

Summary
Micronuclei are aberrant nuclear compartments that can form as a result of chromosome mis-segregation. Frequent loss of micronuclear envelope integrity exposes DNA to the cytoplasm, leading to chromosome fragmentation and immune activation. Here, we use micronuclei purification to show that the endoplasmic reticulum (ER)-associated nuclease TREX1 inhibits cGAS activation at micronuclei by degrading micronuclear DNA upon micronuclear envelope rupture. We demonstrate that the ER accesses ruptured micronuclei and plays a critical role in enabling TREX1 nucleolytic attack. TREX1 mutations, previously implicated in immune disease, that untether TREX1 from the ER disrupt TREX1 localization to micronuclei, alleviate micronuclear DNA damage, and enhance cGAS activation. These results establish ER-directed resection of micronuclear DNA by TREX1 as a critical regulator of cytosolic DNA sensing in chromosomally unstable cells and provide a mechanistic basis for the importance of TREX1 ER-tethering in preventing autoimmunity.
Keywords: chromosome instability, micronuclei, nuclear envelope, cGAS, TREX1, STING, chromothripsis, endoplasmic reticulum
Graphical Abstract
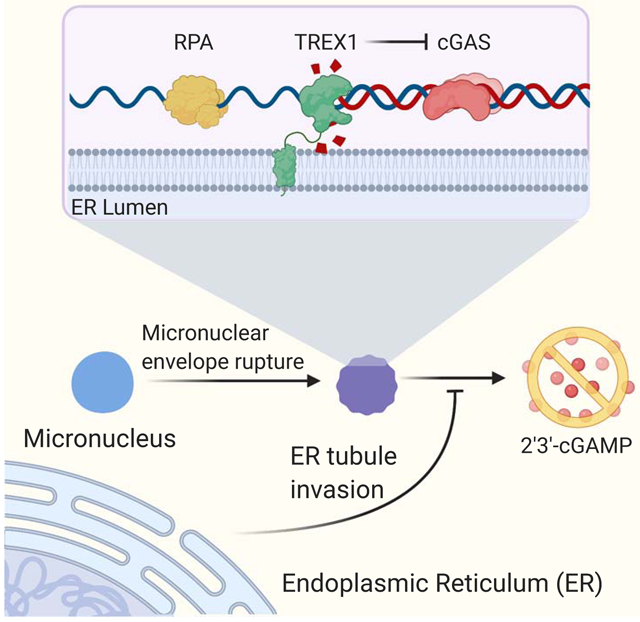
eTOC Blurb
The ER-associated nuclease TREX1 guards against immune disease by clearing immunostimulatory DNA from the cytosol. Mohr et al. identify a key role for the ER in limiting immune activation by directing TREX1 engagement with DNA substrates such as micronuclei.
Introduction
The nuclear envelope (NE) consists of a double membrane bilayer that surrounds the genome and regulates access to DNA. Challenges to NE integrity, such as mechanical compression or defects in the nuclear lamina, can cause NE rupturing and loss of nuclear compartmentalization (Maciejowski and Hatch, 2020). NE ruptures are rapidly repaired in the nucleus, but persist at aberrant, nuclear compartments, termed micronuclei (MN), that form around mis-segregated chromosomes (Hatch et al., 2013). MN with ruptured NEs (i.e. ruptured MN) exhibit defects in transcription and DNA synthesis, as well as extensive DNA damage (Crasta et al., 2012; Hatch et al., 2013). Micronuclear DNA damage is a proposed intermediate in chromothripsis, or chromosome shattering, but the initial cause(s) of damage are unknown (Crasta et al., 2012; Ly et al., 2019; Zhang et al., 2015). In addition to broad nuclear dysfunction, ruptured MN exhibit an aberrant association with ER-derived membranes that is of unknown significance (Hatch et al., 2013; Vietri et al., 2020).
Micronuclear envelope (MNE) rupturing activates the innate immune system by enabling the cytosolic DNA sensor cGAS to be activated by chromosomal DNA (Harding et al., 2017; Mackenzie et al., 2017). Detection of double-stranded DNA (dsDNA) inside ruptured MN stimulates cGAS catalytic activity and the production of 2′3′-cyclic GMP-AMP (cGAMP) (Ablasser et al., 2013; Civril et al., 2013; Diner et al., 2013; Gao et al., 2013; Sun et al., 2013). cGAMP engages with STING, resulting in activation of the kinase TBK1 and the phosphorylation and nuclear translocation of the transcription factors IRF3 and NF-κB (Ishikawa and Barber, 2008; Ishikawa et al., 2009). IRF3 and NF-κB induce the expression of type I interferons and other immunomodulatory proteins (Ablasser and Chen, 2019). Current models of cGAS activation in ruptured MN are consistent with its biochemical preferences: cGAS recognizes dsDNA through sequence-independent interactions, but depends on long (>45 bp) dsDNA for robust activation (Civril et al., 2013; Gao et al., 2013; Sun et al., 2013). ssDNA and small dsDNA fragments do not elicit robust cGAS activity (Civril et al., 2013; Kranzusch et al., 2013; Zhou et al., 2018).
TREX1 is an ER-associated, DNA 3′→5′ exonuclease that protects against chronic cGAS activation and autoimmunity by degrading cytosolic DNA (Ablasser et al., 2014; Gray et al., 2015; Grieves et al., 2015; Mazur and Perrino, 2001; Stetson et al., 2008; Wolf et al., 2016). Although it is clear that TREX1 is a key cGAS antagonist, several important questions remain. First, the mechanisms regulating TREX1 nucleolytic activity are unknown. Second, the DNA substrates that drive disease in the absence of TREX1 are poorly understood. Additionally, although TREX1 is known to associate with the ER, the functional significance of this interaction has been overlooked. Finally, frameshift mutations that eliminate TREX1’s C-terminal extension are associated with systemic lupus erythematosus (SLE) and retinal vasculopathy with cerebral leukodystrophy (RVCL) (Lee-Kirsch et al., 2007; Richards et al., 2007). These mutations compromise TREX1 ER association, but do not affect its catalytic activity (Lee-Kirsch et al., 2007). Therefore, a plausible link between these mutations and potential activation of cytosolic DNA sensing pathways has remained uncertain.
Here, we show that the ER plays a key role in the regulation of cytosolic DNA sensing in chromosomally unstable cells by directing TREX1 nucleolytic activity at ruptured MN. We find that TREX1 inhibits cGAS activation at MN by accessing and degrading micronuclear DNA after MNE rupture. Surprisingly, TREX1 DNA binding function is dispensable for its accumulation in ruptured MN. Instead, we discovered that TREX1 depends on its association with the ER for access to ruptured MN. Together, our results identify TREX1 as a critical regulator of immune sensing in chromosomally unstable cells, define disease-relevant substrates of TREX1, and establish ER-tethering as a critical regulator of TREX1 nucleolytic activity.
Results
TREX1 inhibits cGAS activation in chromosomally unstable cells
To determine if TREX1 inhibits the activation of cytosolic DNA sensing pathways in chromosomally unstable cells, we treated wild-type and TREX1 KO MCF10A cells with the Mps1 inhibitor reversine (Santaguida et al., 2010). Mps1 is a key component of the spindle assembly checkpoint that maintains chromosome stability during mitosis (London and Biggins, 2014). As expected, Mps1 inhibition caused frequent chromosome mis-segregations and generation of MN and DNA bridges in both wild-type and TREX1 KO cells (Figure 1A and 1B; Figure S1A-C; Video S1). To assay cGAS activation in Mps1i-treated cells we measured cGAMP production by ELISA (Figure 1C). This analysis showed that wild-type MCF10A cells accumulated less than 20 fmol of cGAMP per million cells over 3 days of Mps1 inhibition. In contrast, TREX1 KO cells exhibited a substantial increase in cGAMP, accumulating approximately 80–120 fmol of cGAMP per million cells. Similarly, deletion of TREX1 in RPE1-hTERT cells overexpressing GFP-cGAS elicited strong increases in cGAMP production after Mps1i treatment (Figure S1D and S1E). As expected, CGAS deletion compromised cGAMP production, confirming the specificity of the assay.
Figure 1. TREX1 inhibits cGAS activation at MN.

(A) Live-cell imaging of the indicated MCF10A cells expressing H2B-RFP. Insets mark time in minutes since mitotic NE breakdown (t = 0). Arrows mark mis-segregating chromosomes. (B) Quantification of cell divisions resulting in the formation of MN or DNA bridges as shown in (A). Mean and s.d. of n = 3 experiments (>50 cells analyzed per experiment) are shown. (C,D) ELISA analysis of cGAMP production in the indicated cells. Mean and s.d. of n = 3 experiments are shown. (E) Immunoblotting for pIRF3(S386), TREX1 and actin in the indicated MCF10A cells. (F) Quantification of pIRF3(S386) relative to corresponding actin signal as shown in (E). Mean and s.d. of n = 3 experiments are shown. (G) Immunofluorescence for pIRF3(S396) in the indicated MCF10A cells. (H) Quantification of pIRF3(S396) foci as shown in (G). Mean and s.d. of n = 2 experiments are shown (>250 cells quantified per replicate). P values were calculated by Kruskal-Wallis test for multiple comparisons (****P < 0.0001). (I) Nanostring expression analysis in the indicated MCF10A cells. (n = 1 experiment). (J) RT-qPCR of IFNB and ISG56 expression in the indicated MCF10A cells. Mean and s.d. of n = 3 experiments are shown. (K) Immunofluorescence for cGAS in the indicated MCF10A cells. (L) cGAS signal intensity with median as in (K) (n = 3 experiments with >60 MN quantified per experiment). Unless otherwise specified all P values were calculated by one-way ANOVA with Tukey’s multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns = not significant). Scale bars = 10 μm.
We next asked if TREX1 nuclease activity was required for effective cGAS inhibition. ELISA analyses showed that TREX1 KO cells reconstituted with the catalytically-deficient mutant GFP-TREX1-D18N (Lehtinen et al., 2008) accumulated significantly more cGAMP than TREX1 KO cells reconstituted with wild-type GFP-TREX1 after Mps1i treatment, indicating that TREX1 nucleolytic activity is necessary to inhibit cGAS activation in chromosomally unstable cells (Figure 1D).
Following cGAS-STING activation, TBK1 phosphorylates the transcription factor IRF3 at two residues (S386 and S396), to induce IRF3 dimerization and transcription of type I IFN (Liu et al., 2015). Surprisingly, given frequent MN accumulation, IRF3 S386 phosphorylation exhibited only mild to moderate increases in wild-type MCF10A and GFP-cGAS overexpressing RPE1 cells after Mps1 inhibition (Figure 1E and 1F; Figure S1D and S1F). However, TREX1 deletion significantly increased IRF3 S386 phosphorylation in MCF10A and RPE1 hTERT GFP-cGAS cells after Mps1 inhibition. Immunofluorescence analysis of IRF3 S396 phosphorylation showed a subtle increase in nuclear foci after Mps1 inhibition, which was significantly increased in TREX1 KO cells (Figure 1G and 1H). Following Mps1i treatment, TREX1 protein levels were unchanged, indicating that the observed effects occurred in the absence of TREX1 upregulation (Figure 1E; Figure S1G).
In line with the lack of substantial IRF3 phosphorylation, Mps1 inhibition elicited a mild transcriptional response in wild-type MCF10A cells (Figure 1I and 1J). However, expression of IFNB1, ISG56, IFI44, IFIT2, and other Interferon-Stimulated Genes (ISGs) was elevated in TREX1 KO cells after Mps1 inhibition. Consistent with prior observations (Simpson et al., 2020), TREX1 deletion was not sufficient to generate a major inflammatory response in untreated, chromosomally stable MCF10A cells. Deleting CGAS in TREX1 KO cells abolished the observed increases in IRF3 S386 phosphorylation and gene expression, indicating cGAS is the prime sensor active in this system (Figure 1E and 1F and 1I and 1J; Figure S1E).
To further investigate TREX1-mediated regulation of cGAS in chromosomally unstable cells, we adapted a previously described cell model of telomere crisis (Maciejowski et al., 2015). Briefly, we use doxycycline-inducible expression of a dominant negative allele of the Shelterin subunit TRF2 to generate dicentric chromosomes (Figure S1H) (van Steensel et al., 1998). TP53 was deleted to enable cell cycle progression in the presence of telomere dysfunction (Karlseder et al., 1999). Consistent with our prior study (Maciejowski et al., 2015), dicentric chromosomes persisted through mitosis to form long DNA bridges in approximately 80% of cell divisions (Figure S1I and S1J; Video S2).
Loss of Lamin B1 and GFP-cGAS accumulation at DNA bridges indicated likely NE rupture in wild-type and TREX1 KO cells (Figure S1K-N; Video S3). cGAMP production was not increased during telomere crisis (Figure S1O). However, TREX1 KO cells accumulated nearly 20 fmol of cGAMP per million cells during telomere crisis, a >2-fold increase over their TREX1-proficient counterparts. Limited cGAMP production in the telomere crisis model compared to Mps1i-treated cells may reflect differences in the relative amounts of cytosolic DNA produced in these models or tension-induced disruption of B-form DNA structure at DNA bridges (Civril et al., 2013). Taken together, these data suggest that TREX1 limits cGAS activation in chromosomally unstable cells.
Increased cGAMP levels in TREX1 KO cells could not be explained by decreased cGAS signal intensity or localization to MN or increased MN-independent, cytosolic dsDNA levels after TREX1 deletion or Mps1 inhibition (Figure 1K and 1L; Figure S1P-S). cGAS exists in nuclear and cytosolic fractions, associating with chromatin following mitotic NE breakdown and accumulating in the cytosol during the subsequent cell cycle (Gentili et al., 2019; Volkman et al., 2019; Yang et al., 2017). We reasoned that unexpected alterations in cGAS subcellular distribution may increase cGAMP in the TREX1 KO cells. However, cGAS exhibited similar nuclear localization in wild-type and TREX1 KO cells (Figure S1T and S1U).
Purification of cGAS positive MN reveals association with TREX1
To investigate how TREX1 inhibits cGAS in chromosomally unstable cells, we developed a strategy to isolate MN based on their association with cGAS. Existing methods use differential centrifugation to separate MN from primary nuclei, but cannot distinguish between distinct types of MN (Shimizu et al., 1996). Our strategy uses genetically-encoded markers and flow cytometry to isolate MN (Figure 2A). HEK293T cells were labeled with H2B-mCherry and GFP-cGAS to mark chromatin and MN (Figure 2B). GFP-cGAS is expected to localize to ruptured MN within minutes of NE disruption (Harding et al., 2017; Mackenzie et al., 2017). Indeed, nearly 100% of ruptured MN, defined by loss of an NLS-3×mCherry marker, were marked with GFP-cGAS (Figure S2A and S2B). Similarly, the protein BAF, which accumulates at sites of NE rupture (Denais et al., 2016; Halfmann et al., 2019; Raab et al., 2016; Young et al., 2020), was also enriched at GFP-cGAS marked MN (Figure S2C and S2D).
Figure 2. Purification of cGAS-positive MN.

(A) Schematic of MN purification. (B) Immunofluorescence for mCherry and GFP in HEK293T + H2B-mCherry + GFP-cGAS cells. Green arrows mark cGAS-positive MN, red arrows mark cGAS-negative MN. Scale bar = 10 μm. (C) Flow profiles of cGAS-negative and cGAS-positive MN isolated by FACS. (D) Immunofluorescence for GFP (cGAS) in the indicated fractions after FACS. Scale bar = 25 μm. (E) Measurement of the area of sorted primary nuclei (PN) and MN sorted by FACS. (F) Quantification of the GFP-cGAS signal intensity as shown in (D). (G) Immunoblotting for the indicated proteins in MN sorted from parental and 3×FLAG-TREX1 overexpressing HEK293T cells. (H) Quantification of relative cGAS and TREX1 signals normalized to loading control (SMC1 or histone H3) as shown in (G). Mean and s.d. of n = 3 experiments are shown. All P values were calculated by one-way ANOVA with Tukey’s multiple comparisons test (****P < 0.0001, ns = not significant).
Flow cytometric analysis of the MN-containing fractions isolated following density gradient centrifugation revealed two populations that could be distinguished by distinct forward scatter and DAPI-staining intensity (Figure 2C). Image analysis of flow-purified particles identified these populations as primary nuclei and MN (Figure 2D). Further inspection of the MN population by flow cytometry revealed two populations marked by differences in GFP-cGAS signal intensity. All purified MN exhibited a similar distribution of surface area, however, the average GFP-cGAS signal intensity differed more than two-fold across the two populations (Figure 2D-F). Immunoblotting confirmed that GFP-cGAS exhibited marked changes across the two populations (Figure 2G and 2H, Figure S2E). Additionally, BAF and the ESCRT-III component CHMP2A, which localize to ruptured MN (Vietri et al., 2020; Willan et al., 2019; Young et al., 2020), were also present in the GFP-cGAS positive MN population (Figure S2E-G). Taken together, these data validate the utility of this method for purifying cGAS-positive MN and show that ruptured MN are enriched in the GFP-cGAS-positive MN population.
We reasoned that TREX1 may regulate cytosolic DNA sensing pathways in chromosomally unstable cells by inhibiting cGAS activation at MN. To investigate this, we asked whether TREX1 co-purified with MN. Immunoblotting analysis of MN purified from HEK293T cells reconstituted with 3×FLAG-TREX1 showed that TREX1 was indeed enriched in GFP-cGAS-positive MN (Figure 2G and 2H; Figure S2H). These data indicate that TREX1 associates with MN after MNE rupture.
TREX1 rapidly accumulates in ruptured MN
To better understand TREX1 association with purified cGAS-positive MN, we analyzed TREX1 localization in chromosomally unstable cells. Live-cell imaging showed that GFP-TREX1, expressed in MCF10A TREX1 KO cells, displayed perinuclear localization consistent with its ER association (Figure 3A) (Stetson et al., 2008; Wolf et al., 2016). GFP-TREX1 localized to approximately 40% of MN within 24 h of mitotic exit (Figure 3A-C, Video S4). Analysis of GFP-TREX1 in a panel of chromosomally unstable breast cancer cell lines revealed similar localization to MN (Figure S3A).
Figure 3. TREX1 localizes to MN after MNE rupture.

(A) Live-cell imaging of MCF10A cells expressing GFP-TREX1 and H2B-mCherry. Arrows mark MN. (B) Quantification of the cumulative percentage of TREX1-positive MN as shown in (A). Error bars show s.d. of 45 MN quantified across n = 3 experiments. (C) Quantification of the relative fluorescence signal intensities of GFP-TREX1 and H2B-mCherry at MN (t = 0, initial TREX1 localization) as shown in (A). Relative mean fluorescence intensities and s.d. of 11 MN from n = 2 experiments are shown. (D) Live-cell imaging of MCF10A cells expressing GFP-TREX1, H2B-mCherry and NLS-3×mTurquoise2 (t = 0, MNE rupture). (E) Quantification of the relative fluorescence signal intensities of GFP-TREX1 and NLS-3×mTurquoise2 as shown in (D). Mean and s.d. of 15 MN from n = 3 experiments are shown. (F) Immunofluorescence for TREX1 and LSD1 in the indicated cells. Insets show magnified regions. (G) Quantification of the percentage of TREX1-positive ruptured and intact MN as shown in (F). Mean of n = 2 experiments are shown (>90 MN quantified per replicate and cell line). (H) Schematic of full-length TREX1, TREX1 R128A R174A (RA), and TREX1ΔN (amino acids 2-235 deleted). (I) Immunofluorescence for GFP (TREX1) and H3K9ac in the indicated MCF10A cells. Arrows denote MN. (J) Quantification of TREX1 signal intensity at ruptured (rMN) and intact MN (iMN) relative to TREX1 signal intensity at the ER. Data represent >30 MN quantified from n = 3 experiments. Mean and s.d. are indicated. P values were calculated by one-way ANOVA with Tukey’s multiple comparisons test (****P < 0.0001, ns = not significant). All scale bars = 10 μm.
We reasoned that TREX1 may localize to MN after MNE rupture. To test this, we assayed for MNE rupture using a previously described NLS-3×mTurquoise2 marker (Vargas et al., 2012). This analysis showed that GFP-TREX1 localization to MN was coincident with MNE rupture, as measured by loss of NLS-3×mTurquoise2 from MN (Figure 3D and 3E, Video S5). In addition, analysis of MNE rupture using mTurquoise2-tagged cGAS showed that TREX1 and cGAS localize to MN with similar timing (Figure S3B and S3C, Video S6). Immunofluorescence with an anti-TREX1 antibody confirmed the presence of endogenous protein at 40%-70% of ruptured MN and less than 5% of intact MN in cancer cell lines (Figure 3F and 3G). Additionally, transgenic TREX1 was present at 70% of cGAS-positive MN in HEK293T cells (Figure S3D and S3E). GFP-TREX1 was similarly enriched on DNA bridges (Figure S3F and S3G). Based on these data, we conclude that TREX1 localizes to MN following MNE rupture.
ER-tethering is necessary for TREX1 targeting to ruptured MN
We next sought to understand how TREX1 is targeted to ruptured MN. TREX1 possesses high affinity for ssDNA and dsDNA (Lehtinen et al., 2008; Mazur and Perrino, 2001). We therefore reasoned that TREX1 is likely attracted to MN after MNE rupture exposes micronuclear DNA to the cytosol. To test this, we monitored the localization of a mutant protein, GFP-TREX1 R128A R174A (TREX1-RA), which exhibits compromised DNA binding (Fye et al., 2011). Unexpectedly, GFP-TREX1-RA localization to MN was comparable to the wild-type protein suggesting that TREX1 DNA binding function is dispensable for its association with MN (Figure 3H-J and S3H). Indeed, deletion of the entire TREX1 nuclease domain (TREX1ΔN) did not affect localization to ruptured MN (Figure 3H-J). As expected, TREX1-RA and TREX1ΔN maintained ER localization (Figure S3I). These data suggest that the C-terminal region of TREX1 may play a role in localization to MN.
TREX1 possesses a single-pass transmembrane helix at its C-terminus that anchors the protein in the ER and positions the nuclease domain in the cytosol (Lee-Kirsch et al., 2007; Wolf et al., 2016). Deleting this C-terminal extension compromises TREX1 association with the ER but does not affect its catalytic activity (De Silva et al., 2007; Lee-Kirsch et al., 2007). We observed that the ER luminal protein Calreticulin was enriched at TREX1-positive MN (Figure 4A and 4B), suggesting that TREX1 localization to ruptured MN may depend on its ER association. Live-cell imaging of ER dynamics in HeLa and MCF10A cells expressing GFP-TREX1 and NLS-3×Turq using an RFP-KDEL transgene or an ER tracker dye showed that the ER was excluded from intact MN, but rapidly accumulated in MN upon MNE rupture (Figure 4C-F; Figure S4A-D). In rare instances we observed examples of apparent MNE repair in HeLa cells. In these events, ER tracker dye and GFP-TREX1 were co-depleted from MN as compartmentalization was restored (Figure S4E). These data indicate that TREX1 and ER membranes exhibit similar localization dynamics to ruptured MN.
Figure 4. ER-tethering of TREX1 mediates recruitment to ruptured MN.

(A) Immunofluorescence for TREX1 and Calreticulin in the indicated cells. Arrows denote intact or ruptured MN. (B) Quantification of TREX1 and Calreticulin signal intensity at MN (Pearson r = 0.72 HeLa; r = 0.59 MCF10A). (C) Live-cell imaging of HeLa cells expressing GFP-TREX1, RFP-KDEL, and NLS-3×mTurquoise2. Arrow marks MN. (D) Quantification of the relative fluorescence signal intensities of GFP-TREX1, RFP-KDEL, and NLS-3×mTurquoise2 at MN (t = 0 MNE rupture) as shown in (C). Relative mean fluorescence intensities and s.d. are shown (n = 19 MN imaged in cells expressing NLS-3×mTurquoise2, GFP-TREX1, RFP-KDEL). (E) Live-cell imaging of HeLa cells expressing GFP-TREX1 and NLS-3×mTurquoise2. (F) Quantification of the relative fluorescence signal intensities of GFP-TREX1, ER tracker dye, and NLS-3×mTurquoise2. (t = 0 MNE rupture) as shown in (E). Relative mean fluorescence intensities and s.d. are shown (n = 28 MN imaged in cells expressing NLS-3×mTurquoise2, GFP-TREX1, and stained with ER tracker). (G) Schematic of full length TREX1, TREX1ΔC, and TREXΔC-Sec61-TMD. (H,I) Immunofluorescence for GFP (TREX1) and (H) Calreticulin or (I) LSD1 in the indicated cell lines. Arrows mark ruptured MN. (J) Quantification of the percentage of GFP-TREX1-positive ruptured and intact MN as shown in (E). Mean and s.d. of n = 3 experiments are shown (>180 MN quantified per replicate and cell line). P values were calculated by one-way ANOVA with Tukey’s multiple comparisons test (****P < 0.0001). All scale bars = 10 μm.
To test if the ER directs TREX1 localization in chromosomally unstable cells, we deleted the C-terminal 79 residues that contain the predicted ER transmembrane domain (TMD) (TREX1ΔC) (Figure 4G). As previously reported (Stetson et al., 2008), GFP-TREX1ΔC was localized throughout the cell, indicating apparent dissociation from the ER (Figure 4H; Figure S4F and S4G). GFP-TREX1 was present at approximately 75% of ruptured MN, while GFP-TREX1ΔC was never present at MN, even after MNE rupture, indicating a severe defect in normal localization (Figure 4I and 4J; Figure S4G and S4H). To determine if loss of ER association could explain the TREX1ΔC localization defect, we re-tethered TREX1ΔC to the ER via fusion to the TMD of Sec61 (TREX1ΔC-Sec61-TMD), a component of the ER translocon (Figure 4G and S4F). Similar to GFP-TREX1, GFP-TREX1ΔC-Sec61-TMD localized to 75% of ruptured MN (Figure 4I and 4J). Taken together, these data show that TREX1 localization to ruptured MN is dependent on its association with the ER.
TREX1 resects micronuclear DNA
We reasoned that TREX1 may inhibit cGAS activation by partially degrading micronuclear DNA. Indeed, RPA32 foci were visible in 40-60% of ruptured MN compared to less than 5% of intact MN in MCF10A, RPE1-hTERT, and HEK293T + 3×FLAG-TREX1 cells (Figure 5A-C; Figure S5A-D; Video S7). Deletion of TREX1 caused nearly complete loss of RPA32 foci at ruptured MN in all three cell lines. TREX1 CGAS double KO cells exhibited similar defects in the levels of ssDNA, showing that the observed defects are not due to indirect consequences from increased cGAS activation. Therefore, these data demonstrate that TREX1 generates ssDNA in ruptured MN.
Figure 5. TREX1 degrades micronuclear DNA after MNE rupture.

(A) Live-cell imaging of the indicated MCF10A cells. Arrows mark MN. (B) Immunofluorescence for RPA32 and LSD1. Arrows mark ruptured MN. (C) Quantification of the percentage of ruptured and intact MN positive for RPA32 foci as shown in (B). Mean and s.d. from n = 3 experiments are shown (>200 MN quantified per experiment and cell line). (D) Immunofluorescence for BrdU and cGAS. Arrows mark ruptured MN. (E) Quantification of the percentage of BrdU-positive ruptured and intact MN as shown in (D). Mean and s.d. from n = 3 experiments are shown (>180 MN quantified per replicate and cell line). P value was calculated by Student’s t-test (***P = 0.0002). (F) Immunoblotting for the indicated proteins in MN sorted from parental and 3×FLAG-TREX1 overexpressing HEK293T cells. (G) Quantification of relative RPA32 signal intensity normalized to loading control (SMC1) as shown in (F). Mean and s.d. of n = 3 experiments are shown. (H) Immunofluorescence for RPA32 in the indicated cells. Arrows mark MN. (I) Quantification of the percentage of RPA32-positive MN as shown in (H). Mean and s.d. from n = 3 experiments are shown (>220 MN quantified per replicate and cell line). (J) Immunofluorescence for γH2AX and LSD1 in the indicated MCF10A cells. Arrows mark ruptured MN. (K) Quantification of the percentage of ruptured and intact MN with detectable γH2AX foci as shown in (J). Mean and s.d. from n = 3 experiments are shown (>180 MN quantified per replicate and cell line). (L) Representative images of comet assays from MN purified from the indicated cells. Scale bars = 25 μm. (M) Quantification of tail moment (% tail DNA x tail length) as shown in (L). Mean and s.d. are shown (n = 63-104, from n = 4 experiments). (N) Immunofluorescence for RPA32 and LSD1 in the indicated cells. Arrows mark MN. (O) Quantification of the percentage of RPA32-positive ruptured and intact MN as shown in (N). Mean and s.d. from n = 3 experiments are shown (>160 MN quantified per replicate per cell line). P value was calculated by Student’s t-test (*P < 0.05). (P) Immunofluorescence for GFP, RPA32 and LSD1 in indicated cells. Arrows mark ruptured MN. (Q) Quantification of the percentage of RPA32-positive ruptured and intact MN as shown in (P). Mean and s.d. from n = 3 experiments are shown. Unless specified otherwise all P values were calculated by two-way ANOVA with Sidak’s multiple comparisons test (****P < 0.0001). All scale bars = 10 μm.
In a complementary approach, we monitored ssDNA in MN by quantifying 5-bromo-2'-deoxyuridine (BrdU) levels in MN under non-denaturing conditions (Figure 5D and 5E). The BrdU assay is based on the principle that incorporated BrdU cannot be detected under native conditions unless exposed by resection (Tkáč et al., 2016). This analysis showed that the frequency of BrdU-positive ruptured MN was reduced 5-fold in TREX1 KO cells, further supporting a model of TREX1-dependent DNA resection in ruptured MN. In addition, immunoblotting of cGAS-positive MN purified from TREX1-expressing HEK293T cells showed an approximately 50-fold increase in RPA32 relative to cGAS-negative MN indicating the presence of TREX1-dependent ssDNA in cGAS-positive MN (Figure 5F and 5G). Reintroduction of wild-type TREX1, but not the catalytically deficient TREX1-D18N, into TREX1 KO cells restored RPA32 accumulation in MN (Figure 5H and 5I, Figure S5E). Consistent with prior reports (Lehtinen et al., 2008; Maciejowski et al., 2015; Xia et al., 2018), TREX1-D18N exhibited a dominant-negative effect, significantly reducing RPA32 signal in MN.
Analysis of additional DNA damage and repair markers yielded cell type-specific results. γH2AX foci were diminished in ruptured MN in TREX1 KO and TREX1-D18N reconstituted MCF10A cells, albeit to a lesser extent than RPA32 or native BrdU foci (Figure 5J and 5K, Figure S5F and S5G). In contrast, TREX1 loss did not result in a measurable decrease in γH2AX signal in ruptured MN in HEK293T or RPE1 hTERT cells or in cGAS-positive, purified MN (Figure S5H-M). RPA32 phosphorylation, which is induced in response to DNA damage (Ciccia and Elledge, 2010), showed a TREX1-dependent increase at ruptured MN in HEK293T cells, but was not increased at MN in MCF10A cells (Figure S5N-Q).
Markers of DNA damage that depend on an active DNA damage response may not be suitable to monitor DNA damage at MN, where the relevant pathways may function abnormally. To obtain a more direct measure of DNA damage, we performed Comet assays using purified MN to visualize DNA breaks (Figure 5L and 5M). This analysis showed that ruptured MN purified from TREX1-proficient HEK293T cells exhibited significantly longer tail moments when compared to MN isolated from TREX1-deficient cells or to intact MN isolated from TREX1-proficient cells. Taken together these data show that TREX1 generates ssDNA and DNA breaks in ruptured MN.
APE1 primes TREX1 nucleolytic activity at ruptured MN
TREX1 resides in a large, ER-associated macromolecular complex together with the endonucleases NM23-H1 and APE1, which work in concert with TREX1 to degrade nuclear DNA during granzyme A-mediated cell death (Chowdhury et al., 2006; Fan et al., 2003; Yan et al., 2010). We asked if either of these nucleases play a role in the digestion of micronuclear DNA. Deletion of NM23H1 did not affect accumulation of RPA32 foci suggesting that NM23-H1 does not cleave micronuclear DNA (data not shown). However, deletion of APEX1 diminished RPA32 foci at ruptured MN suggesting that APE1 may prime TREX1 nucleolytic activity by nicking or cleaving micronuclear DNA (Figure 5N and 5O; Figure S5R). APEX1 deletion did not affect γH2AX labeling at ruptured MN (Figure S5S and S5T).
ER-tethering directs TREX1 nucleolytic activity at ruptured MN
We next sought to investigate the mechanisms underlying TREX1 activity at ruptured MN. As expected, despite maintaining localization to ruptured MN, neither the TREX1-RA nor the TREX1ΔN mutant was capable of generating RPA32 foci at ruptured MN (Figure 5P and 5Q). To test if ER-tethering was necessary for TREX1 nucleolytic activity at ruptured MN we examined the activities of TREX1ΔC and TREX1ΔC-Sec61-TMD at ruptured MN (Figure 4G). Consistent with its inability to localize to ruptured MN, TREX1ΔC was unable to resect DNA in ruptured MN as measured by RPA32 accumulation. TREX1ΔC was also defective in generating γH2AX foci in ruptured MN (Figure S6A and S6B). In contrast, introducing TREX1ΔC-Sec61-TMD into TREX1 KO cells restored RPA32 and γH2AX signals to nearly normal levels in ruptured MN. These results show that ER-tethering is necessary to direct TREX1 nucleolytic activity at ruptured MN.
TREX1 ER-association is necessary for cGAS inhibition in chromosomally unstable cells
We next asked if TREX1 ER-tethering was required for cGAS inhibition at MN. Analysis of cGAS activity via cGAMP ELISA showed that Mps1i-treated TREX1 KO and TREX1 KO cells reconstituted with TREX1ΔN or TREX1ΔC accumulated 50-60 fmol of cGAMP per million cells (Figure 6A). In contrast, TREX1 KO cells reconstituted with full-length TREX1 or TREX1ΔC-Sec61-TMD accumulated 20-25 fmol of cGAMP after Mps1 inhibition (Figure 6A and S6C). These data indicate that TREX1 tethering to the ER is critical for cGAS inhibition in chromosomally unstable cells.
Figure 6. TREX1 ER-tethering is essential for inhibiting cGAS activation at MN.

(A) ELISA analysis of cGAMP production in the indicated cells. Mean and s.d. of n = 3 experiments are shown. (B) Nanostring analysis of gene expression in the indicated MCF10A cells (n = 3 experiments). (C) ELISA analysis of cGAMP production in the indicated cells. Mean and s.d. of n = 3 experiments are shown. All P values were calculated by one-way ANOVA with Tukey’s multiple comparisons test (****P < 0.0001, **P ≤ 0.01, ns = not significant). (D) MCF10A TREX1 KO cells reconstituted with the indicated GFP-TREX1 constructs were transfected with the indicated ISDs and GFP-TREX1 ISD-association was assessed by immunoblotting.
Gene expression analysis further highlighted a key role for TREX1 ER-tethering in the inhibition of the cGAS-STING pathway in chromosomally unstable cells (Figure 6B). Similar to our initial results, Mps1 inhibition elicited a mild transcriptional response that was further enhanced in TREX1 KO cells. Reintroducing full length TREX1, but not TREX1ΔC or the nuclease deficient TREX1-D18N, into TREX1 KO cells restored normal gene expression. These results indicate that ER-directed TREX1 localization and activity at ruptured MN is critical for inhibiting cGAS activation and the pro-inflammatory transcriptional response.
To test whether ER-tethering is required for TREX1-dependent regulation of cGAS sensing of nucleosome-free DNA, we measured cGAMP levels after transfection with plasmid DNA or a 45 bp immune stimulatory DNA (ISD). TREX1ΔC-reconstituted cells behaved similarly to cells reconstituted with wild-type TREX1 accumulating approximately 300 fmol of cGAMP per million cells after plasmid DNA transfection, while TREX1 KO cells contained nearly 600 fmol of cGAMP per million cells (Figure S6C and S6D). TREX1ΔC was also effective at inhibiting cGAMP accumulation after ISD transfection albeit to a lesser degree than wild-type TREX1 (Figure 6C). Restoring TREX1ΔC ER-tethering via fusion to the Sec61-TMD did not further diminish cGAMP production after ISD transfection. These results suggest that ER localization is dispensable for TREX1 to engage transfected dsDNA substrates. To test this more directly, we transfected cells with biotin labeled ISD and recovered biotinylated DNA protein complexes from cell lysates using streptavidin beads (Figure 6D). Consistent with prior work (Stetson et al., 2008), TREX1 was present in the recovered biotinylated DNA/protein complexes. TREX1 association with biotinylated DNA depended on its N-terminal exonuclease domain, but not its C-terminus. Taken together, these results indicate that ER-tethering is critical for TREX1 action at MN, but dispensable for TREX1 association with and degradation of transfected, nucleosome-free dsDNA.
RVCL and SLE-associated TREX1 mutations disrupt ER association, micronuclear DNA degradation, and cGAS regulation
Mutations in TREX1 are associated with a spectrum of autoimmune and inflammatory diseases including Aicardi-Goutières Syndrome (AGS), SLE, and RVCL (Crow and Manel, 2015). Missense mutations that compromise TREX1 nucleolytic activity are predominantly associated with AGS, while frame-shift mutations that truncate the C-terminus preserve nucleolytic activity and are associated with RVCL and SLE (Yan, 2017). The mechanisms linking TREX1 frame-shift mutations to immune disease are poorly understood and a potential link between altered subcellular localization and engagement with dsDNA substrates has not been explored.
To determine if TREX1 C-terminal frameshift mutations might affect nuclease activity in cells we reconstituted TREX1 KO MCF10A cells with mutants previously associated with RVCL (TREX1-V235fs, −L287fs) and SLE (TREX1-D272fs, P290L, Y305C, G306A) (Lee-Kirsch et al., 2007; Richards et al., 2007; Yan, 2017) (Figure 7A; Figure S7A). In addition, we analyzed an L309* mutant, which is predicted to delete the TREX1 ER luminal peptide while preserving the TMD. Consistent with previous reports, GFP-TREX1-V235fs and −L287fs exhibited pancellular localization while GFP-TREX1-D272fs was predominantly nuclear (Lee-Kirsch et al., 2007) (Figure S7B). The L309* mutant and the P290L, Y305C, and G306A missense mutations, which map to the predicted TMD, did not affect ER localization.
Figure 7. RVCL and SLE-associated TREX1 mutations disrupt ER association, micronuclear DNA degradation, and cGAS regulation.

(A) Schematic of full length TREX1 indicating mutations associated with FCL (purple), RVCL (green), and SLE (blue). fs = frameshift. (B) Immunofluorescence for GFP (TREX1) and H3K9ac in the indicated cells. Arrows denote MN. (C) Quantification of TREX1 signal intensity at ruptured and intact MN relative to TREX1 signal intensity at the ER. Data represent mean and s.d. from >30 MN quantified from 3 experiments. (D) Quantification of the percentage of ruptured and intact MN positive for RPA32 foci in the indicated cells. Mean and s.d. from n = 3 experiments are shown (>120 MN quantified per replicate and cell line). P values were calculated by two-way ANOVA with Tukey’s multiple comparisons test (****P < 0.0001, ns = not significant). (E,F) ELISA analysis of cGAMP production in the indicated cells. Mean and s.d. of n = 3 experiments are shown. All P values were calculated by two-way ANOVA with Tukey’s multiple comparisons test (****P < 0.0001, ***P < 0.001, ns = not significant).
Following Mps1 inhibition GFP-TREX1-V235fs and −L287fs failed to accumulate at ruptured or intact MN (Figure 7B and 7C; Figure S7C). Interestingly, GFP-TREX1-D272fs was enriched at intact MN, but undetectable at ruptured MN. The missense TMD and L309* mutants did not affect TREX1 localization to ruptured MN. Analysis of RPA32 foci formation at intact and ruptured MN showed that, in agreement with a defect in localization to ruptured MN, GFP-TREX1-V235fs, −L287fs, and −D272fs were unable to degrade micronuclear DNA (Figure 7D). The TREX1 TMD missense mutations partially diminished RPA32 foci formation at ruptured MN suggesting a modest defect in TREX1 engagement with micronuclear DNA. TREX1 presence in the ER lumen appeared to be dispensable for directing its nucleolytic activity as the GFP-TREX1-L309* mutant restored RPA32 foci formation at ruptured MN. Strikingly, despite ectopic localization to intact MN, GFP-TREX1-D272fs was incapable of degrading micronuclear DNA. These results indicate that ER-tethering may play a critical role in licensing TREX1 nucleolytic activity that goes beyond enabling access to ruptured MN.
As expected, diminished TREX1 localization and nucleolytic activity at ruptured MN correlated with increased cGAS activation (Figure 7E). In contrast, GFP-TREX1-V235fs and −L287fs were nearly as effective as GFP-TREX1-wt in suppressing cGAMP production after ISD transfection (Figure 7F). GFP-TREX1-272fs was unable to restrict cGAS activation by ISD transfection, presumably because it lacked access to the transfected ISD in the cytosolic compartment. These results further confirm that the ER plays key roles in directing TREX1 localization and nucleolytic activity at ruptured MN and inhibiting cGAS activation and suggest that MN or other sources of escaped nuclear chromatin may contribute to disease etiology in carriers of TREX1 frame-shift mutations that disrupt ER association.
Discussion
Here, we identify the ER as a critical regulator of cytosolic DNA sensing in chromosomally unstable cells. Our data support a model, in which the ER invades ruptured MN and enables TREX1 to access and degrade micronuclear DNA. TREX1 is capable of restricting cGAS activation at MN without completely eliminating MN or dsDNA from the cytosol. Instead, TREX1 appears to target limited regions of micronuclear DNA for resection. It is tempting to speculate that TREX1 may preferentially engage the nucleosome-free DNA most likely to activate cGAS (Zierhut et al., 2019). Observations of MN in healthy tissues (Guo et al., 2019) suggest that TREX1-dependent processing of micronuclear DNA may represent an important checkpoint against autoimmunity. We further speculate that TREX1 may depend on ER-tethering to engage additional DNA substrates. Additional targets with similarity to MN may include engulfed apoptotic chromatin fragments and cytosolic chromatin fragments introduced into the cytosol during senescence (Blander, 2017; Dou et al., 2017; Glück et al., 2017).
In addition to playing a key role in the initial recruitment of TREX1 to ruptured MN, ER tethering also appears to be critical for licensing TREX1 digestion of micronuclear DNA. The TREX1-D272fs mutation positions TREX1 inside nuclei and intact MN. Nevertheless, despite access to DNA inside these compartments, TREX1-D272fs appears to be unable to exercise its nucleolytic activity. Although we do not know the specific mechanism that accounts for TREX1’s dependency on ER-tethering, the distinct requirements for TREX1 action at MN and transfected DNA point towards several possibilities. First, micronuclear DNA is packaged into chromatin while the transfected DNA is nucleosome free. Since chromatin inhibits cGAS activation and TREX1 activity (Chowdhury et al., 2006; Zierhut et al., 2019), TREX1 may be uniquely dependent on ER-tethering to overcome nucleosome-mediated inhibition at MN. Second, it is possible that the ER acts as a catalytic platform that facilitates TREX1 interaction with priming factors such as the ER-associated APE1 that may be necessary to effectively fragment or degrade longer chromosomal DNA substrates (Fan et al., 2003). Finally, transfected DNA is present at quantities that likely exceed the quantity of cytosolic micronuclear DNA. It is conceivable that these artificial quantities do not always represent suitable models of in vivo targets.
The mechanisms promoting TREX1-ER accumulation at ruptured MN are not clear at present. Delivery of new sheets of ER-derived membrane by cytosolic BAF is proposed to be a critical step in the repair of ruptured NEs (Halfmann et al., 2019; Young et al., 2020). BAF also recruits the ESCRT-III complex to reseal ruptured NEs (Halfmann et al., 2019). NE repair generally does not occur in MN, however, BAF and ESCRT-III localize to MN after MNE rupture (Liu et al., 2018; Vietri et al., 2020; Willan et al., 2019). Therefore, TREX1 localization to MN may depend on BAF-dependent recruitment of ER-associated membranes or remodeling of membranes by ESCRT-III.
Unlike TREX1, cGAS accumulates on MN independently of the ER. One potential mechanism to explain these differing requirements is that cGAS condensate formation (Du and Chen, 2018) creates a selective environment that impedes passive diffusion of cytosolic proteins into MN. Despite prior access to nuclear and micronuclear compartments, cGAS exhibits a striking enrichment on chromatin that is exposed to the cytosol through NE rupture. Live-cell imaging of mTurquoise2-tagged transgenic cGAS suggests that this hyper-accumulation of cGAS may draw from micronuclear or cytosolic pools of the protein. These data suggest that cGAS distinguishes between cytosol-exposed chromatin and chromatin protected within a NE compartment.
Our work has implications for understanding clustered chromosome rearrangements. MNE rupture is associated with DNA damage, chromothripsis, and the acquisition of mutation clusters termed kataegis (Crasta et al., 2012; Hatch et al., 2013; Ly et al., 2017, 2019; Umbreit et al., 2020; Zhang et al., 2015). Our report is the first to identify an enzymatic source of DNA damage in MN after MNE rupture. These findings are consistent with recent work showing that TREX1 is the primary source of the DNA damage that occurs in the primary nucleus after compression-induced NE rupture (Nader et al., 2020). Consideration of this data, together with the work presented here, suggests the existence of similar TREX1-dependent mechanisms underlying NE rupture-associated DNA damage at the primary nucleus and at MN.
Finally, our work suggests that TREX1-mediated inhibition of cGAS sensing of ruptured MN may be one mechanism that chromosomally unstable tumor cells rely on to suppress anti-tumor immunity and cell autonomous growth inhibition resulting from chronic cGAS-STING pathway activation. Therefore, TREX1-mediated DNA degradation in MN may enable cancer cells to benefit from chromosomal instability without the potentially lethal effects of cGAS-STING activation.
Limitations
Our results argue against broad functional classification of mutations that truncate the TREX1 C-terminus and suggest a potential mechanism that may guide distinct clinical courses for these mutants. Failure to digest micronuclear DNA does not seem to be related to a proposed role for TREX1 in regulation of the ER oligosaccharyltransferase complex (Fermaintt et al., 2019; Hasan et al., 2015; Kucej et al., 2017) as deletion of the small peptide of TREX1 predicted to access the ER lumen does not perturb RPA32 foci accumulation at ruptured MN (Figure 7). However, our data do not disregard a role for TREX1 in the ER lumen. Indeed, our work at present cannot explain the dominant nature of these frameshift mutations. In the future, it will be interesting to further assess how truncated fragments of TREX1 may interact with the intact protein to affect DNA engagement and nucleolytic activity and to identify DNA substrates that drive disease in the absence of TREX1.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John Maciejowski (maciejoj@mskcc.org).
Materials Availability
Cell lines generated in this study and listed in the Key Resources Table are available from Dr. John Maciejowski. Plasmids used in this study and listed in the Key Resources Table are available on Addgene (www.addgene.org).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APE1 | abcam | Cat#ab194; RRID:AB_302694 |
| BAF | abcam | Cat#ab129184; RRID:AB_11150422 |
| BrdU | Millipore | Cat#MAB3510; RRID:AB_94897 |
| Calreticulin | Invitrogen | Cat#PA3-900; RRID:AB_325990 |
| cGAS | Cell signaling technology | Cat#15102; RRID:AB_2732795 |
| CHMP2A | ProteinTech | Cat#10477-1-AP; RRID:AB_2079470 |
| dsDNA | Abcam | Cat#ab27156; AB_470907 |
| GFP | Santa Cruz | Cat#sc-9996; RRID:AB_627695 |
| GFP | Invitrogen | Cat#A-11120; AB_221568 |
| H3K9ac | Cell signaling technology | Cat#9649; RRID:AB_823528 |
| Histone H2A | Abcam | Cat#ab18255; RRID:AB_470265 |
| LaminB1 | Abcam | Cat#ab16048; RRID:AB_443298 |
| LSD1 | Cell signaling technology | Cat#2184; RRID:AB_2070132 |
| mCherry | Abcam | Cat#ab213511; RRID:AB_2814891 |
| pIRF3 (S386) | Abcam | Cat#ab76493; RRID:AB_152383 |
| IRF3 (pS396) | Cell signaling technology | Cat#29047; RRID:AB_2773013 |
| pRPA32 (pT21) | Abcam | Cat#ab109394; RRID:AB_10860648 |
| Rb | BD | Cat#554136; RRID:AB_395259 |
| RPA32 | Abcam | Cat#ab2175; RRID:AB_302873 |
| SMC1 | Bethyl laboratories | Cat#A300-055A; RRID:AB_2192467 |
| TREX1 | Abcam | Cat#ab185228 |
| TREX1 | Santa Cruz | Cat#sc-271870; RRID:AB_10708266 |
| TRF2 (Rb. #647) | de Lange Lab | N/A |
| γH2AX | Millipore | Cat#056361 |
| α-tubulin | Abcam | Cat#ab7291; RRID:AB_2241126 |
| β-actin (m) | Abcam | Cat#ab8224; RRID:AB_449644 |
| β-actin (rb) | Abcam | Cat#ab8227; RRID:AB_2305186 |
| γ-tubulin | Abcam | Cat#ab11316; RRID:AB_297920 |
| Goat anti-Mouse IgG Alexa Fluor 488 | Invitrogen | Cat#A11001; RRID:AB_2534069 |
| Goat anti-Rabbit IgG Alexa Fluor 488 | Invitrogen | Cat#A11034; RRID:AB_2576217 |
| Goat anti-Mouse IgG Alexa Fluor Plus 555 | Invitrogen | Cat#A32727; RRID:AB_2633276 |
| Goat anti-Rabbit IgG Alexa Fluor Plus 555 | Invitrogen | Cat#A32732; RRID:AB_2633281 |
| Goat anti-Mouse IgG Alexa Fluor 647 | Invitrogen | Cat#A21236; RRID:AB_2535805 |
| Goat anti-Rabbit IgG Alexa Fluor 647 | Invitrogen | Cat#A21245; RRID:AB_2535813 |
| Goat anti-Mouse IgG HRP | Thermo Fisher Scientific | Cat#31432; RRID:AB_228302 |
| Donkey anti-rabbit IgG HRP | SouthernBiotech | Cat#6441-05; RRID:AB_2796374 |
| Goat anti-Mouse IgG Alexa Fluor Plus 680 | Invitrogen | Cat#A32729; RRID:AB_2633278 |
| Goat anti-Rabbit IgG Alexa Fluor Plus 800 | Invitrogen | Cat#A32735; RRID:AB_2633284 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Aqua Hold Pap Pen 2 | Thermo Fisher Scientific | Cat#23-769-533 |
| Cholera Toxin | Sigma-Aldrich | Cat#C8052-2mg |
| Cytochalasin B | Cayman Chemical Company | Cat#11328 |
| Cytofunnel | Shandon | Cat#5991039 |
| Dynabeads MyOne Streptavidin T1 | invitrogen | Cat#65601 |
| Dynabeads Protein G | invitrogen | Cat#10030 |
| ER tracker | Thermo Fisher Scientific | Cat#E34250 |
| Fugene HD Transfection Reagent | Promega | Cat#E2311 |
| Glass bottom dishes | Cellvis | Cat#D35-20-1.5-N |
| Glass bottom dishes (4 chamber) | Cellvis | Cat#D35C4-20-1.5-N |
| Horse Serum | Thermo Fisher Scientific | Cat#26050088 |
| human EGF | Sigma-Aldrich | Cat#E9644-.2mg |
| Hydroxyurea (HU) | Sigma-Aldrich | Cat#H8627-5G |
| Insulin | Sigma-Aldrich | Cat#I9278-5ml |
| ISD (Interferon Stimulatory DNA) | Invivogen | Cat#NC0432595 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L3000075 |
| Ministrainer 40 μm | pluriSelect | Cat#43-10040-46 |
| Nitrocellulose membrane | Thermo Fisher Scientific | Cat#10600002 |
| Novex WedgeWell Tris Glycine Mini gels | Thermo Fisher Scientific | Cat#XP08160BOX |
| Odyssey Blocking Buffer (TBS) | LI-COR Biosciences | Cat#927-50000 |
| ProLong Gold antifade reagent | Invitrogen | Cat#P36934 |
| Protease Inhibitor Tablets, EDTA-free | Thermo Scientific | Cat#A32965 |
| Quick-RNA Miniprep Kit | Zymo | Cat#R1054 |
| Reversine | Cayman Chemical Company | Cat#10004412 |
| SYBR Green Master Mix | Applied Biosystems | Cat#A25742 |
| Spermidine | Thermo Fisher Scientific | Cat#AC132740010 |
| Spermine | Thermo Fisher Scientific | Cat#AC132750010 |
| SuperScript IV First-Strand Synthesis System | Invitrogen | Cat#18091200 |
| Critical Commercial Assays | ||
| 2'3'-Cyclic GAMP Direct EIA Kit | Arbor Assays | Cat#K067-H5 |
| BCA Protein Assay | Thermo Fisher Scientific | Cat#23227 |
| nCounter Human Inflammation V2 Panel | NanoString Technologies | Cat#XT-CSO-HIN2-12 |
| custom nCounter gene expression code set, see Table S1 | NanoString Technologies | N/A |
| Deposited Data | ||
| Raw Western blot, imaging and gene analysis data | this paper; Mendeley Data | http://dx.doi.org/10.17632/d8p53cv3ry.1 |
| Experimental Models: Cell Lines | ||
| BT474 | Michael Stratton Lab; Petljak et al., 2019 | N/A |
| BT549 | Hironori Funabiki Lab; Zierhut et al., 2019 | N/A |
| CAL-51 | Hironori Funabiki Lab; Zierhut et al., 2019 | N/A |
| HCC1143 | Hironori Funabiki Lab; Zierhut et al., 2019 | N/A |
| HEK293T | ATCC | Cat#ACS-4500 |
| HEK293T H2B-mCherry #4 | this paper | cET1 |
| HEK293T H2B-mCherry #4 GFP-cGAS #1 | this paper | cET2 |
| HEK293T H2B-mCherry #4 GFP-cGAS #1 3×FLAG-TREX1 #1 | this paper | cET3 |
| HEK293T GFP-cGAS 3xmCherry-NLS | this paper | cET4 |
| HeLa | ATCC | Cat#CRM-CCL-2 |
| HeLa (iCAS9) GFP-TREX1 NLS-3xmTurq | this paper | cPvM1 |
| HeLa (iCAS9) GFP-TREX1 NLS-3xmTurq RFP-KDEL | this paper | cPvM2 |
| HT1080 | ATCC | Cat#CCL-121 |
| HT1080 TREX1 KO | this paper | cJM1 |
| M2p1 (MCF10A TP53−/− doxi::TRF2-DN) | this paper | cLM1 |
| M2p1 H2B-mCherry GFP-cGAS | this paper | cJM2 |
| M2p1 H2B-mCherry GFP-BAF | this paper | cJM3 |
| M2p1 H2B-mCherry GFP-LaminB1 | this paper | cJM4 |
| M2p1 H2B-mCherry GFP-TREX1 | this paper | cJM5 |
| M2p1 TREX1 KO #1 | this paper | cLM2 |
| M2p1 TREX1 KO #1 H2B-mCherry GFP-cGAS | this paper | cJM6 |
| M2p1 TREX1 KO #1 H2B-mCherry GFP-BAF | this paper | cJM7 |
| M2p1 TREX1 KO #2 | this paper | cLM3 |
| M2p1 TREX1 KO #2 H2B-mCherry GFP-cGAS | this paper | cJM8 |
| M2p1 TREX1 KO #2 H2B-mCherry GFP-BAF | this paper | cJM9 |
| M2p1 TREX1 KO #2 H2B-mCherry GFP-LaminB1 | this paper | cJM10 |
| M2p1 TREX1 KO CGAS KO | this paper | cLM4 |
| MCF10A | Maria Jasin lab | N/A |
| MCF10A H2B-mCherry | this paper | cJM11 |
| MCF10A H2B-mCherry GFP-RPA70 NLS-3xmTurq | this paper | cJM12 |
| MCF10A Turq-3× FLAG-TREX1 | this paper | cLM5 |
| MCF10A Turq-3× FLAG-TREX1-D18N | this paper | cLM6 |
| MCF10A GFP-cGAS | this paper | cJM13 |
| MCF10A TREX1 KO #1 | this paper | cJM14 |
| MCF10A TREX1 KO #1 H2B-mCherry | this paper | cJM15 |
| MCF10A TREX1 KO #1 H2B-mCherry GFP-TREX1 | this paper | cJM16 |
| MCF10A TREX1 KO #1 H2B-mCherry GFP-TREX1 NLS-3xmTurq | this paper | cJM17 |
| MCF10A TREX1 KO #1 H2B-mCherry GFP-TREX1 cGAS-mTurqoise2 | this paper | cJM18 |
| MCF10A TREX1 KO #1 GFP-TREX1 | this paper | cLM7 |
| MCF10A TREX1 KO #1 GFP-TREX1 H2B-iRFP670 | this paper | cLM8 |
| MCF10A TREX1 KO #1 GFP-TREX1 H2B-iRFP670 NLS-3xmTurq | this paper | cPvM3 |
| MCF10A TREX1 KO #1 GFP-TREX1 H2B-iRFP670 NLS-3xmTurq RFP-KDEL | this paper | cPvM4 |
| MCF10A TREX1 KO #1 GFP-TREX1-ΔC (aa1-235) | this paper | cLM9 |
| MCF10A TREX1 KO #1 GFP-TREX1-ΔC H2B-iRFP670 | this paper | cLM10 |
| MCF10A TREX1 KO #1 GFP-TREX1-ΔC-SEC61-TMD | this paper | cLM11 |
| MCF10A TREX1 KO #1 GFP-TREX1-D18N | this paper | cLM12 |
| MCF10A TREX1 KO #1 GFP-TREX1-R128A R174 | this paper | cLM13 |
| MCF10A TREX1 KO #1 GFP-TREX1-ΔN (aa236-314) | this paper | cLM14 |
| MCF10A TREX1 KO #1 GFP-TREX1-309* | this paper | cLM15 |
| MCF10A TREX1 KO #1 GFP-TREX1-V235fs | this paper | cLM16 |
| MCF10A TREX1 KO #1 GFP-TREX1-D272fs | this paper | cLM17 |
| MCF10A TREX1 KO #1 GFP-TREX1-L287fs | this paper | cLM18 |
| MCF10A TREX1 KO #1 GFP-TREX1-P290L | this paper | cLM19 |
| MCF10A TREX1 KO #1 GFP-TREX1-Y305C | this paper | cLM20 |
| MCF10A TREX1 KO #1 GFP-TREX1-G306A | this paper | cLM21 |
| MCF10A TREX1 KO #1 Turq-3× FLAG-TREX1 | this paper | cLM22 |
| MCF10A TREX1 KO #1 Turq-3× FLAG-TREX1-D18N | this paper | cLM23 |
| MCF10A TREX1 KO #1 GFP-cGAS | this paper | cJM19 |
| MCF10A TREX1 KO #2 | this paper | cJM20 |
| MCF10A TREX1 KO #2 H2B-mCherry | this paper | cJM21 |
| MCF10A TREX1 KO #2 GFP-cGAS | this paper | cJM22 |
| MCF10A TREX1 hypomorph | this paper | cJM23 |
| MCF10A TREX1 KO #1 MB21D1 (cGAS) KO | this paper | cLM24 |
| MDA-MB-453 | Michael Stratton lab; Petljak et al., 2019 | N/A |
| Phoenix | Titia de Lange lab | N/A |
| RPE1 hTERT | ATCC | Cat#CRL-4000 |
| RPE1 hTERT TREX1 KO | this paper | cJM24 |
| T2p1 (RPE1 hTERT TP53−/− doxi::TRF2-DN) | Titia de Lange lab | N/A |
| T2p1 APEX1 KO | Titia de Lange lab | N/A |
| U2OS | ATCC | Cat#HTB-96 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| ISD (biotinylated or naked for pulldown experiments); (TACAGATCTACTAGTGATCTATGACTGATCTGTACATGATCTACA) | IDT | N/A |
| IFNβ F for qPCR (AAACTCATGAGCAGTCTGCA) | Carr et al., 2017 | N/A |
| IFNβ R for qPCR (AGGAGATCTTCAGTTTCGGAGG) | Carr et al., 2017 | N/A |
| ISG56 F for qPCR (AAGGCAGGCTGTCCGCTTA) | Diner et al., 2015 | N/A |
| ISG56 R for qPCR (TCCTGTCCTTCATCCTGAAGCT) | Diner et al., 2015 | N/A |
| actin F for qPCR (CAACCGCGAGAAGATGAC) | Bakhoum et al., 2018 | N/A |
| actin R for qPCR (ATCACGATGCCAGTGGTACG) | Bakhoum et al., 2018 | N/A |
| hTREX1; guide RNA #1 (TCAACGCTTCGATGACAACC) | this paper | N/A |
| hTREX1; guide RNA #2 (GCATCTACACCCGCCTGTAC) | this paper | N/A |
| hTREX1; guide RNA #3 (CCACTGGAACAACCAACCTA) | this paper | N/A |
| hCGAS; guide RNA #2 (GGCCCCCATTCTCGTACGGA) | this paper | N/A |
| hCGAS; guide RNA #3 (GTTCGGCCCCGCCAGGAAGT) | this paper | N/A |
| TP53; guide RNA (GGCAGCTACGGTTTCCGTC) | this paper | N/A |
| Recombinant DNA | ||
| pTK puro HA-GFP-cGAS | gift from Zhijian Chen | N/A |
| pQCXIZ 3×FLAG-TREX1 | this paper | Addgene #164243 |
| pQCXIB H2B-mCherry | this paper | Addgene #164244 |
| pQCXIP GFP-TREX1 | this paper | Addgene #164245 |
| pQCXIP NLS-3×mTurquoise2 | this paper | Addgene #164246 |
| pLenti CMV GFP Neo | Campeau et al., 2009 | Addgene #17447 |
| pLenti CMV GFP Blast | Campeau et al., 2009 | Addgene #17445 |
| pLenti CMV GFP Puro | Campeau et al., 2009 | Addgene #17448 |
| pLenti 3xmCherry-NLS | gift from Emily Hatch | N/A |
| pLenti CMV GFP-TREX1 BLAST | this paper | Addgene #164228 |
| pLenti CMV GFP-TREX1-ΔC BLAST | this paper | Addgene #164229 |
| pLenti CMV GFP-TREX1-ΔC-SEC61-TMB BLAST | this paper | Addgene #164230 |
| pQCXIP GFP-RPA70 | this paper | Addgene #164231 |
| pQCXIZ Turq-3×FLAG-TREX1 | this paper | Addgene #164232 |
| pQCXIZ Turq-3×FLAG-TREX1 D18N | this paper | Addgene #164233 |
| pLenti CMV GFP-TREX1 D18N BLAST | this paper | Addgene #164225 |
| pLenti CMV GFP-TREX1 R128A R174 BLAST | this paper | Addgene #164234 |
| pLenti CMV GFP-TREX1 ΔN BLAST | this paper | Addgene #164235 |
| pLenti CMV GFP-TREX1 309* BLAST | this paper | Addgene #164242 |
| pLenti CMV GFP-TREX1 V235fs BLAST | this paper | Addgene #164236 |
| pLenti CMV GFP-TREX1D272fs BLAST | this paper | Addgene #164237 |
| pLenti CMV GFP-TREX1 L287fs BLAST | this paper | Addgene #164238 |
| pLenti CMV GFP-TREX1 P290L BLAST | this paper | Addgene #164239 |
| pLenti CMV GFP-TREX1 Y305C BLAST | this paper | Addgene #164240 |
| pLenti CMV GFP-TREX1 G306A BLAST | this paper | Addgene #164241 |
| pMaxGFP | Lonza | Cat#V4SC-9096 |
| pQCXIZ cGAS-mTurquoise2 | this paper | Addgene #164247 |
| pLentiPGK DEST H2B-iRFP670 | Kudo et al., 2018 | Addgene #90237 |
| pQCXIP GFP-BAF | this paper | Addgene #164248 |
| pQCXIP GFP-LmnB1 | this paper | Addgene #164249 |
| pU6 TREX1-g1 Cas9-T2A-mCherry | this paper | Addgene #164250 |
| pU6 sgTREX1-g2 Cas9-T2A-mCherry | this paper | Addgene #164251 |
| pU6 sgTREX1-g3 Cas9-T2A-mCherry | this paper | Addgene #164252 |
| pU6 cGAS-g2 Cas9-T2A-mCherry | this paper | Addgene #164253 |
| pU6 cGAS-g3 Cas9-T2A-mCherry | this paper | Addgene #164254 |
| pU6 p53-gRNA Cas9-T2A-mCherry | this paper | Addgene #164256 |
| pHP138-FLAG-TRF2-ΔBΔM | this paper | Addgene #164255 |
| p2K7 bsd-UBI-tagRFP-KDEL | Nunes-Hasler et al., 2017 | Addgene #114179 |
| psPAX2 | gift from Didier Trono | Addgene #12260 |
| pMD2.G | gift from Didier Trono | Addgene #12259 |
| Software and Algorithms | ||
| Other | ||
Data and Code Availability
The datasets and original source data generated during this study are available at Mendeley Data [http://dx.doi.org/10.17632/d8p53cv3ry.1].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MCF10A cells were cultured in 1:1 mixture of F12:DMEM media supplemented with 5% horse serum (Thermo Fisher Scientific), 20 ng/ml human EGF (Sigma), 0.5 mg/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma) and 10 μg/ml recombinant human insulin (Sigma). BT474 cells were cultured in RPMI 1640 supplemented with 10% FBS, 1% glucose, 1% sodium pyruvate. HCC1143 cells were cultured in RPMI 1640 supplemented with 10% FBS. RPE1-hTERT and MDA-MB-453 cells were cultured in F12:DMEM supplemented with 10% FBS. HEK293T, Phoenix, 293FT, HeLa, HT1080, CAL51, BT549, and U2OS were grown in DMEM supplemented with 10% FBS. All media was supplemented with 1% penicillin-streptomycin. Unless otherwise noted, all media and supplements were supplied by the MSKCC Media Preparation core facility.
METHOD DETAILS
Viral Transduction
For retroviral transduction, open reading frames were cloned into pQCXIN, pQCXIP, pQCXIB (Clontech), or pQCXIZ, which confer resistance to G418, puromycin, blasticidin, and zeocin. Constructs were transfected into Phoenix amphotropic packaging cells using calcium phosphate precipitation. Cell supernatants containing retrovirus were filtered, mixed 1:1 with target cell media and supplemented with 4 μg /ml polybrene. Successfully transduced cells were selected using G418 (Corning), puromycin (Fisher), blasticidin (Fisher), or zeocin (Life Technologies). Clones were isolated by limiting dilution or flow sorting.
For lentiviral transduction, open reading frames were cloned into pLenti CMV GFP Neo/Blast/Puro (Addgene). Constructs were transfected into 293FT cells together with psPAX2 and pMD2.G (Addgene) using calcium phosphate precipitation. Supernatants containing lentivirus were filtered and supplemented with 4 μg/ml polybrene. Successfully transduced cells were selected as above.
Immunofluorescence microscopy
For immunofluorescence microscopy of micronucleated cells, MCF10A cells, seeded on coverslips 24 h before, were treated with 0.5 μM of the Mps1 inhibitor reversine for 72 h. Parental and 3×FLAG-TREX1 overexpressing HEK293T (+H2B-mCherry +GFP-cGAS) cells, as well as RPE1 hTERT were treated for 48 h, while HeLa, RPE1 hTERT, HT10080, and U2OS cells (Figure 3F) were treated for 24h. Cells were treated with HU (2 mM) for 24 h (Figure S5P and S5Q).
For immunofluorescence staining for BAF, cGAS, Calreticulin, dsDNA, γH2AX, H3K9ac, GFP, LSD1, RPA32, pRPA32 or TREX1, cells were carefully washed with PBS prior to fixation in 2% paraformaldehyde in PBS for 12 min. For staining of pIRF3, cells were incubated with TBS-TX (TBS supplemented with 0.1% Triton X-100) for 5 min and fixed with −20° C methanol and kept at −20° C for at least 1 h. Coverslips were washed with TBS, incubated in TBS with 0.5% Triton X-100 for 5 min and washed again with TBS. Coverslips were incubated in blocking buffer (1 mg/ml BSA, 3% goat serum, 0.1% Triton X-100, 1 mM EDTA in PBS) for 1 h and primary antibodies (see Key Resources Table), diluted in blocking buffer, were added for 2 h. After 4 washes with TBS-TX, coverslips were incubated with secondary antibodies (see Key Resources Table), diluted in blocking buffer, for 1 h, then washed 2 times with TBS-TX. DNA was stained with DAPI or Hoechst (both at 1 μg/ml) for 10 min, before coverslips were washed 2 times with TBS-TX and once with TBS. Coverslips were mounted in ProLong Gold Antifade Mountant (Life Technologies). IF images shown in Figures 1,2, 3F, 4H,I, 5, S1, S3A,D, S5 (excluding S5N) and S6 were acquired on a DeltaVision Elite system equipped with a DV Elite CMOS camera, microtiter stage, and ultimate focus module (Z-stacks through the cells at 0.2 μm increments). For Figures 1G, 5P and S6A images were processed by digital deconvolution of Z-stack image series using softWoRx software. Images shown in Figures 3I, 4A, 7, S2, S3I, S5N and S7 were acquired on a Nikon Eclipse Ti2-E equipped with a CSU-W1 spinning disk with Borealis microadapter, Perfect Focus 4, motorized turret and encoded stage, polycarbonate thermal box, 5 line laser launch [405 (100 mw), 445 (45 mw), 488 (100 mw), 561 (80 mw), 640 (75 mw)], PRIME 95B Monochrome Digital Camera and 100x 1.45 NA objective. Images were further edited with Adobe Photoshop CS5.1.
For staining of cytosolic dsDNA (Figure S1P-S), cells were treated with TBS with 0.02% saponin for 5 min after fixation to selectively permeabilize the plasma membrane as previously described (Bakhoum et al., 2018).
Native BrdU staining
For detection of ssDNA in MN, cells seeded on coverslips were preincubated with BrdU (10 μM) for 24 h, washed twice with media and treated with reversine (0.5 μM) for 48 h. Before BrdU and cGAS IF, cells were carefully washed with PBS, incubated in ice cold extraction buffer (10mM Pipes-NaOH pH 7, 100mM NaCl, 300mM sucrose, 3mM MgCl2, 1mM EGTA, 0.5% Triton X-100) for 10 min. Fixation and staining steps, as well as quantification were performed as described above.
Live-cell imaging
Cells were plated onto 35 mm glass bottom dishes (Cellvis) 48 h before imaging. Where indicated cells were treated overnight with Mps1i (reversine, 0.5 μM) to induce MN. ER tracker (Thermo Fisher Scientific) was used at a final concentration of 1μM and added directly before imaging. Live-cell imaging was performed at 37° C and 5% CO2 using a Nikon Eclipse Ti2-E equipped with a CSU-W1 spinning disk with Borealis microadapter, Perfect Focus 4, motorized turret and encoded stage, polycarbonate thermal box, 5 line laser launch [405 (100 mw), 445 (45 mw), 488 (100 mw), 561 (80 mw), 640 (75 mw)], PRIME 95B Monochrome Digital Camera, and environmental enclosure (Tokai Hit). Objective lenses included CFI Plan Apo Lambda 40x 0.95 NA and CI Plan Apo Lambda 60x 1.40 NA. Images were acquired using NIS-Elements Advanced Research Software on a Dual Xeon Imaging workstation. Maximum intensity projection of z-stacks and adjustment of brightness and contrast were performed using Fiji software. Images were cropped and assembled into figures using Photoshop CS5.1 (Adobe).
MN purification
Micronucleus purification protocol was adapted from a previously described method (Shimizu et al., 1996). Parental and 3×FLAG-TREX1 overexpressing HEK293T (+H2B-mCherry +GFP-cGAS) cells were treated with 0.5 μM reversine 48 h prior to harvesting.108-109 cells were harvested and washed twice in DMEM without serum. Washed cells were resuspended in pre-warmed (37° C) DMEM without serum supplemented with cytochalasin B (Cayman) at 10 μg/ml at a concentration of 107 cells/ml DMEM and incubated at 37° C for 30 minutes. Cells were centrifuged at 300 x g for 5 minutes and the cell pellet was resuspended in cold lysis buffer (10 mM Tris-HCl, 2 mM Mg-acetate, 3 mM CaCl2, 0.32 M sucrose, 0.1 mM EDTA, 0.1% (v/v) NP-40, pH 8.5) freshly complemented (with 1 mM dithiothreitol, 0.15 mM spermine, 0.75 mM spermidine, 10 μg/ml cytochalasin B and protease inhibitors) at a concentration of 2 × 107 cells/ml lysis buffer. Resuspended cells were then dounce homogenized by 10 strokes with a loose-fitting pestle. At this point a verification step was performed by mixing a small volume of lysed cells with an equal volume of PBS + 0.5 μg/ml DAPI to confirm MN release. Cell lysates were then mixed with an equal volume of ice cold 1.8 M sucrose buffer (10 mM Tris-HCl, 1.8 M sucrose, 5 mM Mg-acetate, 0.1 mM EDTA, pH 8.0) freshly complemented (with 1 mM dithiothreitol, 0.3% BSA, 0.15 mM spermine, 0.75 mM spermidine) before use. 10 ml of this mixture (lysed cells + 1.8 M sucrose buffer) was then layered on top of a two-layer sucrose gradient (prepared by slowly adding 20 ml of 1.8 M sucrose buffer slowly on top of 15 ml 1.6 M sucrose buffer in a 50 ml conical tube). This mixture was then centrifuged in a JS-5.2 swinging bucket rotor (Beckman) at 944 × g for 20 min at 4° C. Generally, fractions were collected as follows: upper 2 ml typically contains debris and is discarded; next 5–6 ml contains MN and is collected; final 38 ml contains primary nuclei and is discarded. At this point a verification step was often included to most accurately identify fractions containing MN and lacking cell debris and primary nuclei. Fractions containing MN were then pooled and diluted 1:5 with FACS buffer (ice cold PBS supplemented with 0.3% BSA, 0.1% NP-40 and protease inhibitors). Diluted MN were then centrifuged at 944 × g in JS-5.2 swinging bucket rotor for 20 min at 4° C. Supernatant was then removed by aspiration and MN were resuspended in 2–4 ml of FACS buffer supplemented with 2 μg/ml DAPI. Resuspended samples were filtered through a 40 μm ministrainer (PluriSelect) into FACS tubes. MN were then sorted by FACSAria (BD Biosciences) into FACS buffer at the MSKCC Flow Cytometry Core Facility. Default FSC and DAPI thresholds were lowered and a log scale was used to visualize MN. Sorted MN were centrifuged at 4000 × g in JS-5.2 swinging bucket rotor for 20 min at 4° C and the pellets were stored at −80° C before lysis for Western blotting or used directly for immunofluorescence microscopy.
Immunofluorescence of purified MN
For immunofluorescence microscopy of purified micronuclei, samples were fixed in 2% paraformaldehyde for 10 minutes by adding the appropriate volume of 16% paraformaldehyde to the samples collected in FACS buffer (ice cold PBS supplemented with 0.3% BSA, 0.1% NP-40 and protease inhibitors). Then, samples were loaded in cytofunnels (Thermo Fisher), centrifuged on microscopy slides using a Cytospin 4 centrifuge (Thermo Fisher) and dried overnight at room temperature. Each sample was circled with an aqua-hold pap pen (Fisher Scientific) prior to incubation in blocking buffer, antibody and DAPI staining (see above).
Comet Assay
The alkaline comet assay was performed as described previously (Tice et al., 1990). Microscopic slides were covered with 1% normal agarose prepared in PBS and left to solidify overnight at room temperature. The first agarose layer was covered with 3-5 x105 purified MN resuspended in 10 μl PBS mixed with 75 μl of 1% low melting point agarose at 37°C and immediately covered with a coverslip. Slides were placed on ice to solidify the agarose. Then, a third layer of 1% low melting point agarose at 37°C was added and covered with a coverslip. After solidification on ice, positive control slides were immersed in 25 μM KMnO4 solution, to induce oxidative damage (Koizume et al., 1998; Kouzine et al., 2017) for 20 min at 4°C. Then, all the slides were immersed in the lysing solution (2.5 M NaCl, 100 mM EDTA, 10 mM TRIS, pH 10) for 2h at 4°C. The slides were then transferred to an electrophoresis solution (300 mM NaOH, 1 mM EDTA) for 20 min. Next, electrophoresis was carried out at 21 V, 300 mA for 20 min. The slides were then washed in a neutralization solution (0.4 M Tris, pH 7.5) three times for 5 min and dehydrated in 70%, 90% and 100% ethanol successively, 3 min each. Slides were left to dry for 2h at RT before staining with 2.0 μg/ml DAPI for 20 min at RT. Finally, the slides were imaged with a DeltaVision Elite system.
Immunoblotting
For the Western blots shown in Figures S1C,H and S5E cells were harvested by trypsinization and lysed in 1× Laemmli buffer (50 mM Tris, 10% glycerol, 2% SDS, 0.01% bromophenol blue, 2.5% β-mercaptoethanol) at 107 cells/ml. Lysates were denatured at 95° C and DNA was sheared with a 28 ½ gauge insulin needle. Lysate equivalent to 1-2 x 105 cells was resolved by SDS-PAGE (Life Technologies) and transferred to nitrocellulose membranes (Amersham). Membranes were blocked in 5% milk in TBS with 0.1% Tween-20 (TBS-T) and incubated with primary antibody (see Key Resources Table) overnight at 4° C, washed 4 times in TBS-T, and incubated for 1 h at room temperature with horseradish-peroxidase-conjugated secondary antibody (see Key Resources Table). After four washes in TBS-T, membranes were rinsed in TBS and imaging was performed using enhanced chemiluminescence (Thermo Fisher).
For quantitative Western blotting (Figures 1E, S1D, S3H, S4F, S5R, S6C, S7A), cells were lysed in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS), supplemented with phosphatase inhibitors (10 mM NaF, 20 mM β-glycerophosphate) and protease inhibitor (Thermo Scientific) at ~ 107 cells/ml and incubated on ice for 30 min. After centrifugation (21,000 x g, 30 min, 4°C), protein concentration of the supernatant was determined using BCA protein assay (Thermo Fisher) and 50 μg (Figures 1E, S1D) or 20 μg (Figures S3H, S4F, S5R, S6C, S7A) protein was loaded per sample. Membranes were blocked in Odyssey blocking buffer in TBS (Li-COR). Primary antibodies were diluted in blocking buffer supplemented with 0.2% Tween and incubated with membranes overnight at 4° C. Secondary antibodies (Alexa Fluor 680 and 800; Key Resources Table) were used at 1:20,000 dilutions in blocking buffer supplemented with 0.2% Tween. Fluorescence was measured using an infrared imaging scanner (Odyssey; LI-COR) according to the manufacturer’s instructions.
For Western blots of purified micronuclei (Figures 2G, 5F, S2E,G,H and S5L), samples were lysed in 1× Laemmli buffer at 2.5 × 107 MN/ml. Lysates were denatured at 95° C and DNA was sonicated using a Bioruptor 300 (Diagenode) on the high setting for 8 cycles 30 sec ON / 30 sec OFF. Lysate equivalents to 0.5×106 micronuclei were loaded in each lane and processed as described above for quantitative Western blotting.
Gene expression analysis
Total RNA was isolated from 2 × 106 cells using the Quick-RNA miniprep kit (Zymo) according to the manufacturer’s instructions.
cDNA was generated from 500 ng RNA using the Superscript IV first-strand synthesis system (Life Technologies) with random hexamer priming. qPCR was performed with gene specific primers (see Key Resources Table) using Sybr green detection on a Light Cycler 480 (Roche) or Quantstudio6 (Applied Biosystems) cycler. Relative transcription levels were calculated by normalizing to ACT expression.
NanoString was used to directly quantify mRNA transcripts. Isolated RNA was hybridized with reporter and capture probes of the nCounter Human Inflammation V2 Panel (Figure 1G and S2H, see Key Resources Table) or a custom made nCounter gene expression code set (Figure 5E, see Table S1) according to the manufacturer’s instructions (NanoString Technologies). Data was analyzed using nSolver Analysis software.
2′3′-cGAMP quantification
4 × 106 of MCF10A or M2p1 cells were seeded into 15-cm dishes, and 24 h later cells were treated with reversine (0.5 μM) or doxycycline (1 μg/ml), respectively. For stimulation with ISD and dsDNA, 1.5 × 106 of MCF10A cells were seeded into 10-cm dishes, and 24 h later transfected with 8 μg of 45 bp ISD (Invivogen) using Lipofectamine 3000 transfection reagent (Invitrogen) or 4 μg of pMaxGFP plasmid (Lonza) using Fugene HD transfection reagent (Promega) per manufacturer’s instructions. 72 h after reversine and doxycycline addition or 24 h after transfection, cells were harvested, washed with PBS, pelleted, and stored at −80°C. To quantify 2′3′-cGAMP levels, 8 × 106 cells (reversine) or 2 × 106 cells (DNA) were thoroughly resuspended in 120 μL lysis buffer (20 mM Tris-HCl pH 7.7, 100 mM NaCl, 10 mM NaF, 20 mM β-glycerophosphate, 5 mM MgCl2, 0.1% Triton X-100, 5% glycerol) and lysed with a 28 ½ gauge needle. Lysates were incubated on ice for 30 min, centrifuged at 16,000 × g, 4°C for 10 min and 2′3′-cGAMP levels were quantified using the 2′3′cGAMP ELISA Kit (Arbor Assays) according to the manufacturer’s instructions.
Biotin-ISD pulldown
7x106 of MCF10A cells expressing GFP-TREX1-FL, ΔN or ΔC were seeded on 2 15-cm dishes for each cell line and 24 h later, cells were transfected with 18 μg biotinylated or naked ISD (IDT, see Key Resources Table) using Lipofectamine 3000 transfection reagent (Invitrogen) per manufacturer’s instructions (DNA:Lipofectamine, 1:3). 2 h after transfection, cells were harvested by trypsinization, washed with PBS and resuspended in lysis buffer (50 mM Tris pH 7.5, 200 mM NaCl, 0.075% NP-40, protease inhibitors) at 107 cells/ml. Cells were then dounce homogenized by 10 strokes with a tight-fitting pestle. Lysates were incubated on ice for 20 min, centrifuged at 16,000 × g, 4°C for 20 min and input samples were taken for immunoblotting. To reduce non-specific binding of proteins to the beads, lysates were precleared by incubation with Protein G dynabeads (Invitrogen) for 30 min at room temperature. To pull down the biotinlylated ISD, the cleared lysate was transferred onto streptavidin dynabeads (75 μl per sample, Invitrogen) and again incubated for 30 min at room temperature. The beads were washed 8 times with lysis buffer and then eluted with 2× Laemmli buffer (100 mM Tris, 20% glycerol, 4% SDS, 0.02% bromophenol blue, 5% β-mercaptoethanol).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of micronuclear DNA damage
Quantification of RPA32-, γH2AX- or BrdU-positive MN was performed as follows: After deconvolution of Z-stack image series and maximum intensity projections, MN were identified by Hoechst signal. Then the LSD1, H3K9ac, Rb or cGAS signal was used to distinguish between intact (LSD-, H3K9ac-, Rb-positive or cGAS-negative) and ruptured MN (LSD-, H3K9ac-, Rb-negative or cGAS-positive). Finally, MN were called RPA32-, γH2AX- or BrdU-positive, if there were more than 2 clear foci visible per MN.
Statistical Analysis
Information regarding biological replicates, sample size, and statistical testing is supplied in the Figure Legends.
Supplementary Material
Video S1. Mps1 inhibition results in mitotic errors in wt and TREX1 KO MCF10A cells, Related to Figure 1A. Live-cell imaging of DMSO or Mps1i treated wt and TREX1 KO MCF10A cells expressing H2B-RFP to mark chromatin.
Video S2. GFP-BAF localizes to DNA bridges, Related to Figure 1 and Figure S1I. Live-cell imaging of M2p1 (MCF10A TP53 KO doxi::TRF2-DN) cells expressing H2B-mCh (red) and GFP-BAF (green). Cells were treated with doxycycline to induce telomere fusion 48 h before imaging.
Video S3. GFP-cGAS localizes to DNA bridges, Related to Figure 1 and Figure S1M. Live-cell imaging of M2p1 (MCF10A TP53 KO doxi::TRF2-DN) cells expressing H2B-mCh (red) and GFP-cGAS (green). Cells were treated with doxycycline to induce telomere fusion 48 h before imaging.
Video S4. GFP-TREX1 accumulation at MN, Related to Figure 3A. Live-cell imaging of MCF10A cells expressing H2B-mCh (red) and GFP-TREX1 (green). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Video S5. GFP-TREX1 localizes to micronuclei after MNE rupture, Related to Figure 3D. Live-cell imaging of MCF10A cells expressing H2B-mCh (red), NLS-3×mTurquoise2 (blue), and GFP-TREX1 (green). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Video S6. GFP-TREX1 and cGAS-mTurq2 localization to MN, Related to Figure 3 and Figure S3B. Live-cell imaging of MCF10A cells expressing H2B-mCh (blue), cGAS-mTurq2 (red), and GFP-TREX1 (green). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Video S7. GFP-RPA accumulates in ruptured MN, Related to Figure 5A. Live-cell imaging of MCF10A cells expressing H2B-mCh (red), GFP-RPA70 (green), and NLS-3xmTurquoise2 (blue). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Highlights.
TREX1 limits cGAS activation at micronuclei
TREX1 accesses and degrades micronuclear DNA upon micronuclear envelope rupture
ER-tethering is critical for TREX1 activity and cGAS regulation at micronuclei
Disruption of TREX1 ER-tethering may contribute to the etiology of immune diseases
Acknowledgements
We thank T. de Lange, P. Kranzusch, W. Zhou, C. Zierhut, A. Sfeir, E. Hatch, and Z. Chen for advice and reagents, the Integrated Genomics Operation at MSKCC for assistance with Nanostring analysis, M. Piel and G. Nader for sharing unpublished data, and support from the NCI (R00CA212290), the Pew Charitable Trusts, the Starr Cancer Consortium, the Geoffrey Beene and Ludwig Centers at MSKCC, and MSKCC core grant P30-CA008748.
Footnotes
Declaration of interests
J.M. has received consulting fees from Ono Pharmaceutical Co.. His spouse is an employee of and has equity in Bristol Myers Squibb.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363. [DOI] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K-P, Ludwig J, and Hornung V (2013). cGAS produces a 2’-5'-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, and Hornung V (2014). TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol 192, 5993–5997. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM (2017). The many ways tissue phagocytes respond to dying cells. Immunol. Rev 277, 158–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr JM, Ashander LM, Calvert JK, Ma Y, Aloia A, Bracho GG, Chee SP, Appukuttan B, and Smith JR. (2017). Molecular Responses of Human Retinal Cells to Infection with Dengue Virus. Mediators Inflamm. 2017, 3164375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one, 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung J-S, Demple B, Perrino FW, and Lieberman J (2006). The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142. [DOI] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, and Hopfner K-P (2013). Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, and Pellman D (2012). DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, and Manel N (2015). Aicardi–Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, and Lammerding J (2016). Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, and Hollis T (2007). The crystal structure of TREX1 explains the 3′ nucleotide specificity and reveals a polyproline II helix for protein partnering. J. Biol. Chem 282, 10537–10543. [DOI] [PubMed] [Google Scholar]
- Diner BA, Lum KK, Javitt A, and Cristea IM (2015). Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell. Proteomics 14, 2341–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, and Vance RE (2013). The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, Pommier Y, and Lieberman J (2003). Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat. Immunol 4, 145–153. [DOI] [PubMed] [Google Scholar]
- Fermaintt CS, Sano K, Liu Z, Ishii N, Seino J, Dobbs N, Suzuki T, Fu Y-X, Lehrman MA, Matsuo I, et al. (2019). A bioactive mammalian disaccharide associated with autoimmunity activates STING-TBK1-dependent immune response. Nat. Commun 10, 2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fye JM, Orebaugh CD, Coffin SR, and Hollis T (2011). Dominant mutations of the TREX1 exonuclease gene in lupus and Aicardi-Goutières syndrome. J. Biol. Chem 286, 32373–32382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. (2013). Structure-function analysis of STING activation by c [G (2′, 5′) pA (3′, 5′) p] and targeting by antiviral DMXAA. Cell 154, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentili M, Lahaye X, Nadalin F, Nader GPF, Lombardi EP, Herve S, De Silva NS, Rookhuizen DC, Zueva E, Goudot C, et al. (2019). The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep. 26, 3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasser A (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol 19, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, and Stetson DB (2015). Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi–Goutières Syndrome. The Journal of Immunology 195, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, and Perrino FW (2015). Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc. Natl. Acad. Sci. U. S. A 112, 5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Ni J, Liang Z, Xue J, Fenech MF, and Wang X (2019). The molecular origins and pathophysiological consequences of micronuclei: New insights into an age-old problem. Mutat. Res 779, 1–35. [DOI] [PubMed] [Google Scholar]
- Halfmann CT, Sears RM, Katiyar A, Busselman BW, Aman LK, Zhang Q, O’Bryan CS, Angelini TE, Lele TP, and Roux KJ (2019). Repair of nuclear ruptures requires barrier-to-autointegration factor. J. Cell Biol 218, 2136–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, Li Q-Z, Atkinson JP, Morse HC 3rd, Lehrman MA, et al. (2015). Cytosolic Nuclease TREX1 Regulates Oligosaccharyltransferase Activity Independent of Nuclease Activity to Suppress Immune Activation. Immunity 43, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, and Hetzer MW (2013). Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, and Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, and de Lange T (1999). p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283, 1321–1325. [DOI] [PubMed] [Google Scholar]
- Koizume S, Inoue H, Kamiya H, and Ohtsuka E (1998). Neighboring base damage induced by permanganate oxidation of 8-oxoguanine in DNA. Nucleic Acids Res. 26, 3599–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F, Wojtowicz D, Baranello L, Yamane A, Nelson S, Resch W, Kieffer-Kwon K-R, Benham CJ, Casellas R, Przytycka TM, et al. (2017). Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst 4, 344–356.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS-Y, Berger JM, and Doudna JA (2013). Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucej M, Fermaintt CS, Yang K, Irizarry-Caro RA, and Yan N (2017). Mitotic Phosphorylation of TREX1 C Terminus Disrupts TREX1 Regulation of the Oligosaccharyltransferase Complex. Cell Rep. 18, 2600–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Jeknić S, Macklin DN, Akhter S, Hughey JJ, Regot S, and Covert MW (2018). Live-cell measurements of kinase activity in single cells using translocation reporters. Nat. Protoc 13, 155–169. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee Y-A, de Silva U, Bailey SL, Witte T, Vyse TJ, et al. (2007). Mutations in the gene encoding the 3’-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet 39, 1065–1067. [DOI] [PubMed] [Google Scholar]
- Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, and Perrino FW (2008). The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J. Biol. Chem 283, 31649–31656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu Y-T, Grishin NV, et al. (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Liu S, Kwon M, Mannino M, Yang N, Renda F, Khodjakov A, and Pellman D (2018). Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, and Biggins S (2014). Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell Biol 15, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, and Cleveland DW (2017). Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol 19, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, Pyntikova T, Flanagan AM, Behjati S, Page DC, Campbell PJ, et al. (2019). Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet 51, 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, and Hatch EM (2020). Nuclear Membrane Rupture and Its Consequences. Annu. Rev. Cell Dev. Biol 36, 85–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Chatzipli A, Dananberg A, Chu K, Toufektchan E, Klimczak LJ, Gordenin DA, Campbell PJ, and de Lange T (2020). APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet 52, 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur DJ, and Perrino FW (2001). Excision of 3′ Termini by the Trex1 and TREX2 3′→ 5′ Exonucleases characterization of the recombinant proteins. J. Biol. Chem 276, 17022–17029. [DOI] [PubMed] [Google Scholar]
- Nader GPF, Agüera-Gonzalez S, Routet F, Gratia M, Maurin M, Cancila V, Cadart C, Gentili M, Yamada A, Lodillinsky C, et al. (2020). Compromised nuclear envelope integrity drives tumor cell invasion. bioRxiv. 10.1101/2020.05.22.110122 [DOI] [PubMed] [Google Scholar]
- Nunes-Hasler P, Maschalidi S, Lippens C, Castelbou C, Bouvet S, Guido D, Bermont F, Bassoy EY, Page N, Merkler D, et al. (2017). STIM1 promotes migration, phagosomal maturation and antigen cross-presentation in dendritic cells. Nat. Commun 8, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, Dawson KJ, Ju YS, Iorio F, Tubio J, et al. (2019). Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell, 176, 1282–1294.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Duménil AM, Manel N, et al. (2016). ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362. [DOI] [PubMed] [Google Scholar]
- Richards A, van den Maagdenberg AMJM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-LaBarca M-L, Terwindt GM, Kasai Y, et al. (2007). C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet 39, 1068–1070. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D’Alise AM, Taylor SS, and Musacchio A (2010). Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol 190, 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Kanda T, and Wahl GM (1996). Selective capture of acentric fragments by micronuclei provides a rapid method for purifying extrachromosomally amplified DNA. Nat. Genet 12, 65–71. [DOI] [PubMed] [Google Scholar]
- Simpson SR, Rego SL, Harvey SE, Liu M, Hemphill WO, Venkatadri R, Sharma R, Grayson JM, and Perrino FW (2020). T Cells Produce IFN-α in the TREX1 D18N Model of Lupus-like Autoimmunity. J. Immunol 204, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, and de Lange T (1998). TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401–413. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, and Medzhitov R (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice RR, Andrews PW, and Singh NP (1990). The Single Cell Gel Assay: A Sensitive Technique for Evaluating Intercellular Differences in DNA Damage and Repair Sutherland BM, Woodhead AD (eds) DNA Damage and Repair in Human Tissues. Basic Life Sciences, vol 53 Springer. [DOI] [PubMed] [Google Scholar]
- Tkáč J, Xu G, Adhikary H, Young JTF, Gallo D, Escribano-Díaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. (2016). HeLb Is a Feedback Inhibitor of DNA End Resection. Mol. Cell 61, 405–418. [DOI] [PubMed] [Google Scholar]
- Umbreit NT, Zhang C-Z, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, Sun L, Almubarak HF, Judge K, Mitchell TJ, et al. (2020). Mechanisms generating cancer genome complexity from a single cell division error. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JD, Hatch EM, Anderson DJ, and Hetzer MW (2012). Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 3, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri M, Schultz SW, Bellanger A, Jones CM, Petersen LI, Raiborg C, Skarpen E, Pedurupillay CRJ, Kjos I, Kip E, et al. (2020). Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol 22, 856–867. [DOI] [PubMed] [Google Scholar]
- Volkman HE, Cambier S, Gray EE, and Stetson DB (2019). Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willan J, Cleasby AJ, Flores-Rodriguez N, Stefani F, Rinaldo C, Pisciottani A, Grant E, Woodman P, Bryant HE, and Ciani B (2019). ESCRT-III is necessary for the integrity of the nuclear envelope in micronuclei but is aberrant at ruptured micronuclear envelopes generating damage. Oncogenesis 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, Kretschmer S, König N, Kurth T, Wieczorek D, et al. (2016). RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun 7, 11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Ivanovska IL, Zhu K, Smith L, Irianto J, Pfeifer CR, Alvey CM, Ji J, Liu D, Cho S, et al. (2018). Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J. Cell Biol 217, 3796–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N (2017). Immune Diseases Associated with TREX1 and STING Dysfunction. J. Interferon Cytokine Res 37, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, and Lieberman J (2010). The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol 11, 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. U. S. A 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AM, Gunn AL, and Hatch EM (2020). BAF facilitates interphase nuclear membrane repair through recruitment of nuclear transmembrane proteins. Mol. Biol. Cell 31, 1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, and Pellman D (2015). Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, and Kranzusch PJ (2018). Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 174, 300–311.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo J-D, Carroll T, and Funabiki H (2019). The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 178, 302–315.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Mps1 inhibition results in mitotic errors in wt and TREX1 KO MCF10A cells, Related to Figure 1A. Live-cell imaging of DMSO or Mps1i treated wt and TREX1 KO MCF10A cells expressing H2B-RFP to mark chromatin.
Video S2. GFP-BAF localizes to DNA bridges, Related to Figure 1 and Figure S1I. Live-cell imaging of M2p1 (MCF10A TP53 KO doxi::TRF2-DN) cells expressing H2B-mCh (red) and GFP-BAF (green). Cells were treated with doxycycline to induce telomere fusion 48 h before imaging.
Video S3. GFP-cGAS localizes to DNA bridges, Related to Figure 1 and Figure S1M. Live-cell imaging of M2p1 (MCF10A TP53 KO doxi::TRF2-DN) cells expressing H2B-mCh (red) and GFP-cGAS (green). Cells were treated with doxycycline to induce telomere fusion 48 h before imaging.
Video S4. GFP-TREX1 accumulation at MN, Related to Figure 3A. Live-cell imaging of MCF10A cells expressing H2B-mCh (red) and GFP-TREX1 (green). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Video S5. GFP-TREX1 localizes to micronuclei after MNE rupture, Related to Figure 3D. Live-cell imaging of MCF10A cells expressing H2B-mCh (red), NLS-3×mTurquoise2 (blue), and GFP-TREX1 (green). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Video S6. GFP-TREX1 and cGAS-mTurq2 localization to MN, Related to Figure 3 and Figure S3B. Live-cell imaging of MCF10A cells expressing H2B-mCh (blue), cGAS-mTurq2 (red), and GFP-TREX1 (green). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Video S7. GFP-RPA accumulates in ruptured MN, Related to Figure 5A. Live-cell imaging of MCF10A cells expressing H2B-mCh (red), GFP-RPA70 (green), and NLS-3xmTurquoise2 (blue). Cells were treated with reversine (0.5 μM) 24 h before imaging.
Data Availability Statement
The datasets and original source data generated during this study are available at Mendeley Data [http://dx.doi.org/10.17632/d8p53cv3ry.1].