


Abstract
The construction of microbial cell factories for sustainable production of chemicals and pharmaceuticals requires extensive genome engineering. Using Saccharomyces cerevisiae, this study proposes synthetic neochromosomes as orthogonal expression platforms for rewiring native cellular processes and implementing new functionalities. Capitalizing the powerful homologous recombination capability of S. cerevisiae, modular neochromosomes of 50 and 100 kb were fully assembled de novo from up to 44 transcriptional-unit-sized fragments in a single transformation. These assemblies were remarkably efficient and faithful to their in silico design. Neochromosomes made of non-coding DNA were stably replicated and segregated irrespective of their size without affecting the physiology of their host. These non-coding neochromosomes were successfully used as landing pad and as exclusive expression platform for the essential glycolytic pathway. This work pushes the limit of DNA assembly in S. cerevisiae and paves the way for de novo designer chromosomes as modular genome engineering platforms in S. cerevisiae.
INTRODUCTION
Microbial cell factories have an important role to play in the development of a sustainable and environmentally friendly biobased economy, as already exemplified by the ca. 100 billion litres of bioethanol (1) and half of the world's insulin (2) annually produced by Saccharomyces cerevisiae, more commonly known as baker's yeast. However, the construction of novel microbial production hosts for chemicals and pharmaceuticals is impeded by the poor economic viability of bio-based processes. Improving microbial process profitability and competition with current (petrochemical based) production methods requires powerful microbial cell factories that produce (novel) chemicals from non-native feedstock (3) with high product yields and productivity in harsh industrial conditions. This can only be accomplished by extensive genome engineering that not only adds new functionalities to the host microbe, but also deeply rewires its native cellular and metabolic processes (4,5).
Although the construction of synthetic cell factories with designer genomes, tailor-made for the optimum production of certain chemicals, might become feasible in the future, it is currently far from reach. One reason is our still limited understanding of the absolute requirements of life, as demonstrated by the recent reconstruction of a minimal Mycoplasma genome in which one third of the essential genes are not functionally characterized (6). The second major hurdle in the synthesis of designer genomes is DNA synthesis. The maximum size for the chemical synthesis of ssDNA of a quality that can be used for biotechnological application is 200 bp (7,8). Despite recent developments in stitching together these ssDNA parts into genes, routine synthesis of designer genomes would be too cumbersome and expensive (8,9). Indeed, although numerous fast (seamless) in vitro assembly methods (10) have been developed such as: Gibson assembly, Golden Gate, ligase cycling reaction, seamless ligation cloning extract and circular polymerase extension cloning, these methods are intrinsically limited in the number of fragments that can be assembled as well as the final size of the assembled DNA. Furthermore, Escherichia coli, used as a propagation host (11), cannot faithfully replicate genome-size DNA constructs (12). It is S. cerevisiae that ultimately enabled the complete assembly and replication of the 580 kb Mycoplasma genome (12). The homologous recombination machinery of S. cerevisiae was able to assemble 25 overlapping DNA fragments of ∼24 kb into the Mycoplasma genome as well as assemble 38 overlapping ssDNA pieces of 200 bp (13,14). This high efficiency and fidelity of homologous recombination has undoubtedly contributed to S. cerevisiae popularity as eukaryotic model and cell factory. Yet, the full extent of in vivo assembly capabilities in terms of size and number of fragments has not been explored. Introduction of large genetic constructs in S. cerevisiae typically takes place by integration in existing chromosomes, without much considerations for potential impact on the host chromosome architecture. The recent introduction of the noscapine pathway, for example, required introduction of 31 (heterologous) genes in nine different chromosomal loci (4). Integration of large constructs might alter the chromosome structure and affect the expression of genes surrounding the newly added construct, as well as replication of the host chromosome. It might also trigger unwanted chromosomal rearrangements between native chromosomes. It is long known that S. cerevisiae can stably replicate and segregate Yeast Artificial Chromosomes (YACs) (15). In the present study, we explore the potential of in vivo assembled, supernumerary chromosomes for modular genome engineering in S. cerevisiae.
To enable easy remodelling of existing functions, Kuijpers et al. (16) developed the pathway swapping concept in S. cerevisiae. The genetic reduction and relocalization of the entire Embden–Meyerhoff–Parnas pathway of glycolysis to a single chromosomal locus enabled to swap this essential pathway to any new (heterologous) design in two simple steps (16,17). Combined to an orthogonal expression platform, in the form of a supernumerary synthetic neochromosome, pathway swapping would offer the possibility to make the introduction and expression of large product pathways in combination with the rewiring of essential native pathways, more efficient and predictable. Unlike the construction of chromosomes based on an existing structure for the Mycoplasma and S. cerevisiae genome projects (Sc2.0) (18,19), the neochromosomes assembly design should be based on modularity to allow for easy change in makeup and configuration of native and heterologous pathways.
The present study explores the possibility for a modular assembly of a designer, supernumerary neochromosome for rational engineering of the yeast genome. A pipeline was developed for easy and rapid design, in vivo assembly and verification of neochromosomes. The limits of in vivo assembly in terms of number and size of assembled DNA fragments, efficiency and fidelity were explored. The stability of the neochromosomes and their impact on yeast physiology were probed. Finally the ability of supernumerary synthetic chromosomes to serve as landing pad for metabolic pathways and to carry essential pathways was investigated. This study paves the way for the implementation of supernumerary, synthetic chromosomes as modular expression platforms for native and heterologous functions.
MATERIALS AND METHODS
A detailed description of the Materials and Methods employed in this study can be found in the Supplementary Data. All tables and figures pertaining to Material and Methods are referred to in the Supplementary Data and can be found in the Supplementary Materials and Methods Data file.
Strains, maintenance and growth media
All S. cerevisiae strains used in this study are derived from the CEN.PK family (20). For non-selective growth and propagation, the yeast strains were grown on Yeast extract-Peptone (YP) medium containing: 10 g l−1 Bacto yeast extract and 20 g l−1 Bacto peptone. For selective growth, Synthetic Medium (SM) was used (21). 20 g l−1 glucose was used as carbon source. Liquid yeast culture were grown in 500 ml shake flask with 100 ml medium at 30°C and 200 rpm in an Innova incubator (New Brunswick Scientific, Edison, NJ, USA), unless stated otherwise.
Escherichia coli XL1-blue was used for propagation and isolation of plasmids. E. coli was grown in Lysogeny Broth (10 g l−1 Bacto tryptone, 5 g l−1 Bacto yeast extract and 5 g l−1 NaCl). Cultivation was performed in 15 ml Greiner tubes or 25 ml shake flasks at 37°C and 200 rpm in an Innova 4000 shaker (New Brunswick Scientific).
For storage of S. cerevisiae and E. coli strains, the cultures were mixed with glycerol (30% v/v) and stored in 1 ml vials at −80°C.
Molecular biology techniques
Genomic DNA from S. cerevisiae used for amplification of integrative cassettes or fragments to build synthetic chromosomes was isolated with the YeaStar genomic DNA kit (Zymo Research, Irvine, CA, USA) or the QIAGEN Blood & Cell Culture Kit with 100/G Genomic-tips (Qiagen, Hilden, Germany). Genomic DNA of E. coli was isolated by 1 h incubation at 37°C in lysis buffer (10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.6% SDS (v/v), 0.12 g l−1 proteinase K), followed by the YeaStar genomic DNA kit (Zymo research) Plasmids were isolated from E. coli using the Sigma GenElute Plasmid DNA miniprep kit (Sigma-Aldrich, St. Louis, MO, USA). All DNA fragments for transformation in S. cerevisiae or E. coli of <10 kb were amplified by PCR using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA), all fragments of 10 kb or longer were amplified with LongRange PCR (Qiagen) according to the manufacturer's instructions. PCR fragments were purified with the Zymoclean Gel DNA Recovery kit (Zymo Research), the GenElute PCR Clean-Up kit (Sigma-Aldrich) or using the AMPure XP beads (Beckman Coulter, Brea, CA, USA). Transformation in S. cerevisiae was executed using the lithium acetate/polyethylene glycol method (22).
Plasmid construction
Plasmids carrying the fluorescent markers mRuby2 (pUDC191), mTurquoise2 (pUDC192) and Venus (pUDC193) were constructed by Golden Gate BsaI assembly using the plasmid parts provided in the Yeast Toolkit (23). The transcriptional units of the major glycolytic and fermentation enzymes FBA1, TPI1, PGK1, ADH1, PYK1, TDH3, ENO2, HXK2, PGI1, PFK1, PFK2, GPM1 and PDC1 were also cloned in plasmids by Golden Gate assembly to facilitate amplification by PCR. The promoter (800 bp)-gene-terminator (300 bp) sequences were amplified from their own native location in the genome of CEN.PK113-7D using primers containing BsaI sites. Guide RNA (gRNA) plasmids for introducing Cas9-mediated double strand breaks were constructed as described by Mans et al. (24).
Strain construction
The strains constructed in this study are derived from SwYG described in Kuipers et al. (16) (Figure 7). SwYG is characterized by the genetic reduction and relocalization to the SGA1 locus of the set of genes encoding the glycolytic and fermentative pathways. As host strain for synthetic chromosome assembly, IMX1338 was constructed by integration in SwYG’s genome of an inducible expression cassette for the meganuclease I-SceI (25).
Figure 7.

Strain construction overview. The parental strain IMX1338 is framed by a filled grey box from which its direct decedents are indicated with black arrows. The schematic representations of linear or circular chromosomes indicate the relevant native or synthetic chromosomes that were modified in this study, and the significance of the symbols is explained in the top left box. Strains characterized in this study are framed by a dashed gray box.
For the construction of strains carrying in vivo assembled synthetic chromosomes (IMF1, IMF2, IMF6, IMF23-IMF26), the amount of DNA fragments transformed was kept constant for all experiments with 200 fmol of each E. coli filler fragment, 200 fmol of each fluorescent marker and 100 fmol of the CEN6/ARS4, ARS and selectable markers. The transformants were checked by fluorescent microscopy, FACS, CHEF and Whole Genome Sequencing (WGS) as described below. Moreover strains IMF2 (50 kb), IMF6 (100 kb) and IMF23-IMF26 (100 kb with improved design) were verified by long-read nanopore sequencing.
The synthetic chromosomes in IMF2 and IMF6 were used as landing pads for in vivo assembly and integration of two types of DNA constructs, both 35 kb long. Both constructs were flanked by ARS sequences and carried the KanMX selectable marker, but differed by the presence of non-coding E. coli filler fragments for one, and 13 functional glycolytic and fermentative genes from yeast for the other. Integration of these constructs was facilitated by Cas9-mediated editing of the neochromosomes at the mTurquoise2 locus. For the construction of the glycolytic control strain IMX2109, the glycolytic and fermentative genes were inserted at the CAN1 locus using CRISPR/Cas9.
When required, the glycolytic and fermentative genes in the SGA1 locus on chromosome IX were excised using CRISPR/Cas9. Similarly, deletion of mRuby2 and Venus from strain IMF6 was accomplished using CRISPR/Cas9. To make prototrophic control strains, URA3 and HIS3 were inserted at the X2 locus.
IMX2224, the mRuby2 fluorescent control strain was constructed by integrating mRuby2 at the YPRCTau3 locus using Cas9-mediated editing.
Fluorescence detection by microscopy and flow cytometry
To validate the expression of the fluorescent proteins mRuby2, Venus and mTurquoise2, from the neochromosomes, the transformants were checked by fluorescent microscopy and/or by flow cytometry. Fluorescence was observed by the ZEISS Axio Imager Z1 microscope (Carl Zeiss AG, Oberkochen, Germany), with three different filter sets. mRuby2 was detected by Filter set 14 with excitation at 535 nm and emission at 590 nm. Venus was visualized using filter set 9, with excitation at 470 nm and emission at 515 nm. Finally filter set 47 was used to detect mTurquoise2 at an excitation of 436 nm and an emission of 480 nm. The BDFACSAria™ II Cell Sorter in combination with the BDFACSDiva software (BD Biosciences, Franklin Lakes, NJ, USA) was used for flow cytometry analysis. The machine was equipped with 355, 445, 488, 561 and 640 nm lasers and a 70 μm nozzle, and operated with filtered FACSFlow™ (BD Biosciences).
Contour-clamped homogeneous electric field (CHEF) electrophoresis
The size of the synthetic chromosomes was checked by CHEF. Agarose plugs containing genomic DNA were made using the CHEF Yeast Genomic DNA Plug Kit (Bio-Rad laboratories, Hercules, CA, USA) following the manufacturer's instructions. For in-plug linearization of the synthetic chromosomes, I-SceI digestion (Thermo Fischer Scientific) was performed in the plugs. The chromosomes were separated using a Bio-Rad Electrophoresis Cell in combination with a CHEF-DR® II Control Module and a CHEF-DR® II Drive module (Bio-Rad laboratories). The gel was visualised with an InGenius LHR gel Imaging System (Syngene, Bangalore, India).
Sequencing
The sequence of synthetic chromosomes was checked by short-read and long-read sequencing. For whole genome short-read sequencing, the libraries were sequenced in-house using an Illumina MiSeq sequencer (Illumina, San Diego, CA, USA). For whole genome long-read sequencing, MinION technology was performed in-house (Oxford Nanopore, sequencing kit SQK-LSK109, expansion kit EXP-NBD104). Sanger short-read sequencing was performed on the original purified E. coli chunks used for assembly of the neochromosomes (Baseclear, Leiden, The Netherlands).
RNA-Sequencing was performed on strains IMF17 and IMX2109 grown to mid-exponential phase in shake-flask culture, in biological triplicate, using Illumina technology (Macrogen Europe B.V., Amsterdam, The Netherlands).
Physiological characterisation
Physiological characterisation was performed in shake-flask in 100 ml culture volume or in microtiter plate in a Growth Profiler (EnzyScreen BV, Heemstede, The Netherlands). A maximum specific growth rate (μmax) was calculated from at least five data points in the exponential phase. Each experimental condition was performed in at least biological duplicate.
In vitro enzyme activities
Activity of the glycolytic enzyme was measured from cultures in shake-flask. Cell extracts were prepared as described by Postma et al. (26). Assays were performed as previously described (27,28).
Statistical analysis
If replicates were performed, data are presented as mean ± s.d. and the number of replicates is indicated. Statistical significance was determined either by using a t-test (paired or unpaired is indicated) for comparison of two samples using excel or for comparison of multiple samples by ANOVA with Post-Hoc Tukey-Kramer using GraphPad Prism 4 (Graphpad, San Diego, CA, USA). For both tests, differences were considered significant when P < 0.05.
RESULTS
Design considerations and proof of concept for the modular, de novo assembly of supernumerary synthetic neochromosomes
Establishment of a workflow for fast and flexible construction of neochromosomes should include a set of rules to enable easy design, assembly and screening of neochromosomes with different configurations of genes and auxiliary parts. This flexibility, required for pathway optimization in future strain construction programs, gave the possibility to explore structural requirements for the assembly and maintenance of the neochromosomes in the present study. Indeed, in this nascent field, very little is known about neochromosome design requirements and even less is known about neochromosome stability and impact on physiology. The second most important construction criterion was the minimization of experimental steps to construct the neochromosomes. Accordingly, the chosen construction workflow exploits the strengths of in vitro and in vivo assembly, by first using Golden gate cloning for stitching functional DNA parts together (e.g. construction of transcription units from promoter, gene and terminator), then assembling de novo these in vitro stitched parts using S. cerevisiae highly efficient homologous recombination (Figure 1A). This workflow also minimized the number of PCR amplification steps, that are notoriously error-prone, to a single one.
Figure 1.

Modular genome engineering strategy. Schematic representation of the design and construction of neochromosomes (A), as well as screening based on fluorescence, size and sequencing (B). The data in panel B represent the results pertaining to the assembly of the 100 kb neochromosome in strain IMF6. The red bar in the top left of the fluorescence microscopy pictures (40x) represents 10 μm. The CHEF gel used to determine the size of the neochromosomes, contains two transformants (#3 and #4) of the 100 kb neochromosome transformation. The neochromosomes are either undigested (undig.) by I-SceI and therefore in their circular form, or in-plug linearized by I-SceI digestion (dig.) and therefore in linear form, which can be visualised on CHEF gel.
In vivo neochromosome assembly was promoted by framing the DNA fragments transformed to yeast with short overhangs with no homology with the yeast genome (60 bp long called SHRs (29)), that can be easily and cheaply added by PCR. The neochromosomes were designed to carry replicative fragments, including a centromere and autonomously replicating sequences (ARS) spaced every 30–40 kb (30), and markers to facilitate the selection of transformants with correctly assembled neochromosomes. To evaluate the efficiency of our neochromosome construction workflow, two test chromosomes of 50 and 100 kb were designed and assembled. To mimic assembly of pathways, typically composed of transcription units of ca. 2–3 kb, but to avoid potential interference by gene expression from the neochromosomes, the test neochromosomes were assembled from 2.5 kb E. coli DNA fragments. Although these DNA fragments contain E. coli genes, they are not expected to be expressed in S. cerevisiae due to fundamental differences in prokaryotic and eukaryotic transcription machineries (31,32). Specifically, the test neochromosomes comprised: CEN6/ARS4 (33), ARS1 (and also ARS417 for 100 kb neochromosome), two auxotrophic selection markers (HIS3 and URA3), three fluorescent proteins (Venus, mRuby2 and mTurquoise2), and 2.5 kb non-coding E. coli DNA fragments used to reach the desired chromosome size (called filler fragments) and the telomerator (34). The telomerator allows for in vivo chromosome linearization and was included for potential future use. This resulted in 23 fragments for the assembly of the 50 kb neochromosome and 44 fragments for the 100 kb neochromosome (Figure 2), which were transformed in fixed molar amounts per fragment (therefore approximately twice as much DNA was transformed for the 100 kb neochromosome with respect to the 50 kb neochromosome). Despite the large number of DNA fragments, transformation on selective growth medium (histidine and uracil deficient) resulted in a large number of colonies (ca. 2000 colonies and 275 colonies, for the 50 and 100 kb neochromosome respectively). Four colonies of each transformation were checked by fluorescence microscopy and all expressed the three fluorescent markers (Figure 1B and Supplementary Figure S1). For both test chromosomes, one of these four colonies was randomly selected and tested by diagnostic PCR of the assembly junctions (Supplementary Figure S2), which revealed the correct assembly of the 50 and 100 kb neochromosomes. The genome of two transformants harbouring the 100 kb neochromosome (IMF1 and IMF6) and one transformant harbouring the 50 kb neochromosome (IMF2) were sequenced. While IMF2 and IMF6 were faithfully assembled, harbouring all fragments of the in silico design in the correct configuration, IMF1 was not. According to the sequencing data, IMF1 carried all expected fragments but was harbouring an internal deletion in a filler fragment (6C) and a duplication of a 35 kb region (Supplementary Figure S3). This duplication, covering the middle of mTurquoise2 to the middle of Venus, was confirmed by karyotyping using in-plug linearization of the neochromosome (Supplementary Figures S4 and S5) and most likely resulted from a recombination event between the mTurquoise2 and the Venus genes, that have a nucleotide identity of 97% (35–37). Long-read sequencing of IMF2 and IMF6 further confirmed that in these strains no duplication or recombination events had occurred (Supplementary Figure S6). Neochromosomes can therefore be efficiently and faithfully assembled from as many as 44 fragments, despite the presence of an internal homologous region of approximately 700 bp, as well as regions homologous to the native chromosomes (pTEF1, pTEF2, pCCW12, tSSA1, ARS, HIS3 and URA3 expression cassette). The 60 bp SHRs were therefore sufficient to promote the efficient assembly of the neochromosomes.
Figure 2.

50 and 100 kb neochromosome designs. The outer circle depicts the in vivo assembly of the 100 kb neochromosome from 44 fragments present in strain IMF6. The inner circle depicts the in vivo assembly of the 50 kb neochromosome from 23 fragments present in strain IMF2.
Exploring optimal fragment size and number for efficient, modular in vivo assembly of neochromosomes
Although the assembly of the 50 and 100 kb neochromosomes resulted in a large number of colonies, doubling the number of transformed fragments caused a seven-fold decrease in colony count (ca. 2000 colonies and 275 colonies, respectively). This suggested that the number of fragments in the neochromosome design might become a limiting factor for the construction of larger neochromosomes harbouring many native and non-native pathways. To gain more insight into transformation efficiency (i.e. CFU/108 transformed cells) and to test to what extent efficiency might be influenced by fragment size and/or the number of transformed fragments, new 50 and 100 kb neochromosomes were designed and assembled using DNA filler fragments of three different sizes: 2.5, 5 and 10 kb, transformed in equimolar amounts. The transformations resulted in a large number of colonies (103–104 upon plating of the whole transformation mixture), and was most efficient with 5 kb fragments. On average transformation with 5 kb fragments was four-fold more efficient than 2.5 kb fragments and three-fold more efficient than with 10 kb fragments (one-tailed paired t-tests P ≤ 0.05), but fragments of 2.5 and 10 kb led to the same number of transformants (one-tailed paired t-test P > 0.05) (Figure 3A). The number of fragments transformed to yeast can affect in vivo assembly in different ways. A large number of fragments can negatively affect the efficiency of in vivo assembly as the number of repair events that cells have to perform increases with the number of fragments. Additionally, the probability that all transformed fragments access the nucleus is negatively correlated to the number of fragments (14). Fragment size is also expected to affect in vivo assembly efficiency, considering that cell entry might be hindered for large DNA parts. The intermediate fragment size of 5 kb might present a good compromise between these antagonistic factors. For each specific fragment size, doubling the number of fragments to assemble significantly decreased transformation efficiency by ca. 3-fold (2-tailed paired t-test P < 0.01) (Figure 3A).
Figure 3.

Transformation efficiency and fidelity. (A) Transformation efficiencies for the in vivo assembly of 100 and 50 kb synthetic chromosomes. Both neochromosomes have been assembled with 2.5 kb (dark blue), 5 kb (lighter blue) and 10 kb (lightest blue) fragments. The number above each bar indicates the number of assembled fragments. Data were normalized to 108 cells using controls on YPD plates, performed in biological duplicates (replicate 1 and 2) in technical triplicates for replicate 1 and technical duplicates for replicate 2. The asterisks indicates that transformation with 5 kb fragments resulted in a significantly different transformation efficiency compared to both neighbouring efficiencies (two-tailed paired homoscedastic t-test P< 0.05). (B) Assembly fidelity. In vivo assembly of the 100 kb NeoChr12 from 43 fragments. Eleven colonies that grew on selective medium were tested for mRuby2 and mTurquoise2 fluorescence. Neochromosomes were subsequently screened based on their size on CHEF gel and lastly by Illumina sequencing. The percentage of correct colonies for each screening round is indicated.
Next to transformation efficiency, fidelity of assembly (i.e. percentage of the transformants harbouring all transformed fragments in the correct order) is of paramount importance for functional pathway construction. Verification of the neochromosome assembly by PCR, as performed in most other synthetic neochromosome assemblies (14,18), is not only too labour intensive considering the large number of assembled fragments, but it is also unreliable. Indeed, PCR erroneously indicated a correct configuration for IMF1. Sequencing dozens of chromosomes by next generation sequencing is also time-consuming and costly. We therefore set up a screening pipeline consisting in a first selection based on the presence of auxotrophic markers distributed on the neochromosome, followed by fluorescence-activated cell sorting (FACS) of transformants expressing the fluorescent markers also distributed evenly on the neochromosome, the subsequent filtering based on chromosome size by karyotyping and a final confirmation of correct assembly by sequencing (Figure 1B).
To test the fidelity of assembly and assess the ease of our screening pipeline a novel neochromosome design was made (Supplementary Figure S7). The 100 kb NeoChr12 chromosome was similar to the 100 kb neochromosome carried by IMF6, except that only two fluorescent markers, mRuby2 and mTurquoise2, were included, to prevent unwanted recombinations between the highly similar mTurquoise2 and Venus genes, and the strongly expressed mRuby2 was relocated away from the centromere. From the transformation of 43 fragments of ∼2.5 kb, eleven colonies were screened for correct neochromosome assembly. Eight (73%, Figure 3B and Supplementary Figure S8) showed double fluorescence, of which five displayed the correct neochromosome size by karyotyping (45% of all transformants, Supplementary Figure S9). Of these five correctly sized neochromosomes, Illumina whole genome sequencing revealed that four contained all expected fragments (Supplementary Figure S3). Therefore a remarkably high assembly fidelity of 36% was reached when assembling 43 fragments in S. cerevisiae. It should be noted however that although sequencing data showed coverage for all fragments in the four correct colonies, two showed uniform coverage of all fragments (NeoChr12.4 and NeoChr12.8) while in the other two, the coverage of a small number of fragments differed from the rest of the neochromosome (four and two E.coli filler fragments with lower coverage for NeoChr12.1 and NeoChr12.3 respectively). This coverage variation most likely results from population heterogeneity, with subpopulations carrying different neochromosome configurations. This heterogeneity might have occurred early during neochromosome assembly, even though the strains were selected after single colony isolation, or later by recombination events occurring during strain propagation. Sequencing of IMF2 and IMF6, carrying the 50 and 100 kb original neochromosome design, confirmed a uniform coverage for all fragments in these strains. To investigate the possibility of recombination events during strain propagation, a single colony of IMF6 (100 kb original neochromosome) and of IMF23 (100 kb improved neochromosome), was inoculated in liquid selective medium and transferred in biological triplicate to fresh medium every day for fourteen days. Long-read sequencing from the first and last liquid cultures revealed that for both strains the neochromosome sequence from starting and end population was identical and faithful to the in silico design (Supplementary Figures S6 and S10). In conclusion, neochromosomes are genetically stable during propagation and population heterogeneity most likely occurs from neochromosome assembly and insufficient pure culture isolation.
It is important that assembly of the synthetic chromosomes does not result in a large number of mutations in the native genome or in the assembled synthetic chromosomes. Sequencing data revealed that the assembly of the neochromosomes resulted in a low number of mutations in the native genome as well as in the assembled neochromosome. NeoChr12.1 (IMF24), NeoChr12.3 (IMF25), NeoChr12.4 (IMF26) and NeoChr12.8 (IMF23) contained 3, 1, 13 and 12 SNPs in the neochromosome, respectively (between 0% and 58% of all mutations identified in the neochromosomes were located in SHRs, most likely originating from amplification primers (29)); and the native genome contained 1, 0, 2 and 0 mutations in coding regions, respectively (Supplementary Tables S1 and S2). This was similar to the previously constructed IMF2 (50 kb) and IMF6 (100 kb) strain which only harbored no and a single mutation in coding regions of the native genome; and 5 and 16 mutations in the neochromosomes respectively (Supplementary Tables S1 and S2).
Application of synthetic chromosomes as DNA landing pads
Supernumerary neochromosomes can be attractive landing pads to add, remove or modify functionalities. While recent chromosome engineering efforts have demonstrated that native S. cerevisiae chromosomes are extremely robust to large changes in their size and number (38,39), very little is known about S. cerevisiae’s tolerance to in vivo synthetic chromosome editing (19). The 50 and 100 kb neochromosomes carried by strains IMF2 and IMF6 respectively, were used as landing pads for the integration of an additional 35 kb of non-coding DNA (Figure 4). mTurquoise2 was targeted by CRISPR/Cas9 for introducing a double strand break and replaced by the 35 kb construct in vivo-assembled from 16 DNA fragments, resulting in two chromosomes of 85 and 135 kb (Supplementary Figure S11). FACS (loss of mTurquoise2 fluorescence), diagnostic PCR, karyotyping and short- and long-read sequencing revealed correct integration of all fragments (Supplementary Figures S3, S6, S12, S13).
Figure 4.
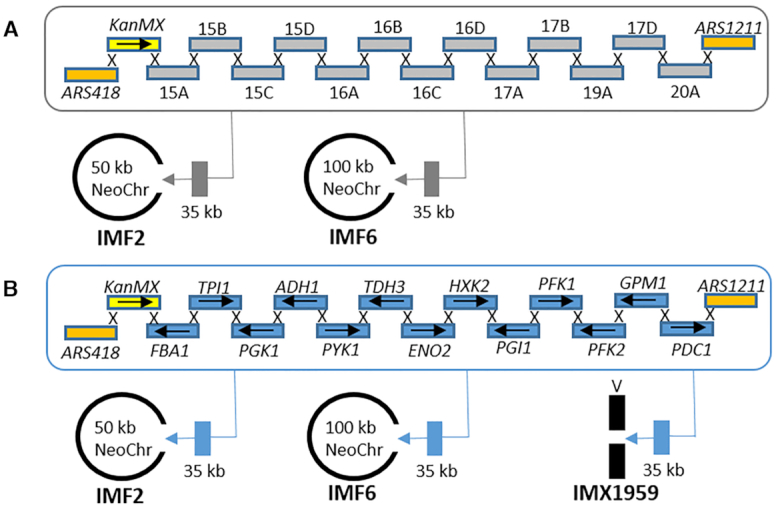
Neochromosomes as landing pads. (A) 35 kb of non-coding DNA was simultaneously in vivo assembled and inserted by CRISPR/Cas9 at the mTurquoise2 locus of the 100 kb neochromosome in IMF6 and of the 50 kb neochromosome in IMF2, resulting in 135 kb (IMF11) and 85 kb (IMF12) neochromosomes, respectively. (B) 35 kb of glycolytic genes were simultaneously in vivo assembled and inserted using CRISPR/Cas9 at the mTurquoise2 locus of the 100 kb neochromosome in IMF6 and of the 50 kb neochromosome in IMF2 resulting in 135 kb (IMF13) and 85 kb (IMF14) neochromosomes, respectively. As control, the same glycolytic cassettes were integrated at the native CAN1 locus of chromosome V resulting in strain IMX1959.
Physiological characterization on selective SMD medium of the initial strains harbouring the four differently sized neochromosomes (50 kb in strain IMF2, 85 kb in strain IMF12, 100 kb in strain IMF6 and 135 kb in strain IMF11) revealed that they grew on average 8% slower than their prototrophic parental strain (IMX2059), irrespective of neochromosome size (Figure 5). Genome sequencing did not reveal mutations that could explain the decrease in growth rate (Supplementary Tables S1 and S2). Conversely, IMF23 (100 kb NeoChr12.8), with the improved neochromosome design, in which the highly expressed mRuby2 was re-located from its place adjacent to the centromere and in which Venus (highly homologous to mTurquoise2) was removed, showed the same growth rate as the parental strain. In addition, in comparison to the prototrophic control strain IMX2059, IMF23 showed no or very little variation in growth rate under a range of carbon sources (galactose, maltose and sucrose) and stress conditions (pH-, oxidative-, osmotic-, salt- and temperature-stress) (Supplementary Figure S14).
Figure 5.

Physiological characterization of strains carrying synthetic chromosomes. (A and B) Specific growth rate of strains carrying empty neochromosomes (Panel A: IMF2, IMF12, IMF6, IMF11 and IMF23) and glycolytic neochromosomes (Panel B: IMF18 and IMF17). Growth rates represent the average and mean deviation of biological duplicates except for the parental strain which cultures were performed in biological quadruplicate. (C and D) Viability measured as number of colonies on YPD (C) or SMD (D) sorted by FACS, divided by the total number of possible colonies (96), for the empty and glycolytic neochromosomes. Data represent biological duplicates and are averaged from 4 days of measurement for all strains except for the 100 kb improved design (two samples at day 1 and day 4). (E) Stability measured as the number of transformants on selective plates (SMD) divided by the number of colonies on non-selective plates (YPD). For each strain, the stability represents the average of 4 days of measurement in biological duplicates, except for 100 kb improved design (IMF23) for which 2 days of measurement were used (day 1 and day 4). (F) Transcript levels of the glycolytic genes from IMF17 (135 kb glycolysis neochromosome) and control strain IMX2109 expressing glycolysis from native chromosome V, grown in aerobic batch cultures. Transcript levels and standard deviations are from biological triplicates. (G) Specific activity of glycolytic enzymes in IMF17 and control strain IMX2109 from aerobic batch cultures. Activities were measured at least in biological duplicates. For panels A, B, C, D and E all significant differences with respect to the first bar are indicated with an asterisk (one-way ANOVA with Post-Hoc Tukey–Kramer, P< 0.05). For panels F and G, the asterisk indicates whether transcript levels or enzyme activities of IMF17 are significantly different with respect to IMX2109 (two-tailed paired homoscedastic t-test P< 0.05).
As S. cerevisiae is tolerant to variations in the number of native chromosomes (38–40), the observed slower growth phenotype for four of the neochromosome strains might be specific to the neochromosome design. It has been previously shown that the size of artificial chromosomes affect their stability (41–44). In selective medium, neochromosome loss would result in cells unable to grow and thereby in reduced growth rate. To evaluate the mitotic stability of the neochromosomes, IMF2, IMF12, IMF6, IMF11 and IMF23 were sequentially transferred in selective media and plated on both non-selective (YPD) and selective media (SMD). Along four successive culture transfers (ca. 25 generations), the viability of the strains on non-selective medium was similar to that of the prototrophic control strain CEN.PK113–7D (Figure 5), or a control strain containing a 6.5 kb centromeric plasmid (IMC153) (one-way ANOVA with Post-Hoc Tukey–Kramer, P>0.05).
Plating on selective medium revealed that for the four neochromosomes with the initial design (strains IMF2, IMF12, IMF6, IMF11), 14% to 30% of the population lost their neochromosome (Figure 5). This percentage remained stable over 25 generations (Supplementary Figure S15), and most likely explained the reduced growth rate measured for neochromosome-bearing strains. Indeed the neochromosome with the improved design (IMF23), which did not show a reduced growth phenotype was highly stable with 92% of the population retaining the neochromosome, which is the same as the control strain IMC153, carrying a 6.5 kb centromeric plasmid with the same auxotrophic markers as located on the neochromosomes, and with the same parental strain as the neochromosome strains (Figure 5). Contrary to earlier reports (41,44) no correlation was observed between neochromosome size and stability (strains IMF2, IMF12, IMF6 and IMF11 with increasing neochromosome size showed stability of 80%, 86%, 70% and 81%, respectively). Therefore, empty neochromosomes can be very stable.
The strains with the four neochromosomes (IMF2, IMF12, IMF6, IMF11) with the initial design showed decreased neochromosome stability as well as a decreased growth rate. In contrast, the improved neochromosome design was stable and resulted in a strain (IMF23) with a growth rate identical to that of the control strain (IMX2059). Deletion of mRuby2 (IMF37) or Venus (IMF38), or both mRuby2 and Venus (IMF39) from the 100 kb neochromosome in IMF6 did not increase the growth rate (Supplementary Figure S16). Therefore homology between mTurquoise2 and Venus (potential cause of recombination and loop-out of genetic elements essential for neochromosome maintenance) and/or strong expression of mRuby2 close to the centromere (potentially leading to centromere destabilisation), cannot account for the reduced growth rate of IMF2, IMF12, IMF6 and IMF11. It is noteworthy that mRuby2 and Venus are not the only differences between the stable and unstable neochromosome designs, as they carried different filler fragments. One or more of these E. coli filler fragments might cause neochromosome instability. This could result in a mother cell retaining one copy of the neochromosome and the daughter cell zero copies (1:0 segregation) or the mother retaining two copies and the daughter zero (2:0 segregation) (Figure 6C). From the coverage of reads belonging to the neochromosomes, we estimated that average copy numbers of 0.6, 1.5, 1.5, 1.7, for the 50 kb (IMF2), 85 kb (IMF12), 100 kb (IMF6) and 135 kb (IMF11) neochromosomes, respectively (Figure 6A and Supplementary Figure S3). Since reads coverage was uniform along the neochromosomes, the most likely explanation for these non-integer average neochromosome copy numbers far from one, is an heterogeneous population with cells carrying none, one, two, or more neochromosome copies. Remarkably, the improved neochromosome design (strain IMF23) showed an average copy number of one, confirming its stable design. Copy number variations were confirmed by monitoring mRuby2 fluorescence, which is present on all neochromosomes. For the neochromosomes with less optimal design, the larger the neochromosome, the higher the fraction of cells with high fluorescence (indicating multiple copies, Figure 6B). For the new design (IMF23) most cells showed the same fluorescence intensity as the control in which mRuby2 (with the same promoter and terminator) was integrated in single copy in the genome of the neochromosome parental strain (IMX2224) (Figure 6B).
Figure 6.

Quantification of neochromosome copy number. (A) neochromosome copy number as determined by whole genome sequencing. (B) neochromosome copy number estimation based on mRuby2 fluorescence. CEN.PK113-7D with no copies of mRuby2 was used as negative control and IMX2224 with a single copy of mRuby2 integrated in the genome as positive control. (C) Schematic overview of the potential segregation of circular neochromosome upon cell division.
Synthetic chromosomes as platforms for exclusive expression of (essential) pathways
Neochromosomes have the potential to become powerful platforms for genome engineering, to rewire yeast native gene networks as well as to act as versatile expression hubs for heterologous pathways and processes. The transplantation of the complete human purine pathway to a neochromosome (45) is an encouraging demonstration, however the potential of neochromosomes as orthogonal expression platforms hasn’t been fully explored yet. Using the pathway swapping concept, we used neochromosomes as exclusive expression platform for the essential glycolytic pathway, and evaluated the impact of neochromosome-borne glycolysis expression on yeast physiology. The 50 kb (IMF2) and 100 kb (IMF6) ‘empty’ neochromosomes were assembled in the SwYG (16) ancestor strain IMX1338, in which the set of 13 genes coding for the glycolytic pathway has been relocalized to a single genomic locus. Using CRISPR/Cas9 editing combined with in vivo assembly of 13 glycolytic transcriptional units (Figure 4), the 35 kb glycolytic cassette was introduced in the mTurquoise2 locus of the 50 and 100 kb neochromosomes. In these strains harbouring two glycolytic gene sets, the single locus glycolysis from chromosome IX was successfully removed by induction of double strand DNA breaks (DSBs) at the flanks of the glycolytic cassette and repaired with a 120 bp DNA fragment (16). The resulting strain with a single neochromosome-borne glycolytic gene set demonstrated that neochromosomes can serve as exclusive expression hubs for essential pathways (strain construction represented in Figure 7). The strains carrying ‘glycolytic neochromosomes’ IMF18 (glycolysis on 85 kb neochromosome) and IMF17 (glycolysis on 135 kb neochromosome) were grown in aerobic batch cultures in chemically defined medium with glucose as sole carbon source, and compared to IMX2109, a control strain carrying an identical genetic configuration of glycolysis on chromosome V. Both IMF17 and IMF18 grew significantly slower than the control strain (Figure 5), IMF17 growing 14% slower than IMF18. Several hypotheses could explain this decreased growth rate of strains with glycolytic neochromosomes, (i) the unfortunate occurrence of deleterious mutations in the native or synthetic chromosomes during transformation, (ii) the low expression of neochromosome-borne glycolytic genes and resulting decreased glycolytic flux, (iii) the loss of neochromosomes or (iv) stalled replication due to transcription-replication clashes at the clustered glycolysis. Both IMF17 and IMF18 carried the same mutation in the GPM1 promoter, in addition IMF17 carried non-synonymous mutations in TPI1 and PFK2 ORFs (Supplementary Table S2). However none of these mutations caused a decrease in the specific activity of the corresponding enzymes tested in vitro comparing IMF17 and the control strain IMX2109 (Figure 5), demonstrating that mutations in the glycolytic genes was most likely not responsible for the decreased growth rate of IMF17 and IMF18. A few mutations were observed in coding regions of other genes, and it cannot be excluded that they contributed to decreasing the growth rate of IMF17 and IMF18 (Supplementary Table S1). Secondly, the similar or slightly higher RNA expression (except PFK1) and specific activity in glycolytic enzymes between IMF17 and control strain IMX2109 expressing glycolysis from native chromosome V (Figure 5) indicated that low expression of neochromosome-borne glycolytic genes was not involved in the reduced growth rate of IMF17 (and IMF18). Furthermore, previous studies have shown that an increase in specific activity of glycolytic enzymes does not result in a decrease in growth rate (46,47). Alternatively, this decreased growth rate might result from neochromosome instability and potential loss in a fraction of the yeast population. Because the loss of glycolytic neochromosomes leads to lethality in selective as well as in non-selective medium (e.g. YPD or YP ethanol), testing their stability is challenging. However, the viability of glycolytic neochromosome strains on YPD could be used as indicator of neochromosome loss when comparing with the viability on YPD of control strains and empty neochromosome strains IMF12 and IMF11. IMF12 and IMF11 can survive neochromosome loss on YPD medium, in which they display a viability similar to that of the control strain (85–92%). Conversely, the viability of IMF18 (85 kb glycolytic neochromosome) and IMF17 (135 kb glycolytic neochromosome), was significantly reduced on YPD to 73% and 68% respectively and on SMD medium to 77% and 70% respectively (Figure 5). Assuming that this decreased viability results from neochromosome loss, ca. 20% of the IMF17 and IMF18 population would lose their glycolytic neochromosome. The neochromosome loss would therefore be similar for neochromosomes with and without the glycolytic genes. The reduced growth rate of IMF17 and IMF18 does therefore most probably not result from a lower stability of glycolytic neochromosomes as compared to empty neochromosomes. Lastly, while stalled replication of the neochromosomes due to transcription-replication clashes might cause the decreased specific growth rate, transcriptome analysis did not support this hypothesis as no genetic markers of DNA replication stress and damage (e.g. RNR1 to RNR4 (48)) were differentially expressed between IMF17 and its control strain IMX2109 (Supplementary Table S3). Differential gene expression analysis by RNA sequencing of IMF17 and its control strain IMX2109 did not provide further leads on the cause of the reduced growth rate. Few transcriptional differences were observed between the two strains, most likely originating from technical differences in the shake flask grown cultures. This analysis additionally confirmed that E.coli DNA used as filler fragments for the construction of the neochromosomes was, as expected, non-coding. With the exception of a few small regions for which transcripts with low abundance could be detected, there was virtually no expression from the E. coli DNA (Supplementary Figure S17). It is highly unlikely that these transcripts from E. coli DNA lead to translation of (functional) peptides in S. cerevisiae.
DISCUSSION
The present study explores the potential of de novo assembled synthetic chromosomes to operate as large, modular expression islands in the popular workhorse and model strain S. cerevisiae. The design and construction workflow of neochromosomes developed in this study are highly versatile, enabling the facile construction of chromosomes with a wide variety of configurations.
Probing the limits of in vivo assembly, the present results demonstrate that a large number of DNA fragments of the size of expression cassettes can be efficiently assembled de novo in yeast. While other studies have reported this remarkable feature of S. cerevisiae (e.g. assembly of 38 fragments of 200 bp (13), and 25 fragments of 24 kb (14)), quantitative information on the transformation and assembly efficiencies and fidelity was so far lacking. The neochromosome assembly from 44 fragments, the record to date, showed a remarkably high efficiency, with 36% of faithfully assembled neochromosomes. This efficiency is substantially higher than previously reported, for instance by Annaluru et al. (49) where assembly of 27 fragments into chromosome III resulted in 0.5% of clones with the expected marker combination. Still, increasing the number of fragments was accompanied by a decrease in transformation efficiency and in the total number of clones on plates, which can become a limitation for the modular assembly of large neochromosomes. The construction of neochromosomes involves several cellular mechanisms, many of which are yet poorly understood but need to be considered to further enhance the neochromosomes construction efficiency. For instance, access of all required DNA fragments to the nucleus is essential for neochromosome assembly, but the mechanisms involved in DNA translocation during transformation are not fully resolved and are poorly investigated. Once all DNA fragments have reached the nucleus, the Homologous Recombination (HR) machinery has to be mobilized to repair a large number of DSBs. While the number of simultaneous DNA breaks that S. cerevisiae tolerates is yet undefined, hundreds of simultaneous repairs most probably present a formidable challenge. Additionally, while HR is the preferred mechanism for DSB repair in S. cerevisiae, other, non-homologous repair mechanisms exist (50,51), and might take over when the HR machinery is inactive or saturated, thereby promoting erroneous assemblies. Inactivation of non-homologous end joining (NHEJ) is unfortunately not an option in S. cerevisiae as, due to the pleiotropic role of the NHEJ proteins, it leads to deleterious consequences (52–54). Last but not least, assembly in a complete neochromosome is not a guarantee for cell survival, as they have to recover from the transformation stress, which is yet another poorly explored phenomenon. The combination of complex and often poorly understood events that have to take place for DNA to be assembled largely explains why typically a small fraction only of the transformed cell harbour the expected constructs (ca. one cell out of millions). One thing is however certain: the full potential of yeast in vivo assembly hasn’t been reached yet. While intensive efforts have been made to turn E. coli into a universal molecular biology tool, the power of S. cerevisiae as DNA assembly platform has so far been largely untapped. Despite the pivotal role played by S. cerevisiae in the last decade in the assembly of genomes (14,55–58) there is so far relatively little effort invested in turning this yeast into a universal and powerful DNA assembly platform (59,60), a situation that we expect will change in the future.
Neochromosome stability and impact on the host physiology is a critical feature for the applicability of neochromosomes for synthetic biology. In the present study, introduction of four differently sized empty neochromosomes caused a decrease in specific growth rate and in neochromosome loss in 20% of the cell population. In-depth analysis by Illumina sequencing and flow cytometric analysis showed a lower average neochromosome copy number at population level and per cell for the smallest, 50 kb chromosome. For larger neochromosomes the average copy number was above one, considering that part of the population did not carry a neochromosome, it means that a substantial fraction of the cells carried more than one neochromosome. This phenotype is most likely explained by aberrant segregation of the neochromosomes. However this was not caused by strong expression of the gene located next to the centromere (61), possibly impairing the centromere function, nor the possible recombination between highly homologous DNA on the neochromosome. Another hypothesis that remains to be tested is that particular E. coli filler fragments cause chromosome destabilisation. This would result from DNA architecture and not from transcription, since there was no or very little transcription from the E. coli DNA. The lower average copy number of the smallest neochromosome could also be caused by failed replication or loss of the neochromosome. Still, the average percentage of cells in a population (70–86%) containing at least one neochromosome copy was similar between chromosomes of 50–135 kb. Comparing the stability of the empty neochromosomes with published data is not straightforward as earlier work with YACs typically evaluates plasmid loss in non-selective media. However, the absence of size-dependency of neochromosome stability contrasts with earlier work showing that circular chromosomes increase in stability up to ∼100 kb but decrease in stability above this size, a phenomenon tentatively attributed to the formation of dicentric dimeric circles (41,44,62). Considering future applications in synthetic biology, population homogeneity might be further enhanced by linear neochromosome design, either by linearization of the circular neochromosomes (for instance with the telomerator carried by the neochromosomes (34)) or direct, de novo assembly of linear neochromosomes. Alternatively, other strategies to improve segregation such as equipping chromosomes with synthetic kinetochores can be considered (63).
The present study demonstrates for the first time that synthetic chromosomes, fully de novo assembled from transcription-unit sized DNA parts can serve as exclusive, orthogonal expression platforms for essential metabolic pathways. There are few reports using synthetic and artificial chromosomes to express genes in yeast using heterologous genes or, in the case of the tRNA neochromosome, expressing a second, complete set of tRNAs, which understandably lead to a decrease in growth rate (45,62,64–67). In the present study, the native glycolytic and fermentative pathways were transplanted from a native chromosome to a neochromosome with an identical genetic configuration (Figure 4), a unique experimental set-up that enables the direct comparison of native and neochromosome-borne expression of an entire pathway. This approach revealed that neochromosome-based expression of an essential pathway reduced the host specific growth rate by ca. 14–24% and suggested a negative correlation between neochromosome size and growth rate. Although this decrease in growth rate could be partly explained by aberrant segregation as observed for some of the empty neochromosomes strains, other factors most probably play a role. Reduced expression and capacity of the glycolytic and fermentative enzymes or population heterogeneity in term of neochromosome copy number are most likely not involved. One important aspect to consider is that the expression of the clustered glycolytic and fermentative genes is driven by some of the most highly transcriptionally active promoters, resulting in a transcriptional hotspot. An alternative explanation might therefore be a clash between the transcription and replication machineries, causing delayed replication (68). In IMX2109, carrying the same glycolytic cassette but on a native chromosome, such a conflict might be attenuated by the native genetic context (e.g. different chromatin structure, ARS sequences). Future neochromosome architecture should therefore take into account directionality of transcription with respect to neighbouring ARSs and localization of highly transcribed genes.
Altogether this study highlights the potential of synthetic chromosomes to serve as platforms for modular assembly of native and heterologous pathways and paves the way towards modular genomes for synthetic biology.
DATA AVAILABILITY
All processed data are available in the manuscript or as supplementary data. Upon request, raw data that support the findings of this paper will be made available. All genomic data for this paper have been deposited in the NCBI database (https://www.ncbi.nlm.nih.gov/) under the BioProjectID PRJNA596648. All RNA-seq sequences have been deposited at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) under GEO accession number: ‘GSE159524’.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Marijke Luttik for assistance with assaying the glycolytic enzymes activity, Mark Bisschops for technical assistance with FACS analysis, Melanie Wijsman for assistance with the neochromosome stability assays, Jordi Geelhoed for performing the growth profiler experiment and Pilar de la Torre for (RNA) sequencing of strains. We thank Anna Wronska and Thomas Perli for sharing the plasmids pGGkd017 and pUDR514, respectively. This project was funded by the AdLibYeast European Research Council (ERC) consolidator 648141 grant awarded to Pascale Daran-Lapujade.
Author contribution: E.P., S.D., J.M.D. and P.D.-L. designed the experiments and wrote the manuscript. E.P., S.D., L.vB. and S.T.P. performed the experiments. M.vdB. performed the bioinformatics analysis.
Contributor Information
Eline D Postma, Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2627HZ Delft, The Netherlands.
Sofia Dashko, Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2627HZ Delft, The Netherlands.
Lars van Breemen, Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2627HZ Delft, The Netherlands.
Shannara K Taylor Parkins, Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2627HZ Delft, The Netherlands.
Marcel van den Broek, Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2627HZ Delft, The Netherlands.
Jean-Marc Daran, Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2627HZ Delft, The Netherlands.
Pascale Daran-Lapujade, Department of Biotechnology, Delft University of Technology, van der Maasweg 9, 2627HZ Delft, The Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
AdLibYeast ERC consolidator [648141 to P.D.L.]; European Union's Horizon 2020 Framework Programme for Research and Innovation. Funding for open access charge: ERC consolidator [648141].
Conflict of interest statement. None declared.
REFERENCES
- 1. Walker G.M., Walker R.S.K.. Gadd G.M., Sariaslani S.. Chapter three - Enhancing yeast alcoholic fermentations. Advances in Applied Microbiology. 2018; 105:Academic Press; 87–129. [DOI] [PubMed] [Google Scholar]
- 2. Meehl M.A., Stadheim T.A.. Biopharmaceutical discovery and production in yeast. Curr. Opin. Biotechnol. 2014; 30:120–127. [DOI] [PubMed] [Google Scholar]
- 3. Chubukov V., Mukhopadhyay A., Petzold C.J., Keasling J.D., Martín H.G.. Synthetic and systems biology for microbial production of commodity chemicals. Npj Syst. Biol. Applic. 2016; 2:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Y., Li S., Thodey K., Trenchard I., Cravens A., Smolke C.D.. Complete biosynthesis of noscapine and halogenated alkaloids in yeast. Pro. Natl. Acad. Sci. U.S.A. 2018; 115:E3922–E3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paddon C.J., Keasling J.D.. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014; 12:355–367. [DOI] [PubMed] [Google Scholar]
- 6. Hutchison C.A., Chuang R.-Y., Noskov V.N., Assad-Garcia N., Deerinck T.J., Ellisman M.H., Gill J., Kannan K., Karas B.J., Ma L.et al.. Design and synthesis of a minimal bacterial genome. Science. 2016; 351:aad6253. [DOI] [PubMed] [Google Scholar]
- 7. Veneziano R., Shepherd T.R., Ratanalert S., Bellou L., Tao C., Bathe M.. In vitro synthesis of gene-length single-stranded DNA. Sci. Rep. 2018; 8:6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hughes R.A., Ellington A.D.. Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb. Perspect. Biol. 2017; 9:a023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kosuri S., Church G.M.. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods. 2014; 11:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Casini A., Storch M., Baldwin G.S., Ellis T.. Bricks and blueprints: methods and standards for DNA assembly. Nat. Rev. Mol. Cell Biol. 2015; 16:568–576. [DOI] [PubMed] [Google Scholar]
- 11. Kok S.D., Stanton L.H., Slaby T., Durot M., Holmes V.F., Patel K.G., Platt D., Shapland E.B., Serber Z., Dean J.et al.. Rapid and reliable DNA assembly via ligase cycling reaction. ACS Synth. Biol. 2014; 3:97–106. [DOI] [PubMed] [Google Scholar]
- 12. Gibson D.G., Benders G.A., Andrews-Pfannkoch C., Denisova E.A., Baden-Tillson H., Zaveri J., Stockwell T.B., Brownley A., Thomas D.W., Algire M.A.et al.. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008; 319:1215–1220. [DOI] [PubMed] [Google Scholar]
- 13. Gibson D.G. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res. 2009; 37:6984–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gibson D.G., Benders G.A., Axelrod K.C., Zaveri J., Algire M.A., Moodie M., Montague M.G., Venter J.C., Smith H.O., Hutchison C.A.. One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:20404–20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruschi C., Gjuracic K., Tosato V.. Yeast Artificial Chromosomes. Encyclopedia of Life Sciences. 2006; Chichester: John Wiley & Sons Ltd. [Google Scholar]
- 16. Kuijpers N.G., Solis-Escalante D., Luttik M.A., Bisschops M.M., Boonekamp F.J., van den Broek M., Pronk J.T., Daran J.-M., Daran-Lapujade P.. Pathway swapping: Toward modular engineering of essential cellular processes. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:15060–15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Solis-Escalante D., Kuijpers N.G., Barrajon-Simancas N., van den Broek M., Pronk J.T., Daran J.-M., Daran-Lapujade P.. A minimal set of glycolytic genes reveals strong redundancies in Saccharomyces cerevisiae central metabolism. Eukaryot. Cell. 2015; 14:804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richardson S.M., Mitchell L.A., Stracquadanio G., Yang K., Dymond J.S., DiCarlo J.E., Lee D., Huang C.L.V., Chandrasegaran S., Cai Y.et al.. Design of a synthetic yeast genome. Science. 2017; 355:1040–1044. [DOI] [PubMed] [Google Scholar]
- 19. Rideau F., Le Roy C., Descamps E.C.T., Renaudin H., Lartigue C., Bébéar C.. Cloning, stability, and modification of Mycoplasma hominis genome in Yeast. ACS Synth. Biol. 2017; 6:891–901. [DOI] [PubMed] [Google Scholar]
- 20. Entian K.-D., Kötter P.. Stansfield I., Stark M.J.R.. 25 Yeast Genetic Strain and Plasmid Collections. Methods in Microbiology. 2007; 36:Academic Press; 629–666. [Google Scholar]
- 21. Verduyn C., Postma E., Scheffers W.A., Van Dijken J.P.. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992; 8:501–517. [DOI] [PubMed] [Google Scholar]
- 22. Gietz R.D., Woods R.A.. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002; 350:87–96. [DOI] [PubMed] [Google Scholar]
- 23. Lee M.E., DeLoache W.C., Cervantes B., Dueber J.E.. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol. 2015; 4:975–986. [DOI] [PubMed] [Google Scholar]
- 24. Mans R., van Rossum H.M., Wijsman M., Backx A., Kuijpers N.G., van den Broek M., Daran-Lapujade P., Pronk J.T., van Maris A.J., Daran J.M.. CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 2015; 15:fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boonekamp F.J., Dashko S., Duiker D., Gehrmann T., van den Broek M., den Ridder M., Pabst M., Robert V., Abeel T., Postma E.D.et al.. Design and experimental evaluation of a minimal, innocuous watermarking strategy to distinguish Near-Identical DNA and RNA sequences. ACS Synth. Biol. 2020; 9:1361–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Postma E., Verduyn C., Scheffers W.A., Van Dijken J.P.. Enzymic analysis of the crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 1989; 55:468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansen M.L., Diderich J.A., Mashego M., Hassane A., de Winde J.H., Daran-Lapujade P., Pronk J.T.. Prolonged selection in aerobic, glucose-limited chemostat cultures of Saccharomyces cerevisiae causes a partial loss of glycolytic capacity. Microbiology. 2005; 151:1657–1669. [DOI] [PubMed] [Google Scholar]
- 28. Cruz L.A., Hebly M., Duong G.H., Wahl S.A., Pronk J.T., Heijnen J.J., Daran-Lapujade P., van Gulik W.M.. Similar temperature dependencies of glycolytic enzymes: an evolutionary adaptation to temperature dynamics. BMC Syst. Biol. 2012; 6:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuijpers N.G., Solis-Escalante D., Bosman L., van den Broek M., Pronk J.T., Daran J.-M., Daran-Lapujade P.. A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb. Cell Fact. 2013; 12:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dhar M.K., Sehgal S., Kaul S.. Structure, replication efficiency and fragility of yeast ARS elements. Res. Microbiol. 2012; 163:243–253. [DOI] [PubMed] [Google Scholar]
- 31. Kanhere A., Bansal M.. Structural properties of promoters: similarities and differences between prokaryotes and eukaryotes. Nucleic Acids Res. 2005; 33:3165–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griesenbeck J., Tschochner H., Grohmann D.. Harris J.R., Marles-Wright J.. Structure and Function of RNA Polymerases and the Transcription Machineries. Macromolecular Protein Complexes: Structure and Function. 2017; Cham: Springer International Publishing; 225–270. [Google Scholar]
- 33. Sikorski R.S., Hieter P.. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989; 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitchell L.A., Boeke J.D.. Circular permutation of a synthetic eukaryotic chromosome with the telomerator. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:17003–17010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lambert T.J. FPbase: a community-editable fluorescent protein database. Nat. Methods. 2019; 16:277–278. [DOI] [PubMed] [Google Scholar]
- 36. Nagai T., Ibata K., Park E.S., Kubota M., Mikoshiba K., Miyawaki A.. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002; 20:87. [DOI] [PubMed] [Google Scholar]
- 37. Goedhart J., von Stetten D., Noirclerc-Savoye M., Lelimousin M., Joosen L., Hink M.A., van Weeren L., Gadella T.W.J. Jr, Royant A.. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012; 3:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shao Y., Lu N., Wu Z., Cai C., Wang S., Zhang L.-L., Zhou F., Xiao S., Liu L., Zeng X.et al.. Creating a functional single-chromosome yeast. Nature. 2018; 560:331–335. [DOI] [PubMed] [Google Scholar]
- 39. Luo J., Sun X., Cormack B.P., Boeke J.D.. Karyotype engineering by chromosome fusion leads to reproductive isolation in yeast. Nature. 2018; 560:392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gorter de Vries A.R., Pronk J.T., Daran J.G.. Industrial relevance of chromosomal copy number variation in Saccharomyces yeasts. Appl. Environ. Microbiol. 2017; 83:e03206-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Murray A.W., Schultes N.P., Szostak J.W.. Chromosome length controls mitotic chromosome segregation in yeast. Cell. 1986; 45:529–536. [DOI] [PubMed] [Google Scholar]
- 42. Surosky R.T., Newlon C.S., Tye B.-K.. The mitotic stability of deletion derivatives of chromosome III in yeast. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sleister H.M., Mills K.A., Blackwell S.E., Killary A.M., Murray J.C., Malone R.E.. Construction of a human chromosome 4 YAC pool and analysis of artificial chromosome stability. Nucleic Acids Res. 1992; 20:3419–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hieter P., Mann C., Snyder M., Davis R.W.. Mitotic stability of yeast chromosomes: a colony color assay that measures nondisjunction and chromosome loss. Cell. 1985; 40:381–392. [DOI] [PubMed] [Google Scholar]
- 45. Agmon N., Temple J., Tang Z., Schraink T., Baron M., Chen J., Mita P., Martin J.A., Tu B.P., Yanai I.et al.. Phylogenetic debugging of a complete human biosynthetic pathway transplanted into yeast. Nucleic Acids Res. 2020; 48:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hauf J., Zimmermann F.K., Muller S.. Simultaneous genomic overexpression of seven glycolytic enzymes in the yeast Saccharomyces cerevisiae. Enzyme Microb. Technol. 2000; 26:688–698. [DOI] [PubMed] [Google Scholar]
- 47. Schaaff I., Heinisch J., Zimmermann F.K.. Overproduction of glycolytic enzymes in yeast. Yeast. 1989; 5:285–290. [DOI] [PubMed] [Google Scholar]
- 48. Tkach J.M., Yimit A., Lee A.Y., Riffle M., Costanzo M., Jaschob D., Hendry J.A., Ou J., Moffat J., Boone C.et al.. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 2012; 14:966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Annaluru N., Muller H., Mitchell L.A., Ramalingam S., Stracquadanio G., Richardson S.M., Dymond J.S., Kuang Z., Scheifele L.Z., Cooper E.M.et al.. Total synthesis of a functional designer eukaryotic chromosome. Science. 2014; 344:55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aylon Y., Kupiec M.. DSB repair: the yeast paradigm. DNA Repair (Amst.). 2004; 3:797–815. [DOI] [PubMed] [Google Scholar]
- 51. Heyer W.D., Ehmsen K.T., Liu J.. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010; 44:113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barnes G., Rio D.. DNA double-strand-break sensitivity, DNA replication, and cell cycle arrest phenotypes of Ku-deficient Saccharomycescerevisiae. Proc. Natl. Acad. Sci. 1997; 94:867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Laroche T., Martin S.G., Gotta M., Gorham H.C., Pryde F.E., Louis E.J., Gasser S.M.. Mutation of yeast Ku genes disrupts the subnuclear organization of telomeres. Curr. Biol. 1998; 8:653–656. [DOI] [PubMed] [Google Scholar]
- 54. Gravel S., Larrivee M., Labrecque P., Wellinger R.J.. Yeast Ku as a regulator of chromosomal DNA end structure. Science. 1998; 280:741–744. [DOI] [PubMed] [Google Scholar]
- 55. Benders G.A., Noskov V.N., Denisova E.A., Lartigue C., Gibson D.G., Assad-Garcia N., Chuang R.-Y., Carrera W., Moodie M., Algire M.A.et al.. Cloning whole bacterial genomes in yeast. Nucleic Acids Res. 2010; 38:2558–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tagwerker C., Dupont C.L., Karas B.J., Ma L., Chuang R.-Y., Benders G.A., Ramon A., Novotny M., Montague M.G., Venepally P.et al.. Sequence analysis of a complete 1.66 Mb Prochlorococcus marinus MED4 genome cloned in yeast. Nucleic Acids Res. 2012; 40:10375–10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Karas B.J., Molparia B., Jablanovic J., Hermann W.J., Lin Y.C., Dupont C.L., Tagwerker C., Yonemoto I.T., Noskov V.N., Chuang R.Y.et al.. Assembly of eukaryotic algal chromosomes in yeast. J. Biol. Eng. 2013; 7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Venetz J.E., Del Medico L., Wölfle A., Schächle P., Bucher Y., Appert D., Tschan F., Flores-Tinoco C.E., van Kooten M., Guennoun R.et al.. Chemical synthesis rewriting of a bacterial genome to achieve design flexibility and biological functionality. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:8070–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DiCarlo J.E., Conley A.J., Penttila M., Jantti J., Wang H.H., Church G.M.. Yeast oligo-mediated genome engineering (YOGE). ACS Synth. Biol. 2013; 2:741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu S.C., Kuemmel F., Skoufou-Papoutsaki M.N., Spanu P.D.. Yeast transformation efficiency is enhanced by TORC1- and eisosome-dependent signaling. MicrobiologyOpen. 2019; 8:e00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hill A., Bloom K.. Genetic manipulation of centromere function. Mol. Cell. Biol. 1987; 7:2397–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Walker R.S.K. 2017; Doctoral thesis from the University of Edinburgh, de novo biological engineering of a tRNA neochromosome in yeasthttps://era.ed.ac.uk/handle/1842/28921.
- 63. Lacefield S., Lau D.T., Murray A.W.. Recruiting a microtubule-binding complex to DNA directs chromosome segregation in budding yeast. Nat. Cell Biol. 2009; 11:1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Naesby M., Nielsen S.V., Nielsen C.A., Green T., Tange T.O., Simon E., Knechtle P., Hansson A., Schwab M.S., Titiz O.et al.. Yeast artificial chromosomes employed for random assembly of biosynthetic pathways and production of diverse compounds in Saccharomyces cerevisiae. Microb. Cell Fact. 2009; 8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Klein J., Heal J.R., Hamilton W.D., Boussemghoune T., Tange T.O., Delegrange F., Jaeschke G., Hatsch A., Heim J.. Yeast synthetic biology platform generates novel chemical structures as scaffolds for drug discovery. ACS Synth. Biol. 2014; 3:314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Essani K., Glieder A., Geier M.. Combinatorial pathway assembly in yeast. 2015; 2:423–436. [Google Scholar]
- 67. Hughes S.R., Cox E.J., Bang S.S., Pinkelman R.J., López-Núñez J.C., Saha B.C., Qureshi N., Gibbons W.R., Fry M.R., Moser B.R.. Process for assembly and transformation into Saccharomyces cerevisiae of a synthetic yeast artificial chromosome containing a multigene cassette to express enzymes that enhance xylose utilization designed for an automated platform. J. Lab. Autom. 2015; 20:621–635. [DOI] [PubMed] [Google Scholar]
- 68. Hamperl S., Cimprich K.A.. Conflict resolution in the genome: how transcription and replication make it work. Cell. 2016; 167:1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All processed data are available in the manuscript or as supplementary data. Upon request, raw data that support the findings of this paper will be made available. All genomic data for this paper have been deposited in the NCBI database (https://www.ncbi.nlm.nih.gov/) under the BioProjectID PRJNA596648. All RNA-seq sequences have been deposited at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) under GEO accession number: ‘GSE159524’.