


Abstract
Human DNA ligase I (LIG1) is the main replicative ligase and it also seals DNA breaks to complete DNA repair and recombination pathways. Immune compromised patients harbor hypomorphic LIG1 alleles encoding substitutions of conserved arginine residues, R771W and R641L, that compromise LIG1 activity through poorly defined mechanisms. To understand the molecular basis of LIG1 syndrome mutations, we determined high resolution X-ray structures and performed systematic biochemical characterization of LIG1 mutants using steady-state and pre-steady state kinetic approaches. Our results unveil a cooperative network of plastic DNA-LIG1 interactions that connect DNA substrate engagement with productive binding of Mg2+ cofactors for catalysis. LIG1 syndrome mutations destabilize this network, compromising Mg2+ binding affinity, decreasing ligation efficiency, and leading to elevated abortive ligation that may underlie the disease pathology. These findings provide novel insights into the fundamental mechanism by which DNA ligases engage with a nicked DNA substrate, and they suggest that disease pathology of LIG1 syndrome could be modulated by Mg2+ levels.
INTRODUCTION
The three mammalian DNA ligase genes encode highly conserved enzymes with essential functions in DNA replication, repair and recombination. The ligases share a conserved catalytic core but differ in their N- and C-terminal domains that mediate distinct protein–protein interactions and target each ligase to different cellular pathways (1,2). DNA ligase 1 (LIG1) is recognized to be the primary replicative ligase and it interacts with proliferating cell nuclear antigen (PCNA) via an N-terminal PCNA- interacting protein-box (PIP-box). LIG1 associates with the replisome to finalize Okazaki fragment maturation, completing >50 million ligation events during each cycle of DNA replication. In addition, LIG1 ligates single-strand DNA breaks during long-patch base excision repair (BER) and can participate in the alternative end-joining (A-EJ) pathway (3,4). DNA ligase III (LIG3) interacts with X-ray cross-complementing protein 1 (XRCC1) which localizes LIG3 to the nucleus and mediates a network of protein–protein interactions (5–7). LIG3 is also translated as a mitochondria targeted isozyme that is not bound to XRCC1 and its mitochondrial partners are not yet known (8). LIG3 catalyzes both single-strand and double-strand break repair, but its nuclear function is apparently redundant with the other two ligases (3,4,9). The C-terminus of DNA ligase IV (LIG4) interacts strongly with X-ray cross-complementing protein 4 (XRCC4) and other partners in the non-homologous end-joining (NHEJ) pathway, which is specialized for repair of double-strand breaks (10,11).
Both LIG1 and LIG4 have been implicated in immunological diseases (12–14). LIG4 syndrome, defined by deficiency of LIG4, has been shown to severely impact the adaptive immune response, due to its role in NHEJ, which is needed to complete both V(D)J and class-switch recombination (15). In addition to immune system defects, patients with loss of function mutations in LIG4 show increased radiation sensitivity and/or developmental defects (16,17). In contrast, LIG1 syndrome is poorly understood and the severity of disease varies greatly (18–20). To date, all individuals with LIG1 syndrome harbor biallelic mutations with at least one hypomorphic allele that retains some DNA ligase activity. Patients exhibit varying degrees of deficiencies in antibody production, decreased abundance of B and T cells, and macrocytic anemia (18,19).
Previous work identified two LIG1 syndrome alleles, R641L and R771W (18). Although these single amino acid substitutions are located in different domains of the protein and far from the enzyme active site (Figure 1A, B) both variants exhibit markedly decreased DNA ligation activity (18). LIG1−/− HEK293T cells complemented with LIG1 syndrome alleles exhibit increased sensitivity to ionizing radiation (IR) and DNA alkylating agents that cause DNA damage, consistent with a LIG1-dependent DNA repair deficiency contributing to LIG1 syndrome pathology (18). However, the molecular basis for how these variants effect LIG1 structure and activity leading to catastrophic disease outcomes remains poorly defined.
Figure 1.

Mapping of LIG1 syndrome mutations and impact on DNA binding. (A) Schematic of human LIG1 showing disease mutations R641L (pink) and R771W (red) map to the AdD (teal) and OBD (blue) respectively. (B) Structure of LIG1 bound to a nicked DNA substrate (PDB 6P0C). R641 scaffolds a key DNA binding loop (pink) that is connected to the active site. R771 is located in a DNA binding loop in the OBD (red) at the OBD-DBD domain junction. (C) Sequence alignment of LIG1 homologs generated by Clustal Omega shows stringent conservation of R641 and R771. (D) Equilibrium binding of WT and mutant LIG1 proteins to a 28mer nicked DNA substrate was monitored using fluorescence quenching of a TFAM reporter in the absence of Mg2+ to prevent ligation. Mean values ± SD (n ≥ 3) were fit by a simple binding hyperbola and the KD values are provided in the inset. The R641L and R771W mutations do not cause large defects in substrate binding.
Here, we report the impacts that the R641L and R771W mutations have on the structure and biochemical activity of human LIG1. Comprehensive kinetic characterization exposes defects in Mg2+ binding that result in decreased catalytic efficiency and increased abortive ligation. X-ray structures reveal that both LIG1 syndrome mutations disrupt a cooperative network of protein–DNA–metal interactions that link the DNA-binding surfaces of LIG1 with the organization of the active site. LIG1 employs a unique metal-mediated fidelity mechanism whereby rigidity imposed by metal binding allows for discrimination against base mismatches on the 3′-hydroxyl side of the nick. We demonstrate that mutations disabling this high-fidelity organization partially rescues the defects in DNA and active site metal binding of the LIG1 syndrome variants. Overall, our results underscore how ligase encirclement of its DNA substrate is regulated by a network of DNA binding surfaces, and how dynamic DNA binding dictates the precise organization of the ligase active site. This network is integral to efficient DNA ligation. The similar biochemical defects observed for LIG1 pathological variants suggests that a common mechanism for LIG1 inactivation underpins distinct disease states.
MATERIALS AND METHODS
Protein expression and purification
The plasmid for expression of the catalytic domain of human LIG1 (232–919) used for kinetic analysis has been previously described (21). Mutant Δ232 LIG1 vectors were constructed using site-directed mutagenesis with synthetic primers and confirmed by sequencing of the coding region. Expression and purification were performed as previously described (18). WT and mutant Δ232 LIG1 proteins were dialyzed into 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM DTT and 0.1 mM EDTA and stored at −80°C. Protein concentrations were estimated using absorbance at 280 nm. Active enzyme concentrations were determined using single-turnover ligation reactions, in which increasing concentrations of LIG1 were incubated with a fixed concentration of the fluorescein-labeled nicked 28mer substrate (22). For crystallization, LIG1 proteins (262–904) were expressed and purified as previously described (23). Freshly purified proteins were concentrated and used immediately in crystallization experiments.
Crystallization and structure determination
LIG1–DNA complex crystals were grown by hanging drop. 1 μl of protein•DNA complex solution (20 mg ml−1 LIG1, 1.5:1 DNA:protein molar ratio, in 150 mM NaCl, 20 mM Tris–HCl, pH 7.5, 1 mM TCEP) with an equal volume of precipitant solution [100 mM MES, pH 6, 100 mM lithium acetate, 15% (w/v) polyethylene glycol PEG3350]. Nicked DNA used in crystallization was from annealing oligos 1, 2 and 3 (Supplementary Table S1). All crystals grew in 1–2 days and they were washed in cryoprotectant (40% PEG3350 in precipitant solution supplemented with 2 mM EDTA) and flash frozen in liquid nitrogen for data collection. X-ray diffraction data was collected on Beamline 22-ID of the Advanced Photon Source at a wavelength of 1.000 Å. X-ray diffraction data were processed and scaled using the HKL2000 (24). All structures were solved by molecular replacement using PDB entry 6P0A as a search model with PHASER (25). Iterative rounds of model building in COOT (26) and refinement with PHENIX (27) were used to produce the final models. The crystallization and refinement statistics are summarized in Supplementary Table S2.
SYPRO differential scanning fluorometry assay
LIG1 proteins were used at a final concentration of 12.5 μM for this assay. SYPRO Orange (40× stock) (Sigma) was used at a final concentration of 10X. Experiments in the absence of ligand were carried out in 6.25 mM Tris–HCl, pH 7, 37.5 mM NaCl, 1.25% glycerol and to determine the effect of substrate the same solutions were supplemented with 125 μM nicked DNA generated by annealing oligos 1, 2 and 3 (Supplementary Table S1). Fluorescent intensity was recorded starting at 25°C and increasing to 95°C (2°C/min) with the QuantStudio 7 Flex Real Time PCR System (ThermoFisher Scientific) using excitation and emission wavelengths of 492 and 610 nm, respectively. Melting curves were analyzed using the Protein Thermal Shift Software (ThermoFisher Scientific) and then exported directly from the instrument. Tm values are reported as the average ± standard deviation for three independent experiments.
Steady-state fluorescence binding assays
Fluorescence intensity measurements were collected using a FluoroMax 3 fluorometer (Horiba) controlled by the DataMax software. Data were collected using excitation and emission wavelengths of 495 and 515 nm, respectively. Binding assays were performed at 37°C by titration of increasing amounts of LIG1 against a 2.5 ml solution containing 1–2 nM of the TFAM-44mer and the standard reaction buffer supplemented with 0.1 mM EDTA. After addition of LIG1, samples were allowed to equilibrate for 30 s to 1 min before measurements were taken. Relative fluorescence is plotted as a function of added [LIG1] and the data was fit to the following hyperbolic binding curve to determine the LIG1 binding affinity (KD) for the DNA substrate:
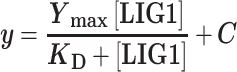 |
In which y is fluorescence intensity, Ymax is the fluorescence intensity at the plateau, and C is a constant.
Gel-based ligation assays
Oligonucleotides substrates were purchased from IDT, Midland Certified Reagent Company, or the Keck Center at Yale University and purified by denaturing PAGE as previously described (22). Ligation assays were carried out at 37°C in a standard reaction buffer consisting of 50 mM NaMOPS at pH 7.5, 1 mM dithiothreitol, 0.05 mg/ml BSA, and sufficient NaCl to maintain a constant ionic strength of 150 mM. Concentrations of ATP, MgCl2, 28mer substrate, and LIG1 were varied as indicated below. The free magnesium concentration for all reactions with ATP were calculated using the dissociation constants for the Mg2+•ATP complex (22). Reactions were quenched in the standard loading buffer (50 mM EDTA/90% formamide/0.01% xylene cyanol/0.01% bromophenol blue). Quenched samples were heated to 95°C and separated on either a 15 or 20% (w/v) polyacrylamide gel containing 8 or 6.6 M urea, respectively. The fluorescein-labeled oligonucleotide was detected using an Amersham Typhoon 5 imager (GE), and gel images were analyzed using ImageQuant TL software (GE). Rate constants are reported as the average ± standard deviation for at least three independent experiments.
Determining the steady-state dependences for ligation
Steady-state dependences were performed using the standard reaction buffer and 0.1–10 nM LIG1. For both the Mg2+ and ATP dependences, 500 nM of the 28mer substrate was used along with either 200 μM ATP or 20 mM MgCl2, respectively. For the DNA substrate dependences at saturating levels of MgCl2 and ATP, reactions contained 20 mM MgCl2 and 200 μM ATP. DNA substrate dependences were also performed under physiological conditions (2 mM MgCl2 and 1 mM ATP). Initial rates for all steady-state reactions were determined by linear regression, and the rates were fit with the Michaelis–Menten equation:
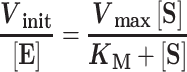 |
For reactions in which an accumulation of both intermediate and product was observed, the initial rates of total substrate disappearance were used. The fraction of observed abortive ligation events was determined by dividing the rate of intermediate accumulation (Vintermediate) by the rate of total substrate disappearance (Vproduct + Vintermediate):
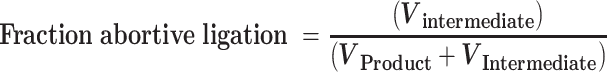 |
Single-turnover ligation assays
To determine the individual rates of the adenylyl-transfer and nick-sealing steps, single-turnover reactions were performed with 800 or 1600 nM Δ232 LIG1 and 80 or 160 nM DNA substrate. Reactions to determine Mg2+ dependence of the chemical steps were performed in a KinTek RQF-3 quench-flow apparatus, where LIG1 was loaded in a separate sample loop from the 28mer, both at double the desired final concentrations to account for dilution upon mixing. Both ligase and DNA solutions were prepared with the standard reaction buffer supplemented with desired concentrations of MgCl2. Drive syringes were loaded with the standard reaction buffer and reactions were quenched using the standard loading buffer. Ligation in the presence of ≤2 mM MgCl2 was slow enough that reactions were set up by hand and incubated in a heat-block set at 37°C in place of the quench-flow apparatus. Rates were obtained from fits to the single-turnover data using a two-step irreversible model in Berkeley-Madonna as previously described (22).
RESULTS
LIG1 syndrome mutations have mild impacts on DNA binding affinity and protein folding
To understand the potential impacts of LIG1 syndrome mutations, we first examined the molecular environments of the substitutions. R641 scaffolds a key DNA binding loop in the LIG1 Adenylylation domain (AdD) (Figure 1A, B, teal) whereas R771 maps to the C-terminal OB-fold domain (OBD) (Figure 1A, B, blue). Both arginine residues are stringently conserved among LIG1 homologs (Figure 1C) and they participate in a network of protein-DNA and interdomain interactions including the DNA binding domain (DBD) that stabilize a closed ring conformation of LIG1 bound to its DNA substrate. Given the placement of these residues, we hypothesized the mutants may impact equilibrium DNA binding affinity. To probe LIG1 binding to DNA, we used a reporter DNA substrate containing a fluorescein-conjugated deoxythymidine at the nick (TFAM; Supplementary Figure S1), a strategy that has been previously used to characterize DNA binding by T4 DNA ligase (28). In the absence of Mg2+, equilibrium binding of LIG1 to DNA could be monitored by either changes in fluorescence anisotropy or the quenching of TFAM that occurs upon binding of LIG1 to the labeled nick (Figure 1D; see Supplementary Figure S1d for changes in anisotropy). We determined a KD value of 3.2 ± 0.4 nM for wild-type (WT) LIG1 (Figure 1D). DNA binding is specific to the presence of a ligatable nick, as TFAM DNA lacking a 5′-phosphate moiety was bound very weakly with a Kd value of 4 ± 1 μM (Supplementary Figure S1). KD values for binding of the R641L and R771W mutants to the ligatable substrate were both within 1.6-fold of the WT enzyme, indicating that the mutations do not have a large impact on substrate binding in the absence of Mg2+ (Figure 1D).
We next evaluated whether the mutations impact protein stability and folding. Thermal shift protein denaturation assays performed in the absence of ligands yielded a melting temperature (Tm) of 47.9 ± 0.05°C for the WT enzyme (LIG1WT) that is significantly increased to 55.7 ± 0.01 with the addition of substrate DNA (Supplementary Figure S2). The mutant proteins gave only slightly reduced Tm values in the absence of DNA, LIG1R641L (45.6 ± 0.05°C; ΔTm = 2.3°C) and LIG1R771W (46.1 ± 0.05°C; ΔTm = 1.8°C) compared to LIG1WT (Supplementary Figure S2). Moreover, the Tm values are indistinguishable between WT and mutant proteins in the presence of DNA substrate which supports the finding that R641L and R771W substitutions do not appreciably weaken DNA binding affinity. We further probed stability of mutant and WT LIG1 under the standard ligase assay conditions and all proteins were stable and retained full activity for 2 h at 37°C (Supplementary Figure S1). Taken together, these studies demonstrate that the R641L and R771W mutations do not cause substantial defects in the thermodynamics of protein folding or DNA binding. This suggests that the mutations exert their effect on steps downstream of the initial engagement of the DNA substrate.
Remodeling of the LIG1 AdD DNA-binding interface by R641L
To more precisely define the impacts of the LIG1 syndrome mutations, we crystallized and determined X-ray structures of mutant LIG1•DNA complexes using a truncated LIG1 construct that contains the catalytic core of the protein (262–904) (23). The mutant proteins were extensively dialyzed in EDTA and were crystallized with a ligatable nicked DNA substrate bearing 3′-hydroxyl and 5′-phosphate moieties at the nick site (Supplementary Table S1). This crystallization method yields a DNA-adenylate crystal form in which Mg2+ is not bound in the active site (23).
The 1.90 Å X-ray structure of LIG1 R641L reveals an extensive reorganization of the R641 DNA binding loop [Figure 2A, Movie S1)]. Overall, the electron density for the R641L mutation is well defined (Figure 2B). Structural overlays highlight how the mutated R641L AdD DNA-binding loop is restructured compared to the WT enzyme (Figure 2A, B, Supplementary Figure S3a). In LIG1WT, R641 participates in a key scaffolding interaction through a buried salt bridge with D600 (Figure 2C). This scaffold supports an array of basic amino acid–DNA interactions from the R641 AdD DNA binding loop. Residues K642 and K644 form salt bridge interactions to the phosphodiester backbone of the DNA substrate, and R643 penetrates the major groove of the DNA to contact the C4′ oxygen of a deoxyribose sugar (Figure 2C).
Figure 2.

The R641L mutation grossly remodels the LIG1 AdD–DNA binding interface. (A) Overlay of the WT (PDB 6P0C, light blue) and R641L (purple) LIG1 X-ray structures. The R641L mutation causes the R641 DNA-binding loop to collapse and move 11 Å away from the DNA backbone, resulting in the loss of protein–DNA contacts and distortion of the DNA helix. (B) Final 2Fo – Fc electron density contoured at 1.0 σ for the R641L mutant structure. (C) R641 scaffolds a key DNA binding loop of the AdD (purple loop) by forming a salt bridge to D600. Neighboring basic residues K642 and K644 on the loop make contacts with the phosphodiester backbone of the DNA substrate, while R643 contacts the C4′ oxygen of a deoxyribose sugar. (D) In R641L, the leucine substitution causes the loss of the salt-bridge interaction with D600, resulting in loop arrangements and disengagement from the DNA substrate.
The remodeled AdD DNA-binding loop of R641L shows disengagement of the K642–R643–K644 DNA binding contacts, and coincident distortions in the DNA backbone (Figure 2A, D). In the absence of the R641–D600 salt bridge anchor, a complete reorganization of the secondary structure of the R641L DNA binding loop is observed and the Cα position of residue 641 is displaced by ∼11 Å away from the phosphodiester backbone (Figure 2A, D). The R641–D600 salt bridge is replaced by the packing of the mutant L641 sidechain into a new hydrophobic pocket molded by A625, V653 and V655 (Figure 2D). Unlike the DNA binding loop rearrangements, there are no large scale alternations to the enzyme active site or in the alignment of the nicked DNA ends in the R641L structure. We note, however, that the absence of Mg2+ ions in the R641L–DNA crystal structure leaves open the possibility that additional dynamic structural rearrangements could occur upon engagement with the Mg2+ cofactors.
Disruption of DNA-binding and interdomain interactions by R771W
The R771W mutant was also crystallized and the X-ray structure was determined at 2.0 Å resolution (Figure 3A, B). As was observed for the R641L mutant structure, there was clear electron density for structural rearrangements caused by the R771W substitution (Figure 3B, Supplementary Figure S3b). The R771W substitution induces a cascade of local structural rearrangement that significantly remodels protein-protein interdomain contacts at the juncture of the OBD and DBD, as well as protein-DNA contacts (Figure 3A, Movie S2). In LIG1WT, R771 forms a salt bridge to D802 which supports a critical OBD DNA binding loop (Figure 3C). The R771 DNA binding loop normally intercalates into the DNA minor groove and makes water-mediated contacts with the DNA. An additional salt-bridge interaction between R774 and E806 and DNA base contacts by K770 further stabilize the protein-DNA contacts in LIG1WT. Proximal to R771, salt-bridging contacts between R768 (OBD) and D351 (DBD) secure the interdomain OBD-DBD interface, reinforcing enzyme encirclement of the DNA substrate (Figure 3C). Structural equivalents to the R771–D802 salt bridge and the R771 loop are found among the three human DNA ligases (Supplementary Figure S4) pointing to key roles for these structural elements in mediating DNA ligase function.
Figure 3.
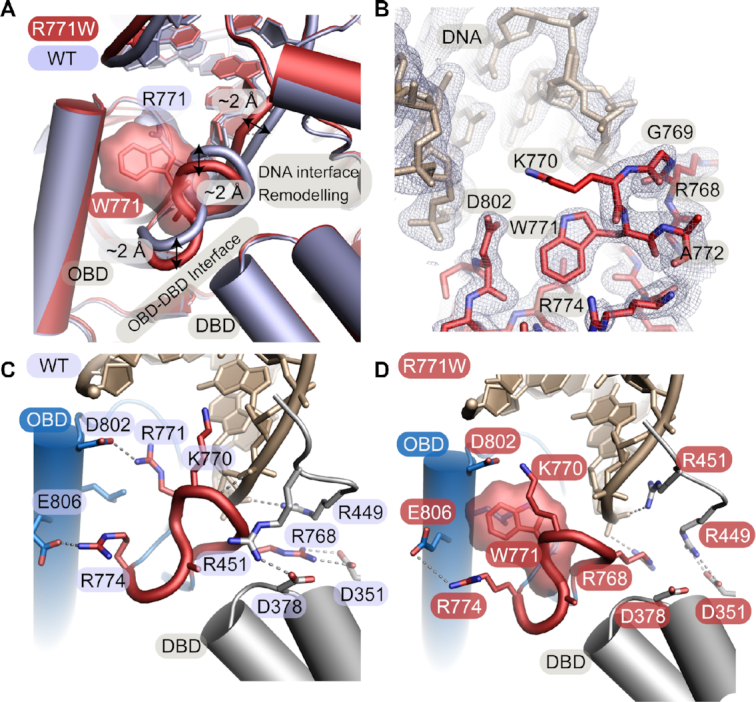
The R771W mutation disrupts DNA-binding and interdomain interactions. (A) Overlay of the WT (PDB 6P0C, light blue) and R771W (red) LIG1 X-ray structures. The R771W mutation causes local conformational changes in the OBD DNA binding loop with a ∼2 Å movement away from the minor groove of the DNA helix and distortions in the DNA backbone. The altered loop structure results in the loss of protein-DNA and interdomain contacts. (B) Final 2Fo‐Fc electron density contoured at 1.0 σ for the R771W mutant structure. (C) R771 is located on a DNA binding loop of the OBD (red loop) where it intercalates into the minor groove of the DNA helix and makes water-mediated contacts with the DNA. The neighboring K770 makes a direct DNA base contact. R771 forms a salt bridge with D802 and there is an additional salt bridge interaction between R774 and E806 that further stabilizes this topology. The interdomain ring closure between the OBD and the DBD is stabilized by a salt bridge between R768 of the OBD loop and D351 and G377 (not shown) of the DBD. (D)The crystal structure of R771W LIG1 highlights the movement of the R771 loop away from the DNA helix and the loss of the salt bridge with D351 of the DBD.
In the mutant structure, the R771W loop rotates and shifts away from the DNA helix with displacements of up to ∼2 Å (Figure 3A). Coincident with Van der Waals-driven steric displacement of the neighboring side chains by W771, a cascade of rearrangements in protein–protein and protein–DNA interactions are observed in the vicinity of the mutated residue. R768 is shifted away from D351 resulting in a loss of an OBD–DBD interdomain salt bridge. K770 rotates away from the DNA and becomes disordered, resulting in loss of DNA contacts (Figure 3B, D). Structural reorganization of the R771W loop extends to an adjacent DNA binding loop in the DBD that is also remodeled, with residue R451 in the DBD shifting toward the DNA, effectively reorganizing the protein–DNA interface.
R641L and R771W mutations decrease catalytic efficiency and increase abortive ligation
Akin to the R641L crystal structure, little change is observed in the active site of the R771W mutant. However, given roles for the DNA 3′-OH strand in mediating dynamic active site Mg2+ binding (23), and that R641L and R771W mutations impact local DNA binding structure, we hypothesized that the mutants could impact the formation of the reactive LIG1 complex upon binding of Mg2+. Consistent with this, initial biochemical characterization of these mutants indicated that the Mg2+ dependence for multiple turnover ligation is shifted to require higher Mg2+ to saturate the R641L and R771W LIG1 mutants (18). To better understand impacts on metal cofactor binding of the mutants, multiple turnover ligation kinetics were determined using a gel-based ligation assay at a physiological Mg2+ concentration. Under these conditions with 1 mM ATP and 2 mM total Mg2+ (1 mM Mg2+free), the mutant enzymes exhibit both a reduction in kcat and an increase in KM,DNA relative to WT LIG1 (Figure 4A, B and Table 1). These changes lead to an overall reduction in the catalytic efficiency for the DNA substrate (kcat/KMDNA) of 22-fold for R641L and 36-fold for R771W, relative to WT LIG1 (Figure 4C, Table 1). The reduction in kcat/KM,DNA at 1 mM Mg2+free concentration is significantly more pronounced than the previously determined defects of 5- and 10-fold, respectively, for kcat/KMDNA values determined with saturating amounts of 20 mM Mg2+ (18). This underscores the unexpected sensitivity of the mutant enzymes to Mg2+ concentration, and the partial rescue of activity by increasing the Mg2+ concentration.
Figure 4.

LIG1 syndrome mutations decrease catalytic efficiency and increase abortive ligation. (A) Representative time course from multiple turnover reactions containing 10 nM enzyme, 500 nM DNA, 2 mM MgCl2 (1 mM Mg2+free) and 1 mM ATP. Initial rates from multiple turnover reactions were determined from the slope of linear fits to the amount of substrate reacted. (B) The steady-state DNA dependence was determined with 0.1–10 nM LIG1 and increasing amounts of the DNA substrate. The resulting data were fit by the Michaelis–Menten equation to obtain kcat and KM,DNA values for WT and mutant LIG1 enzymes. (C) The R641L and R771W mutants have substantially reduced catalytic efficiency (kcat/KM values from the fits in panel b). (D) Representative denaturing gel image from a ligation assay containing 5 nM R641L, 500 nM DNA, 2 mM MgCl2 (1 mM Mg2+free) and 1 mM ATP highlighting buildup of the aborted intermediate. (E) The fraction of abortive ligation events observed in the steady-state magnesium dependences. The physiologically-relevant [Mg2+]free (∼1 mM) is marked by a dashed line. R641L and R771W LIG1 exhibit enhanced rates of abortive ligation compared to WT at low [Mg2+]free. All values are mean ± SD (n ≥ 3).
Table 1.
Steady-state kinetic parameters for LIG1-catalyzed ligation
| WT | R641L | R771W | |
|---|---|---|---|
| k cat (s−1) | 0.36 ± 0.07 | 0.066 ± 0.009 | 0.041 ± 0.002 |
| K M (nM) | 50 ± 18 | 211 ± 67 | 220 ± 74 |
| k cat/KM (M−1s−1) | 7.8 ± 1.7 × 106 | 0.36 ± 0.02 × 106 | 0.22 ± 0.08 × 106 |
Steady-state kinetics were determined at 37°C with saturating ATP (0.2 mM) and 1 mM Mg2+free (Figure 4B). Values are the mean ± SD (n ≥ 3).
Both R641L and R771W are susceptible to abortive ligation at 1 mM Mg2+, which is close to the expected physiological Mg2+ concentration (18). Abortive ligation occurs prior to the final nick-sealing step of ligation and results in the release of the adenylylated DNA intermediate from the enzyme. The aborted ligation intermediate cannot be rebound by LIG1 when ATP is present as rapid enzyme adenylylation occurs. Since an increased abortive ligation burden is potentially cytotoxic and could be relevant to the onset of LIG1 syndrome, we further investigated the abortive ligation burden caused by the R641L and R771W LIG1 variants. Steady-state ligation reactions were performed with WT and mutant LIG1 enzymes under a range of magnesium concentrations to observe the magnesium dependence of abortive ligation (Figure 4E). A representative time course for R641L is shown in Figure 4D, showing roughly equal amounts of ligated product and released adenylylated DNA at near-physiological concentrations of Mg2+ (1 mM). At the lowest Mg2+ concentration tested (50 μM Mg2+free) the mutants could only perform abortive ligation, whereas WT LIG1 aborted only 10% (Figure 4E).
Single turnover ligation kinetics demonstrate that R641L and R771W are compromised for both adenylyl transfer and nick sealing
The substantial decrease in catalytic efficiency and the prevalence of abortive ligation at lower Mg2+ concentrations prompted us to investigate the effect of the LIG1 syndrome mutations on the DNA-dependent steps of the DNA ligation reaction, adenylyl transfer and nick sealing (Figure 5A). Reaction time courses are shown for the mutant enzymes at a 10-fold excess of enzyme over DNA substrate and with 1 mM Mg2+free (Figure 5B). To uncover any defects in the individual chemical steps of ligation, we performed single-turnover ligation assays using a rapid-quench apparatus over a range of magnesium concentrations. The mutants exhibit lower KMg2+ values for the adenylyl-transfer step than for nick sealing showing that, like WT LIG1, the mutants bind the catalytic magnesium ion tighter during the first DNA-dependent step of ligation. However, the affinity of the magnesium ion during adenylyl transfer is ∼40–50-fold weaker for both R641L and R771W relative to WT enzyme (Figure 5C, Table 2). A defect is also apparent in the KMg2+ value for nick sealing, with the affinity for the catalytic magnesium ion being ∼2–3-fold weaker for the mutants as compared to WT (Figure 5D, Table 2). These results indicate that the structural changes resulting from the R641L and R771W mutations are somehow impacting the binding of the catalytic magnesium ion, causing a weakening of Mg2+ binding during both adenylyl transfer and nick sealing (Table 2). Additionally, the maximal rates of the two chemical steps are reduced by ∼3-fold for the mutants relative to the WT enzyme even at saturating concentration of Mg2+ (Table 2). Thus, the mutant enzymes bind the catalytic Mg2+ cofactor less tightly than the WT enzyme. This suggests that dynamics of LIG1 ring closure and concomitant active site alignment for catalysis is impaired in LIG1 syndrome mutants.
Figure 5.

LIG1 clinical mutations weaken catalytic metal affinity in both the adenylyl transfer and nick-sealing steps. (A) After the adenylylated LIG1 enzyme binds to a nick site, the AMP moiety is transferred to the 5′ phosphate during a step known as adenylyl transfer. Nick-sealing follows with attack of the 3′ hydroxyl on the 5′ phosphate, releasing AMP and the sealed DNA product. (B) Representative single-turnover ligation time courses for R641L (lavender) and R771W (red) in the presence of 1 mM Mg2+. Data were fit to a two-step irreversible model in Berkeley-Madonna to determine the microscopic rates of ligation. The rates of adenylyl transfer (C) and nick sealing (D) were determined from single-turnover experiments containing 800 nM LIG1, 80 nM DNA and varying concentration of Mg2+. The maximal rates and KMg values for both steps were determined from hyperbolic fits to the single-turnover data and the values are summarized in Table 2. All values are reported as the mean ± S.D. (n ≥ 3).
Table 2.
Maximal rates and magnesium affinities for the individual chemical steps of ligation
| WT | R641L | R771W | |
|---|---|---|---|
| k cat (s−1) | 0.69 ± 0.10 * | 0.27 ± 0.05 * | 0.18 ± 0.03 * |
| k transfer (s−1) | 3.0 ± 0.6 † | 0.8 ± 0.1 | 0.46 ± 0.07 |
| k seal (s−1) | 16 ± 4 † | 5.3 ± 0.3 | 7.0 ± 1.4 |
| K Mg (mM) | 0.86 ± 0.03 * | 4.3 ± 1.5 * | 3.5 ± 0.4 * |
| K Mg,transfer (mM) | 0.10 ± 0.03 † | 3.9 ± 0.4 | 6.1 ± 1.0 |
| K Mg,seal (mM) | 4.9 ± 2.1 † | 13 ± 6 | 17 ± 8 |
Rate constants for single-turnover ligation were measured using rapid chemical quench (Figure 5). Values are the mean ± SD (n ≥ 3).*Values for kcat and KMg are from (18). †Values for WT LIG1 are from (22).
Removal of the HiFi Mg2+-binding site partially rescues the defects of the LIG1 syndrome mutations
Given the evidence for structural rearrangements far from the mutated residues R641L and R771W, and the defects in cofactor binding and catalysis, it appears that LIG1 engages its substrate via a cooperative network of interactions that couple conformational rearrangements in both protein and DNA to achieve the active enzyme structure. A unique feature of the reactive LIG1 structure is the HiFi Mg2+ binding site, a structural Mg2+-binding site that enforces high fidelity ligation by providing structural rigidity between the protein and the 3′-hydroxyl strand of the DNA (23). Structures of the R641L and R771W mutants show perturbations in the DNA conformation in the region of the HiFi Mg2+ binding site that is formed by E346 from the DBD and E592 from the AdD (Figure 6A, B, Supplementary Figure S3c–f). We hypothesized that these propagated changes in DNA conformation would create additional strain once Mg2+ is bound in the HiFi site and therefore that removal of the HiFi site may rescue the catalytic defects in the LIG1 syndrome mutants. Consistent with this idea, it was previously demonstrated that the reduced fidelity LIG1 mutant (E346A/E592A), lacking the HiFi Mg2+ site, suppresses abortive ligation and enhances ligase activity on improperly base-paired oxidatively damaged DNA substrates (23).
Figure 6.

Removal of the HiFi Mg2+ site partially rescues the defects in the LIG1 syndrome variants. (A) Overlay of the DNA substrate from the WT LIG1 crystal structure (blue) and the substrate from either the R641L (lavender) or R771W (red) crystal structure. R641L and R771W both cause helical distortions that impact the positioning of the phosphodiester backbone proximal to the MgHiFi site. (B) Schematic depicting the relationship between the catalytic Mg2+ and the DNA binding loops and the role of the HiFi Mg2+ ion. (C) Representative steady-state ligation assays containing 5 nM [LIG1], 500 nM [DNA], 2 mM MgCl2 (1 mM Mg2+free) and 1 mM ATP highlight the suppression of abortive ligation caused by the clinical mutants when the MgHiFi ligands (E346 and E592) are absent. The E346A/E592A variants of both R641L and R771W demonstrate decreased abortive ligation burden (D) and enhanced catalytic efficiency (E) compared to LIG1 enzymes bearing the individual clinical mutations. (F) The addition of the E346A/E592A mutations to the R641L and R771W LIG1 enzymes causes a decrease in the KMg values for both steady-state ligation and the individual chemical steps, indicating that the triple mutants have a higher affinity for Mg2+ compared to singly mutated LIG1 enzymes. Values are from Supplementary Table S3 and reflect the mean ± S.D. (n ≥ 3).
To test this idea, we created triple mutants that combined each LIG1 syndrome mutation with the previously characterized E346A/E592A double mutation that ablates the HiFi magnesium-binding site (Supplementary Figure S5). Multiple turnover ligation kinetics demonstrate that the triple mutants exhibit a significant decrease in the burden of abortive ligation (Figure 6C, D) and 20–30-fold increased catalytic efficiency (Figure 6E, Supplementary Table S3) compared to the singly mutated enzymes. Whereas it is easy to rationalize how the HiFi Mg2+ site contributes to the cooperativity between the rearranged R641 loop as they are both on the 3′-hydroxyl side of the nick, we also observed very similar rescue for the R771W mutation which is located across the nick on the 5′-phosphate side. Therefore, structural rearrangements in the LIG1 complex are able to propagate across the nick and potentially tie recognition of both the 3′-hydroxyl end and the 5′-phosphate end together in assembling the active site for catalysis.
We next examined the Mg2+ dependence for multiple and single turnover ligation kinetics (Supplementary Figure S5 and Table S3). Consistent with the emerging model for cooperativity between DNA binding and active site assembly, we observed 2–3-fold increases in the affinity for the catalytic Mg2+ in the triple mutants, relative to the LIG1 syndrome single mutations (Figure 6F). This suggests that the structural rigidity of LIG1–DNA engagement enforced by the HiFi site contributes to propagating rearrangements along the DNA binding surface with catalytic interactions in the active site, and further helps to explain why the triple mutants almost completely suppress abortive ligation (Figure 6D). As previously shown, removal of the HiFi site did not have a significant effect on the catalytic rates themselves (23) and both ktransfer and kseal are similar for the single and triple mutants (Supplementary Table S3). In summary, the connectivity between the HiFi-mediated recognition of the 3′-hydroxyl strand and LIG1 syndrome mutations at the DNA-binding interface of the AdD (R641L) and the OBD (R771W) emphasizes the cooperative remodeling of enzyme and substrate that occurs in the final stages of DNA ligation.
DISCUSSION
DNA ligase 1 deficiency in humans was first reported in 1992 with an individual who suffered from developmental delays and immune deficiency (19,20). The immortalized fibroblast cell line 46BR.1G1 was established from this patient with homozygous R771W alleles of LIG1 (19), and these cells show sensitivity to DNA alkylating agents and delayed Okazaki fragment ligation consistent with inadequate DNA ligase activity (19,29–31). More recently, biallelic LIG1 mutations were identified in individuals with a range of clinical symptoms from an early-onset Ig production defect to severe combined B and T cell deficiency requiring hematopoietic stem cell transplantation (18). It is not yet clear why LIG1 mutations cause immune deficiencies or the degree to which specific alleles affect the severity of disease symptoms. In the current work we have probed the structural and biochemical consequences of two of the LIG1 mutations identified in immunodeficient patients, including the R771W mutation associated with the first case of LIG1 deficiency syndrome. Although R641L and R771W map to distinct regions of LIG1, they confer markedly similar defects in enzyme function. Biochemical characterization of these two variants reveal defects in Mg2+ cofactor binding, decreased catalytic efficiency, and increased frequency of abortive ligation by the enzyme that have critical implications for LIG1 syndrome patients.
High-resolution X-ray structures of the two variants allowed us to identify altered conformations in key DNA binding loops. The R641L mutation results in a marked rearrangement of an AdD DNA binding loop structure. The R641 loop shifts ∼11 Å away from the phosphodiester backbone of the DNA substrate disrupting protein-DNA contacts. Although this loop is far from the active site, the R641L mutation has a large impact on both catalytic Mg2+ binding and ligation efficiency, as reflected by a 30-fold reduction in catalytic efficiency for the DNA substrate. It is intriguing to note that the recently reported X-ray structures of the LIG4-DNA complexes show how the topologically equivalent R641 loop undergoes metamorphic conformational changes to mediate interactions with the DNA substrate during the various catalytic events of the ligation reaction (32). The corresponding loop of human LIG4 is disordered in a DNA-bound, open enzyme conformation in which the OBD partially disengages the DNA substrate (Supplementary Figure S4a). This loop becomes structured in an enzyme closed catalytically competent conformation in which the core domains encircle the DNA substrate. In LIG1, conformational remodeling of the R641 loop may therefore influence the conformational changes associated with DNA substrate binding and/or product release. However, we note that this loop adopts a distinct fold compared to loops in LIG3 and LIG4 (Supplementary Figure S4a), suggesting that the three mammalian ligases acquired distinct modes of DNA substrate interactions through evolution that mediate their specialized functions in distinct biological contexts, such as ligation of single-strand versus double-strand DNA breaks.
The R771W mutation also causes perturbations in the DNA binding surface, but compared to R641L, these are more localized structural rearrangements. This region of the OBD is structurally conserved among the human DNA ligases (Supplementary Figure S4b), and thus is expected to be critical for DNA ligase activity for LIG3 and LIG4. The R771 loop located in the OBD is involved in the opening and closure of the ring formed by the core enzyme domains by engaging salt-bridging interactions with DBD residues. This tryptophan substitution eliminates a key salt-bridge interaction with D802, resulting in an intricate cascade of protein–DNA complex alternations, including rearrangements of a network of salt-bridge interactions that reinforce the OBD–DBD junction of LIG1 in the encirclement of the DNA substrate. Our data suggest that these rearrangements compromise the stability of the ring-closed complex that is necessary for tight DNA binding in the adenylylation and nick sealing steps.
The substantial biochemical defects that we documented for the R641L and R771W variants are belied by relatively minor structural perturbations at the active site of the crystallized LIG1•DNA complexes solved in the absence of Mg2+. We suggest that binding of Mg2+ to the HiFi site strengthens the connectivity between the active site and distal parts of the DNA binding surface. Consistent with this model, DNA rearrangements occur near the unoccupied HiFi site in the structures of both variants (Figure 6A, Supplementary Figure S3c–f). We further explored this connectivity by combining mutations that eliminate the HiFi Mg2+ binding site with the LIG1 syndrome variants. The biochemical parameters of these triple mutants reveal substantial (≥20-fold)) rescue of the catalytic efficiency with more modest, but significant (2–3-fold) rescue of catalytic Mg2+ binding (Figure 6E, F). It is reasonable to speculate that the inherent flexibility in DNA ligases that require large conformational rearrangements through the catalytic cycle also permits remodelling in response to disruptive mutations such as these clinical mutations. Indeed, a bacterial DNA ligase has been shown to readily develop drug resistance mutations that weaken binding of an active-site directed inhibitor while retaining substantial DNA ligase activity (33). If this speculative hypothesis is true, then we can expect that more examples will be forthcoming as the eukaryotic DNA ligases and their disease-associated variants are studied.
The implications of this work for LIG1-catalyzed ligation overall and for the molecular basis of LIG1 syndrome specifically are summarized in Figure 7. In contrast to the seamless remodelling of WT LIG1 that occurs upon engagement of a ligatable nick (Figure 7A, middle complex), the R641L and R771W mutants disrupt this process (Figure 7B). In WT LIG1 the encirclement of the DNA is stabilized by interdomain interactions as well as protein-DNA interactions both upstream and downstream of the nick. The structural HiFi Mg2+ is important for remodelling of the 3′-hydroxyl strand toward an A-form-like structure and discrimination against ligation of damaged or mismatched substrates (23). In the case of a proper ligatable nick, the enzyme holds tightly onto the adenylylated intermediate and achieves very high efficiency of ligation (22). In the case of the R641L and R771W mutation, the closed conformation is destabilized which provides greater opportunity for abortive ligation (Figure 7B). It is notable that both mutants can be partially rescued by super-physiological concentrations of Mg2+ which suppresses abortive ligation and leads to higher catalytic efficiency. The common biochemical features of these LIG1 syndrome mutations has immediate and long-term implications for the patients. Most directly, we have shown that the R641L and R771W defects are strongly sensitive to Mg2+ in the physiological range. Therefore, it is important that patients avoid dietary Mg2+ deficiency which is rare and can be easily treated by supplemental Mg2+ intake.
Figure 7.

Cooperative engagement of DNA and impact of LIG1 syndrome mutations on ligase efficiency and abortive ligation. (A) LIG1 encircles a DNA nick, remodeling the DNA substrate and forming a network of DNA binding interactions and interdomain contacts. Tight binding of the adenylylated intermediate (central complex) prevents premature release via an open complex (top) and ensures high efficiency ligation (bottom). (B) The R641L and the R771W mutations disrupt DNA-binding interactions and remodel the DNA-binding surface and interdomain contacts. This destabilizes the closed, high affinity complex as demonstrated by weaker Mg2+ binding and increased frequency of abortive ligation (top). These defects can be overcome by saturation of the active site Mg2+ cofactor (bottom). Abortive ligation, which is a shared feature of the LIG1 syndrome variants, may contribute to the deleterious cellular effects which may be further modulated by pathways for repair of adenylylated DNA, including strand-displacement and flap cleavage by FEN1 or direct hydrolytic removal by APTX.
More generally, we have shown that both clinical variants release high levels of adenylylated intermediates and it is notable that all recognized cases of LIG1 syndrome have one of these two alleles represented. This raises the possibility that the physiological symptoms could be specific to the abortive ligation itself, and are not simply explained by diminished level of LIG1 catalytic activity. Adenylated DNA adducts are presumably cytotoxic because they can block DNA repair and replication (Figure 7B). Normally adenylated DNA can be repaired by two distinct pathways. Aprataxin (APTX) catalyzes the hydrolysis of adenylylated DNA to directly restore the 5′-phosphate terminus (34,35) and Flap endonuclease 1 (FEN1) flap cleavage can generate a new ligatable DNA nick after strand displacement synthesis (22). Elevated abortive ligation products generated by LIG1 syndrome mutants would require a high level of APTX or FEN1 activity to maintain genomic stability. Thus, the specific activity of a patient's APTX and FEN1 repair pathways could be important modifiers of the severity of LIG1 deficiency syndromes. Currently the identified LIG1 syndrome patients are relatively young and it will be important to monitor whether there is any connection between the primary immune deficiency experienced in youth and the neurodegenerative symptoms caused by APTX deficiency that typically manifest later in life (18). This hypothesis also has implications for heterozygous individuals with a single copy of the R771W or R641L variant, as even a single hypomorphic allele may increase the risk for immune diseases. More research is required to address this new hypothesis that abortive ligation could be an important factor in primary immune deficiency.
DATA AVAILABILITY
Coordinates for LIG1-R641L-DNA complex (7L34) and LIG1-R771W-DNA complex (7L35) have been submitted to the RCSB Protein Data Bank.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lars Pedersen of the NIEHS collaborative crystallography group, and the Advanced Photon Source (APS) Southeast Regional Collaborative Access Team (SER-CAT) staff for assistance with crystallographic data collection. We thank Joseph Dahl and Kasia Bebenek for comments.
Notes
Present address: Matthew J. Schellenberg, Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, MN, 55905, USA.
Contributor Information
Thomas J Jurkiw, Department of Biological Chemistry, University of Michigan School of Medicine, Ann Arbor, MI, 48109, USA.
Percy P Tumbale, Structural Cell Biology group, Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, US National Institutes of Health, Department of Health and Human Services, Research Triangle Park, NC, 27709, USA.
Matthew J Schellenberg, Structural Cell Biology group, Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, US National Institutes of Health, Department of Health and Human Services, Research Triangle Park, NC, 27709, USA.
Charlotte Cunningham-Rundles, Division of Clinical Immunology, Departments of Medicine and Pediatrics, and Graduate School of Biomedical Sciences, Immunology Institute, Icahn School of Medicine at Mount Sinai, New York, NY, 10029, USA.
R Scott Williams, Structural Cell Biology group, Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, US National Institutes of Health, Department of Health and Human Services, Research Triangle Park, NC, 27709, USA.
Patrick J O’Brien, Department of Biological Chemistry, University of Michigan School of Medicine, Ann Arbor, MI, 48109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Health Intramural Program, National Institute of Health Sciences [1Z01ES102765 to R.S.W.]; National Institutes of General Medical Studies [R01GM130763 to P.O.]; National Institutes of Allergy and Infectious Disease [R21AI142144 to C.C.R. and P.O.]; Cellular Biotechnology Training Program at the University of Michigan [T32GM008353 to T.J., in part]; Rogel Cancer Center at Michigan Medicine [P30CA046592]; X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory; Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences [W-31–109-Eng-38]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tomkinson A.E., Vijayakumar S., Pascal J.M., Ellenberger T.. DNA ligases: structure, reaction mechanism, and function. Chem. Rev. 2006; 106:687–699. [DOI] [PubMed] [Google Scholar]
- 2. Ellenberger T., Tomkinson A.E.. Eukaryotic DNA ligases: structural and functional insights. Annu. Rev. Biochem. 2008; 77:313–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu G., Duan J., Shu S., Wang X., Gao L., Guo J., Zhang Y.. Ligase I and ligase III mediate the DNA double-strand break ligation in alternative end-joining. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:1256–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Masani S., Han L., Meek K., Yu K.. Redundant function of DNA ligase 1 and 3 in alternative end-joining during immunoglobulin class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simsek D., Furda A., Gao Y., Artus J., Brunet E., Hadjantonakis A.K., Van Houten B., Shuman S., McKinnon P.J., Jasin M.. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. 2011; 471:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao Y., Katyal S., Lee Y., Zhao J., Rehg J.E., Russell H.R., McKinnon P.J.. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. 2011; 471:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caldecott K.W., McKeown C.K., Tucker J.D., Ljungquist S., Thompson L.H.. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol. 1994; 14:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lakshmipathy U., Campbell C.. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol. 1999; 19:3869–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han L., Masani S., Hsieh C.L., Yu K.. DNA ligase I is not essential for mammalian cell viability. Cell Rep. 2014; 7:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilson T.E., Grawunder U., Lieber M.R.. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997; 388:495–498. [DOI] [PubMed] [Google Scholar]
- 11. Grawunder U., Wilm M., Wu X., Kulesza P., Wilson T.E., Mann M., Lieber M.R.. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997; 388:492–495. [DOI] [PubMed] [Google Scholar]
- 12. Sun D., Urrabaz R., Nguyen M., Marty J., Stringer S., Cruz E., Medina-Gundrum L., Weitman S.. Elevated expression of DNA ligase I in human cancers. Clin. Cancer Res. 2001; 7:4143–4148. [PubMed] [Google Scholar]
- 13. Harrison C., Ketchen A.M., Redhead N.J., O'Sullivan M.J., Melton D.W.. Replication failure, genome instability, and increased cancer susceptibility in mice with a point mutation in the DNA ligase I gene. Cancer Res. 2002; 62:4065–4074. [PubMed] [Google Scholar]
- 14. Muvarak N., Kelley S., Robert C., Baer M.R., Perrotti D., Gambacorti-Passerini C., Civin C., Scheibner K., Rassool F.V.. c-MYC generates repair errors via increased transcription of alternative-NHEJ factors, LIG3 and PARP1, in tyrosine kinase-activated leukemias. Mol. Cancer Res. 2015; 13:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grawunder U., Zimmer D., Fugmann S., Schwarz K., Lieber M.R.. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell. 1998; 2:477–484. [DOI] [PubMed] [Google Scholar]
- 16. O’Driscoll M., Cerosaletti K.M., Girard P.M., Dai Y., Stumm M., Kysela B., Hirsch B., Gennery A., Palmer S.E., Seidel J.et al.. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell. 2001; 8:1175–1185. [DOI] [PubMed] [Google Scholar]
- 17. Altmann T., Gennery A.R.. DNA ligase IV syndrome; a review. Orphanet. J. Rare. Dis. 2016; 11:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maffucci P., Chavez J., Jurkiw T.J., O’Brien P.J., Abbott J.K., Reynolds P.R., Worth A., Notarangelo L.D., Felgentreff K., Cortes P.et al.. Biallelic mutations in DNA ligase 1 underlie a spectrum of immune deficiencies. J. Clin. Invest. 2018; 128:5489–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barnes D.E., Tomkinson A.E., Lehmann A.R., Webster A.D., Lindahl T.. Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell. 1992; 69:495–503. [DOI] [PubMed] [Google Scholar]
- 20. Webster A.D., Barnes D.E., Arlett C.F., Lehmann A.R., Lindahl T.. Growth retardation and immunodeficiency in a patient with mutations in the DNA ligase I gene. Lancet. 1992; 339:1508–1509. [DOI] [PubMed] [Google Scholar]
- 21. Pascal J.M., O’Brien P.J., Tomkinson A.E., Ellenberger T.. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004; 432:473–478. [DOI] [PubMed] [Google Scholar]
- 22. Taylor M.R., Conrad J.A., Wahl D., O’Brien P.J.. Kinetic mechanism of human DNA ligase I reveals magnesium-dependent changes in the rate-limiting step that compromise ligation efficiency. J. Biol. Chem. 2011; 286:23054–23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tumbale P.P., Jurkiw T.J., Schellenberg M.J., Riccio A.A., O’Brien P.J., Williams R.S.. Two-tiered enforcement of high-fidelity DNA ligation. Nat. Commun. 2019; 10:5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 25. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Echols N., Headd J.J., Hung L.W., Jain S., Kapral G.J., Grosse Kunstleve R.W.et al.. The Phenix software for automated determination of macromolecular structures. Methods. 2011; 55:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bauer R.J., Jurkiw T.J., Evans T.C. Jr, Lohman G.J. Rapid time scale analysis of T4 DNA Ligase-DNA binding. Biochemistry. 2017; 56:1117–1129. [DOI] [PubMed] [Google Scholar]
- 29. Prigent C., Satoh M.S., Daly G., Barnes D.E., Lindahl T.. Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol. Cell. Biol. 1994; 14:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teo I.A., Broughton B.C., Day R.S., James M.R., Karran P., Mayne L.V., Lehmann A.R.. A biochemical defect in the repair of alkylated DNA in cells from an immunodeficient patient (46BR). Carcinogenesis. 1983; 4:559–564. [DOI] [PubMed] [Google Scholar]
- 31. Soza S., Leva V., Vago R., Ferrari G., Mazzini G., Biamonti G., Montecucco A.. DNA ligase i deficiency leads to replication-dependent DNA damage and impacts cell morphology without blocking cell cycle progression. Mol. Cell. Biol. 2009; 29:2032–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaminski A.M., Tumbale P.P., Schellenberg M.J., Williams R.S., Williams J.G., Kunkel T.A., Pedersen L.C., Bebenek K.. Structures of DNA-bound human ligase IV catalytic core reveal insights into substrate binding and catalysis. Nat. Commun. 2018; 9:2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Podos S.D., Thanassi J.A., Pucci M.J.. Mechanistic assessment of DNA ligase as an antibacterial target in Staphylococcus aureus. Antimicrob. Agents Chemother. 2012; 56:4095–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tumbale P., Williams J.S., Schellenberg M.J., Kunkel T.A., Williams R.S.. Aprataxin resolves adenylated RNA-DNA junctions to maintain genome integrity. Nature. 2014; 506:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahel I., Rass U., El-Khamisy S.F., Katyal S., Clements P.M., McKinnon P.J., Caldecott K.W., West S.C.. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006; 443:713–716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates for LIG1-R641L-DNA complex (7L34) and LIG1-R771W-DNA complex (7L35) have been submitted to the RCSB Protein Data Bank.