

Abstract
Fuchs endothelial corneal dystrophy (FECD) is an age-related disease whereby progressive loss of corneal endothelial cells (CEnCs) leads to loss of vision. There is currently a lack of therapeutic interventions as the etiology of the disease is complex, with both genetic and environmental factors. In this study, we have provided further insights into the pathogenesis of the disease, showing a causal relationship between senescence and endothelial-mesenchymal transition (EMT) using in vitro and in vivo models. Ultraviolet A (UVA) light induced EMT and senescence in CEnCs. Senescent cells were arrested in G2/M phase of the cell cycle and responsible for the resulting profibrotic phenotype. Inhibiting ATR signaling and subsequently preventing G2/M arrest attenuated EMT. In vivo, UVA irradiation induced cell cycle re-entry in post mitotic CEnCs, resulting in senescence and fibrosis at 1- and 2-weeks post-UVA. Selectively eliminating senescent cells using the senolytic cocktail of dasatinib and quercetin attenuated UVA induced fibrosis, highlighting the potential for a new therapeutic intervention for FECD.
Keywords: Fuchs endothelial corneal dystrophy, senescence, endothelial-mesenchymal transition, fibrosis, cell cycle
Graphical Abstract

1. Introduction
The cornea, the eyes outermost lens, is a transparent window primarily responsible for refraction of incident light. Corneal transparency is largely governed by the spatial arrangement of collagen fibrils in the stroma (Meek et al., 2003), a layer constituting approximately 90% of the corneal thickness. The most posterior layer of the cornea consists of a monolayer of hexagonal endothelial cells that border the anterior chamber of the eye. Corneal endothelial cells (CEnCs) play a vital role in maintaining stromal transparency by controlling fluid and solute transport into the cornea. These cells are in a post-mitotic state with limited ability to proliferate in vivo, whereby they are maintained in G0/G1 phase of the cell cycle (Joyce et al., 1996b, Joyce et al., 1996a). There is a gradual loss of CEnCs with age that can be partly attributed to oxidative stress, incurred by high metabolic activity and lifelong exposure to ultraviolet (UV) light. This CEnC loss is accelerated in Fuchs Endothelial Corneal Dystrophy (FECD), a condition thought to affect around 4% of people over the age of 40 in the United States. Significant loss of CEnCs in FECD ultimately results in functional impairment leading to corneal edema, decreased visual clarity, and the need for approximately 20,000 corneal transplantations annually in the US (Eye Bank Association of America, 2017).
FECD is a complex disorder with both genetic and environmental factors contributing to disease progression. Given that the precise molecular pathophysiology of the disease is unclear, there are currently no therapeutic interventions besides transplantation. In addition to cell death, another hallmark of the disease is fibrosis in the form of dome-shaped extracellular deposits, termed guttae, protruding from the thickened basement membrane (Descemet’s membrane) to which CEnCs are attached. Extracellular matrix (ECM) components including collagen III, XVI, transforming growth factor-β induced (TGFβI) and clusterin are upregulated in late-onset FECD (Weller et al., 2014, Jurkunas et al., 2009, Jurkunas et al., 2008); however, the molecular mechanisms of excessive/abnormal accumulation of ECM resulting in guttae formation and subsequent Descemet’s membrane thickening in FECD are poorly understood. Tissue remodeling and ECM accumulation during fibrosis is often a result of persistent Endothelial-mesenchymal transition (EMT)-inducing signals. EMT is a repair-associated process whereby endothelial cells lose their cell-cell adhesion, develop migratory and invasive properties leading to tissue fibrosis. We previously identified evidence for EMT in ex vivo FECD specimens where diseased cells showed an increase in Snail1 protein, a transcriptional regulator of EMT, and loss of organized junctional staining of plasma-membrane bound N-Cadherin (Katikireddy et al., 2018). Similarly, EMT-inducing genes ZEB1 and SNAI1 have been shown to be highly expressed in FECD cells in vitro and associated with excessive production of various ECM proteins through TGFβ−1 signaling pathway (Okumura et al., 2015).
In addition to EMT, accelerated mechanisms of ageing such as premature cellular senescence have been implicated in FECD pathogenesis (Matthaei et al., 2012). Cellular senescence is a biological process associated with a wide range of age-related diseases (Childs et al., 2015), whereby cells lose their proliferative capacity through permanent exit from the cell cycle, and is characterized by distinct metabolic activity and changes in cell morphology (Herranz and Gil, 2018). The mechanisms by which post-mitotic CEnCs are maintained in a non-replicative state are thought to include: lack of growth factors (Tripathi et al., 1991), the presence of TGFβ in the aqueous humor (Chen et al., 1999), and active suppression through contact inhibition within the monolayer (Joyce et al., 1998, Senoo et al., 2000). Breaking the cell-cell contacts and providing CEnCs with growth media stimulates proliferation ex vivo (Senoo et al., 2000), indicating that CEnCs are in a state of quiescence in vivo, as opposed to being senescent and incapable of replication. Both nuclear p21 and CDKN2A (p16) have been shown to be elevated in human FECD tissue compared to normal corneal endothelium (Matthaei et al., 2012, Matthaei et al., 2014). The molecular mechanism of cellular senescence and its subsequent involvement in FECD pathogenesis remains to be elucidated. It is possible that this impaired cellular function may subsequently contribute to other detrimental processes including EMT and fibrosis.
Guttae are typically present in the central cornea of FECD patients, with the periphery appearing markedly less severe. Similarly, CEnC loss begins in the center before progressing to the periphery as the disease progresses. Exposure to sunlight UVA light (320–400nm wavelength) has been linked to the onset of ocular pathologies (Yam and Kwok, 2014). Previous studies have shown that corneal endothelium is susceptible to UV light-induced oxidative stress (Zinflou and Rochette, 2017), UV transmission is increased in the central cornea compared to the periphery (Doutch et al., 2012), and that central CEnCs exhibit increased oxidative DNA damage (Joyce et al., 2011). Furthermore, we have shown that lifelong exposure to solar UV radiation plays a role in FECD pathophysiology and guttae formation and have developed an in vivo mouse model for FECD, whereby UVA light recapitulates the morphological and molecular changes observed in humans (Liu et al., 2020). The aim of our current study is to investigate the molecular mechanism of UVA-induced senescence in post-mitotic cells, and how this is associated with the fibrotic response.
Herein, we show that UVA light induces a paradoxical cell cycle re-entry and shift from G0/G1 to arrest in G2/M phase, causing post-mitotic CEnCs to undergo premature senescence and EMT as seen in FECD. Cells in G2/M phase were shown to up-regulate profibrotic factors compared to respective G0/G1 phase cells, indicating a mechanistic link between senescent cells and fibrotic response. In vivo, UVA light induced cell cycle re-entry in post-mitotic CEnCs, leading to senescence and fibrosis. These findings aligned with observations in FECD cell lines and tissue specimens. Importantly, we were able to selectively eliminate UVA-induced senescent cells and subsequently alleviate EMT/fibrosis using a senolytic cocktail of dasatinib and quercetin (DQ). Therefore, given that transplantation is the only intervention for FECD, senescent cells could be targeted to improve to improve cornea endothelial function and slow the progression of the disease. Our findings provide important insights into premature ageing of CEnCs in FECD, outlining a new potential therapeutic target.
2. Methods
2.1. Human Tissue
This study was conducted according to the tenets of the Declaration of Helsinki and approved by the Massachusetts Eye and Ear Institutional Review Board. Written and informed consent was obtained from patients undergoing surgical treatment for FECD. Post-keratoplasty specimens were obtained from surgeries performed at Price Vision Clinic (Indianapolis, IN) and Massachusetts Eye and Ear Hospital (Boston, MA). Normal donor corneas recovered and preserved in Optisol-GS (Bausch & Lomb, Rochester, NY) within 24-hours of death were obtained from Eversight Eye Bank (Ann Arbor, MI) based on criteria reported in our previous studies (Jurkunas et al., 2008, Miyai et al., 2019). See supplement table 1 for further details on human specimens used in this study.
2.2. Cell Culture
Telomerase (normal - HCEnC-21T) and SV40 T antigen (Normal 67F and FECD 67F, 54F, 74F) immortalized CEnC lines used in this study were previously generated in our laboratory (Schmedt et al., 2012). All cells were cultured in Chen’s medium (Konomi et al., 2005) (OptiMEM-I; Invitrogen, Carlsbad, CA), containing 8% fetal bovine serum (HyClone, Rockford, IL), 5 ng/mL epidermal growth factor (Millipore, Billerica, MA), 100 mg/mL bovine pituitary extract (Invitrogen), 200 mg/L calcium chloride (Sigma-Aldrich, St. Louis, MO), 0.08% chondroitin sulfate (Sigma-Aldrich), 50 mg/mL gentamicin (Invitrogen), and 1:100 diluted antibiotic/antimycotic solution (Sigma-Aldrich) at 37 degrees and 5% CO2. For UVA irradiation, two 19.5-inch UVA tubes (XX-15L; Analytik Jena US LLC) emitting 365 nm light (irradiance: 14.77 mW/cm2) were used to irradiate normal CEnCs (HCEnC-21T) at a fluence of 25J/cm2, followed by a recovery period of 24 hours (unless otherwise stated) in Opti-MEM. ATR inhibition was achieved by pre-treatment for 2-hours, (as well as 24-hour recovery following UVA) with 5μm AZD6738 (Selleckchem, Houston, TX). Cells were arrested in G2/M using 10µM RO3306 (RO) (Sigma-Aldrich) in opti-MEM for 24-hours. For DQ, 24-hours post-UVA irradiation, cells were treated with a combination of 30µM quercetin (Sigma-Aldrich) and 200nm dasatinib (Sigma-Aldrich) for 5-hours.
2.3. Mouse Eye UVA Irradiation
C57BL/6 wild-type mice (male and female, 7- to 15-week-old; The Jackson Laboratory or Charles River) were used for this study. See Liu et al. (2020) for full description of the UVA irradiation protocol for mouse eyes, as well as in vivo imaging and cell density analysis. Briefly, A UVA LED light source was focused to illuminate a 4-mm-diameter spot onto the mouse cornea. The time of UVA exposure was adjusted to deliver a fluency of 500J/cm2 (21 minutes), with the other untreated eye serving as a control. Irradiated eyes were given a recovery period of 24-hours, 1-week, or 2-weeks.
2.4. ROS assay
Cells were pre-treated with 30µM quercetin for 2-hours before UVA irradiation. ROS production was measured immediately after UVA using Image-iT LIVE Green Reactive Oxygen Species Detection Kit (Molecular Probes, Inc., Eugene, OR). Cells were stained with 25µM 5-(and-6)-carboxy-2’, 7’-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) for 30 minutes at 37°C and transferred to a flat-bottomed 96-well plate. Fluorescence of carboxy-DCDFA stained cells (485/520nm) was measured using a fluorescence microplate reader (BioTek-Synergy 2; BioTek Instruments, Inc., Winooski, VT). Relative fluorescent units were normalized to the cell number.
2.5. SA-β-GAL Stain
Human FECD specimens and all cell lines included were stained for SA-β-GAL using a senescence histochemical staining kit (Sigma-Aldrich) according to the manufacturer’s protocol, with an incubation period of 16 hours at 37°C. Images of stained cells (10 images acquired at x20 magnification for each well) were captured using bright field microscopy (EVOS XL Core). For quantification, positively stained cells were manually counted and reported as a percentage of total cells.
2.6. Cell Cycle Analysis
CEnCs were fixed with 70% ethanol for 20 minutes, treated with 100µg/ml RNase, and stained with 50µg/ml propidium iodide. Cell cycle data was acquired using a BD LSR II flow cytometer. Single cells were identified by measuring forward and side scatter, and cell doublets excluded using pulse area vs pulse width. The combined gates were applied to a forward scatter vs propidium iodide signal (PE channel) to produce a histogram plot. Quantification of cells in each phase of the cell cycle was carried out using FlowJo cell cycle analysis (v10.6.2, FlowJo, LLC). For FACS (Fluorescence-Activated Cell Sorting), G0/G1 cells were sorted from cells in G2/M phase of the cell cycle, directly into RLT lysis buffer, using Cytomation MoFlo cell sorter. G0/G1 and G2/M phase gates were selected manually based on the cell count vs propidium iodide intensity histogram. FECD cells were detached from native tissue with EDTA (Ethylenediaminetetraacetic) solution for 20 minutes at 37°C before processing for cell cycle analysis.
2.7. Western Blot
HCEnC-21T cells were lysed in RIPA buffer containing HALT protease inhibitor cocktail (PIC) (Thermo Fisher Scientific) and protein concentration was determined using the BCA assay kit (Thermo Fisher Scientific). Mouse or human corneal endothelial tissue consisting of Descemet’s membrane with CEnCs was homogenized with ReadyPrep’s Sequential Extraction Reagent 3 containing 10µM Tributyl Phosphine reducing agent and HALT PIC. Lysates were loaded onto a 4–12% gradient bis-tris gel under reducing conditions for SDS-PAGE, followed by blotting onto polyvinylidene difluoride membrane (Millipore). The membrane was blocked with 5% nonfat dry milk or 5% Bovine Serum Albumin in Tris Buffered Saline with 0.1% Tween-20 (TBST) for 1 hour and incubated overnight at 4°C with primary antibodies (See Table S2). Blots were exposed to HRP-conjugated anti-mouse or –rabbit IgG (Santa Cruz), for 1 hour and developed with SuperSignal West Pico Plus or SuperSignal West Femto Maximum Sensitivity chemiluminescent substrates. Densitometry was analyzed using Image J software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD) and relative protein expression levels were determined by normalizing band intensities to that of β-Actin.
2.8. RT-PCR
RNA was extracted from cultured CEnCs using Trizol (Invitrogen) by following the manufacturer’s protocol. For RNA extraction from smaller quantities of cells (cell sorting experiments), RNeasy Micro Kit (Qiagen, Valencia, CA) was used according to manufacturer’s protocol. RNA quality and quantity were measured using NanoDrop (LabTech International, Uckfield, UK). iScript cDNA synthesis kit (Bio- Rad, Hercules, CA) was used to reverse-transcribe RNA. RT-PCR was performed by TaqMan gene expression assays (Applied Biosystems, Foster City, CA) in a Mastercycler Realplex 2 (Eppendorf, Hamburg, Germany) as with Probe Fast Master Mix (Kapa Biosystems, Wilmington, MA) for detection of mRNA expression of all genes. Results were normalized to GAPDH internal control. Relative expression expressed as 2^ΔΔ(-CT).
2.9. Immuno-Fluorescent Labelling
For cell lines, CEnCs were fixed with 4% paraformaldehyde (PFA) for 10 minutes at RT and permeabilized/blocked in a buffer containing 0.1% triton x-100 and 3% bovine serum albumin (BSA) in PBS. Primary antibodies (See Table S2), including mouse anti-cyclin B1 (1/100), mouse anti-p21 (1/250), and rabbit anti-Ki67 (1/50) were diluted in blocking buffer and applied overnight at 4°C. Goat anti-mouse/rabbit tagged with Alexa Fluor 488 (1/1000) secondary antibodies were applied for 1-hour at RT. Slides were mounted with vecta shield (Vector Laboratories, CA), containing DAPI and imaged with Leica DMi8 live cell imaging inverted microscope. For staining of whole mount mouse corneas, enucleated eyes were dissected to obtain the corneal cup, which was fixed in 4% PFA for 10 minutes, permeabilized with 1% triton x-100, and blocked for 2-hours with a buffer containing 3% BSA/10% goat serum/0.1% triton x-100 in PBS. Primary antibodies including rabbit anti-ki67 (1/1000), mouse anti-ZO (Zona Occludens)-1(1/200, Invitrogen), mouse anti-αSMA (1/50), mouse anti-H3k9Me2 (1/100) were applied overnight. Secondary antibodies goat anti-mouse Fluorescein Isothiocyanate (FITC) (1/200–400), goat anti-rabbit Alexa Fluor 568 (1/1000) were applied overnight. Whole mount corneas were imaged using confocal microscopy with Z-stacks used to create a maximum projection image (DM6000S with LCS 1.3.1 software, Leica).
2.10. Statistical Analysis.
Statistical analysis was carried out using Graphpad Prism v5 (Graphpad Software Inc, CA) using T-Test and one- or two-way analysis of variance (ANOVA), with Tukey’s post hoc test for ANOVAs. All data is reported as the mean ±SEM. Significance for all measures was set at P < 0.05.
3. Results
3.1. UVA light induces EMT and senescence in CEnCs as seen in FECD
Since we previously showed that menadione-induced oxidative stress induces EMT in CEnCs, which is potentiated with the loss of antioxidant NQO1 (Katikireddy et al., 2018), we first sough to determine whether the environmental stressor UVA light causes EMT and senescence in CEnCs, using an established protocol from our previous studies (Liu et al., 2014). CEnCs were grown to a confluent hexagonal monolayer before being irradiated with UVA light and given a recovery time of 0–24-hours in reduced serum media. Phase-contrast microscopy revealed changes in cellular morphology starting at six-hours after sublethal dose of UVA, manifesting in cells clustering around acellular centers in the form of rosettes, which have previously been described in FECD (Halilovic et al., 2016). Rosette-like structures increased in size and number with time, becoming more difficult to identify at 24-hours post-UVA as cellular morphology continued to change (Fig 1A). UVA irradiation resulted in cell elongation, junctional disruption, and the formation of long processes, indicative of EMT. This cellular phenotype is comparable to that seen in FECD, where primary cells appear fibroblastic in vitro, and surround the guttae on ex vivo specimens (Fig 1B).
Figure 1.
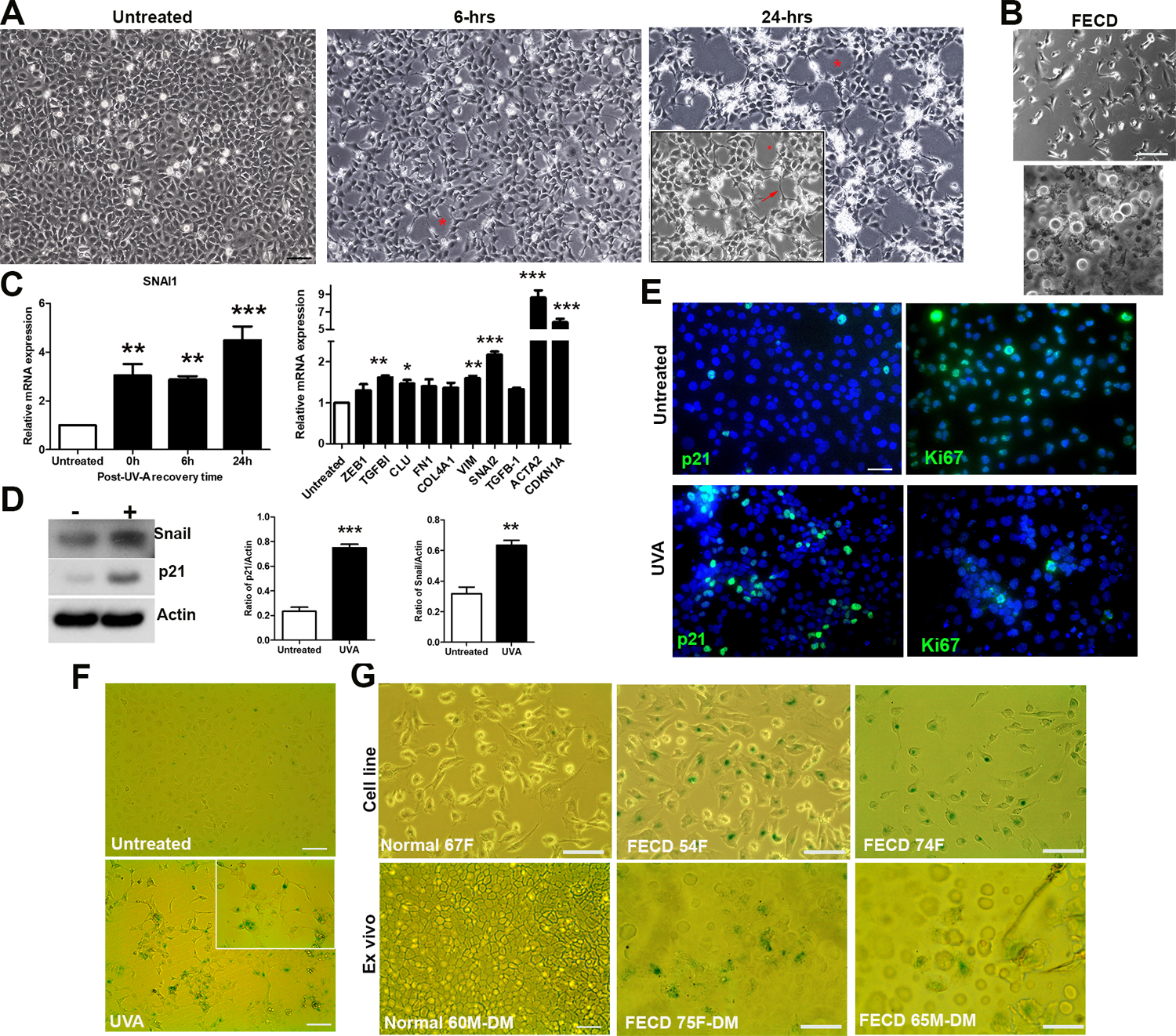
In vitro model of FECD where UVA light induces EMT and senescence in CEnCs. A. Phase-contrast microscopy images of CEnCs before, 6-hours post, and 24-hours post UVA irradiation highlighting rosette (Asterix) and fibrotic process (arrow) formation B. FECD cells: primary culture (p0, top), and attached to native Descemet’s membrane (bottom). C. SNAI1 mRNA levels as a function of time post-UVA (left), and gene expression 24 hours post-UVA (right) expressed as fold increase versus untreated. *P < 0.05, ** P < 0.01, ***P < 0.001 (n=3). D. Western blot of Snail and p21 +/− UVA (left) with densitometry analysis relative to actin (right). ** P < 0.01, ***P < 0.001 (n=3). E. Immunostaining of p21 and Ki67 +/− UVA. F. SA-β -GAL staining +/− UVA. G. SA-β-GAL staining of untreated cell lines (1 normal, 2 FECD) (top), and ex vivo CEnCs attached to Descemet’s membrane (1 normal, 2 FECD) (bottom). Data presented as mean values ± SE. Scale bars = 100µm.
Snail plays a key role in EMT (Cano et al., 2000). Gene expression analysis correlated with cell morphology, whereby UVA up-regulated SNAI1 at all time points, with the highest expression seen at 24-hours post-UVA (Fig 1C). Based on cellular morphology and gene expression data, it was decided that all subsequent in vitro UVA experiments would be carried out using a 24-hour post-UVA recovery period. At this time point, several other genes associated with EMT and FECD were up-regulated, most notably SNAI2 (encoding Snail2, associated with EMT), ACTA2 (encoding α-SMA, a cytoskeletal protein that is highly expressed in fibroblasts), and CDKN1A (encoding p21, associated with cell cycle arrest) (Fig. 1C). In addition to mRNA, UVA treated cells produced higher amounts of Snail and p21 protein (Fig 1D), prompting further analysis into cellular senescence. An increase in p21 (a cell cycle inhibitor), and the contrary decrease in Ki67 (indicator of cell proliferation) in UVA irradiated cells compared to untreated cells inferred cell cycle arrest (Fig 1E). Indeed, this cell cycle arrest was permanent, i.e. cells became senescent, as shown by positive Senescence-associated β-Galactosidase (SA-β-GAL) staining (Fig 1F). This observation of cellular senescence corresponded with untreated FECD cell lines, and FECD ex vivo specimens, both of which showed a significant proportion of senescent cells compared to controls obtained from normal cells/tissue (Fig 1G). These SA-β-GAL positive cells were located centrally surrounding guttae, and to a lesser extent in the periphery of the tissue.
3.2. Activation of ATR signaling causes senescence via G2/M phase arrest
Cellular senescence is defined as permanent exit from the cell cycle. It is thought that in vivo, CEnCs are arrested in G0/G1 phase of the cell cycle due to mechanisms including contact inhibition. As we detected evidence for UVA-induced cellular senescence in vitro, we next sought to investigate the cell cycle status of these cells compared to untreated FECD cell line (Fig. 2A). Cells in each stage of the cell cycle were quantified using flow cytometry based on fluorescence intensity of propidium iodide labelled DNA content (Fig. 2B). Cells irradiated with UVA light showed a marked increase in G2/M phase cells compared to untreated (13% vs 40%, P<0.01). Interestingly, untreated immortalized FECD cells (61F) showed a similar cell cycle profile to UVA treated cells, with a high percentage of cells in G2/M phase (40% and 48%, respectively). Cell cycle progression is tightly regulated by various cyclins and cyclin dependent kinase (CDK) enzymes. G2/M phase of the cell cycle is regulated by the Cyclin B1/CDK1 complex which plays a role in chromosome condensation and nuclear envelope breakdown in preparation for mitosis. Increased Cyclin B1 expression was evident in UVA-treated cells, with accumulation in the cytoplasm indicting that cells had not entered mitosis (Fig 2C). UVA light induced double stranded DNA breaks indicated by the phosphorylation of H2AX, followed by the activation of ATR, which in turns phosphorylates CHK1, resulting in downstream cell cycle arrest in G2/M as seen by increased Cyclin B1 and decreased Cyclin D1 levels (Fig 2D).
Figure 2.
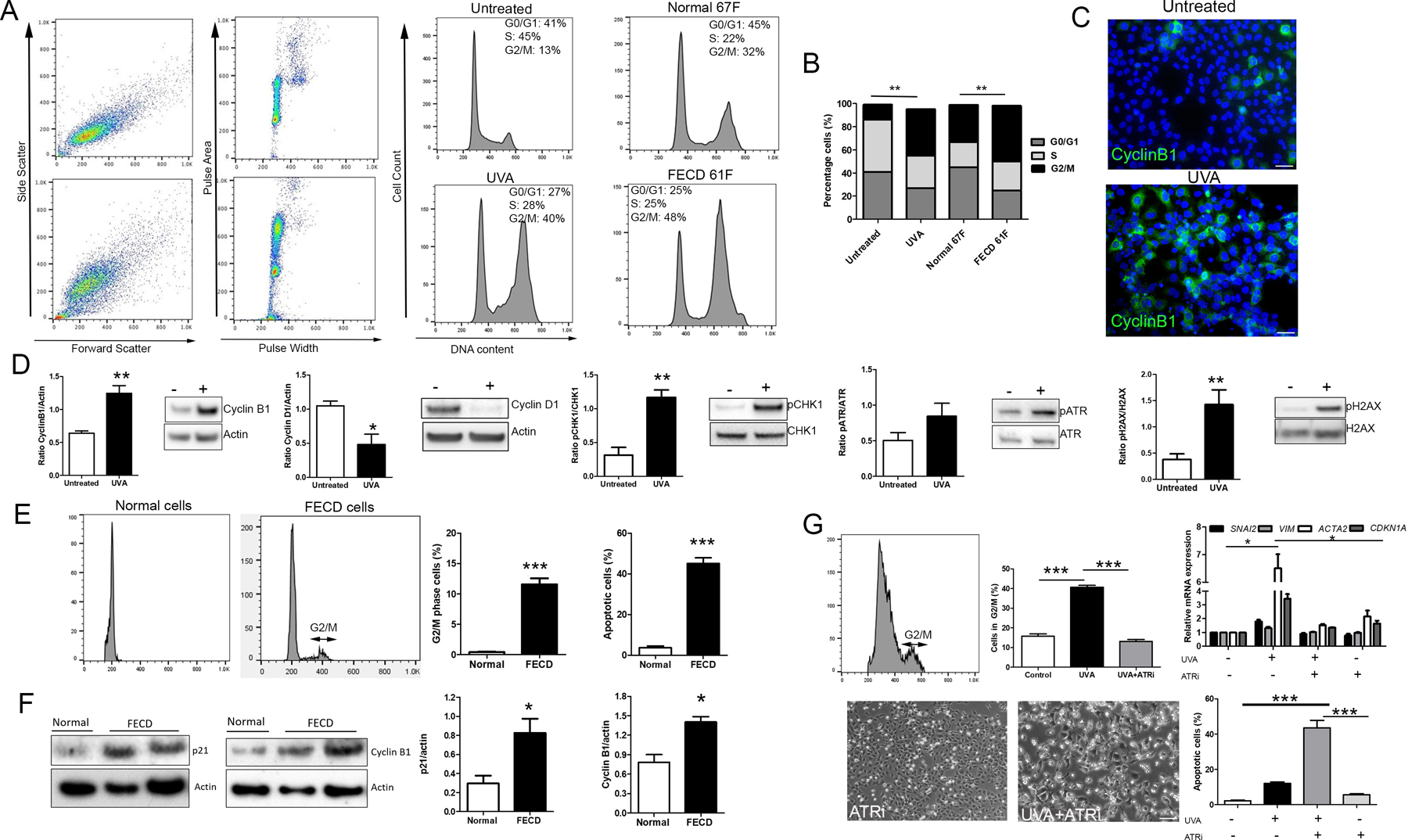
UVA light induced G2/M phase arrest is attenuated by ATR inhibition. A. Cell cycle analysis by propidium iodide staining and flow cytometry for CEnCs (HCEnC-21T) +/− UVA (middle), and untreated normal and FECD cell lines (SV40 immortalized) (right). B. Percentage of cells in each stage of the cell cycle. **P < 0.01, untreated vs UVA, and normal untreated vs FECD untreated, respectively (n = 6 for each condition). C. Immunostaining of Cyclin B1 +/− UVA. D. Western blot analysis of cell cycle proteins and components of the ATR signaling pathway +/− UVA. ** P < 0.01 (n=3). E. Cell cycle analysis of normal (n=3) and FECD (n=17, pooled) CEnCs detached from Descemet’s membrane and stained with propidium iodide. Percentage of cells in G2/M phase, and percentage apoptotic cells were calculated from the cell cycle profiles. ***P < 0.001. F. Western blot analysis of p21 (n=5 for normal, n=9 for FECD) and Cyclin B1 (n=7 for normal, n=13 for FECD) in cells from normal and FECD tissue specimens with densities standardized against actin. *P < 0.05, ***P < 0.001 (n=3). G. Cell cycle profile of CEnCs treated with ATR inhibitor +UVA light, with percentage of cells in G2/M phase (***P < 0.001, n=3), and mRNA levels relative to control (*P < 0.05, comparison vs UVA treatment, n=3) for each condition (top). Phase-contrast microscopy and quantification of apoptotic cells (bottom). ***P < 0.001 (n=3). Data presented as mean values ± SE. Scale bars = 50µm.
We then sought to relate these findings back to non-immortalized, post-mitotic CEnCs as seen in vivo. Cells were stripped from Descemet’s membrane of both normal and FECD human tissue specimens to analyze the cell cycle status. Due to the low cell count on FECD tissue, samples were pooled together (n=17) to obtain sufficient cell numbers (Table S1). As expected, normal cells were in G0/G1 phase, whereas FECD cells produced a G2/M peak with approximately 11% cells in this phase, as opposed to no detectable G2/M peak in normal samples corroborating the notion of G0/G1 arrest in normal CEnCs (Fig 2E, Fig S1). FECD cells also produced a larger sub-G0/G1 peak (data not shown), indicative of increased apoptotic cells (3% normal vs 40% FECD). Further evidence for senescence and accumulation of cells in G2/M in FECD in human tissue was provided by analyzing protein levels, with diseased cells showing higher p21 and Cyclin B1 compared to normal cells (Fig 2F).
To confirm the role of ATR signaling in G2/M arrest/senescence and subsequent EMT/fibrosis as seen in UVA-treated cells in vitro, we inhibited ATR by pre-treating and recovering CEnCs in the presence of ATR inhibitor (ATRi) AZD6738. Combining ATRi with UVA irradiation markedly reduced cells in G2/M back to untreated levels (40% reduction vs UVA, P<0.001), with a similar reduction in CDKN1A, SNAI2, and ACTA2 mRNA expression, indicating a causal relationship between G2/M arrest and profibrotic phenotype (Fig 2G). ATR is activated as part of the DNA damage response to halt cell cycle progression and provide time for DNA repair. By inhibiting this pathway during UVA irradiation, CEnCs showed a considerable increase in apoptosis compared to UVA alone (43% vs 12%, respectively, P<0.001) (Fig G).
3.3. Cells in G2/M phase up-regulate fibrosis-associated genes
We next aimed to discover if the EMT gene expression profile in G2/M arrested cells differed to that of G0/G1 phase cells. Firstly, we separated normal and UVA-treated cells into their respective G0/G1 and G2/M phases using FACS and extracted RNA (Fig 3A). Transcription of various genes associated with EMT/fibrosis were quantified for each phase of the cell cycle (Fig 3B). In untreated cells, there was no difference between G0/G1 and G2/M. UVA light upregulated gene expression in both G0/G1 and G2/M phases compared to corresponding phases of untreated cells. Calculating the untreated/UVA ratio of mRNA expression showed significant increases in CDKN1A (2-fold), SNAI2 (2-fold), and ACTA2 (1.5-fold) in G2/M phase relative to G0/G1 phase (Fig 3C).
Figure 3.
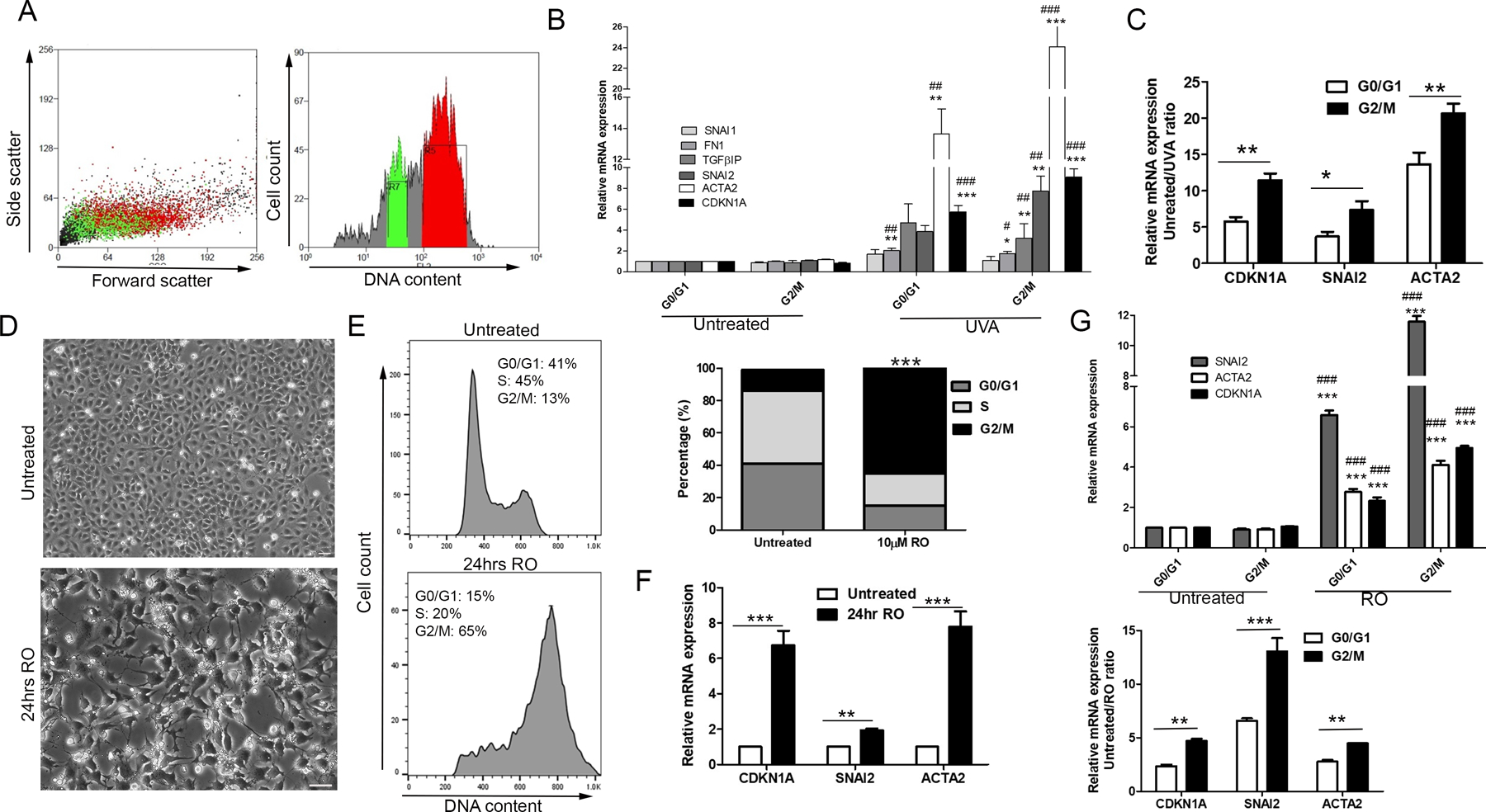
Prolonged arrest in G2/M causes profibrotic phenotype. A. FACS was used to separate G0/G1 phase cells (green) from G2/M cells (red) based on propidium iodide staining. Representative image shows UVA treated cells. B. Gene expression analysis of G0/G1 and G2/M cells, from both untreated and UVA treated CEnCs. Data is expressed as fold change vs untreated G0/G1 cells. ***P < 0.001, **P < 0.01, *P < 0.05 vs untreated G0/G1; ###P < 0.001, ##P < 0.01, #P < 0.05 vs untreated G2/M (n=3). C. CDKN1A, SNAI2, and ACTA2 mRNA expression displayed as untreated/UVA treated ratio. **P < 0.01, *P < 0.05, G0/G1 vs G2/M (n=3). D. Morphological changes induced by 24-hours treatment with 10µM RO. E. Cell cycle analysis of untreated cells (top) and RO treated cells (bottom), with each phase quantified (right). ***P < 0.001, RO G2/M vs untreated G2/M (n=3). F. Changes in total gene expression after 24 hours RO treatment displayed as fold change vs untreated. ***P < 0.001, **P < 0.01 (n=3). G. Untreated and RO treated cells sorted into G0/G1 and G2/M phases using FACS and analysis of gene expression (top). ***P < 0.001 vs untreated G0/G1; ###P < 0.001 vs untreated G2/M (n=3). Same data expressed as untreated/RO treated ratio (bottom). ***P < 0.01, **P < 0.05, G0/G1 vs G2/M (n=3). Data presented as mean values ± SE. Scale bars = 50µm.
To confirm that the profibrotic phenotype originates from cells sustained in G2/M phase, we administered RO3306 (RO), a CDK1 inhibitor that arrests cells in G2/M phase, to CEnCs for 24-hours. RO led to morphological changes, with a loss of hexagonal shape, loss of cell-cell contacts and formation of fibrotic processes (Fig 3D). Cell cycle analyses revealed that around 65% of these cells were arrested in G2/M phase after 24-hours, with only 13% in untreated (Fig 3E). Collecting RNA of RO treated cells revealed gene expression profiles remarkably like UVA-treated cells. Gene expression analysis showed upregulation of CDKN1A, SNAI2, and ACTA2 in RO treated cells (Fig 3F), with significant higher expression of these genes in G2/M phase arrested cells compared to corresponding G0/G1 cells (Fig 3G), highlighting that UVA-induced cellular changes can be induced by holding cells in G2/M, irrespective of DNA damage.
3.4. UVA light causes cell cycle re-entry leading to senescence and fibrosis in vivo
The next aim was to determine if UVA light induces senescence and EMT through G2/M arrest in vivo, where CEnCs are quiescent with minimal proliferative capacity. Mice eyes were irradiated with 500J/cm2 UVA light and analyzed following three recovery times points: 24-hours, 1-week, and 2-weeks post-UVA. Based on unpublished observations, 500J/cm2 was deemed an optimal intensity to induce damage without significant apoptosis.
We hypothesized that UVA induced DNA damage activates the cell cycle within the first 24-hours, resulting in G2/M phase arrest/senescence, followed by EMT and subsequent fibrosis at later time points. To check for cell cycle activation, we looked for Ki67 staining in CEnCs using whole mount corneas (+/− UVA light). Indeed, 24 hours-post UVA irradiation, mouse CEnCs showed signs of proliferative activity through nuclear expression of Ki67, suggesting that the cell cycle machinery had been activated (Fig 4A). Additionally, UVA irradiated mouse CEnCs contained significantly higher levels of p21 and Cyclin B1 protein compared to untreated eyes, indicative of G2/M arrest (Fig 4B).
Figure 4.
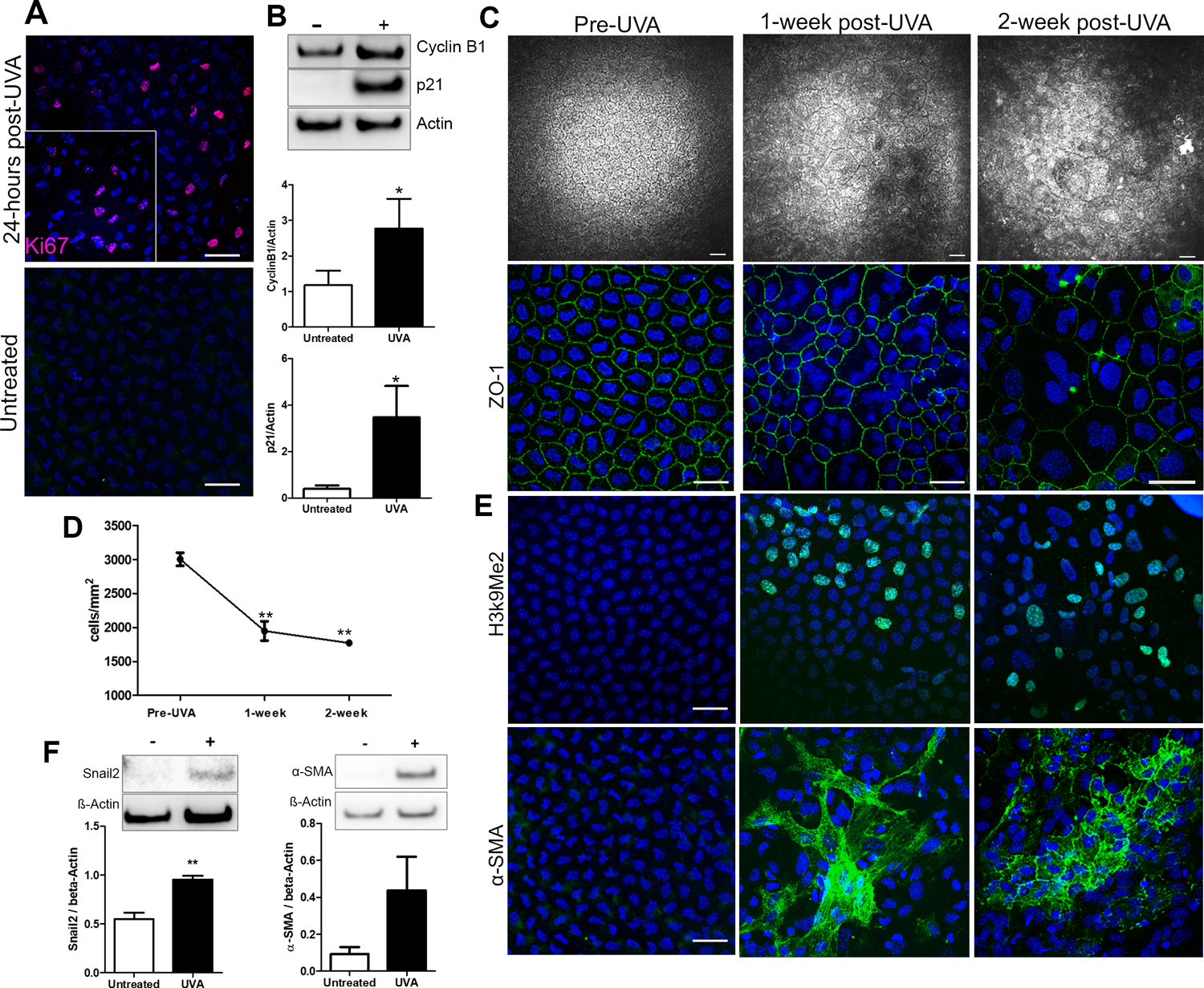
UVA light activates the cell cycle leading to senescence and fibrosis in vivo. A. Ki67 expression in mouse CEnCs 24-hours post-UVA irradiation (top) and untreated (bottom). B. Representative western blots and densitometry analysis of Cyclin B1 and p21 levels in mouse CEnCs untreated vs 24-hours post UVA irradiation (n=5). *P < 0.05. C. Progressive changes in mouse CEnC morphology from untreated (Pre-UVA) to 1- and 2-week post-UVA irradiation. Top panel displays HRT images, bottom panel shows ZO-1 staining. D. Cell density analysis from pre-UVA to 2-weeks post-UVA (n=3). **P<0.001 comparison between post-UVA groups and pre-UVA. E. H3k9Me2 (top) and α-SMA (bottom) staining in mouse CEnCs in untreated eyes, 1-, and 2-weeks post-UVA irradiation. F. Representative western blots and densitometry analysis of Snail2 and α- SMA levels in mouse CEnCs untreated vs 2-weeks post-UVA irradiation (n=5). **P < 0.001. Scale bars = 50µm.
By using HRT (Heidelberg Retinal Tomography) imaging to view cellular morphology in vivo, it was evident that UVA irradiation disrupted the normal hexagonal mosaic of corneal endothelial cells, where larger cells seen 1-week post-UVA, characteristic of senescence, which progressively worsened at 2 weeks post-UVA (Fig 4C), whereas CEnCs in the untreated eye were unaffected (Fig S2). This was accompanied by a significant decrease in CEnC density from pre-UVA (3000 ±97 cells/mm2) to 1-week post-UVA (1950 ±140 cells/mm2, P<0.01), without further significant reduction at 2-weeks post-UVA (Fig 4D). To further investigate cellular morphology, we fluorescently labelled the endothelium with junctional marker ZO-1 and imaged cellular borders using confocal microscopy. At 1-week post-UVA, the hexagonal mosaic seen prior to UVA was disrupted with the presence of large multi-nucleated cells that was more extensive at 2-weeks post-UVA (Fig 4C), corresponding with HRT images. Additionally, greater variability in nuclear size was observed at the 2-week time point. Interestingly, UVA light did not affect to cell morphology in the periphery of the endothelium, similar to findings in FECD patients (Liu et al., 2020) (Fig S3).
Next, we checked if cell cycle activation proceeded to senescence and fibrosis and 1- and 2-weeks post-UVA. Senescence was detected by the presence of senescence associated heterochromatic foci (SAHF), visualized by the presence of di-methylation of Lys9 on histoneH3 (H3k9Me3) (Fig 4E). α-SMA, a cytoskeletal protein expressed by myofibroblasts, was clearly present at 1- and 2-weeks post-UVA, suggesting that a portion of cells had already undergone EMT at these time points (Fig 4E). This was confirmed with western blot, where the EMT markers Snail2 and α-SMA showed increased levels at 2-weeks post-UVA compared to untreated CEnCs (Fig 4F). Interestingly, untreated CEnCs in aged mice (12–14 months) showed characteristics of UVA-treated young mouse corneal endothelium, including multi-nucleated cells and positive H3k9Me2 staining (Fig S4), highlighting how UVA exposure accelerates the ageing process in CEnCs.
3.5. Anti-oxidants and senolytics alleviate UVA-induced fibrosis
It is widely accepted that the presence of senescent cells is detrimental to tissues, and many therapies aim to selectively remove these cells. Given that our results imply a mechanistic link between G2/M arrested (senescent) cells and EMT leading to fibrosis, the next aim was to attenuate this fibrotic response by reducing the number of senescent cells. For the first treatment, we pre-treated cells with the anti-oxidant quercetin before UVA-irradiation to reduce reactive oxygen species (ROS) induced DNA damage and subsequent senescence. For the second treatment we used the senolytic cocktail of quercetin combined with dasatinib (DQ), following UVA irradiation to selectively target senescent cells (Fig 5A). We believe that these two models represent both early and late-stage therapeutic intervention for FECD.
Figure 5.
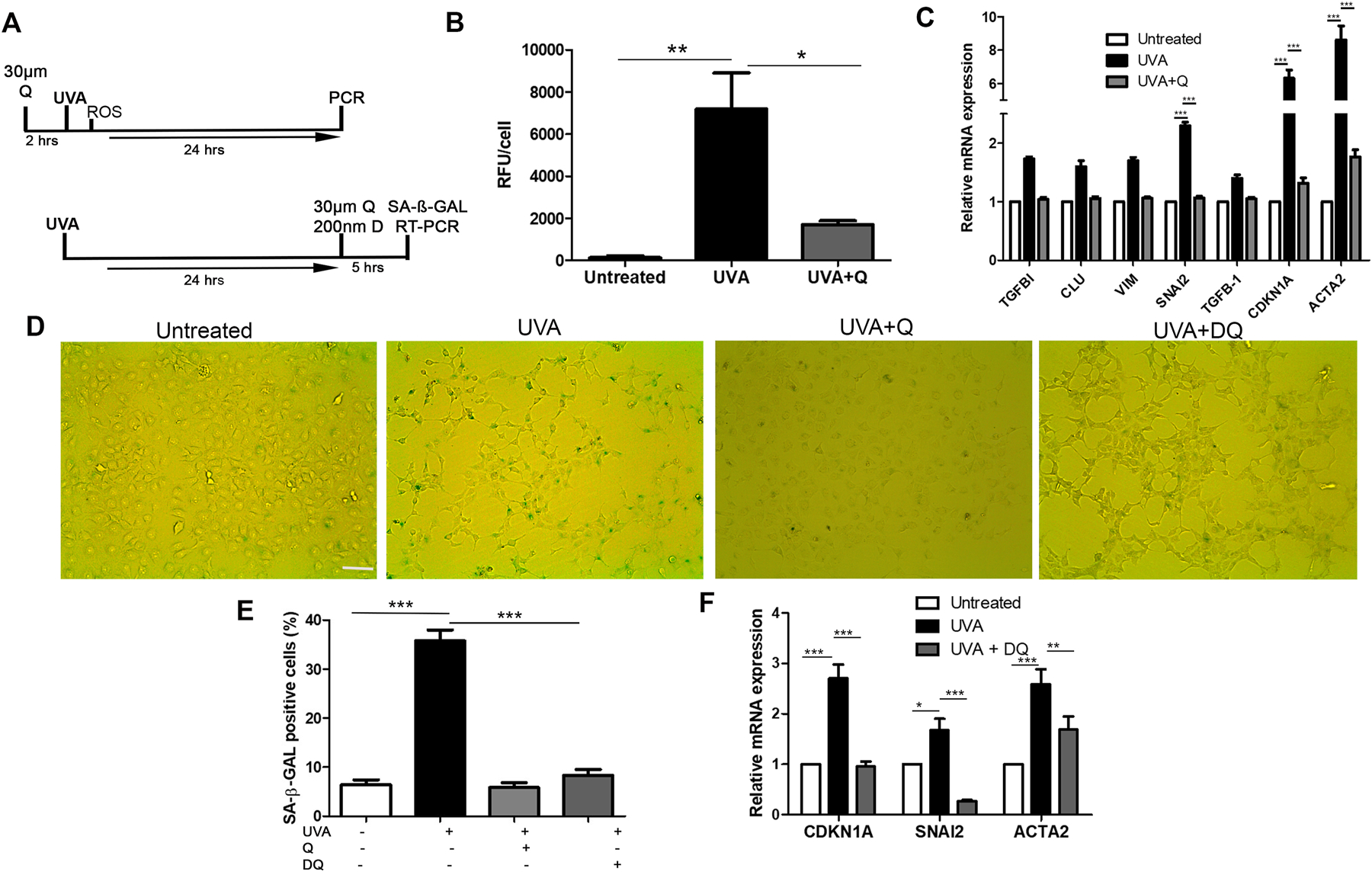
Preventing accumulation of senescent cells mediates fibrosis. A. Timelines of two treatment protocols used. 2-hours pre-treatment with 30µm quercetin prior to UVA (top), and 5-hours 200nm dasatinib + 30µm quercetin (DQ) treatment 24-hours post-UVA (bottom). B. Intracellular ROS analysis for untreated, UVA treated, and quercetin + UVA. **P < 0.01, *P < 0.05 (n=3). C. mRNA levels for untreated, UVA treated, and UVA + quercetin pre-treatment, expressed as fold change vs untreated. ***P < 0.001 (n=3). D. SA-β-GAL staining for each treatment group. E. Quantification of SA-β-GAL positive cells in each treatment group. ***P < 0.001 (n=3, with 10 random areas analyzed in each sample). F. mRNA levels for untreated, UVA treated, and UVA + DQ treatment, expressed as fold change vs untreated. ***P < 0.01, **P < 0.01, *P < 0.05. Data presented as mean values ± SE. Scale bars = 100µm.
Immediately after UVA irradiation, ROS levels were significantly elevated in comparison to untreated cells. By pre-treating the cells for 2-hours with quercetin, we were able to drastically reduce the UVA induced ROS (Fig 5B). This correlated with mRNA expression, where pre-treatment with the ani-oxidant returned EMT-associated gene expression back to baseline (Fig 5C). Furthermore, quercetin prevented the formation of fibrotic phenotype and senescence, as indicated by SA-β-GAL staining, with cells appearing like untreated controls (Fig 5D). After inducing the fibrotic phenotype with UVA light, treatment with DQ for 5 hours was able to substantially reduce the number of SA-β-GAL positive cells from 35% (±2.2) to 8% (±1.2)(P<0.001) (Fig 5E). In similar fashion to quercetin treatment, reduction in senescent cells following DQ treatment led to down regulation of CDKN1A, SNAI2, and ACTA2. Given the causal relationship between senescence and EMT, both therapeutic approaches alleviate the fibrotic phenotype in CEnCs.
4. Discussion
FECD is a complex, multifactorial disease involving interplay between various genetic defects and environmental factors, leading to endothelial cell loss, fibrosis in the form of extracellular deposits (guttae), and ultimately loss of vision (Ong Tone et al., 2020). Many of the observable phenotypic characteristics of FECD can be seen in a recently developed mouse model by exposing the corneas to UVA light (Liu et al., 2020). We previously reported evidence of oxidative stress induced EMT in CEnCs both in vitro and ex vivo using menadione as a stressor to induce intracellular ROS production (Katikireddy et al., 2018). In FECD, excessive deposition of ECM in the form of guttae leads to further loss of adjacent endothelial cells which leads to cellular remodeling, disruption the hexagonal monolayer, and edema due to functional impairment of the endothelium to pump excess fluid out of the stroma. Although a role for EMT in FECD has been established, the molecular pathophysiology is poorly understood. Furthermore, it is widely accepted that CEnCs are arrested in G0/G1 phase in vivo, with minimal proliferative capacity (Joyce et al., 1996a); however, this is the first study showing that in FECD paradoxical G2/M cell cycle arrest leads to changes in the phenotypic footprint of corneal endothelium during the degenerative changes.
Using UVA light as a physiologic stressor of the eye, we have shown a strong correlation between cell cycle status and EMT, with senescent cells arrested in G2/M phase showing increased profibrotic phenotype. In vivo, senescence and fibrosis are preceded by cell cycle re-entry, which has not previously been reported in post-mitotic CEnCs. The senolytic cocktail DQ was able to reduce the percentage of UVA-induced senescent cells and subsequently attenuate EMT, highlighting the potential for an anti-fibrotic therapy for FECD.
During EMT, cells lose contact with their neighbors and gain mesenchymal properties. In vitro, UVA irradiation simulated fibrotic phenotypic changes as seen previously with menadione treatment (Katikireddy et al., 2018, Halilovic et al., 2016, Miyajima et al., 2020) and up-regulated various genes associated with EMT. Throughout, we focused on SNAI2, and ACTA2. The Snail superfamily are prominent inducers of EMT that are known to be upregulated by oxidative stress and TGFβ, the latter of which has been shown to be elevated in the aqueous humor of FECD patients (Matthaei et al., 2015). Furthermore, the role of oxidative stress induced TGFβ and its downstream effectors in FECD is well established (Jurkunas, 2018, Katikireddy et al., 2018). The role of Snail1 has been implicated in excessive ECM production in FECD (Okumura et al., 2015), and has been shown to be up-regulated in FECD ex vivo cells (Katikireddy et al., 2018). Our data suggests that UVA light induces Snail driven EMT in CEnCs which proceeds to fibroblast formation and fibrotic response.
UVA light simultaneously induced cellular senescence in cycling CEnCs in vitro, which corresponded with FECD endothelial cells from surgical specimens. Normal CEnCs are quiescent in vivo, although they have the ability to proliferate in culture. In contrast, primary FECD cells are difficult to culture, likely due to the presence of a high proportion of senescent cells. It has been shown that normal primary CEnCs lose their proliferative capacity in culture following DNA damage (Joyce et al., 2011), and that FECD endothelium has a oxidant-antioxidant imbalance, which in turn leads to oxidative DNA damage (Jurkunas et al., 2010), indicating a role for senescence in FECD. Senescence was originally thought to occur at the G1 checkpoint, until the discovery that p21 could also induce G2/M arrest. In our study, the arrest in G2/M is triggered by activation of DNA damage response protein ATR and downstream phosphorylation target CHK1, ultimately resulting in inactivation of the key mitotic regulator, cyclin B1/CDK1 complex. This purported protective mechanism prevents the cell from replicating damaged DNA. In CEnCs, UVA light induced senescence in vitro via upregulation of p21 and subsequent G2/M phase arrest 24 hours post-irradiation. We also detected significantly higher G2/M phase cells in both untreated FECD cell line and cells from FECD tissue specimens. Surgical tissue specimens often contain a more central region of confluent guttae with low number of cells, and a ‘healthier’ peripheral region with often a normal looking hexagonal monolayer. The low percentage of cells in G2/M from FECD specimens may account for the analysis including ‘healthier’ peripheral cells in addition to centrally located diseased cells. These observations can be related to UVA-irradiated mice corneas, which showed normal endothelial structure at the periphery, likely due to increased absorption of UVA light in the central cornea (Doutch et al., 2012).
Senescent UVA irradiated cells arrested in G2/M showed upregulation of EMT-associated genes compared to G0/G1 phase cells. These observations could be simulated by holding cells in G2/M by inhibiting CDK1, indicating that G2/M arrest mediates EMT and the fibrotic response in CEnCs. Associations between G2/M and fibrosis have been made in proliferative kidney epithelial cells. In proximal tubular cells, G2/M arrested cells upregulated profibrotic cytokine production, which could be prevented by bypassing G2/M arrest (Yang et al., 2010). Conversely, in kidney tubular epithelial cells, it has been suggested that EMT precedes G2/M arrest, whereby deletion of Snail1 restored cell proliferation (Lovisa et al., 2015). We propose that persistent DNA damage to corneal endothelium leads to DNA damage response and permanent G2/M arrest, resulting in senescent cells that secrete pro-inflammatory cytokines/growth factors that affect both themselves (paracrine) and/or neighboring cells (paracrine), leading to profibrotic phenotypic changes and subsequent ECM deposition. This feature of senescent cells is best known as senescence associated secretory phenotype (SASP). It is likely that growth factors secreted from senescent cells, including TGFβ, act of the surrounding environment to positively re-enforce growth arrest and fibrotic response (Wu et al., 2013, Salama et al., 2014). Interestingly, in idiopathic pulmonary fibrosis, SMA-expressing fibroblasts responsible for ECM production exhibit senescent phenotype, such as increased p21 and positive SA-β-GAL staining (Álvarez et al., 2017), suggesting that EMT and senescence may not be mutually exclusive.
Our in vivo UVA mouse model has been shown to produce similar degenerative endothelial changes to those seen in FECD (Liu et al., 2020). Building on this previous data, we have provided evidence of UVA light-induced cell cycle re-entry for the first time in post mitotic CEnCs, whereby cells transition beyond G0/G1 phase. This was followed by senescence and fibrosis at later timepoints. Comparable to our findings in CEnCs, there is an abundance of evidence suggesting that post mitotic neurons re-enter the cell cycle following damage and contribute to the progression of various neurodegenerative conditions (Fielder et al., 2017, Frade and Ovejero-Benito, 2015), where persistent DNA damage leads to a senescence-like phenotype (Jurk et al., 2012). As seen in neurons, CEnCs may have to activate the cell cycle to both initiate DNA repair and apoptosis (Schwartz et al., 2007, Kruman et al., 2004). We suspect that persistent DNA damage from UVA light in CEnCs leads to cell cycle activation, transition from G0/G1 phase, and subsequent arrest in G2/M. If DNA damage cannot be repaired, cells undergo apoptosis accounting for cell loss seen both in FECD and our mouse model (Liu et al., 2020), or survive in a senescence-like state (large, polyploid cells) instigating onset of fibrosis. Further studies are needed to determine whether senescent cells secrete cytokines that induce EMT and fibroblast formation, or whether damaged cells simultaneously become senescent-like fibroblasts that both secrete ECM (leading to guttae formation) and have SASP-like characteristics. The latter is most likely the case in CEnCs, as cells arrested in G2/M upregulate both EMT and senescence markers.
The in vitro therapeutic models used in our study aimed at preventing senescence and apoptosis at early stage (Q pre-treatment) and clearing senescent cells at late stage (DQ cocktail), showed potential to mediate fibrosis. It is thought that DQ cocktail transiently disables networks of senescent cell anti-apoptotic pathways that enable senescent cells to survive (Schafer et al., 2017). Similar interventions are being tested in fibrotic lung disease, with studies showing that elimination of senescent cells in idiopathic pulmonary fibrosis attenuates fibrosis and restores lung function in mice with experimental lung fibrosis (Pan et al., 2017, Schafer et al., 2017, Lehmann et al., 2017). Interestingly, senolytic therapy for pulmonary fibrosis has been tested for the first time in a pilot clinical study, where treatment significantly and clinically-meaningfully improved physical function (Justice et al., 2019). Future studies will test this treatment in our UVA-induced mouse fibrosis model reported here to determine if cornea function can be improved by clearing senescent cells.
5. Conclusions
In conclusion, this study reports a novel mechanism involved in FECD pathogenesis that is applicable to other degenerative conditions, where stress-induced premature ageing of post mitotic cells in associated with fibrotic response. Selective elimination of senescent cells is a promising new therapeutic intervention to improve tissue function and slow down disease progression.
Supplementary Material
Supplement Figure 1. Cell cycle analysis of CEnCs obtained from normal and FECD tissue specimens.
Supplement Figure 2. HRT images of mouse corneal endothelium from the corresponding untreated eye before UVA treatment, plus 1- and 2-weeks later.
Supplement Figure 3. Mouse CEnCs from the periphery and center of the cornea 2-weeks post-UVA irradiation, stained with DAPI (blue) and ZO-1 (green). Scale bar = 50µm.
Supplement Figure 4. Aged (12-months), untreated mouse CEnCs stained with H3k9Me3 and ZO-1, showing the presence of multi-nucleated cells (white arrow). Scale bar = 50µm.
Supplement Table 1. Details of the human FECD and normal specimens used for each experiment
Supplement Table 2. Details of antibodies used for western blot and immuno-labelling
Highlights.
UVA light induced EMT and senescence in CEnCs in vitro and in vivo
In vivo, UVA light activated the cell cycle in post-mitotic CEnCs
Senescent cells arrested in G2/M phase were responsible for the resulting profibrotic phenotype
Eliminating senescence cells with a senolytic cocktail of DQ mediated UVA-induced fibrosis
Senolytic therapy is a potential therapeutic intervention for FECD
Acknowledgements
This work was supported by NIH/National Eye Institute Grant R01EY020581 (to U.V.J.), an Alcon Young Investigator Grant (to U.V.J.), Core Grant P30EY003790, a Research to Prevent Blindness Award (to U.V.J.), an Alcon Research Institute Young Investigator Award (to U.V.J.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors report no conflict of interest.
References
- Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D’cunha H, Sembrat J, Nouraie M, Shanker S, Caufield C, Shiva S, Armanios M, Mora AL & Rojas M 2017. Ipf lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol, 313, L1164–l1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Pérez-Moreno M, Rodrigo I et al. 2000. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol, 2, 76–83. [DOI] [PubMed] [Google Scholar]
- Chen KH, Harris DL & Joyce NC 1999. Tgf-beta2 in aqueous humor suppresses s-phase entry in cultured corneal endothelial cells. Invest Ophthalmol Vis Sci, 40, 2513–9. [PubMed] [Google Scholar]
- Childs BG, Durik M, Baker DJ & Van Deursen JM 2015. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat Med, 21, 1424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doutch JJ, Quantock AJ, Joyce NC & Meek KM 2012. Ultraviolet light transmission through the human corneal stroma is reduced in the periphery. Biophys J, 102, 1258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eye Bank Association of America. Eye Banking Statistical Report. Washington, DC: EBAA; 2017 [Google Scholar]
- Fielder E, Von Zglinicki T & Jurk D 2017. The DNA damage response in neurons: Die by apoptosis or survive in a senescence-like state? J Alzheimers Dis, 60, S107–s131. [DOI] [PubMed] [Google Scholar]
- Frade JM & Ovejero-Benito MC 2015. Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle, 14, 712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halilovic A, Schmedt T, Benischke AS, Hamill C, Chen Y, Santos JH & Jurkunas UV 2016. Menadione-induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in fuchs endothelial corneal dystrophy. Antioxid Redox Signal, 24, 1072–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N & Gil J 2018. Mechanisms and functions of cellular senescence. J Clin Invest, 128, 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce NC, Harris DL & Zhu CC 2011. Age-related gene response of human corneal endothelium to oxidative stress and DNA damage. Invest Ophthalmol Vis Sci, 52, 1641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce NC, Harris DL & Zieske JD 1998. Mitotic inhibition of corneal endothelium in neonatal rats. Invest Ophthalmol Vis Sci, 39, 2572–83. [PubMed] [Google Scholar]
- Joyce NC, Meklir B, Joyce SJ & Zieske JD 1996a. Cell cycle protein expression and proliferative status in human corneal cells. Invest Ophthalmol Vis Sci, 37, 645–55. [PubMed] [Google Scholar]
- Joyce NC, Navon SE, Roy S & Zieske JD 1996b. Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Invest Ophthalmol Vis Sci, 37, 1566–75. [PubMed] [Google Scholar]
- Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron K & Von Zglinicki T 2012. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell, 11, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkunas UV 2018. Fuchs endothelial corneal dystrophy through the prism of oxidative stress. Cornea, 37 Suppl 1, S50–s54. [DOI] [PubMed] [Google Scholar]
- Jurkunas UV, Bitar M & Rawe I 2009. Colocalization of increased transforming growth factor-beta-induced protein (tgfbip) and clusterin in fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci, 50, 1129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkunas UV, Bitar MS, Funaki T & Azizi B 2010. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am J Pathol, 177, 2278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkunas UV, Rawe I, Bitar MS, Zhu C, Harris DL, Colby K & Joyce NC 2008. Decreased expression of peroxiredoxins in fuchs’ endothelial dystrophy. Invest Ophthalmol Vis Sci, 49, 2956–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice JN, Nambiar AM, Tchkonia T, Lebrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N & Kirkland JL 2019. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine, 40, 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katikireddy KR, White TL, Miyajima T, Vasanth S, Raoof D, Chen Y, Price MO, Price FW & Jurkunas UV 2018. Nqo1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in fuchs endothelial corneal dystrophy. Free Radic Biol Med, 116, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konomi K, Zhu C, Harris D, Joyce NC. 2005. Comparison of the proliferative capacity of human corneal endothelial cells from the central and peripheral areas. Invest Ophthalmol Vis Sci. 46, 4086–91. [DOI] [PubMed] [Google Scholar]
- Kruman Ii, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R Jr., Gorospe M & Mattson MP 2004. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron, 41, 549–61. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Günther A & Königshoff M 2017. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen Y, Kochevar IE & Jurkunas UV 2014. Decreased dj-1 leads to impaired nrf2-regulated antioxidant defense and increased uv-a-induced apoptosis in corneal endothelial cells. Invest Ophthalmol Vis Sci, 55, 5551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Miyajima T, Melangath G, Miyai T, Vasanth S, Deshpande N, Kumar V, Ong Tone S, Gupta R, Zhu S, Vojnovic D, Chen Y, Rogan EG, Mondal B, Zahid M & Jurkunas UV 2020. Ultraviolet a light induces DNA damage and estrogen-DNA adducts in fuchs endothelial corneal dystrophy causing females to be more affected. Proc Natl Acad Sci U S A, 117, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovisa S, Lebleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M & Kalluri R 2015. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med, 21, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaei M, Gillessen J, Muether PS, Hoerster R, Bachmann BO, Hueber A, Cursiefen C & Heindl LM 2015. Epithelial-mesenchymal transition (emt)-related cytokines in the aqueous humor of phakic and pseudophakic fuchs’ dystrophy eyes. Invest Ophthalmol Vis Sci, 56, 2749–54. [DOI] [PubMed] [Google Scholar]
- Matthaei M, Hu J, Kallay L, Eberhart CG, Cursiefen C, Qian J, Lackner EM & Jun AS 2014. Endothelial cell microrna expression in human late-onset fuchs’ dystrophy. Invest Ophthalmol Vis Sci, 55, 216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaei M, Meng H, Meeker AK, Eberhart CG & Jun AS 2012. Endothelial cdkn1a (p21) overexpression and accelerated senescence in a mouse model of fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci, 53, 6718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek KM, Leonard DW, Connon CJ, Dennis S & Khan S 2003. Transparency, swelling and scarring in the corneal stroma. Eye (Lond), 17, 927–36. [DOI] [PubMed] [Google Scholar]
- Miyai T, Vasanth S, Melangath G, Deshpande N, Kumar V, Benischke AS, Chen Y, Price MO, Price FW Jr. & Jurkunas UV 2019. Activation of pink1-parkin-mediated mitophagy degrades mitochondrial quality control proteins in fuchs endothelial corneal dystrophy. Am J Pathol, 189, 2061–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima T, Melangath G, Zhu S, Deshpande N, Vasanth S, Mondal B, Kumar V, Chen Y, Price MO, Price FW Jr., Rogan EG, Zahid M & Jurkunas UV 2020. Loss of nqo1 generates genotoxic estrogen-DNA adducts in fuchs endothelial corneal dystrophy. Free Radic Biol Med, 147, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Minamiyama R, Ho LT, Kay EP, Kawasaki S, Tourtas T, Schlötzer-Schrehardt U, Kruse FE, Young RD, Quantock AJ, Kinoshita S & Koizumi N 2015. Involvement of zeb1 and snail1 in excessive production of extracellular matrix in fuchs endothelial corneal dystrophy. Lab Invest, 95, 1291–304. [DOI] [PubMed] [Google Scholar]
- Ong Tone S, Kocaba V, Böhm M, Wylegala A, White TL & Jurkunas UV 2020. Fuchs endothelial corneal dystrophy: The vicious cycle of fuchs pathogenesis. Prog Retin Eye Res, 100863. [DOI] [PMC free article] [PubMed]
- Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, Wang Y, Hauer-Jensen M, Zhou D & Meng A 2017. Inhibition of bcl-2/xl with abt-263 selectively kills senescent type ii pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys, 99, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama R, Sadaie M, Hoare M & Narita M 2014. Cellular senescence and its effector programs. Genes Dev, 28, 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H & Lebrasseur NK 2017. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun, 8, 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmedt T, Chen Y, Nguyen TT, Li S, Bonanno JA & Jurkunas UV 2012. Telomerase immortalization of human corneal endothelial cells yields functional hexagonal monolayers. PLoS One, 7, e51427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EI, Smilenov LB, Price MA, Osredkar T, Baker RA, Ghosh S, Shi FD, Vollmer TL, Lencinas A, Stearns DM, Gorospe M & Kruman Ii 2007. Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle, 6, 318–29. [DOI] [PubMed] [Google Scholar]
- Senoo T, Obara Y & Joyce NC 2000. Edta: A promoter of proliferation in human corneal endothelium. Invest Ophthalmol Vis Sci, 41, 2930–5. [PubMed] [Google Scholar]
- Tripathi RC, Borisuth NS, Tripathi BJ & Fang VS 1991. Analysis of human aqueous humor for epidermal growth factor. Exp Eye Res, 53, 407–9. [DOI] [PubMed] [Google Scholar]
- Weller JM, Zenkel M, Schlötzer-Schrehardt U, Bachmann BO, Tourtas T & Kruse FE 2014. Extracellular matrix alterations in late-onset fuchs’ corneal dystrophy. Invest Ophthalmol Vis Sci, 55, 3700–8. [DOI] [PubMed] [Google Scholar]
- Wu CF, Chiang WC, Lai CF, Chang FC, Chen YT, Chou YH, Wu TH, Linn GR, Ling H, Wu KD, Tsai TJ, Chen YM, Duffield JS & Lin SL 2013. Transforming growth factor β−1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol, 182, 118–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam JC & Kwok AK 2014. Ultraviolet light and ocular diseases. Int Ophthalmol, 34, 383–400. [DOI] [PubMed] [Google Scholar]
- Yang L, Besschetnova TY, Brooks CR, Shah JV & Bonventre JV 2010. Epithelial cell cycle arrest in g2/m mediates kidney fibrosis after injury. Nat Med, 16, 535–43, 1p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinflou C & Rochette PJ 2017. Ultraviolet a-induced oxidation in cornea: Characterization of the early oxidation-related events. Free Radic Biol Med, 108, 118–128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure 1. Cell cycle analysis of CEnCs obtained from normal and FECD tissue specimens.
Supplement Figure 2. HRT images of mouse corneal endothelium from the corresponding untreated eye before UVA treatment, plus 1- and 2-weeks later.
Supplement Figure 3. Mouse CEnCs from the periphery and center of the cornea 2-weeks post-UVA irradiation, stained with DAPI (blue) and ZO-1 (green). Scale bar = 50µm.
Supplement Figure 4. Aged (12-months), untreated mouse CEnCs stained with H3k9Me3 and ZO-1, showing the presence of multi-nucleated cells (white arrow). Scale bar = 50µm.
Supplement Table 1. Details of the human FECD and normal specimens used for each experiment
Supplement Table 2. Details of antibodies used for western blot and immuno-labelling