


Abstract
Sensing of environmental cues is crucial for cell survival. To adapt to changes in their surroundings cells need to tightly control the repertoire of genes expressed at any time. Regulation of translation is key, especially in organisms in which transcription is hardly controlled, like Trypanosoma brucei. In this study, we describe the shortening of the bulk of the cellular tRNAs during stress at the expense of the conserved 3′ CCA-tail. This tRNA shortening is specific for nutritional stress and renders tRNAs unsuitable substrates for translation. We uncovered the nuclease LCCR4 (Tb927.4.2430), a homologue of the conserved deadenylase Ccr4, as being responsible for tRNA trimming. Once optimal growth conditions are restored tRNAs are rapidly repaired by the trypanosome tRNA nucleotidyltransferase thus rendering the recycled tRNAs amenable for translation. This mechanism represents a fast and efficient way to repress translation during stress, allowing quick reactivation with a low energy input.
INTRODUCTION
Organisms are constantly subjected to environmental challenges to which they need to adequately adapt. To do that they have to adjust the repertoire of genes that are expressed at any given time. When these environmental changes happen slowly the cell can gradually adapt but sudden changes in growth conditions or the encounter of specific stress situations require a more immediate response. Examples for the latter are faced by organisms that transit through different hosts during their life cycle like the parasitic protozoon Trypanosoma brucei. This parasite lives in the bloodstream of mammalian hosts, where it feeds on glucose, has to evade a mammalian immune response and experiences a constant temperature of 37°C. The living conditions change drastically inside the tsetse fly (the insect vector), where T. brucei uses amino acids as energy source and experiences fluctuations in the growth temperature (1).
Most organisms are able to modulate gene expression by regulating the transcription of individual genes (2). Trypanosomes though face a special challenge as all their genes are organized in long polycistronic units and transcribed as pre-mRNAs that are processed by coupled trans-splicing and polyadenylation (3). Therefore they rely mainly on posttranscriptional mechanisms to control gene expression (reviewed in 4). A crucial step in gene expression is translation, in which the ribosome decodes the information contained in the mRNA to drive the synthesis of proteins. Important players in this process are tRNAs, small non-coding RNA molecules that serve as adaptors for translation (5). tRNAs are highly conserved molecules that have a very defined secondary and tertiary structure (6). Even though individual tRNA species that are able to decode specific mRNA codons have different primary as well as secondary structures, they all adopt a characteristic L-shaped three-dimensional architecture capable of binding to and transiting through the three tRNA binding sites on the ribosome. While the anticodon loop on one end of the L-shaped tRNA molecule pairs with the mRNA codon the acceptor stem on the other contains the universally conserved CCA-tail that serves as platform for amino acid attachment. Because translation is one of the most energy consuming processes in the cell, many strategies have evolved to inhibit global translation during stress conditions thus preserving crucial cellular energy resources (recently reviewed in 7). Among them the sequestration of mRNAs and ribosomal subunits in stress granules and the inhibition of cap-dependent translation initiation by phosphorylation of eIF2a are the most widely studied and thus best understood ones. In the last years also the direct deactivation of tRNAs by removal of the CCA-tail has been reported in mammalian cells exposed to oxidative stress (8,9). Although in vitro data analysis indicated that angiogenin was involved in CCA-tail removal a recent study suggests that this is not the case in vivo (DOI 10.1101/811174). Therefore, the identity of the involved nuclease(s) and its regulation remain unknown.
tRNAs are transcribed as precursors containing extensions at both ends that need to be processed. The 5′ extension is removed by the conserved activity of RNaseP (10), while the 3′ trailer can be removed by different RNases, the main one in eukaryotes being RNaseZ (11). Upon removal of the 3′ trailer terminal nucleotidyl transferases (also known as CCA-adding enzymes) add the non-templated CCA-tail to all eukaryal tRNAs (12,13). In most bacteria tRNA genes already encode the CCA-tails and therefore these enzymes are only involved in repairing damaged tRNAs (14).
Here, we report the 3′ CCA shortening of the bulk of the tRNAs in T. brucei parasites exposed to nutritional stress. This shortening is due to the trimming of the conserved 3′ CCA-tail, which renders tRNAs unsuitable substrates for translation. Depending on the tRNA the CCA-tail can be trimmed in two consecutive steps resulting in the complete removal of the trinucleotide tail. In this study, we revealed LCCR4 (Tb927.4.2430), the homologue of the conserved deadenylase Ccr4, to be responsible for the observed tRNA 3′ trimming in T. brucei. Once normal growth conditions are restored the shortened tRNAs are rapidly repaired by the CCA-adding enzyme thus converting them again into vital substrates for a productive translation machinery.
MATERIALS AND METHODS
Cell lines and culture
Trypanosoma brucei Lister 427 wild-type, 29-13 or NYSM (New York single markers) cell lines were used in all experiments. Procyclic stage Lister 427 wild-type and 29-13 cells were grown at 27°C in SDM-79 media containing 5% or 10% Fetal bovine serum (FBS), respectively. Exponentially growing T. brucei cells were harvested at a density of 10–20 × 106 cells per ml, whereas T. brucei cells in stationary phase were harvested at a density of 60–70 × 106 cells/ml. Stress conditions included heat shock, cold shock and nutritional stress. For heat shock, exponentially growing cells were pelleted and resuspended in 41°C SDM-79 containing 5% FCS followed by incubation for 30 min at 41°C. For cold shock, exponentially growing cells were harvested as mentioned before and resuspended in 13°C SDM-79 containing 5% FCS and incubated for 30 min at the same temperature. In the case of nutritional stress, exponentially growing T. brucei cells were centrifuged, washed in chilled 1× PBS (phosphate buffered saline) and resuspended in pre-warmed 1× PBS at 27°C. After 2 h of nutritional stress, T. brucei cells were harvested.
The bloodstream form of T. brucei strain NYSM was cultured in HMI-9 supplemented with 10% FBS at 37°C/5% CO2. Exponentially growing cells of the T. brucei bloodstream form were harvested at a density of 106 cells per ml and nutritional stress was applied by growing the cells for 1 h in 1× PBS.
Northern blot analysis
For northern blot analysis 5–20 μg of total RNA extracted using the TRI Reagent (Zymo Research) according to the manufacturer's instructions was mixed with RNA loading buffer and separated in denaturing polyacrylamide gels (7 M urea, 1× TBE and 8 or 12% polyacrylamide, depending on the resolution required). Afterwards RNA was blotted to nylon membranes (Amershan Hybond N+, GE Healthcare) and hybridized as described (15). The sequences of the oligonucleotides used are described in Supplementary Table S1.
Western blot analysis
For western blot analysis total cell lysates or fractions obtained during affinity purification were separated in 10% SDS-PAGE gels.
To investigate the levels of CCA-adding enzyme (CAE) during exponential growth or upon stress cells were pelleted, washed with 1× PBS and cell pellets directly resuspended in Laemmli buffer. The corresponding volume to 2 × 106 cells was run per lane. For analysis of affinity purifications fractions the corresponding to 2–3 × 106 cells of input lysates and flow-through fractions and 2 × 107 cells of final beads were run per lane. Gels were transferred onto a nitrocellulose membrane (Amersham Biosciences) and blocked for 1 h in 1× PBS/0.1% Tween-20 containing 5% nonfat milk. The membranes were incubated with mouse anti–HA antibody (1:5000, Covane) at 4°C overnight, washed (3 × 10 min each with 1× PBS/0.1% Tween-20) and horseradish peroxidase-conjugated secondary antibodies were added (1:3000; Roche). The membranes were washed as before and results were visualized using an enhanced chemiluminescence SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific). As loading control EF1A was detected as before using specific antibodies (1:10 000, Santa Cruz).
Constructs for protein tagging and RNAi
Tetracycline-inducible expression of C-terminally HA-tagged LCCR4 (Tb927.4.2430) and CAF1 (Tb927.6.600) was done using pLew100-derived constructs carrying the puromycin resistance marker and three copies of the HA epitope (16,17). The complete LCCR4 ORF was amplified using oligos 133 and 139, while the complete CAF1 ORF was amplified with oligos 143 and 144. The constructs were transfected in 427-derived cells expressing the T7 RNA polymerase and the Tet repressor from a stably integrated plasmid.
For N-terminal in situ tagging of the CCA-adding enzyme (Tb927.9.8780) the most 5′ 356 nt of the ORF were amplified using oligos 40 and 41 and cloned in frame downstream of a single HA epitope in vector pPURO-HA containing a Puromycin resistance cassette (18). The final construct was digested using EcoRV and transfected into 29–13 procyclic T. brucei cells. Homologous recombination results in tagging one allele of the CCA-adding enzyme.
Generation of the cell lines allowing inducible expression of the tagged tRNAGlu and RNAi of the CCA-adding enzyme have been previously described (19,20).
All RNAi cell lines were stem-loop constructs based on pLew100 carrying the blasticidin resistance gene (16,17). For the LCCR4 RNAi construct the region comprising bases 731 to 1235 were amplified using oligonucleotides 99 and 100 and used as RNAi target, while for CAF1 RNAi the region between bases 416 to 914 were targeted by using oligos 151 and 152.
To generate the LCCR4 N454A mutant codon AAC was changed to GCC by overlap extension PCR. For that purpose amplification was done using genomic DNA as template and primer pairs 133/166 and 139/165. The products of both PCRs were purified, combined and reamplified using oligos 133/139.
Pulse chase of endogenous RNA using 32P-UTP
For labelling of endogenous RNA molecules cells were grown in SDM-79 media containing 3 mCi/ml of α32P-UTP (6000 Ci/mmol) and let grow for 2 days. After this time cells were washed with normal media and resuspended in media containing a 1000-fold excess of cold UTP (277 nM). After 2 h of incubation a sample was taken and the rest was stressed by incubation in 1× PBS for 2 h. Another sample was taken and remaining cells were recovered in normal media containing an excess of cold UTP (277 nM) for 2 h. Cells were harvested, RNA was extracted from all samples and separated in a 8% polyacrylamide/urea gel. After drying, the gel was exposed to a phosphorimager screen that was scanned using a Typhoon system.
Production of tRNAVal transcripts
To generate the different tRNAValin vitro the gene was first amplified from genomic DNA using forward oligonucleotide 43 in combination with reverse oligonucleotide 51 (no CCA tail), 124 (CC tail) or 45 (complete CCA tail). The clean PCR products were used as template for in vitro transcription as described in (21). For 5′ radiolabeling the tRNA containing the CCA tail was dephosphorylated using alkaline phosphatase and finally phosphorylated using polynucleotide kinase and γ32P-ATP. The presence of double bands in polyacrylamide gels (Supplementary Figure S4A) is most probably due to the untemplated addition of nucleotides by the T7 RNA polymerase at the 3′ termini as reported previously (22,23).
In vitro tRNA cleavage
To test tRNA cleavage activity in total cell lysates 1–2 × 109 cells growing exponentially or subjected to nutritional stress were harvested and frozen in liquid nitrogen. After thawing on ice cell pellets were resuspended in 1ml of ribosome buffer A (20 mM Tris pH 7.6, 120 mM KCl, 2 mM MgCl2) and frozen and thawed as before. Cells were passed 10 times through a G25 needle, 10 times through a G27 needle and finally cell debris removed by centrifugation at maximal speed for 15 min.
The volume corresponding to 3 × 107 cells was incubated with the 5′ radiolabeled tRNAVal (15 000–20 000 cpm) at 30°C for the indicated times and finally the RNA was extracted using phenol/chloroform and ethanol precipitated. The RNA samples were then separated on 8% polyacrylamide/urea gels, dried and exposed to phosphorimager screens for detection.
For RNase inhibition tests in vitro cleavage reactions were supplemented with 80 U of Ribolock (ThermoFischer Scientific), 80 U of Protector (Roche) or 20 mM ribonucleoside vanadyl complex (RVC) and processed as described above.
Purification and in vitro activity of the CCA-adding enzyme
Cells were grown either under exponential or stress conditions, harvested, washed with 1× PBS and resuspended in lysis buffer (20 mM Tris pH 7.7, 3 mM MgCl2, 0.1% NP40, 1 mM DTT and 20 mM KCl) containing protease inhibitors (Roche) at a density of 109 cells/ml of lysis buffer. After 5 min incubation on ice KCl was added to a final concentration of 150 mM, the lysate was cleared by centrifugation for 15 min at maximal speed and then incubated with pre-washed anti HA beads (Roche). After 2 h of rotation the beads were washed with lysis buffer containing 150 mM KCl, then with the same buffer containing 500 mM KCl and finally washed and equilibrated with CCA-adding enzyme reaction buffer (30 mM HEPES/KOH, pH 7.6, 6 mM MgCl2, 30 mM KCl and 2 mM DTT). To determine the amount of bead-bound protein to be used in the reaction a fraction of the beads was boiled in Laemmli buffer and tested by western blot using anti HA antibodies as described. The volume of beads containing the same amounts of CCA-adding enzyme from exponentially or stressed cells was incubated with 700 ng of gel extracted total tRNAs (app. 30 pmol) from stressed cells as substrate and 50 pmol of CTP supplemented with 5 μCi of α 32P-CTP in a total volume of 20 μl in CCA-adding enzyme reaction buffer. Reactions were incubated at 30°C and after the indicated times the reactions were stopped by addition of phenol/chloroform. Finally, RNA was precipitated using ethanol, resuspended in RNA loading buffer, separated on polyacrylamide/urea gels, dried, exposed to phosphorimager screens and imaged.
RIP
Cells expressing HA-tagged LCCR4 were induced for 3 days and allowed to grow exponentially or stressed. For crosslinking paraformaldehyde was added to the cells to a final concentration of 0.1% and incubated 8 min at 27°C with shaking. Finally, glycine was added to 125 mM, cells were incubated for 5 min and harvested. Cell pellets were resuspended in buffer containing 20 mM Tris–HCl pH 7.7, 140 mM, 1.8 mM MgCl2, 0.1% NP40, 10% glycerol, 1 mM DTT, protease inhibitor (Roche) and 2 mM ribonucleoside vanadyl complex (RVC) and lysed by sonicaton. Cell debris was removed by centrifugation and HA-tagged LCCR4 purified on anti HA beads as described. After washing the beads were treated with RNase-free DNaseI for 10 min on ice followed by de-crosslinking in buffer containing 20 mM Tris–HCl pH 7.7, 5 mM EDTA, 50 mM NaCl and 0.1% SDS and proteins degraded by treatment with proteinase K (50 μg/ml) at 70°C for 40 min. Before proteolytic treatment samples were taken for western blot to assess the success of the purification. RNA was finally isolated using phenol/chloroform, ethanol precipitated and resuspended in water. RNA (800 ng for input and flow through and 50 ng for final sample from LCCR4 from exponential growing cells and the sample equivalents for the rest) were reverse transcribed using Superscript IV (ThermoFischer Scientific) following the manufacturer's instructions using oligos specific for tRNAVal and M6 rRNA (51 and 39 respectively). Finally, 2 μl of the reverse transcription reaction were used as template for PCR (using oligos 51/43 for tRNAVal and 39/153 for M6 rRNA).
In vitro cleavage activity of LCCR4
Cells that allow overexpression of LCCR4 carrying a C-terminal HA tag were induced or left untreated for 2 days. After this, they were either stressed by incubation on 1× PBS for 2 h or grown exponentially. Cells were then lysed and the tagged LCCR4 purified as for the CCA-adding enzyme. To test its activity the beads containing the LCCR4 enzyme still attached were washed with ribosome buffer A, resuspended in the same buffer and small aliquots incubated with 5 μg of total RNA purified either from exponentially growing cells or from LCCR4 RNAi cells that were induced for 2 days and stressed. Reactions were incubated at 30°C and after the indicated time points beads were removed by centrifugation. Finally, RNA was extracted using phenol/chloroform, precipitated, resuspended in RNA loading buffer, separated on 12% polyacrylamide/urea gels, and analyzed by northern blot.
Aminoacylation
To generate S100 fractions cell lysates from exponentially growing or nutritionally stressed cells were prepared as for in vitro tRNA cleavage in ribosome buffer A containing 20 mM RVC. The volume of lysate corresponding to 1.5 × 109 cells (∼500 μl) was loaded onto 800 μl of a 1.1 M sucrose cushion prepared on ribosome buffer A and centrifuged for 2.5 h at 200 000 × g and 4°C. The supernatant corresponds to S100 fractions. Aminoacylation reactions were prepared in aminoacylation buffer (50 mM HEPES pH 7.5, 1 mM DTT, 0.5 mM EDTA, 10 mM KCl, 10 mM Mg acetate and 4 mM ATP) containing 0.4 mM 14C labeled arginine (l-arginine, [14C(U)], 312 mCi/mmol) and 5 μl of S100. After 30 min incubation at 30°C samples were put on a glass fiber filter and sequentially washed for 5 min with 10% trichloroacetic acid (TCA), 2 min with 5% TCA and 2 min with ethanol. Filters were air dried, mixed with scintillation liquid and the radioactive signal counted in a scintillation counter.
Metabolic labeling of proteins
Analysis of in vivo translation was done as described before (15). Briefly, cells (107/ml) were grown in SDM-79 media supplemented with 35S-methionine (2 μl/ml of cell culture). After incubation for the indicated times at 27°C cells were washed with 1× PBS and resuspended in Laemmli buffer for PAGE analyses.
RESULTS
Nutritional stress causes shortening of most tRNAs
Our recent studies have shown that in T. brucei tRNA fragments accumulate upon cellular stress (15). This observation was made by analyzing cDNA libraries encoding small RNAs associated with ribosomes. In these libraries, though, we were unable to detect any full length tRNA, a common phenomenon mainly due to the fact that tRNAs are highly structured and post-transcriptionally modified and therefore not susceptible to efficient reverse transcription (24). To study whether stress also causes other alterations to full length tRNAs we performed comprehensive northern blot analyses using probes that allow the detection of as many tRNA isoacceptors as possible. Many of the tRNAs analyzed showed a faster migration in denaturing polyacrylamide gels in samples isolated from cells exposed to nutritional stress (Figure 1A). This effect was not only detected in the procyclic stage of T. brucei but also when the bloodstream form of the parasites was exposed to nutritional stress, suggesting a central underlying mechanism in both life stages (Supplementary Figure S1A). To test if this faster migration was due to a shortening of the tRNAs we performed 3′ RACE analyses and investigated one of the most affected tRNAs, tRNAVal. Our results indicate that the faster migration is due to the shortening of the tRNAs at the 3′ end, therefore affecting the universally conserved CCA-tail. It is worth mentioning that although the tRNAThr seems to behave differently upon nutritional stress this is only due to an aberrant faster migration of the tRNA containing the full CCA-tail, as previously described in (15). The fact that the fast migration of the tRNAs can already be visualized by staining gels with ethidium bromide indicates that this is a comprehensive phenomenon affecting most tRNA species (Figure 1B, Supplementary Figure S1A). In order to have a more quantitative insight on the shortening process we made use of the ability of T4 DNA ligase to ligate a short fluorescent RNA-DNA hybrid to full length tRNAs by complementarity to complete CCA-tails (25). Supplementary Figure S1B shows the appearance of an extra band in ethidium bromide stained gels that corresponds to the ligated fluorescent full length tRNAs. Quantification of the intensity of the bands shows that upon nutritional stress 70% of the tRNAs were not susceptible to ligation, suggesting that their CCA-tails are not intact. This observation was specific to nutritional stress and was not apparent during other stress conditions investigated (Supplementary Figure S1C). For the following investigations we decided to use tRNAVal as proxy for tRNA shortening as it is one of the tRNAs that is trimmed most efficiently.
Figure 1.

tRNAs are trimmed upon stress and repaired once normal growth conditions are restored. (A) Northern blot analyses of tRNAs on total RNA extracted from T. brucei. RNA was isolated from procyclic T. brucei cells either during the exponential growth phase (Exponential) or after heat shock, nutritional deprivation (Nutr. stress), oxidative stress, during the stationary phase or from exponentially growing bloodstream form (Exponential BSF). Full length tRNAs and 3′ trimmed tRNAs are indicated with filled and open arrowheads, respectively. (B) Total RNA extracted from cells growing exponentially (expo), subjected to nutritional stress (stress), or allowed to recover in normal media for 15 or 30 min after stress, was analyzed by northern blot for tRNAVal and tRNAArg (NB, upper panel). Full length tRNAs and 3′ trimmed tRNAs are indicated with filled and open arrowheads, respectively. Ethidium bromide staining (EtBr, lower panel) shows the migration of the bulk of the tRNAs. (C) Decay of total endogenous RNA was investigated by a 2-day pulse using α32P-UTP followed by washes and chase with cold UTP. Total RNA extracted from cells growing exponentially (expo), stressed for 2h in PBS (stress) or allowed to recover for 2 h in normal media (recov) was separated on 8% polyacrylamide/urea gels. (D) Cells were induced for expression of a tagged tRNA followed by removal of the inducer from the culture media. RNA was extracted from cells growing exponentially (expo), after nutritional stress (stress) or after 1 or 2 h of recovery in normal media and the tagged RNA investigated by northern blot. As control the same treatment was applied to uninduced cells (left part).
Trimmed tRNAs can be repaired
The accumulation of shortened tRNAs is completely reversible. When stressed cells were allowed to recover in fresh media the migration of tRNAVal and tRNAArg was almost fully restored after 30 min (Figure 1B). This might be due to repairing of the shortened tRNAs or by new transcription of most of the tRNA genes. To differentiate between these scenarios, we followed two different approaches. In the first one, newly synthesized RNAs were monitored by pulse chase using α 32P-UTP. Cells were labeled for 2 days, chased for 2 h, then stressed and after stress finally allowed to recover in normal media. By tracing the radioactive tRNA signal during these phases on a denaturing gel we observed a downshift during nutritional stress followed by a subsequent upshift to the original position upon stress recovery (Figure 1C). Importantly, the intensity of the 32P signal did not change strongly suggesting that the truncated tRNAs were repaired upon restitution of normal growth conditions and were thus not replaced by newly transcribed tRNAs. In the second approach, cells harboring a tetracycline-inducible mutated version of the endogenous tRNAGlu that can be specifically detected by northern blot analysis (19) were briefly induced for expression, then stressed and subsequently allowed to recover in the absence of tetracycline. Like the endogenous tRNAs, also the tagged tRNAGlu was truncated during starvation (Figure 1D). Importantly, during stress recovery the trimmed and tagged tRNA resumed its normal migration behavior in gel electrophoresis. This can only be due to repairing of the trimmed tRNAs as new synthesis of the tagged tRNA does not take place in the absence of tetracycline-mediated induction. These data clearly show that the tRNAs that are trimmed during stress are repaired and therefore become available for translation once normal growth conditions are restored.
The CCA-adding enzyme is not regulated during stress
In trypanosomes tRNA genes do not encode the CCA-tail and therefore this sequence has to be added posttranscriptionally by the tRNA nucleotidyltransferase, also referred to as CCA-adding enzyme (CAE) (20). The observed accumulation of tRNAs lacking the CCA-tail could thus be the consequence of a normal CCA-tail turnover but a reduced activity of the CAE during stress. To investigate this, the levels of the endogenous CAE were investigated. To that end one gene copy of the CAE was tagged in situ at the 5′ end using an HA epitope. This gene still bears the endogenous 3′ untranslated region and is therefore expected to be regulated as the wild type copy. Western blot analyses, however, did not reveal any differences in the abundance of the enzyme between exponentially growing or starved cells (Figure 2A). This data shows that the total protein levels of the CAE are not changed during stress but its specific activity could still be regulated. To test this, the tagged CAE was purified from cells grown under exponential or stress conditions and its activity tested in vitro. A time course experiment was performed using the same amounts of enzyme purified from each condition and an excess of substrate (tRNA and CTP). Our results show that the specific activity of the enzyme is not reduced during stress (Figure 2B and C). As a matter of fact, a slightly increased activity is detected for the enzyme purified from stressed cells. Many different stress conditions trigger the formation of stress granules (SGs), membrane-less intracellular compartments where translation initiation complexes as well as proteins involved in mRNA decay and stabilization are stored (26). To investigate if the CAE is recruited to SGs during nutritional stress and therefore unable to repair the shortened tRNAs, cells expressing a GFP-tagged inducible copy of the CAE were subjected to stress and the localization of the CAE analyzed by immunofluorescence. Supplementary Figure S1D shows that nutritional stress does not trigger the accumulation of the CAE in SGs and that the enzyme remains ubiquitously distributed as previously described (20). Altogether these data demonstrate that the amount as well as the enzymatic activity and localization of the CAE are not affected during nutritional stress.
Figure 2.
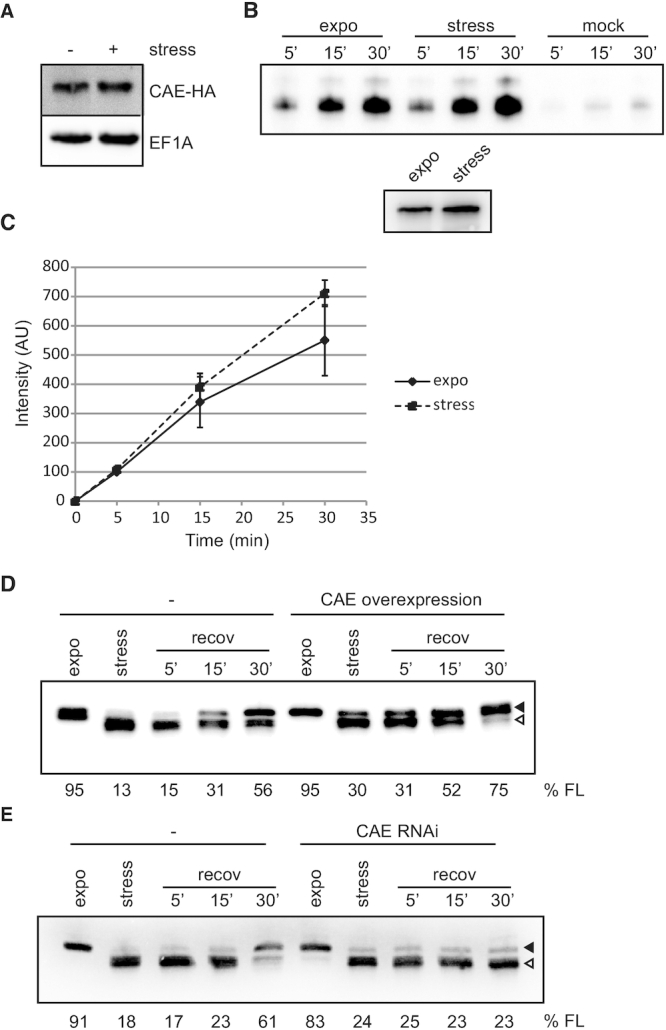
The CCA-adding enzyme repairs trimmed tRNAs. (A) The levels of an in situ HA-tagged CCA-adding enzyme (CAE-HA) were investigated by western blot on cells growing exponentially or upon nutritional stress. EF1A serves as loading control. (B) The tagged CAE was purified from parasites growing exponentially (expo) or subjected to nutritional stress (stress) and its activity was tested using tRNAs purified from stressed cells and α32P-CTP after 5, 15 and 30 min of incubation (upper panel). A mock purification was done using cells not expressing the tagged CAE. A representative gel is shown. The levels of the CAE used were analyzed by western blot (lower panel). (C) Signals from experiments in B were quantified and normalized to the intensity of the product generated by the CAE purified from exponentially growing cells and incubated for 5 min. Depicted is the mean of 3 independent experiments ± SD. (D, E) Cells allowing inducible overexpression (D) or depletion (E) of the CAE were stressed and then allowed to recover in normal media. Uninduced cells (−) exposed to the same conditions serve as control. RNA was extracted at the indicated time points and shortening of tRNAVal was analyzed by northern blot. Full length tRNAs and 3′ trimmed tRNAs are indicated with filled and open arrowheads, respectively. The percentage of full length tRNAVal present is indicated (% FL).
The CCA-adding enzyme repairs shortened tRNAs
Stress-induced shortened tRNAs could represent genuine substrates for CCA-addition and therefore might be repaired by the CAE to be recycled for translation. To test this hypothesis the CAE was affinity purified and its activity tested in vitro on two different substrates: either total tRNAs purified from cells grown exponentially or from cells exposed to stress. Supplementary Figure S1E shows that the CAE is indeed able to repair tRNAs isolated from both growth conditions but addition of radiolabeled CTP was more efficient on tRNAs from stressed cells. This can only be explained by the fact that more tRNAs lacking the CCA-tails, and therefore more substrates for CCA-tail addition, were available in this sample. When repairing was assessed solely in the presence of radiolabeled ATP, no incorporation was observed (Supplementary Figure S1E). These data indicate that the 3′ terminal CA or CCA sequence was removed during nutritional stress, therefore requiring that addition of cytidine nucleotide(s) precedes adenosine nucleotide incorporation.
To test if the CAE is also able to repair trimmed tRNAs in vivo we generated a cell line that allows inducible overexpression of the T. brucei CAE. When these cells were exposed to nutritional stress lower amounts of shortened tRNAs were observed compared to the control cells expressing normal levels of the endogenous CAE and complete tRNA repair was achieved sooner during stress recovery (Figure 2D). To provide direct evidence that the CAE is responsible for in vivo tRNA repair a cell line was used that allows inducible depletion of the enzyme by RNAi (20). Figure 2E shows that cells depleted of the CAE are unable to fully repair the trimmed tRNAs even after 30 min of recovery. All these data demonstrate that the CAE is responsible for repairing the trimmed tRNAs in vivo.
Shortening of tRNAs is mediated by a nuclease that is constitutively expressed in cells
The genome of trypanosomes does not code for homologues of any of the already described enzymes involved in the production of either tRNA halves or in the CCA-tail removal from tRNAs. Some of these enzymes (namely angiogenin in mammals and Rny1 in yeast) have been shown to be present in exponentially growing cells but are only activated in the cytosol under stress conditions, thus granting them regulated access to their substrates (27–29). To test if this is also the case in trypanosomes we investigated whether the trimming activity is constitutively present in cells. For that we incubated 5′ radiolabeled full-length tRNAVal with total lysates of cells grown exponentially or from starved cells. Figure 3A shows that both lysates are able to trim the CCA-tail of this tRNA to similar extents, suggesting the nuclease being constitutively present but kept inactive in exponentially growing cells. In order to get insights into the identity of the enzyme involved in CCA trimming we made use of commercially available inhibitors known to specifically block the activity of certain families of RNases. The data indicate that the CCA-trimming nuclease does not belong to the RNase families A, B, C or T2 (Figure 3B). Only the general RNase inhibitor ribonucleoside vanadyl complex (RVC) was able to abolish tRNA shortening. Similar experiments performed in the presence of cations chelators also indicate that the nuclease uses Mg2+ as a cofactor for its nucleolytic activity on tRNAs (Figure 3C) and that aminoacylation retards tRNA shortening (Figure 3D).
Figure 3.
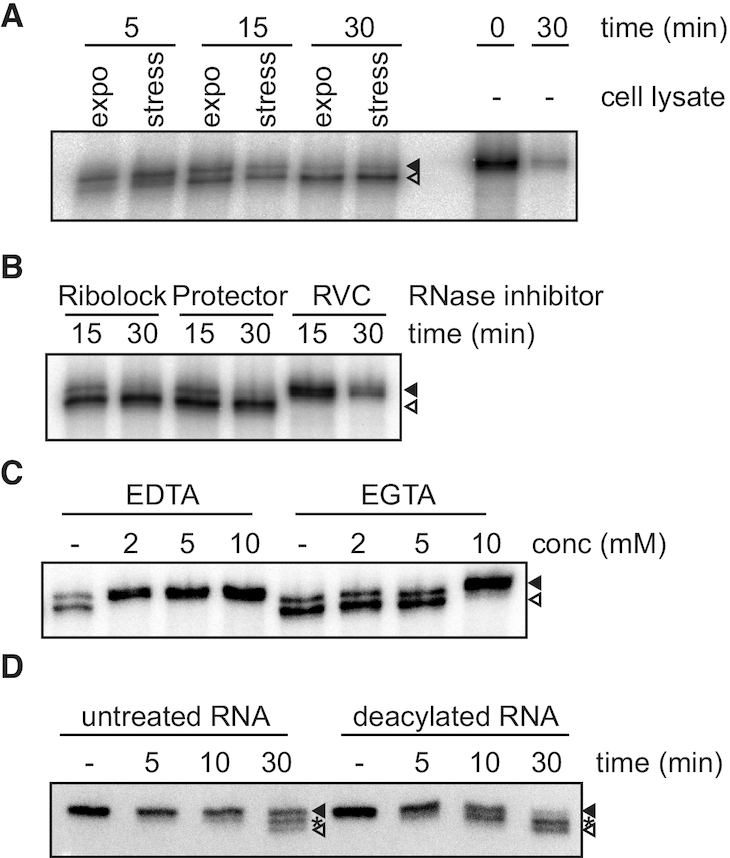
The nuclease trimming the tRNAs is constitutively expressed in cells and can be inhibited by ribonucleoside-vanadyl complex and Mg2+ chelators. (A) The tRNA trimming activity was tested in vitro by incubating total cell lysates from cells grown exponentially (expo) or nutritionally stressed (stress) with 5′ 32P-labeled tRNAVal. After the indicated times total RNA was precipitated, separated on 8% polyacrylamide/urea gels, dried and the radioactivity detected by exposure to phosphoimager screens. Incubation of the substrate under the same conditions in the absence of cell lysate (−) serves as control. (B, C) Cell lysates were treated with specific RNase inhibitors (Ribolock, Protector or RVC; B) or different concentrations of divalent cations chelators (C) and the cellular tRNA trimming activity tested as before. (D) The effect of aminoacylation on tRNA trimming was tested using RNA extracted from cells expressing a tagged tRNAGlu. After incubation of total cell lysates with total RNA either aminoacylated (untreated) or subjected to alkaline deacylation RNA was extracted and tRNA shortening investigated by northern blot against tRNAVal. Full length tRNAVal and fully 3′ trimmed tRNAVal are indicated with filled and open arrowheads, respectively. Asterisks indicate intermediate cleaved tRNAVal.
LCCR4 is involved in tRNA shortening
In order to identify the nuclease responsible for tRNA shortening a classical biochemical fractionation approach was performed. Briefly, whole cell lysates were subjected to sequential rounds of size exclusion chromatography and the obtained fractions were tested for trimming activity on an in vitro transcribed tRNA (Supplementary Figure S2). Mass spectrometry analyses of the active fractions revealed several candidates among them LCCR4 (Tb927.4.2430), a homologue of the yeast deadenylase Ccr4 (human CNOT6, Supplementary Table S2 and ProteomeXchange Consortium identifier PXD021533, 30). In contrast to observations in other species trypanosomal LCCR4 does not have deadenylase activity, neither does it assemble in a complex with Not1 and Caf1 (31,32). One of these studies also showed that a tagged version of LCCR4 was present in the cytosol as well as in the nucleus of cells (31). To investigate a putative role of LCCR4 in tRNA shortening we generated cells that allow inducible knock-down of LCCR4 and exposed them to nutritional stress. After LCCR4 RNAi induction cells showed a mild growth phenotype (Supplementary Figure S3A and B) but no changes in the tRNA length during exponential growth were observed (Figure 4A). In contrast, upon nutritional stress a very clear difference in the shortening pattern of tRNAVal could be observed. Although the same amounts of tRNA were trimmed, complete CCA-tail removal was markedly inhibited upon LCCR4 RNAi (Figure 4A). Size comparison using in vitro transcripts bearing different CCA-tail lengths suggests that this incomplete shortened species correspond to the tRNAVal carrying only a single C and that the fully trimmed tRNA corresponds to tRNAs that lost the full CCA-tail (Supplementary Figure S4A). This indicates that tRNAVal shortening happens in two steps and that LCCR4 might be involved in the second phase of shortening. Another plausible explanation is that LCCR4 is responsible for the complete tRNA trimming but does it in two steps, an initial more rapid cleavage of the terminal CA sequence, followed by a more difficult and therefore slower removal of the last C. Thus, the reduced levels of LCCR4 remaining after RNAi would be still capable of performing the first step of trimming but unable to complete the process. Furthermore, when LCCR4-depleted cells were allowed to recover in fresh medium tRNAs were repaired faster than in uninduced cells (Figure 4B). To further characterize the role of LCCR4 in tRNA shortening we created a cell line expressing an inducible HA-tagged copy of LCCR4. As shown in Supplementary Figure S3C, induced cells showed a slight decrease in their growth rate. Cells expressing the HA-tagged LCCR4 were exposed to nutritional stress and total RNA was analyzed by northern blot. Upon stress no shortening intermediate was detected and only the completely trimmed tRNAVal was present in induced cells (Figure 4C). Furthermore, repairing of trimmed tRNAs was drastically delayed in cells overexpressing LCCR4 compared to uninduced cells. This delay in repairing was not restricted to tRNAVal but applied to the bulk of tRNAs as can be appreciated in the ethidium bromide staining (Figure 4C, bottom panel). All these data strongly suggest that LCCR4 is involved in the observed tRNA shortening in vivo and that it is kept inactive during exponential growth by a yet unknown mechanism.
Figure 4.

LCCR4 is involved in stress-triggered tRNA shortening. (A) Cells induced for LCCR4 RNAi or kept uninduced were grown exponentially or under nutritional stress and tRNA shortening analyzed by northern blot detecting tRNAVal. tRNAs containing a complete CCA-tail are marked with a full arrowhead while incomplete and totally shortened species are marked with an asterisk or open arrowheads, respectively. (B) As in (A) but cells were allowed to recover in complete media for the indicated times after nutritional stress. (C) Cells induced for LCCR4-HA overexpression or left uninduced and growing exponentially or under nutritional stress were allowed to recover in normal media for the indicated times. The upper panel shows a northern blot where tRNAVal was investigated while the lower panel corresponds to the same gel stained with ethidium bromide (EtBr). The region corresponding to the bulk cellular tRNAs is indicated. The percentage of full length tRNAVal present is indicated (%FL).
LCCR4 interacts with tRNA and is active in vitro
To investigate if LCCR4 interacts with tRNAs we followed an RNA immuno-precipitation (RIP) approach, in which cells expressing the HA-tagged LCCR4 were crosslinked, the protein immunoprecipitated (IP) and the presence of co-purifying tRNAs investigated by RT-PCR. Figure 5A shows that LCCR4-HA was successfully purified under these conditions. Furthermore, tRNAVal was found associated with LCCR4 (Figure 5B). At the same time the endogenous M6 rRNA failed to co-IP with the HA-tagged LCCR4 (Figure 5B) thus highlighting the specificity of the obtained data. Subsequently the activity of LCCR4 was investigated in vitro. HA-tagged LCCR4 was purified from parasites and its trimming activity investigated on full length tRNAs isolated from cells. As observed in Figure 6A, affinity purified LCCR4 was able to trim full length tRNAs in vitro. Surprisingly, LCCR4 purified from cells growing exponentially or under stress were equally active on trimming tRNAs (Figure 6A and B) again suggesting a mechanism of trimming inhibition acting in vivo on normally growing cells. LCCR4 is not only active on full length tRNAs but also on 3′-shortened tRNAs isolated from stressed LCCR4-depleted cells (Supplementary Figure S4B). The shorter tRNAs species accumulating in stressed LCCR4-depleted cells contain a single C residue at the 3′ end and this C is removed by LCCR4 to generate completely trimmed tRNAs (Supplementary Figure S4A and B). Furthermore, deacylated tRNAs seem to be a better substrate for shortening by LCCR4, in agreement with our previous observations using total cell lysates (Supplementary Figure S4C).
Figure 5.
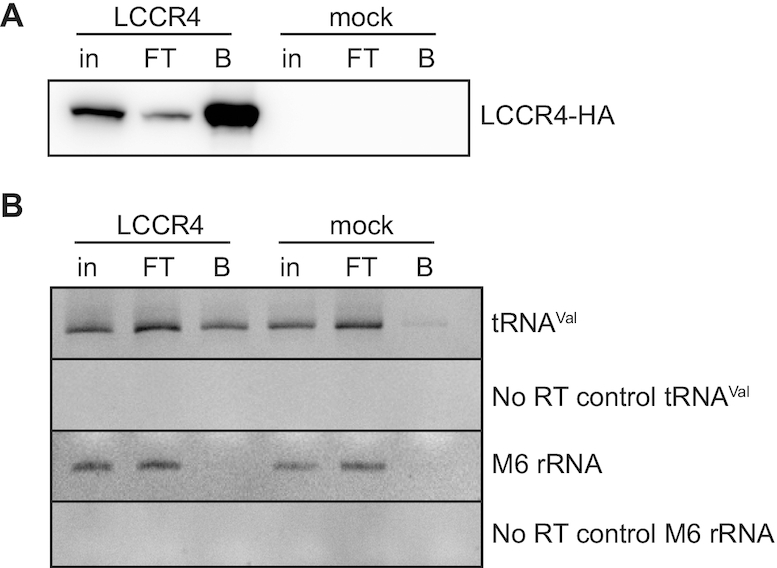
LCCR4 interacts with tRNAs. (A) Inducibly expressed LCCR4-HA was immunoprecipitated and samples obtained from each purification step were analyzed by western blot using antiHA antibodies. (B) LCCR4-HA was immunoprecipitated and its interaction with tRNAVal was investigated by RT-PCR. M6 rRNA serves as specificity control. in: input, FT: flow-through; B: bead-bound.
Figure 6.
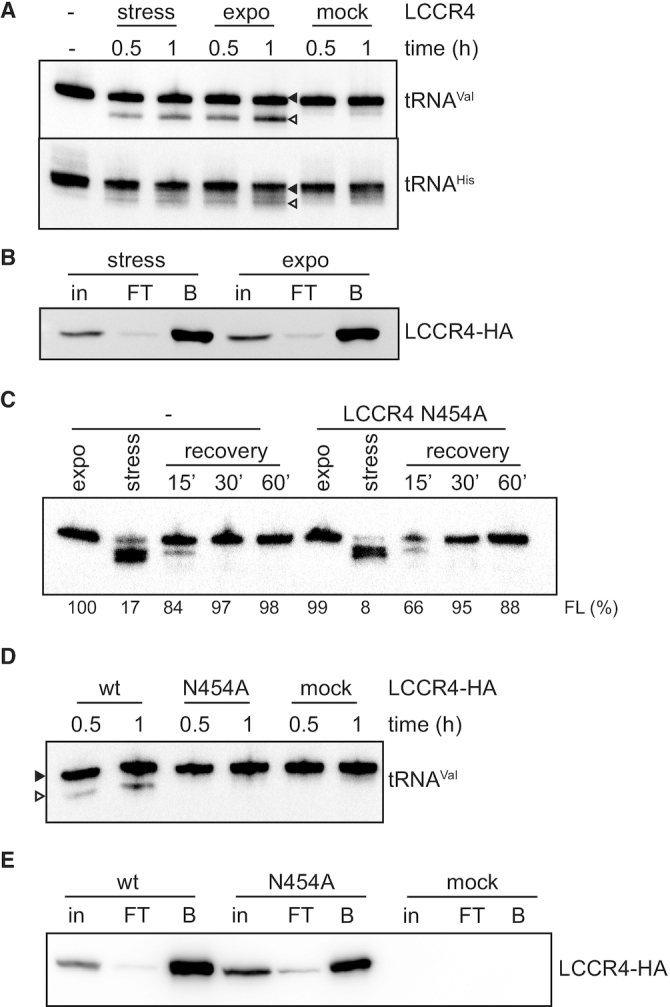
LCCR4 can cleave tRNAs in vitro. (A) LCCR4-HA expression was induced and the protein was purified from cells growing exponentially (expo) or subjected to nutritional stress (stress) and its activity tested on beads using total RNA as substrate. After incubation for the indicated times the RNA was extracted and analyzed by northern blot for tRNAVal and tRNAHis. Samples for the mock control were obtained from uninduced cells. Full length tRNAs are marked with a full arrowhead while shortened species are marked with open arrowheads. (B) Western blot analysis of samples used in (A). (C) Cells overexpressing the mutant LCCR4 N454A were subjected to stress and then allowed to recover in fresh media for the indicated times. RNA was extracted at each time point and tRNA shortening analyzed by northern blot against tRNAVal. (D) Wild type and N454A mutant LCCR4-HA were purified and their activity tested in vitro as in (A). (E) Western blot analysis of samples used in (D). in: input, FT: flow-through; B: bead-bound.
The observed activity of LCCR4 in vitro might be either due to its inherent enzymatic activity or the activity of associated proteins. In order to identify putative interacting partners, we performed mass spectrometry analysis of co-purifying proteins using a SILAC approach (full data deposited in ProteomeXchange Consortium identifier PXD021540). Data in Supplementary Table S3 shows that only four proteins were significantly and specifically detected in LCCR4-HA immunoprecipitations and none of them have predicted domains usually present in known RNases. In addition no assembly of LCCR4 in higher molecular weight complexes was detected when cell lysates were investigated on native gels (Supplementary Figure S5A) strongly supporting the mass spectrometry analysis and previous reports (31). In most organisms, Ccr4 interacts with the deadenylase Caf1 forming the Ccr4/Caf1/Not complex, the main deadenylation complex in cells (33). To investigate a role of Caf1 in the observed stress-induced tRNA shortening we followed different approaches, including overexpression of CAF1, tRNA interaction studies and downregulation of CAF1 or CAF1 and LCCR4 simultaneously. Altogether, data shown in Supplementary Figure S5B-H exclude that CAF1 plays a role in tRNA trimming in T. brucei.
The data presented before strongly indicate that LCCR4 is the sole enzyme responsible for tRNA shortening in vitro. To further test this hypothesis we generated a cell line that allows inducible overexpressing of an inactive version of HA-tagged LCCR4 with Asn454 mutated to Ala. This residue has been shown in the human orthologue CNOT6L to be part of the active site and essential for substrate binding (34), corresponds to Asn412 in human CNOT6L; Supplementary Figure S6). Overexpression of this mutant followed by nutritional stress and stress recovery shows no effect on tRNA repair over time (Figure 6C). This is in marked contrast to what was observed when overexpressing wild type LCCR4 (Figure 4C) indicating that in T. brucei LCCR4 Asn412 is essential for tRNA trimming in vivo. When the activity of the purified mutant protein was tested in vitro no tRNA shortening could be detected, in contrast to the wild-type LCCR4 (Figure 6D and E). Cumulatively, these results show that LCCR4 is the sole responsible enzyme for the tRNA CCA-tail removal.
tRNA repair is essential for proper translation during stress recovery
Stress-induced shortened tRNAs are expected to be unsuitable substrates for translation as the CCA-tail is essential for attachment of the amino acid by the corresponding aminoacyl tRNA synthetase (aaRS). To investigate if this is indeed the case we performed in vitro aminoacylation analysis using cell lysates either from exponentially grown or stressed cells in which ribosomes have been removed by ultracentrifugation (S100 fractions, Supplementary Figure S7). As shown in Figure 7A the efficiency of aminoacylation of tRNAArg from stressed cells is drastically reduced (to ∼28% compared to exponentially growing cells). This is not due to a lower activity of the aaRS present in stressed cells as aminoacylation of supplemented yeast tRNAs was equally efficient in both conditions (Figure 7A). This clearly shows that shortened tRNAs are not adequate substrates for aminoacylation.
Figure 7.

Repairing of shortened tRNAs is crucial for translation during stress recovery. (A) In vitro aminoacylation was tested on S100 cell fractions from cells grown exponentially or stressed for 2 h in PBS, using as substrate either the endogenous tRNAs (endog. tRNAs) or after supplementation with total yeast tRNA (yeast tRNA). Dark and light grey bars correspond to signals from exponential or stressed samples, respectively. The percentage of aminoacylation was calculated by normalization to the signal for S100 from exponentially growing cells. Depicted is the mean of three experiments ± SD. (B) Cells induced for LCCR4-HA overexpression or not were stressed and translation efficiency investigated during stress recovery by metabolic labeling with 35S-Methionine. After the indicated times cells were pelleted, proteins separated by SDS-PAGE and incorporated radioactivity detected by exposing to phosphorimager screens. Unstressed cells labeled for 30min were also tested (two most right lanes). The upper panel shows a representative autoradiogram of the metabolic labeling experiment while the lower panel is the corresponding gel stained with coomassie. (C) Quantification of 35S-Methionine incorporation of 4 independent experiments. Dark and light grey bars correspond to the percentage of 35S-incorporation in uninduced and induced cells, respectively. Signals were normalized to the intensity corresponding to uninduced cells recovered for 1h. Data are represented as mean ± SD. (D) RNA was extracted from cells treated as in (B) and tRNA repair was investigated by northern blot against tRNAVal. The percentage of full length tRNAVal present is indicated (%FL).
Overexpression of LCCR4 leads to a delay in the repairing of tRNAs during stress recovery with no effects on tRNA integrity under normal growth conditions (Figure 4C). We therefore wanted to investigate whether this delayed repairing has an impact on the translational competence of cells. For that purpose we performed metabolic labeling of cells overexpressing LCCR4 during recovery from nutritional stress. Since overexpression of LCCR4 causes a mild growth defect we decided to limit the time of induction to one day when no growth defect was observed yet (Supplementary Figure S3C). The first observation was that overexpression of LCCR4 for one day does not cause per se a change in the translational activity of cells grown under normal conditions (Figure 7B, lanes 7 and 8). However, the translational activity during stress recovery was markedly reduced in cells overexpressing LCCR4 (Figure 7B). This effect was particularly strong when we analyzed translation efficiency for 2 h after stress removal with ∼45% reduction upon LCCR4 overexpression (Figure 7B, lanes 3 and 6 and Figure 7C). Analysis of tRNA integrity during these recovery times showed that although tRNA repair was delayed, most of the tRNAs were in their full-length form after 2 h of recovery (Figure 7D).
DISCUSSION
Here, we analyze the effect of nutritional stress on tRNA integrity in the pathogenic protozoon Trypanosoma brucei. Our studies show the deactivation of tRNAs by removal of the essential and universally conserved 3′ CCA-tail. This event is not restricted to a single tRNA species but affects most tRNAs (Figure 1A and B; Supplementary Figure S1B and C). Furthermore, the shortening of the tRNAs is specifically observed upon nutritional stress and not when parasites are exposed to other stress conditions (cold and heat shock, oxidative stress and stationary phase, Figure 1A, Supplementary Figure S1B and C). Furthermore, tRNA shortening happens in both procyclic as well as bloodstream forms of the parasite, suggesting a physiologically important mechanism of tRNA inactivation for T. brucei biology (Supplementary Figure S1A). The removal of the CCA-tail renders tRNAs unsuitable for protein biosynthesis as they cannot be aminoacylated thus excluding them from the productive translational machinery (Figure 7A).
Enzymes capable of cleaving tRNAs have been described in both prokaryotes and eukaryotes. Examples of prokaryotic enzymes are colicins E5 and D, both produced by bacteria to trigger contact-dependent growth inhibition of competitive sensitive bacteria (35). These RNases cleave tRNAs in the anticodon loop depleting the cells from full length tRNAs and therefore preventing protein synthesis. Another example of a tRNA cleaving enzyme is the recently characterized CdiA-CT toxin from E. coli (36). This RNase is able to cleave the single stranded 3′ ends of tRNAs only on tRNAs that contain G as discriminator base, removing the CCA-tail plus one extra base. The nucleases angiogenin and Rny1 cleave tRNAs in the anticodon loop generating tRNA halves in humans and yeast, respectively (28,37). Because all the enzymes mentioned above cleave within the tRNA body the resulting molecules cannot be repaired by common cellular enzymes. In marked contrast, the process we describe here does not affect the tRNA body and therefore trimmed tRNAs can be repaired in order to be reused for translation (Figure 1).
In this study we revealed the Ccr4 homologue LCCR4 as a tRNA 3′-end removing enzyme, by biochemical (Figure 6) and genetic (Figure 4) means. The Ccr4-Not complex is a highly conserved multisubunit complex present in all eukaryotes studied so far (33). It is composed of several proteins all of them assembled around the scaffold protein Not1. Among them Caf1 and Ccr4 function as catalytic subunits responsible for the deadenylation activity of the complex (38,39). Several reports have shown that each of these proteins display preferential deadenylase activity depending on the nature of the substrate and the organism analyzed (40,41). In trypanosomes a homologue of Caf1 assembles in a complex with Not1 and several other Not subunits and is responsible for mRNA deadenylation (31). A homologue of Ccr4 (LCCR4) is also encoded in the genome of the parasite but previous studies have shown that the protein does not interact with the Caf1-Not complex (31). T. brucei LCCR4 does not contain the N-terminal leucine-rich repeat region (LRR) involved in the interaction with Caf1 in the Ccr4/Not complex (42,43). Several homologues of Ccr4 lacking the LRR have been described in other organisms, in which the sequence similarity is restricted to the endonuclease/exonuclease/phosphatase family domain (44). Among them we find nocturnin, a deadenylase involved in cellular metabolism (45), and protein angel 1 and 2, recently shown to be 2′,3′-cyclic phosphatases (46). Here we discovered a novel role for the trypanosome LCCR4: stress-induced cleavage of the 3′-CCA end of tRNAs. We were able to demonstrate that LCCR4 not only interacts with tRNAs in vivo (Figure 5) but it is also capable of shortening tRNAs when overexpressed in stressed cells (Figure 4). In vitro studies employing purified LCCR4 show that this enzyme is indeed capable of removing the full CCA-tail from tRNAs and that this activity is dependent on a conserved residue (Asn454) present in the active site of this family of deadenylases (Figure 6). Our studies further showed that the conserved Ccr4-interacting protein Caf1 does not play any role in tRNA trimming in T. brucei. Overexpression as well as depletion of CAF1 by RNAi and RIP analysis did not reveal a role of CAF1 in tRNA shortening during stress (Supplementary Figure S5). Furthermore, we found no indications that LCCR4 assembles in any complex neither were we able to detect relevant interacting partners by immunoprecipitation followed by MS (Supplementary Table S3; Supplementary Figure S5A), strongly suggesting that LCCR4 is the sole nuclease involved in tRNA 3′-end trimming.
Analysis of the crystal structure of the human Ccr4 homologue CNOT6L bound to poly(A) DNA explains its magnesium-dependent activity and its strict poly(A) substrate specificity (34). Despite the fact that all the residues involved in magnesium coordination and substrate binding are conserved in T. brucei LCCR4 and that structure predictions locate them in the same architectural context as in human CNOT6L (Supplementary Figure S6) our data shows that the substrate specificity is altered. Further structural analysis would help clarifying how this is possible.
In mammals, the endonuclease angiogenin is responsible for the generation of certain tRNA halves by cleaving in the anticodon region (29,37,47). Angiogenin shows preference for CA sequences and it has been reported that it is capable of cleaving the CCA-tail of tRNAs in vitro (8). Whether the same is true in vivo still awaits future investigations. This enzyme belongs to the RNaseA family and is kept inactive in cells by interacting with its inhibitor RNH1. Upon oxidative stress RNH1 is degraded, allowing angiogenin to cleave tRNAs (48). To date no tRNA trimming has been reported in yeast. In agreement with this, we did not detect any tRNA shortening in S. cerevisiae exposed to nutritional stress neither in total cell lysates of wt cells nor in cells overexpressing Ccr4-HA (Supplementary Figure S8). These data indicate that no such activity is present in S. cerevisiae cells and suggest that the canonical Ccr4 does not possess tRNA trimming activity. In trypanosomes LCCR4 is a ubiquitous protein (31) and our data shows that even when the protein is overexpressed no shortening of tRNAs is observed during exponential growth (Figure 4C). In contrast, once cells overexpressing LCCR4 are exposed to nutritional stress an increase in tRNA shortening is observed, and more importantly there is a delay in tRNA repair once stress is removed. All these data point to a very efficient cellular mechanism keeping LCCR4 inactive during normal growth conditions even when the levels of the protein are artificially increased. Once cells encounter nutritional stress, which requires rapid downregulation of metabolic activities, this inactivation is abrogated and LCCR4 is liberated to execute its tRNA-trimming activity. A similar deregulation of LCCR4 activity is observed when cells are lysed and tRNA shortening is assessed in vitro (Figure 3). We speculate that the unknown LCCR4 inhibitor is unstable in our lysis conditions leaving LCCR4 to display its full activity. Future efforts will be devoted to the identification of this LCCR4 inhibitor.
3′-Trimming of tRNAs seems to be a very efficient and fast process. Even overexpression of the CCA-adding enzyme, the tRNA nucleotidyltransferase responsible for tRNA repair (Figure 2), is not enough to abolish tRNAs shortening. Only after removal of nutritional stress, and therefore inactivation of the LCCR4 nuclease, can the tRNAs be repaired. As mentioned above, oxidative stress triggers tRNA shortening in mammalian cells (8,9). These tRNAs were reported to possess a 2′,3′-cyclic phosphate that needs to be resolved by other so far unknown enzyme(s) in order for the tRNA nucleotidyl transferase to repair the tRNAs. A similar situation is observed during ribosome associated quality control (RQC). In this process stalled ribosomes induce the recruitment of ANKZF1 that releases the nascent peptides from the peptidyl-tRNA by cleaving the CCA-tail (49). While the nascent peptide is tagged for proteasome degradation the shortened tRNA is repaired by TRNT1 (the human CAE) after the cyclic phosphate is removed by ELAC1 (50). In contrast to the E. coli enzyme the trypanosome CAE does not contain an HD domain and should therefore be unable to resolve any 2′,3′-cyclic phosphate left behind by the nuclease that trims the tRNAs (51). Our analysis shows that the trypanosome CAE is capable of repairing the shortened tRNAs without the action of any accessory protein (Figure 2B, Supplementary Figure S1E). Furthermore, ligation of an RNA adaptor molecule to the 3′ end of shortened tRNAs was possible without any prior treatment. These results strongly suggest that LCCR4 removes the CCA-tail without generating a cyclic phosphate therefore allowing the CAE to directly repair the trimmed ends. One of the steps in RQC involves the cleavage of the tRNA CCA-tail and therefore highly resembles the process described in this study. Although both mechanisms follow the same main principle, the strongest difference is the magnitude of the process. While RQC only affects a minor fraction of the cellular tRNAs (those found in the P-site at the moment of ribosome stalling) and therefore their trimming should not be detrimental for global translation, shortening of tRNAs triggered by nutritional stress in T. brucei affects a major fraction of the cellular tRNA pool (70%, Supplementary Figure S1B and C). This dramatic reduction in the translationally competent tRNA pool has therefore major consequences on global translation efficiency and their fast repair is crucial for growth recovery once favorable conditions are restored (Figure 7).
Recently, the retrograde transport of tRNAs from the cytoplasm to the nucleus has been reported in mammalian cells exposed to oxidative stress (9). This retrograde transport seems to be selective for certain tRNA species and preferentially for 3′-shortened tRNAs. It is tempting to speculate that these tRNAs are imported to undergo a possible nuclear quality control mechanism (52). Considering that in trypanosomes around 70% of all tRNAs are trimmed upon nutritional stress it is hard to conceive that all shortened species are imported back into the nucleus but we cannot exclude that retrograde import exists.
The removal of the CCA-tail from tRNAs is an energetically efficient way to abolish translation. Since the tRNA backbones remain intact they can be quickly repaired by the CCA-adding enzyme with minimum energetic input. In contrast, if tRNAs were completely degraded they would have to be newly transcribed, processed and modified in order to be available for translation. This would involve a much larger energy investment as the mere addition of the three nucleotides required to recycle the non-functional trimmed tRNAs.
As we report here, in T. brucei stress-triggered shortening of tRNAs is a massive phenomenon affecting most of the tRNA pool (Figure 1A). As the probes used for northern blot analysis were designed to detect as many isoacceptors as possible it is not clear to which extent each individual tRNA isoacceptor is trimmed. One possibility is that a certain isoacceptor is completely cleaved while another is kept unaffected. Previous reports have shown that in most organisms tRNA abundance correlates with codon usage (53,54). It has also been reported that in T. brucei tRNA gene copy number directly correlates with codon usage and protein expression levels (55,56). Therefore, differential inactivation of tRNAs by 3′ shortening could represent a fast and elegant way to achieve translation reprogramming during stress conditions. In bacteria, amino acid starvation triggers the specific degradation of tRNAs (57). The authors suggested that this selective tRNA degradation would help the cells to adapt to stress by reducing possible mistranslation due to competition between cognate and near-cognate tRNAs. It is tempting to speculate that the tRNA shortening phenomenon we describe here could also result in a differential inactivation of tRNAs therefore fulfilling a similar function in nutritionally-stressed T. brucei.
The shortening of tRNAs by means of CCA-tail removal has so far only been described in mammalian cells exposed to oxidative stress and shown to affect around 30% of the tRNA population (8). Trypanosomes belong to the excavata super-group (58) and therefore they are evolutionarily speaking far away from the ophistokont group to which metazoa and yeast belong. Our studies add one more grain of sand in the understanding of cellular processes during eukaryotic evolution. Trypanosomes, which are known for their peculiar RNA biology (including fragmentation of 28S rRNA, extensive RNA editing in the mitochondria, trans-splicing of all mRNAs, transcription of protein coding genes by RNA PoI I and many more), seem to once again make use of a conserved mechanism and take it to its extreme. They engage a highly conserved protein, the deadenylase LCCR4, but use it for a completely different purpose, namely tRNA 3′-trimming.
Originally described as rather passive adaptor molecules involved in translation, tRNAs and especially tRNA-derived RNAs have been shown recently to play roles in a plethora of additional cellular functions (59,60). In particular, tRNA halves and fragments have been described in all kingdoms of life and were also shown to promote survival in cells exposed to non-favorable environmental conditions (61). In trypanosomes exposed to nutritional stress many tRNA halves are produced and our previous studies identified the tRNAThr 3′ half as a promoter of translation during stress recovery (15). Together with the data presented here we can clearly state that nutritional stress, a physiological challenge the parasite regularly encounters in the insect host, triggers the processing of tRNAs into structural and functional diverse molecules that play essential roles in regulating translation and concomitantly cell survival.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Andreas Hämmerle for help during chromatography, Shikha for sharing constructs, Martine Collart for constructs and Yulia Gonskikh for insightful discussions. Manfred Heller from the Proteomics & Mass Spectrometry Core Facility (University of Bern) is acknowledged for help during MS data analyses. Laura Varsky (lauravarsky.com.ar) is acknowledged for cover art.
Author contributions: M.C. performed the majority of the experiments and coordinated the supporting experiments performed by R.B., O.J. and B.S. A.S. contributed experimental advice and provided constructs. M.C. and N.P. design all experiments, supervised the study and wrote the final manuscript. All authors contributed to data interpretation, analysis and commented on the manuscript.
Contributor Information
Marina Cristodero, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
Rebecca Brogli, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, Bern, Switzerland.
Oliver Joss, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
Bernd Schimanski, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
André Schneider, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
Norbert Polacek, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NCCR ‘RNA & Disease’ funded by the Swiss National Science Foundation; Swiss National Science Foundation [31003A_188969, 31003A_166527 to N.P.]. Funding for open access charge: Swiss National Science Foundation [NCCR ‘RNA & Disease’].
Conflict of interest statement. None declared.
REFERENCES
- 1. Fenn K., Matthews K.R.. The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 2007; 10:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roeder R.G. 50+ Years of eukaryotic transcription: an expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol. 2019; 26:783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Preußer C., Jaé N., Bindereif A.. mRNA splicing in trypanosomes. Int. J. Med. Microbiol. 2012; 302:221–224. [DOI] [PubMed] [Google Scholar]
- 4. Clayton C. Regulation of gene expression in trypanosomatids: living with polycistronic transcription. Open Biol. 2019; 9:190072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoagland M.B., Stephenson M.L., Scott J.F., Hecht L., Zamecnik P.C.. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 1958; 231:241–257. [PubMed] [Google Scholar]
- 6. Robertus J.D., Ladner J.E., Finch J.T., Rhodes D., Brown R.S., Clark B.F.C., Klug A.. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 1974; 250:546–551. [DOI] [PubMed] [Google Scholar]
- 7. Advani V.M., Ivanov P.. Translational control under stress: reshaping the translatome. Bioessays. 2019; 41:1900009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Czech A., Wende S., Mörl M., Pan T., Ignatova Z.. Reversible and rapid transfer-RNA deactivation as a mechanism of translational repression in stress. PLoS Genet. 2013; 9:e1003767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwenzer H., Jühling F., Chu A., Pallett L.J., Baumert T.F., Maini M., Fassati A.. Oxidative stress triggers selective tRNA retrograde transport in human cells during the integrated stress response. Cell Rep. 2019; 26:3416–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robertson H.D., Altman S., Smith J.D.. Purification and properties of a specific Escherichia coli ribonuclease which cleaves a tyrosine transfer ribonucleic acid precursor. J. Biol. Chem. 1972; 274:5243–5251. [PubMed] [Google Scholar]
- 11. Maraia R.J., Lamichhane T.N.. 3′ processing of eukaryotic precursor tRNAs. Wiley Interdiscip. Rev. RNA. 2011; 2:362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aebi M., Kirchner G., Chen J.Y., Vijayraghavan U., Jacobson A., Martin N.C., Abelson J.. Isolation of a temperature-sensitive mutant with an altered tRNA nucleotidyltransferase and cloning of the gene encoding tRNA nucleotidyltransferase in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1990; 265:16216–16220. [PubMed] [Google Scholar]
- 13. Xiong Y., Steitz T.A.. A story with a good ending: tRNA 3′-end maturation by CCA-adding enzymes. Curr. Opin. Struct. Biol. 2006; 16:12–17. [DOI] [PubMed] [Google Scholar]
- 14. Deutscher M.P., Evans J.A.. Transfer RNA nucleotidyltransferase repairs all transfer RNAs randomly. J. Mol. Biol. 1977; 109:593–597. [DOI] [PubMed] [Google Scholar]
- 15. Fricker R., Brogli R., Luidalepp H., Wyss L., Fasnacht M., Zywicki M., Schneider A., Cristodero M., Polacek N.. A tRNA half modulates translation as stress response in Trypanosoma brucei. Nat. Commun. 2019; 10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wirtz E., Leal S., Ochatt C., Cross G.A.. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999; 99:89–101. [DOI] [PubMed] [Google Scholar]
- 17. Bochud-Allemann N., Schneider A.. Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. J. Biol. Chem. 2002; 277:32849–32854. [DOI] [PubMed] [Google Scholar]
- 18. Schimanski B., Nguyen T.N., Günzl A.. Characterization of a multisubunit transcription factor complex essential for spliced-leader RNA gene transcription in trypanosoma brucei. Mol. Cell. Biol. 2005; 25:7303–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bruske E.I., Sendfeld F., Schneider A.. Thiolated tRNAs of Trypanosoma brucei are imported into mitochondria and dethiolated after import. J. Biol. Chem. 2009; 284:36491–36499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shikha S., Schneider A.. The single CCA-adding enzyme of T. brucei has distinct functions in the cytosol and in mitochondria. J. Biol. Chem. 2020; 295:6138–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Erlacher M.D., Chirkova A., Voegele P., Polacek N.. Generation of chemically engineered ribosomes for atomic mutagenesis studies on protein biosynthesis. Nat. Protoc. 2011; 6:580–592. [DOI] [PubMed] [Google Scholar]
- 22. Milligan J.F., Groebe D.R., Whherell G.W., Uhlenbeck O.C.. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987; 15:8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krupp G. RNA synthesis: strategies for the use of bacteriophage RNA polymerases. Gene. 1988; 72:75–89. [DOI] [PubMed] [Google Scholar]
- 24. Motorin Y., Muller S., Behm-Ansmant I., Branlant C.. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol. 2007; 425:21–53. [DOI] [PubMed] [Google Scholar]
- 25. Czech A. Deep sequencing of tRNA’s 3′-termini sheds light on CCA-tail integrity and maturation. RNA. 2020; 26:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ivanov P., Kedersha N., Anderson P.. Stress granules and processing bodies in translational control. Cold Spring Harb. Perspect. Biol. 2019; 11:a032813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shapiro R., Vallee B.L.. Human placental ribonuclease inhibitor abolishes both angiogenic and ribonucleolytic activities of angiogenin. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:2238–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thompson D.M., Parker R.. The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J. Cell Biol. 2009; 185:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamasaki S., Ivanov P., Hu G.-F., Anderson P.. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 2009; 185:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deutsch E.W., Bandeira N., Sharma V., Perez-Riverol Y., Carver J.J., Kundu D.J., García-Seisdedos D., Jarnuczak A.F., Hewapathirana S., Pullman B.S.et al.. The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020; 48:D1145–D1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwede A., Ellis L., Luther J., Carrington M., Stoecklin G., Clayton C.. A role for Caf1 in mRNA deadenylation and decay in trypanosomes and human cells. Nucleic Acids Res. 2008; 36:3374–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Erben E., Chakraborty C., Clayton C.. The CAF1-NOT complex of trypanosomes. Front. Genet. 2013; 4:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Collart M.A. The Ccr4-Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip. Rev. RNA. 2016; 7:438–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H., Morita M., Yang X., Suzuki T., Yang W., Wang J., Ito K., Wang Q., Zhao C., Bartlam M.et al.. Crystal structure of the human CNOT6L nuclease domain reveals strict poly(A) substrate specificity. EMBO J. 2010; 29:2566–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ogawa T. tRNA-targeting ribonucleases: molecular mechanisms and insights into their physiological roles. Biosci. Biotechnol. Biochem. 2016; 80:1037–1045. [DOI] [PubMed] [Google Scholar]
- 36. Michalska K., Gucinski G.C., Garza-Sánchez F., Johnson P.M., Stols L.M., Eschenfeldt W.H., Babnigg G., Low D.A., Goulding C.W., Joachimiak A.et al.. Structure of a novel antibacterial toxin that exploits elongation factor Tu to cleave specific transfer RNAs. Nucleic Acids Res. 2017; 45:10306–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fu H., Feng J., Liu Q., Sun F., Tie Y., Zhu J., Xing R., Sun Z., Zheng X.. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009; 583:437–442. [DOI] [PubMed] [Google Scholar]
- 38. Tucker M., Valencia-Sanchez M.A., Staples R.R., Chen J., Denis C.L., Parker R.. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell. 2001; 104:377–386. [DOI] [PubMed] [Google Scholar]
- 39. Tucker M., Staples R.R., Valencia-Sanchez M.A., Muhlrad D., Parker R.. Ccr4p is the catalytic subunit of a Ccr4p/Pop2p/Notp mRNA deadenylase complex in Saccharomyces cerevisiae. EMBO J. 2002; 21:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Viswanathan P., Ohn T., Chiang Y.C., Chen J., Denis C.L.. Mouse CAF1 can function as a processive deadenylase/3′-5′-exonuclease in vitro but in yeast the deadenylase function of CAF1 is not required for mRNA poly(A) removal. J. Biol. Chem. 2004; 279:23988–23995. [DOI] [PubMed] [Google Scholar]
- 41. Webster M.W., Chen Y.H., Stowell J.A.W., Alhusaini N., Sweet T., Graveley B.R., Coller J., Passmore L.A.. mRNA deadenylation is coupled to translation rates by the differential activities of Ccr4-Not nucleases. Mol. Cell. 2018; 70:1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Clark L.B., Viswanathan P., Quigley G., Chiang Y.C., McMahon J.S., Yao G., Chen J., Nelsbach A., Denis C.L.. Systematic mutagenesis of the Leucine-rich Repeat (LRR) domain of CCR4 reveals specific sites for binding to CAF1 and A separate critical role for the LRR in CCR4 deadenylase activity. J. Biol. Chem. 2004; 279:13616–13623. [DOI] [PubMed] [Google Scholar]
- 43. Wahle E., Winkler G.S.. RNA decay machines: deadenylation by the Ccr4-Not and Pan2-Pan3 complexes. Biochim. Biophys. Acta - Gene Regul. Mech. 2013; 1829:561–570. [DOI] [PubMed] [Google Scholar]
- 44. Godwin A.R., Kojima S., Green C.B., Wilusz J.. Kiss your tail goodbye: the role of PARN, Nocturnin, and Angel deadenylases in mRNA biology. Biochim. Biophys. Acta - Gene Regul. Mech. 2013; 1829:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baggs J.E., Green C.B.. Nocturnin, a deadenylase in Xenopus laevis retina: a mechanism for posttranscriptional control of circadian-related mRNA. Curr. Biol. 2003; 13:189–198. [DOI] [PubMed] [Google Scholar]
- 46. Pinto P.H., Kroupova A., Schleiffer A., Mechtler K., Jinek M., Weitzer S., Martínez J.. ANGEL2 is a member of the CCR4 family of deadenylases with 2′,3′-cyclic phosphatase activity. Science (80-.). 2020; 530:524–530. [DOI] [PubMed] [Google Scholar]
- 47. Su Z., Kuscu C., Malik A., Shibata E., Dutta A.. Angiogenin generates specific stress-induced tRNA halves and is not involved in tRF-3-mediated gene silencing. J. Biol. Chem. 2019; 53:1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blázquez M., Fominaya J.M., Hofsteenge J.. Oxidation of sulfhydryl groups of ribonuclease inhibitor in epithelial cells is sufficient for its intracellular degradation. J. Biol. Chem. 1996; 271:18638–18642. [DOI] [PubMed] [Google Scholar]
- 49. Yip M.C.J., Keszei A.F.A., Feng Q., Chu V., McKenna M.J., Shao S.. Mechanism for recycling tRNAs on stalled ribosomes. Nat. Struct. Mol. Biol. 2019; 26:343–349. [DOI] [PubMed] [Google Scholar]
- 50. Yip M.C.J., Savickas S., Gygi S.P., Shao S.. ELAC1 repairs tRNAs cleaved during Ribosome-Associated quality control. Cell Rep. 2020; 30:2106–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lizano E., Scheibe M., Rammelt C., Betat H., Mörl M.. A comparative analysis of CCA-adding enzymes from human and E. coli: differences in CCA addition and tRNA 3′-end repair. Biochimie. 2008; 90:762–772. [DOI] [PubMed] [Google Scholar]
- 52. Chatterjee K., Nostramo R.T., Wan Y., Hopper A.K.. tRNA dynamics between the nucleus, cytoplasm and mitochondrial surface: location, location, location. Biochim. Biophys. Acta. 2018; 1861:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Novoa E.M., Ribas de Pouplana L.. Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. 2012; 28:574–581. [DOI] [PubMed] [Google Scholar]
- 54. Gingold H., Tehler D., Christoffersen N.R., Nielsen M.M., Asmar F., Kooistra S.M., Christophersen N.S., Christensen L.L., Borre M., Sørensen K.D.et al.. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014; 158:1281–1292. [DOI] [PubMed] [Google Scholar]
- 55. Horn D. Codon usage suggests that translational selection has a major impact on protein expression in trypanosomatids. BMC Genomics. 2008; 9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jeacock L., Faria J., Horn D.. Codon usage bias controls mRNA and protein abundance in trypanosomatids. Elife. 2018; 7:e32496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Svenningsen S.Lo., Kongstad M., Stenum T.S., Muñoz-Gómez A.J., Sørensen M.A.. Transfer RNA is highly unstable during early amino acid starvation in Escherichia coli. Nucleic Acids Res. 2017; 45:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Adl S.M., Simpson A., Lane C., Lukes J., Bass D., Bowser S.S., Brown M., Burki F., Dunthorn M., Hampl V.et al.. The revised classification of eukaryotes. J. Eukaryote Microbiol. 2013; 59:429–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cristodero M., Polacek N.. The multifaceted regulatory potential of tRNA-derived fragments. Non-coding RNA Investig. 2017; 1:7. [Google Scholar]
- 60. Polacek N., Ivanov P.. The regulatory world of tRNA fragments beyond canonical tRNA biology. RNA Biol. 2020; 17:1057–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Soares A.R., Santos M.. Discovery and function of transfer RNA-derived fragments and their role in disease. WIREs RNA. 2017; 8:doi:10.1002/wrna.1423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.