

Abstract
Homologous recombination dominates as the major form of DNA repair in Trypanosoma brucei, and is especially important for recombination of the subtelomeric variant surface glycoprotein during antigenic variation. RAD50, a component of the MRN complex (MRE11, RAD50, NBS1), is central to homologous recombination through facilitating resection and governing the DNA damage response. The function of RAD50 in trypanosomes is untested. Here we report that RAD50 and MRE11 are required for RAD51-dependent homologous recombination and phosphorylation of histone H2A following a DNA double strand break (DSB), but neither MRE11 nor RAD50 substantially influence DSB resection at a chromosome-internal locus. In addition, we reveal intrinsic separation-of-function between T. brucei RAD50 and MRE11, with only RAD50 suppressing DSB repair using donors with short stretches of homology at a subtelomeric locus, and only MRE11 directing DSB resection at the same locus. Finally, we show that loss of either MRE11 or RAD50 causes a greater diversity of expressed VSG variants following DSB repair. We conclude that MRN promotes stringent homologous recombination at subtelomeric loci and restrains antigenic variation.
INTRODUCTION
Trypanosoma brucei is a protozoan parasite and the causative agent of human African trypanosomiasis, or sleeping sickness, and nagana in cattle. Trypanosomes cycle between their insect vector, the tsetse fly and mammalian hosts, where they colonise the blood, fat (1) and skin (2) and eventually cross the blood brain barrier in late stage infection. If left untreated, trypanosomiasis is normally fatal (3). In the mammalian host, each trypanosome cell is covered in a dense layer of a single species of variant surface glycoprotein (VSG). The highly immunogenic VSG layer (4,5) acts as a barrier, concealing other surface components from the host immune response (6). Trypanosomes maintain a persistent infection by continuously escaping the host's immune response through antigenic variation (7). Central to this survival strategy is monoallelic expression of the VSG from a subtelomeric locus, known as an expression site (VSG-ES), and stochastic VSG switching. The ∼15 VSG-ESs in the trypanosome genome share a high degree of sequence and structure conservation (8), each being an RNA polymerase-I (RNA Pol-I) polycistronic transcription unit with a single VSG gene found adjacent to the telomere, up to 60 kb downstream of the promoter (8). The VSG gene is flanked by two sets of repetitive sequence: downstream is the telomere, and upstream is a block of repetitive sequence, known as the 70-bp repeats, which modulates VSG switching (8,9). Characteristic of a trypanosome infection are recrudescent waves of parasitemia, each of which is composed of a diverse VSG expressing population, with between 7–79 VSGs detected in each peak of parasitemia (10–12). VSG diversity arises through altering the single VSG-ES that is transcribed or, more commonly, by recombination of silent VSGs into the active VSG-ES. The seemingly unrestricted use of VSG genes might be expected to result in a rapid exhaustion of the VSG gene repertoire. However, the parasite's ability to sustain an infection appears to lie in an enormous repertoire of >2000 VSG genes and pseudogenes (13–15), mainly found in subtelomeric VSG arrays, and a capacity for generation of novel ‘mosaic’ VSG genes through segmental gene conversion of multiple (pseudo) VSGs, in particular late in an infection (10,11,14). Importantly, almost all of the array VSGs are associated with upstream tracts of 70-bp repeats, providing the necessary substrate needed for homologous recombination mediated antigenic variation (16).
A DNA double-strand break (DSB) is an extremely toxic lesion in any cell, which if left unrepaired can lead to cell death. In T. brucei RAD51-dependent homologous recombination (HR) dominates as the major DNA repair and recombination pathway, with microhomology mediated end-joining (MMEJ) playing a minor role (17–20). HR is important for VSG switching, and though it is not clear how MMEJ acts in this reaction, repair of induced DSBs can occur by coupled HR and MMEJ, and MMEJ is more frequently used for repair of DSBs induced within the VSG-ES (21). Unrepaired DSBs appear to persist throughout the cell cycle without inhibiting the trypanosome's ability to replicate their DNA (22), but whether HR or MMEJ are regulated is unknown. In addition, non-homologous end-joining (NHEJ) appears to be absent in trypanosomes (21,23,24). These features of trypanosome DSB repair contrast with mammalian cells, where NHEJ is highly active, HR predominates in S and G2 phase cells and MMEJ is considered a minor reaction (25). In trypanosomes both transcriptionally active and silent subtelomeres are fragile (26,27), and accumulate natural breaks. Within the active VSG-ES specifically, a DSB between the VSG and 70-bp repeats acts as a potent driver of antigenic variation and precipitates VSG switching (27). Several DNA repair and recombination proteins have been shown to be important for antigenic variation in trypanosomes, thus linking VSG switching with this process: loss of RAD51 (18), the RAD51–3 paralogue (28), or the RAD51-interacting protein BRCA2 (29,30) results in impaired VSG switching, while loss of RECQ2 (31), TOPO3α or RMI1 increases VSG switching (32,33), as does loss of the histone variants H3.V and H4.V (15). Loss of ATR, which is involved in DNA damage signalling, impairs monoallelic VSG expression and increases VSG switching through localized DNA damage (34). Histone Acetyltransferase (HAT3) is required for recombination repair of a chromosome-internal DSB, but suppresses DSB repair within the VSG-ES which suggests repair is compartmentalised in trypanosomes (35).
The DNA damage response (DDR) is an orchestrated cellular response to many different genome lesions, including DSBs, which most commonly form via stalled replication forks (36). Critical to DSB repair is the MRE11–RAD50–NBS1 (MRN) complex (in yeast MRE11–RAD50–XRS1, MRX), which acts as a DNA damage sensing complex and is responsible for recognizing the free DNA ends, where it is one of the first complexes to bind and initiate HR (37,38). MRE11–RAD50 forms the core of this complex and is conserved across all domains of life, whereas NBS1 only forms part of the complex in eukaryotes (37). MRN consists of two molecules of each component protein, and diffuses along homoduplex DNA searching for free DNA ends—a process that is driven by RAD50 (39). The MRE11 subunit is a nuclease with both 5′ flap endonuclease activity and 3′→5′ exonuclease activity and catalyses resection through cleaving the 5′ strand, internal to the DSB, which is then resected using its exonuclease function to generate the short 3′ single-strand (ss) DNA overhangs (40). These overhangs are further resected by Exonuclease 1 (EXO1), forming long tracts of 3′ ssDNA on either side of the DSB (39). Thus, although MRE11 is important for end resection, it is not essential for this process: processing of HO-induced DSBs (41) and breaks in both DT40 cell and human TK6 B cells (42) can be efficiently processed in the absence of MRE11 via nuclease redundancy within each system. NBS1, the eukaryote-specific component, is responsible for binding multiple phosphorylated proteins and recruiting MRE11 and RAD50 to DSB sites (43) through its interaction with MRE11, CtIP, which is also required for initiating resection, and the ATM kinase (44). End recognition and DSB processing by MRN is an ATP dependent process: here, ATP binding to RAD50 acts to switch the complex from an open to a closed conformation (45), which facilitates DSB recognition, tethering and ATM activation (45). In yeast, the MRX complex also acts in telomere maintenance by binding the end of short telomeres and recruiting TEL1, which then recruits telomerase to extend the telomere (46). Conversely, in mammalian cells, MRN regulates an ATM dependent response at dysfunctional telomeres (47). The MRN/X complex exists primarily as a heterohexamer, but several separation-of-function allele mutants have pointed to the specific functions each protein. Mutations in MRE11 do not result in disassembly of the complex itself, but affect aspects of DSB repair, such as resection (48,49). Conversely, point mutations that lead to disruption of MRE11–RAD50 interaction, or mutations that prevent a change to the conformational state of the MRN complex, result in impaired checkpoint activation, DNA tethering and end-resection (50,51).
RAD50 (Tb.927.11.8210), MRE11 (Tb927.2.4390) (52,53) and NBS1 (Tb 927.8.5710) homologues are present in the trypanosome genome and previous studies have shown that MRE11 is required for HR but its inactivation did not lead to telomere shortening or changes in VSG switching (52,53), despite the dominance of HR in repair in trypanosomes and requirement for the reaction in antigenic variation. MRE11 and RAD50 have also been studied in the related kinetoplastid Leishmania infantum (54,55). Paradoxically, while L. infantum MRE11 nulls can be generated, RAD50 is only dispensable in the absence of MRE11 (55). Both MRE11 and RAD50 are required for Leishmania genome stability, while MRE11 has a specific role in genome amplification in response to environmental changes, and RAD50 in subtelomeric stability (54,55). What roles these proteins play in the trypanosome DDR is largely unexplored. In addition, though we know that DSBs accumulate at the subtelomeres (26,27), it is unclear how they are sensed or how they contribute to antigenic variation. Given the central, early role of the MRN complex in DSB recognition and in telomere maintenance we set out to characterise its role in HR and VSG switching in trypanosomes. We found that RAD50, like MRE11, is required for efficient HR, and in its absence MMEJ dominated as the major form of repair. We also provide evidence of separation-of-function between these proteins in T. brucei, with differing effects of MRE11 and RAD50 loss on DSB survival, MMEJ levels and DSB resection at a telomeric VSG locus. Finally, we reveal that RAD50 and MRE11 play a perhaps surprising role in VSG switching, where they restrict HR substrate selection in the VSG repertoire and may act to preserve the VSG archive during long-term infections.
MATERIALS AND METHODS
Trypanosoma brucei growth and manipulation
Lister 427, MITat1.2 (clone 221a), bloodstream stage cells were cultured in HMI-11 medium (56) at 37.4°C with 5% CO2. Cell density was determined using a haemocytometer. For transformation, 2.5 × 107 cells were spun for 10 min at 1000g at room temperature and the supernatant discarded. The cell pellet was resuspended in prewarmed cytomix solution (57) with 10 μg linearised DNA and placed in a 0.2 cm gap cuvette, and nucleofected (Lonza) using the X-001 program. The transfected cells were placed into one 25 cm2 culture flask per transfection with 36 ml warmed HMI-11 medium only and placed in an incubator to allow the cells to recover for ∼6 h. After 6 h, the medium was distributed into 48-well plates with the appropriate drug selection. Strains expressing TetR and I-SceI with I-SceI recognition-sites at a chromosome-internal locus (17) and an active VSG-ESs (27) have been described previously. G418, and blasticidin were selected at 10 and 2 μg ml−1 respectively. Puromycin, phleomycin, G418, hygromycin and blasticidin and tetracycline were maintained at 1 μg ml−1. Clonogenic assay were set up as previously published (17,27), but briefly, cells were plated out at either 32 cells per plate under non-inducing conditions for 1HR and VSGup cell lines, 32 cells per plate under inducing conditions for 1HR, and 480 cells per plate for VSGup, rad50 and mre11 null cell lines under inducing conditions. Plates were counted 5–6 days later and subclones selected for further analysis. Clonogenic assays were done using one biological clone for 1HR and VSGup, these are published and validated cell lines (17,27). For the rad50 and mre11 null mutants, two biological clones were used.
Plasmid construction
For native C-terminal epitope tagging of Tb927.5.1700/RPA2 a 765-bp fragment was amplified using primers RPA28F:GATCAAGCTTATGGAAGGAAGTGGAAGTAA; and RPA28R:GATCTCTAGAAATGCCAAACTTACAATCATG and cloned in pNATxTAG (58) using the HindIII and XbaI sites (underlined). The construct was linearized with XhoI prior to transfection. MRE11F5 (GATCgcggccgcATGGCCGAGAGGGCATC), MRE11R5 (GATCtctagaCAACGAAGATGTATGCCC), MRE11F3 (GATCgggcccCGATGGATAGTGGTAAT) and MRE11R3 (GATCggtaccCTAATAGTTATCTGGCA) were used to clone in target regions to generate pMRE11KOBLA and pMRE11KONEO. For transfection, 20 μg pMRE11KO Blasticidin (BLA) and Neomycin (NEO) plasmids were sequentially digested with Acc65I and NotI and cleaned by phenol- chloroform extraction and ethanol precipitation after each digestion. Strains were validated using MRE11F5 and MRE11R3 in a PCR assay. Heterozygous (+/−) and homozygous (+/−) knockout mutants of RAD50 were generated by replacing most of the gene's open reading frame with either BLA and NEO. The strategy used is as described in (59); briefly, two modified versions of the plasmid pmtl23 were used to allow PCR-amplified 5′ and 3′ flanking untranslated regions of RAD50 to be inserted around BLA and NEO cassettes (where the antibiotic resistance genes’ ORF were flanked by tubulin and actin intergenic regions). The selective drug markers, flanked by RAD50 5′ and 3′ untranslated regions, were then excised using NotI and transfected into T. brucei, and clones selected using 10 μg ml−1 blasticidin or 5 μg ml−1 G418. To generate the RPAMyc tagged 1HRrad50 nulls in the 1HR strain we employed multi-step transfection strategy (60) that recycled a Neomycin phosphotransferase gene (NEO) in order to rescue one marker (here Blasticidin – BLA). Briefly, an I-SceI recognition sites was inserted into the pRAD50-BLA knock-out cassette between the 5′UTR and BLA ORF (Figure 1C) in the 2T1 cell line (61) with a tetracycline inducible Sce ORF. Induction of Sce induces a break in the BLA cassette and subsequent repair, using homology in the NEO modified allele, replaces BLA with NEO. The 1HR cells line was then generated as in (17) and the RPA tagging as in (22).
Figure 1.

RAD50 is essential for DSB response and repair at a chromosome-internal locus. (A) Upper panel: Schematic of TbRAD50 with protein domains. Amino acid position of conserved domains are: ATPase-N, 4–170; MRE11 binding site (MBS), 182–205; Zn hook, 690–693; MRE11 binding site, 1158–1181; ATPase-C, 1243–1333. Lower panel: the structure of the T. brucei RAD50 was modelled using Phyre2 showing the SMC head domain with a coiled coil. (B) Schematic of the chromosome 11 Tb927.11.4530/40 locus (upper panel: wild type) and the modified 1HR chromosome-internal DSB cell line with the I-SceI recognition site, SceR, highlighted (lower panel: 1HR). (C) A clonogenic assay reveals survivors following a DSB at a chromosome-internal locus in the parental and 1H Rrad50 cell lines. 96–480 cells were plated out into media with or without tetracycline and assessed after 7 days of growth. Box-plot centre lines show the medians; box limits indicate one standard deviation. The proportion of survivors was calculated by dividing the number of induced survivors by uninduced. SD from seven or more 96-well plates: 1HR uninduced 7, 1HR induced 8 and for 1HRrad50 uninduced 9, 1HRrad50 induced 8. P-value calculated using a Student t-test. White boxes, genes; RFP:PAC, red fluorescent protein: puromycin fusion gene; black boxes UTR.
Immunofluorescence microscopy
Immunofluorescence analysis was carried out using standard protocols as described previously (17). Mouse α-Myc was used at 1:400 and rabbit α-γH2A (62) was used at 1:250. Fluorescein-conjugated goat α-rabbit and goat α-mouse secondary antibodies (Pierce) were used at 1:2000. Samples were mounted in Vectashield (Vector Laboratories) containing 4,6-diamidino-2-phenylindole (DAPI). In T. brucei, DAPI-stained nuclear and mitochondrial DNA can be used as cytological markers for cell cycle stage (63); one nucleus and one kinetoplast (1N:1K) indicate G1, one nucleus and an elongated kinetoplast (1N:eK) indicate S phase, one nucleus and two kinetoplasts (1N:2K) indicate G2/M and two nuclei and two kinetoplasts (2N:2K) indicate post-mitosis. Images were captured using a ZEISS Imager 72 epifluorescence microscope with an Axiocam 506 mono camera and images were processed and in ImageJ. Foci counts were done by two people.
ssDNA qPCR resection assay
1 × 106 cells were collected at 0, 6 and 12 h post I-SceI induction. Genomic DNA was extracted using the Qiagen Blood and Tissue kit. 500 ng of DNA was digested overnight at 37°C with HindIII or a mock digest was set up (64). HindIII was heat inactivated by heating to 85°C for 10 min. 500 ul of dH2O was added to each digest to give a 1 ng/ul sample. 5 ng was analysed by qPCR using Luna Universal qPCR MasterMix (NEB) with 300 pmol of primers for the 1HR analysis or 600 pmol of primers for the VSG2 analysis. For each pair of primers (below), triplicates of each sample were run per plate (Hard-shell PCR Plates 96-well, then wall; Bio-Rad), which were sealed with Microseal ‘B’ seal Seals (BioRad). All experiments were run on a CFX96 Touch Real-time Detection system with a C1000 Touch Thermal cycler (Bio-Rad), using the following PCR cycling conditions: 95°C for 3 min, followed by 40 cycles of 95°C for 10 s and 55°C for 30 s and fluorescence intensity data collected at the end of the last step. Data was then analysed by relative quantification using the ΔΔCt method (CFX Maestro software – Bio-Rad). ssDNA at each time was quantified by subtracting the mock digested from the HindIII digested sample to give the ΔCt and then using the following formula from (65) where f is the fraction cut by I-SceI: % resected = [100/[(1 + 2ΔCt)/2]]/f. For the 1HR cell lines primer pairs to detect ssDNA in the RFP:PAC fusion gene are RFPF.2 (CTCGCGGTGCTGATTTCTG) and RFPR.2 (TGAAGCGCATGAACTCCTTG). For the VSGup cell lines, the primer pairs used are VSG2F1b (AGGCCAAGAAAGCGCTAACA) and VSG2R1b (CCACTGGCTGCTCGGATATG)
DNA analysis
PCR analysis of RAD50 nulls to confirms knock-out were done using standard PCR conditions with the following primers; a 402 bp product for RAD50 using RAD50KOF (CGTGAGAAACAGGAACAGCA) and RAD50KOR (AACACGTTTTTCCAACTCGG); a 399 bp product for Blasticidin ORF using BlaF (GATCGAATTCATGGCCAAGCCTTTGTCT) and BlaR (GATCCCATGGTTAGCCCTCCCACACATAA); and a 795 bp product for Neomycin Phosphotransferase ORF using NPTF (ATGATTGAACAAGATGGATTG) and NPTR (TCAGAAGAACTCGTCAAGAA). Analysis of subclones was previously described (21,27,35) and used the following primers VSG221F (CTTCCAATCAGGAGGC), VSG221R (CGGCGACAACTGCAG), RFP (ATGGTGCGCTCCTCCAAGAAC), PAC (TCAGGCACCGGGCTTGC), ESAG1F (AATGGAAGAGCAAACTGATAGGTTGG), ESAG1R (GGCGGCCACTCCATTGTCTG). PCR analysis of VSG pseudo gene and VSG2 or VSG pseudo gene and VSG8 was done using LongAmp Taq DNA polymerase (NEB) and the following PCR conditions: 95°C for 30 s; 30 cycles of 95°C for 30 s, 55°C for 30 s, 65°C for 12 min 30 s; 65°C for 10 min with the following primers PseudoF (GTACCCCGGGGCGCCGAATTTAATGCAAATATG), VSG221R (CGGCGACAACTGCAG), VSG8R (TACCCTGGAAACTACCGTCG).
VSG sequencing analysis
For the RT-PCR, the reaction mix were as following; 1 μg of cDNA, 1× PCR buffer, 0.2 mM dNTPs, 1 μl each of SL (ACAGTTTCTGTACTATATTG) and SP6–14mer (GATTTAGGTGACACTATAGTGTTAAAATATATC) primers, H2O to 50 μl and 0.5 μl Phusion polymerase (NEB). For the PCR conditions. Five cycles were carried out at 94 °C for 30 s, 50 °C for 30 s and 72°C for 2 min; followed by 18 cycles at 94°C for 30 s, 55°C for 30 s and 72°C for 2 min. DNA concentration was measured using a Nanodrop. Libraries were prepared from VSG PCR products and sequenced on a BGI-Seq (BGI500) with a 150 bp paired-end read length with BGI Genomics Hong Kong.
Replicate libraries for VSGUP uninduced and VSGUP induced, VSGUPrad50 uninduced and VSGUPrad50 induced, and triplicate libraries for VSGUPmre11 uninduced and VSGUPmre11 induced were sequenced on the BGIseq500 platform producing 8.03 and 9.01 million reads for VSGUP uninduced, 7.60 and 7.22M for VSGUP induced, 6.66, 7.07M for VSGUPrad50 uninduced, 6.90 and 6.98M for VSGUPrad50 induced, 4.6M reads for each VSGUPmre11 uninduced and 4.6, 4.6 and 3.9M reds for VSGUPmre11 induced. Reads were aligned to the T. brucei Lister 427 genome (15) with the cohort of minichromosomal VSGs added from the Lister 427 VSGnome (13) using bowtie2 (66) with the parameters—very-sensitive and BAM files created with samtools (67), aligning (VSGUP uninduced) 97.76, 98.42, (VSGUP induced) 97.69, (VSGUPrad50 uninduced) 97.57, 98.60, (VSGUPrad50 induced) 97.63, 97.80, (VSGUPmre11 uninduced) 99.04, 99.01, 98.77 and (VSGUPmre11 induced) 98.57%, 98.52%, 98.53% of reads successfully. Reads counts per transcript were obtained using featureCounts (68). Differential expression analysis was performed using EdgeR (69) on all genes, followed by filtering for VSG genes (1848 VSG sequences in total). An R script (https://github.com/LGloverTMB/DNA-repair-mutant-VSG-seq) was used to perform differential expression analysis, and generate Volcano and genome scale plots. BLAST analysis was performed locally using a database containing all VSG genes, including 2 kb of sequence upstream and downstream of the start and stop codons, respectively (except where sequences in the contigs 5′ or 3′ to the CDS were shorter than 2 kb excluding). The resulting database of 2,153VSGs was queried using the VSG2 sequence including 2 kb of sequence upstream of the CDS and all sequence between the stop codon and end of contig (1658 nt). The BLASTn algorithm was used query the database using default parameters except allowing 1 hit per subject sequence. Alignments were plotted using a custom R script (https://github.com/LGloverTMB/DNA-repair-mutant-VSG-seq). Lengths of average alignments were calculated for cohorts of VSGs up-regulated in each VSGup, VSGuprad50 or VSGupmre11.
RESULTS
RAD50 is required for normal cell growth and DSB repair
RAD50, the largest component of the MRN complex, belongs to the structural maintenance of chromosomes (SMC) family of proteins (70) and has not been examined in T. brucei, though the gene has been reported to be essential in Leishmania infantum (55). The domain architecture of RAD50 is approximately palindromic (Figure 1A) and characterized by the presence of ATP-binding cassette (ABC)-ATPase domains at the N- and C- termini, each followed by an MRE11 binding site (MBS), and then by anti-parallel coiled-coil regions, which form linker structures that enable the MRN complex to act as a tethering scaffold to hold broken chromosomes together for repair (71). Between the antiparallel coiled-coils, a central Zn hook, a CxxC motif, facilitates Zn2+ dependent RAD50-RAD50 subunit interactions and is presumed to be important for tethering (72). A conformational change is invoked through binding of RAD50 to two ATP molecules, which then allows for binding to DNA (45). Primary sequence comparison suggested all RAD50 domains are recognisably conserved in the putative T. brucei RAD50 homologue (Tb.927.11.8210; Supplementary Figure S1). Within the ATPase domains, the ABC nucleotide binding domain is defined by the conserved presence of Walker A, Q-loop, Signature, Walker B, D-loop, and H-loop motifs required to form the active ATPase site (73). Furthermore, structure prediction using Phyre2 (74) modeled 503 residues (37% of the sequence) of the T. brucei protein, revealing a SMC head domain and antiparallel coiled coil regions (Figure 1A).
To test the function of RAD50 in DSB repair, we used a previously validated T. brucei cell line, referred to as 1HR (17) (Figure 1B). In this cell line, a single I-SceI recognition site was inserted into an RFP-PAC (red fluorescent protein – puromycin N-acetyltransferase) fusion cassette and targeted to the intergenic region between Tb427.11. 4530/4540 in the core of chromosome 11 (17) (Figure 1B; upper panel: WT and lower panel: 1HR). Previous validation of this cell line (17) revealed that the fusion cassette was integrated into one allele of this locus (here designated ‘b’; Figure 1B; lower panel: 1HR). In the 1HR cell line induction of I-SceI using tetracycline, was shown to result in cutting of the RFP:PAC fusion gene in the majority of the cells and repair mainly by homologous allelic recombination (17). We generated rad50 null mutants (referred to as 1HRrad50) in these cells by sequentially replacing the two gene alleles with neomycin phosphotransferase (NEO) and blasticidin (BLA) resistance cassettes: PCR analysis of double antibiotic resistant clones confirmed RAD50 loss and replacement (Supplementary Figure S2A and S2B) and demonstrates RAD50 is not essential in T. brucei. To determine the role RAD50 plays in DSB repair, we set up clonogenic survival assays. In this assay, under inducing conditions, only cells that are able to repair the I-SceI break will survive. Previous work has shown that in this cell line repair occurs mainly via homologous recombination (17,35) (Supplementary Figure S3), which will resolve the break back to the WT allele state (Figure 1B; upper Panel: WT ‘a’ and ‘b’; and Supplementary Figure S3), or via MMEJ (17,21) (Supplementary Figure S3). Cells were distributed across 96-well plates under both I-SceI non-inducing (–tet) and inducing (+tet) conditions, and wells with live cells scored after 5 -7 days. This revealed a significant growth defect in the unperturbed (–tet) 1HRrad50 null cells (Figure 1C Left panel and Table 1): 95% of the WT 1HR cells survived cloning compared with ∼35% of the 1HRrad50 cells, revealing a 2.6-fold decrease in cell survival. This growth impairment is likely due to impaired ability to repair spontaneous DSBs. Consistent with previous findings, in the WT 1HR strain ∼48% of cells are able to repair the DSB and survive (Figure 1C, left panel and (17) and Table 1), whereas in the 1HRrad50 cells, a severe growth defect was seen following a DSB, with less than 3% survival (a 16-fold reduction), suggesting a significant (P = 8.7e–07) defect in DSB repair (Figure 1C, left panel and Table 1). This was recapitulated when assessing the normalised survival efficiency (induced compared to uninduced survival) following an I-SceI induced DSB (Figure 1C, right panel and Table 1). These data reveal a significant reduction in the ability of the 1HRrad50 cells to survive a DSB.
Table 1.
Details of clonogenic assays of strains described in this manuscript following a DSB
| Strain | Uninduced (%) | SD | Induced (%) | SD | Proportion | SD | Strain | Uninduced (%) | SD | Induced (%) | SD | Proportion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1HR | 95.09 | ±17.1 | 47.9 | ±11.93 | 0.52 | ±0.1 | 1HR | 95.10 | ±17.2 | 47.10 | ±11.94 | 0.52 |
| 1HRrad50 | 35.76 | ±6.83 | 2.91 | ±4.36 | 0.08 | ±0.12 | 1HRrad51 | 35.77 | ±6.83 | 2.92 | ±4.37 | 0.08 |
| 1HRmre11 | 47.27 | ±6.97 | 1.79 | ±1.65 | 0.04 | ±0.03 | 1HRmre12 | 47.28 | ±6.97 | 1.80 | ±1.65 | 0.04 |
| VSGup | 85.42 | ±15.1 | 5.45 | ±1.81 | 0.07 | ±0.02 | VSGup | 85.43 | ±15.2 | 5.45 | ±1.81 | 0.07 |
| VSGuprad50 | 51.04 | ±16.3 | 5.81 | ±1.64 | 0.11 | ±0.03 | VSGuprad51 | 51.05 | ±16.4 | 5.81 | ±1.64 | 0.11 |
| VSGupmre11 | 73.44 | ±27.86 | 1.75 | ±0.37 | 0.02 | ±0.005 | VSGupmre12 | 73.45 | ±27.87 | 1.76 | ±0.38 | 0.02 |
We next asked what effect loss of RAD50 had on the mechanisms by which trypanosomes recognize a DSB lesion and initiate a signalling cascade resulting in DNA repair (75). In T. brucei, several markers have been used to visualise the DDR after an I-SceI induced DSB. These markers include an increase of cells in G2 (17), the phosphorylation of histone H2A (62) and accumulation of RAD51 foci at the site of the DSB (17). We therefore used these as markers to assess the DDR in the rad50 null cell lines. In trypanosomes, the position and the timing of division of the nucleus and the kinetoplast (mitochondrial genome) can be used to determine individual cells’ position in the cell cycle (76,77). We specifically assessed the number of cells in G2 (one nucleus and two kinetoplasts), as T. brucei shows an accumulation of these cells in response to a DSB (17). In WT, 1HR ∼13% of the population cells were in G2 which increased to ∼21% 9 h after I-SceI induction (Figure 2A). In contrast, no increase in G2 cells was seen after DSB induction in the 1HRrad50 cells (Figure 2A), suggesting RAD50 is required for eliciting the G2/M checkpoint. In mammals the MRN complex recruits the ATM kinase to a DSB, where it phosphorylates H2AX (37). Using an antibody specific to the Thr130 phosphorylated form of T. brucei H2A (γH2A) (62), background staining levels of ∼13% of nuclei with foci (See Figure 2B inset for an example of a γH2A foci) was seen in unperturbed WT 1HR cells, similar to what has previously been reported (35,62), which increased to ∼ 48% at 9 h post I-SceI induction (Figure 2B). In the 1HRrad50 cells, the background level of γH2A foci was reduced to 4%, and no increase was seen following a DSB (Figure 2B), with only 6% of cells containing γH2A foci: an 8-fold reduction compared to WT 1HR. Repair of a DSB at this locus is predominately via RAD51-dependent homologous recombination (17), and so we next assessed RAD51 foci assembly following DSB induction. In the WT 1HR strain, the number of detectable foci increased from 0 to 34% within 9 h after I-SceI induction. In contrast, in 1HRrad50 cells the background level of RAD51 foci seen before I-SceI induction was higher at 6%, and only increased to 9% (∼3-fold reduced) in response to a DSB (Figure 2C). Prior to RAD51 loading on to ssDNA, the trimeric RPA (replication protein A) complex binds the ssDNA and is subsequently displaced by RAD51 (78). Rescue of the BLA selectable marker in this strain (Supplementary Figure S2) allowed tagging of RPA2 with the myc epitope for localization studies. In WT 1HR cells, the number of nuclei with RPA foci increased 5-fold (from 10% to 50%) following an I-SceI break (Figure 2D). The 1HRrad50 cells showed a pronounced increase in RPA foci prior to induction, and only a marginal increase at 12 h post DSB (∼30–55%; Figure 2D). In the WT 1HR cells, a single RPA focus is most commonly seen in response to an I-SceI break (22). However, we observed multiple RPA foci in the 1HRrad50 null cells (Figure 2E and F). We therefore tentatively conclude that most RPA signal in the 1HRrad50 cells (22) represents persistent, widespread genome damage.
Figure 2.
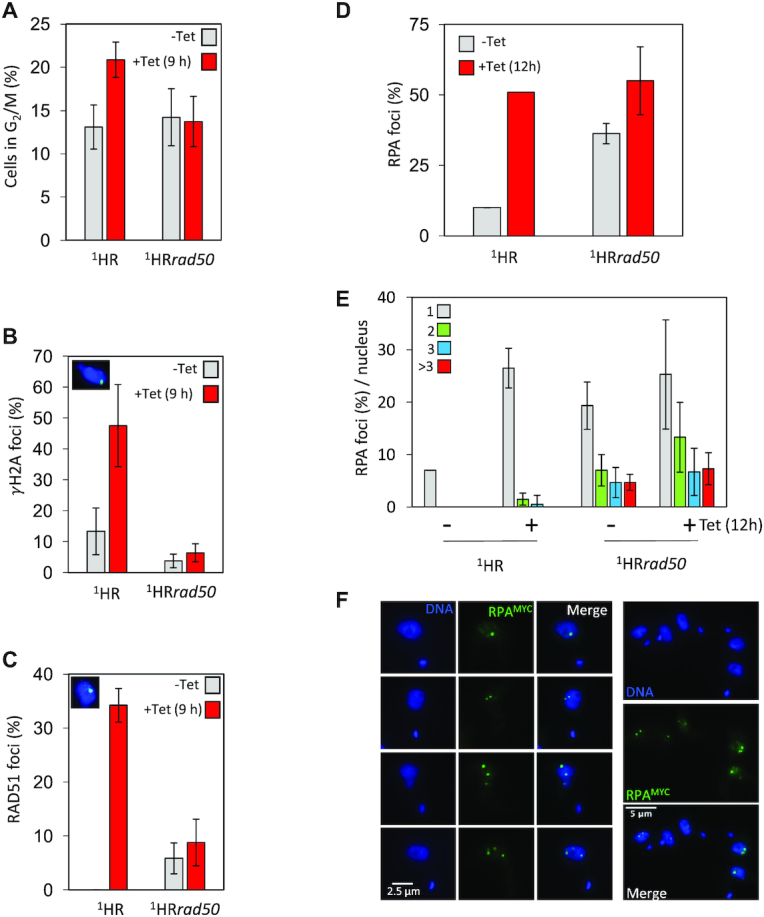
DNA damage response is compromised in 1HRrad50 cells. (A) The number of cells in G2/M phase cells was counted by DAPI staining at several points following induction of an I-SceI break in. G2 cells contain one nucleus and two kinetoplasts. Error bars, SD, for 1HR n = 3, and with 1HRrad50; n = 3. (B) Immunofluorescence assay to monitor γH2A foci. The number of positive nuclei were counted in uninduced cells and 12 h post DSB. Inset showing a nucleus with a γH2A focus. n ≥ 600 for each time point in the 1HR cell line and n≥ 600 for the 1HRrad50 strain. Error bars, SD, for 1HR n = 3, and with 1HRrad50; n = 3. (C) Immunofluorescence assay to monitoring RAD51 foci. The number of positive nuclei were counted in uninduced cells and 12 h post DSB. Inset showing a nucleus, with a single RAD51 focus. n ≥ 600 for each time point in the 1HR cell line and n ≥ 600 for the 1HRrad50 strain. Error bars, SD, for 1HR n = 3, and with 1HRrad50; n = 3. (D) Immunofluorescence assay to monitoring RPA foci. The numbers of positive nuclei were counted in uninduced cells and 12 h post DSB. n = 200 for each time point in the 1HR cell line and n = 400 for the 1HRrad50 strain. Error bars, SD, for 1HRrad50 biological replicates for the strains; n = 3. (E) Immunofluorescence assay to monitor the number of RPA foci per nucleus. The number of RPA foci was counted in uninduced cells and 12 h post DSB. n = 200 nuclei for each time point in the 1HR cell line and n = 400 nuclei for the 1HRrad50 cells. Error bars, SD, for 1HRrad50 biological replicates for the strains; n = 3. (F) The gallery of representative immunofluorescence images showing cells with RPA foci 12 h after I-SceI induction in the 1HRrad50 null cell line.
In parallel to the above experiments, we also tested the function of MRE11, as it forms a complex with RAD50, by generating null mutants through sequentially replacing the two gene alleles with NEO and BLA resistance cassettes; PCR analysis confirmed MRE11 loss and replacement (Supplementary Figure S2C). The mre11 nulls (referred to as 1HRmre11) also showed a growth defect in the unperturbed cells, with only 47% of uninduced cells surviving compared to the WT1HR cells (where close to 100% of the uninduced cells survived; Figure 3A). Induction of the I-SceI meganuclease resulted in a severe growth defect in 1HRmre11 cells, with less than 2% survival, suggesting a significant impairment (P = 1.8e–06) in DSB repair (Figure 3A and Table 1). This was similar to the 1HRrad50 cells, consistent with them acting together in a complex (37,38). We then assessed the normalised survival efficiency, which again showed a significant reduction in the mre11 null cells ability to repair a DSB (Figure 3A). We next looked at the DDR in these cells. Like in the 1HRrad50 cells, we detected loss of the G2/M checkpoint (8.5% in the mre11 nulls compared with 20% in WT following induction of a DSB, Figure 3B), loss of γH2A foci (4% of cells with γH2A foci detected compared to 48% in the WT 1HR, Figure 3B), and a significant reduction in the number of RAD51 foci (9% compared with 34% in WT, Figure 3B) following a DSB. These results reveal an important role for RAD50 and MRE11 in the DDR to a DSB in trypanosomes at a single copy locus and suggest wider roles in tackling spontaneous DNA damage.
Figure 3.

MRE11 is essential for DSB response and repair at a chromosome-internal locus. (A) Clonogenic assay reveals survivors following a DSB at a chromosome-internal locus in the parental and 1HRmre11 null cell lines. Details as in Figure 1. SD from seven or more 96-well plates: 1HR uninduced 7, 1HR induced 8 and for 1HRmre11 uninduced 8, 1HRmre11 induced 12. (B) Left panel: The number of cells in G2/M phase cells was counted by DAPI staining at several points following induction of an I-SceI break in. G2 cells contain one nucleus and two kinetoplasts. Middle panel: Immunofluorescence assay to monitoring γH2A foci. The number of positive nuclei were counted in uninduced cells and 12 hours post DSB. Right panel: Immunofluorescence assay to monitoring RAD51 foci. The number of positive nuclei were counted in uninduced cells and 12 h post DSB. Error bars, SD, n ≥ 600 for each time point in the 1HR cell line and n≥ 600 for the 1HRmre11 strain. Error bars, SD, 1HR n = 3 and with 1HRrad50; n = 3.
RAD50 is required for efficient homologous recombination in T. brucei
An early step in the DSB repair cycle is the formation of extensive 3′ ssDNA overhangs, initiated by MRE11 3′–5′ nuclease activity, which are a substrate for RAD51 nucleoprotein filament formation and act as a template for homology-directed repair (37). In light of the reduced accumulation of RAD51 foci, but increase in the number of RPA foci after DSB induction in rad50 nulls, we sought to determine whether the formation of ssDNA at the I-SceI target locus was compromised. To do this we used a quantitative resection assay used in both yeast and mammalian cells (64,65). Briefly, following an I-SceI DSB, genomic DNA is purified and digested with HindIII or mock digested. HindIII will specifically cut dsDNA and not ssDNA generated, in these circumstances, via resection. Only the resected ssDNA can then be used as a template for amplification, the dsDNA template will be destroyed by the HindIII digestion (Figure 4A). In the WT 1HR cells, ssDNA accumulated up to 12 h after I-SceI induction (Figure 4B), mirroring the phosphorylation of H2A and accumulation of RAD51 (Figure 2A-D) (17). Resection in the 1HRrad50 and the 1HRmre11 cells matched that of the WT 1HR cells, with a slight increase in at 12 h (WT 1HR—35%; 1HRrad50—44%; 1HRmre11—46%) (Figure 4B). We conclude that DNA resection is not impaired in either the 1HRrad50 or 1HRmre11 cells, despite the DDR being compromised (Figure 2 and 3). Given these two apparently conflicting results we decided to explore how trypanosomes repair a DSB in the rad50 and mre11 nulls. For this we used the DSB-survivors generated from the clonogenic assay. In the WT 1HR cell line, previous work has shown that 85% of repair is via allelic HR and ∼5% by MMEJ (17). By using sets of primers that flank the RFP-PAC cassette, the presence or absence of the PCR product cassette can be used as a marker for repair by MMEJ (Supplementary Figure S3). Prior to the PCR assay, all sublcones were tested for sensitivity to puromycin, since I-SceI cleavage followed by loss of the RFP:PAC cassette or error-prone repair within it will render the subcloned survivors sensitive to puromycin. All the uninduced (–tet) subclones were puromycin resistant (indicating no I-SceI cleavage (17); 5 subclones for each cell line tested), while all the induced (+tet) subclones were puromycin sensitive, indicating cutting by I-SceI (17) (data not shown, WT1HR, 15 subclones tested; 1HRrad50, 23 subclones tested; 1HRmre11, 15 sublcones tested). In the WT1HR, 1HRrad50 and 1HRmre11 cell lines, all tested uninduced subclones gave the expected PCR product of a 1389 bp fragment, corresponding to the size of the full-length RFP:PAC orf. In the WT1HR cell line, 2 out of 15 induced subclones gave a PCR product that was reduced in size, which in conjunction with the puromycin sensitivity assay suggests cutting by I-SceI and infrequent repair by MMEJ (as shown previously) (17,21) (Figure 4C and F). In the 1HRrad50, 13 out of 23 induced subclones showed a reduced RFP:PAC PCR product size (Figure 4D), revealing that 56% of survivors had repaired the DSB by MMEJ (Figure 4F). MMEJ repair in the RFP:PAC cassette was confirmed by sequencing (Supplementary Figure S3B). In contrast, in the 1HRmre11 cells, 3 out of 15 subclones showed a smaller RFP:PAC PCR product (Figure 4E), indicating only 20% had undergone DSB repair by MMEJ (Figure 4F). For those subclones where no PCR product could be amplified, cells could have repaired by MMEJ outside of the cassette or by HR. These data show a shift in the pathway used to repair a DSB in the 1HRrad50 null cells at a chromosome-internal locus, with repair by MMEJ dominating (Figure 4F), compared with the pronounced predominance of homologous recombination in WT 1HR cells and in 1HRmre11 null cells (17).
Figure 4.
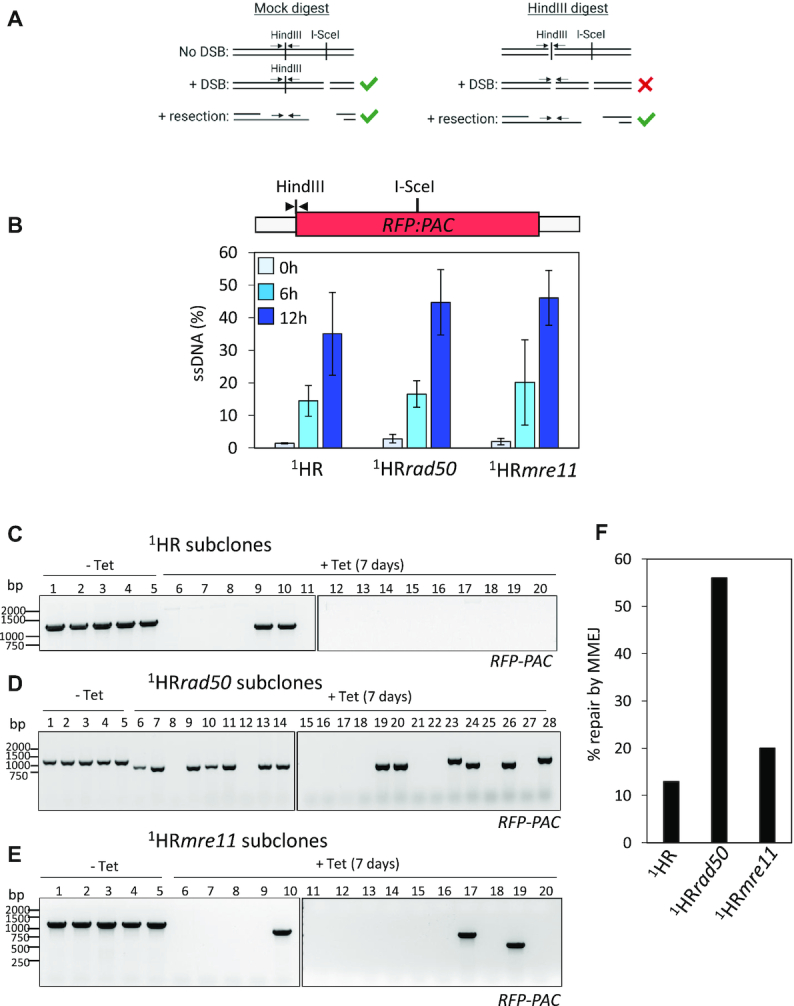
RAD50 is required for homologous recombination. (A) Schematic showing the Quantitative qPCR assay to measure DNA resection in trypanosomes. Design of qPCR primers (black arrow) to measure ssDNA at a HindIII site upstream of the I-SceI recognition site on Chromosome 11. The mock HindIII digestion is used as a control. The primer pairs flank the HindIII site. Green arrow depicts where a PCR product will be formed and a red cross where no PCR product will be formed. Schematic created with BioRender.com (B) Schematic shows the position of the HindIII site in the RFP:PAC fusion gene (red box) and UTRs (grey box) and qPCR primers (black arrow). Histogram showing % ssDNA in the 1HR, 1HRrad50 and 1HRmre11 cell lines. I-SceI cleavage was induced with tetracycline and genomic DNA extracted at 0, 6 and 12 h for qPCR analysis of end resection. Error bars = SD n = 3 for 1HR and n = 4 for 1HRrad50 and 1HRmre11 (C–E) PCR analysis of repaired subclones showing RFP:PAC presence or absence. (C) 1HR, (D) 1HRrad50, (E) 1HRmre11, (F) percentage of repair pathway by MMEJ in the 1HR, 1HRrad50 and 1HRmre11 cell lines based on the PCR analysis of the repaired subclones.
Loss of RAD50 but not MRE11 increases survival following a DSB at the active VSG-ES
T. brucei relies on homologous recombination to facilitate antigenic variation. We therefore wanted to test the role of RAD50 in VSG switching. We generated RAD50 nulls in a previously validated (27) cell line where the I-SceI recognition site is fused to a puromycin selectable marker and inserted immediately downstream of the major block of 70-bp repeats and upstream of VSG2 (also known as VSG221) in Bloodstream form Expression site 1 (BES1) (on chromosome 6a), the active VSG-ES in this strain (Figure 5A). The resulting cell line is known as VSGup (27). A DSB at this site is severely detrimental to cell growth (26,27). All cells that are able to repair and survive were shown to have switched their VSG, making this cell line a valuable tool for studying VSG switching (26,27). We generated rad50 and mre11 null VSGup strains (VSGuprad50 and VSGupmre11), by replacing the two gene alleles with NEO and BLA resistance cassettes; PCR analysis of double antibiotic resistant clones confirmed RAD50 and MRE11 loss and replacement (Supplementary Figure S2A–C). Using a clonogenic assay, cells were distributed across 96-wells plates under both I-SceI non-inducing (–tet) and inducing (+tet) conditions. Wells with live cells were scored after 5–7 days. We found that, like in the 1HR strain, there was a growth defect (1.6-fold reduction) in the VSGuprad50 nulls compared with VSGup WT cells in the absence of I-SceI induced damage (Figure 5B and Table 1). This effect again suggests an impaired ability to repair spontaneous damage in the absence of RAD50. As seen before, a DSB in the active BES is highly toxic (27,34), with both the VSGup and VSGuprad50 nulls showing a dramatic reduction in survival following an I-SceI break (5% and 5.8%, respectively (Figure 5B). However, in contrast to 1HR cells, when we assessed induced cell survival as a proportion of the uninduced, there was a significant (P = 0.0053) increase in survival of the VSGuprad50 nulls (Figure 5B and Table 1). These data indicate that RAD50 suppresses DSB repair at a VSG-ES, the opposite of its role at a chromosome-internal DSB. We then assessed the DDR in the VSGup cell lines. As in the 1HR cells, following a DSB, the number of cells that accumulate in G2/M increased to ∼25% in the VSGup WT cell line (27), and this cell cycle checkpoint was lost in the VSGuprad50 cells (Figure 5C). A similar pattern was seen with γH2A, with the number of foci increasing from 9% to 35% after I-SceI induction in the VSGup WT cell line (27), but in the VSGuprad50 nulls, only increasing from 5% to 12% following a DSB (Figure 5D). We then assessed the VSGupmre11 nulls, here clonal survival was reduced relative to WT, with only 1.75% of cells able to survive a DSB compared to 5% in VSGup WT (Figure 6A and Table 1). This reduction became more pronounced when comparing the proportion of survivors in the VSGupmre11 nulls (Figure 6A), to the VSGuprad50 nulls (Figure 5A). Despite this difference in survival, the DDR response in the mre11 nulls showed a similar phenotype to the rad50 nulls: loss of the G2/M checkpoint and reduced γH2A foci (Figure 6B). Thus, while it appears that loss of RAD50 or MRE11 diminishes the capacity of T. brucei cells to phosphorylate H2A in response to a DSB, the increased survival in the VSGuprad50 cells suggests that while RAD50 is required for an efficient DDR, in its absence the cells are more adept at repair.
Figure 5.
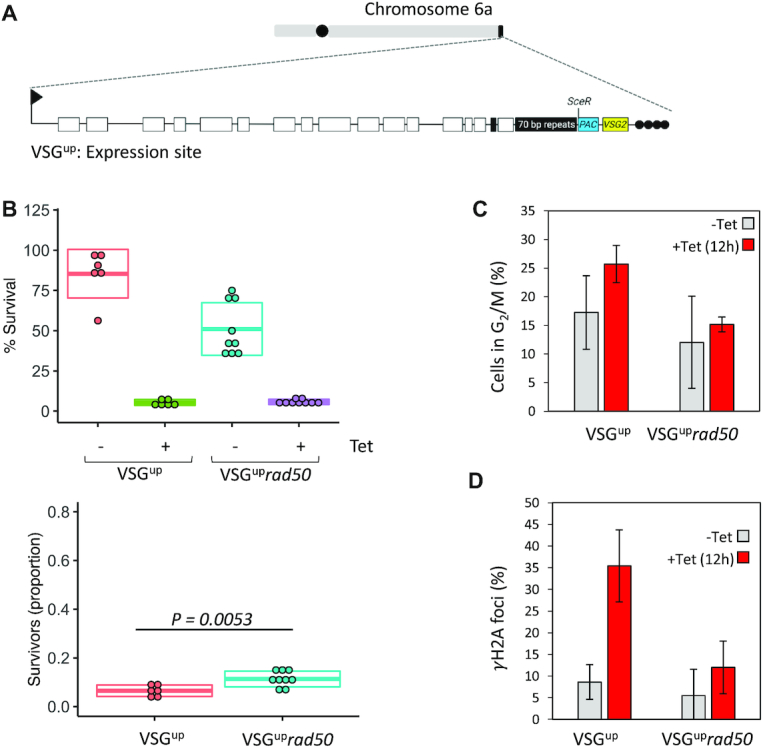
RAD50 suppresses repair at a subtelomeric locus. (A) A schematic of the active expression site DSB cell line with the I-SceI recognition site, SceR, highlighted. Schematic created with BioRender.com (B) A clonogenic assay reveals the survivors following a DSB at the active expression site in the parental and VSGUPrad50 cell lines. Cells were plated out into media with or without tetracyline and counted after seven days. Other details as in Figure 1. (C) The number of cells in G2/M phase cells was counted by DAPI staining at 0 and 12 h following induction of an I-SceI break in. G2 cells contain one nucleus and two kinetoplasts. (D) Immunofluorescence assay to monitoring γH2A foci. The nuclei with γH2A foci were counted in uninduced cells and 12 h post DSB. Error bars, SD, n ≥ 600 for each time point in the VSGup cell line n≥ 600 for the RAD50 null. 1HR n = 3 and with 1HRrad50; n = 3.
Figure 6.

MRE11 is essential for DSB response and repair at an expression site. (A) Clonogenic assay reveals survivors following a DSB in the VSGup strain in the parental and VSGUPmre11 cell lines. Details as in Figure 1 (B) Upper panel: The number of cells in G2/M phase cells was counted by DAPI staining at several points following induction of an I-SceI break in. G2 cells contain one nucleus and two kinetoplasts. Lower panel: Immunofluorescence assay to monitoring γH2A foci. The number of positive nuclei were counted in uninduced cells and 12 hours post DSB. Error bars, SD, n ≥ 600 for each time point in the VSGup cell line n ≥ 600 for the mre11 null. 1HR n = 3, and with 1HRrad50; n = 3.
Using our quantitative resection assay, we next looked at resection in the VSGup, VSGuprad50 and VSGupmre11 nulls. ssDNA accumulated to ∼ 20% 12 hrs after DSB formation in both the VSGup and VSGuprad50 nulls (Figure 7A). In contrast, in the VSGupmre11 nulls, less ssDNA was detected, with only 10% accumulating at 12 h (Figure 7A). This reduction in the amount of ssDNA in the mre11 nulls suggests compromised processing of the DSB, which may impair break repair, explaining the reduced survival in the clonogenic assays (Figure 6A), which is not seen in the absence of RAD50 (Figure 5A). Using a series of assays (Figure 7B and Supplementary Figure S4 and S5) we next looked at DNA changes in the VSG-ES to determine how the cells repair a DSB. As previously described, all the subclones were tested for sensitivity to puromycin, and all the uninduced (–tet) subclones from the three cell lines were puromycin resistant, indicating no I-SceI cleavage (17) (5 subclones for each cell line tested), while all the induced (+tet) subclones were puromycin sensitive, indicating cutting by I-SceI (17) (WT1HR, 20 subclones tested; 1HRrad50, 25 subclones tested; 1HRmre11, 20 sublcones tested), except for one VSGuprad50 induced subclone that was puromycin resistant (Figure 7B). This suggests that I-SceI cutting efficiency is comparable across all three cell lines. We then looked at surface expression of VSG2 by immunofluorescence. All uninduced subclones tested were VSG2 positive (data not show) as expected, as no DNA break had been induced, whereas in the induced panels, all VSGup (20 tested) and VSGupmre11 (20 tested) sublcones were VSG2 negative (Figure 7C). Of the 24 puromycin sensitive VSGuprad50 subclones, 23 were VSG2 negative (Figure 7C); a single puromycin resistant clone was VSG2 positive, suggesting I-SceI did not cut (Supplementary Figure S4, subclone 14). These data suggest that loss of neither RAD50 nor MRE11 affected the cell's ability to undergo VSG switching. We then used PCR to look at DNA rearrangements in the BES using primers specific to VSG2 and ESAG1. ESAG1 is found upstream of the 70-bp repeats in the active VSG-ES (Figure 7B), and by using primers specific to ESAG1 (32) in BES1 (the BES containing VSG2 and the I-SceI site) we tested for the gene's presence, since cells that retain ESAG1 are presumed to have repaired by gene conversion using the 70-bp repeats (9,26,27,79) to insert a new VSG gene into the BES. Cells that retain both the VSG2 and ESAG1 genes, but are shown to no longer express VSG2 by immunofluorescence assay, are expected to have switched by in situ switching, the transcriptional deactivation of the active BES and concomitant activation of another BES. By PCR, both VSG2 and ESAG1 were retained in all the uninduced control subclones (Figure 7D and Supplementary Figure S4). Additionally, using primers specific to the VSG pseudo gene in BES1 and VSG2, we could show linkage between the two genes in the uninduced samples at a population level (Supplementary Figure S5B). In the induced samples, following 196 h of induction, the pseudo-VSG2 PCR product was either diminished or lost suggesting cleavage of I-SceI (Supplementary Figure S5B). In the WT VSGup induced set, all subclones had retained ESAG1 and lost VSG2, suggesting switching by gene conversion (Figure 7D and Supplementary Figure S4 and S5A). This finding agrees with previously published data (26,27,79). Recent analysis of VSG switching the WT VSGup cell line, where a GFP:BLA (Green Fluorescent Protein: Blasticidin resistance marker) fusion cassette had been inserted downstream of the BES1 promoter, confirmed that the majority of the VSG switching events in the cell line were via gene conversion events (switched survivors had replaced VSG2 but maintained GFP:BLA expression) (79). Amongst the 25 VSGuprad50 induced subclones, 22 had retained ESAG1 and 23 had lost VSG2, suggesting the majority had switched by gene conversion (Figure 7D and Supplementary Figure S4), as seen in VSGup. One VSGuprad50 subclone was found to be puromycin sensitive, VSG2 negative by immunofluorescence, and ESAG1 positive and VSG2 positive by PCR. This pattern suggests that this single clone had undergone in situ switching and activated another BES (Figure 7C and Supplementary Figure S4), although we cannot rule out a gene conversion event that retained VSG2. In the 20 VSGupmre11 induced subclones, 18 had retained ESAG1 and all the subclones had lost the VSG2 gene (Figure 7C and Supplementary Figure S4). To determine whether VSG genes from silent BES's could be recombined into BES1 following an I-SceI break we used a primer specific to VSG8 in BES12. No product was amplified in the uninduced samples, as expected, but multiple products could be amplified in the 196 h induced samples in the VSGup, VSGuprad50 and VSGupmre11 cell lines (Supplementary Figure S5B). The presence of multiple bands could be indicative of gene conversion events using different lengths of 70 bp repeats for homology. This suggests that silent BES VSG genes can be recombined into BES1 following a DSB. These data suggest that loss of RAD50 or MRE11 does not impair the T. brucei's ability to undergo switching by DNA recombination.
Figure 7.
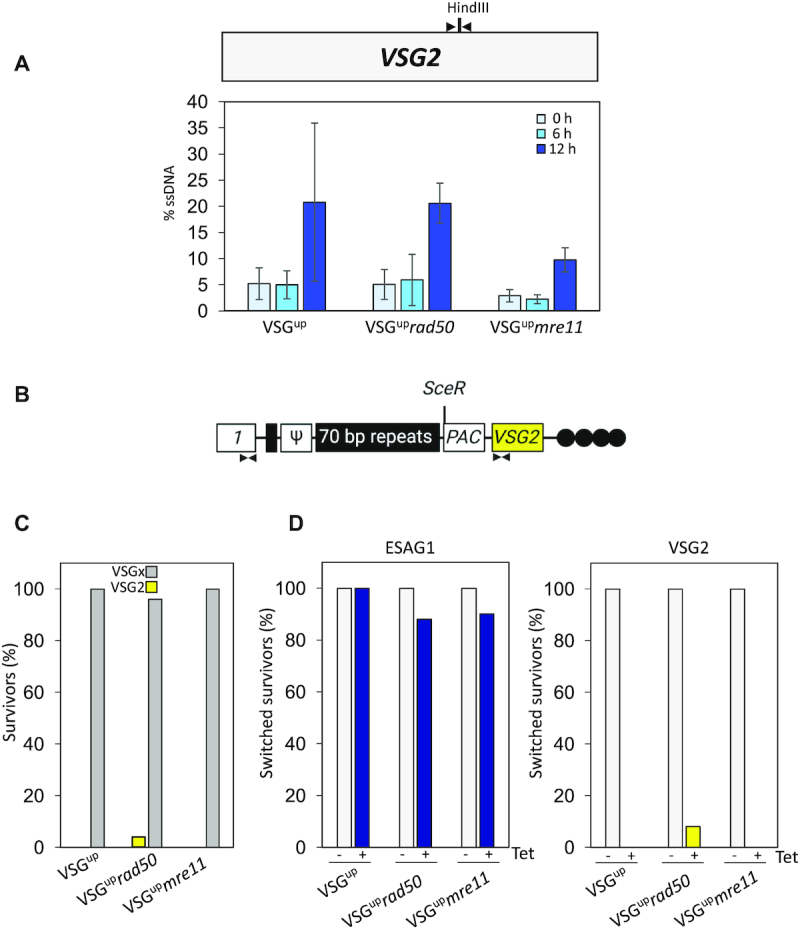
RAD50 is not required for VSG switching. (A) Schematic shows the position of the HindIII site in the VSG2 gene (grey box) and qPCR primers (black arrow). Histogram showing % ssDNA in the VSGup, VSGuprad50, VSGupmre11 cell lines. I-SceI cleavage was induced with tetracycline and genomic DNA extracted at 0, 6 and 12 h for qPCR analysis of end resection. Error bars = SD, n = 3 for VSGup and n = 4 for VSGuprad50, VSGupmre11. (B) A schematic map shows the primer position at the active expression site. Schematic created with BioRender.com (C) Immunofluorescence assay for VSG2, showing the percentage of switched survivors in the VSGup, VSGuprad50, VSGupmre11 cell lines. Gray bars or ‘VSGx’ represent the number of clones that had switched to an unknown VSG. (D) PCR analysis shows the percentage of switched survivors that retained ESAG1 and VSG2 in the VSGup, VSGuprad50, VSGupmre11 cell lines. –Tet represents the uninduced clones and +Tet the induced clones. Arrows indicate position of primers; box with diagonal lines, 70-bp repeats; vertical lines, telomere.
RAD50 and MRE11 restrict antigenic variation
We next asked whether the increased survival of the VSGuprad50 nulls after induction of a DSB, and the resection defect seen in VSGupmre11 nulls, lead to changes in the VSG switching profile compared to the WT parental cell line. In the T. brucei genome there are in excess of 2000 VSG genes found at the subtelomeric arrays (13,15,16,80), 90% of which are associated with a stretch of 70-bp repeat sequence (14) that can be used for homology during repair (9). RAD50 and MRE11 knock-outs have been shown to be hyper-recombinogenic (46,81), and so to ask if antigenic variation is affected in our rad50 and mre11 nulls we used VSG-seq (10), which allows for the amplification of VSG mRNA using primers specific to the 5′ and 3′ UTRs. In the VSGup WT cell line, 74 VSG gene transcripts were significantly enriched in the induced cells compared with uninduced (Figure 8A) (>2 log2 fold change, and P value of <0.05). In the VSGuprad50 cell line, a greater number of VSGs were detected: here 187 VSG transcripts were significantly enriched after I-SceI induction (Figure 8A). Surprisingly, given the resection defect, 230 VSG transcripts were detected in the mre11 nulls, suggesting a similar alteration in VSG switching to that seen in the rad50 null cells (Figure 8A and B). To understand if the increased VSG transcripts in the rad50 and mre11 nulls arose from the same change in VSG switching, we looked at the genomic position of the VSG cohorts. In the VSGup cells, we found that approximately equal numbers of enriched VSGs mapped to the VSG-ES (Figure 8B, and inset showing the silent BES VSGs) and minichromosomes relative to the megabase VSG arrays, despite the much greater number of genes in the latter component of the archive (Figure 8B, Supplementary Figure S6A). In the VSGuprad50 and VSGupmre11 nulls cell lines, the enriched VSG genes also mapped to the VSG-ESs, minichromosomes and megabase arrays, but a significantly higher proportion (62.5% and 67% compared with 27% in WT) were from subtelomeric arrays (Figure 8B, Supplementary Figure S6A). We then looked at the proportion of total RPKM in the all three cell lines in both the uninduced and induced samples (Supplementary Figure S6B). In all the uninduced samples VSG2 dominated, suggesting that the VSG switching frequency did not increase in the absence of either RAD50 or MRE11 (Supplementary Figure S6B). In the induced samples, VSG2 no longer dominated as the major VSG in the population, rather the VSG diversity was increased (Supplementary Figure S6B). This mapping suggests that the VSGuprad50 and VSGupmre11 nulls cells are able to access a greater proportion of the silent VSG archive for repair and VSG switching. Furthermore, these data suggest that the MRN complex constrains a hyper-recombinogenic mechanism in subtelomeric loci, through which the cells would otherwise be able to switch VSGs at a higher rate.
Figure 8.
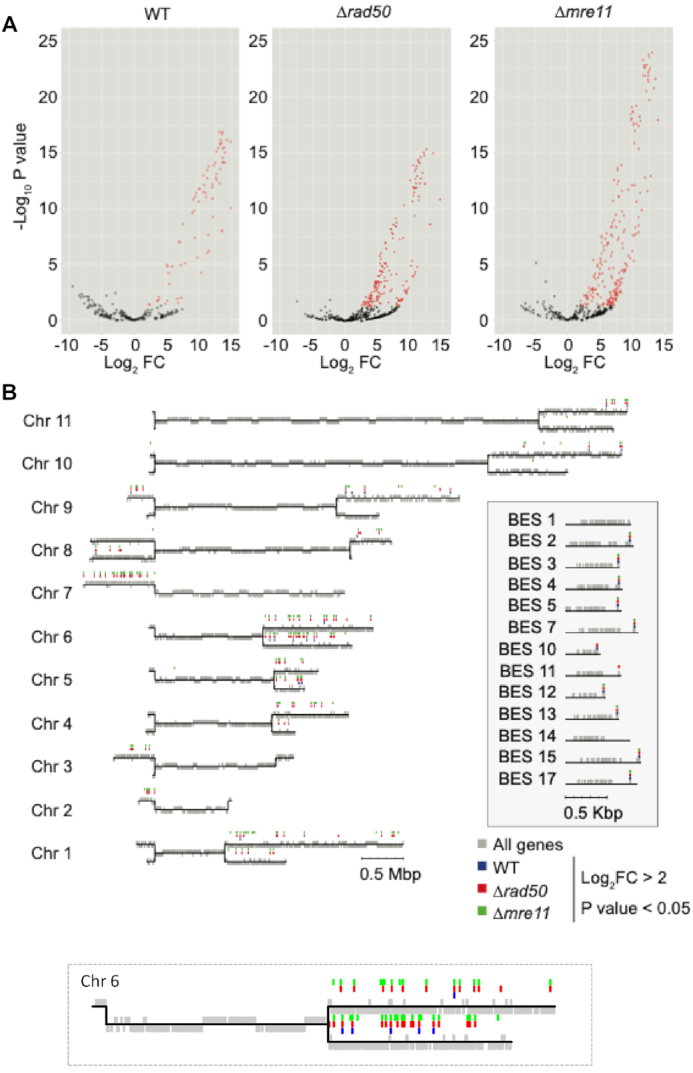
RAD50 restricts antigenic variation. (A) Volcano plots of VSG-seq showing log2 fold change versus log10P value for uninduced vs induced for all three cells lines—VSGup and VSGuprad50 and VSGupmre11 cell lines following an I-SceI DSB. Red genes are significantly up-regulated (P value < 0.05 and log2 FC > 2. (B) T. brucei 427 Genome map showing all 11 megabase chromosomes in black lines and all genes in gray. Significantly up-regulated VSG genes from either VSGup (blue), VSGuprad50 (red) or VSGupmre11 (green). Inset box shows all 13 BES with significantly up-regulated VSG genes from either VSGup (blue), VSGuprad50 (red) or VSGupmre11 (green). Expanded Chromosome 6 insert to show VSG genes up-regulated in greater detail. Grey boxes, genes. Replicate libraries were generated for VSGup and pairs of clones in the VSGuprad50 and triplicate libraries for VSGupmre11 cell lines.
To ask if increased VSG switching in the absence of either RAD50 or MRE11 could be explained by changes in the mechanism of recombination, we looked at the length of homology used for repair. Using BLAST analysis, we queried the significantly enriched VSGs against VSG2 or the telomeric end of BES1, searching for regions of homology (Supplementary Figure S6C and D). This analysis revealed that when all VSG genes were compared to VSG2, they shared short stretches of homology (Supplementary Figure S6C and D). We then compared the VSG genes activated in the VSGup WT cells to the VSG2 locus and found that the cells shared longer stretches of homology—>400 bp (34% of survivors) (Supplementary Figure S6C and D). In contrast, in both the VSGuprad50 and VSGupmre11 nulls the VSG genes activated shared shorter stretches of homology with the active VSG2 locus. We observed more a than 2.5- and 4-fold increase in the number of genes with 0–100 bp of alignment in the VSGuprad50 and VSGupmre11 null cell lines respectively (Supplementary Figure S6C and D). Thus, in the absence of RAD50 or MRE11 greater access to archival VSGs could be facilitated by using shorter stretches of homology.
DISCUSSION
Central to the DDR and subsequent repair is the MRN complex, where the MRE11–RAD50 heterodimer forms the conserved catalytic core (82). In T. brucei the severe growth defect seen in both the mre11 (52,53) and rad50 null suggests these proteins are critical for cell survival. Previous characterization of MRE11 has revealed a role in HR and genomic stability in T. brucei but detected no role in VSG switching (52,53). In Leishmania, paradoxically, RAD50 is essential in the presence of MRE11, with mutants of each gene showing specific roles in genomic and subtelomeric stability potentially driving rapid adaptation to new environments (54,55). RAD51-dependent homology-based recombination appears to be the major form of repair in kinetoplastids, and dominates during VSG switching (17,26,27,79,83), being more prevalent than MMEJ and non-homologous end joining (NHEJ) which appears to be entirely absent (24). These pathway preferences are in contrast to mammalian cells where NHEJ accounts for a significant proportion of repair (84). Studies in T. brucei using I-SceI induction of a DSB at both chromosome-internal (17) or telomeric loci showed repair to be predominantly by HR, whereas a CRISPR-Cas9 induced DSB in Leishmania, T. cruzi or T. brucei appears to be frequently repaired via MMEJ (85–87). What dictates the preference for HR or MMEJ in these reactions in T. brucei and related kinetoplastids is unknown. Here we show that T. brucei RAD50 and MRE11 are critical for normal cell growth and efficient homology-based DNA repair after induction of a DSB, which is consistent with a conserved role for the MRN complex in DSB recognition and repair. We demonstrate that in the absence of RAD50 or MRE11 the DDR is severely compromised, as evidenced by the loss of both γH2A accumulation and the G2/M cell-cycle checkpoint at both subtelomeric and chromosome-internal DSBs. We also show that HR is dependent on RAD50, as null mutants displayed compromised RAD51 foci formation, and that loss of RAD50 switched repair pathway choice from HR to MMEJ at a chromosome-internal DSB, an effect not seen in MRE11 mutants. We also reveal a MRE11-specific resection defect, not seen in RAD50 mutants, and limited to a VSG-ES DSB, which most likely leads to an increased defect in survival after DSB formation. In contrast, we demonstrate that RAD50 restrains the DDR at a VSG-ES DSB, as in its absence cell survival improved following break induction. Thus, this work provides insight into DSB repair pathway choice in T. brucei, and reveals a separation-of-function between components of the MRN complex, which may have relevance for the differing effects of MRE11 and RAD50 loss in Leishmania and, perhaps, wider kinetoplastids.
In T. brucei RAD51-dependent HR accounts for ∼85% of DSB repair at a chromosome-internal locus, and a DSB triggers a conventional DDR (17). The data shown here indicate that the role of T. brucei RAD50 and MRE11in the DDR is consistent with other eukaryotes (88,89). The MRN complex is a DNA damage sensor and recruit's ATM to the site of the damage (90). ATM, in return, phosphorylates H2Ax to give γH2Ax (91). In the 1HR cell lines, following a DSB, loss of RAD50 or MRE11 results in reduced γH2A and RAD51 foci, as well as loss of the G2 cell cycle checkpoint. This suggests that, as in other eukaryotic systems, the RAD50-MRE11 complex in T. brucei is required for damage sensing and recruitment of factors that facilitate HR. DNA resection following a DSB is one of the early steps in the DDR and MRN is an early acting complex in DSB processing, initiating resection (39,92) and recruiting the SGS2 nuclease, DNA2 helicase/nuclease and EXO1 exonuclease for long-range resection thereafter (39,93). The lack of resection defect in the rad50 and mre11 mutants in the 1HR mutants suggests that in trypanosomes there is flexibility in initiation of resection at a DSB, allowing unknown nucleases to act in an unrestricted manner, which has also been reported in other systems (41,42). Although these results show that RAD50 and MRE11 both sense damage, our data suggest that separation-of-function between these two proteins in T. brucei is, surprisingly, built into the function of the complex. In 1HRrad50 nulls, DSB repair no longer favours HR but instead uses MMEJ, suggesting RAD50 is required to direct repair via the HR pathway. This is not seen in the mre11 nulls. RAD50 is thought to form a ‘hook’ structure that tethers the broken ends of the chromosomes together in order to facilitate repair (45,70). In its absence, only those cells that are able to repair rapidly, via a process that does not require extensive resection, such as MMEJ, survive. In the mre11 nulls, RAD50 is still active and HR utilised, although at a much-reduced efficiency. This type of separation of function between T. brucei RAD50 and MRE11 may then be an adaptation in the parasite's MRN complex to allow MMEJ to gain prominence in genome repair, perhaps because of the absence of NHEJ. If so, such an adaptation may be widespread in kinetoplastids and other parasites that appear to lack NHEJ (24). The persistence of RPA foci in the 1HRrad50 cells is further evidence of defective repair in these cells, explained by RPA remaining associated with ssDNA at sites of damage. This observation may also reflect a damage tolerance strategy employed by T. brucei (22). In T. cruzi, damage tolerance following ionizing radiation is dependent on RAD51 (94), though this seems not to be the case in T. brucei where, in the rad50 nulls, γH2A and RAD51 are not recruited to the break.
DNA DSBs are a potent trigger for T. brucei antigenic variation and it has been shown that the subtelomeric VSG-ESs are fragile and prone to DSBs (26,27). A DSB upstream of the active VSG triggers antigenic variation, and subsequent repair by recombination is facilitated by stretches of 70-bp repeat that provide homology (9,26,27), via either a RAD51-dependent or independent pathway (27). Loss of RAD50 in T. brucei revealed a striking difference in the response to a DSB at a chromosome-internal location versus the active VSG-ES, with dramatically impaired survival at the former and improved survival at the latter. Loss of MRE11, in contrast, did not cause any difference in terms of DSB survival between the chromosomal-internal versus VSG-ES locus, but did show a locus-specific resection deficiency at a VSG-ES DSB, which might have been predicted to influence the cell's ability to undergo VSG switching. However, loss of either protein increased the range of VSGs activated after DSB formation in the VSG-ES, suggesting a concerted role for the MRN complex in managing VSG substrate choice during DSB-induced switching. How might these apparently conflicting data be explained? One explanation may be found in the data we present showing that, in trypanosomes, RAD50 appears to play a central role in driving the DDR, at both chromosomal-internal and VSG-ES loci, channelling repair via RAD51-dependent HR at a chromosomal-internal locus. At a VSG-ES 40% of repair is RAD51-independent (27), whereas MMEJ only accounts for 5% of repair at a chromosomal-internal locus (17). Therefore, at a VSG-ES, MMEJ and potentially other RAD51-independent gene conversion events may rapidly compensate for the loss of the RAD51-dependent repair pathway. The distinct effects of MRE11 and RAD50 loss during VSG-ES DSB repair may reach the same outcome in terms of VSG switching by different routes, and due to flexibility in the reactions that can lead to a change in expressed VSG. It is also possible that such flexibility is not available at a DSB in the chromosome core: MMEJ repair is more error-prone (such as by mutation or translocation) than HR (95), and the use of error-prone MMEJ after RAD50 loss may have a greater impact at a DSB in 1HR cells, since inaccurate repair could affect housekeeping genes. To date, the machinery that directs MMEJ in T. brucei or any kinetoplastid has not been characterised, and so direct tests of these suggestions are not yet possible. In addition, it remains to be shown that MMEJ directs the activation of silent array VSGs after a DSB in the VSG-ES, since the length of homologies we describe here (predominantly ∼100 bp) appear longer than the ∼5–15 bp microhomologies so far detailed in experimental characterization of T. brucei MMEJ (21,96). Nonetheless, previous work has shown considerable flexibility in the substrate requirements of recombination in T. brucei (96), and linked MMEJ and HR-mediated repair of a DSB has been detailed (21). Further work should be possible to ask if and how MRN determines the routing of DNA repair throughout the T. brucei genome.
More broadly, the increased survival and corresponding VSG switching data may also suggest an unconventional signalling pathway following a DSB at the active VSG-ES that allows the cells to bypass the conventional MRN/ATM pathway. One way that might occur is that the DSBs in the VSG-ES do not arise directly, but are generated by routes such as clashes between replication and transcription (31), or the formation of RNA-DNA hybrids (97–99), which might not elicit recruitment of MRN or ATM (100,101). Such a scenario may explain why previous work did not detect a change in VSG switching after loss of MRE11 (52). Nonetheless, our data reveals that VSGuprad50 and mre11 null cells are able to access more of the genomic VSG archive for repair, and the repair reaction in the absence of RAD50 or MRE11 occurs as a result of recombination using shorter stretches of homology. The MRN complex, therefore, appears to suppress antigenic variation by committing T. brucei to RAD51-dependent repair pathway that is reliant on long stretches of donor homology. We also observed a striking number of donor VSG genes used for repair on Chromosome 6, which is where BES1, used in these experiments, is found. Indeed, several other factors have also been shown to suppress antigenic variation: HAT3 (35), TOPO3α (32) and the telomere-interacting factor TIF2 (102). Antigenic variation following a DSB is driven by break-induced repair (26,27) and, in yeast, RAD51-independent BIR has been shown to require as little as 30 bp of homology for repair (103). Thus, loss of RAD50 or MRE11 may allow T. brucei to enlist RAD51-independent BIR to respond to a DSB in the VSG-ES, using shorter stretches of homology, rather than MMEJ. Irrespective, the question arises as to why T. brucei might limit the range of VSGs that can be used in antigenic variation, since this appears counter to the observed diversity of VSGs seen during infections (10,11). It is possible that MRN-mediated restraint of VSG recombination is needed to preserve the VSG archive, saving genes for use in prolonged infections. A more radical possibility is that control by the MRN complex underlies the hierarchy of VSG expression, directing DSB repair to telomeric VSG substrates early in an infection and then giving way to a distinctly signalled reaction, such as MMEJ, that allows access to the whole VSG archive.
DATA AVAILABILITY
All scripts are hosted on GitHub: https://github.com/LGloverTMB/DNA-repair-mutant-VSG-seq. All data has been deposited onto the ENA under study accession number: PRJEB37290 and unique study name: ena-STUDY-INSTITUT PASTEUR-15–03-2020–20:11:26:661-115. Sanger sequencing of 1HRrad50 MMEJ PCR products on Zenodo: 10.5281/zenodo.4109616.
Supplementary Material
ACKNOWLEDGEMENTS
A-K.M., M.P, A.D.-H. and L.G. performed the experiments, S.H. performed the VSG-Seq data analysis, R.M. and L.G. discussed the results and wrote the manuscript. A.-K.M., S.H., R.M. and L.G. edited the manuscript. We would also like to that David Horn for discussions and support.
Notes
Present address: Ann-Kathrin Mehnert, Centre for Infectious Diseases, Virology, Heidelberg University Hospital, Im Neuenheimer Feld 344, 69120 Heidelberg, Germany.
Contributor Information
Ann-Kathrin Mehnert, Trypanosome Molecular Biology, Department of Parasites and Insect Vectors, Institut Pasteur, 75015, Paris, France.
Marco Prorocic, Wellcome Center for Integrative Parasitology, Sir Graeme Davis Building, 120 University Place, Glasgow G12 8TA, UK.
Annick Dujeancourt-Henry, Trypanosome Molecular Biology, Department of Parasites and Insect Vectors, Institut Pasteur, 75015, Paris, France.
Sebastian Hutchinson, Trypanosome Cell Biology Unit, Department of Parasites and Insect Vectors, Institut Pasteur & INSERM U1201, 75015 Paris, France.
Richard McCulloch, Wellcome Center for Integrative Parasitology, Sir Graeme Davis Building, 120 University Place, Glasgow G12 8TA, UK.
Lucy Glover, Trypanosome Molecular Biology, Department of Parasites and Insect Vectors, Institut Pasteur, 75015, Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A-K.M. was supported by the Erasmus + program of the European Union; work in the L.G. laboratory has received financial support from the Institut Pasteur (G5 Junior group) and the National Research Agency [ANR – VSGREG]; aspects of the work in this grant was support by the Wellcome Trust [089172, 206815]; further work in R.M.’s lab was supported by the BBSRC [BB/K006495/1, BB/N016165/1]; Wellcome Centre for Integrative Parasitology is supported by core funds from the Wellcome Trust [104111]; S.H. is a Marie Curie fellow, this project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie [794979]. Funding for open access charge: Institut Pasteur G5 funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Trindade S., Rijo-Ferreira F., Carvalho T., Pinto-Neves D., Guegan F., Aresta-Branco F., Bento F., Young S.A., Pinto A., Van Den Abbeele J.et al.. Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe. 2016; 19:837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Capewell P., Cren-Travaille C., Marchesi F., Johnston P., Clucas C., Benson R.A., Gorman T.A., Calvo-Alvarez E., Crouzols A., Jouvion G.et al.. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016; 5:e17716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jamonneau V., Ilboudo H., Kabore J., Kaba D., Koffi M., Solano P., Garcia A., Courtin D., Laveissiere C., Lingue K.et al.. Untreated human infections by Trypanosoma brucei gambiense are not 100% fatal. PLoS Negl Trop Dis. 2012; 6:e1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baltz T., Baltz D., Pautrizel R., Richet C., Lamblin G., Degand P.. Chemical and immunological characterization of specific glycoproteins from Trypanosoma equiperdum variants. FEBS Lett. 1977; 82:93–96. [DOI] [PubMed] [Google Scholar]
- 5. Diffley P. Trypanosoma brucei: immunogenicity of the variant surface coat glycoprotein of virulent and avirulent subspecies. Exp. Parasitol. 1985; 59:98–107. [DOI] [PubMed] [Google Scholar]
- 6. Schwede A., Jones N., Engstler M., Carrington M.. The VSG C-terminal domain is inaccessible to antibodies on live trypanosomes. Mol. Biochem. Parasitol. 2011; 175:201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sima N., McLaughlin E.J., Hutchinson S., Glover L.. Escaping the immune system by DNA repair and recombination in African trypanosomes. Open Biol. 2019; 9:190182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hertz-Fowler C., Figueiredo L.M., Quail M.A., Becker M., Jackson A., Bason N., Brooks K., Churcher C., Fahkro S., Goodhead I.et al.. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One. 2008; 3:e3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hovel-Miner G., Mugnier M.R., Goldwater B., Cross G.A., Papavasiliou F.N.. A conserved DNA repeat promotes selection of a diverse repertoire of Trypanosoma brucei surface antigens from the genomic archive. PLoS Genet. 2016; 12:e1005994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mugnier M.R., Cross G.A., Papavasiliou F.N.. The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science. 2015; 347:1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall J.P., Wang H., Barry J.D.. Mosaic VSGs and the scale of Trypanosoma brucei antigenic variation. PLoS Pathog. 2013; 9:e1003502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jayaraman S., Harris C., Paxton E., Donachie A.M., Vaikkinen H., McCulloch R., Hall J.P.J., Kenny J., Lenzi L., Hertz-Fowler C.et al.. Application of long read sequencing to determine expressed antigen diversity in Trypanosoma brucei infections. PLoS Negl. Trop. Dis. 2019; 13:e0007262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cross G.A., Kim H.S., Wickstead B.. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol. Biochem. Parasitol. 2014; 195:59–73. [DOI] [PubMed] [Google Scholar]
- 14. Marcello L., Barry J.D.. Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res. 2007; 17:1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muller L.S.M., Cosentino R.O., Forstner K.U., Guizetti J., Wedel C., Kaplan N., Janzen C.J., Arampatzi P., Vogel J., Steinbiss S.et al.. Genome organization and DNA accessibility control antigenic variation in trypanosomes. Nature. 2018; 563:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D.C., Lennard N.J., Caler E., Hamlin N.E., Haas B.et al.. The genome of the African trypanosome Trypanosoma brucei. Science. 2005; 309:416–422. [DOI] [PubMed] [Google Scholar]
- 17. Glover L., McCulloch R., Horn D.. Sequence homology and microhomology dominate chromosomal double-strand break repair in African trypanosomes. Nucleic Acids Res. 2008; 36:2608–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCulloch R., Barry J.D.. A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev. 1999; 13:2875–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson N.P., Burman N., Melville S.E., Barry J.D.. Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol. Cell. Biol. 1999; 19:5839–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conway C., Proudfoot C., Burton P., Barry J.D., McCulloch R.. Two pathways of homologous recombination in Trypanosoma brucei. Mol. Microbiol. 2002; 45:1687–1700. [DOI] [PubMed] [Google Scholar]
- 21. Glover L., Jun J., Horn D.. Microhomology-mediated deletion and gene conversion in African trypanosomes. Nucleic Acids Res. 2011; 39:1372–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Glover L., Marques C.A., Suska O., Horn D.. Persistent DNA damage Foci and DNA replication with a broken chromosome in the African Trypanosome. MBio. 2019; 10:e01252-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burton P., McBride D.J., Wilkes J.M., Barry J.D., McCulloch R.. Ku heterodimer-independent end joining in Trypanosoma brucei cell extracts relies upon sequence microhomology. Eukaryot Cell. 2007; 6:1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nenarokova A., Zahonova K., Krasilnikova M., Gahura O., McCulloch R., Zikova A., Yurchenko V., Lukes J.. Causes and effects of loss of classical nonhomologous end joining pathway in Parasitic Eukaryotes. MBio. 2019; 10:e01541-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shibata A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 2017; 803–805:51–55. [DOI] [PubMed] [Google Scholar]
- 26. Boothroyd C.E., Dreesen O., Leonova T., Ly K.I., Figueiredo L.M., Cross G.A., Papavasiliou F.N.. A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature. 2009; 459:278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Glover L., Alsford S., Horn D.. DNA break site at fragile subtelomeres determines probability and mechanism of antigenic variation in African trypanosomes. PLoS Pathog. 2013; 9:e1003260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Proudfoot C., McCulloch R.. Distinct roles for two RAD51-related genes in Trypanosoma brucei antigenic variation. Nucleic Acids Res. 2005; 33:6906–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trenaman A., Hartley C., Prorocic M., Passos-Silva D.G., van den Hoek M., Nechyporuk-Zloy V., Machado C.R., McCulloch R.. Trypanosoma brucei BRCA2 acts in a life cycle-specific genome stability process and dictates BRC repeat number-dependent RAD51 subnuclear dynamics. Nucleic Acids Res. 2013; 41:943–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hartley C.L., McCulloch R.. Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination. Mol. Microbiol. 2008; 68:1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Devlin R., Marques C.A., Paape D., Prorocic M., Zurita-Leal A.C., Campbell S.J., Lapsley C., Dickens N., McCulloch R.. Mapping replication dynamics in Trypanosoma brucei reveals a link with telomere transcription and antigenic variation. Elife. 2016; 5:e12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim H.S., Cross G.A.. TOPO3alpha influences antigenic variation by monitoring expression-site-associated VSG switching in Trypanosoma brucei. PLoS Pathog. 2010; 6:e1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim H.S., Cross G.A.. Identification of Trypanosoma brucei RMI1/BLAP75 homologue and its roles in antigenic variation. PLoS One. 2011; 6:e25313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Black J.A., Crouch K., Lemgruber L., Lapsley C., Dickens N., Tosi L.R.O., Mottram J.C., McCulloch R.. Trypanosoma brucei ATR links DNA damage signaling during antigenic variation with regulation of RNA polymerase I-transcribed surface antigens. Cell Rep. 2020; 30:836–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Glover L., Horn D.. Locus-specific control of DNA resection and suppression of subtelomeric VSG recombination by HAT3 in the African trypanosome. Nucleic Acids Res. 2014; 42:12600–12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Syed A., Tainer J.A.. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018; 87:263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lisby M., Barlow J.H., Burgess R.C., Rothstein R.. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004; 118:699–713. [DOI] [PubMed] [Google Scholar]
- 39. Myler L.R., Gallardo I.F., Soniat M.M., Deshpande R.A., Gonzalez X.B., Kim Y., Paull T.T., Finkelstein I.J.. Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol. Cell. 2017; 67:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paull T.T., Gellert M.. The 3' to 5' exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell. 1998; 1:969–979. [DOI] [PubMed] [Google Scholar]
- 41. Llorente B., Symington L.S.. The Mre11 nuclease is not required for 5' to 3' resection at multiple HO-induced double-strand breaks. Mol. Cell. Biol. 2004; 24:9682–9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoa N.N., Akagawa R., Yamasaki T., Hirota K., Sasa K., Natsume T., Kobayashi J., Sakuma T., Yamamoto T., Komatsu K.et al.. Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells. 2015; 20:1059–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Y., Zhou J., Lim C.U.. The role of NBS1 in DNA double strand break repair, telomere stability, and cell cycle checkpoint control. Cell Res. 2006; 16:45–54. [DOI] [PubMed] [Google Scholar]
- 44. Paudyal S.C., You Z.. Sharpening the ends for repair: mechanisms and regulation of DNA resection. Acta Biochim. Biophys. Sin. (Shanghai). 2016; 48:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hopfner K.P., Karcher A., Shin D.S., Craig L., Arthur L.M., Carney J.P., Tainer J.A.. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000; 101:789–800. [DOI] [PubMed] [Google Scholar]
- 46. Oh J., Symington L.S.. Role of the Mre11 complex in preserving genome integrity. Genes. 2018; 9:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Longhese M.P. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008; 22:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Limbo O., Moiani D., Kertokalio A., Wyman C., Tainer J.A., Russell P.. Mre11 ATLD17/18 mutation retains Tel1/ATM activity but blocks DNA double-strand break repair. Nucleic Acids Res. 2012; 40:11435–11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yin Y., Smolikove S.. Impaired resection of meiotic double-strand breaks channels repair to nonhomologous end joining in Caenorhabditis elegans. Mol. Cell. Biol. 2013; 33:2732–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cassani C., Vertemara J., Bassani M., Marsella A., Tisi R., Zampella G., Longhese M.P.. The ATP-bound conformation of the Mre11-Rad50 complex is essential for Tel1/ATM activation. Nucleic Acids Res. 2019; 47:3550–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deshpande R.A., Williams G.J., Limbo O., Williams R.S., Kuhnlein J., Lee J.H., Classen S., Guenther G., Russell P., Tainer J.A.et al.. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014; 33:482–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robinson N.P., McCulloch R., Conway C., Browitt A., Barry J.D.. Inactivation of Mre11 does not affect VSG gene duplication mediated by homologous recombination in Trypanosoma brucei. J. Biol. Chem. 2002; 277:26185–26193. [DOI] [PubMed] [Google Scholar]
- 53. Tan K.S., Leal S.T., Cross G.A.. Trypanosoma brucei MRE11 is non-essential but influences growth, homologous recombination and DNA double-strand break repair. Mol. Biochem. Parasitol. 2002; 125:11–21. [DOI] [PubMed] [Google Scholar]
- 54. Laffitte M.C., Genois M.M., Mukherjee A., Legare D., Masson J.Y., Ouellette M.. Formation of linear amplicons with inverted duplications in Leishmania requires the MRE11 nuclease. PLos Genet. 2014; 10:e1004805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Laffitte M.C., Leprohon P., Hainse M., Legare D., Masson J.Y., Ouellette M.. Chromosomal translocations in the parasite leishmania by a MRE11/RAD50-independent microhomology-mediated end joining mechanism. PLos Genet. 2016; 12:e1006117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hirumi H., Hirumi K.. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989; 75:985–989. [PubMed] [Google Scholar]
- 57. van den Hoff M.J., Moorman A.F., Lamers W.H.. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Res. 1992; 20:2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alsford S., Horn D.. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol. Biochem. Parasitol. 2008; 161:76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Devlin R., Marques C.A., Paape D., Prorocic M., Zurita-Leal A.C., Campbell S.J., Lapsley C., Dickens N., McCulloch R.. Mapping replication dynamics in Trypanosoma brucei reveals a link with telomere transcription and antigenic variation. eLife. 2016; 5:e12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jeacock L., Baker N., Wiedemar N., Maser P., Horn D.. Aquaglyceroporin-null trypanosomes display glycerol transport defects and respiratory-inhibitor sensitivity. PLoS Pathog. 2017; 13:e1006307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alsford S., Horn D.. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol. Biochem. Parasitol. 2008; 161:76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Glover L., Horn D.. Trypanosomal histone gammaH2A and the DNA damage response. Mol. Biochem. Parasitol. 2012; 183:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Woodward R., Gull K.. Timing of nuclear and kinetoplast DNA replication and early morphological events in the cell cycle of Trypanosoma brucei. J. Cell Sci. 1990; 95:49–57. [DOI] [PubMed] [Google Scholar]
- 64. Zhou Y., Caron P., Legube G., Paull T.T.. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014; 42:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zierhut C., Diffley J.F.. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008; 27:1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 69. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hopfner K.P., Karcher A., Craig L., Woo T.T., Carney J.P., Tainer J.A.. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001; 105:473–485. [DOI] [PubMed] [Google Scholar]
- 71. Hopfner K.P., Craig L., Moncalian G., Zinkel R.A., Usui T., Owen B.A., Karcher A., Henderson B., Bodmer J.L., McMurray C.T.et al.. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002; 418:562–566. [DOI] [PubMed] [Google Scholar]
- 72. Williams R.S., Williams J.S., Tainer J.A.. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell. Biol. 2007; 85:509–520. [DOI] [PubMed] [Google Scholar]
- 73. De la Rosa M.B., Nelson S.W.. An interaction between the Walker A and D-loop motifs is critical to ATP hydrolysis and cooperativity in bacteriophage T4 Rad50. J. Biol. Chem. 2011; 286:26258–26266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jackson S.P., Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Siegel T.N., Hekstra D.R., Cross G.A.. Analysis of the Trypanosoma brucei cell cycle by quantitative DAPI imaging. Mol. Biochem. Parasitol. 2008; 160:171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Woodward R., Gull K.. Timing of nuclear and kinetoplast DNA replication and early morphological events in the cell cycle of Trypanosoma brucei. J. Cell Sci. 1990; 95:49–57. [DOI] [PubMed] [Google Scholar]
- 78. Stauffer M.E., Chazin W.J.. Physical interaction between replication protein A and Rad51 promotes exchange on single-stranded DNA. J. Biol. Chem. 2004; 279:25638–25645. [DOI] [PubMed] [Google Scholar]
- 79. McWilliam K.R., Ivens A., Morrison L.J., Mugnier M.R., Matthews K.R.. Developmental competence and antigen switch frequency can be uncoupled in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:22774–22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Glover L., Hutchinson S., Alsford S., McCulloch R., Field M.C., Horn D.. Antigenic variation in African trypanosomes: the importance of chromosomal and nuclear context in VSG expression control. Cell. Microbiol. 2013; 15:1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Moreau S., Ferguson J.R., Symington L.S.. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol. 1999; 19:556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Connelly J.C., Leach D.R.. Tethering on the brink: the evolutionarily conserved Mre11-Rad50 complex. Trends Biochem. Sci. 2002; 27:410–418. [DOI] [PubMed] [Google Scholar]
- 83. McCulloch R., Morrison L.J., Hall J.P.. DNA recombination strategies during antigenic variation in the African Trypanosome. Microbiol. Spectr. 2015; 3:doi:10.1128/microbiolspec.MDNA3-0016-2014. [DOI] [PubMed] [Google Scholar]
- 84. Weterings E., Chen D.J.. The endless tale of non-homologous end-joining. Cell Res. 2008; 18:114–124. [DOI] [PubMed] [Google Scholar]
- 85. Rico E., Jeacock L., Kovarova J., Horn D.. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in African trypanosomes. Sci. Rep. 2018; 8:7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Waldrip Z.J., Byrum S.D., Storey A.J., Gao J., Byrd A.K., Mackintosh S.G., Wahls W.P., Taverna S.D., Raney K.D., Tackett A.J.. A CRISPR-based approach for proteomic analysis of a single genomic locus. Epigenetics. 2014; 9:1207–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang W.W., Matlashewski G.. CRISPR-Cas9-mediated genome editing in Leishmania donovani. MBio. 2015; 6:e00861-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Grenon M., Gilbert C., Lowndes N.F.. Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat. Cell Biol. 2001; 3:844–847. [DOI] [PubMed] [Google Scholar]
- 89. Marechal A., Zou L.. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013; 5:a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lee J.H., Paull T.T.. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005; 308:551–554. [DOI] [PubMed] [Google Scholar]
- 91. Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J.. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001; 276:42462–42467. [DOI] [PubMed] [Google Scholar]
- 92. Westmoreland J., Ma W., Yan Y., Van Hulle K., Malkova A., Resnick M.A.. RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma-induced double-strand break ends. PLoS Genet. 2009; 5:e1000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G.. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008; 134:981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gomes Passos Silva D., da Silva Santos S., Nardelli S.C., Mendes I.C., Freire A.C.G., Repoles B.M., Resende B.C., Costa-Silva H.M., da Silva V.S., Oliveira K.A.et al.. The in vivo and in vitro roles of Trypanosoma cruzi Rad51 in the repair of DNA double strand breaks and oxidative lesions. PLoS Negl. Trop. Dis. 2018; 12:e0006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Seol J.H., Shim E.Y., Lee S.E.. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2018; 809:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Barnes R.L., McCulloch R.. Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases. Nucleic Acids Res. 2007; 35:3478–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Briggs E., Crouch K., Lemgruber L., Lapsley C., McCulloch R.. Ribonuclease H1-targeted R-loops in surface antigen gene expression sites can direct trypanosome immune evasion. PLoS Genet. 2018; 14:e1007729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Briggs E., Crouch K., Lemgruber L., Hamilton G., Lapsley C., McCulloch R.. Trypanosoma brucei ribonuclease H2A is an essential R-loop processing enzyme whose loss causes DNA damage during transcription initiation and antigenic variation. Nucleic Acids Res. 2019; 47:9180–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chang E.Y., Tsai S., Aristizabal M.J., Wells J.P., Coulombe Y., Busatto F.F., Chan Y.A., Kumar A., Dan Zhu Y., Wang A.Y.et al.. MRE11-RAD50-NBS1 promotes Fanconi anemia R-loop suppression at transcription-replication conflicts. Nat. Commun. 2019; 10:4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Balestrini A., Nicolas L., Yang-Lott K., Guryanova O.A., Levine R.L., Bassing C.H., Chaudhuri J., Petrini J.H.. Defining ATM-Independent functions of the Mre11 complex with a novel mouse model. Mol. Cancer Res. 2016; 14:185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M.. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011; 145:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jehi S.E., Nanavaty V., Li B.. Trypanosoma brucei TIF2 and TRF suppress VSG switching using overlapping and independent mechanisms. PLoS One. 2016; 11:e0156746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ira G., Haber J.E.. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell. Biol. 2002; 22:6384–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scripts are hosted on GitHub: https://github.com/LGloverTMB/DNA-repair-mutant-VSG-seq. All data has been deposited onto the ENA under study accession number: PRJEB37290 and unique study name: ena-STUDY-INSTITUT PASTEUR-15–03-2020–20:11:26:661-115. Sanger sequencing of 1HRrad50 MMEJ PCR products on Zenodo: 10.5281/zenodo.4109616.