


Abstract
Replication protein A (RPA) binds to single-stranded DNA (ssDNA) and interacts with over three dozen enzymes and serves as a recruitment hub to coordinate most DNA metabolic processes. RPA binds ssDNA utilizing multiple oligosaccharide/oligonucleotide binding domains and based on their individual DNA binding affinities are classified as high versus low-affinity DNA-binding domains (DBDs). However, recent evidence suggests that the DNA-binding dynamics of DBDs better define their roles. Utilizing hydrogen–deuterium exchange mass spectrometry (HDX-MS), we assessed the ssDNA-driven dynamics of the individual domains of human RPA. As expected, ssDNA binding shows HDX changes in DBDs A, B, C, D and E. However, DBD-A and DBD-B are dynamic and do not show robust DNA-dependent protection. DBD-C displays the most extensive changes in HDX, suggesting a major role in stabilizing RPA on ssDNA. Slower allosteric changes transpire in the protein–protein interaction domains and linker regions, and thus do not directly interact with ssDNA. Within a dynamics-based model for RPA, we propose that DBD-A and -B act as the dynamic half and DBD-C, -D and -E function as the less-dynamic half. Thus, segments of ssDNA buried under the dynamic half are likely more readily accessible to RPA-interacting proteins.
Graphical Abstract
Graphical abstract.

RPA binds to DNA as two dynamic halves.
INTRODUCTION
In eukaryotes, the single-strand DNA (ssDNA)-binding protein replication protein A (RPA) is essential for most DNA metabolic processes including DNA replication, repair and recombination. RPA binds to ssDNA with high affinity (KD <10−10M) (1) and protects it from degradation by exo- and endonucleases. The formation of RPA-ssDNA complexes triggers the ATM/ATR cellular DNA damage checkpoint response (2,3). RPA physically interacts with over three dozen DNA processing enzymes and recruits several of them onto DNA (4). Finally, RPA hands-off the DNA to these enzymes and correctly positions them to facilitate their respective catalytic activity (5). Plasticity for such multiplexed functional roles for RPA can be ascribed to its flexible structure and its corresponding context-dependent interactions with DNA (6,7).
RPA is a heterotrimer composed of RPA70, RPA32 and RPA14 subunits. Structurally, it can be further partitioned into six oligonucleotide/oligosaccharide-binding folds (OB-folds; denoted A-F in Figure 1A and Supplemental Figure S1). RPA70, the largest subunit, is composed of OB-folds F, A, B and C. RPA32 consists of an N-terminal phosphorylation region, OB-fold D and a C-terminal winged-helix domain (wh). RPA14 harbors OB-fold E (Figure 1A). The heterotrimer is constitutively held together by the trimerization core (Tri-C) through interactions between domains C, D and E (Figure 1A) (8–10). RPA mediates protein–protein interactions with over three dozen proteins primarily through two dedicated protein-interaction domains (PIDs) (11–13). For clarity, here we partition the OB-folds as DNA-binding domains (DBDs) and protein interaction domains (PIDs) based on their established biochemical functions. Domains A, B, C, D and E are DBDs whereas F and wh are PIDs. The F-domain in RPA70 (PID70N) and the ‘wh’ domain in RPA32 (PID32C) mediate direct physical interactions to most RPA-interacting proteins (RIPs). DBDs and PIDs are connected by flexible linkers of varying lengths and thus allow RPA to adopt a multitude of configurations on ssDNA (6,14,15). The DNA-bound crystal structure of Ustilago maydis RPA shows how the DBDs of RPA are arranged on ssDNA (16). DBD-A, -B, -C and -D engage the DNA and parlay 5′-3′ directionality with DBD-A residing toward the 5′ end (8,17–21). PID70N and PID32C are not present in this structure, and DBD-E does not contact the DNA. Most of the intervening linker regions are also disordered (i.e. flexible; electron density not visualized) in the structure.
Figure 1.

Sequence coverage of the DNA binding and protein interaction domains of RPA. (A) Top: Domain composition of human RPA70, RPA32 and RPA14 subunits and their respective linkers. The red dotted line in the N-terminus of RPA32 denotes the region of phosphorylation. Bottom: The crystal structures of the individual domains of RPA are depicted and color coded to match the domains on the top panel. For domain C, DNA is modeled onto DBD-C using SWISS-MODEL with the Ustilago maydis structure as reference. Peptides identified in the MS analysis corresponding to (B) RPA70, (C) RPA32 and (D) RPA14 are shown. 56 peptides from RPA70 (59% sequence coverage), 17 peptides from RPA32 (50% sequence coverage) and 11 peptides from RPA14 (49% sequence coverage) were identified. The y-axis indicates the peptide fragment number, and the x-axis denotes peptides fragments arranged in order of increasing residue numbers from the N-terminus of each subunit as denoted in Supplementary Table S1.
Although RPA binds to ssDNA with high affinity, it must be removed by RIPs that bind ssDNA with much lower affinity. This is achieved by the differential DNA-binding properties of individual DBDs (22). In solution, human RPA displays multiple NaCl concentration-dependent DNA-binding modes denoted by the occluded site-size (23). This varies from 22 nucleotides (nts) in conditions below 50 mM NaCl to 28–30 nts at higher salt concentrations of up to 1.5 M NaCl. These binding modes are propagated by selective binding of one or more DBDs. Furthermore, transitions between the DNA-binding modes likely drive the diffusion and remodeling of RPA on ssDNA (24). Thus, multiple configurations of RPA on ssDNA can be formed by using one or a subset of the DBDs. Two models have been proposed for the utilization of DBDs: a sequential DBD-binding model posits that smaller occluded site-sizes are likely due to the higher affinity ssDNA-binding properties of DBD-A and -B that bind first onto DNA (25). Subsequent binding of the other DBDs in the trimerization core drive the 30 nt mode. An alternate dynamic DBD engagement model is built on the intrinsic on–off properties of each DBD with no particular emphasis on the order of DBD binding (22). Both models are built on decades of DNA-binding studies of isolated DBDs, where DBD-A and -B are classically defined as the high affinity binders with DBD-C and -D as low affinity ssDNA-binding domains (26–28). A few studies have also reported DBD-E and PID70N to possess very weak DNA-binding properties (29,30). In both scenarios, a canonical bias exists that DBD-A and -B are drivers of stable or robust complex formation between RPA and ssDNA. Therefore, many models also invoke that RIPs will have to remove the interactions between DBD-A and -B and DNA to displace or remodel RPA.
Recent cryoEM and single molecule studies present a more nuanced view of the DBDs of RPA (22,31,32). CryoEM structures of Saccharomyces cerevisiae RPA on longer ssDNA (100 nts) reveals an interaction between the DBDs when multiple RPA molecules are bound (32). In these structures, the resolution of DBD-A and -B are poor compared to Tri-C. Furthermore, DBD-A from one RPA molecule physically interacts with DBD-E of the neighboring RPA molecule. Such an arrangement opens up segments of ssDNA bound between two RPA molecules, likely acting as DNA-binding regions for RIPs (32). In the single-molecule experiments, DBD-A and DBD-D have four binding states on DNA with the lifetime of each state differing between the two DBDs (22,31). Associated bulk fluorescence kinetic experiments have shown conditional displacement of DBD-A by Tri-C (31). These findings contradict the high-affinity binding label ascribed to DBD-A and -B as these domains are either not bound to DNA or readily outcompeted by Tri-C. The dynamics and arrangement of individual DBDs can also be altered by post-translational modifications and interactions with RIPs. Phosphorylation at S178 in S. cerevisiae RPA70, a position on the FA-linker adjacent to DBD-A, alters its positioning with respect to the neighboring DBDs (32). Similarly, the presence of Rad52, a mediator protein that functions in homologous recombination, selectively alters the number of states formed by DBD-D on ssDNA (31). This alteration enables Rad52 to load onto the 3′ end of the RPA-coated DNA by remodeling DBD-D. In contrast to classical DNA-binding studies of RPA, these new tools provide information on individual DBDs whilst investigating full-length RPA. Nevertheless, information on the DNA-binding dynamics of DBD-B, -C, -E and the PIDs are lacking due to technical limitations.
Here, we use time-resolved hydrogen–deuterium exchange mass spectrometry (HDX-MS) and full-length human RPA to investigate the protein solvent accessibility and kinetic stability of each DBD on ssDNA. HDX-MS is ideal for studying protein folding and its conformational dynamics (33–36). HDX-MS measures the rate of amide-H exchange to report on changes in the local environment. D2O exchange rates are shaped by dynamics, H-bonding, secondary structure and solvent exposure. Increased rates of D2O exchange result from greater solvent accessibility, disruption in backbone H-bonding, or increased dynamics. Decreased D2O exchange rates suggest less solvent accessibility or local stabilization (i.e. H-bonding and/or formation of secondary structure). As such, exchange rate disruptions are valuable indicators of protein allostery, dynamics and interfaces (37). In recent years, HDX-MS has gained attention as a powerful tool to study protein–DNA binding interactions (33,36,38,39). Here, using HDX-MS we identify the effects on solvent accessibility, which are interpreted in terms of protein dynamics caused by changes in the contacts between the various DBDs, linkers, and PIDs of human RPA upon complex formation with ssDNA. Based on the data, in the context of the dynamics-based model for RPA, we propose that RPA can be envisioned to function as two halves: DBD-A, DBD-B and PID70N (all in RPA70) serve as the dynamic half of RPA. Tri-C, consisting of DBD-C (RPA70), DBD-D (RPA32) and DBD-E/RPA14, functions as the less dynamic half with DBD-C serving as the anchor that likely controls the residence time of RPA on ssDNA.
MATERIALS AND METHODS
HDX-MS
Human RPA was purified as described (23). RPA and DNA (dT)30 were mixed as 1:1 molar equivalents (3.9 mg/ml RPA) and diluted 1:10 into deuterated reaction buffer (30 mM HEPES, 200 mM KCl pD 7.8 in D2O). Undeuterated controls (zero time points) were diluted 1:10 into nondeuterated buffer. About 10 μl of the reaction was removed at each time point (0.008, 0.05, 0.5, 3 and 24 h) and quenched on ice by adding 60 μl of 0.75% formic acid (FA, Sigma) pH 2.5 containing 0.25 mg/ml porcine pepsin (Sigma). Each sample was digested for 2 min with vortexing every 30 s and then flash frozen in liquid nitrogen. Samples were stored in liquid nitrogen until liquid chromatography mass spectrometry (LC-MS) analysis. All reactions were performed in triplicate.
LC-MS analysis of RPA was performed on a 1290 UPLC series chromatography stack (Agilent Technologies) coupled directly to a 6538 UHD Accurate-Mass QTOF LC/MS mass spectrometer (Agilent Technologies). Before electrospray−time-of-flight (ESI-TOF) analysis, peptides were separated on a reverse phase (RP) column (Phenomenex Onyx Monolithic C18 column, 100 × 2 mm) at 1°C using a flow rate of 500 μl/min under the following conditions: 1.0 min, 5% B; 1.0−9.0 min, 5−45% B; 9.0−11.8 min, 45–95% B; 11.80−12.0 min, 5% B; solvent A = 0.1% FA in water (Thermo-Fisher) and solvent B = 0.1% FA in acetonitrile (ACN, Thermo-Fisher). Data were acquired at 2 Hz over the scan range 50−1700 m/z in positive mode. Electrospray settings were as follows: nebulizer set to 3.7 bar, drying gas at 8.0 l/min, drying temperature at 350°C, and capillary voltage at 3.5 kV.
Peptides were identified as outlined in Berry et al. (34) using MassHunter Qualitative Analysis version 6.0 (Agilent Technologies), Peptide Analysis Worksheet (PAWs, ProteoMetrics LLC), and Peptide Shaker version 1.16.42 paired with Search GUI version 3.3.16 (Compomics). Deuterium uptake was determined using HDExaminer version 2.5.1 (Sierra Analytics).
Data analysis
Relative deuterium uptake difference (ΔHDX) summed over all HDX incubation periods was calculated using Equation (1) (40).
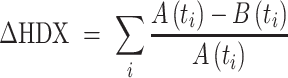 |
The ΔHDX between bound and free RPA was calculated with the above equation, where A is the deuterium uptake for sample A (RPA+DNA) at one time point (ti), and B is the deuterium uptake for sample B (free RPA) at the same time point (ti). Summation of ΔHDX at all five time points together gives ΔHDX for a peptide (Figure 2). Mean of the deuterium uptake value of a peptide was used to generate a heat map and plotted using GraphPad Prism 8.4.1. Crystal structures were rendered using PyMOL (Schrödinger, LLC). The uptake curve for each peptide was plotted using a nonliner/nonlogarithmic scale using MS-Excel. Each data point represents the mean value, and error bars represent the ±SD of mean for three biological replicates. Peptides with a SD> 0.5 were excluded from this study. To compare the difference at time points with ±DNA statistical significance were determined. *, the first incubation time point that shows statistical difference is indicated in the linear plot. No significance is indicated as ‘ns’ and P < 0.05 was considered statistically significant (* P-value < 0.05, ** P < 0.01, ***P < 0.001, two-tailed, unpaired t test). In the full-length RPA plasmid, RPA32 is his-tagged, thus HDX peptide 17 has an extra 6 His residues at its C-terminus, i.e. TDAEHHHHHH.
Figure 2.

Global deuterium uptake profiles and DNA induced changes in RPA. Heat map representing the deuterium content of (A), RPA70, (B) RPA32 and (C) RPA14 peptides in unbound and DNA bound states as a function of time. Each row represents a timepoint and each column represents a peptide. The heat map gradients are color coded yellow (low deuterium uptake) to red (high deuterium uptake). The white boxes depict absence of a corresponding dataset for specific peptides under one condition (presence or absence of DNA). • denotes peptides in DNA-binding sites, and + marks peptides from the linker regions. To map the peptide location in RPA70, RPA32 and RPA14, peptides in same region were grouped and assigned roman numerals. Binding to ssDNA leads to varying patterns of protection by HDX-MS and the differences are denoted as bar graphs below the respective heat maps. The arrows denote whether the peptides were protected or exposed to solvent upon DNA binding. The data show the cumulative difference in deuterium uptake (ΔHDX) in RPA over all five HDX timepoints.
RESULTS
HDX-MS generates a conformational landscape for DNA-induced changes in human RPA
To determine the ssDNA binding associated conformational changes in RPA, we performed HDX-MS experiments with human RPA in the absence and presence of ssDNA. A (dT)30 oligonucleotide was used to provide adequate binding sites for all the domains of RPA as the binding site-size for RPA is ∼22–28 nt (23). Since RPA binds to ssDNA stoichiometrically, we used an equimolar concentration of RPA and (dT)30. Although RPA is a large (1007 amino acids), we observe excellent sequence coverage (Figure 1B–D and Supplementary Table S1). We identified 56, 17 and 11 different peptides for the three subunits, respectively (Supplementary Table S1). This provided an overall 51% sequence coverage (RPA70, 363 of 616 aa ∼59%; RPA32, 139 of 276 aa ∼50%; RPA14, 59 of 121 aa ∼49%) with the majority of the unreported sequence in the FA-linker (Figure 1B). For much of the complex, multiple overlapping peptides were identified. These were grouped and assigned Roman numerals for clarity (Figure 2). There are 14, 8, and 5 groups for three RPA subunits, respectively (Figure 2A, B and C).
RPA binds rapidly to ssDNA (kon = 1.1 ± 0.6 × 108 M−1s−1) (31). Therefore, we reasoned direct DNA-binding induced changes would occur rapidly, while diffusion and DBD rearrangement associated deuterium exchange would occur on a longer timescale. To get a direct readout of the kinetics of deuterium exchange in the absence and presence of ssDNA, we performed the experiments as a function of time. The data are described in terms of the kinetics and patterns of HDX. In terms of the HDX kinetics, two prominent deuterium exchange patterns are observed which we classify as fast and slow exchange. Fast exchanges are deuteration that has occurred before the first time point captured in our experiments. RPA binding to ssDNA is diffusion limited; thus, the initial DNA-induced rapid conformational changes occur faster than our ability to capture them. Differences in exchange kinetics that occur only on a fast timescale suggest both rapid and stable changes on binding to ssDNA. If exchange is only observed at early time points, and the amount of deuterium taken up by the peptide does not increase over time, the exchange is consistent with lasting H-bond networks and a stable structure. Slow exchange is used here to describe deuterium exchange occurring at later time points. Slow exchange is a result of dynamic H-bond networks leading to solvent exposure. This infers slower on/off rates due to conformational changes and dynamics. Differences in HDX rates that occur over a 3–30 min interval are likely due to rearrangements of the domains’ organization and other allosteric effects, which we have classified as slow exchange.
With respect to HDX patterns, we either observe a protection or exposure of certain regions of RPA upon binding to ssDNA. In our experiments, data are collected after mixing RPA with ssDNA. Since the binding is diffusion limited, a change in uptake at the first time point (30 s) likely reflects a direct DNA-induced change. If a domain forms a long-lived stable complex on DNA, a corresponding long-lived difference in deuterium exchange for a particular peptide will be observed compared with RPA alone. We classify such a signal as protection. This can be thought of as protein surfaces that become buried upon DNA binding and do not come apart. If exchange continues at the minutes to hours time points, then those peptide regions are transiently exposed to solvent and are therefore ‘less’ protected, hence more exposed or dynamic.
Data for all the peptides are summarized as a heat map where red and yellow indicate high and low deuterium uptake, respectively (Figure 2A–C). White boxes denote absence of data as one of the corresponding peptides were not identified in the MS analysis. Peptides 1–5, and 10–56 show the highest degree of deuteration and correspond to PID70N, DBD-A, -B and -C in RPA70, respectively (Figure 2A). For RPA32, peptides 7–8 and 11–14 in DBD-D show larger changes in deuteration when bound to DNA. Peptides 15 and 17, which are in the winged-helix domain (PID32C), show less exchange (Figure 2B). Thus, both protein interaction domains in RPA (PID70N and PID32C) show minimal changes that transpire on a slower timescale, likely reflecting conformational changes driven indirectly after DNA binding to the DBDs. Peptides 5, 6 and 8–11 in DBD-E show moderate deuterium exchange, but the fast exchange kinetics coincide with the rate of DNA binding (Figure 2C). This is consistent with interactions between DBD-E and DNA as suggested by crosslinking experiments (29).
To construct a global landscape of the DNA-induced deuterium exchange in RPA, we quantified the relative deuterium uptake difference (ΔHDX) in the absence and presence of DNA (Figures 2). ΔHDX for each DNA binding experiment relative to apo RPA is summed over time for every peptide. The scale of the bars denotes the net change in deuterium uptake. A change in the upward direction denotes increased deuterium uptake (less protected or exposed) in the presence of DNA. Conversely, change in the downward direction denotes less uptake (protected) upon DNA binding. ΔHDX shows robust deuterium exchange in DBD-C with additional changes in DBD-A and DBD-B as expected from known DBD–ssDNA interactions (Figure 2A). Similarly, ΔHDX are observed for DBD-D (RPA32; Figure 2B) and DBD-E (Figure 2C). DBD-E binding to DNA has been observed in a few biochemical studies, but not in the X-ray or cryoEM structures of RPA-DNA complexes. Our data show DNA-driven fast exchanges in DBD-E and support interactions with DNA.
To best represent the ΔHDX data, we mapped the peptides identified in the MS analysis onto the available crystal structures of human RPA (Figures 3–5 and Supplementary Figures S2–S8). Since a structure of full-length human RPA is not available, we constructed one for reference using the information available for the two halves of human RPA. We used the ssDNA-bound structure of DBD-A and -B (PDB ID:1JMC (41)) and RPA Tri-C (PDB ID: 1L10 (8); Supplementary Figure S1C). However, the Tri-C structure is not in complex with DNA. Thus, we used the DNA-bound structure of Ustilago maydis RPA as reference (Supplementary Figure S1A) and modeled DNA onto human RPA Tri-C (PDB ID: 1L10) using SWISS-MODEL (42). Our data below are presented from the perspective of two dynamic halves in RPA: PID70N, DBD A and DBD-B constituting the ‘FAB half’, and the ‘Tri-C half’ made of DBD-C, DBD-D, PID32C and DBD-E.
Figure 3.

Time-resolved HDX-MS analysis of DNA-induced conformational changes in the FAB half of RPA. (A). Peptides identified in the MS analysis are mapped onto the crystal structure of PID70N, DBD-A and DBD-B and colored in red. The F-AB linker and A-B linkers are denoted by dotted lines. DNA is depicted as black sticks. (B–Q) Deuterium exchange profiles of individual peptides from the FAB half of RPA. Error bars reflect standard deviations, calculated as described in ‘Material and Methods’ section. */**/*** are P-values calculated using a two-tailed unpaired t-test between the respective time points for the RPA and RPA+DNA samples. Each star denotes t-test values that differ by an order of magnitude. P-values are denoted in Supplementary Table S2. No significance indicated as ‘ns’.
Figure 4.

Time-resolved HDX-MS analysis of DNA-induced conformational changes in DBD-C. (A) Crystal structure of the Tri-C core of human RPA composed of DBD-C, RPA32 and RPA14. The peptides identified in the MS analysis corresponding to DBD-C are denoted in red. DNA is denoted as black sticks. (B–M) Deuterium exchange profiles of individual peptides from DBD-C are shown. Error bars reflect standard deviations, calculated as described in ‘Material and Methods’. */**/*** are P-values calculated using a two-tailed unpaired t-test between the respective time points for the RPA and RPA+DNA samples. Each star denotes t-test values that differ by an order of magnitude. P-values are denoted in Supplementary Table S2. No significance indicated as ‘ns’.
Figure 5.

Time-resolved HDX-MS analysis of DNA-induced conformational changes in RPA32 and RPA14. (A) Crystal structure of the Tri-C core of human RPA composed of DBD-C, RPA32 and RPA14. The peptides identified in the MS analysis corresponding to RPA32 and RPA14 are denoted in red. DNA is depicted as black sticks. (B–Q) Deuterium exchange profiles of individual peptides from RPA32 and RPA14 are shown. Error bars reflect standard deviations, calculated as described in ‘Material and Methods’. */**/*** are P-values calculated using a two-tailed unpaired t-test between the respective time points for the RPA and RPA+DNA samples. Each star denotes t-test values that differ by an order of magnitude. P-values are denoted in Supplementary Table S2. No significance indicated as ‘ns’.
The FAB half of RPA shows highly dynamic ssDNA-dependent changes in HDX
PID70N, DBD-A and DBD-B reside in RPA70 and are connected by flexible linkers. The 79 aa FA-linker connects DBD-A to the PID70N and the shorter 10 aa long AB-linker links DBD-A and -B (Figure 3A). DBD-A and -B are well-established DNA binding domains (KD = 2 μM DBD-A; 20 μM DBD-B; 50 nM DBD-A+B) (22,27,28). Canonically, these DBDs are considered as the high affinity DNA-binding domains of RPA. For PID70N, we observe changes on longer time scales accompanied by a slow decrease in protection upon DNA binding (Figures 3B–E and Supplementary Figures S2A–C). Peptides 4 and 5 are part of the basic cleft of the proposed weak DNA-binding site of PID70N. Both peptides show similar total exchange with or without DNA, but the uptake rate is slower without DNA (Figure 3C and Supplementary Figure S2C). Peptide 6 is protected without DNA, becoming more dynamic (exchange over a long timescale), with slow conformational changes when DNA bound (Figure 3D). These observations suggest that the conformational changes in PID70N in the presence of DNA occur on a timescale well past the initial formation of the RPA–DNA complex. Thus, PID70N does not likely interact directly with DNA.
For DBD-A, six peptides were observed with varying HDX changes. Peptide15 is one of two loops (L12) at the cleft of the DNA-binding pocket (Figure 3F). This region does not show rapid HDX but slower exposure to solvent is observed. The regions away from the cleft in the α2/β4 loop do not show changes in HDX (peptides 16, and 17; Figure 3G, H and Supplementary Figure S3B). Rapid DNA-induced protection is observed behind the DNA-binding cleft, and the tail-end of this region feeds into the linker that connects to DBD-B (peptides 19 and 20; Figure 3I, J and Supplementary Figure S3D–E). In the absence of DNA, peptides 21 and 22 from the AB-linker show slow and fast deuterium exchange overtime, respectively. Upon DNA binding an increase deuterium uptake is observed for both the peptides; however, the pattern was different (Figure 3K and L). The rate of exchange for peptide 21 was impacted at the first time point, whereas peptide 22 was impacted at later time points. This suggests that protection and dynamics are altered by DNA binding. Presumably, since the linker is located closer to the DNA-binding cleft of DBD-B, greater deuterium uptake in these two peptides is likely directly tied to DNA binding (Supplementary Figures S3F and G). Overlay of apo and DNA bound DBD-A shows overall conformational changes upon DNA binding (Supplemental Figure S3H).
MS coverage for DBD-B yielded a total of 14 peptides (Figure 1B and Supplementary Table S1). The HDX changes in this domain were minimal upon DNA binding. Most peptides in this region (peptides 23, 24, 27, 29) do not show significant changes in HDX (Supplementary Figure S4A–D). Peptides 25 and 26 showed slower conformational changes upon DNA binding (Figure 3M and N). Peptide 26 in DBD-B (β3-strand) shows slow deuterium uptake kinetics with significantly decreased uptake (protection) at later time points with DNA. Regions within the DNA-binding cleft (peptides 27, 28, 29, 30) have similar D2O exchange patterns, with a minor decrease initially that is more pronounced at later time points upon DNA binding (Figure 3O and P and Supplementary Figure S4C and D). In the C-terminus of DBD-B (α2-helix; peptide 31), DNA binding leads to protection from D2O exchange (Supplemental Figure S4E). Peptide 32 shows significant DNA-induced protection at the first time point (Figure 3Q). Peptides corresponding to the BC-linker (peptides 32, 33 and 34) display dynamic changes with DNA-dependent protection (Supplementary Figure S4F and G). Along with the DNA-induced conformational changes, HDX kinetics in several regions of DBD-B fluctuate between time points, especially within the DNA-binding cleft. Comparison of apo (PDB:1FGU) and DNA-bound (PDB:1JMC) DBD-B structures reveals several regions undergoing conformational changes. Specifically, large scale changes occur in the L12 and L45 loop regions (Supplementary Figure S4H).
In the context of the full-length RPA, our data support a more dynamic model for DBD-A and -B engagement in agreement with recent single molecule fluorescence studies (31). As expected, binding to DNA is observed as rapid HDX changes are observed for both DBDs; however, the extent of protection is not consistent with a stably bound configuration. DBD-A and -B can adopt an array of configurations due to flexible and rotational effects of the AB linker (14). Our HDX data are consistent with conformational flexibility of these domains. Residues 314–316 (peptides 24–25) and 395–401 (peptides 29–30) are at the interface between DBD-A and -B in the DNA bound structure and are solvent occluded. These residues show protection upon DNA binding (Figures 2A, 3M and P and Supplementary Figure S4B and D). Similarly, peptide 20 in DBD-A that includes the linker to DBD-B also supports similar DNA binding induced protection (Figure 3J). Thus, our observations agree with the existence of various configurational states of DBD-A and -B (14,43). There are limited contacts between DBD-A and -B with a flexible linker in apo structures that are brought together upon DNA binding. Time-resolved deuterium exchange in the FAB half of RPA shows a gradual increase in deuterium uptake that is not linear. Furthermore, the amplitude of deuterium uptake does not reach saturation (Figure 3; Supplemental Figures S2–S4). Such deuterium exchange profiles are characteristic of protein–DNA interactions that are dynamic with rapid on and off rates (38,44). The dynamic DNA-binding properties of the FAB half are in agreement with recent single molecule studies that showcase multistep dynamic interactions among DBD-A, DBD-B and ssDNA (22,31).
Tri-C shows less dynamic ssDNA-dependent changes in HDX
The trimerization core (Tri-C) is composed of DBD-C (RPA70), DBD-D (RPA32), PID32C and DBD-E (Figure 4A). Structural and biochemical evidence suggests that Tri-C works as a single unit and binds to 24 nts (8). DBD-C is connected to DBD-B by the BC-linker (13 residues) and can be envisioned as a hinge holding the two halves of the RPA together (Figure 1A). HDX analysis of DBD-C, the largest DNA-binding domain in RPA, yields excellent coverage with total 24 peptides (Figure 1B and Supplementary Table S1). The HDX changes in DBD-C align well with expectations for a stable DNA-binding domain. Most differences in deuterium uptake upon DNA binding in DBD-C occurred on fast timescales and were of significant magnitude (Figures 2A and 4; Supplementary Figure S5). Part of the BC-linker and the N-terminus of DBD-C (peptides 35, 36, 37 and 39) consistently show deuteration differences at every time point with a noticeable decrease in deuterium uptake upon DNA binding (Figure 4B–E). The N-terminus is less protected on DNA binding (peptide 38, Supplementary Figure S5A). The BC-linker in the U. maydis RPA structure assumes two β-turn structures (16). The first β-turn inserts into the DNA-binding groove of DBD-B and is proposed to preclude DNA from adopting conformations observed in the 8 nt mode. In human RPA, this linker is shorter (13 residues) compared to U. maydis RPA (28 residues). However, the residues in the first β-turn are conserved (G435,435, V436,436, G437, G438). Our data likely reflect this structural transition from a flexible loop into a two β-turn upon binding to ssDNA. Furthermore, the loss in deuterium uptake amplitude upon DNA binding suggests occlusion of this region from solvent.
Upon DNA binding, major changes in the DBD-C structure are also observed around the Zn2+-finger motif. Four cysteines (C481, C486, C500 and C503) that coordinate zinc binding match peptides 41–43 and 45–46 (Figure 4F–H). Correspondingly, in our HDX analysis, peptides 43–48 have major changes in deuterium uptake (Figure 4F–J and Supplementary Figure S5). The inner surface of DBD-C containing the DNA-binding cleft shows significant decrease in deuterium exchange levels suggesting protection upon DNA binding and displays rapid exchange kinetics (peptides 43, 45; Figure 4F and G, and peptide 44; Supplementary Figure S5B). The decrease in D2O uptake reflects occlusion of this region from solvent upon DNA binding and the rapid kinetics denotes direct DNA-induced changes. Similarly, the solvent exposed outer surface region of the Zn2+-finger region showed a robust increase in deuterium uptake upon DNA binding with fast exchange kinetics (peptides 46, 47; Figures 4H and I).
Significant differences in HDX was also observed in the helix-turn-helix motif (Supplementary Figure S6, HTH; α2a- α2b- α2c; 533 to 566 aa), a region underneath the DNA-binding cleft of DBD-C (peptides 49–54, Figure 4K and L, Supplemental Figure S5C-E). Peptide 48 is protected in presence of DNA, becoming more dynamic (exchange on a long timescale), having slow movements when DNA is absent (Figure 4J). Upon DNA binding, the N-terminal half of HTH motif (peptides 49–51) showed reduction in deuterium uptake levels. The first incubation time point that shows statistical difference in deuterium uptake from the RPA alone are indicated for peptides 49–51 (Supplementary Table S2) respectively (Figure 4K and Supplementary Figure S5C and D). In contrast, the C-terminal half of HTH motif (peptides 52–54) displayed increased deuterium uptake and faster exchange kinetics (Figure 4L and M; Supplementary Figure S5E). The first time point post DNA addition that shows statistical difference in deuterium uptake are indicated for peptides 52–54 respectively (Supplementary Table S2; Figure 4L and M; Supplementary Figure S5E). Simultaneous protection and exposure in D2O exchange within this region suggests correlated conformational movements. That is, occlusion from solvent on the N-terminal side and exposure to solvent on the C-terminal side of HTH motif. It can be envisioned that DNA binding occludes the N-terminal end from deuteration by compaction that leads to conformational change that extends the C-terminal half. Indeed, in our modelled structure, we do see an extended conformation of HTH C-terminal half that can now contact DBD-D and DNA through van der Waals interactions (Supplementary Figure S6B) (16). A similar interaction between DBD-C and -D is observed in the cryoEM structure of S. cerevisiae RPA (Supplementary Figure S1B) (32). The L45 loop, connecting β-strand 4 and β-strand 5, is a flexible region in DBD-C and harbors Y581, a conserved residue that base-stacks with DNA. Another conserved aromatic residue F532 resides in β3-strand of DBD-C. Other residues in this region (R575, K577, Y581 and R586) also make extensive contacts with ssDNA (Supplementary Figure S6C). Electron density for L45 in the structure of U. maydis Tri-C is poorly defined and suggests a dynamic region that is likely ordered upon binding to DNA. Peptide 55 corresponds to the L45 loop and is protected on DNA binding at early time points with a steady increase in HDX (Supplementary Figure S5F). Our data for DBD-C showcase robust and stable changes in deuteration levels upon interaction with DNA. In comparison to the highly dynamic FAB-half of RPA, DBD-C appears to stably interact with DNA with fewer dynamic changes.
RPA32 harbors a disordered N-terminal region that can be heavily phosphorylated, DBD-D and PID32C. Seventeen total peptides for RPA32 were identified and analyzed: peptides 1–5 cover the N-terminal region and show small changes in HDX upon DNA binding. Peptides 1 and 5 show rapid DNA binding dependent changes whereas peptides 2 and 4 do not (Figure 5B–D and Supplemental Figure S7A). These data suggest that this region is undergoing conformational changes upon DNA binding, but it is interesting to note that this region is not part of the DNA-binding domain in the structures. Peptides 9–13 encompass DBD-D and correspondingly show fast exchange kinetics at early time points, indicative of direct DNA-binding induced changes in the structure (Figure 5F–J and Supplemental Figure S7). The data are consistent with DNA-dependent occlusion/compaction of the DNA-binding site. One key structural motif in DBD-D is the β4-β5 hairpin (i.e. β4-L45-β5) which is an extension of the DNA-binding site and displays rapid exchange (peptides 9 and 10) with a two-phase deuterium exchange profile observed for the β5 peptide (peptide 10). Upon DNA binding, the C-terminal α-helix shows exposed (peptide 11) and protected (peptide 12) profiles (Figure 5H–I). These changes suggest that the α-helical bundle of Tri-C is dynamic and propagates DNA-induced conformational changes.
Interestingly, PID32C (peptides 15, 16 and 17) and its linker region (peptide 13) also show changes in deuteration in the presence of DNA (Figure 5J–M and Supplementary Figure S7C). Peptide 13 corresponds to the linker connecting DBD-D and PID32C (Figures 2B and 5A) and shows a significant increase in deuterium uptake suggesting solvent exposure (Figure 5J). One possibility is that interactions exist between PID32C and DBD-D and these interactions are perturbed upon DNA binding. Altered uptake patterns are also observed in the tail-end of the helix in DBD-D (described above) and the linker. The deuteration profile of the PID32C peptides do not show robust changes (peptides 15–17; Figure 5K and L; Supplementary Figure S7C) similar to the observations for PID70N (Figure 3B–E). An overlay of apo (PDB:1L10) and DNA bound (PDB:4GNX) DBD-D structures reveals mild changes (Supplemental Figure S7D). Thus, we conclude that both the protein interaction domains in human RPA do not interact directly with ssDNA.
DBD-E is the third component of Tri-C. In the crystal structures, no direct interactions are observed between DNA and DBD-E. However, crosslinking experiments have suggested DNA interactions in this domain (29). Interestingly, in the cryoEM structure of multiple yeast RPA molecules on ssDNA, DBD-A of one RPA molecule interacts with DBD-E of the neighboring RPA (32). This interaction is further stabilized upon phosphorylation at position S178 in the FA-linker closer to DBD-A. Thus, DBD-E is an integral component of cooperative binding of RPA molecules upon phosphorylation. Eleven peptides were tracked in DBD-E (Supplementary Table S1). Our data show both fast (peptides 5–6, 8, 10) and slow (peptides 3–4, 7, 9, 10–11) D2O exchange for DBD-E. Several of the corresponding peptides (4-6, and 8–9) show DNA dependent solvent exposure (Figure 5M–Q and Supplementary Figure S8). Peptides 3 and 4 are in the β1-strand region, and significant solvent exposure was observed for peptide 4 (Figure 5M). Peptide 5 is part of L34 loop in DBD-E and also displays significant solvent exposure kinetics at 3 h (Figure 5N). There are two overlapping peptides for the β4−β5 hairpin; where peptide 6 shows rapid and robust deuterium uptake while significant change was observed for peptide 7 (Figure 5O and Supplementary Figure S8B). This suggests solvent exposure of the β4 and L45 regions in DBD-E. Analogous regions in other DBDs (DBD-B peptides 18–19; DBD-C peptides 41–44; DBD-D peptides 9–10) show large conformational changes upon DNA binding. The loop between β5 and α2 region in DBD-E was covered by peptides 8, 9 and 10. Peptide 8 that overlaps both 9 and 10 showed significant and delayed deuterium uptake (Figure 5P). Interestingly, peptides 9 and 10 showed greater exchange reflecting exposure and protection respectively (Figure 5Q and Supplementary Figure S8C). Thus, the entire DBD-E is exposed and the beginning of α2 (peptide 10) is protected (Figure 2B and Supplementary Figure S8C). However, most of the α2-helical region in DBD-E (peptide 11) is dynamic without any significant change in deuteration on DNA binding (Supplementary Figure S8D). The dynamic characteristics of the C-terminal α-helix in DBD-E is similar to the C-terminal α-helix in DBD-D (Figure 5H–I). Both these α-helices are part of the α-helix bundle in Tri-C.
Taken together, our data show that the Tri-C region of RPA is more stable compared to the FAB region. The Tri-C region of RPA shows rapid deuterium uptake in the presence of DNA and the deuteration levels suggest that the structures (H-bonding networks) are stable compared to the FAB region. Much of this stability arises from the interaction of DBD-C and DNA, with dynamic changes observed in DBD-D and -E. Thus, within Tri-C, DBD-D and DBD-E are relatively dynamic in comparison to DBD-C but are less dynamic compared to DBD-A and -B. Our data support a model for RPA where DBD-A and -B form the dynamic half and DBD-C, -D and -E form a less dynamic half of RPA. These conclusions do not suggest a loss in DNA binding for the FAB half but likely reflect faster intrinsic on-off rates for ssDNA interactions.
DISCUSSION
One of the primary roles of RPA is to protect ssDNA from nucleases and act as a platform to recruit other proteins. This is an inherently dynamic process in which RPA hands off the DNA to the incoming enzyme. The flexibility in RPA structure arises from the compartmentalization of its DBDs and PIDs that are tethered together by flexible linkers. The modular architecture allows the domains of RPA to employ both independent and coordinated DNA interactions that are tailored for specific functional outcomes (6,7,22).
Canonical models for RPA–DNA interactions primarily arose from biochemical investigations of isolated or select pairs of DBDs. DBD-A and -B are defined as high affinity DNA binding domains. Tri-C is considered the weaker DNA-binding portion of RPA. Recent single molecule and structural data suggest that these high-affinity DBDs might be more dynamic. DBD-A and -B dissociate from DNA and interact with DBD-E from the neighboring molecule when multiple RPA molecules are visualized using cryoEM (32). The Tri-C is more ordered in these structures as well. In single-molecule experiments where the DNA binding dynamics of individual DBDs are monitored, DBD-A and -D can form four distinct configurations on DNA and transition between these states. In the presence of an RPA interacting protein, one of these states is not formed (22,31). These studies provide direct support for the dynamics-based model wherein the dynamics of individual DBDs provides a mechanism to access RPA-bound ssDNA while not completely dissociating RPA (6,7,15). The single molecule experiments do not provide such information for the other DBDs or PIDs (due to technical limitations). Thus, a complete picture of the dynamics of all the domains of RPA and how they are rearranged in the presence of DNA is lacking.
The HDX data presented here provide a coarse-grain structural and kinetic landscape of the DNA-driven conformational changes (Figure 6A). The HDX profiles of all five DBDs (A, B, C, D, E) have altered exchange kinetics on a fast timescale in the presence of DNA. This result is consistent with rapid on-rates for the individual DBDs binding to DNA. However, the exchange patterns of the HDX signal varies for the individual DBDs. The HDX of DBD-A and -B rapidly re-equilibrate suggesting rapid on–off kinetics. Thus, these domains and their interactions are more dynamic in nature as denoted by the early timepoints in the HDX analysis. DBD-C, -D and -E also show large scale deuterium uptake profiles and show slow re-equilibration. HDX data at the later timepoints reveal that deuterium uptake levels off over a period of 24 h for many of their peptides suggesting formation of stable H-bonding networks (Figures 4 and 5). Thus, the DBDs in the Tri-C region are less dynamic compared to the ones in the FAB half of RPA.
Figure 6.
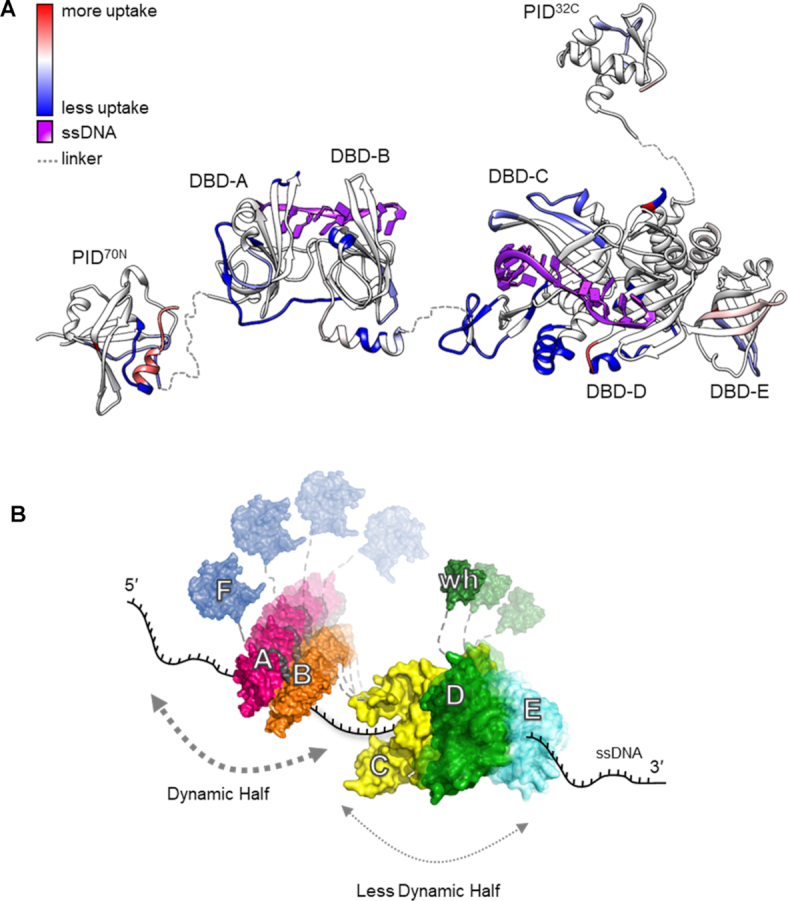
RPA binds to ssDNA using dynamic and less dynamic halves. (A) Global HDX changes in human RPA upon binding to DNA are mapped onto a composite structure. Blue and red denote the difference in scale of deuterium uptake for the unbound (RPA) versus DNA-bound (RPA+DNA) forms of RPA. DNA binding primarily drives protection of regions from deuterium uptake as they become occluded from solvent. (B) An updated dynamics-based model for RPA+DNA interactions is depicted. RPA is proposed to function as two halves (FAB and CDE/Tri-C). PID70N (F domain), DBD-A and DBD-B form the dynamic half and have short residence on time. DBD-C, RPA32 (DBD-D) and RPA14 (DBD-E) form the less dynamic half and have longer residence time on DNA owing to the stability of DNA binding arising from the larger DBD-C domain. The refined model allows RPA to be stably bound to DNA using all DBDs while providing access to both ends of the DNA due to the dynamics of the individual DBDs. PID70N and PID32C primarily serve to bind and recruit RPA-interacting proteins onto DNA.
We do not observe fast exchange kinetics or robust deuterium changes in either PID70N or PID32C suggesting that the primary roles of these domains are to interact with RIPs. Binding of p53 to the cleft in PID70N has been shown to be outcompeted by DNA, and thus proposed to possess very weak DNA-binding properties (30). But more importantly, PID70N serves as a high affinity binding site for more than two dozen proteins including p53 (30) and Dna2 (45). In addition, PID70N interacts with the N-terminal phosphorylation motif of RPA32 (46). Thus, the HDX changes we observe in the later timescales likely originate from the release of such intra-RPA:PID70N interactions upon DNA binding to DBD-C and -D, and not from direct DNA binding to PID70N.
Biophysical characterization of full-length RPA suggests that DNA binding is highly dynamic and that the interaction of individual DBDs is not sequential as previously envisioned (25). The correlated movements/dynamics of the DBDs (and likely PIDs) enable rapid RPA diffusion on ssDNA (∼5000 nt2 s−1 at 37°C) and efficiently melt small hairpins and duplex regions as it diffuses (23). RPA–DNA complexes have also been shown to exchange rapidly in the presence of free RPA (47,48). Transition between specific DNA-binding modes are thought to promote such mechanical movements of RPA. An initial 8–10 nt is formed first followed by transition to a 20 nt mode and finally resulting in the 30 nt mode where all DBDs are DNA-bound (49). RPA dynamically forms at least two kinetically distinct complexes on DNA with different stabilities that are microscopically dissociating and re-associating (50).
Our data support a DNA binding dynamics-based model for RPA–DNA interactions. We envision RPA to function as two dynamic halves partitioned as the FAB half and the Tri-C half (Figure 6B). The FAB half is dynamic with rapid on–off kinetics on ssDNA and the Tri-C half is less-dynamic with more stable interactions with DNA, primarily driven by the stability of DBD-C. These data are not to interpreted as the FAB half having poor DNA-binding activity as the domains likely maintain high affinity interactions while possessing rapid on–off kinetics. These findings do suggest that the DNA buried under the FAB half of RPA should be transiently accessible to RIPs.
The HDX data do not support a role for the PIDs in mediating direct DNA interactions, but intra-domain interactions exist between the PIDs and the DBDs. These interactions are released as conformational changes are driven upon DNA binding as slow exchange HDX is observed for both PIDs. Such changes would allow RPA to maintain high affinity interactions through stable binding by DBD-C and contributions from other parts of Tri-C. The dynamic interactions of DBD-A and -B would enable RIPs that interact with PID70N to gain access to the 5′ end of the RPA-coated DNA. Similarly, even the less-dynamic DBD-D and -E can be remodeled by RIPs that interact with PID32C and bind to the 3′ of the RPA-coated DNA as previously observed for Rad52 (22,31). When multiple RPA molecules are bound on the DNA, the dynamic nature of the DBDs would enable RIPs to bind between multiple RPA molecules bound to the DNA.
Supplementary Material
Notes
Present address: Nilisha Pokhrel, New England Biolabs, Ipswich, MA 01938, USA.
Contributor Information
Faiz Ahmad, Department of Biochemistry, Saint Louis University, School of Medicine, St. Louis, MO 63104, USA.
Angela Patterson, Department of Chemistry and Biochemistry, Montana State University, Bozeman, MT 59717, USA.
Jaigeeth Deveryshetty, Department of Biochemistry, Saint Louis University, School of Medicine, St. Louis, MO 63104, USA.
Jenna R Mattice, Department of Chemistry and Biochemistry, Montana State University, Bozeman, MT 59717, USA.
Nilisha Pokhrel, Department of Biological Sciences, Marquette University, Milwaukee, WI 53201, USA.
Brian Bothner, Department of Chemistry and Biochemistry, Montana State University, Bozeman, MT 59717, USA.
Edwin Antony, Department of Biochemistry, Saint Louis University, School of Medicine, St. Louis, MO 63104, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01GM130746, R01GM133967 to E.A.]. Funding for Proteomics, Metabolomics and Mass Spectrometry Facility used in this publication was made possible in part by the MJ Murdock Charitable Trust and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM103474. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Kim C., Paulus B.F., Wold M.S.. Interactions of human replication protein A with oligonucleotides. Biochemistry. 1994; 33:14197–14206. [DOI] [PubMed] [Google Scholar]
- 2. Namiki Y., Zou L.. ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zou L., Elledge S.J.. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003; 300:1542–1548. [DOI] [PubMed] [Google Scholar]
- 4. Marechal A., Zou L.. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015; 25:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Topolska-Wos A.M., Sugitani N., Cordoba J.J., Le Meur K.V., Le Meur R.A., Kim H.S., Yeo J.E., Rosenberg D., Hammel M., Scharer O.D.et al.. A key interaction with RPA orients XPA in NER complexes. Nucleic Acids Res. 2020; 48:2173–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fanning E., Klimovich V., Nager A.R.. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006; 34:4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sugitani N., Chazin W.J.. Characteristics and concepts of dynamic hub proteins in DNA processing machinery from studies of RPA. Prog. Biophys. Mol. Biol. 2015; 117:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bochkareva E., Korolev S., Lees-Miller S.P., Bochkarev A.. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J. 2002; 21:1855–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bochkareva E., Belegu V., Korolev S., Bochkarev A.. Structure of the major single-stranded DNA-binding domain of replication protein A suggests a dynamic mechanism for DNA binding. EMBO J. 2001; 20:612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bochkareva E., Korolev S., Bochkarev A.. The role for zinc in replication protein A. J. Biol. Chem. 2000; 275:27332–27338. [DOI] [PubMed] [Google Scholar]
- 11. Nakaya R., Takaya J., Onuki T., Moritani M., Nozaki N., Ishimi Y.. Identification of proteins that may directly interact with human RPA. J. Biochem. 2010; 148:539–547. [DOI] [PubMed] [Google Scholar]
- 12. Dueva R., Iliakis G.. Replication protein A: a multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer. 2020; 2:zcaa022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mer G., Bochkarev A., Gupta R., Bochkareva E., Frappier L., Ingles C.J., Edwards A.M., Chazin W.J.. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell. 2000; 103:449–456. [DOI] [PubMed] [Google Scholar]
- 14. Brosey C.A., Soss S.E., Brooks S., Yan C., Ivanov I., Dorai K., Chazin W.J.. Functional dynamics in replication protein A DNA binding and protein recruitment domains. Structure. 2015; 23:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brosey C.A., Yan C., Tsutakawa S.E., Heller W.T., Rambo R.P., Tainer J.A., Ivanov I., Chazin W.J.. A new structural framework for integrating replication protein A into DNA processing machinery. Nucleic Acids Res. 2013; 41:2313–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan J., Pavletich N.P.. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012; 26:2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iftode C., Borowiec J.A.. 5′ → 3′ molecular polarity of human replication protein A (hRPA) binding to pseudo-origin DNA substrates. Biochemistry. 2000; 39:11970–11981. [DOI] [PubMed] [Google Scholar]
- 18. de Laat W.L., Appeldoorn E., Sugasawa K., Weterings E., Jaspers N.G., Hoeijmakers J.H.. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 1998; 12:2598–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bae S.H., Bae K.H., Kim J.A., Seo Y.S.. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature. 2001; 412:456–461. [DOI] [PubMed] [Google Scholar]
- 20. Sugawara N., Haber J.E.. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol. Cell. Biol. 1992; 12:563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bastin-Shanower S.A., Brill S.J.. Functional analysis of the four DNA binding domains of replication protein A. The role of RPA2 in ssDNA binding. J. Biol. Chem. 2001; 276:36446–36453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caldwell C.C., Spies M.. Dynamic elements of replication protein A at the crossroads of DNA replication, recombination, and repair. Crit. Rev. Biochem. Mol. Biol. 2020; 55:482–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nguyen B., Sokoloski J., Galletto R., Elson E.L., Wold M.S., Lohman T.M.. Diffusion of human replication protein A along single-stranded DNA. J. Mol. Biol. 2014; 426:3246–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Antony E., Lohman T.M.. Dynamics of E. coli single stranded DNA binding (SSB) protein-DNA complexes. Semin. Cell Dev. Biol. 2019; 86:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai L., Roginskaya M., Qu Y., Yang Z., Xu Y., Zou Y.. Structural characterization of human RPA sequential binding to single-stranded DNA using ssDNA as a molecular ruler. Biochemistry. 2007; 46:8226–8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walther A.P., Gomes X.V., Lao Y., Lee C.G., Wold M.S.. Replication protein A interactions with DNA. 1. Functions of the DNA-binding and zinc-finger domains of the 70-kDa subunit. Biochemistry. 1999; 38:3963–3973. [DOI] [PubMed] [Google Scholar]
- 27. Wyka I.M., Dhar K., Binz S.K., Wold M.S.. Replication protein A interactions with DNA: differential binding of the core domains and analysis of the DNA interaction surface. Biochemistry. 2003; 42:12909–12918. [DOI] [PubMed] [Google Scholar]
- 28. Arunkumar A.I., Stauffer M.E., Bochkareva E., Bochkarev A., Chazin W.J.. Independent and coordinated functions of replication protein A tandem high affinity single-stranded DNA binding domains. J. Biol. Chem. 2003; 278:41077–41082. [DOI] [PubMed] [Google Scholar]
- 29. Salas T.R., Petruseva I., Lavrik O., Saintome C.. Evidence for direct contact between the RPA3 subunit of the human replication protein A and single-stranded DNA. Nucleic Acids Res. 2009; 37:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bochkareva E., Kaustov L., Ayed A., Yi G.S., Lu Y., Pineda-Lucena A., Liao J.C., Okorokov A.L., Milner J., Arrowsmith C.H.et al.. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15412–15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pokhrel N., Caldwell C.C., Corless E.I., Tillison E.A., Tibbs J., Jocic N., Tabei S.M.A., Wold M.S., Spies M., Antony E. Dynamics and selective remodeling of the DNA-binding domains of RPA. Nat. Struct. Mol. Biol. 2019; 26:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yates L.A., Aramayo R.J., Pokhrel N., Caldwell C.C., Kaplan J.A., Perera R.L., Spies M., Antony E., Zhang X.. A structural and dynamic model for the assembly of Replication Protein A on single-stranded DNA. Nat. Commun. 2018; 9:5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Erp P.B.G., Patterson A., Kant R., Berry L., Golden S.M., Forsman B.L., Carter J., Jackson R.N., Bothner B., Wiedenheft B.. Conformational dynamics of DNA binding and Cas3 recruitment by the CRISPR RNA-guided cascade complex. ACS Chem. Biol. 2018; 13:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berry L., Poudel S., Tokmina-Lukaszewska M., Colman D.R., Nguyen D.M.N., Schut G.J., Adams M.W.W., Peters J.W., Boyd E.S., Bothner B. H/D exchange mass spectrometry and statistical coupling analysis reveal a role for allostery in a ferredoxin-dependent bifurcating transhydrogenase catalytic cycle. Biochim. Biophys. Acta Gen. Subj. 2018; 1862:9–17. [DOI] [PubMed] [Google Scholar]
- 35. Knox R., Lento C., Wilson D.J.. Mapping conformational dynamics to individual steps in the TEM-1 beta-Lactamase catalytic mechanism. J. Mol. Biol. 2018; 430:3311–3322. [DOI] [PubMed] [Google Scholar]
- 36. Zheng J., Chang M.R., Stites R.E., Wang Y., Bruning J.B., Pascal B.D., Novick S.J., Garcia-Ordonez R.D., Stayrook K.R., Chalmers M.J.et al.. HDX reveals the conformational dynamics of DNA sequence specific VDR co-activator interactions. Nat. Commun. 2017; 8:923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masson G.R., Burke J.E., Ahn N.G., Anand G.S., Borchers C., Brier S., Bou-Assaf G.M., Engen J.R., Englander S.W., Faber J.et al.. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods. 2019; 16:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Graham B.W., Tao Y., Dodge K.L., Thaxton C.T., Olaso D., Young N.L., Marshall A.G., Trakselis M.A.. DNA interactions probed by Hydrogen-Deuterium Exchange (HDX) fourier transform ion cyclotron resonance mass spectrometry confirm external binding sites on the minichromosomal maintenance (MCM) helicase. J. Biol. Chem. 2016; 291:12467–12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J., Li J., Craig T.A., Kumar R., Gross M.L.. Hydrogen-Deuterium Exchange Mass Spectrometry reveals calcium binding properties and allosteric regulation of downstream regulatory element antagonist modulator (DREAM). Biochemistry. 2017; 56:3523–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Q., Willison L.N., Tripathi P., Sathe S.K., Roux K.H., Emmett M.R., Blakney G.T., Zhang H.M., Marshall A.G.. Epitope mapping of a 95 kDa antigen in complex with antibody by solution-phase amide backbone hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2011; 83:7129–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bochkarev A., Pfuetzner R.A., Edwards A.M., Frappier L.. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997; 385:176–181. [DOI] [PubMed] [Google Scholar]
- 42. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L.et al.. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pretto D.I., Tsutakawa S., Brosey C.A., Castillo A., Chagot M.E., Smith J.A., Tainer J.A., Chazin W.J.. Structural dynamics and single-stranded DNA binding activity of the three N-terminal domains of the large subunit of replication protein A from small angle X-ray scattering. Biochemistry. 2010; 49:2880–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Corrales-Guerrero L., He B., Refes Y., Panis G., Bange G., Viollier P.H., Steinchen W., Thanbichler M.. Molecular architecture of the DNA-binding sites of the P-loop ATPases MipZ and ParA from Caulobactercrescentus. Nucleic Acids Res. 2020; 48:4769–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou C., Pourmal S., Pavletich N.P.. Dna2 nuclease-helicase structure, mechanism and regulation by Rpa. Elife. 2015; 4:e09832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Binz S.K., Lao Y., Lowry D.F., Wold M.S.. The phosphorylation domain of the 32-kDa subunit of replication protein A (RPA) modulates RPA-DNA interactions. Evidence for an intersubunit interaction. J. Biol. Chem. 2003; 278:35584–35591. [DOI] [PubMed] [Google Scholar]
- 47. Pokhrel N., Origanti S., Davenport E.P., Gandhi D., Kaniecki K., Mehl R.A., Greene E.C., Dockendorff C., Antony E.. Monitoring Replication Protein A (RPA) dynamics in homologous recombination through site-specific incorporation of non-canonical amino acids. Nucleic Acids Res. 2017; 45:9413–9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gibb B., Ye L.F., Gergoudis S.C., Kwon Y., Niu H., Sung P., Greene E.C.. Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014; 9:e87922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kumaran S., Kozlov A.G., Lohman T.M.. Saccharomyces cerevisiae replication protein A binds to single-stranded DNA in multiple salt-dependent modes. Biochemistry. 2006; 45:11958–11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen R., Subramanyam S., Elcock A.H., Spies M., Wold M.S.. Dynamic binding of replication protein a is required for DNA repair. Nucleic Acids Res. 2016; 44:5758–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.