


Abstract
DNA double-strand breaks (DSBs) at ribosomal gene loci trigger inhibition of ribosomal DNA (rDNA) transcription and extensive nucleolar reorganization, including the formation of nucleolar caps where rDNA DSBs engage with canonical DSB signaling and repair factors. While these nucleolar responses underlie maintenance of rDNA stability, the molecular components that drive each of these events remain to be defined. Here we report that full suppression of rRNA synthesis requires the DYRK1B kinase, a nucleolar DSB response that can be uncoupled from ATM-mediated DSB signaling events at the nucleolar periphery. Indeed, by targeting DSBs onto rDNA arrays, we uncovered that chemical inhibition or genetic inactivation of DYRK1B led to sustained nucleolar transcription. Not only does DYRK1B exhibit robust nucleolar accumulation following laser micro-irradiation across cell nuclei, we further showed that DYRK1B is required for rDNA DSB repair and rDNA copy number maintenance, and that DYRK1B-inactivated cells are hypersensitised to DSBs induced at the rDNA arrays. Together, our findings not only identify DYRK1B as a key signaling intermediate that coordinates DSB repair and rDNA transcriptional activities, but also support the idea of specialised DSB responses that operate within the nucleolus to preserve rDNA integrity.
INTRODUCTION
Maintenance of ribosomal DNA (rDNA) stability poses immense challenge to the host DNA repair machineries (1). Indeed, its repetitive structure and the high transcriptional activity associated with rRNA synthesis renders it particularly prone to recombination and breakage, deleterious events that could lead to rDNA loss and perturbed cell homeostasis (2). Notably, recent studies are unveiling a collection of specialised nucleolar DNA damage responses (DDRs) that, in concerted effort, swiftly and faithfully repair rDNA damage, although the molecular bases that drive each of these DDRs remain to be defined (1).
DNA double-strand breaks (DSBs) are arguably the most toxic form of DNA damage, and it has become evident that DSBs that target the rDNA loci trigger extensive nucleolar restructuring, and that DSB-inflicted rDNA is shunted to the nucleolar periphery (a.k.a. nucleolar caps) where canonical DSB signaling and repair factors, including γH2AX and 53BP1, accumulate. In this context, the master DDR kinase ATM plays an apical role to orchestrate a wide range of cellular responses to DNA damage; Not only does ATM promote the inhibition of RNA polymerase I (RNAPI) transcription in trans (3–5), but emerging evidence indicates that it also mediates localised nucleolar responses to rDNA DSBs (6,7). Importantly, while mobilization of rDNA DSB to nucleolar caps coincide with inhibition of rDNA transcription, more recent findings suggest that these ATM-dependent events can be uncoupled (8,9), underscoring the complexity of the DDRs in the non-membrane bound organelle.
We recently identified the DYRK1B kinase as a signaling intermediate that regulates transcriptional activity on the damaged chromatin (10). DYRK1B is recruited to laser-induced DNA damage tracks, and is required for transcription silencing on the DSB-flanking chromatin. Although DYRK1B promotes repair of ionizing radiation (IR)-induced DSBs and chromosomal stability, it remains unknown whether the kinase may coordinate rDNA DSB repair by suppressing gene expression in the nucleoli.
MATERIALS AND METHODS
Cell culture
HeLa, U2OS, hTERT RPE-1 and HEK293T cells were obtained from ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) media supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin (Gibco) at 37°C in 5% CO2.
Plasmids
Plasmids used in this study are listed in the Supplementary Table S1.
Antibodies
Antibodies used for immunofluorescence (IF) staining experimentations: Anti-Ki-67 (EMD Millipore, #MAB4190); Anti-C23 (H-250; Santa Cruz, #sc-13057); Anti-γH2AX (11); Anti-53BP1 (11); Anti-UBF (F-9) (Santa Cruz, #sc-13125); Anti-BRCA1 (D9; Santa Cruz, #sc-6954); Anti-Rad51 (14B4; Novus Biologicals; NB100-148); Anti-RNF169 (12); Anti-Flag (M2; Sigma-Aldrich, #F3165); Anti-HA.11 Epitope Tag Antibody (BioLegend, #901514); Alexa Fluor 594 AffiniPure Goat Anti-Mouse IgG (H+L) (Jackson ImmunoResearch, #115-585-166); Alexa Fluor 488 AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch, #111-545-144).
Antibodies used for western blotting: Anti-DYRK1B (H-6; Santa Cruz, #sc-390417); Anti-RNF169 (12); Anti-Flag (M2; Sigma-Aldrich, #F3165); Anti-HA.11 Epitope Tag Antibody (BioLegend, #901514); Anti-β-Actin (Sigma-Aldrich, #A5441); Anti-Human IgG antibody (Abcam, #ab2410); Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 405 (Thermo Fisher Scientific, # A31556); Peroxidase AffiniPure Rabbit Anti-mouse IgG+IgM (H+L) (Jackson ImmunoResearch, #315-035-048); Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch, #111–035-144).
Small molecule inhibitors
ATM inhibitor (KU-55933, Selleckchem, #S1092, 10 μM); DYRK1B inhibitor (AZ191, Selleckchem, #S7338, 10 μM); PARP inhibitor (Olaparib, Selleckchem, #S1060, 10 μM); DMSO (Sigma-Aldrich, #D2650).
Lentivirus packaging and cell line generation
U2OS/HeLa/RPE-1 I-PpoI cells: Lentivirus was generated by co-transfection with pLVX-PTuner-I-PpoI, psPAX2 (Addgene, plasmid#12260) and pMD2.G (Addgene, plasmid#12259) using polyethylenimine (PEI) (Polysciences, #23966) in HEK293T cells. Lentivirus was harvested at 48 h post-transfection and filtered with Syringe Filter (0.45 μM membrane, PALL Lifer Sciences). U2OS/HeLa/RPE-1 I-PpoI cells were infected with lentivirus in DMEM medium supplemented with 10% FBS and 8 μg ml−1 polybrene (Sigma-Aldrich) for 24 h. Cells were selected in normal growth media with 1 μg ml−1 puromycin (Sigma-Aldrich) for 1 week.
CRISPR/Cas9-mediated knockout (KO) cells: non-targeting control gRNA and single guide RNAs (sgRNAs) targeting sequences were cloned into LentiCRISPR v2 vector (Addgene, plasmid#52961) following the target guide sequence cloning protocol from Feng Zhang's Lab. Lentivirus was generated as described above. Cells were incubated with the media containing lentivirus supplemented with 8 μg ml−1 polybrene (Sigma-Aldrich) twice at 24 h intervals. After puromycin selection for 1 week, pooled cells were switched to normal culture medium and the knockout efficiency was validated by western blotting. Sequences of sgRNAs are listed in Supplementary Table S2.
Drug treatment
For the activation of rDNA DSBs, I-PpoI cells were pre-treated with inhibitors for 1 h before the addition of Shield-1 (TaKaRa, #632189, 1 μM) and 4-OHT (Sigma-Aldrich, #H7904, 2 μM) for indicated period of time. For live cell imaging, cells were pre-treated with indicated inhibitors prior to laser microirradiation.
Small interfering RNA (siRNA)-mediated knockdown
Cells were transfected with non-targeting control or gene-specific siRNAs (Dharmacon) using Lipofectamine (Invitrogen) twice at 24 h intervals. Samples were collected and subjected to western blotting to evaluate knockdown efficiency. Sequences of siRNAs are listed in Supplementary Table S3.
5-EU incorporation assay
Transcription activity was measured by Click-iT RNA Alexa FluorTM 594 Imaging Kit (Thermo Fisher Scientific, #C10330) following manufacturer's guidelines. Briefly, pre-treated cells grown on coverslips were incubated with medium containing recommended 5-ethynyl uridine (EU) concentration for 1 h followed by fixation, permeabilization, and 5-EU detection. Cells were immunostained with γH2AX or the indicated antibodies before DAPI staining. After mounting, cells were subjected to imaging using the wide-field microscope (Olympus BX53). 5-EU nucleolar intensity profile was analysed by ImageJ software (NIH) using the ROI manager with a circle tool centered in the nucleoli. γH2AX/53BP1-marked nucleolar caps were used to outline the nucleoli after rDNA DSBs induction and nucleoli with large size and distinct margins were subjected to analysis. The intensity plots were generated by the averaged 5-EU reads in the nucleoli normalised to the averaged reads with the same area selection which was outside of and adjacent to the nucleoli in the same nuclei. More than 200 cells with nucleoli of large size and clear margins were analysed from at least two independent experiments.
Nuclear run-on assay
After the induction of rDNA DSBs, cells were chilled with ice-cold DMEM medium on ice for 10 min and were pulsed-labeled with medium containing 1 mM 5-ethynyl uridine (EU) for 20 min to assess transcription activity. rDNA DSBs were induced with Shield-1 and 4-OHT prior to cell fixation. After fixation, 5-EU detection and analysis were performed as described in the 5-EU incorporation assay.
Western blotting and co-immunoprecipitation
Harvested cells were washed with PBS and lysed with ice-cold NETN buffer [20 mM Tris·HCl (pH 8.0), 100 mM NaCl, 0.5% Nonidet P40, and 1 mM EDTA] with the addition of Benzonase nuclease (ChemCruz) for 30 min on ice. Whole cell lysates were subsequently subjected to SDS-PAGE and transferred onto PVDF membrane. After blocking in 5% milk for 1h, the membranes were incubated with primary antibodies diluted in 3% bovine serum albumin (BSA)/Tris-buffered saline + Tween 20 (TBST) at 4°C overnight followed by incubation with peroxidase-coupled secondary antibodies at room temperature (RT). Proteins were detected by chemiluminescence solutions (SuperSignal West Pico PLUS Stable Peroxide Solution and Luminol/Enhancer Solution, Thermo Scientific) using ChemiDoc MP Imaging System (Bio-Rad). For co-immunoprecipitation, whole cell extracts were centrifuged at 15 000 rpm for 15 min at 4°C after lysis, supernatants were incubated with either streptavidin-conjugated beads (GE Healthcare, Sigma-Aldrich) for 4 h or Protein A beads overnight at 4°C. The beads were washed with ice-cold NETN buffer three times prior to immunoblotting.
Tandem affinity purification
HEK293T cells were transfected with S protein-Flag-Streptavidin binding peptide (SFB)-tagged DYRK1B or RNF169 plasmids and selected in medium containing 1 μg ml−1 puromycin. Cells stably expressing SFB-tagged proteins were validated by western blotting and immunofluorescence. For affinity purification, cells were lysed in ice-cold NETN buffer supplemented with Benzonase nuclease (ChemCruz) for 30 min on ice. After centrifugation, the supernatant was incubated with 200 μl streptavidin-conjugated beads (GE Healthcare, Sigma-Aldrich) at 4°C for 4 h with gentle rotation. Then the pellet was washed by ice-cold NETN buffer twice and subsequently resuspended with 1 ml NETN buffer containing 2 μg ml−1 biotin (Sigma-Aldrich). Incubation at 4°C for 1 h with gentle rotation. The biotin-elutes were then incubated with 60 μl S-protein agarose (Novagen) at 4°C for 2 h. S-protein agarose bounded proteins were washed twice with NETN buffer, resolved by SDS-PAGE and subjected to mass spectrometry analysis.
Immunofluorescence staining
After treatment, cells grown on coverslips in six-well plate were fixed with 3% paraformaldehyde (PFA) for 30 min and permeabilized with 0.5% Triton X-100 for 30 s at RT. After washing twice with 1× PBS, cells were incubated with primary antibodies diluted in 3% BSA/TBST for 45 min at RT. Appropriately diluted secondary antibodies (Alexa Fluor 488 and Alexa Fluor 594 antibodies) were added onto the coverslips for another 45 min at RT followed by 4’,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, #D1306) staining. Coverslips were mounted with fluorescence mounting medium (Dako, Agilent) and were subjected to microscopy using a wide-field microscope (Olympus BX53) equipped with a digital camera (Hamamatsu, ORCA Flash4.0LT). Images were captured using HCImageLive software.
Live cell imaging
Cells cultured on confocal dishes (SPL Life Sciences) were transiently transfected with GFP tagged plasmids using PEI (Polysciences, #23966) and subjected to laser microirradiation 24 h post-transfection. Laser microirradiation was performed in the chamber (37°C, 5% CO2) with Carl Zeiss LSM 780 microscope equipped with laser microbeam system. Time-lapse images were captured at 2-s intervals following laser damage by ZEN 2012 (Carl Zeiss) software and the mean fluorescence intensity (MFI) of the indicated proteins at the laser-microirradiated stripes was quantified by ImageJ (NIH) software. Briefly, the ROI manager with the rectangle tool in ImageJ was used to measure the accumulation of protein at laser-microirradiated stripes both in nucleoli and in nucleoplasm. In nucleoli, the MFI reads of the damaged region were normalized to the corresponding undamaged region with the same area selection in a nucleolus to generate relative MFI plots. Similarly, the intensity reads at laser-induced damaged tracks were normalized to the adjacent undamaged region with same area selection in the same nucleus to create relative MFI plots of the indicated protein in nucleoplasm. A minimum of 10 cells in each group were counted from at least two independent experiments.
Neural comet assay
After treatment, cells were harvested at 5 × 105 cells ml−1 in ice-cold PBS and mixed in 37°C molten LMAgarose (Trevigen) at a 1:10 dilution ratio. The mixture was transferred onto 37°C prewarmed comet slides (Trevigen) and spread evenly. The slides were subsequently incubated in dark at 4°C for 30 min. After incubation, slides were immersed in prechilled lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris–HCl, 1% N-lauroylsarcosine sodium, 1% Triton X-100) for 1 h at 4°C and then in freshly prepared alkaline solution for 30 min at RT. Slides were washed with 1× TBE (Tris–borate–EDTA) buffer twice and subjected to TBE electrophoresis. Samples were fixed in absolute ethanol for 5 min. After air dry, slides were stained with 1 μg ml−1 propidium iodide (Sigma-Aldrich) for 30 min at RT in the dark. Slides were rinsed in distilled water and were subjected to microscopy after complete air dry. Images were processed and analysed with OpenComet plugin by ImageJ (NIH) software.
RNA isolation and RT-qPCR
RNA extraction was performed with Illustra RNAspin Mini (GE Healthcare, #25-0500-72) following the manufacturer's instructions. 1 μg RNA was processed for reverse transcription using SuperScript II Reverse Transcriptase (Invitrogen, #18064-014). The real-time PCR was performed with the diluted cDNA, the indicated primers and iTaq Universal SYBR Green Supermix (Bio-Rad, #172-5124) on CFX96 Real-Time System (Bio-Rad). Data were acquired by Bio-Rad CFX Maestro software and analysed by 2−ΔΔCT method with GAPDH used as the internal reference gene. Primers used for RT-qPCR are listed in Supplementary Table S4.
Genomic DNA extraction and real-time qPCR
Genomic DNA extraction was performed with QIAamp DNA Mini Kit (QIAGEN, #51304) following the manufacturer's recommendation. Genomic DNA was subjected to real time PCR and analysed as described above. Primers used for genomic DNA qPCR are listed in Supplementary Table S5.
Micronuclei measurement
After treatment, cells were fixed with 3% PFA for 15 min followed by permeabilization with 0.5% Triton X-100 at RT. Cells were counterstained with DAPI and were subjected to microscopy for micronuclei quantification. At least 800 cells were counted from three independent experiments.
Clonogenic survival assay
HeLa I-PpoI cells were seeded on 60 mm dishes. At 24 h after seeding, cells were treated with Shield-1 (TaKaRa, #632189, 1 μM) and 4-OHT (Sigma-Aldrich, #H7904, 2 μM) for either 5 min or 10 min to induce rDNA DSBs. After washing with PBS twice, cells were cultured in medium supplemented with 10% FBS for 2 weeks before fixation. Coomassie Blue solutions stained colonies were counted by Quantity One Analysis Software (Bio-Rad). HeLa cells were pre-treated with ionizing radiation (IR), ultraviolet (UV), or the indicated concentrations of cisplatin for 12 h and subjected to clonogenic survival assays as described above.
Statistical analyses
All data are presented as mean ± SEM from three independent experiments unless otherwise noted. Comparisons between indicated groups were analysed by two-tailed Student's t test using Prism 8 (GraphPad) software. Statistical significance is indicated as follows: *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001; ns, not significant.
RESULTS
DYRK1B facilitates transcriptional silencing at rDNA DSBs
To investigate whether DYRK1B mediates DSB-induced transcriptional repression at the rDNA loci, we took advantage of the inducible I-PpoI platform in which the I-PpoI endonuclease can be induced by cell pre-treatment with 4-hydroxytamoxifen (4-OHT) and Shield-1. The I-PpoI endonuclease, fused N-terminally with the estrogen receptor (ER) peptide, generates site-specific DSBs within the 28S sequence of rDNA array (Figure 1A). We established I-PpoI-inducible stable U2OS cells, and validated that the I-PpoI endonuclease generates rDNA DSBs and induces nucleolar cap formation following 4-OHT and Shield-1 treatment. Accordingly, upon I-PpoI induction, we observed robust accumulation of DSB markers γH2AX and 53BP1 at the nucleolar periphery, as well as the redistribution of nucleolin (C23) and upstream binding factor (UBF) at nucleolar caps (Supplementary Figure S1A–C). Using this platform, we found that cells pre-treated with the DYRK1B-specific inhibitor (DYRK1Bi; AZ191), similar to those targeted with the ATM inhibitor KU55933 (ATMi), failed to suppress de novo RNA synthesis in the nucleoli (Figure 1B), as evident by the sustained nucleolar incorporation of the uridine analogue 5-ethynyl uridine (5-EU). This observation suggested that the DYRK1B kinase activity may be required for nucleolar transcription silencing following introduction of rDNA DSBs. Consistently, similar observations were made in cells pre-treated with two independent small interference RNAs (siRNAs) that targeted DYRK1B (Figure 1C). Consistent with the requirement of DYRK1B kinase activity in suppressing rDNA transcription following I-PpoI induction, we found that wildtype DYRK1B, but not its kinase inactive mutants (K140M and D239A), complemented loss of DYRK1B in rDNA DSB-induced transcription silencing (Figure 1D).
Figure 1.

DYRK1B promotes rDNA DSB-induced transcription suppression. (A) Schematic illustration of 45 rDNA repeats with I-PpoI target site. (B) Analysis of transcription activity by 5-Ethynyl Uridine (EU) incorporation assay in DYRK1B inactivated U2OS I-PpoI cells following rDNA DSBs. U2OS I-PpoI cells were pre-treated with ATM inhibitor (ATMi; KU-55933) or DYRK1B inhibitor (DYRK1Bi; AZ191) for 1 h prior to the addition of 1 μM Shield-1 and 2 μM 4-OHT for 4 h. Cells were subsequently cultured in medium supplemented with 5-EU for 1 h before fixation. Fixed cells were labelled with 5-EU and 53BP1. Nuclei were counterstained with DAPI. The dashed circles indicate the edge of the nuclei. The arrowheads indicate nucleoli. Quantification of relative 5-EU nucleolar intensity from two independent experiments is shown in Turkey boxplots. 200 cells with nucleoli of large size and clear margins were analysed. ns, not significant; ****P< 0.0001. (C) HeLa I-PpoI cells transfected with control (siControl) or DYRK1B-targeted siRNAs (siDYRK1B-1 and siDYRK1B-2) were induced rDNA DSBs as described in (B) for 4 h before subjecting to 5-EU incorporation assay. Western blotting was performed to measure siRNA-mediated DYRK1B knockdown efficiency using the indicated antibodies. The squares show the enlarged nucleoli. Asterisk denotes the band of endogenous DYRK1B. Quantification was performed and is shown as Turkey boxplots as in (B). ns, not significant; ****P< 0.0001. (D) RPE-1 I-PpoI cells transduced with DYRK1B gRNA (DYRK1B gRNA#1) were reconstituted with doxycycline (Dox)-inducible vector or DYRK1B alleles (WT, wild type; Kinase-dead mutants, K140M and D239A). Cells were induced with 0.5 μg/ml Dox for 24 h and were subjected to 5-EU incorporation assay after I-PpoI induction. Enlarged images show the details of the nucleoli. Quantification was derived from three independent experiments and is shown in Turkey boxplots. 200 cells with nucleoli of large size and clear margins were analysed. ****P< 0.0001. The expression of DYRK1B was evaluated by western blotting. Asterisk denotes the band of endogenous DYRK1B.
To corroborate that 5-EU incorporation within the non-membrane bound organelle reflected de novo rRNA synthesis, we quantified the expression of pre-rRNA by quantitative real-time polymerase chain reaction (RT-PCR) (Supplementary Figure S2A). In agreement with a role of DYRK1B in promoting rDNA DSB-induced transcriptional repression, we found that DYRK1B inhibition, similar to ATMi cells but albeit to a milder extent, reproducibly led to compromised suppression of pre-rRNA synthesis (Supplementary Figure S2B and C). To further exclude that change in pre-rRNA level may be an indirect result arising from difference in RNA transcript processing, we performed nuclear run-on experimentations (Supplementary Figure S2D), and found that both chemical inhibition as well as RNAi-mediated depletion of DYRK1B compromised rDNA DSB-induced transcription silencing (Supplementary Figure S2E and F). Together, these data suggest that DYRK1B promotes rDNA DSB-induced transcriptional silencing, and that its role in suppressing nucleolar transcription may be effected via a kinase-dependent manner.
DYRK1B-mediated nucleolar transcription silencing is independent of nucleolar segregation
Given the pivotal roles of the master DDR kinase ATM in driving nucleolar responses to rDNA DSBs, including nucleolar segregation and inhibition of rRNA synthesis, we next tested whether DYRK1B may elicit similar functions in the subcellular compartment following I-PpoI induction. To this end, we first examined the distributions of nucleolar proteins, including UBF, C23 and Ki-67 following I-PpoI induction (Figure 2). In stark contrast to those observed in ATM-inhibited cells, we found that chemical inhibition of DYRK1B did not noticeably affect the I-PpoI-elicited redistribution of UBF, C23 or Ki-67 (Figure 2A–C), suggesting that DYRK1B may specifically promote nucleolar transcription repression, and that it may facilitate one of the many ATM-dependent pathways in the nucleolar DDR (Figure 2D). In support of this notion, accumulation of DDR factor BRCA1 and 53BP1 at nucleolar caps was not affected following DYRK1B depletion (Supplementary Figure S3A–C). Similar to cells pre-treated with the DYRK1B inhibitor AZ191 (Figure 2A), while CRISPR/Cas9-mediated DYRK1B inactivation did not noticeably affect redistribution of UBF onto I-PpoI-induced nucleolar caps (Supplementary Figure S3D), UBF is not completely depleted from the nucleoli.
Figure 2.

Uncoupling of ATM-dependent nucleolar cap formation. (A–C) U2OS I-PpoI cells were pre-treated with ATM inhibitor (ATMi; KU-55933) or DYRK1B inhibitor (DYRK1Bi; AZ191) for 1 h before supplementation with Shield-1 and 4-OHT for 4 hr. After fixation, cells were processed for immunofluorescence with anti-UBF (A), anti-C23 (B), anti-Ki-67 (C) or anti-γH2AX antibodies, respectively. The squares show the enlarged nucleoli and the dashed circles indicate margins of the enlarged nucleoli. Percentages of cells with the indicated nucleolar caps were quantified from at least two experiments and 300 cells from each condition were analysed. Bars represent mean ± SEM; ns, not significant; ***P< 0.001; ****P< 0.0001. (D) Graph illustrates rDNA DSBs-induced nucleolar reorganization following ATM or DYRK1B inhibition.
RNF169 is a bona fide DYRK1B-interacting protein
To further explore how DYRK1B participates in nucleolar responses to DSBs, we profiled the DYRK1B interactome. To this end, we affinity-purified DYRK1B protein complexes from HEK293T cells that stably express S protein-Flag-Streptavidin binding peptide (SFB)-tagged DYRK1B, and subjected DYRK1B co-purifying proteins to liquid chromatography with tandem mass spectrometry (LC–MS/MS) for protein identification (Figure 3A). We compared the list of DYRK1B-interacting proteins to those that resided in the RNF169 macromolecular complexes (Figure 3B) (12), and decided to examine in more details the putative interaction between DYRK1B and RNF169, as RNF169 plays an established role in the DDR, including an ability to localize at DSBs (12–16). Consistently, we validated an endogenous DYRK1B-RNF169 interaction by co-immunoprecipitation (co-IP) (Figure 3C). To better understand the DYRK1B-RNF169 complex formation, we took advantage of an existing panel of RNF169 deletion mutants (12), and mapped the DYRK1B-binding domain on the RNF169 polypeptide (Figure 3D). Domain-mapping results indicated that DYRK1B binds on the RNF169 C-terminus (Figure 3E). We further narrowed down the DYRK1B-binding region, and showed that deleting the 334–408 residues on RNF169 was sufficient to abolish its interaction with DYRK1B (Figure 3F and G). Taken together, these data suggest that the Ring finger protein RNF169 is a bona fide DYRK1B-interacting factor.
Figure 3.

RNF169 is a bona fide DYRK1B-interacting protein. (A) Affinity purification of DYRK1B complex identified a list of DYRK1B-interacting proteins. Names and total peptides of the top hits are listed. RNF169 is highlighted in red. (B) RNF169-associated proteins were identified from tandem affinity purification of RNF169 protein. DYRK1B is shown in red. (C) Co-immunoprecipitation (IP) in HEK293T cells. Whole cell lysates incubated with protein A beads were supplemented with either IgG antibody or RNF169 antibody at 4°C overnight. Protein A-conjugated proteins were subjected to western blotting using anti-DYRK1B and anti-RNF169 antibodies. (D) Schematic illustration of RNF169 protein domains. ‘+’ represents DYRK1B binds to the indicated RNF169 truncated mutants and ‘–’ denotes DYRK1B binding defective mutants. NLS, nuclear localization signal; RING, really interesting new gene; MIU, motif interacting with ubiquitin. (E) HEK293T cells were transiently co-transfected with Strep-Flag tagged DYRK1B and HA tagged (HA-) wildtype (WT) RNF169 or the indicated RNF169-truncated mutants. Cell lysates were incubated with Streptavidin beads and the immunoprecipitates were subjected to western blotting using anti-Flag and anti-HA antibodies. HA tagged RNF168 (HA-RNF168) was used as a DYRK1B-binding defective control. (F) Graph depicting the C-terminal deletion mutants of RNF169 protein. ‘+’ indicates DYRK1B-binding mutants and ‘−’ denotes DYRK1B-binding defective mutants, respectively. (G) HEK293T cells were co-transfected with Strep-Flag tagged DYRK1B and HA tagged RNF169 WT or RNF169 C-terminal deletion mutants. After lysis, the lysates were processed for co-immunoprecipitation using the indicated antibodies.
DYRK1B is recruited to DSBs in PARP-dependent and -independent pathways
Given that DYRK1B can be recruited to laser microirradiated sites, and that PARP mediates early recruitment of many DDR factors to DSBs, we examined the accumulation of DYRK1B at laser-induced DNA damage tracks following PARP inhibition. Interestingly, we found that PARP inhibition abrogated DYRK1B accumulation at laser microirradiated DNA damage tracks in the nucleoplasm, but did not noticeably affect its recruitment within the nucleoli (Figure 4A), suggesting that DYRK1B concentration at rDNA DSBs may be effected via distinct genetic requirements. In line with this idea, we observed robust accumulation and colocalization of mCherry-tagged (mCherry-) DYRK1B and green fluorescence protein (GFP)-tagged RNF169 (GFP-RNF169), but not GFP-53BP1 at the damaged nucleoli (Figure 4B and C, Supplementary Figure S4A and B). These observations highlight potentially dissimilar mechanisms that drive RNF169 and 53BP1 docking onto DSBs in different subcellular compartments. To examine whether DYRK1B mobilization onto laser-induced DNA damage sites may require RNF169, we depleted RNF169 in cells transduced with two independent RNF169-targeting guide RNAs (gRNAs), and found that RNF169 promoted DYRK1B accumulation at laser-induced DSBs in both nucleoplasm and nucleoli (Figure 4D & Supplementary Figure S5A and B). By contrast, DYRK1B depletion did not noticeably affect GFP-RNF169 accumulation at laser-induced DNA damage sites (Supplementary Figure S4C). To consolidate the role of RNF169 in facilitating DYRK1B recruitment at DSBs, we further showed that RNF169 promotes the accumulation of DYKR1B at nucleolar caps following I-PpoI-induced rDNA DSBs (Figure 4E) and IR-induced DSBs (Supplementary Figure S4D).
Figure 4.

Regulation of DYRK1B dynamics in response to DNA damage. (A) U2OS cells transfected with GFP tagged DYRK1B (GFP-DYRK1B) expression construct were pre-treated with PARP inhibitor (PARPi; olaparib) for 1 h before subjecting to live cell imaging. Relative mean fluorescence intensity (MFI) at laser-induced stripes in nucleoli and nucleoplasm were quantified. Arrowheads denote laser-microirradiated tracks. Red rectangle and white rectangle denote the area for GFP-DYRK1B relative MFI in nucleoli and nucleoplasm, respectively. The dashed rectangles show the corresponding referenced areas for relative MFI in nucleoli or nucleoplasm. At least 10 cells from two independent experiments were analysed. Bars represent mean ± SEM. (B, C) U2OS cells co-transfected with mCherry tagged DYRK1B (mCherry-DYRK1B) and GFP-RNF169 (B) or GFP-53BP1 (C) were subjected to laser microirradiation. Arrowheads denote sites of laser-microirradiation. Zoomed images show the details of the nucleoli denoted with white square. (D) U2OS cells transduced with non-targeting gRNA (CTR gRNA) or RNF169-targeting gRNAs (RNF169 gRNA#2 and RNF169 gRNA#3) were transiently transfected with GFP-DYRK1B before live cell imaging. Quantification was processed as described in (A). At least 10 cells from two independent experiments were analysed. Western blotting was performed to evaluate RNF169 expression. (E) U2OS I-PpoI cells co-transfected Flag tagged (Flag-) DYRK1B with either HA tagged (HA-) wildtype (WT) RNF169 or DYRK1B-binding defective HA-RNF169 ΔC1 mutants were treated with Shield-1 and 4-OHT for 4 h to induce rDNA DSBs. Fixed cells were processed for immunofluorescence by labeling HA and Flag tags. Nuclei were visualized by staining with DAPI. The edge of the nuclei was labeled with dotted line. Zoomed images show the details of the nucleoli denoted with white squares. Quantification was derived from three independent experiments. 300 cells from each condition were analysed. Bars represent mean ± SEM; ns, not significant; ****P< 0.0001.
Given the apical role of ATM in mounting nucleolar responses to DSBs, we also examined the requirement of ATM activity in mobilizing DYRK1B following laser-induced DNA damage across cell nuclei. Interestingly, although chemical inhibition of ATM led to attenuated recruitment of GFP-DYRK1B across the nucleoplasm, it did not noticeably affect DYRK1B concentration in the nucleoli (Supplementary Figure S4E). These observations are in stark contrast to the requirement of ATM activity in fueling GFP-RNF169 accumulation in both compartments (Supplementary Figure S5C). Moreover, in line with a dispensable role of PARylation in the early recruitment of DYRK1B within the damaged nucleoli (Figure 4A), GFP-PARP1, which can be seen enriched within nucleolar compartments in unperturbed condition, did not further accumulate following local laser microirradiation (Supplementary Figure S4F). On the contrary, I-PpoI induction led to robust concentration of GFP-PARP1 at the nucleolar caps (Supplementary Figure S4G). Considering that RNF169 deficiency did not compromise rDNA DSB-induced transcription repression (Supplementary Figure S6A–C), these data together suggest that DYRK1B suppresses rDNA transcription in an RNF169-independent manner, and that the DYRK1B-RNF169 pathway may be involved in mediating one of the sub-branches of rDNA DSB responses (see Discussion).
DYRK1B promotes rDNA DSB repair and maintains rDNA stability
Failing to repair rDNA DSBs can lead to compromised rDNA stability (17). To examine whether DYRK1B may be required for rDNA repair, we performed neutral comet assay following cell recovery from I-PpoI induction (Figure 5A). Consistent with a role of DYRK1B in coordinating rDNA repair, we found that DYRK1B inactivation led to sustained DNA damage (Figure 5B & Supplementary Figure S7A). To further investigate whether rDNA DSB repair defect associated with DYRK1B loss may impact genome stability, we analysed micronucleus formation frequency (Figure 5C), and found elevated frequencies of micronuclei formation in DYRK1B-inhibited cells (Figure 5D), suggesting that DYRK1B preserves genome stability by facilitating repair of rDNA DSBs.
Figure 5.

DYRK1B promotes rDNA DSB responses. (A) Schematic diagram illustrates the workflow of neutral comet assay. (B) HeLa I-PpoI cells transduced with control gRNA (CTR gRNA) and DYRK1B-targeting gRNAs (DYRK1B gRNA#1 and DYRK1B gRNA#2) were induced with Shield-1 and 4-OHT for rDNA DSBs. Cells were subjected to neutral comet assay as described in (A). Relative tail moment of at least 800 cells from two independent experiments were analysed and plotted. Bars represent mean ± SEM; ****P<0.0001; ns, not significant. (C) Graph illustrating the workflow of micronuclei counting. Briefly, U2OS I-PpoI cells were pre-treated with 10 μM DYRK1B inhibitor (DYRK1Bi; AZ191) for 1 h prior to the I-PpoI induction. After fixation, nuclei were counterstained with DAPI and processed for microscopy. (D) Representative images from undamaged and I-PpoI activation-induced damaged cells were shown. At least 500 cells from three independent experiments were counted. Error bars represent mean ± SEM; ***P<0.001; ns, not significant. Arrowheads denote micronuclei. (E) Schematic diagram represents 45S rDNA repeats. I-PpoI targeted sequence is shown. The numbers and the coverage indicated by the arrowheads denote the primers used in this study. (F) Workflow illustrates the process of the measurement of rDNA copy number in I-PpoI survivor cells. Briefly, RPE-1 I-PpoI cells were induced with rDNA DSBs for 5 h and cultured for three weeks prior to genomic DNA extraction from the survivor cells. Genomic DNA was processed for real-time qPCR following the standard procedures. (G) The relative rDNA copy number in DYRK1B-deficient RPE-1 I-PpoI survivor cells was quantified as depicted in (F). Data were derived from three independent experiments. Bars represent mean ± SEM; *P< 0.05; **P< 0.01; ***P< 0.001; ****P< 0.0001; ns, not significant. (H) Colony survival of CRISPR–Cas9-mediated DYRK1B knockout (KO) HeLa I-PpoI cells following I-PpoI activation. rDNA DSBs in cells were induced with Shield-1 and 4-OHT for 5 or 10 min. After washing with PBS twice, cells were allowed to grow for two weeks before fixation and Coomassie Blue staining. Error bars indicate mean with 95% confidence interval (CI). *P< 0.05; ns, not significant. DYRK1B KO cells were validated by western blotting using anti-DYRK1B and anti-β-Actin antibodies. The asterisk denotes the band of endogenous DYRK1B.
To more directly examine how DYRK1B deficiency may affect rDNA stability, we next evaluated rDNA abundance following cell recovery from I-PpoI induction. We quantified rDNA copy number using quantitative PCR analyses (Figure 5E and F). Consistent with a role of DYRK1B in genome stability maintenance, we found that DYRK1B deficiency led to loss of rDNA copy number following recovery from nucleolar DSBs (Figure 5G), indicating that DYRK1B maintains rDNA stability. The above observations where DYRK1B promotes rDNA DSB repair prompted us to examine whether DYRK1B is also required for cell tolerance and survival to rDNA damage. Accordingly, we performed clonogenic survival assays and found that DYRK1B deficiency led to cell hypersensitivity to rDNA DSBs (Figure 5H & Supplementary Figure S7B), indicating that DYRK1B promotes cell viability in response to rDNA DSBs. Together, these data suggest that DYRK1B facilitates rDNA DSB repair, cell survival and preserves rDNA stability.
DISCUSSION
In this study, we report a role of the DYRK1B kinase as a suppressor of rDNA transcription during cellular responses to rDNA DSBs. We found that DYRK1B is targeted to laser-induced DNA damage sites within the nucleolar compartment, and is required of pausing of local transcriptional activities. Failing to suppress rDNA transcription resulted in sustained DNA damage and elevated frequencies of chromosomal aberrations, which in turn led to loss of rDNA copy number and cell hypersensitivity to rDNA DSBs. Together, our findings establish DYRK1B as a component of the mammalian nucleolar DSB response network (Figure 6).
Figure 6.
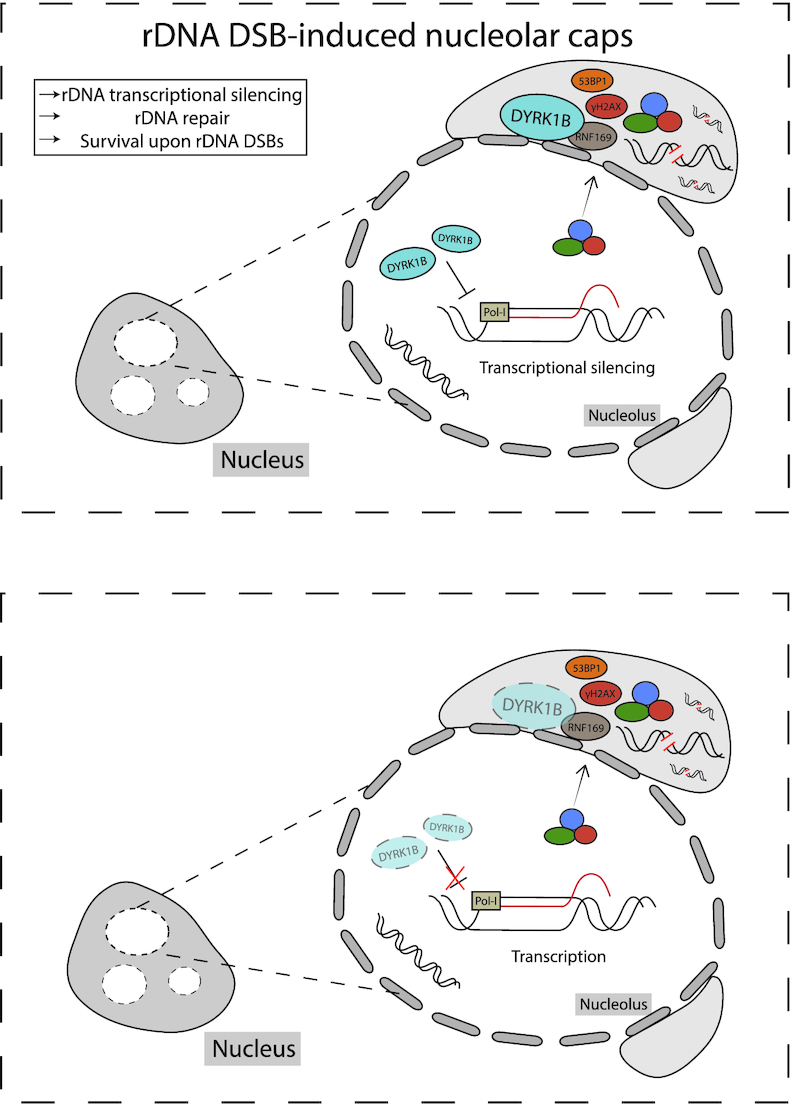
Working Model. Working model on DYRK1B-mediated silencing of rDNA transcription in response to DNA damage.
Emerging evidence suggest that rDNA array carrying DSBs are mobilized to the nucleolar periphery to undergo high-fidelity homologous recombination (HR) repair (9,17,18). Considering that non-homologous end joining (NHEJ) also effects rDNA DSB repair, but within the nucleoli (6), we envisage that a balance of HR and NHEJ may be in place, and that failing to suppress transcription may tilt this balance and compromise timely repair of rDNA DSBs. Thus, our observation wherein loading of RNF169 and RAD51 at nucleolar caps is compromised in DYRK1B-deficient cells (Supplementary Figure S8A–E), and that DDR signaling at nucleolar caps persisted in the absence of DYRK1B (Supplementary Figure S9A–C) are congruent of this idea. While further experimentations will be needed to fully dissect the choice of rDNA DSB repair, our findings that DYRK1B-inactivated cells suffered from sustained DNA damage and elevated micronuclei formation following I-PpoI induction highlight the kinase as a novel activity that participates in nucleolar DSB responses.
Although PARylation drives early responses to DNA damage, including the mobilization of DYRK1B onto nucleoplasmic DSBs (Figure 4A), we found that PARP activity was dispensable for DYRK1B accumulation inside the damaged nucleoli (Figure 4A). Moreover, the fact that 53BP1, but not RNF169, was excluded from concentrating in the subcellular compartment (Figure 4B, C & Supplementary Figure S4A, B) supports the idea that specialized DDRs are in place to preserve rDNA integrity. While the precise molecular bases that preferentially permit docking of DYRK1B over 53BP1 onto rDNA DSBs remain enigmatic, our data suggests that this is also effected via an ATM-independent pathway (Supplementary Figure S4E). That RNF169 is not required for DSB-induced transcription silencing at the local chromatin (Supplementary Figure S6A–C) suggests that the RNF169-DYRK1B complex may participate in a sub-pathway of rDNA DSB responses. Indeed, RNF169 appears dispensable for the formation of rDNA DSB-induced nucleolar caps (Supplementary Figure S10A–C), although ectopically expressed RNF169 is effective in displacing 53BP1 from these sub-nucleolar structures (Supplementary Figure S11A and B) just as it does at IRIF (12,13,15,16). Moreover, in support of a putative role of RNF169 in rDNA DSB responses, inactivation of RNF169 compromised rDNA DSB repair (Supplementary Figure S12A and B), led to loss of rDNA array repeats (Supplementary Figure S12C–F), and hypersensitized cells to rDNA DSBs (Supplementary Figure S12G, H). While DYRK1B and RNF169 may be epistatic in promoting cell survival by mounting rDNA DSB responses (Supplementary Figure S13A and B), further work is warranted to fully dissect the multifunctional roles of DYRK1B in both RNF169-dependent and RNF169-independent DDRs. Detailed analyses of the DYRK1B phospho-proteome (19) will also shed light in the molecular bases via which DYRK1B drives each of these rDNA damage responses.
While rDNA DSB-induced transcription suppression has been associated with formation of nucleolar caps (5,6,17,18), a scenario has emerged in which these nucleolar responses are driven by distinct mechanisms. Congruent with this notion, we found that DYRK1B deficiency compromised rDNA transcription silencing but not nucleolar segregation and DDR signaling at the nucleolar periphery, thus uncoupling the two nucleolar responses to rDNA DSBs. Given that DYRK1B is also required for preserving rDNA integrity following cell recovery from nucleolar DSBs, we propose that rDNA transcription suppression represents a pre-requisite for faithful rDNA DSB repair and for the maintenance of rDNA stability. Our observation that DYRK1B deficiency led to cell hypersensitivity to a wide variety of clastogens (Supplementary Figure S14A-D) also implicates a boarder role of the kinase in mammalian DNA damage responses.
DATA AVAILABILITY
All data are available upon request. Source of all expression constructs are detailed in Supplementary Table S1. Sequence of siRNAs, gRNAs and primer oligonucleotides are in Supplementary Table S2-5.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Roger Greenberg for the I-PpoI expression construct, Dr Simon Bekker-Jensen for GFP-53BP1 and Tina Wang for help with illustrations.
Contributor Information
Chao Dong, School of Biomedical Sciences, LKS Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong S.A.R.
Liwei An, School of Biomedical Sciences, LKS Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong S.A.R.
Cheng-han Yu, School of Biomedical Sciences, LKS Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong S.A.R.
Michael S Y Huen, School of Biomedical Sciences, LKS Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong S.A.R; State Key Laboratory of Brain and Cognitive Sciences, The University of Hong Kong, 5 Sassoon Road, Pokfulam, Hong Kong S.A.R.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Grants Council [C7007-17GF, 17104618, N_HKU722/19 to to M.S.Y.H.]. Funding for open access charge: RGC Hong Kong.
Conflict of interest statement. None declared.
REFERENCES
- 1. van Sluis M., McStay B.. Nucleolar DNA double-strand break responses underpinning rDNA genomic stability. Trends Genet. 2019; 35:743–753. [DOI] [PubMed] [Google Scholar]
- 2. Larsen D.H., Stucki M.. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2016; 44:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Larsen D.H., Hari F., Clapperton J.A., Gwerder M., Gutsche K., Altmeyer M., Jungmichel S., Toledo L.I., Fink D., Rask M.B.et al.. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell Biol. 2014; 16:792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ciccia A., Huang J.W., Izhar L., Sowa M.E., Harper J.W., Elledge S.J.. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:18631–18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kruhlak M., Crouch E.E., Orlov M., Montaño C., Gorski S.A., Nussenzweig A., Misteli T., Phair R.D., Casellas R.. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007; 447:730–734. [DOI] [PubMed] [Google Scholar]
- 6. Harding S.M., Boiarsky J.A., Greenberg R.A.. ATM dependent silencing links nucleolar chromatin reorganization to DNA damage recognition. Cell Rep. 2015; 13:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Korsholm L.M., Gál Z., Lin L., Quevedo O., Ahmad D.A., Dulina E., Luo Y., Bartek J., Larsen D.H.. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. 2019; 47:8019–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Siebenwirth C., Greubel C., Drexler G.A., Reindl J., Walsh D.W.M., Schwarz B., Sammer M., Baur I., Pospiech H., Schmid T.E.et al.. Local inhibition of rRNA transcription without nucleolar segregation after targeted ion irradiation of the nucleolus. J. Cell Sci. 2019; 132:jcs232181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marnef A., Finoux A.L., Arnould C., Guillou E., Daburon V., Rocher V., Mangeat T., Mangeot P.E., Ricci E.P., Legube G.. A cohesin/HUSH- and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev. 2019; 33:1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dong C., West K.L., Tan X.Y., Li J., Ishibashi T., Yu C.H., Sy S.M.H., Leung J.W.C., Huen M.S.Y. Screen identifies DYRK1B network as mediator of transcription repression on damaged chromatin. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:17019–17030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huen M.S., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J.. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007; 131:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J., Feng W., Jiang J., Deng Y., Huen M.S.. Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. J. Biol. Chem. 2012; 287:27715–27722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. An L., Dong C., Li J., Chen J., Yuan J., Huang J., Chan K.M., Yu C.H., Huen M.S.Y.. RNF169 limits 53BP1 deposition at DSBs to stimulate single-strand annealing repair. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E8286–E8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. An L., Jiang Y., Ng H.H., Man E.P., Chen J., Khoo U.S., Gong Q., Huen M.S.. Dual-utility NLS drives RNF169-dependent DNA damage responses. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E2872–E2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poulsen M., Lukas C., Lukas J., Bekker-Jensen S., Mailand N.. Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J. Cell Biol. 2012; 197:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Panier S., Ichijima Y., Fradet-Turcotte A., Leung C.C., Kaustov L., Arrowsmith C.H., Durocher D. Tandem protein interaction modules organize the ubiquitin-dependent response to DNA double-strand breaks. Mol. Cell. 2012; 47:383–395. [DOI] [PubMed] [Google Scholar]
- 17. Warmerdam D.O., van den Berg J., Medema R.H.. Breaks in the 45S rDNA lead to recombination-mediated loss of repeats. Cell Rep. 2016; 14:2519–2527. [DOI] [PubMed] [Google Scholar]
- 18. van Sluis M., McStay B.. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015; 29:1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong C., West K.L., Tan X.Y., Li J., Ishibashi T., Yu C.H., Sy S.M.H., Leung J.W.C., Huen M.S.Y. Screen identifies DYRK1B network as mediator of transcription repression on damaged chromatin. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:17019–17030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available upon request. Source of all expression constructs are detailed in Supplementary Table S1. Sequence of siRNAs, gRNAs and primer oligonucleotides are in Supplementary Table S2-5.