


Abstract
The occurrence of accidental mutations or deletions caused by genome editing with CRISPR/Cas9 system remains a critical unsolved problem of the technology. Blocking excess or prolonged Cas9 activity in cells is considered as one means of solving this problem. Here, we report the development of an inhibitory DNA aptamer against Cas9 by means of in vitro selection (systematic evolution of ligands by exponential enrichment) and subsequent screening with an in vitro cleavage assay. The inhibitory aptamer could bind to Cas9 at low nanomolar affinity and partially form a duplex with CRISPR RNA, contributing to its inhibitory activity. We also demonstrated that improving the inhibitory aptamer with locked nucleic acids efficiently suppressed Cas9-directed genome editing in cells and reduced off-target genome editing. The findings presented here might enable the development of safer and controllable genome editing for biomedical research and gene therapy.
INTRODUCTION
Clustered, regularly interspaced short-palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) gene-editing technology has brought about a biotechnological revolution (1,2). The CRISPR–Cas9 system facilitates gene knockouts (3) gene knockins (4), gene mutations (5) and gene regulation (6). However, serious concerns exist regarding its controllability and safety, given its known toxicity, off-target effects (7,8) and accidental deletions or genomic rearrangements (9). Cleavage and mutation at unexpected sites or homologous genomic sites might interfere with therapeutic analysis or harm an organism via excessive or prolonged Cas9 activity (10). To escape the CRISPR–Cas9 immune system, natural protein antagonists have evolved. Proteins inhibiting the Class 1, type I CRISPR–Cas system were first reported in 2013 (11), and the field of anti-CRISPR proteins (Acrs) has evoked great interest owing to potential applications modulating cleavage activities of various Cas9 proteins. Recently, several protein families were found to inhibit type I, type II, and type V-A (Cpf1) CRISPR–Cas defense systems in different species of bacteria (12). AcrIIA2, AcrIIA4 and AcrIIA5 were shown to bind Streptococcus pyogenes Cas9 (SpCas9), which is extensively utilized for programmable genome editing in a wide range of cells and organisms (13). It is to be noted that AcrIIA2 and AcrIIA4 mimic DNA and occupy the protospacer adjacent motif (PAM)-interacting domain to prevent target DNA binding (14). AcrIIA5 is a homolog of AcrIIA4 and is also functional against SpCas9 (15). Moreover, AcrVAs against Cpf1 have been recently identified, and are being considered to expand the toolkit of the CRISPR-Cas system (16). Here, we developed an inhibitory DNA aptamer against Cas9 with clear advantages over protein-based inhibitors, such as easy and cost-effective chemical synthesis, tolerance to chemical modifications, a longer shelf-life, high stability, and low toxicity and immunogenicity (17).
MATERIALS AND METHODS
Cells
HEK293 FT cell and Flp-In™ –293 cell that were purchased from Thermo Fisher Scientific (Waltham, MA, USA) were grown in DMEM (Dulbecco's modified Eagle's medium, Thermo Fisher Scientific) with 10% FBS and a penicillin/ampicillin (Thermo Fisher Scientific) mix at 37°C under 5% CO2.
Cas9 expression and purification
Cas9 proteins were expressed using Escherichia coli BL21(DE3) strain carrying the plasmid pET-28b-Cas9-His (Addgene #47327). Escherichia coli was cultured at 18°C for 16 h following 1 mM IPTG induction. Cells were collected by centrifugation, lysed with lysis 10 buffer (10 mM Tris–HCl, 500 mM NaCl, 10 mM imidazole, 10% glycerol, pH 8.0) supplemented with 0.1 mM PMSF and 0.2% Triton X-100 and sonicated for 30 min. Cas9 proteins were first purified using Ni-NTA column with 20 mM imidazole wash and 500 mM imidazole elution buffer containing TBSG (10 mM Tris–HCl, 500 mM NaCl, 10% glycerol, pH 8.0). Proteins were next purified by gel filtration column, HiPrep 16/60 Sephacryl S-200 HR, equipped with ÄKTA-start (GE Healthcare, IL, USA) and Buffer A (20 mM HEPES, 500 mM NaCl, 10% glycerol, pH 7.4). Out of two major peaks in gel filtration chart, the fractions with smaller molecular weight were collected, flash-frozen by liquid N2 and kept at −80°C until use. Protein concentration was determined by Coomassie Blue-stained SDS-PAGE with varying concentration of BSA standards.
AcrIIA4 and AcrIIA5 expression and purification
Plasmids were constructed by inserting synthetic sequences corresponding to AcrIIA4 or AcrIIA5 (D1126) into pET-28a to yield pET-AcrIIA4 or pET-AcrIIA5 (D1126), respectively. Protein purification was performed as described above, except that the Ni-NTA eluent was concentrated with Amicon Ultra (10 kDa) and washed with Buffer A supplemented with 1 mM DTT. Protein concentration was determined using Coomassie Blue-stained SDS-PAGE with varying concentrations of BSA standards.
SELEX
The target protein, Cas9-His, was immobilized on Dynabeads His-tag magnetic beads (Thermo Fisher Scientific) in PBS (Wako Chemicals, Japan) buffer with 0.005% tween 20 overnight at 4°C. Four libraries designed by our laboratory consisted of a 17, 19, 21, 23-base random region respectively flanking by 17 bases constant regions for polymerase chain reaction (PCR) amplification. Annealed four libraries (100 pmol) were mixed with protein bound beads in binding buffer (10 mM Tris–HCl, 10 mM MgCl2, 150 mM NaCl, 0.05% tween 20, pH 7.5) with tRNA (0.1–6 mg/ml) for 1 h at 25°C. After incubation, the beads were washed with binding buffer three times. Then, the ssDNA bound to the beads was eluted with 7 M urea and 10 mM EDTA, purified by the phenol/chloroform extraction and ethanol precipitation. The eluted ssDNA libraries were amplified with the following primers: the forward primer, 5′-CCG ATC TAA CCA AGT GGA GAG GTT CTT ACA-3′) and the reverse primer, 5′-TGC GTA GAG CGA TTG GCG GAG AGG CTC TCA CA-3′). The amplified dsDNA was amplified again using primers with barcode sequences for next generation sequencing. Six rounds of selection were carried out to select ssDNA molecules that bind to Cas9. Finally, seven minor populations were found from data of fifth round selection when using the N17 library.
Predictions of secondary structure and search for consensus sequences
Secondary structures were predicted using the prediction program for secondary folding, mfold, developed by M. Zuker (18). Consensus sequence were identified using the MEME Suite 5.0.1 motif analysis tool (http://meme.nbcr.net/meme/tools/meme), applying the criterion of a minimum motif length of 17, 19, 21 or 23 nucleotides according to the length of randomized region of each library (19).
Plasmid construction
To assess the genome editing efficiency in cells and evaluate the inhibitory activity of aptamers, the initiation codon deleted scarlet gene-based reporter system in HEK293 was to be established. Flp-In system was used to generate the stable cell line. Firstly, a fragment with the sgCCR5 target region including a stop codon and the scarlet gene fragment without ATG were artificially synthesized and assembled by PCR using the following primers: the forward primer for a fragment with sgCCR5 target region, 5′-GGA GAC CCA AGC TGG CTA GCG TTT A-3′; the reverse primer for a fragment with sgCCR5 target region, 5′-TTG CTC ACC TCC TCG CCC TTG CTC ACC GT-3′; the forward primer for the scarlet gene (-ATG), 5′-CGA GGA GGT GAG CAA GGG CGA GGC AGT G-3′; the reverse primer for the scarlet gene (-ATG) 5′-CAG CGG GTT TAA ACG GGC CGA ATT CTA CTT GTA CAG CTC-3′. Next, the In-Fusion HD PCR Cloning Kit (TAKARA Bio, Shiga, Japan) was employed to insert the assembled fragment (the sgCCR5 target region and the scarlet (-ATG)) into Nhe I and PspOMI sites of pcDNA5/FRT/TO plasmid (Thermo Fisher Scientific).
To evaluate the cleavage efficiency in vitro, plasmids with target regions were constructed. pCX-EGFP was kindly gifted from Dr. Masaru Okabe. pcEGFR-GFP was generated by subcloning human EGF receptor-GFP fusion DNA fragment into pcDNA3 expression vector (Invitrogen, San Diego, CA, USA). Plasmid with EpCAM target sequence was generated by inserting annealed oligonucleotides of the 22-nucleotide target sequence with BsmBI sticky ends into BsmBI sites of pGuide-it-tomato plasmid (TAKARA). The oligonucleotides are as follows: TsgEpCAM-f, 5′-CCG G GT GCA CCA ACT GAA GTA CAC CG-3′; TsgEpCAM-r: 5′-AAA CCG GTG TAC TTC AGT TGG TGC AC-3′.
To evaluate the on- and off-target genome editing in cells, we constructed a plasmid encoding Cas9 and sgRNA expression cassettes by inserting sgRNA sequences for EMX1 and HBB loci into BsmBI sites of pGedit (20).
For the CRISPR activation (CRISPRa) experiment, we generated two expression plasmids [dCas9-VP64- and sgRNA2.0-expression plasmid and MS2-p65-HSF1-expression plasmid (pdCas9-VP64-sgRNA2.0 and pMS2-p65-HSF1, respectively) and one reporter plasmid (TurboGFP reporter plasmid with 6x TRE including promoter, p6xTRE-TurboGFP). dCas9-VP64- and MS2-p65-HSF1-expression plasmids were purchased from VectroBuilder (Yokohama, Japan). pdCas9-VP64-sgRNA2.0 plasmid was made by inserting U6-driven sgRNA2.0 cassette into the pCas9-VP64-expression plasmid. The p6xTRE-TurboGFP was ordered from Eurofins Genomics (Tokyo, Japan) by inserting TurboGFP coding sequence with PEST tag and the same 6xTRE minimum CMV promoter sequence as reported (21) into a backbone vector (pEX-A2J1).
Generation of ATG initiation codon deleted Scarlet stable cell line
To assess the efficiency of genomic editing in human cells, Flp-In™–293 cell lines were used to generate a disrupted mScarlet stable cell line by Flp recombinase-mediated integration. The mScarlet is a bright monomeric red fluorescent protein for cellular imaging. In the 293-stop-mScarlet plasmid construction, upstream of the mScarlet coding region was followed by a CCR5 DNA fragment including a stop codon. If Cas9/crRNA could cause indels at the stop codon and make an in-frame mScarlet fusion protein, the reporter cells would show fluorescence based on mScarlet expression. The detailed protocol was described in the manufacture's instruction (Themo Fisher Scientific). Adherent cells were seeded onto 10 cm dish and reached 70%-90% confluence at the time of transfection. The pOG44 plasmid (4.5 μg) and the pcDNA5/FRT/disrupted Scarlet vector (0.5 μg) were added to 250 μl Opti-MEM medium (Thermo Fisher Scientific) at 9:1 ratio, Meanwhile, the other 250 μl Opti-MEM medium was added to an Eppendorf tube, followed by addition of 15 μl of Lipofectamine 2000 (Invitrogen). Then, diluted DNA was added to the diluted Lipofectamine 2000 reagent. After incubating 5 min at room temperature, the DNA–lipid complex was added to Flp-In™ –293 cells. Three days after transfection, puromycin (0.5 μg/ml; WAKO chemicals) was added to the medium. After 2 days, the medium was changed by DMEM with 0.1 μg/ml puromycin. Fourteen days later, single colonies were individually picked up and expanded as a disrupted Scarlet HEK293 line.
In vitro cleavage assay
crRNAs were purchased from Integrated DNA Technologies (Coralville, IA, USA). crRNA/tracrRNA was annealed in annealing buffer (50 mM Tris–HCl, 150 mM NaCl, pH 7.5). The cleavage reaction was performed by mixing crRNA/tracrRNA (30 nM) and Cas9 proteins (50 nM) in cleavage buffer (20 mM HEPES, pH 6.5, 100 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA). After 10 min incubation, annealed aptamer was added to the reaction at different concentrations. Ten minutes later, the plasmid containing the target site was added to the reaction mix (10 μl in total). The reaction was performed at 37°C for 20 min and then run in 0.7% TAE agarose gel. After electrophoresis, the gels were visualized by ChemiDoc XRS Plus (Bio-Rad, Hercules, CA, USA) imaging. The percentage of cleavage was quantified using bio-rad image lab 5.0 software.
Three independent assays were done for each in vitro cleavage experiment and error bars shown in all panels are standard deviation (SD). In the quantification of the gel image, the % of the uncleaved plasmid (unit of vertical axis in Figure 1B and C) was calculated using the following formula: percentage of uncleaved plasmid = 100 × ([uncleaved plasmid (supercoiled band)]/[uncleaved plasmid + cleaved plasmid (linear and nicked bands)].
Figure 1.

Analysis of the inhibitory activities of aptamer candidates for Cas9 in in vitro-cleavage assays. (A) Three candidate aptamers (S21, S36, and S40) showed high inhibitory activities for Cas9/crGFP1 at low concentrations. The pCX-EGFP plasmid was cleaved with Cas9/crGFP1 at the GFP1 site in the presence of the aptamer candidate. The mobilities of open circular (nicked), linear and covalently closed circular (supercoiled) plasmids are indicated on the right. SA-Ap: streptavidin aptamer. (B) Mutational analysis of the inhibitory aptamer in the loop region. Thirty nanomolar aptamers were used in the assays. SS21 (a shortened inhibitory aptamer of S21) was used as the parental aptamer. Mutations in the ‘CGCC’ sequence [LM1–LM4, especially at the first or second position (LM1 or LM2)] seriously impaired the capability of the SS21 aptamer. Electrophoresis data for these graphs are presented in Supplementary Figure S2. (C) Mutation analysis of the inhibitory aptamer in the neck region. Aptamers were used at a concentration of 30 nM. (D) Summary of mutation experiments. ‘CGCC’ in the loop region and the sequences in the neck region were invariable regions of S21. The inhibitory activities of mutant aptamers in the neck region were ranked as follows: 5′-T1C2–3>5′-C1C2–3>5′-T1T2–3. (E) Gel mobility shift assay showing the interaction of inhibitory aptamers (S21 and SS21) with Cas9. Loop-NC: negative control where the neck and loop regions of S21 were replaced by tetra-loop, ‘GTAA’. SA-Ap: a streptavidin aptamer. (F) Surface plasmon resonance analysis of the S21–Cas9 interaction. To minimize the mass transport-limited binding effect, a low level of biotinylated inhibitory aptamer or the negative control aptamer was immobilized on the streptavidin chip, and measurements were performed with a high flow rate. To eliminate nonspecific DNA binding to target proteins, the Kd was measured at a high salt condition (250 mM NaCl). Data are presented as means ± SD from three independent experiments. Error bars represent S.D. No response was observed with the Loop-NC negative control.
Electrophoretic mobility shift assay (EMSA)
The purified Cas9 (100 nM) was mixed with inhibitory aptamer, mutant aptamer, or negative control aptamer (2 μM) separately at a molar ratio of 1:20 and then incubated at room temperature for 30 min in reaction buffer (20 mM Tris–HCl, pH 7.5, 250 mM NaCl, 1 mM MgCl2, 2.5% glycerol, 0.05% tween-20, 50 μg/ml tRNA) to form Cas9–aptamer complexes. The samples were run on an 8% (w/v) non-denaturing polyacrylamide gel (40:1 acrylamide/bisacrylamide) at 30°C in 0.5× Tris-borate EDTA electrophoresis buffer. After electrophoresis, the gels were visualized by SYBR Gold gel staining (Thermo Fisher Scientific).
SPR measurements
SPR assays were performed with a BIACORE X (GE Healthcare) apparatus. To minimize the mass transport-limited binding effect, a low level of biotinylated inhibitory aptamer or the negative control aptamer was immobilized on the streptavidin chip. The 5′-biotinylated oligonucleotides N23–7 and Loop-Negative Control (Loop-NC) were immobilized on SA sensor chips (GE Healthcare), by injecting 100 nM DNA solutions in phosphate-buffered saline containing 0.005% Tween-20 (PBS-T) into one of the two flow cells (flow cell 2) at a flow rate of 3 μl/min until 40 RU had been immobilized. The other (flow cell 1) was used as a control for non-specific protein binding to the matrix. SpyCas9 protein was diluted to different concentrations (2.5, 5, 10, 20, 50 and 100 nM) in a running buffer (20 mM Tris–HCl, pH 8.0, 250 nM NaCl, 10 mM MgCl2) and injected successively into the two flow cells of the sensor chip, at a rate of 20 μl/min for 300 s. Then, flow cells were washed by running buffer at the same flow rate and the protein–DNA complex was to be dissociated for another 300 s. Next, the surface of the sensor chip was washed by injection of 1 M NaCl, 50 mM NaOH for 2 s to remove the residual protein for chip regeneration. The responses in flow cell 1 during the injection of the protein solutions were subtracted from those in flow cell 2 to correct the signal drift and unspecific binding. The data were prepared for kinetic analysis. The kinetic constants and dissociation constants for binding were calculated using BIA evaluation software. Kd was calculated by simultaneous fittings of dissociation and association data by BIAevaluation software and 1:1 Langmuir binding model was used.
Transfections
When adherent cells reached 30–70% confluence in 24-well plate, we did transfection experiments. Cas9 protein (0.5–2 μg), annealed crRNA/tracrRNA (0.125–0.5 μg) and Lipofectamine™ Cas9 Plus™ reagent (1 μl) were added to 25 μl Opti-MEM medium in sequence and incubated for 5 min to form Cas9 RNPs. At the same time, 25 μl Opti-MEM medium was added to a tube followed by addition of 3 μl Lipofectamine™ CRISPRMAX™ reagent (Invitrogen). After 5 min incubation, the Cas9 RNPs were then added to the diluted Lipofectamine™ CRISPRMAX™ reagent solution. After mixing well, the sample was incubated at 25°C for 10–15 min to form Cas9 RNPs and CRISPRMAX™ reagent complexes and then added to cells in 300 μl DMEM medium. To detect the suppression activity of the inhibitor, complex of DNA inhibitor and Lipofectamine 2000 reagent was delivered following the addition of Cas9 RNPs. The concentration of the inhibitor was adjusted from 20 to 400 nM according to the experiment. At 72 h post-transfection, the cells were harvested for analysis of genomic editing efficiency using Guide-it Mutation Detection Kit (Clontech). Alternatively, disrupted Scarlet stable cells were analyzed by flow cytometry to determine the percentage of Scarlet positive cells.
Detection of indel mutations by the T7E1 cleavage assay
At 72 h post-transfection of Cas9/crRNA/tracrRNA/inhibitor, the cells were harvested for DNA extraction. For DNA extraction, according to the manufacturer's instructions (Guide-it™ Mutation Detection Kit, Clontech, Cat #631443), harvested cells were incubated in extraction buffer 1 at 95°C for 10 min. Then extraction buffer 2 was added and mixed by gentle pipetting until homogeneous. Extracted DNA was diluted with ddH2O for PCR amplification. PCR was performed by Terra polymerase with primers flanking the targeted site, <30 amplification cycles. For the HPRT1 target, a forward primer: 5′-ACA TCA GCA GCT GTT CTG -3′ and a reverse primer: 5′-GGC TGA AAG GAG AGA ACT-3′ were used. The PCR condition was set at 98°C for 1 min for one cycle, then at 98°C for 15 s, 56°C for 20 s, and 72°C for 30 s for a total of 26 cycles. The final extension was set at 72°C for 5 min. For the EMX1 target, a forward primer: 5′-GCC ATC CCC TTC TGT GAA TGT TAG AC-3′ and a reverse primer: 5′-CGG AAT CTA CCA CCC CAG GCT CT-3′ were used. The annealing temperature of PCR amplification was 62°C. Then, PCR products were purified using gel extraction kit (Qiagen), followed by 2% agarose gel electrophoresis. Purified PCR products were mixed with NEB buffer 2 (New England Biolabs) to be heat-denatured, re-annealed for heteroduplex formation using a thermal cycler according to the manufacturer's instructions. Finally, 200 ng heteroduplex sample was incubated with T7E1 for 20 min at 37°C. Then 1/10 volume of stop solution was added to stop the reaction. The digested product was loaded on 2% TAE agarose gel and quantified using bio-image lab software.
FACS analysis
HEK293 cells were collected 3 days after transfection and directly analysed for fluorescence using BD Accuri C6 Plus Flow Cytometer (BD biosciences). Viable cells were gated on size and shape using forward and side scatter. Scarlet fluorescence was collected in the FL2 channel using a 585/42 band-pass filter. Flow cytometry data were analysed using software supplied by the instrument.
Molecular beacon assay
Fluorescence measurements were performed using a spectrofluorometer (JascoFP-8300, Japan) in binding buffer [20 mM Tris–HCl (pH 7.9), 120 mM NaCl, 5% glycerol, 0.1 mM DTT and 1 mM MgCl2] containing 0.02% Tween 20 at 25°C Three nM of Cas protein (Cas9 or dCas9) was incubated with 5 nM annealed crRNA/tracrRNA for 30 min at 25°C in the binding buffer followed by the addition of DNA inhibitor or anti-CRISPR proteins at various concentrations to the final mixture. DNA probes were synthesized by Integrated DNA Technologies (Coralville, IA) and labeled with fluorescein and Iowa Black FQ at the end of target and non-target strands, respectively. Double-stranded DNA probes were formed by mixing equimolar amounts of synthetic complementary strands in an annealing buffer (50 mM Tris–HCl, 150 mM NaCl, pH 7.5). Fluorescence intensities were recorded with an excitation wavelength of 490 nm and an emission wavelength of 520 nm. Time-dependent fluorescence changes were monitored after the addition of negligible volumes of the beacon (1 nM) and DNA aptamers to a cuvette followed by manual mixing.
Next-generation sequencing and data analysis
HEK 293FT cells plated in 24-well plates were transfected with Cas9 protein, crRNA/tracrRNA, and inhibitory aptamers 72 h before genomic DNA extraction. For the experiment of evaluation of on- and off-target editings, the plasmid harbouring Cas9- and sgRNA-expression cassettes were transfected into 293FT cells. The LNA-modified inhibitory aptamers were delivered into cells 14 h (or 12 h) and 26 h (or 24 h) later after plasmids transfection. After 4 days, cells were harvested for genomic DNA extraction. The genomic region flanking the CRISPR target site for each gene was amplified using primers with adaptor listed in Supplementary Table S3. Products of the first PCR were purified and then amplified in a second ten-cycle PCR using primers with the Illumina P5 and P7 adapters. Barcoded and purified DNA samples were quantified using a Qubit Fluorometer (Life Technologies) and then sequenced with an Illumina MiSeq sequencer. Data were filtered by Phred quality (Q score) of at least 30, and then was analysed using CRISPResso (crispresso.rocks) (22). The amplicon sequences containing an indel with a window of 30 to 40 based around the cleavage site of Cas9/crRNA/tracrRNA were called ‘edited’ and the remaining amplicon sequences were called ‘unedited’. The editing efficiency was calculated with the formula: edited sequence count/(edited sequence count + unedited sequence count).
RESULTS
Selection of DNA aptamers for Cas9
To obtain DNA aptamers against Cas9, we performed systematic evolution of ligands by exponential enrichment (SELEX) (23,24) for His-tagged Cas9 protein with mixed stem-loop type libraries (N17, N19, N21 and N23 libraries) containing random sequences of various lengths. After five rounds of selection, several tens of thousands of read sequences acquired by deep sequencing were analysed, which revealed seven minor populations from the N17 libraries (Supplementary Figure S1A; Table S1), five of which shared a common motif #1, containing 5′-T(C/T)(G/A)-3′ at 5′ end and 5′-(C/T)GG-3′ at 3′ end (Supplementary Figure S1A). For the seven representative candidate aptamers of each population, we evaluated the inhibitory effects on Cas9 nuclease activity by performing an in vitro cleavage assay of a plasmid harboring encoding the green fluorescence protein (GFP) gene. Although the candidates did not show obvious inhibitory activities for Cas9 nuclease at lower concentrations (10 and 30 nM, Supplementary Figure S1B), increasing concentrations of aptamers to 100 nM resulted in a gradually increasing inhibitory effect compared with those of negative control aptamers. To explore more effective inhibitory aptamers, we next arbitrarily selected 55 aptamers (Supplementary Table S2) from minor sequence populations (similar sequence pairs with one nucleotide difference) after five rounds in each library (based on motif 1) and screened them by performing in vitro cleavage assays.
Three aptamers (S21, S36 and S40) showed high inhibitory activities against Cas9/CRISPR RNA against GFP#1 (crGFP1/tracrRNA) at 100 nM, whereas negative controls (a streptavidin aptamer and a sgRNA competitor) did not have any inhibitory activity (Supplementary Figures S1B and S1C). Re-evaluation of these three aptamers at 10 and 30 nM revealed that S21 and S36 had higher inhibitory activities and that S21 better matched the common motif (Motif 1 with ‘TCG’ at 5′ end in Figure 1A and Supplementary Figure S1A). Thus, we used aptamer S21 in further experiments.
Characterization of the inhibitory aptamer for Cas9
The secondary structure of the S21 aptamer was categorized into loop, neck, and stem regions (Supplementary Figure S1D). First, we investigated the effect of shortening the loop region on the inhibitory activity. The inhibitory activity did not appreciably decrease until the length of loop region was six bases; thus, the minimal aptamer with a 7-base loop sequence was referred to as the SS21 aptamer (Supplementary Figure S2A). A single-mutation scanning experiment of the minimized loop sequence showed that the first four nucleotides ‘CGCC’ from the 5′ side of the loop region were critical for inhibiting Cas9/crGFP1 (Cas9/crGFP1/tracrRNA complex) activity (Figure 1B and Supplementary Figure S2B). Deletion and mutation experiments in the stem region revealed that the stem region must be at least seven base pairs in length and its inhibitory activity was sequence-independent (Supplementary Figures S2C and D). In contrast, certain single-base pair substitutions in the neck region severely abrogated the inhibitory activity, indicating that the sequence in the neck region was important for the inhibitory activity (SM7, Supplementary Figure S2D). Next, we generated various mutants in the neck region and analysed the relationship between the sequence and the inhibitory activity in detail. The first nucleotide from the side of the loop region (positions 3 and 4, Figure 1D) was needed for base pairing (G3:C4, Figure 1C, D and Supplementary Figure S2E). Good inhibitory activity was seen with 5′-G5G6–3′ being the second and third nucleotide from the side of the loop region in the antisense strand, or 5′-T1C2–3′> ‘5′-C1C2–3′> 5′-T1T2–3′in the sense strand (Figure 1D). Notably, the T1:G6 mismatched base pair in the third nucleotide position from the side of the loop region was superior to C1:G6, indicating the sequence-dependent structure might influence the inhibitory ability, probably by enhancing the interaction between the aptamer and the Cas9/crRNA/tracrRNA complex.
An electrophoretic mobility shift assay was performed to clarify the binding ability of inhibitory aptamers to Cas9 (Figure 1E). Aptamers S21 and SS21 were obviously retarded after incubation with Cas9, whereas the mutated aptamer (NM2, Figure 1C), Loop-Negative control (Loop-NC; neck and loop regions of S21 replaced with ‘GTAA’), and negative-control aptamer (streptavidin aptamer [SA-Ap]) did not show obvious lagging bands (Figure 1E). These results indicate that the sequence motif in the neck region is important for binding between the aptamer and Cas9.
To quantify the binding ability, the dissociation constant (Kd) value was determined by performing surface plasmon resonance (SPR) assays (25). The aptamer–Cas9 Kd value was calculated as 0.63 nM, whereas the Loop-NC aptamer showed no interaction with Cas9 under the same conditions (Figure 1F). In summary, the physicochemical data demonstrated that the inhibitory aptamer bound Cas9 with a high affinity and specificity, in contrast to the Loop-NC aptamer, inert mutant aptamer, and unrelated control aptamer. We hypothesized that the neck region was crucial for the inhibitory function and may represent a binding site for Cas9.
Design of novel types of inhibitory aptamers for Cas9/crRNA complex
We next examined the target site dependency of the inhibitory aptamer. Although the S21 aptamer showed high inhibitory activity against Cas9/crRNA at the GFP1 site (Cas9/crGFP1), unfortunately, the aptamer exhibited weaker inhibitory activity against Cas9/crGFP2 and very weak inhibitory activities against Cas9/crGFP3 and Cas9/crGFP4 (>10-fold weaker than Cas9/crGFP1, Figure 2A). The most plausible explanation of why S21 did not inhibit Cas9/crRNA complexes at other sites was likely attributable to the interaction between the aptamer and crRNA. We carefully examined the sequence of the aptamer that could possibly interact with crRNA and found 4 nucleotides (5′-GGCG-3′) in a seed region of crRNA (crGFP1), which could potentially form a duplex with the conserved sequence 5′-CGCC-3′ in the aptamer loop region (Figure 2B). Furthermore, we realized that if these sequences form a duplex, considering the topology of structural data (26,27), the neck region of the aptamer should correspond to the PAM sequence. A model describing this possibility is depicted in Figure 2C.
Figure 2.

Aptamer-based inhibition of Cas9/crRNA activity by duplex formation with crRNA. (A) Analysis of the specificity of SS21 for Cas9/crRNAs targeting different sites of GFP. Apparent inhibitory activity was delimited to Cas9/crGFP1. For three other Cas9/crGFPs (crGFP2, crGFP3, and crGFP4), the capability of SS21 was attenuated. (B) Quantification of the ratio of cleaved-to-uncleaved plasmid from (A). The SS21 aptamer (30 nM) was used for the experiments. The four nucleotides in loop region were considered complementary to the seed sequence of crGFP1, as indicated by the black frame. (C) Schematic illustration of interactions between Cas9/crRNA and three types of inhibitory aptamers. The minimal inhibitory aptamer SS21 was the stem-loop type. The loop-A aptamer was derived from the stem-loop type by replacing all nucleotides with ‘As,’ except for ‘CGCC’ in loop region. The flap-type aptamer was shortened by deleting all nucleotides in the loop region except for ‘CGCC’ at 3′end. A stable tetra loop ‘GAAA’ sequence was used to close another end of the aptamer stem region. Cas9 (light green) assembled with the crRNA (blue) and tracrRNA (grey) complex to search for double-stranded DNA with a PAM region (yellow). Four nucleotides (red) in the loop or flap region of the aptamer (purple) complemented the crRNA sequence (blue). The aptamer (purple) inhibited Cas9/crRNA activity by blocking the interaction of Cas9/crRNA with target DNA. (D) Comparison of three types of inhibitory aptamers for Cas9/crGFP1. The flap-type and loop-A aptamers performed better than stem–loop aptamers. (E) Gel electrophoresis data for (D). Three dosages of each inhibitor were assessed for Cas9/crGFP1 by performing in vitro cleavage assays.
Based on this structural speculation, we designed new types of aptamers, which were designated as a loop-A aptamer and a flap-type aptamer (Figure 2C). In the loop-A aptamer, a stretch of 13 nucleotides were all changed to ‘A’ (not including ‘CCGC’ in the loop region). The loop-A aptamer showed the same inhibitory activity as the original aptamer (SS21) (Figure 2D and E), indicating the independence of the loop region sequence on inhibition (except for the first four nucleotides, i.e. ‘CCGC’). To facilitate pairing between the loop region and crRNA, we also designed the flap-type aptamer, wherein the first complementary 4-nucleotide sequence (flap sequence) in the loop region was connected to the 3′end of the neck region, and the remainder of the loop region was deleted (Figure 2C). The flap-type aptamer exhibited slightly better inhibitory activity than the short stem-loop-type aptamer (SS21) (Figure 2D and E). In summary, based on topological speculation and results obtained using newly designed aptamers, our data clearly suggested that the S21 aptamer inhibited the Cas9/crGFP1 complex via duplex formation between the loop region and crRNA in addition to blocking the interaction of the PAM sequence (neck region) with the Cas9 protein.
To further test the hypothesis that the first four nucleotides of the loop region could form a duplex with crRNA, we designed loop-A and flap-type inhibitory aptamers for another two target sites in GFP. The ‘CCGC’ sequence in the loop region or the flap region was replaced with the complement of the 4-nucleotide seed sequence of crGFP2 and crGFP3, respectively, and the inhibitory activity was evaluated by performing in vitro cleavage assays. As expected, all designed aptamers showed high inhibitory activities against each Cas9/crRNA complex, which were comparable to those against Cas9/crGFP1 (Supplementary Figure S2F), supporting our inhibition model for aptamer–crRNA interactions.
To check the specificity of each type of inhibitory aptamer, we examined the inhibitory effects of a stem-loop aptamer (SS21) and a flap-type aptamer (Flap-GFP1) against Cas9/crGFP1 at different target sites (GFP2 and GFP3) (Figure 3A and B). In contrast to the high inhibitory activities of the Flap-GFP2 or Flap-GFP3 aptamers against Cas9/crGFP2 or Cas9/crGFP3, respectively, Flap-GFP1 did not inhibit Cas9/crGFP2 or Cas9/crGFP3, demonstrating the high sequence specificity of the flap-type inhibitory aptamer. The stem-loop-type aptamer at the GFP1 site (SS21) showed moderate inhibitory activity for Cas9/crGFP2 and Cas9/crGFP3, implying less specificity of stem-loop-type aptamers compared with flap-type aptamers.
Figure 3.

Development and target-site specificity of flap-type aptamers. (A) Flap-type aptamers exhibited more efficient activity than stem-loop aptamers for two GFP target sites. Flap (0) was a flap-type aptamer without a flap sequence. (B) A histogram was generated based on the data shown in (A) for each inhibitor at 30 nM. The data shown are presented as means ± SD from three independent experiments. (C) The inhibitory activity depended on the flap sequence length. The flap-type aptamer for Cas9/crGFP1 without a flap sequence [Flap (0)] did not show substantial inhibitory activity, even at 100 nM. The inhibitory activity of the aptamer increased by extending the flap length [Flap (2), Flap (3), and Flap (4) in Supplementary Figure S3A]. Aptamers with 4–6-nucleotide flaps exhibited high inhibitory activities at the same level. Data are presented as means ± SD from three independent experiments. Error bars represent S.D. (D, E) Flap-type inhibitors were competent for inhibiting cleavage at EpCAM and EGFR target sites in vitro.
Next, we found that as the flap sequence was extended, the inhibitory activities of aptamers increased until the flap region reached four nucleotides (Figure 3C and Supplementary Figure S3A). A small difference between Flap (5) and Flap (3) was observed at 10 nM. Of note, the aptamer with the flap region masked by the antisense strand [Flap (4)-masked], DNA antisense for crRNA (AS-DNA), did not have inhibitory activity (Supplementary Figures S3B and C). On the other hand, antisense RNA for crRNA (AS-RNA) exhibited moderate inhibitory activities at 30 nM and above, probably owing to the antisense RNA being able to form a more stable duplex with crRNA than antisense DNA. Regarding the length of the stem region, it could be shortened to six base pairs while retaining the same inhibitory activity as the original flap-type aptamer with a 12 base-pair stem region (Supplementary Figure S3D).
Next, we confirmed the generality of the design concept of the inhibitory aptamers by evaluating flap-type aptamers for Cas9/crRNAs targeting genes encoding the epithelial cell adhesion molecule (EpCAM) and epidermal growth factor receptor (EGFR) (Figure 3D and E). Although the aptamers against EpCAM (Cas9/crEpCAM) were functional, two EGFR aptamers [Flap (4)-EGFR1 and Flap (4)-EGFR2] showed relatively low inhibitory activities at 30 nM (Figure 3E). One possible reason for the deficient inhibitory activity of the aptamers is that the sequences involved in duplex formation between the flap sequences of aptamers and crRNAs are AT-rich (‘AUAA’ and ‘UCAA’ in crEGFR1 and crEGFR2, respectively), resulting in an unstable double-stranded region with a low annealing temperature. We designed the inhibitory aptamers with longer flap sequences (5 nt and 6 nt) to stabilize the duplex between the flap sequence and crRNA, and examined the inhibitory activities for Cas9/crEGFR1 and Cas9/crEGFR2 (Supplementary Figure S3E). As expected, the inhibitory activities of the both aptamers [Flap (6)-EGFR1 and Flap (6)-EGFR2] with longer flap sequences were enhanced. The aptamers with a 6-nt flap sequence in both AT-rich sites exhibited the same inhibitory activities as the aptamers with 4-nt flap sequences for GFP and EpCAM.
LNA-modified flap-type aptamer showed high inhibitory activity for Cas9/crRNA/tracrRNA in human cells
Next, we addressed whether the inhibitory aptamer could repress Cas9/crRNA/tracrRNA-mediated genome editing within cells. To evaluate this, we used 293-stop-mScarlet cells (28), a reporter cell line that expresses a fluorescent protein (mScarlet) when genome editing occurs (Supplementary Figure S4A). In 293-stop-mScarlet cells, a CCR5 DNA fragment that included a stop codon was inserted upstream of the mScarlet fluorescent protein and led to no expression of the mScarlet protein. If Cas9/crRNA could cause indels at the stop codon and make an in-frame mScarlet fusion protein, then the reporter cells would show fluorescence resulting from mScarlet expression. We evaluated the inhibitory aptamer at three different time points [2 h before (B2), at the same time (S), or 2 h after addition to the cells (A2)]. Introducing the Cas9 protein/crRNA complex targeting the stop codon resulted in the appearance of 4–6% fluorescent cells. We noticed that delivering the aptamer before, after, or simultaneously with the Cas9 protein/crRNA complex inhibited Cas9-mediated gene editing by 66%, 70% and 72%, respectively (Figure 4A).
Figure 4.

Flap-type aptamers had efficient inhibitory activities for Cas9 in human cells. (A) Simultaneous delivery of Cas9 RNP and an aptamer markedly interfered with Cas9-mediated gene editing in human cells. Chromosomally integrated Scarlet-293 stable cells were transfected with Cas9 RNP, as well as aptamers (2 h before [B2], simultaneously with [S], or 2 h after [A2] Cas9 RNP transfection). At 72 h post-transfection, flow cytometry was performed to quantify the indel frequencies based on lost Scarlet expression in Scarlet-293 cells. Data are presented as means ± S.D. from three independent experiments. (B) An LNA-modified aptamer for Cas9 RNP markedly inhibited genome editing compared with a normal DNA aptamer at 200 nM. (C)The LNA-modified aptamer interfered with Cas9-mediated gene editing in a dose-dependent manner. The LNA-modified aptamer (50 nM) completely blocked gene editing (Cas9: crRNA/tracrRNA: inhibitor molar ratio, 1:1:1.4). LNA-modified nucleotides are indicated by yellow. Underlining indicates nucleotides with a phosphorothioate backbone. (D) LNA-modified aptamers could turn off Cas9-mediated genome editing for endogenous targets in HEK293FT cells. Indels caused by Cas9 RNP were detected in the T7E1 assay. Cells that were not transfected with Cas9/crRNA/tracrRNA and the aptamer were used as a negative control. Digested bands are indicated with star symbols. (E) Cpf1 editing of GFP1 was blocked in vitro in the presence of the aptamer designed for Cpf1. Since Cpf1 recognizes the PAM sequence (yellow) at the 3′ side of crRNA, a flap-type aptamer designed for Cpf1 is needed for the flap at the 5′ end of the aptamer to form a duplex with crRNA. Three mutants with a G:T mismatch in the PAM region showed decreased inhibitory activities. Schematic illustration of the interaction between crRNA (blue) and the aptamer designed for Cpf1 (purple) at the GFP1 site, with complementary base pairing indicated in red. (F) The inhibitory activities of Cpf1 aptamers were confirmed at three target sites in EGFR in vitro.
Furthermore, to increase the sensitivity and affinity of the aptamer for crRNA binding, we synthesized aptamers with locked nucleic acid (LNA)-modified flap sequences instead of normal DNA nucleotides (29,30). As expected, the inhibitory effect of the LNA-modified aptamer was enhanced compared with the normal aptamer at the same concentration (Figure 4B). Moreover, the minimum effective concentration was 50 nM, which was 8–16 times lower than the normal aptamer (Figure 4C, Supplementary Figures S4C and D). Next, we verified the inhibitory activity of the LNA-modified flap-type aptamers in cells for Cas9/crRNAs targeting the endogenous genes, Hypoxanthine-guanine phosphoribosyl transferase (HPRT) and Empty spiracles homolog 1 (EMX1). The LNA-modified aptamers could almost abrogate Cas9-mediated genome editing at both sites, as determined by T7E1 cleavage assays (Figure 4D), confirming the inhibitory activities.
Design of an inhibitory aptamer for Cpf1/crRNA
Finally, we explored whether the design of inhibitory aptamers for Cas9/crRNA/tracrRNA complexes is also applicable to CRISPR/Cas homologs recognizing different PAM sequences. Recently, Cpf1 which belongs to the class 2 type-V CRISPR/Cas nuclease family is widely used to produce sticky ends for broad application (31). We selected Cpf1 as a target, which recognizes a different PAM sequence (TTTN). We designed an inhibitory aptamer for Cpf1/crRNA within the GFP coding sequence (Figure 4E) and examined the inhibitory activities for Cpf1/crRNA-directed plasmid cleavage. The aptamer targeting Cpf1/crGFP1 exhibited high inhibitory activity at a similar level as the aptamers designed for Cas9/crRNA (Figure 4E). The inhibitory activities of the designed aptamers were verified for Cpf1/crRNAs at three additional sites of EGFR coding genes (Figure 4F).
Kinetic analysis of the interaction between the inhibitory aptamer and the Cas9/crRNA/tracrRNA complex
To better understand the mechanisms underlying the inhibitory effects of the aptamer, a fluorometric molecular beacon assay (32) was performed to examine the kinetic and mechanistic aspects of the inhibitory aptamer. Schematic representations of the beacon assay and structures of beacons used in the experiments are shown in Supplementary Figure S5A. The beacon shows low fluorescence intensity without the Cas9/crRNA/tracrRNA complex. Once incubated with the Cas9/crRNA/tracrRNA complex, the beacon is recognized by the complex as a target DNA. The double-stranded DNA beacon is then separated into a fluorescently labelled strand and a quencher labelled target strand by the formation of a DNA–RNA heteroduplex between the target strand and the crRNA, resulting in an increase in fluorescence intensity and disappearance of the quenching effect.
We used a nuclease-inactive mutant of Cas9 (dCas9) that does not have cleavage activity and maintain sgRNA-directed binding for substrate DNA to monitor only the inhibition effect of the aptamer on the interaction step between Cas9 complex and substrate DNA. As shown in Supplementary Figure S5B, mixing preincubated dCas9/crRNA/tracrRNA complex with 1 nM beacon led to a rapid increase in fluorescence intensity of more than 2-fold that remained constant with the extended response time. In contrast, incubation with Cas9/crRNA/tracrRNA led to a continuously increasing fluorescence intensity, likely owing to cleavage reaction by Cas9 (32).
We added different concentrations of the Flap-type inhibitory aptamer [Flap (4)-GFP1] immediately after mixing dCas9/crRNA/tracrRNA with the beacon and monitored the inhibitory effects on fluorescence intensity derived from the interaction. The inhibitory aptamer exhibited the inhibitory effects in the range from 0.1 nM to 3 nM in dose-dependent manner (Figure 5A). At 3 nM, the fluorescence intensity was almost reduced to background level that was observed without dCas9. In combination with the topological binding speculation mentioned above (Figure 2C), the inhibitory aptamer was considered to competitively inhibit the initial step of binding between the Cas9 complex and the DNA substrate by masking both the Cas9 PAM and crRNA seed regions.
Figure 5.

Comparison of anti-Cas9 protein inhibitor and aptamer-based DNA inhibitor activities. (A) Dose-dependent inhibitory effects of the inhibitory aptamer on the kinetics of beacon binding to dCas9/crRNA/tracrRNA. Fluorescence intensity decreased gradually after inhibition of the interaction between beacon and dCas9/crRNA/tracrRNA with increasing concentrations of the inhibitory aptamer (from 0.03 to 3 nM). (B) Flap-type aptamer [Flap (4)] showed higher inhibitory activities than AcrIIA4 and AcrIIA5 for Cas9/crGFP1 in the in vitro cleavage assays. The molar ratios of Cas9 to the inhibitory aptamer or the protein inhibitor were 1:0.06, 1:0.2, 1:0.6, 1:2, 1:6 at 3, 10, 30,100 and 300 nM of inhibitors, respectively. (C) Quantification of inhibitory data shown in (B). Data are presented as means ± SD from three independent experiments. Error bars represent S.D. (D) LNA-modified Flap-type aptamers were superior to AcrIIA4 and AcrIIA5 in 293-stop-mScarlet cells. The percentage of edited cells were analyzed using FACS. (E) Dose-dependent inhibitory effects of AcrIIA4 on the kinetics of beacon binding to dCas9/crRNA/tracrRNA. (F) Dose-dependent inhibitory effects of AcrIIA5 on the kinetics of beacon binding to dCas9/crRNA/tracrRNA.
Comparison of the inhibitory aptamer and anti-CRISPR proteins
As mentioned above, naturally phage-derived proteins such as AcrIIA4 and AcrIIA5 that can block genomic editing by interacting with Cas9 to prevent target DNA binding have been discovered. The reports of these protein inhibitors prompted us to perform further experiments to compare the nucleic acid inhibitory aptamer and the protein inhibitors. We utilized three different types of experiments, in vitro cleavage assay, in-cell editing assay, and fluorometric molecular beacon assay, to compare various aspects of these inhibitors. In the in vitro cleavage assay, the inhibitory aptamer had inhibitory effect starting at 10 nM and completely abolished the cleavage activity of Cas9 at 30 nM. Nevertheless, neither AcrIIA4 nor AcrIIA5 displayed any effectiveness against Cas9 in the same concentration range. After the concentration was increased to 300 nM, AcrIIA4 showed an inhibitory effect on cleavage activity, whereas AcrIIA5 did not exhibit inhibitory effects (Figure 5B and C). Next, we compared the inhibitory efficiency of the aptamer and Acrs (AcrIIA4 and AcrIIA5) in mammalian cells. 293-stop-mScarlet cells were evaluated by counting the number of fluorescent cells mediated by genomic editing. Addition of the LNA-modified aptamer led to a decrease in the percentage of edited cell from 10% to 0.32% at 50 nM (Figure 5D). Increasing the concentration to 200 nM resulted in genomic editing being almost abrogated. No Acr presented greater effectiveness than the inhibitory aptamer. In contrast to the in vitro results, AcrIIA5 exhibited modest inhibitory activity at 50 nM, whereas AcrIIA4 showed a working concentration of more than 1 μM. Furthermore, the fluorometric molecular beacon assay also demonstrated that the Flap-type aptamer has higher inhibitory activity compared with AcrIIA4 and AcrIIA5 (Figure 5A, E and F). One hundred nM of Acrs was required to abolish the interaction between dCas9 and the substrate DNA (molecular beacon) through the PAM interacting domain. In contrast, the inhibitory aptamer prevented the interaction at 3 nM (Figure 5A). In all three different types of experiments, the inhibitory aptamer exhibited higher inhibitory effects than Acrs, presumably because that the inhibitory aptamer possesses an additional segment for base pairing with crRNA in addition to the PAM-interacting domain binding.
The inhibitory aptamer inhibits genome editing at on- and off-targets and increases reliability of Cas9-mediated genome editing
To test the effects of the aptamer-based inhibitor on on- and off-targeting, we performed a molecular beacon assay using a crRNA with a single point mutation in the seed region (the first 8 nucleotides immediately adjacent to PAM). The second nucleotide (‘C’) upstream of PAM was changed to ‘A’, like similarly mutated substrates, and was regarded as an off-target site. The flap sequence of the inhibitory aptamer for the mutated crRNA was correspondingly changed to the complementary sequence. Using the on- and off-target evaluation assays, we examined the inhibitory activities of the aptamer for on- and off-target sites (Supplementary Figure S5C). In the off-target assay, fluorescence intensity decreased by 13% compared with a perfect match (on-target) after adding the beacon. After addition of the aptamer (1 nM) specific for each crRNA (aptamerm for cRNAm or aptamer for crRNA in Supplementary Figure S5C), the off-target binding was reduced almost to background levels, indicating that the inhibitory aptamer could act on the off-target site as well as on-target site by inhibiting the binding of Cas9/crRNA/tracer complex to substrate DNA (Supplementary Figure S5D). Further evaluation was performed on on- and off-target sites of β-globin (HBB) and EMX 1 genes in mammalian cells. Next-generation sequencing of amplicons that included the target sites showed that the addition of 10 nM specific aptamers resulted in appreciably decreases in both on- and off-target editing (Figure 6A and B). Therefore, genome editing efficiency could be modulated by different concentrations of the aptamer in a dose-dependent manner. Because the inhibitory effects on genome editing at on- and off-target sites were almost at the same level (Supplementary Figure S6A), further efforts were taken to reduce off-target editing only. Finally, we could show off-target reducing effects of the inhibitory aptamer utilizing genome editing system using Cas9- and sgRNA-expression plasmid, by which the long-term expression of Cas9/sgRNA in cells is known to cause more serious off-target genome editing. Specifically, we transfected Cas9- and sgRNA-expression plasmid targeting at EMX1 and HBB loci into 293FT cells. Fourteen and 26 hours after plasmid transfection, we transfected the LNA-modified inhibitory aptamer and evaluated on- and off-target genome editing efficiencies by deep sequencing. As expected, the inhibitory aptamer was able to inhibit off-target editing more efficiently than on-target editing at both EMX1 and HBB loci (Figure 6C). By adjusting the experimental condition (timing of aptamer transfection), the inhibition ratio between off-target editing and on-targeting was improved to 2.4 folds (Supplementary Figure S6B). This result means that the inhibitory aptamer can be used to increase reliability of genome editing in various therapeutic applications using plasmid- or virus vector-based genome editing system.
Figure 6.

The applications of the inhibitory aptamer for reduction of on and off-target genome editing and control of dCas9-based transcription activation. (A) and (B) Editing efficiencies of on- and off-target sites were inhibited by the LNA-modified inhibitory aptamer in a does-depended manner, as determined by deep sequencing. 293FT cells were co-transfected with Cas9/crRNA/tracrRNA and aptamer. (C) Timed delivery of LNA-modified inhibitory aptamer could differentially reduce off-target editing compared to on-target editing. 293FT cells were transfected with plasmids encoding Cas9 and sgRNA targeting EMX1 or HBB locus, respectively. Fourteen hours and 26 hours after the transfection, 100 nM aptamers were delivered into cells. Inhibitions of editing by the inhibitory aptamer at on- and off-target sites of EMX1 and HBB were examined by next-generation sequencing. (D) Schematic diagram of CRISPR-based gene activation experiment. 293FT cells were co-transfected with plasmids encoding dCas9-VP64-sgRNA, Turbo GFP, MS2-P65-HSF1, or dsRed which is used for standardization. (E) Dose-dependent inhibition of CRISPRa by the LNA-modified inhibitory aptamer in 293FT cells. Cells were co-transfected with plasmids (pdCas9-VP64-sgRNA2.0, pMS2-p65-HSF1 and p6xTRE-TurboGFP) and the LNA-modified aptamers at indicated concentrations and incubated for 48 h. The top and bottom panels represent the bright field and GFP channels, respectively. Bar = 200 μm. (F) Flow-cytometric analysis of CRISPRa inhibition assay. (G) Quantification of fluorescence fold changes shown in (F). Data are presented as means ± SD from three independent experiments. Error bars represent SD.
The inhibitory aptamer can control dCas9-based transcription activation
As another application of the inhibitory aptamer, we next addressed whether the aptamer can control CRISPR/Cas9-directed gene regulation—CRISPR activation or CRISPR interference (CRISPRa or CRISPRi). We utilized the CRSPRa system reported by Konermann et al. (6), in which two expression plasmids (dCas9-VP64 and sgRNA2.0 expression plasmid and MS2-p65-HSF1 expression plasmid) and one reporter plasmid (promoter including 6× sgRNA target sites driven TurboGFP reporter plasmid were used (21). As shown in Figure 6D, the transcriptional activation complex consisted of dCas9-VP64/gRNA2.0 and MS2-p65-HSF1 are recruited on the promoter including 6× sgRNA target sites. Consequently, the expression of downstream TurboGFP was induced. The LNA-modified inhibitory aptamer could inhibit the dCas9-directed transactivation of TurboGFP in a dose-dependent manner. The activation was almost blocked to the background level by using 100 nM of the inhibitory aptamer (Figure 6E–G). This result demonstrated the possibility of the inhibitory aptamer as a molecular tool for controlling CRISPR-based gene regulation in mammalian cells.
DISCUSSION
Herein, we described a method for designing DNA inhibitors for the CRISPR/Cas genome-editing enzymes. Firstly, we obtained inhibitory aptamer candidates by in vitro selection against Cas9 protein and subsequent in vitro cleavage assays using the Cas9/crRNA/tracrRNA complex (Figure 1), and through various experiments, speculation, and experimental verifications, we show that the inhibitory aptamer consists of three components, a sequence-independent double-stranded region, a PAM recognition region (neck region), and single-stranded region that forms a duplex with the seed region of crRNA (flap region) (Figure 2). We propose a model of mechanism for the inhibitory aptamer to suppress SpCas9 activities (Figure 7): without aptamer, PAM region in the genome and the base pairing between the crRNA and its target directs the Cas9 nuclease to bind and generate double-strand breaks (DSBs). In the presence of aptamer, the activity of Cas9 was inhibited by aptamer which is competing with target DNA for binding with Cas9 and crRNA. The switch for genomic editing is off. Furthermore, we demonstrate that the inhibitory aptamer aimed at Cpf-1, another member of CRISPR/Cas family, can inhibit its activity (Figure 4). It is therefore likely that this method for designing inhibitory aptamers is generally applicable to a broad range of CRISPR/Cas family members, including those for which natural inhibitory proteins have not been identified.
Figure 7.
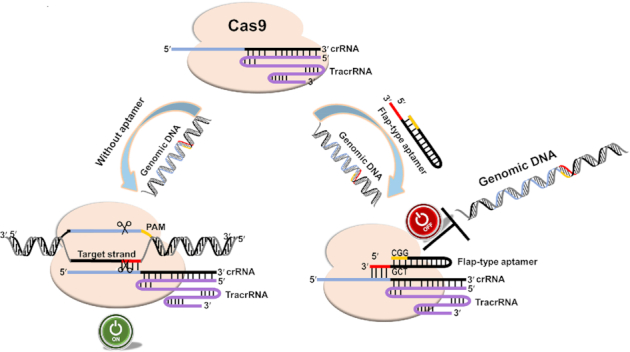
Schematic illustration of on–off regulation of genome editing by a flap-type aptamer. The Cas9 protein (light orange) assembles with the crRNA (blue) and tracrRNA (purple) to bind genomic DNA with a PAM sequence (yellow). In the absence of the inhibitory aptamer, the switch is on, resulting in genomic DNA editing. However, in the presence of the aptamer, the binding site is occupied by the aptamer that also contains a competitive PAM sequence (CGG). Four nucleotides in the flap region (red) of the aptamer form a complementary duplex with the crRNA seed sequence (blue). Consequently, Cas9’s target DNA binding and complementary base pairing between target DNA and crRNA are inhibited, and the switch is turned off.
The LNA-modified inhibitory aptamer for Cas9 drastically blocked genomic editing in cells due to its unprecedented thermal stability when hybridized to a complementary crRNA strand and markedly increased stability to nuclease. (Figure 4). As indicated in recent reports, off-targeting (7), toxic effects (33), and/or occurrence of accidental deletions/genomic rearrangements during genome editing are critical problems (9), especially when genome editing is used for therapeutic purposes. The LNA modified inhibitory aptamer we developed here and further development of related technologies may provide a clue to solve these problems.
For example, in the case of cell therapy using genome-edited cells with AAV- or plasmid-mediated expression of Cas9/sgRNA, the prolonged-expression of the Cas9/sgRNA enzyme would increase the risk of off-targeting, toxic effects, and/or accidental editing (34,35). Based on detailed comparisons between anti-CRISPR proteins and aptamer, our result demonstrates that the aptamer co-transfected with Cas9 exhibited higher inhibitory activity for genome editing (Figure 5) (36). Additionally, it was noted that timed delivery of the inhibitory aptamer limited the off-target editing caused by the long duration of Cas9 and maintained the high frequency of the on-targeting, thus increasing the fidelity of Cas9 at on-target site compared to off-target sites (Figure 6C, Supplementary Figure S6B). Compared with AcrII4 protein and peptide inhibitors (37), the inhibitory effect is in line with the published data suggesting that inhibitory aptamer can work as a molecular tool to reduce the off-target editing with lower perturbation on the on-target editing. It is expected the aptamer would also be a molecular modulator for increasing the fidelity of CRISPR base editors for an on-target region (38). Long activation of Cas9 raises the risk of unintended edits, thus differentially reducing off-target editing sheds a light on safer and more accurate cell therapies for pharmaceutical applications.
The property of the inhibitory aptamer to competitively inhibit DNA recognition by the Cas9/crRNA/tracrRNA complex allows its application to various tools utilizing Cas9-directed DNA recognition. CRISPR-based gene regulation systems, known as CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi), are representative of these tools, where dCas9 fused with a transcriptional activator or repressor is used for up- or down-regulation of gene expression, respectively (39). We revealed that the inhibitory aptamer could efficiently block dCas9-mediated transcription activation (Figure 6E–G), which demonstrates controllable applications on CRISPR-based genomic regulation. The result was consistent with a recent report by Nakamura et. al. who demonstrated that Acrs can regulate CRISPR-mediated activation and repression of the reporter or endogenous genes in various cell types, and that it can also be used to control CRISPR-based gene circuits via an Acrs-mediated feedback loop (40). The inhibitory aptamer developed in this study, which has higher and crRNA-dependent inhibitory activities, could regulate CRISPRa and CRISPRi as well, and would allow the generation of more complex gene circuits.
To date, anti-CRISPR proteins, small molecules and bacteriophage-derived peptides were discovered to impede activity of CRISPR systems in gene-editing by preventing target DNA binding, cleaving or sgRNA loading (36,37,41). The advantage of protein inhibitors is that they are genetically encoded and can be expressed in cells. This is feasible to continually protect cells from editing by long duration of Cas activity. However, risks of potential toxicity and immunogenicity accompany with expression of anti-CRISPR proteins or peptide. In addition, inhibition event caused by anti-CRISPR proteins is not readily reversible. Nevertheless, light, tamoxifen-inducible systems and small molecules have been engineered to improve reversibility, temporal and quantitative control (41–43). Compared with anti-CRISPR proteins, the DNA inhibitor is compact in size and can be synthesized in a large scale. Furthermore, DNA inhibitory aptamer has high inhibitory activities, broad spectrum for other Cas families and is capable of crRNA sequence-dependent repression of the Cas9/crRNA/tracrRNA complex, which are advantages over the small molecules and the inhibitory peptides. Although our system is difficult to control genome editing reversibly, it is excellent in regulating the editing efficiency quantitatively with a small amount, and therefore it is considered that it can be used properly depending on applications. The anti-CRISPR protein inhibitor and the DNA aptamer inhibitor have different characteristics. Taking advantage of the characteristics of each inhibitor, both protein and DNA inhibitors may be efficiently utilized to control the CRISPR/Cas system in various situations. Finally, the application of the inhibitors to phage medicine by inhibiting the immunity to phage, which is an essential function of the CRISPR/Cas system (44), may also be possible.
DATA AVAILABILITY
Secondary structures were predicted using the prediction program for secondary folding, mfold (http://unafold.rna.albany.edu/?q=mfold). Consensus sequence were identified using the MEME Suite 5.0.1 motif analysis tool (http://meme.nbcr.net/meme/tools/meme). CRISPResso2 was used for analysis of genome editing outcomes from Illumina MiSeq sequencing data (http://crispresso.pinellolab.partners.org/).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Yamazaki K. for helpful comments and for sharing Biacore instrument. We also thank Ms. Murakami T. for protein purification and Furuhata Y. for helpful comments on CRISPRa experiments.
Contributor Information
Jing Zhao, Molecular Composite Physiology Research Group, Health and Medical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba Science City 305-8566, Japan; Department of Plant Pathology, Nanjing Agricultural University, Nanjing 210095, China.
Rika Inomata, Molecular Composite Physiology Research Group, Health and Medical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba Science City 305-8566, Japan; Program in Life Science Innovation, School of Integrative and Global Majors, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8577, Japan.
Yoshio Kato, Molecular Composite Physiology Research Group, Health and Medical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba Science City 305-8566, Japan.
Makoto Miyagishi, Molecular Composite Physiology Research Group, Health and Medical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba Science City 305-8566, Japan; Program in Life Science Innovation, School of Integrative and Global Majors, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8577, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online. The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
FUNDING
New Energy and Industrial Technology Development Organization (NEDO) program, which is a part of ‘Development of Production Techniques for Highly Functional Biomaterials Using Smart Cells of Plants and Other Organisms’ project; internal grant from National Institute of Advanced Industrial Science and Technology (AIST). Funding for open access charge: AIST.
Conflict of interest statement. None declared.
REFERENCES
- 1. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A.et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G.et al.. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014; 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M.et al.. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014; 159:440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R.. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013; 153:910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H.et al.. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015; 517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D.. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P.et al.. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015; 33:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kosicki M., Tomberg K., Bradley A.. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018; 36:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R.. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013; 31:839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bondy-Denomy J., Pawluk A., Maxwell K.L., Davidson A.R.. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013; 493:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pawluk A., Davidson A.R., Maxwell K.L.. Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 2018; 16:12–17. [DOI] [PubMed] [Google Scholar]
- 13. Pawluk A., Amrani N., Zhang Y., Garcia B., Hidalgo-Reyes Y., Lee J., Edraki A., Shah M., Sontheimer E.J., Maxwell K.L.et al.. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell. 2016; 167:1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu L., Yin M., Wang M., Wang Y.. Phage AcrIIA2 DNA Mimicry: Structural basis of the CRISPR and Anti-CRISPR Arms race. Mol. Cell. 2019; 73:611–620. [DOI] [PubMed] [Google Scholar]
- 15. Hynes A.P., Rousseau G.M., Lemay M.L., Horvath P., Romero D.A., Fremaux C., Moineau S.. An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nat Microbiol. 2017; 2:1374–1380. [DOI] [PubMed] [Google Scholar]
- 16. Watters K.E., Fellmann C., Bai H.B., Ren S.M., Doudna J.A.. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science. 2018; 362:236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keefe A.D., Pai S., Ellington A.. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010; 9:537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic. Acids. Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagasaki A., Kato Y., Meguro K., Yamagishi A., Nakamura C., Uyeda T.Q.P.. A genome editing vector that enables easy selection and identification of knockout cells. Plasmid. 2018; 98:37–44. [DOI] [PubMed] [Google Scholar]
- 21. Furuhata Y., Nihongaki Y., Sato M., Yoshimoto K.. Control of adipogenic differentiation in mesenchymal stem cells via endogenous gene activation Using CRISPR-Cas9. ACS Synth. Biol. 2017; 6:2191–2197. [DOI] [PubMed] [Google Scholar]
- 22. Pinello L., Canver M.C., Hoban M.D., Orkin S.H., Kohn D.B., Bauer D.E., Yuan G.C.. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016; 34:695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 24. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 25. Hart D.J., Speight R.E., Cooper M.A., Sutherland J.D., Blackburn J.M.. The salt dependence of DNA recognition by NF-kappaB p50: a detailed kinetic analysis of the effects on affinityand specificity. Nucleic Acids Res. 1999; 27:1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anders C., Niewoehner O., Duerst A., Jinek M.. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014; 513:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O.. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014; 156:935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bindels D.S., Haarbosch L., van Weeren L., Postma M., Wiese K.E., Mastop M., Aumonier S., Gotthard G., Royant A., Hink M.A.et al.. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods. 2017; 14:53–56. [DOI] [PubMed] [Google Scholar]
- 29. Cromwell C.R., Sung K., Park J., Krysler A.R., Jovel J., Kim S.K., Hubbard B.P.. Incorporation of bridged nucleic acids into CRISPR RNAs improves Cas9 endonuclease specificity. Nat. Commun. 2018; 9:1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Veedu R.N., Wengel J.. Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 2010; 7:536–542. [DOI] [PubMed] [Google Scholar]
- 31. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A.et al.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mekler V., Minakhin L., Semenova E., Kuznedelov K., Severinov K.. Kinetics of the CRISPR-Cas9 effector complex assembly and the role of 3′-terminal segment of guide RNA. Nucleic Acids Res. 2016; 44:2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J.. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018; 24:927–930. [DOI] [PubMed] [Google Scholar]
- 34. Hsu PD., Scott DA., Weinstein JA., Ran FA., Konermann S., Agarwala V., Cradick T.J.. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim S., Kim D., Cho S.W., Kim J., Kim J.S.. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014; 24:1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin J., Jiang F., Liu J.J., Bray N.L., Rauch B.J., Baik S.H., Doudna J.A.. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv. 2017; 3:e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cui Y.R., Wang S.J., Chen J., Li J., Chen W., Wang S., Jiang B.. Allosteric inhibition of CRISPR-Cas9 by bacteriophage-derived peptides. Genome Biol. 2020; 21:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H.. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019; 364:289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perez-Pinera P., Kocak D.D., Vockley C.M., Adler A.F., Kabadi A.M., Polstein L.R., Thakore P.I., Glass K.A., Ousterout D.G., Leong K.W.et al.. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods. 2013; 10:973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakamura M., Srinivasan P., Chavez M., Carter M.A., Dominguez A.A., La Russa M., Lau M.B., Abbott T.R., Xu X., Zhao D.et al.. Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun. 2019; 10:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maji B., Gangopadhyay S.A., Lee M., Shi M., Wu P., Heler R., Dančík V.. A high-throughput platform to identify small-molecule inhibitors of CRISPR-Cas9. Cell. 2019; 177:1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Polstein L.R., Gersbach C.A.. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015; 11:198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu K.I., Ramli M.N.B., Woo C.W.A., Wang Y., Zhao T., Zhang X., Jung J.. A chemical-inducible CRISPR–Cas9 system for rapid control of genome editing. Nat. Chem. Biol. 2016; 12:980–987. [DOI] [PubMed] [Google Scholar]
- 44. Levin B.R., Bull J.J.. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004; 2:166–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Secondary structures were predicted using the prediction program for secondary folding, mfold (http://unafold.rna.albany.edu/?q=mfold). Consensus sequence were identified using the MEME Suite 5.0.1 motif analysis tool (http://meme.nbcr.net/meme/tools/meme). CRISPResso2 was used for analysis of genome editing outcomes from Illumina MiSeq sequencing data (http://crispresso.pinellolab.partners.org/).