alaynick WA, Jessell TM, and Pfaff SL (2011). Snapshot: spinal cord development. Cell 146, 178–178.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp 2008, P10008. [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner M, Miao Z, Wolf FA, Teichmann SA, and Theis FJ (2019). A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49. [DOI] [PubMed] [Google Scholar]
- Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, Hollinger HC, Hartley JA, Brockington A, Burness CE, et al. (2010). Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PloS One 5, e9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delile J, Rayon T, Melchionda M, Edwards A, Briscoe J, and Sagner A (2019). Single cell transcriptomics reveals spatial and temporal dynamics of gene expression in the developing mouse spinal cord. Dev. Camb. Engl 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies J-E, Shkreta L, Moszczynski AJ, Sidibé H, Semmler S, Fouillen A, Bennett ER, Bekenstein U, Destroismaisons L, Toutant J, et al. (2018). TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain J. Neurol 141, 1320–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bonito M, Glover JC, and Studer M (2013). Hox genes and region-specific sensorimotor circuit formation in the hindbrain and spinal cord. Dev. Dyn. Off. Publ. Am. Assoc. Anat 242, 1348–1368. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, Bazett-Jones DP, Le Grice S, McKay RDG, Buetow KH, et al. (2008). Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2, 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori K, Ishikawa M, Otomo A, Atsuta N, Nakamura R, Akiyama T, Hadano S, Aoki M, Saya H, Sobue G, et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med 24, 1579–1589. [DOI] [PubMed] [Google Scholar]
- Gaublomme JT, Li B, McCabe C, Knecht A, Yang Y, Drokhlyansky E, Van Wittenberghe N, Waldman J, Dionne D, Nguyen L, et al. (2019). Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nat. Commun 10, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, Pedraza O, Lucas JA, Murray ME, Desaro P, et al. (2015). Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. (Berl.) 130, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, and Marioni JC (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol 36, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks SC, Townes FW, Teng M, and Irizarry RA (2018). Missing data and technical variability in single-cell RNA-sequencing experiments. Biostat. Oxf. Engl 19, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, Higginbottom A, Raman R, Ferraiuolo L, Cooper-Knock J, et al. (2014). Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol 40, 670–685. [DOI] [PubMed] [Google Scholar]
- Ho R, Sances S, Gowing G, Amoroso MW, O’Rourke JG, Sahabian A, Wichterle H, Baloh RH, Sareen D, and Svendsen CN (2016). ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci 19, 1256–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph RM (2014). Neuronatin gene: Imprinted and misfolded: Studies in Lafora disease, diabetes and cancer may implicate NNAT-aggregates as a common downstream participant in neuronal loss. Genomics 103, 183–188. [DOI] [PubMed] [Google Scholar]
- Keenan AB, Jenkins SL, Jagodnik KM, Koplev S, He E, Torre D, Wang Z, Dohlman AB, Silverstein MC, Lachmann A, et al. (2018). The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 6, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang Y-D, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-Y, Kim E, Yoon SK, and Yoon J-B (2008). Herp enhances ER-associated protein degradation by recruiting ubiquilins. Biochem. Biophys. Res. Commun 369, 741–746. [DOI] [PubMed] [Google Scholar]
- Kirby J, Ning K, Ferraiuolo L, Heath PR, Ismail A, Kuo S-W, Valori CF, Cox L, Sharrack B, Wharton SB, et al. (2011). Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain J. Neurol 134, 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev VY, Andrews TS, and Hemberg M (2019). Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet 20, 273–282. [DOI] [PubMed] [Google Scholar]
- Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci 22, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kovel CGF, Lisgo S, Karlebach G, Ju J, Cheng G, Fisher SE, and Francks C (2017). Left-Right Asymmetry of Maturation Rates in Human Embryonic Neural Development. Biol. Psychiatry 82, 204–212. [DOI] [PubMed] [Google Scholar]
- Krach F, Batra R, Wheeler EC, Vu AQ, Wang R, Hutt K, Rabin SJ, Baughn MW, Libby RT, Diaz-Garcia S, et al. (2018). Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol. (Berl.) 136, 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzak M, Raykov Y, Boukouvalas A, Cutillo L, and Angelini C (2019). Benchmark and Parameter Sensitivity Analysis of Single-Cell RNA Sequencing Clustering Methods. Front. Genet 10, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Luo R, Oldham MC, and Horvath S (2011). Is my network module preserved and reproducible? PLoS Comput. Biol 7, e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer CW, Torrisi A, Pantelidou M, Santama N, and Cavallaro S (2007). Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Song M-R, Xu Z, Lanuza GM, Liu Y, Zhuang T, Chen Y, Pfaff SL, Evans SM, and Sun Y (2011). Isl1 is required for multiple aspects of motor neuron development. Mol. Cell. Neurosci 47, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Williams CE, Ruhl DA, Estevez-Silva MC, Chapman ER, Coon JJ, and Ashton RS (2015). Deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem Cell Rep. 4, 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Niu T, and Alaynick WA (2015). Molecular and cellular development of spinal cord locomotor circuitry. Front. Mol. Neurosci 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecken MD, and Theis FJ (2019). Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury Y, Côme J, Piskorowski RA, Salah-Mohellibi N, Chevaleyre V, Peschanski M, Martinat C, and Nedelec S (2015). Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 33, 89–96. [DOI] [PubMed] [Google Scholar]
- Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci 22, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzis V, Steinhauser S, Pakanavicius E, Gouti M, Stamataki D, Ivanovitch K, Watson T, Rayon T, Mousavy Gharavy SN, Lovell-Badge R, et al. (2018). Nervous System Regionalization Entails Axial Allocation before Neural Differentiation. Cell 175, 1105–1118.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montibeller L, and de Belleroche J (2018). Amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD) are characterised by differential activation of ER stress pathways: focus on UPR target genes. Cell Stress Chaperones 23, 897–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardo G, Iennaco R, Fusi N, Heath PR, Marino M, Trolese MC, Ferraiuolo L, Lawrence N, Shaw PJ, and Bendotti C (2013). Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain J. Neurol 136, 3305–3332. [DOI] [PubMed] [Google Scholar]
- Philippidou P, and Dasen JS (2013). Hox genes: choreographers in neural development, architects of circuit organization. Neuron 80, 12–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pla P, Hirsch M-R, Le Crom S, Reiprich S, Harley VR, and Goridis C (2008). Identification of Phox2b-regulated genes by expression profiling of cranial motoneuron precursors. Neural Develop. 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma Y-A, and Trapnell C (2017). Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin SJ, Kim JMH, Baughn M, Libby RT, Kim YJ, Fan Y, Libby RT, La Spada A, Stone B, and Ravits J (2010). Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum. Mol. Genet 19, 313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragagnin AMG, Shadfar S, Vidal M, Jamali MS, and Atkin JD (2019). Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci 13, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med 5, 208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saris CGJ, Horvath S, van Vught PWJ, van Es MA, Blauw HM, Fuller TF, Langfelder P, DeYoung J, Wokke JHJ, Veldink JH, et al. (2009). Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC Genomics 10, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, and Levine AJ (2018). Massively Parallel Single Nucleus Transcriptional Profiling Defines Spinal Cord Neurons and Their Activity during Behavior. Cell Rep. 22, 2216–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang W-H, Hung S-T, Hendricks E, Linares GR, Wang Y, Son EY, et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, and Lengauer T (2005). ROCR: visualizing classifier performance in R. Bioinforma. Oxf. Engl 21, 3940–3941. [DOI] [PubMed] [Google Scholar]
- Stein JL, de la Torre-Ubieta L, Tian Y, Parikshak NN, Hernández IA, Marchetto MC, Baker DK, Lu D, Hinman CR, Lowe JK, et al. (2014). A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang W-Y, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, et al. (2014). Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 83, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, and Robberecht W (2014). The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol 10, 661–670. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH, and Cleveland DW (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MR, Brockington A, Scaber J, Hollinger H, Marsden R, Shaw PJ, and Talbot K (2010). Pattern of spread and prognosis in lower limb-onset ALS. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis 11, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umahara T, Uchihara T, Shibata N, Nakamura A, and Hanyu H (2016). 14–3-3 eta isoform colocalizes TDP-43 on the coarse granules in the anterior horn cells of patients with sporadic amyotrophic lateral sclerosis. Brain Res. 1646, 132–138. [DOI] [PubMed] [Google Scholar]
- Volpato V, and Webber C (2020). Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Model. Mech 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltman L, and van Eck NJ (2013). A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, 471. [Google Scholar]
- Wang M, Zhao Y, and Zhang B (2015). Efficient Test and Visualization of Multi-Set Intersections. Sci. Rep 5, 16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, and Macosko EZ (2019). Single-Cell Multiomic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 177, 1873–1887.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ, Ngo HD, Rosowski KA, Schein PA, Ackeifi CA, et al. (2013). A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12, 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Subject cell line data, Related to Figure 1. Subject clinical and experimental attributes.
Table S2. HOX expression, VZ, MZ, expression, and cell meta data and annotations, Related to Figures 2 and 3.
A. Unit expression matrix of HOX genes as they are expressed along the rostrocaudal axis of the developing embryo.
B. Unit expression matrix of ventricular and mantle zone developmental marker genes as they are expressed along the dorsoventral and medial lateral axes of the developing embryonic spinal cord.
C. Cell meta data and annotations based on rostrocaudal, VZ, and MZ gene expression and global clustering.
Table S3. Differentially expressed genes in MN, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within MN for each experimental batch, separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison.
Table S4. Differentially expressed genes in V1 Renshaw, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within V1 Renshaw for each experimental batch, separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison.
Table S5. Differentially expressed genes in V2a, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within V2a for each experimental batch, separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison.
Table S6. Differentially expressed genes in MNs, V1 Renshaw, and V2a interneurons, Related to Figure 4. Tables of genes called significantly differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Each tab lists significant genes determined from sample to sample comparisons within each of three populations: MN (A and B), V1 Renshaw (C and D), and V2a (E and F) interneurons, respectively separated by upregulated and downregulated categories. ALS to CTR or ISO comparisons were only analyzed between samples within each experimental batch, indicated above each comparison. Genes called significant are highlighted blue. The number of instances each gene was called significant within each category is listed to the right: comparisons, ALS cell lines, CTR or ISO cell lines, and batches. The ratio of each number to total number of comparisons, ALS cell lines, CTR or ISO cell lines, or batches is also displayed. The sum of ratios is calculated to the far right of each table. Genes are displayed based on ranking their ratio sums from highest to lowest. Genes determined to be significant in at least two comparisons were grouped into each set used for subsequent analysis in Figures 4, 5, and 6.
Table S7. Enriched Gene Ontology terms among genes differentially expressed in MNs, V1 Renshaw, and V2a interneurons, Related to Figure 4.
A. Differentially expressed genes (DEG) table shows the number of comparisons in which genes were significantly, differentially expressed using the bimodal expression likelihood ratio test with log fold-change threshold greater than 0.1 at a Bonferroni-adjusted P-value less than 0.05. Genes observed to be differentially expressed in two or more comparisons were deemed to be reproducibly dysregulated and thereby kept for subsequent GO analysis.
B - G. Tables of Gene Ontology (GO) terms enriched among genes that are reproducibly dysregulated by ALS within each neuronal subtype in the upregulated or downregulated categories.
H - M. Tables of Gene Ontology (GO) terms enriched among genes that are reproducibly dysregulated by ALS uniquely within each neuronal subtype in the upregulated or downregulated categories.
GO terms are displayed based on ranking Benjamini-Hochberg-corrected P-values from lowest to highest. Benjamini-Hochberg-corrected P-values < 0.05 are highlighted blue. Bonferroni-corrected P-values < 0.01 are highlighted yellow.
Data Availability Statement
The scRNA-seq source data have been deposited at Gene Expression Omnibus and are publicly available under the accession number: GSE138121.
The original codes used for the analyses reported in this study are publicly available at https://github.com/ritchieho/2020_scRs_iPSC_ALS.
The scripts used to generate the figures reported in this paper are available at https://github.com/ritchieho/2020_scRs_iPSC_ALS.
Any additional information required to reproduce this work is available from the Lead Contact.