


Abstract
Sequence-specific protein-DNA interactions are at the heart of the response of the tumor-suppressor p53 to numerous physiological and stress-related signals. Large variability has been previously reported in p53 binding to and transactivating from p53 response elements (REs) due, at least in part, to changes in direct (base) and indirect (shape) readouts of p53 REs. Here, we dissect p53 REs to decipher the mechanism by which p53 optimizes this highly regulated variable level of interaction with its DNA binding sites. We show that hemi-specific binding is more prevalent in p53 REs than previously envisioned. We reveal that sequences flanking the REs modulate p53 binding and activity and show that these effects extend to 4–5 bp from the REs. Moreover, we show here that the arrangement of p53 half-sites within its REs, relative to transcription direction, has been fine-tuned by selection pressure to optimize and regulate the response levels from p53 REs. This directionality in the REs arrangement is at least partly encoded in the structural properties of the REs. Furthermore, we show here that in the p21-5′ RE the orientation of the half-sites is such that the effect of the flanking sequences is minimized and we discuss its advantages.
INTRODUCTION
The current view on protein–DNA interactions is that proteins achieve DNA binding specificity by forming direct contacts with the bases using hydrogen bonds, or hydrophobic interactions (‘direct’ or ‘base’ readout) that is complemented by ‘indirect’ or ‘shape’ readout, which depends on the structural and dynamical (conformational flexibility) properties of the specific DNA targets (1). Both types of recognition contribute to the interaction between p53 and its response elements (REs) (2–8). p53 is one of the most central regulatory hubs in human cells, connected to a complex network in the living cell, activating numerous genes, leading to diverse cellular outcomes (9–11). At the molecular level p53 primarily functions as a transcription factor (TF) that binds p53 REs in its target–gene promoters. The importance of sequence-specific binding of p53 to its REs is highlighted by the large number of mutations in p53 that are observed in cancer (∼50% of all cancers), most of which occur in the p53 DNA binding domain (DBD) (∼86% of tumorigenic mutations are in the DBD, 12,13). The consensus binding site of p53 REs consists of two decameric repeats of the form RRRCWWGYYY (R = A,G;W = A,T;Y = C,T), separated by 0–18 bp (14). Full-length p53 as well as shorter protein constructs that incorporate the DBD bind to their DNA targets cooperatively as tetramers, composed of a dimer of dimers (15). As the outcome of p53 activation depends on the function of the p53-induced target genes, discrimination of its numerous target sites is crucial for p53 to trigger specific cell-fate decisions. Two alternative models have been proposed (16,17): the selective binding model assumes that p53 REs are not equal, and discrimination is achieved at the level of DNA binding through differences in binding affinity (and hence affected by p53 cellular level), in p53 post-translational modifications (PTMs), or in the binding of other p53 co-regulators. The selective context model assumes that p53 binds to all accessible genomic binding sites more or less equally, and selectivity is conferred by the cellular and genomic ‘context’, i.e. binding of independent TFs, histone modifications, nucleosome positioning, target gene core promoter architecture, or stimulation of RNAPII activity at the initiation or the elongation steps (16,18–20), as well as dependent on other genes expressed in the cell at the same time, the cell type, or the stress signal (21). The truth is probably somewhere between these two alternative models, but the extent to which each process contributes to p53 functional response is currently unknown. p53 is currently considered to act only as a transcription activator, following direct binding to its REs. Transrepression by p53 is mediated by indirect transcription, where p21 activation by p53 directs transrepression through the p53-p21-DREAM pathway leading to downregulation of many cell-cycle genes (22–25).
In our previous publications, we have offered several rules pertaining to p53 target discrimination and its dependence on the structural characteristics of p53 REs (3,4,26,27). In these studies we have shown experimentally that the major structural change between consensus-like p53 REs, that differ in the uncontacted WW motif at the center of each decameric repeat (i.e. p53 half-site), is the torsional (twist) flexibility (the ease of opening and closing of the DNA double helix), and that it leads to significant changes in the DNA binding affinity and especially the binding cooperativity (3). In particular, we showed that p53 REs containing the CATG central motif are more torsionally flexible than CAAG and CTAG containing REs. REs containing the CATG motif are bound with high affinity and low cooperativity by p53. The more torsionally rigid CTAG- and CAAG-containing REs are bound with lower affinity and higher cooperativity (3). Moreover, we found that torsional flexibility of REs is the key determinant of gene activation when p53 levels are low, whereas transactivation at high p53 levels correlates with binding affinity instead (4). Since the prevalent p53 species at low protein concentration are dimers of p53 (28–30), binding at such conditions can be accomplished only when p53 dimers are kinetically stable on the REs, to assure the continuous presence of the first dimer on the DNA until a second dimer comes along and interacts with the first dimer to form a stable transactivation-capable tetramer. Such kinetic stability is provided by optimizing protein–protein interaction through re-orientation of the two monomers within the bound dimer, facilitated by DNA torsional flexibility (4). Furthermore, recently we showed that p53 binds to REs containing long spacers in two different modes: fully specific and hemi-specific. In the latter, only one p53 dimer is specifically bound to a DNA half-site, whereas the other dimer is bound to the spacer DNA. Nonetheless, the two modes have comparable binding affinity and specificity, as well as similar in vivo transactivation potential (27). Using computational and experimental methods we dissect here the role played by p53 full-sites, half-sites, quarter-sites and flanking sequences in the mechanism of recognition between p53 and its target sites, and its implication to transactivation from these sites. We show the intricate ways in which apparent evolutionary selection pressure has optimized the sequence and orientation of the RE’s half and quarter sites to modulate the outcome of p53 binding to, and transactivation from, p53 target sites.
MATERIALS AND METHODS
Protein and DNA
Human p53 core domain (residues 94–293, referred to as p53DBD) was overexpressed and purified as described by Kitayner et al. (2). Protein concentration was determined spectroscopically, as previously described (3). Prior to the binding studies, the fraction of p53DBD molecules active for DNA binding was determined as described previously (3,31). All DNA sequences used in the electrophoretic mobility shift assay (EMSA) (except Flex-RL+DAP, synthesized by Sigma UK) were synthesized by Sigma Genosys (Israel) and purified by a reverse-phase cartridge. The sequences were designed as intramolecular hairpin constructs with 30 DNA bases in the stem and five cytosines in the loop, as described previously (3). The DNA test sequences for cyclization experiments (bottom PCR templates) were synthesized by Sigma Genosys (Israel), and contained three copies of the decameric half-site of the studied sequences (Table 2), whereas the library DNA sequences (top PCR templates) and the fluorescein- and tetramethylrhodamine (TAMRA)-labeled oligonucleotide primers (32) were synthesized by the Keck foundation Laboratory at Yale University, and purified as previously described (32).
Table 2.
Best-fit parameters from cyclization kinetics measurements for the sequences studied here
| Name | Sequence | Bend angle (°)a,b | Twist angle (°)b | Roll and tilt fluctuations (°)b,c | Twist fluctuations (°)b,d | Torsional force constant × 1019 erg*cme |
|---|---|---|---|---|---|---|
| p21-5′ left HS | GAACATGTCC | 0.36 ± 0.11 | 34.33 ± 0.13 | 4.40 ± 0.12 | 4.43 ± 0.11 | 2.31 |
| p21-5′ right HS | CAACATGTTG | −0.17 ± 0.07 | 34.22 ± 0.08 | 4.51 ± 0.09 | 5.45 ± 0.08 | 1.53 |
| p21-3′ left HS | GAAGAAGACT | 0.23 ± 0.11 | 34.29 ± 0.12 | 4.46 ± 0.13 | 4.37 ± 0.11 | 2.37 |
| p21-3′ right HS | GGGCATGTCT | 0.80 ± 0.08 | 34.09 ± 0.09 | 4.63 ± 0.10 | 5.41 ± 0.08 | 1.55 |
| Range of values observedf | −10.0 to 2.4 | 34.04 to 34.85 | 4.30 to 6.40 | 3.9 to 5.80 | 2.98–1.34 |
All measurements were made at 21°C.
aBending is by roll and its center is located at the fourth or fifth step of all sequences.
bThe simulation errors (±), were calculated as described in (33).
cRoll and tilt fluctuations are the thermal fluctuations of the roll and tilt angle between adjacent base pairs (32,33).
eThe torsional force constant, C, is given by C = lkBT/σt2 (33), where l is the helical rise of one DNA base pair, kB is Boltzmann constant, T is the Kelvin's temperature and σt is the twist fluctuations in radians.
Cyclization kinetics and simulation of cyclization data
Constructs were synthesized using the PCR scheme developed by Zhang and Crothers (32), as described previously (3). In short, PCR reactions (50 μl) contained 1× PrimeSTAR buffer, 0.2 mM dNTP mix, 2 μM of each of the primers, 30 nM of the top and bottom templates and 0.04 units of PrimeSTAR DNA polymerase (Takara, Japan). Cyclization kinetics measurements were carried out as described before (3,32). Ligase concentration was varied as a function of phasing length (0.3 U/μl for the in-phase 156L14 and 156L16 constructs and 1.2 U/μl for all other phasing constructs) and total length (from 0.2 U/μl for the 157 constructs, to 0.8 U/μl for the 154–155 and 158–160 constructs and 2.2 U/μl for the 150–153 constructs). Quantitative data on the conformational properties of the tested DNA molecules was derived by the simulation program developed by Zhang and Crothers (33), as previously described (3). The outcome of the simulations are the bend angle, the twist angle, the roll and tilt fluctuations (defined as the thermal fluctuations of the roll and tilt angle between adjacent base pairs, 32,33), and the twist fluctuations (defined as the thermal fluctuations of the twist angle between adjacent bases, 32,33), per DNA sequence (p53 half-site).
Binding affinity and stability measurements
Binding affinity measurements were conducted as previously described (3,4). In short, radiolabeled and gel-purified DNA hairpin duplexes (concentration < 0.1 nM) and increasing amounts of p53DBD were incubated on ice for 2 h in a buffer containing 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 1 mM ATP, 25 μg/ml BSA, 13% glycerol, 10 mM DTT, 150 mM KCl and 0.05% Nonidet P-40. By considering the salts added to the reaction mix by adding the protein with its protein dilution buffer (6 mM Tris–HCl (pH 7.5) and 60 mM NaCl) the total ionic strength of the binding buffer was 296 mM. Complexes were resolved from free DNA by electrophoresis on 6% native gel (37.5:1 acrylamide: bisacrylamide ratio). Samples were loaded on running gels run at 550 V and 4°C in 1× TG running buffer (25 mM Tris–HCl, 190 mM glycine, pH 8.3) and run until the bromophenol blue dye migrated 8 cm. Dried gels were quantified using a GE Typhoon7000 phosphoimager. We analyzed the system separately for each bound band using Cliqs version 1.1 (TotalLab Ltd., UK). Association binding constants were calculated using nonlinear least-squares methods of parameter estimation (SigmaPlot, Jandel Scientific, CA, USA). The apparent basic binding unit of p53DBD to DNA is a dimer (3), and therefore we used a regular two binding-site model for all p53 binding sites (34). For gel patterns in which only the bound tetramer species was observed, we add initial zeros to account for the unobserved dimer band (3). Thus, the following equations were used:
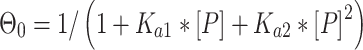 |
(1) |
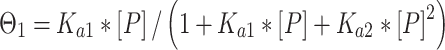 |
(2) |
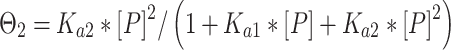 |
(3) |
Θi, the fraction of DNA molecules with i protein molecules bound, was calculated from the equation: Θi = (PSL – bg)i/Σi(PSL – bg)i, where PSL is the photostimulated luminescence and bg = background, and the summation is over all the bands in a given lane. P is the protein and the macroscopic association binding constants are Ka1 and Ka2, for the dimeric and the tetrameric species, respectively. The macroscopic binding constants are related to the intrinsic, microscopic binding constant as follows:
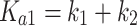 |
(4) |
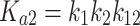 |
(5) |
where k1 and k2 are the intrinsic microscopic binding constants for each half-site and k12 is the cooperativity constant (34). k12 measures the increase (or decrease) in the binding affinity of the second dimer relative to that of the first dimer due to the cooperativity of the binding interaction. For targets with identical half-sites (k1 = k2), and assuming equipartition of binding free energies, an apparent dimer-equivalent association constant can be calculated by taking the square root of the Ka2 value. Even when k1 is not equal to k2 this measure gives an estimation for an averaged dimer equivalent tetramer KD also for these sites. The reported values (Table 3) are the dissociation binding constants (KD), which are the reciprocal of the association binding constants. The values in parentheses in all tables and text are the standard error of the mean (SEM).
Table 3.
Structural and binding parameters for p53 REs studied here
| Name | 5′ V(B), 4-bp flanks (°3Å3)a | 5′ Bridge dinuc. V(B) (°3Å3)a | Sequence b | 3′ Bridge dinuc. V(B) (°3Å3)a | Total V(B), RE & 4-bp 5′ flanks (°3Å3)a,d,e | Average dimer KD (nM) c | Macroscopic tetramer KD (× 1015 M2)c | Average dimer equiv. tetramer KD (nM)c | Average cooperativityc |
|---|---|---|---|---|---|---|---|---|---|
| Rig-LR | 0.900 | 4.7 | ttctt GAACATGTCC CAACATGTTG gcaga | 2.6 | 2.191 | 799 ± 65 | 4.7 ± 0.5 | 50 ± 3 | 255 ± 26 |
| LR | 2.167 | 4.7 | tacat GAACATGTCC CAACATGTTG gcaga | 2.6 | 2.357 | 838 ± 59 | 4.5 ± 0.2 | 50 ± 1 | 292 ± 41 |
| Flex-LR | 2.633 | 2.3 | tatac GAACATGTCC CAACATGTTG gcaga | 2.6 | 2.313 | 942 ± 30 | 5.8 ± 0.3 | 56 ± 1 | 285 ± 22 |
| Flex-LR &GC | 2.800 | 2.3 | ccacc GAACATGTCC CAACATGTTG gcaga | 2.6 | 2.385 | 682 ± 43 | 5.5 ± 0.4 | 55 ± 2 | 157 ± 17 |
| 5′ Nat-LR | 2.267 | 2.6 | gtcag GAACATGTCC CAACATGTTG gcaga | 2.6 | 2.278 | 530 ± 28 | 4.1 ± 0.4 | 47 ± 2 | 132 ± 16 |
| 5′3′ Nat-LR | 2.267 | 2.6 | gtcag GAACATGTCC CAACATGTTG agctc | 1 | 2.278 | 424 ± 34 | 3.0 ± 0.5 | 38 ± 4 | 146 ± 30 |
| Rig-RL | 0.900 | 1.0 | ttctt CAACATGTTG GAACATGTCC gcaga | 2.3 | 2.030 | 474 ± 32 | 4.5 ± 0.2 | 50 ± 1 | 92 ± 11 |
| RL | 2.167 | 1.0 | tacat CAACATGTTG GAACATGTCC gcaga | 2.3 | 2.196 | 678 ± 52 | 4.4 ± 0.6 | 49 ± 3 | 199 ± 29 |
| Flex-RL | 2.633 | 2.6 | tatac CAACATGTTG GAACATGTCC gcaga | 2.3 | 2.326 | 822 ± 38 | 5.7 ± 0.4 | 56 ± 2 | 230 ± 34 |
| Flex-RL &GC | 2.800 | 2.6 | ccacc CAACATGTTG GAACATGTCC gcaga | 2.3 | 2.348 | 659 ± 43 | 5.0 ± 0.2 | 52 ± 1 | 160 ± 16 |
| Flex-RL+DAPf | 2.6 | txtxc CAACATGTTG GAACATGTCC gcaga | 2.3 | 362 ± 28 | 1.6 ± 0.3 | 29 ± 3 | 168 ± 31 | ||
| RLminus | 2.167 | 1.0 | tacat CAACATGTTG GGACATGTTC gcaga | 2.3 | 2.196 | 620 ± 30 | 3.2 ± 0.3 | 42 ± 2 | 231 ± 27 |
| Dimer KD (nM)c | Macroscopic tetramer KD × 1015 (M2)c | Dimer equiv. tetramer KD (nM)c | Cooperativityc | ||||||
| LL | 2.167 | 4.7 | tacat GAACATGTCCGAACATGTCC gcaga | 2.3 | 2.109 | 371 ± 31 | 1.8 ± 0.3 | 31 ± 3 | 149 ± 15 |
| RR | 2.167 | 1.0 | tacat CAACATGTTGCAACATGTTG gcaga | 2.6 | 1900 ± 70 | 11 ± 1 | 77 ± 3 | 617 ± 49 |
aThese values represent the calculated deformability (°3Å3) of the 4-bp abutting the specific RE from the 5′ or the 3′ side, that of the bridge dinucleotide between the flanks and the specific RE, or the total deformability of the specific RE and the flanks. See text for details.
bThe sequences are embedded in the stems of hairpin constructs with a 5-bp loop (3). The distal and proximal half-sites, with respect to transcription direction of the wild-type sequence, are bold and underlined, respectively. Sequences outside the specific 20-bp REs are in lower case.
cThe values are average of 6–10 experiments conducted with each sequence ± the standard error of the mean. See Materials and Methods for definitions and details.
dSee text for details.
eThe V(B) of the 4-bp constant 3′-flank is 2.53°3 Å3 and that of the natural 3′-flank is 1.33°3 Å3.
fX = DAP.
Binding stability (dissociation kinetics) measurements were conducted as previously described (4,35,36). Radiolabeled hairpin duplexes (0.4 nM) and p53DBD (160 nM active protein) were incubated for 2 h at 4°C in the same binding buffer used for KD measurements (except for Nonidet P-40), before adding unlabeled competitor DNA of the same sequence (1.6 μM), in linear double-stranded DNA form. F(t), the fraction of bound tetrameric complex at the different time points, was calculated from the equation: F(t) = (PSL – bg)complex(t)/[(PSL – bg) complex(t) + (PSL – bg)free(t)], where PSL is the photostimulated luminescence and bg is the background. ln[F(t)/F(0)] was plotted as a function of time (t) after the addition of the unlabeled competitor. The data was fitted to a two-phase first-order kinetic equation: F(t)/F(0) = Ae−k1*t + Be–k2*t, where A and B are fractions of molecules dissociating with rate constants k1 and k2, respectively. Half-life of complexes dissociating by the B process was calculated from: t1/2B = ln 2/k2.
Transactivation experiments of p21-5′ RE variants
A yeast-based functional assay was exploited, starting from strain yLFM-ICORE (37,38) that enables targeting of single-stranded oligonucleotides containing the chosen RE sequence at a chromosomal location containing a minimal promoter and the Firefly luciferase reporter gene. This homology driven process is facilitated by the production of a site-specific double-strand break and the oligonucleotide targeting results in the replacement of the ICORE cassette, an event that is identified exploiting the URA3 counter-selectable gene (39). This approach enabled us to generate a panel of strains where the chosen REs are inserted at the same site, i.e. flanked by the same surrounding sequence and located at the same distance from the transcription start site (TSS). Correct targeting was confirmed by colony PCR across the RE-containing sequence followed by Sanger sequencing. The reporter strains were then transformed with human wild type p53 expression plasmid based on the pRS314 or pRS315 vectors (40) and containing the GAL1,10 promoter to achieve regulatable transcription of the p53 cDNA by varying the amount of galactose in the culture medium (41). Transformants were selected on glucose plates and luciferase assays were performed in a 96-well plate format (42). An empty expression vector was used to measure and subtract out the p53-independent expression of the reporter at each condition. Each independent experiment consisted of between five and seven replicates and tested the entire panel of p215′-derivative strains. Six independent experiments were carried out.
A curated list of spacer-less p53 REs associated with p53-dependent up-regulated target genes
We assembled a set of p53 REs that contain sites validated for binding in cellular context and for p53-dependent gene expression. A flow chart (Supplementary Figure S1) describes the steps taken to assemble the list. We started with the 943 p53 cistrome targets from the study by Nguyen et al. (43, Supplementary Table ST8). First, we deleted REs near target genes shown to be down-regulated by the adjacent sites (down to 650 REs). We then deleted REs with more than 20-bp (i.e. those having spacer sequences between the two half-sites). However, it is known that p53 can regulate its dependent genes from p53 RE clusters, that is several consecutives p53 REs (14,44). In such cases part of the cluster can be REs with spacer sequences and some not. We looked for such REs in another study that included p53 RE sequence in the reported results (45), and ended up with 373 REs. From those we deleted REs found in front of pseudogenes, and REs located near ncRNA genes (thought to be involve in p53 repressive functions, 46,47). REs that were found to be potentially bidirectional were taken as pointing to the more highly expressed gene, or twice (sense and antisense strand sequences) if genes on both sides were equally transcribed. We thus ended with 338 p53 REs. REs with cistrome score of 2 and above (i.e. those with occupancy and differentially expressed genes in at least two cell types) were all collected. REs with cistrome score of 1 (43, Supplementary Table ST8), were collected when they were supported by an expression score in the recent meta-analysis of Fischer (23,24), or appeared in Supplementary Tables ST2 and ST6 in Nguyen (43). We also added 41 non-cistrome REs, based on similar considerations. The last step was pruning REs that were located in intragenic regions, because for these sites there is no meaning to being aligned with transcription direction, and this reduced the 274 REs list to a final dataset that comprises 250 REs (Supplementary Table S1).
Discrimination energy analysis to the curated p53 REs list
To evaluate the conservation properties of p53 REs within our curated p53 REs list, we used discrimination energy (DE) analysis. Berg and von Hippel (48,49) defined the mononucleotide discrimination energy per base pair (ϵlB, a dimensionless positive number) as the reduction in binding free energy (in units of kT) when a consensus base pair (denoted by ϵl0 and defined as the most commonly observed base pair at position l) is replaced by another base pair. An estimation for ϵlB is given by (48,49):
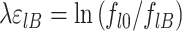 |
(6) |
where 0 is the consensus base and B any other base, and thus fl0 and flB are the frequencies of occurrence of the consensus base, or any other base, in position l in the population of all possible binding sites, respectively. The constant λ is a dimensionless scaling factor, which has values mostly near 1, that relate population of base-pair choices to binding free energies. The ϵlB values are not usually known a priori, and hence are estimated from equation (6). Thus, the total discrimination energy for a specific DNA sequence, 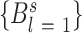 (where s is the site size) is given by:
(where s is the site size) is given by:
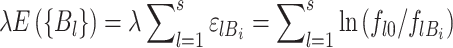 |
(7) |
where E expresses the reduction in favorable binding free energy (in units of kT) from that of the consensus sequence. To account for small sample properties, ln (fl0/flBi) is usually replaced by (49):
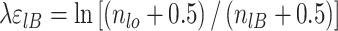 |
(8) |
since flB values are the base frequencies in the population of all sequences that are recognized by a particular protein, whereas the known sites constitute probably only a fraction of this population.
Information score analysis to the studied sequences using the curated p53 REs list
We used ‘information score’ (‘ISc’) analysis (50,51) as a measure for the calculated binding energy of the studied sequences. To that end we calculated a mononucleotide position-specific weight matrix from our set of 250 p53 REs:
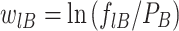 |
(9) |
where flB is defined as in the previous section, and PB of base B is the frequency in the whole genome, which is usually taken to be 0.25 for each base. The frequencies are corrected for small sample errors, as described previously (48). To score individual sequences, the weight matrix is multiplied element-wise by a matrix (slB) containing only 0’s and 1’s, corresponding to sequences for which binding data was experimentally determined in this study. The summation of this multiplication yields an individual information score (ISc) for each sequence (50–52):
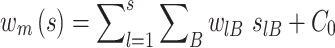 |
(10) |
We scale the obtained sum by adding a constant (C0), chosen such that the best binding site scores zero and poorer sites score positively.
To test for nearest-neighbor non-additive effects we need to calculate dinucleotide information scores, for which we need to add to mononucleotide information scores the following term (48,51,52):
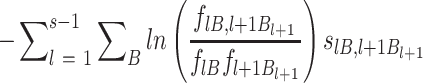 |
(11) |
where the numerator is the observed frequencies of the doublet BlBl + 1 in positions l and l + 1 and the denominator is the observed frequencies of its monomeric components. Here again we correct for small sample errors in the mononucleotide frequencies. The results were scaled by the same C0 chosen for the mononucleotide term. When calculating the information score for p21-5′ RE variants it was omitted from the 250 RE training set. Error bars for the obtained individual information scores were derived by the n – 1 Jackknife method (53), where ISc for p21-5′ RE variants was estimated by conducting 249 times the calculation of information scores using 248 REs, each time omitting one RE of the 249 REs set and then calculating the variance from:
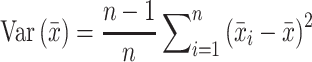 |
(12) |
where 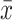 i is the information score estimated from each n – 1 subset, and
i is the information score estimated from each n – 1 subset, and 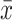 is the result when calculated from the whole set.
is the result when calculated from the whole set.
Other computational and statistical tests
In testing the strength of relationships between variables, we calculated Spearman's rank correlation coefficient (denoted by ρ) as a non-parametric measure of correlation. In these tests we used ties whenever the values were within ±SEM of each other. In testing for statistically significant differences between means of sequence-conservation categories we used Kruskal–Wallis test for non-parametric measures of the different between ranked sums. We used the Steel–Dwass method as a post-hoc test, which is adjusted for multiple comparisons, to determine the specific groups that significantly differed from each other. All analyzes were conducted using JMP®, Version 14 (SAS Institute Inc., Cary, NC, 1989–2019). The values for the deformability (V in units of °3Å3) of each DNA base-pair step is from Balasubramanian et al. (54, part viii of Supplementary Table S3). Here we used the values derived only from DNA sequences having the B-DNA conformation (V(B) °3Å3).
RESULTS AND DISCUSSION
A curated list of spacer-less p53 REs associated with p53-dependent up-regulated target genes
To evaluate p53 REs for binding and structural properties genome wide, one need first to assemble a set of p53 REs, that contain sites that are validated for p53 binding and that ends up in p53-dependent gene expression. Information on binding and upregulation by p53 were based on a meta-analysis by Nguyen et al. (43), carried out on data from 41 genome-wide ChIP-seq experiments, 16 of which included also associated gene expression data, to establish a genome-wide p53 cistrome. In addition, we used the meta-analysis of Fischer (23,24), based on data from 20 genome-wide TP53-dependent gene expression studies, and 15 genome-wide TP53 ChIP-seq studies. Our gathered data set (Supplementary Table S1) included only p53 REs that transactivate a nearby gene (excluding transrepressed nearby genes), based on the indications that p53 acts in gene regulation mostly as a direct transactivator protein, whereas p53-dependent transrepression is mostly carried out indirectly (22,24,25). Moreover, we excluded p53 REs containing spacers sequences between the two half-sites, because p53 half-sites (HSs) separated by spacer sequences may have unique structural properties (Anna Zivan and T.E.H., unpublished results). Thus, the compiled data set (Supplementary Table S1) consists of 250 p53 REs, with validated ChIP-seq peaks, that also show differentially expressed genes (DEGs) that are solely upregulated (see Material and Methods and Supplementary Figure S1). The 250 RE sequences were aligned with transcription direction (that is the 5′ HS is upstream and the 3′ HS is downstream in the strand being transcribed, though not necessarily upstream to the transcription start site (TSS), here they are termed ‘left’ and ‘right’ HS, respectively, see below).
The buildup of our curated list of p53 REs is comprehensive for REs with cistrome score of two and above (Supplementary Figure S1). There are 156 REs with cistrome score of 1 in Nguyen et al. (43). Such REs can, on the one hand, be a false positive signal (55), and on the other hand, indicate a truly functional but weakly binding RE, that needs special conditions to mediate p53-dependent transcriptional activation of the associated gene. To balance these two opposing possibilities, we inserted ∼20% of cistrome score = 1 sites, where we chose both high expression scores as well as low expression scores from Fischer (24), as well as low and high values from p53 associated transcriptome DEG (43, Supplementary Table ST6) and activated p53 peaks (43, Supplementary Table ST2). This same procedure was taken on 41 non-cistrome REs, with expression score of four and above (24), most of which were validated in individual studies (Supplementary Table S1).
Hemi-conservation of p53 response elements
The consensus sequence of p53 is extremely degenerate (Figure 1, the sequence logo of our curated list of 250 p53 REs, and (14,56,57)). Nevertheless, most p53 REs deviate from this consensus in at least one position (14,56). To study the conservation properties of p53 REs genome wide (that is their likelihood to be near or far from the consensus), we calculated the discrimination energy (DE) of all 250 p53 REs in our curated list (Supplementary Table S1). The discrimination energy per base pair denotes the change in binding free energy when a consensus base pair is replaced by any other base pair (48,49), and this is summed over the whole sequence. By convention, the consensus sequence has DE = 0 and those further away from the consensus have positive values (48,49). When we sum the DE values on p53 full-sites (FSs), we do not find any sequence with FS DE = 0. This reiterate our previous finding that sequences that resemble the consensus in both HSs [such as (GGGCATGTCC)2, (4)], are supertransactivator (STA) sequences, that transactivate p53 to high levels when there are few p53 molecules around and are thus un-regulatable. The range of DE values observed in p53 HSs spans from 0.00 (GGGCATGTCC, the 5′ HS of TNFRSF10A, B, or C, and the 3′ HS of ZFPM1) to 14.1 (CCTGAAGCCC, the 3′ HS of SERTAD1). The mean DE value for the left and right HSs is not significantly different from each other (3.60 ± 0.12 and 3.74 ± 0.13, for the left HS and right HS, respectively). Interestingly, as can be observed from Figure 2, the DE of the two HSs are inversely correlated (ρ = –0.396, P < 0.0001). When one HS is conserved (having low DE values), the other HS is less conserved (having high DE values). All 250 REs, except 11, are within the 95% confidence ellipse (Figure 2).
Figure 1.
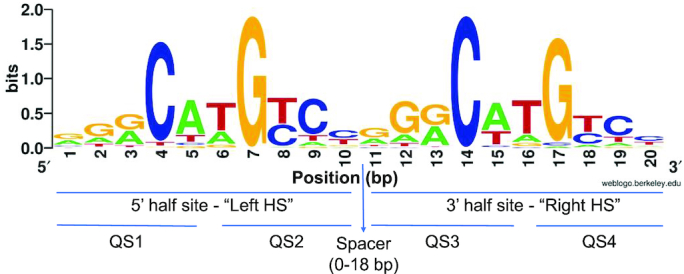
Sequence logo of our curated set of 250 activating p53 REs without spacers, aligned by transcription direction. Sequence logo was generated by weblogo (96). HS, half-site. QS1, QS2, QS3 and QS4 are the first, second, third and fourth quarter sites from the 5′ to the 3′ end of the sites.
Figure 2.
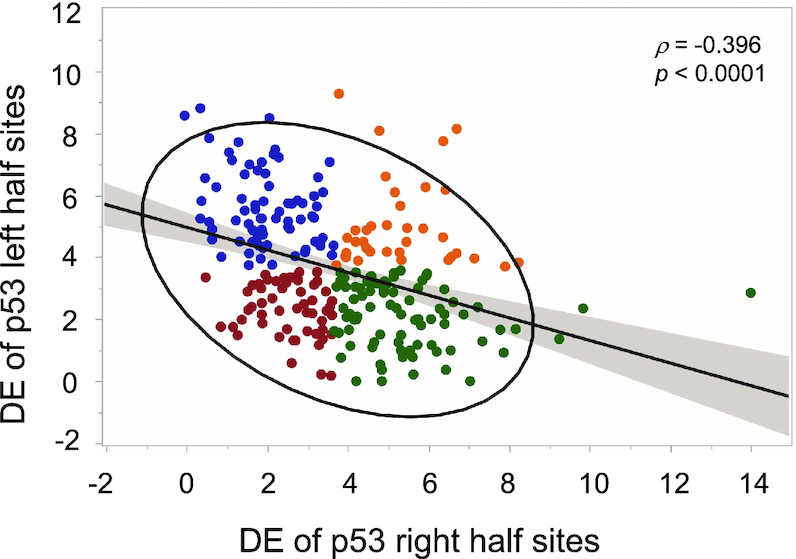
Discrimination energy analysis to p53 half-sites. The discrimination energy (DE) of the left (5′) half sites of p53 REs anticorrelate with the discrimination energy of right (3′) half sites. Analysis was conducted on our curated set of 250 activating p53 REs without spacers. Blue, DC category; green, CD category; red, CC category; orange, DD category. See text for details.
To be able to further discuss p53 REs that differ in their conservation, we turned the DE data into categorical data, where each HS can be either ‘conserved’ (C), or ‘degenerate’ (D). This is done by dividing the DE value of each decamer HS by half of the mean value of all 250 full-sites (each being a 20-bp sequence), and noting whether its value now is below 1 (C) or above 1 (D). Thus, we have created four categories: CC, DD, CD, and DC (colored differently in Figure 2). Using Chi-squared test for categories of data, we get χ2 (3, N = 250) = 22.35 (P = 0.00006), rejecting the assumption of random division to these four categories. Accordingly, the conservation of p53 HSs is not random. More importantly, the results show that full-sites (FSs) composed of two degenerate HSs, category DD, are underrepresented (36 REs), whereas FSs belonging to the CD and DC categories are overrepresented (85 and 74 REs, respectively, Figure 2).
This categorization of the REs data enables us to clearly see that there are many p53 FSs where only one HS has high enough sequence conservation to be contributing significantly to specific binding. By our definition, all REs of the DC and CD categories have one degenerate HS and one conserved HS, but they are not necessarily very different in their conservation level. However, by looking at ‘very’ degenerate HSs from the CD or DC categories (HSs with DE values > 5.89, which is the average DE value per HS plus one SD), we note that above average degenerate HS from these two groups are linked with above average conserved second HS (Supplementary Figure S2). Altogether there are 60 such outlying sites, which are 38% of the combined CD and DC category. This pattern, in which two HSs within one FS differ significantly in their conservation level, resembles the hemi-specific mode of binding that we observed previously, when we studied p53 REs that contain non-sequence specific spacer sequences between HSs (27). Thus, the hemi-specific mode of binding through spacer sequences may be an extreme case of the hemi-specificity phenomenon, which as we show here, is prevalent to lesser degrees in many p53 REs.
Conservation properties of inner versus outer p53 quarter-sites
p53 binds to a 20-bp sequence composed of four quarter-sites (QSs), each 5-bp long (Figure 1). Crystallographic studies of p53/DNA complexes (e.g. 2,5–7,58,59–61), show that p53-DNA tetramer has a shape of a parallelogram, because each monomeric subunit interacts with sequential 5-bps, and hence sequential p53 monomers interacts with bases on opposite strands on the DNA double helix, related by a 2-fold symmetry to each other. p53 monomers bound to the internal QSs (the ones closest to the center of the parallelogram, QS2 and QS3 in Figure 1) are therefore closer to each other, than those bound to the outer QSs, and thus play an important role in stabilizing the tetramer on the DNA, by providing the anchor or the nucleus of the tetramer. This may be the reason why the internal G7 and C14 are more conserved than the outer C4 and G17, as suggested by Nguyen et al. (43). This trend is clearly visible in the logo presented in Figure 1, which is based on our curated list of 250 spacer-less REs, associated with upregulation of p53-dependent target genes. Quantitating this observation by DE analysis (Supplementary Table S2) yields a large and significant difference in conservation between the inner and outer C/G bases (paired t = 4.15, P < 0.0001). However, as can be appreciated from Figure 1, and Supplementary Table S2, this difference in conservation level extends also to the bases on either side of the central C and G bases. The difference in conservation between the outer RRC/GYY motifs versus the inner ones is a bit lower but significant (paired t = 3.50, P = 0.0005). Moreover, the difference in conservation between external and internal quarter sites extends also to the non-contacted W5W6 positions, and hence to entire quarter sites (paired t = 3.06, P = 0.002). Surprisingly, here too, as we observed for the two HSs, the conservation (DE values) of each QS is inversely correlated to the neighboring QS within each HS (ρ = –0.33, 0.37, P < 0.00001, for QS1 to QS2 and QS3 to QS4, respectively), and similar in strength to the correlation between HSs.
As for FSs, we have corroborated these observations by turning the DE data into categorical data. When we turn each HS into four categories (CC, DD, CD and DC), the chi-squared test [χ2 (3, N = 250) = 31.0 for left HS and 30.9 for right HS, P < 0.0001] is not very informative, as the only category that deviates from the expected (by being under-represented) is the DD category. A more detailed picture can be achieved when we calculate all possible combinations between the four QSs in each FS leading to 16 categories [χ2 (15, N = 250) = 89.6, P < 0.00001, Table 1]. Here three categories are markedly overrepresented (>10%), CCCD (30 FSs), DCCC (29 FSs) and DCCD (26 FSs), illustrating the tendency of outer QSs to be of a degenerate character, as discussed above. The notably (<1%) underrepresented categories are DDDD (2 FS), DDCD (2 FSs), and DCDD (5 FSs) where inner QSs are degenerate. Interestingly, CCCC, i.e. fully conserved sites, are markedly underrepresented (8 FSs only, Table 1). The conclusion emerging from the detailed QSs analysis is that the pattern of FSs conservation is 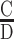 CC
CC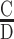 , but avoiding CCCC. Based on the anticorrelation between QSs that are within the same HS, we suggest that even though p53 monomers may enter the nucleus linked by their tetramerization domain (62), they form core-domain protein-protein contacts only on the DNA double helix and not prior to that. To control binding, the first p53 binds to the more conserved QS and the second p53 monomer binds to the adjacent QS, facilitated by core-domain protein-protein interactions. Hence, we call this novel mode of binding the ‘Cherry pair’ model, to illustrate that binding of monomers through their tetramerization domain does not entail their preforming protein-protein interactions through their DBDs.
, but avoiding CCCC. Based on the anticorrelation between QSs that are within the same HS, we suggest that even though p53 monomers may enter the nucleus linked by their tetramerization domain (62), they form core-domain protein-protein contacts only on the DNA double helix and not prior to that. To control binding, the first p53 binds to the more conserved QS and the second p53 monomer binds to the adjacent QS, facilitated by core-domain protein-protein interactions. Hence, we call this novel mode of binding the ‘Cherry pair’ model, to illustrate that binding of monomers through their tetramerization domain does not entail their preforming protein-protein interactions through their DBDs.
Table 1.
Chi-squared analysis to p53 quarter site
| Category | CCCC | CCCD | CCDC | CCDD | CDCC | CDCD | CDDC | CDDD | DCCC | DCCD | DCDC | DCDD | DDCC | DDCD | DDDC | DDDD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Observed counts | 8 | 30 | 20 | 12 | 22 | 23 | 23 | 6 | 29 | 26 | 21 | 5 | 13 | 2 | 8 | 2 |
| Estimated probability | 0.032 | 0.120 | 0.080 | 0.048 | 0.088 | 0.092 | 0.092 | 0.024 | 0.116 | 0.104 | 0.084 | 0.020 | 0.052 | 0.008 | 0.032 | 0.008 |
| Expected counts | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 | 15.6 |
| (O-E)2/E | 3.7 | 13.2 | 1.2 | 0.8 | 2.6 | 3.5 | 3.5 | 5.9 | 11.5 | 6.9 | 1.9 | 7.2 | 0.4 | 11.9 | 3.7 | 11.9 |
Bold values show over and under representation categories that extremely deviated from expected, (O – E)2/E ≥ 6.9, P< 0.0001.
Global conformation characteristics of naturally occurring p53 REs
Here we experimentally determined, by cyclization kinetics of DNA mini-circles, the global conformation of two parts (p21-5′ and p21-3′) of the well-known natural p53 binding site, located in the promoter of the p21 gene (also known as CDKNA1, WAF1, or p21CIP1, ref. 63). As the sequence of each half site in natural p53 sites is different, and to enhance the signal to noise ratio, the 30-bp test sequences were designed as three repetitions of each half-site decamer, as in our previous study on consensus-like sites (3). To avoid confusion between the 5′ and 3′ in the full-site name, and those of the 5′ and 3′ HSs, 5′ HSs are here termed ‘left HS’, and the 3′ HSs are here termed ‘right HS’. Experimental J factors were measured as previously described (3,27). The relationship between the experimental J factors and DNA structure was obtained by minimizing the differences between experimental J factors and calculated J factors, based on a model for the structural properties of the tested sequences (Supplementary Figure S3), using the method of Zhang and Crothers (33). The outcome of the simulations are quantitative measures for DNA structure and flexibility (Table 2). From Table 2, it is clear that the only significant difference between the sequences of the HSs is the twist fluctuations, indicative of twisting (torsional) flexibility, as observed previously by us for p53-consensus sequences (3,27). The differences in twist fluctuations between the different HSs studied here appear to be small. However, as can be noted from the last row of Table 2, they cover a range of ∼60% of all twist fluctuations measurements made to date, for DNA in any protein–DNA system, not only those of p53 REs. The limited variation in the twist and bending (roll and tilt) fluctuations, as a function of the base sequence, was noted previously (64,65). It was estimated that the range of variation of the torsional and bending force constants are 2- and 4-fold, respectively (64,65). Moreover, from the range of observed values for bend angles presented in the last row of Table 2 [from other studies, (3,32,66)] it is clear that there are no significant changes in global bend angles among the sequences studied here.
Analyzing flexibility of p53 REs genome-wide
Since cyclization kinetics is a laborious technique that is time consuming, it is not feasible to experimentally measure the flexibility properties of the entire set of 250 p53 REs. Therefore, to assess the flexibility properties of our curated set of p53 REs, we need to be able to calculate it, assuming that we can represent DNA sequences based on nearest-neighbor interactions only (i.e. base-pair steps have independent structural properties). Previously we used ‘twist disp’ values, which are the standard deviation of ‘twist angle’ values derived from crystallographic studies of protein–DNA complexes (67), to signify the ‘twist flexibility’ of DNA molecules that have not been measured experimentally (3). However, now that our data set of experimentally measured p53 binding sites have grown, we find that the twist fluctuations of the experimentally measured sites are nicely correlated (ρ = 0.90, P < 0.000001, Supplementary Figure S4) to a more rigorously defined parameter, termed ‘deformability’ (54,67). Deformability is a set of sequence-dependent empirical ‘energy’ functions, derived from crystal structures of protein-DNA complexes that denote the volume of conformational space within common energy contours (54,67). It is thus a measure of the cost of deforming DNA molecules of a particular base sequence. As it is derived from all six structural parameters that relate two successive base-pairs (‘base-pair step’), it is a more comprehensive measure for base-pair steps flexibility. The observation that calculated deformability correlates well with our experimentally measured twist-fluctuation values (Supplementary Figure S4), may mean that indeed, as we have observed previously, the main contribution to flexibility of p53 binding sites is twist (torsional) flexibility.
The values of the deformability parameter (V(B)) ranged from 0.96 °3Å3 (left HS of p21-3′) to 2.91 °3Å3 (right HS of CPE). When the sites are arranged with transcription direction, we find that the differences in the average deformability between the HSs is not significant (2.14 ± 0.02 °3Å3, 2.12 ± 0.02 °3Å3 for left HS and right HS, respectively). The average value of V(B) for all FS REs is 2.20 ± 0.02 °3Å3, which means that FS REs are on average more deformable than each separate HS, indicating the contribution of the base-pair step connecting the two HSs (average V(B) value 3.3 ± 0.1 °3Å3) to the increase in overall deformability of FS REs. Indeed, the structural properties of the dinucleotide connecting the two HSs have been shown to be unique, relative to those of the base-pair steps within the two HSs (5,6).
We then checked whether the conservation properties of p53 full-site REs are related to the calculated deformability of these sites. DE values of p53 full-sites have a weak but significant negative correlation with their deformability (ρ = –0.22, P = 0.0004, Supplementary Figure S5A). This means that full sites that are more conserved (low DE values) tend also to be more deformable. We then analyzed the relationship between each QS DE and deformability (Supplementary Figure S5B–E). A significant negative correlation exists between DE and V(B) for QS1 and QS4 (Supplementary Figure S5 B, C, ρ = –0.32 and –0.21, P < 0.0001, and 0.0009, respectively), both being outer QSs (Figure 1), but not for the two inner quarter sites (QS2 and QS3, Supplementary Figure S5 D, E). This we suggest is due to the outer QSs being more variable in their conservation level than the inner QSs.
Transactivation from switched p53 half-sites of p21-5′
To experimentally probe the importance of HSs and QSs conservation and orientation for p53 binding and transactivation, we designed two binding sites with alternative sequence exchanges. The first switch, which preserved the inner versus outer locations of the QSs, was made by changing the transcribed strand to the antisense strand (termed here ‘vertical’ switch), thus converting wild type p21-5′ (henceforth called LR) to RLminus (Figure 3). In the second exchange the inside/outside location of the HSs were switched within each strand (LR to RL, termed here ‘horizontal’ switch, Figure 3). Switching the strands, with respect to the strand being transcribed (LR to RLminus), can also assess whether there is directionality in p53 binding to its REs, as this change should not matter for in vitro binding of a symmetric molecule such as p53, but could matter for ‘functional binding’, i.e. for transactivation from a nearby reporter gene. Two scenarios can be envisioned here. First, such a change in vivo could possibly change the direction of transcription. This option is not considered here. Second, the directionality (relative positioning) of QSs and HSs, with respect to the TSS affect transactivation levels. Moreover, since we are switching only the 20-bp RE, the horizontal and vertical switches can also test whether the flanking sequence matter for binding and transactivation, as the sequence context of each half site is changed upon these exchanges (i.e. after the exchanges the left HS is near the 3′ flanks and the right HS is near the 5′ flanks).
Figure 3.
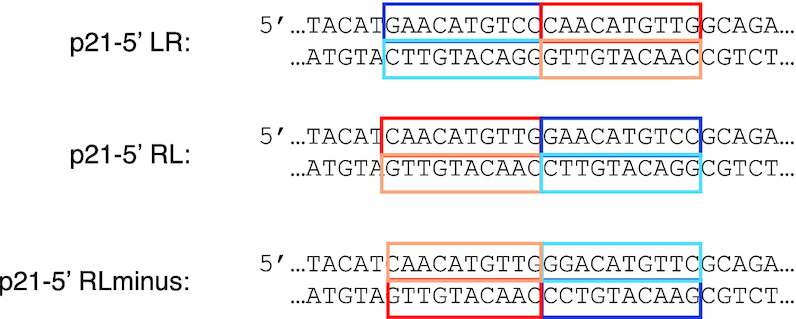
Switching p53 half-sites of p21-5′. Horizontal (LR to RL) and vertical (LR to RLminus) switching of p21-5′ half sites. Half sites are boxed and color coded. The flanking sequences are those of the original plasmid construct. See text for details.
We have used a tightly defined experimental system in yeast to measure transactivation from LR, RL and RLminus (42). Using this system one can place by homologous recombination a desired variant of the p53 RE at the same chromosomal location that had previously been engineered to contain a minimal promoter and the Firefly luciferase reporter gene (37). At low p53 level [0.002% galactose, corresponding to ∼440 p53 molecules, ∼0.5 nM, per yeast cell, (4)], there is a small but significant difference between LR and RL (t = 3.32, P = 0.0015), which is significant also when taking into account multiple comparisons (Figure 4, Supplementary Tables S3 and S4). Relative to LR and RL, the RLminus site has the highest transactivation level at all p53 concentrations. The difference between LR and RLminus, and between RL and RLminus, is large, highly significant, and exists at all p53 concentration levels (Figure 4, Supplementary Tables S3 and S4), though, as with LR versus RL, the difference is higher at low p53 concentration (0.002% galactose, t = 10.99 and 6.92, for LR-RLminus and RL-RLminus, respectively, P < 0.000001).
Figure 4.
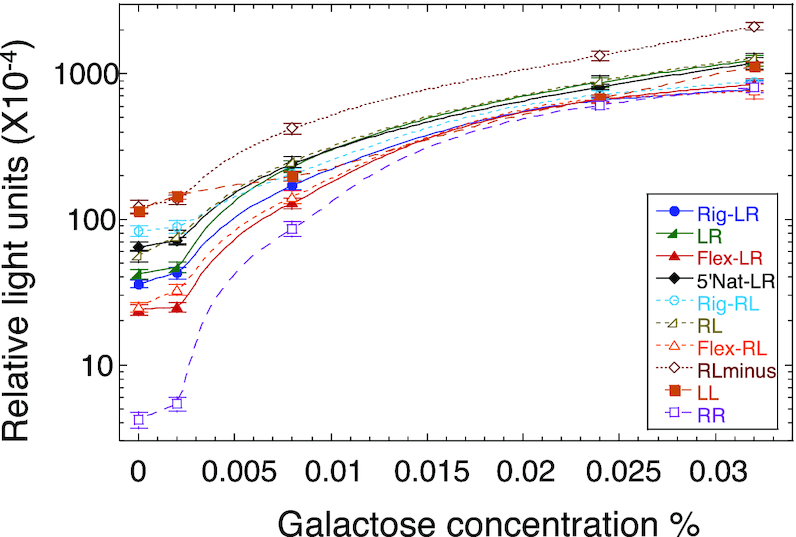
Transactivation level from variants of the p21-5′ RE as a function of p53 protein levels. Raffinose (marked as 0% galactose) and increasing concentrations of galactose were used to achieve variable cellular concentration of p53. Transactivation is represented as relative light units (RLU). Each value is an average of 4–6 independent experiments. Each independent experiment contained 5–7 replicas of each RE. Error bars are SEM values. Cells were grown overnight in a glucose containing medium, were washed and resuspended in a fresh medium containing raffinose (2%) or increasing concentration of galactose (at time zero). Transactivation was measured after 6 h.
Effects of flanking sequences on p53 transactivation level
To explore the extent of the effect that the structural properties of the flanking sequences have on transactivation level from p53 sites, we designed two 10-bp DNA motifs that differ in their deformability, as a measure for total flexibility of the DNA double helix. Thus, we designed two DNA motifs having extreme deformability values: AACTTTTCTT, termed ‘rigid flank’ (V(B) = 0.81°3Å3) and AGTAGTATAC, termed ‘flexible flank’ (V(B) = 2.72°3Å3). The 10-bp flanking sequences of the original reporter construct is AATAATACAT (V(B) = 2.37°3Å3). We inserted these motifs only at the 5′ side of the REs, since we did not want to alter the distance of the REs from the TSS. When comparing transactivation levels between REs with flanks possessing opposing structural characteristics (‘Flex’ versus ‘Rig’ in Figure 4 and Supplementary Table S3), one observes that the differences within the RL series are larger than within the LR series, and are especially high when transactivation was measured at basal p53 concentration (at 2% raffinose, t = 7.9 and 4.8, P < 0.000001 and <0.0001, for RL versus LR series, respectively, Supplementary Table S4). Thus, at 2% raffinose there is a significant 232% change between Rig-RL and Flex-RL, but only 50% change between Rig-LR and Flex-LR. Moreover, the influence of the flexible flank on transactivation from the two types of REs (RL versus LR) is similar (Supplementary Table S3) and thus not statistically different (Supplementary Table S4), but the effect of the rigid flank on transactivation level is double in Rig-RL than in Rig-LR at basal p53 concentrations (Supplementary Table S3), and therefore highly statistically different (t = 6.7, P < 0.000001, at 2% raffinose, Supplementary Table S4). Surprisingly, none of the studied sites (except LL at basal p53 levels) approached the level of transactivation level induced by the RLminus site (having the original medium deformability flanks). The difference between transactivation levels of the various p21-5′ sites at highly induced p53 levels (0.024% and 0.032% galactose) is significantly smaller than in basal levels, and resembles the small range of binding affinity differences between p53 tetramers (as discussed below). It should be noted that upon switching the RE HSs, the dinucleotide bridging the RE to the flanks has changed as well. The 5′ bridge nucleotide is very rigid in Rig-RL and RL, versus very flex in Rig-LR and LR. This can be another factor contributing to the higher transactivation level in the RL series versus the LR series.
Effect of half-site doubling on p53 transactivating levels
Transactivation from a site composed of double left HS of p21-5′, in the original flexibility context (LL, Figure 4, Supplementary Table S3), started as a site from which there was high p53 transactivation level at basal p53 concentration, similar in value to RLminus, but p53 transactivation level from this site declined to about 40% relative to that from RLminus from medium p53 levels onwards. On the other hand, a site composed of double right HS of p21-5′ with the original flexibility flanks (RR, Figure 4, Supplementary Table S3) started, at basal p53 concentration, as the site with the lowest transactivation capacity from all studies REs, but at the highest p53 concentration it reached a transactivation level comparable to some other sites studied here. These observations can be ascribed to interdimer core-domain interactions in highly induced p53 levels in yeast cells [at least 0.024% galactose, estimated to be ∼33 500 p53 molecules, amounting to ∼40 nM p53 (4)], leading to either rapid formation of tetramers upon binding to DNA, or possibly to pre-formation of tetramers prior to DNA binding. Hence, these observations imply that when p53 levels are high, p53 sites devoid of even a single fully-canonical p53 HS (such as those belonging to the DD category) can be transactivated at a level comparable to transactivation from sites with at least one consensus-like half-site. In human cells basal p53 levels range from 17 000 to 200 000 molecules, which is ∼60–500 nM p53 (68–70), and they can increase up to 20-fold upon cellular damage signals (69,71), whereas in yeast cells, induced p53 levels are defined as those where galactose levels are >0.002% (<0.4 nM p53). Hence, the definition of basal versus induced p53 levels are different in yeast versus human cells.
Binding affinity and stability of switched p21-5′ variant sites
Differences in transactivation levels between REs studied here were shown to be more pronounced at basal p53 concentrations (Figure 4, Supplementary Table S3), where wild-type p53 exists mainly as dimers (4,28). To assess the binding affinities of the same sites measured for transactivation capacity, we used the p53DBD construct and EMSA experiments (Figure 5, Supplementary Figure S6). The use of the p53DBD construct is to facilitate the observation of p53 dimers during EMSA, and their quantification. Transiently bound dimers are important binding species for deciphering the mechanism of p53 binding to its REs (28). Only when using the isolated DBD, protein-protein interactions originating from the tetramerization domain do not obscure those originating from protein-protein contacts within DBDs. Moreover, as shown previously, the relationship between transactivation and p53 tetramer binding affinity is independent of the p53 construct used in the binding experiments (p53DBD versus p53CT, 4). The length of the flanking region in sequences used for binding affinity measurements were kept short, at 5-bp each (and identical to the flanking sequences used in the transactivation assay), to avoid the hemi-specific binding mode (27).
Figure 5.

Binding affinity measurements by EMSA of p53DBD to variants of the p21-5′ RE. The number bellow each gel is the concentration of p53DBD monomers active for DNA binding. DNA targets were embedded in DNA hairpin constructs (concentration < 0.1 nM). The gels are representative examples of six to eight experiments conducted with each site.
Measuring the binding parameters (Table 3) revealed that the averaged dimer KD (averaged over the two non-identical HSs) is more variable than the averaged dimer equivalent tetramer KD values. The extreme values for both parameters belong to the LL and RR sites. Considering the large difference between the dimer KD values of LL and RR (371 nM and 1900 nM, respectively, Table 3, Supplementary Figure S6), it is reasonable to assume that the first dimer to bind to the p21-5′ site, or any of its variants, is the one with the higher binding affinity, i.e. ‘left HS’. To assess whether LL is also the site with the higher binding stability, we determined the kinetic off-rate of p53DBD from LL and RR, using EMSA (Supplementary Figure S7). Analysis of the tetrameric band dissociation constant (Figure 6, Supplementary Table S5) revealed that the dissociation of p53 from both sites is biphasic, as previously observed (4). Analysis of the slow and reproducible fraction (‘B’ in Supplementary Table S5) showed that the LL site has a lower koff relative to RR, and thus has a longer half-life (46 ± 5 min versus 12.1 ± 0.3 min, respectively). It is interesting to further compare the binding of the RR site to that of the LL site. Both sites contain the flexible CATG center, but their overall torsional flexibility is significantly different (5.45° and 4.43° for RR and LL respectively, Table 2). LL, the more rigid RE, binds p53 with a significantly higher tetramer affinity than RR (77 and 31 for RR and LL respectively, Table 3). The only departure of the RR site from the consensus is in the non-contacted bases at the first and last position of each decameric HS. In our previous study (3), the variation between the studied p53 consensus sequences was at the non-contacted W5W6 step of the central CWWG region and then the observation was that the more flexible site bound with higher binding affinity. Here, the variation is at the non-contacted R1 and Y10 bases of p21-5′ RE variants and now a more rigid site has higher binding affinity. Hence, it seems that it matters where within the RE are the flexible/rigid base-pair steps. One needs more such analyses to reach a more detailed conclusion.
Figure 6.
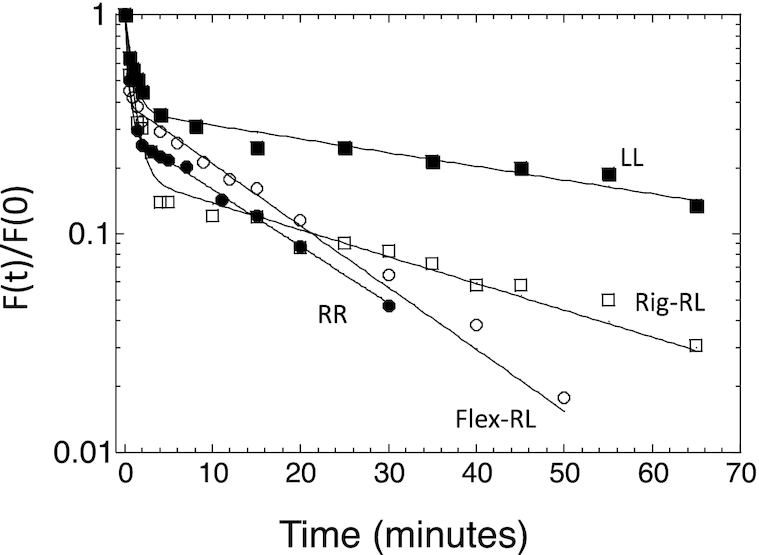
Kinetic off-rate analysis of p21-5′ RE variants from p53DBD. Shown is a plot of the fraction of molecules bound to p53DBD at time (t) divided by the fraction of molecules bound at time 0 as a function of time. The lines are from the best fit to a double-exponential curve. Solid squares, LL; solid circles, RR; open squares, Rig-RL; open circles, Flex-RL. The shown experimental points are those from one experiment, out of 3–5 independent experiments conducted with each DNA target. Hence, they may deviate slightly from the averaged values presented in Supplementary Table S5.
Comparing the pattern of changes observed in transactivation levels, at basal p53 levels (2% raffinose, 0.002% galactose, Figure 4, Supplementary Table S3) to those observed in measuring binding parameters (Figure 5, Table 3) shows several similarities. First, LL is the binding site with the highest binding affinity and RR is the site with the lowest binding affinity. Second, Rig-RL is the site with the lowest dimer KD (highest binding affinity) from the LR or RL groups, comparable to that from 5′Nat-LR, as in the transactivation assays. Third, the influence of the flanks on dimer binding affinity is similar to the transactivation capacity of the LR versus the RL groups at basal p53 concentration. There is ∼70% change in dimer binding affinity between Rig-RL and Flex-RL, but Rig-LR binding affinity is only ∼18% higher than that of Flex-LR. Thus, the difference in binding affinity between Rig-RL and Flex-RL is significant (t = 7.05, P < 0.0001), whereas that between Rig-LR and Flex-LR is not significant (t = 1.82, P = 0.09).
Dimer KD is strongly (ρ = 0.97, 0.98) and significantly (P < 0.0001) correlated with transactivation at basal p53 concentrations (2% raffinose and 0.002% galactose, respectively). Since here all REs are similar in that they all contain a CATG center, transactivation is also significantly correlated with dimer equivalent tetramer KD, again only at basal p53 concentration (ρ = 0.94, P < 0.0001, for both 2% raffinose and 0.002% galactose). Previously we showed that transactivation at basal p53 concentration is linked to the binding stability (off-rate kinetics), whereas here we show that the correlation is between transactivation and dimer binding affinity. Hence, we measured the binding kinetics from Rig-RL and Flex-RL (Figure 6 and Supplementary Figure S5). Analysis of the gel pattern shows that Rig-RL is the site with a longer half-life than Flex-RL (24 ± 2 min versus 15 ± 3 min, respectively, Supplementary Table S5). This, together with kinetic off-rate measurements of LL and RR, shows that the relationship between basal level transactivation and dimer binding affinity parallels that of basal level transactivation and kinetic of-rate.
RLminus transactivation levels are the only exception to the similarity observed when comparing transactivation experiments to binding assays. RLminus reaches significantly higher transactivation levels, relative to all other sites (except LL at basal p53 concentrations). At basal p53 concentration the differences between RLminus transactivation level to all other sites (except LL) is the highest, but the difference remains significantly higher than for all other sites also at the more induced p53 levels (including LL). This is in contrast to the dimer binding affinity of RLminus, which is not significantly different from that of the other sites studied here.
Likewise, the dimer equivalent tetramer KD of RLminus does not have the lowest value from all sites studies here. Differences in transactivation capacity between p53 REs at highly induced levels were shown by us (4) to be linked to p53 binding affinity, whereas differences at basal p53 levels to binding stability (off-rate kinetics, here extended to dimer binding affinity). The RLminus site show differences in transactivation level at both ends of the transactivation spectrum but does not show any linkage to either dimer binding affinity or tetramer binding affinity. Possible origins for the transactivation pattern observed with RLminus site could be packing considerations within inner QSs, which may be more favorable in the RLminus site than in RL (but this cannot explain the difference to LR). Alternatively, it may be related to more optimal interaction of the p53/RLminus complex with other TFs binding nearby, or some other currently unknown effect.
Information score analysis of p21-5′ RE variants
It is instructive to examine the binding potential of p21-5′ variants using information-theory matrix methods (48–51). To that end, we used our curated set of 249 p53 natural REs as a training set (containing all but the studied p21-5′ RE) to construct a mononucleotide as well as dinucleotide position-specific information-content weight matrix (denoted WM, see Materials and Methods). The matrices elements are the maximum probability estimate for the binding energy contribution of each base at each position, assuming independent contribution of each position to the binding energy for a mononucleotide WM, and non-additive nearest-neighbor interactions for the dinucleotide WM (50,51). When we multiply these matrices element-wise by a matrix (containing only 0’s and 1’s as its elements) corresponding to each site studied here, and sum the resultant elements, we get an ‘informational score’ (ISc) for each site, which is the calculated total binding energy for that sequence, either on the mononucleotide level, or on the mononucleotide plus the dinucleotide level, termed ‘dinucleotide ISc’ (50,51). The 5′ flanking sequences were included, so that one can infer their indirect contribution to the total calculated binding energy at any specific flanks length. As the 3′ flanks are identical (except for 5’3’ natural LR), we did not include them in this calculation. The mononucleotide ISc values were scaled such that LL RE only, the best binding site, scored zero and poorer sites score positively. Dinucleotide ISc values were scaled by mononucleotide LL RE only as well, to maintain both sets on the same scale.
Several observations can be made from the ISc values calculated for p21-5′ (Figure 7, Supplementary Table S6, and Supplementary Figure S8). First, on the mononucleotide level, and without the flanking sequences, LR, RL and RLminus have approximately similar calculated binding energies (4.18 ± 0.07, 4.18 ± 0.05 and 3.98 ± 0.06 bits, respectively, Supplementary Figure S8A). Second, information score (calculated binding energy) is significantly improved upon adding a dinucleotide term, which accounts for non-additive nearest-neighbor interaction, to the mononucleotide ISc of the RE itself of LR, RL, and RLmin (3.22 ± 0.07, 1.61 ± 0.04 and 1.57 ± 0.06, respectively, Figure 7 and Supplementary Figure S8B). This demonstrates that for the p21-5′ RE, non-additive effects, which reflect the indirect readout (structural) component of protein-DNA interactions (36,51), operating here at the nearest-neighbor interaction level, improve binding energy with p53. Interestingly, the reduction in calculated binding energy is more significant for RL and RLminus than for LR. This means that the interaction energy of RL and RLminus variants of p21-5′ is more affected by the structural properties of base-pair steps within the RE sequence itself than within the LR variant (Figure 7). This seems counterintuitive, since all sites studied here have the same base-pair step composition within the sequence of the RE itself (including the base-pair step connecting the two HSs). However, it should be noted that the weight matrix used here (like all WMs) is a position-specific weight matrix, and thus determined by the frequency of base-pair steps as observed in the training set of natural REs in specific positions, which may not be the same in left (5′) versus right (3′) HSs. For example, the TG step is about twice more frequently observed at position Y9Y10, as in RL, than the TG step in position Y19Y20 as in LR. Hence, the term for T9G10 in the WM will contribute more favorably to the total calculated binding energy of RL, than the term for T19G20 in LR. Likewise, C1A2 (or equivalently T19G20 on the other strand) is twice more frequent in RLminus than T19G20 (or equivalently C1A2 on the other strand) is, as in LR. The same can be observed for many other dinucleotide and triplets of p53 REs. Berg and von Hippel were the first to propose that changes in base-pair frequency correlate with protein binding energy (48,49). Here we show that the location within the RE (left versus right HS) of these base-pair step frequencies matters as well, and that base-pair doublets can have differing frequencies, even when they are related by a 2-fold helical axis in a binding site of an apparently symmetric protein. This leads us to suggest that p53 REs have directional properties built into them, which are encoded, at least in part, in the structural (indirect readout) properties of the REs. To validate this suggestion, we randomly swapped the left HS with the right HS (within the same RE) of 125 REs from our 249 REs training set. This was carried out 10 000 times. In each combination, of swapped and unswapped REs, we ensured that at least 50 REs were different from any other combination of swapped and unswapped 249 REs. Table 4 shows that the information score on the sequence level (mononucleotide ISc) was not affected by these swapping, whereas the information score on the dinucleotide level was considerably changed, abolishing the significant difference between LR and RL type of REs.
Figure 7.

Information score analysis to p21-5′ RE variants as a function of flanking sequences length. Mononucleotide and dinucleotide information score for (A) LR group, and (B) RL group and RLminus.
Table 4.
Information score of p21-5′ variants before and after horizontal swapping of half sites
| Information scorea | ISc full site original datab | ISc full site after swappingb |
|---|---|---|
| LR mono | −11.40 ± 0.07 | −11.480 ± 0.003 |
| RL mono | −11.40 ± 0.05 | −11.407 ± 0.003 |
| RLminus mono | −11.60 ± 0.06 | −11.170 ± 0.003 |
| LR dinuc – term | 0.96 ± 0.05 | 1.672 ± 0.004 |
| RL dinuc – term | 2.56 ± 0.04 | 1.878 ± 0.004 |
| RLminus dinuc – term | 2.41 ± 0.03 | 1.584 ± 0.004 |
| LR dinuc – total | −12.40 ± 0.07 | −13.15 ± 0.01 |
| RL dinuc – total | −14.00 ± 0.04 | −13.350 ± 0.004 |
| RLminus dinuc – total | −14.00 ± 0.06 | −12.75 ± 0.01 |
aInformation score (ISc) represents the binding energy calculated from our 249 p53 REs training set for the indicated p21-5′ RE variants. 'mono' is ISc on the mononucleotide level; 'dinuc-term' is the term added to the mononucleotide ISc to yield the dinucleotide, total, ISc. See text for details.
bValues of ISc presented here are not scaled to the best binder sequence as in Figure 7 and Supplementary Table S6. The ± are the standard error of the mean values, derived by the n – 1 Jackknife method (53). See text for details.
Upon adding flanking sequences to the mononucleotide ISc sites, rigid flanks and the original reporter-construct flanks (Figure 7, Supplementary Figure S8B) decrease somewhat in favorable binding energy, whereas the flexible flanks remain unchanged, and the site with the natural flanks increase in favorable binding energy (Figure 7, Supplementary Figure S8B). These patterns in the flanks obviously stem from the composite sequence characteristics of the flanking sequences in the training set, and probably belong to those of other TFs, and other sequence signals surrounding p53 REs, that probably change between the various REs of p53. Hence, at the mononucleotide level these small pattern changes in the flanking sequences do not necessarily mean much. However, when including nearest-neighbor interactions, the features of the designed flanking sequences used here can contain useful information on the structural characteristics of p53 flanking sequences. For example, only the rigid flanks do not interfere with p53 binding, when they are 5′ to the LR (or RL) sites (Figure 7), even though they are not the natural flanks of this RE. Moreover, Rig-LR has a flank-dependent pattern that is the closest one to the pattern shown by 5′Nat-LR site, the natural RE (Figure 7A). The flexible and the reporter-construct original flanks on the other hand, decrease the favorable binding energy from the first flanking base onwards. Furthermore, the ISc pattern of the RL site and that of the RLminus site, as a function of the flank length, are highly similar to each other (Figure 7B), which agrees with the similarity in their binding affinity, as measured by EMSA experiments (Table 3).
To gain knowledge on the length to which the flanks influence the properties of the REs, we analyzed the relationship between our measured binding affinity and that calculated from dinucleotide ISc values, as a function of flank length. Five flank lengths were used in the analysis (2–6 bp step length) and three binding parameters (dimer KD, dimer equivalent tetramer KD, and binding cooperativity). Thus, a total of 15 relationships were analyzed, and hence the minimal P value for statistical significance is 0.003. When we used dinucleotide ISc values with 4-bp or 5-bp flanks there was a strong and significant relationship to the measured dimer binding affinity (ρ = 0.96, P < 0.00001). However, the relationship of dinucleotide ISc to tetramer binding affinity is not significant (ρ = 0.69, P = 0.03), given the multiple comparisons. The cooperativity of binding was also significantly correlated to dinucleotide ISc calculated with 4- or 5-bp flanks (ρ = 0.92, P = 0.0002). This demonstrates the important role played by the (transient) binding of dimers in the mechanism of specific binding by p53 to its REs. Moreover, the relationship may mean that the flanking-sequence influence extends also to the binding of dimeric p53 molecules to HSs. Furthermore, as for the correlation between dimer binding affinity and transactivation at basal p53 levels, discussed above, there is a correlation between calculated binding energy (ISc dinucleotide model) and transactivation at basal p53 levels. When we include all ten p21-5′ variants studied here, a significant relationship is observed when the flanks are 3-bp long (ρ = 0.83 and 0.85, P = 0.003 and 0.002, for 2% raffinose and 0.002% galactose, respectively). However, as noted above, RLminus is an unusual site. The transactivation level from this site is exceptionally high, whereas the binding affinity of p53 to this site is average. When we exclude this p21-5′ variant, the significant relationship is again between information score with either 4-bp or 5-bp flanks and transactivation levels (ρ = 0.88 and 0.92, P = 0.002 and 0.0005, for 2% raffinose and 0.002% galactose, respectively). These relationships suggest that the effect of the flanks on RE properties and interactions extends mainly to flanks that are 4-5 bp long. This agrees with our previous study on the TATA-binding protein (TBP), where we observed that the extent of the influence of the flanks on TBP/TATA-box interactions is also 4-bp (35), where we explain it by the observation that 4-bp long DNA is the minimal length for an independent cooperatively built DNA unit (72).
Torsional rigidity of flanking sequences improves p53/DNA interactions
To further elucidate the relationship between rigid flanking sequences and high binding affinity of p53 to these sites, we synthesized a variant of the Flex-RL binding site (the site with the lowest binding affinity in the RL group, the group in which the flanks have a more noticeable effect on binding affinity), where we replaced two T–A base-pair steps with a sequence containing the modified base 2,6-diamino purine (2,6-DAP), thus creating two T-DAP base-pair steps. DAP containing sequences have an unusually long twist persistence length, and thus are more torsionally rigid (73). In addition, this substitution changed the number of hydrogen bonds in these base-pair steps from two hydrogen bonds per base-pair to three hydrogen bonds per base-pair. Figure 5 and Table 3 show that this conversion improved the binding affinity of both the dimeric as well as the tetrameric p53/Flex-RL+DAP complex, relative to all sites with the RL orientation, making it be the best binding sequence of the RL group. This corroborates our observation that rigid flanking sequences improve the binding affinity of p53 REs, both on the dimer as well as the tetramer level. If the improvement in binding affinity is due to DAP-containing sequences having three instead of two hydrogen bonds, then G,C-rich flanks may show a similar trend, as they contain three hydrogen bonds as well. When we synthesized a variant of Flex-RL site with flanking sequences that are both flexible and G,C-rich (Flex-RL&GC, Table 3, Supplementary Figure S6) we observed that the dimer binding affinity somewhat improved relative to Flex-RL, but not as much as in Flex-RL+DAP, whereas the tetramer binding affinity did not improve (Table 3 and Supplementary Figure S6). This is the case also for the flexible and G,C-rich variant of Flex-LR (Flex-LR&GC, Table 3, Supplementary Figure S6). This means that having three hydrogen bonds is beneficial, however, it is the torsional rigidness of the flanking sequences that is the more crucial structural aspect. Moreover, we determined the binding affinity of wild-type p21-5′ that contain the natural flanks both 5′ as well as 3′ to the RE (5′3′ Nat-LR, Table 3, Figure 5). 5′3′ Nat-LR is the best binding site of the whole LR series, for both the dimer and the tetramer species. The constant 3′ flanks of the sequences studied here are G,C-rich (3 out of 5 nucleotides are G or C), but only the 3′-flank of 5′3′ Nat-LR, the fully natural site, that is both G,C-rich and of low deformability (1.33°3Å3 for 4-bp flanks) improves the binding affinity of both dimer and tetramer complexes with p53, relative to 5′ Nat-LR. Thus, it seems that torsionally rigid flanks, whether 5′ or 3′ to p21-5′, improves binding of p53 to this site.
Deformability of flanking sequences of natural p53 REs genome wide
The observation above, that low deformability of flanking sequences of p21-5′ variants improve their binding to p53, prompted us to look at the deformability of all p53 REs in our curated dataset. The values for the 5′ (upstream) and 3′ (downstream) flanks are shown in Supplementary Figure S9, where no difference in the upstream versus the downstream flanking sequences deformability is observed, except for few unique positions (8, 9, 21 and 23 bases from the RE Supplementary Figure S9). We now ask whether the flanks of conserved HSs have different structural properties than those of degenerate HSs, and whether the 5′ flanks differ from the 3′ flanks in that respect. To that end, we grouped together p53 REs belonging to the CC and the CD categories, both having a conserved left HS, as well as the DC and the DD categories, having a degenerate left HS, and compared the grouping as a function of the 5′ flanking sequence. There was a significant difference between these two left HS groups (t = 2.57, P = 0.01), when we took the average of three abutting upstream base-pair steps (four nucleotides adjacent to the 5′ end of the RE). Thus, 5′ flanks bordering a conserved p53 HS from the 5′-side (upstream) are more flexible (V(B) = 2.18 ± 0.05 °3Å3 for 3 bp steps) than those bordering degenerate p53 HSs from this side (V(B) = 1.96 ± 0.07 °3Å3 for 3 bp steps). No significant pattern emerged from a similar analysis of sequences bordering p53 REs from the 3′ side (downstream). As observed for the bridge dinucleotide between HSs, the bridge dinucleotide between the 5′ flanks and the 5′ (left) HS and the 3′ (right) HS have averaged deformability values (2.4 ± 0.1 and 2.5 ± 0.1 °3Å3 for the 5′ and 3′ bridge dinucleotide, respectively, Supplementary Figure S9) that are above those of any other base-pair step within the REs, except for the super deformable positions, CW and WG (3).
Relationship of sequence conservation to p53 functionality in cellular context
To validate the relevance of our RE conservation analysis to in vivo p53 functionality, we first looked at the relationship between p53-level-specific reporter-gene assays in yeast, from our previous study (4) and DE values. In that study there were 12 natural p53 REs and six consensus-like sequences without spacers. We added the six consensus sequences to the list of 250 p53 REs and then recalculated the DE values, because the analysis is always relative to the best binder of the whole set. Looking at the data in Supplementary Table S7, we observe that the DE values of p53 FSs have a modest but statistically significant correlation to expression level from a linked reporter, at most p53 levels. Surprisingly, the DE value of p53 left HSs have a strong and significant correlation at all p53 levels, starting from the most induced level measured (0.024% galactose, ρ = –0.83, P <0.0001) to the basal level of 0.002% galactose (ρ = –0.72, P = 0.002). The DE values of p53 right HSs are not significantly correlated to the expression level from the linked reporter. This is another manifestation of directionality in p53 REs, which is unrelated to the detrimental effects that flanking sequences can have on p53 functionality (flanking sequences are the same in all sites in this study).
We next looked at p53 occupancy in cellular context, as determined from ChIP-seq analyses and its distribution between different HS categorical groups (CC, CD, DC and DD). To that end, we used the data from the meta-analysis of Nguyen et al. (43, Supplementary Table ST2), in which they enumerate activated ChIP-seq p53 peaks in 41 datasets, and noted in how many datasets each ChIP-seq peak appeared (Supplementary Table S1). We carried out one-way non-parametric analysis on the distribution of ChIP-seq peaks between HS conservation categories. The Kruskal–Wallis Chi-squared value (also known as the H statistics) that we get from the analysis of 239 REs that appear both in Supplementary Table ST2 (43) and in our list of p53 REs (Supplementary Table S1) was significant (χ2 (3, N = 239) = 34.3, P < 0.0001). This result indicates that the division of ChIP-seq occupancy data between HS categories is not random (Figure 8). A post-hoc test established that the most significant different categories are CC versus DC (z = –4.19, P = 0.0002), CC versus CD (z = –4.27, P = 0.0001, and CC versus DD (z = –5.11, P < 0.0001), CC appearing in more binding events than any other category, and DD appearing in the least number of binding events (Figure 8). We then carried out the same analysis on the distribution of activated ChIP-seq peaks between 16 QS conservation categories. The Chi-square value was significant in this test as well (χ2 (3, N = 239) = 51.85, P < 0.0001). However, the post-hoc test did not show significant differences between individual QSs categories due to the small number of REs in each QS category.
Figure 8.
The number of ChIP-seq datasets (out of 41 datasets used in the meta-analysis of Nguyen et al. (43)) with p53 peaks versus half-site conservation categories. The occupancy of p53 on fully-conserved REs (CC category) is significantly higher than on partially conserved or fully degenerate p53 sites. P values are marked as follows: P = 0.037 *, P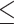 0.001 **, P < 0.0001 ***.
0.001 **, P < 0.0001 ***.
GENERAL DISCUSSION
In this study, we have deconstructed the p53 RE, sequence-wise and structure-wise. This enabled us to evaluate at high resolution the involvement of each specific p53-RE region in p53/DNA interactions, contributing to p53 functional activity in the myriad functions that are regulated by p53. The first conclusion from this study is that the hemi-specific mode of p53 binding is much more prevalent than previously estimated. It now seems to be a plausible mechanism for binding of at least ∼24% of spacer-less REs from our curated p53 REs list, those that are from the CD or DC category and have one very degenerate HS. p21-5′ right HS (DE = 5.59) differs significantly from p21-5′ left HS (DE = 0.99). Thus, even though p21-5′ right HS is not a very degenerate HS (HSs where DE values are > 5.89), dimers of p53 are only weakly bound to this site (Table 3). Hence, even the p21-5′ RE can be considered to be bound by p53 with somewhat of a hemi-specific character. Such RE pattern explains why in many studies p53 looked as if it was functioning from a single HS (74–80), an observation which is at odds with the functional species of p53 being tetrameric (28,81,82), and it re-enforces our previous suggestion (27) that the hemi-specific mode of binding is a functional and important mode of p53 activity.
The second conclusion reached from the present study is that two half-sites within p53 full-site and two quarter-sites within p53 half-sites behave in a mechanistically analogous manner. In this study, we found that two HSs within p53 FS are anticorrelated in their sequence conservation level. Similarly, two QSs within each HS are also anticorrelated, and with similar strength to the correlation between HSs. It has been demonstrated before that p53 tetramers are mostly formed on the DNA after binding of the separated dimers (28,83). p53 dimers have been shown to form concurrently with translation (62), and hence two p53 monomers enter the nucleus attached to each other though the tetramerization domain. This does not contradict however our suggestion here that p53 monomers form intra-dimer core-domain protein-protein contacts only after they are bound to p53 HSs, starting with binding of one monomer to the more conserved quarter site. We coin this model as the ‘Cherry-pair’ model. Our novel model may explain the conflicting results of Gaglia et al. (30) reporting that at rest 28% of p53 molecules are bound as monomers versus the idea of dimers as the minimal p53 species at low p53 concentration.
Focusing on variants of p21-5′, we showed here that there is a strong correlation between the calculated binding energy (dinucleotide ISc of p21-5′ variants) and measured binding affinity by EMSA when the flanks are 4 or 5-bp long. Since transactivation levels at basal p53 concentration also correlated well with calculated binding energy when the flanks are 4 or 5-bp long, we conclude that the influence of the flanks on REs interactions with p53 is limited to the 4- to 5-bp proximal to the RE. This is not a surprising result, since it is known that a 4-bp DNA element is the minimal unit of corporative DNA structure (72,84–86), and it corresponds to the flank length shown by us to modulate TBP/TATA box interactions (35). Thus, the sites used in our in vitro binding assays and those inserted in the 5′-end of the binding sites for transactivation assays capture the entire effect of the flanks. It is the structural properties of the flanks that modulate p53 binding to the REs and transactivation from them. In particular, torsional rigid flanking sequences were demonstrated here to enhance binding affinity and stability considerably. In addition, the dinucleotide bridging the 5′ flanking sequences to the RE can also affect binding and transactivation levels. Here again, it is the rigid bridge dinucleotide that leads to enhanced binding affinity and transactivation levels.
By switching the HSs of p21-5′ (LR to RL) we demonstrated, using a nearest-neighbor model for calculated binding energy (i.e. structural recognition included in the model), that RL has a lower free energy of binding (higher binding affinity), regardless of sequence context (flanking sequences). Using in vitro binding experiments and transactivation assays in yeast, we showed here that the effects of the flanks are larger in the RL that in the LR group (that is the difference between Rig-RL and Flex-RL is larger than between Rig-LR and Flex-LR in binding and transactivation). Combining these two observations, we suggest that HS positioning with respect to the transcription direction (LR versus RL) may have been fine-tuned by an evolutionary selection pressure to regulate the extent of the effect of RE structure on its activity, as well as to minimize the effect of the flanking sequences on the activity from the RE. As pointed out by Berg and von Hippel (48) ‘the connection between sequence variability and binding affinity drives from evolutionary selection constraints’. Thus, we emphasize the apparent positive evolutionary selection pressure that have shaped current p53 sites and brought them to their current fitness state, able to optimally carry out their regulatory role. This is especially important with regard to the effect that flanking sequences can have on the functionality of p53 REs. The apparent evolution of p53 binding sites was towards ‘minimizing the maximum loss of specificity’ (48), by selecting half-site orientation where the influence of flanking sequences on RE activity is minimal, and hence the DNA region where random mutations can influence RE function is minimized.
It has been known for some time that flanking sequences can influence binding and activity from TF binding sites (35,87–92). Our novel finding here is that these effects of the flanking sequences can be detrimental for p53 functionality, and hence are minimized by a judicious use of the positioning of the HSs with respect to transcription direction. Thus, by having directional preferences built into p53 REs several different goals are achieved simultaneously: regulating the level of effect that DNA structure has on RE activity; limiting the influence of the flanks on RE activity; and regulating the effects of TFs bound nearby on activity of p53 REs. Moreover, our observations that the upstream flanking sequences show differences in deformability as a function of HS conservation, but not downstream flanks, as well as the correlation between reporter gene assays in yeast with left HS DE values, but not with right HS DE values, also point to directionality built into p53 RE architecture.
It is now appreciated that even though p53 seems to be able to regulate its target genes without cooperating with additional sequence-specific TFs (93,94), they can nonetheless contribute to p53-dependent transcriptional activation at some genomic binding sites (95). We suggest that the effect of nearby yeast cis-regulatory elements, and the TFs that bind them, may be an answer to the question of the significantly higher transactivation levels from RLminus relative to those from other p21-5′ variants studied here, when its binding characteristics (measured and predicted) are not different from those of the other variants.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ivan Raimondi for help in the initial stages of the yeast transactivation experiments. We thank Michael Shmoish and Guy Horev for help in the initial statistical analysis.
Notes
Present address: Vasundhara Sharma, University Health Network, Princess Margaret Cancer Centre, 610 University Ave, Toronto, ON M5G 2C1, Canada.
Present address: Dmitrij Golovenko, RNA Therapeutics Institute, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, MA, USA.
Contributor Information
Alon Senitzki, Department of Biology, Technion - Israel Institute of Technology, Technion City, Haifa 3200003, Israel.
Jessy Safieh, Department of Biology, Technion - Israel Institute of Technology, Technion City, Haifa 3200003, Israel.
Vasundhara Sharma, Department of Cellular, Computational and Integrative Biology (CIBIO), University of Trento, via Sommarive 9, 38123 Trento, TN, Italy.
Dmitrij Golovenko, Department of Biology, Technion - Israel Institute of Technology, Technion City, Haifa 3200003, Israel.
Yael Danin-Poleg, Department of Biology, Technion - Israel Institute of Technology, Technion City, Haifa 3200003, Israel.
Alberto Inga, Department of Cellular, Computational and Integrative Biology (CIBIO), University of Trento, via Sommarive 9, 38123 Trento, TN, Italy.
Tali E Haran, Department of Biology, Technion - Israel Institute of Technology, Technion City, Haifa 3200003, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Israel Science Foundation [1517/14 to T.E.H.]; Italian Association for Cancer Research (AIRC) [IG #18985 to A.I., in part]. Funding for open access charge: Israel Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kitayner M., Rozenberg H., Kessler N., Rabinovich D., Shaulov L., Haran T.E., Shakked Z.. Structural basis of DNA recognition by p53 tetramers. Mol. Cell. 2006; 22:741–753. [DOI] [PubMed] [Google Scholar]
- 3. Beno I., Rosenthal K., Levitine M., Shaulov L., Haran T.E.. Sequence-dependent cooperative binding of p53 to DNA targets and its relationship to the structural properties of the DNA targets. Nucleic Acids Res. 2011; 39:1919–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jordan J.J., Menendez D., Sharav J., Beno I., Rosenthal K., Resnick M.A., Haran T.E.. Low-level p53 expression changes transactivation rules and reveals superactivating sequences. Proc. Natl. Acad. Sci. USA. 2012; 109:14387–14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kitayner M., Rozenberg H., Rohs R., Suad O., Rabinovich D., Honig B., Shakked Z.. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010; 17:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y., Dey R., Chen L.. Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Structure. 2010; 18:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y., Zhang X., Dantas Machado A.C., Ding Y., Chen Z., Qin P.Z., Rohs R., Chen L.. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013; 41:8368–8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X., Dantas Machado A.C., Ding Y., Chen Y., Lu Y., Duan Y., Tham K., Chen L., Rohs R., Z. Q.P.. Conformations of p53 response elements in solution deduced using site-directed spin labeling and Monte Carlo sampling. Nucleic Acids Res. 2014; 42:2789–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vousden K.H., Prives C.. Blinded by the light: the growing complexity of p53. Cell. 2009; 137:413–431. [DOI] [PubMed] [Google Scholar]
- 10. Levine A.J., Oren M.. The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer. 2009; 9:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kastenhuber E.R., Lowe S.W.. Putting p53 in Context. Cell. 2017; 170:1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leroy B., Fournier J.L., Ishioka C., Monti P., Inga A., Fronza G., Soussi T.. The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013; 41:D962–D969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bouaoun L., Sonkin D., Ardin M., Hollstein M., Byrnes G., Zavadil J., Olivier M.. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum. Mutat. 2016; 37:865–876. [DOI] [PubMed] [Google Scholar]
- 14. Riley T., Sontag E., Chen P., Levine A.. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008; 9:402–412. [DOI] [PubMed] [Google Scholar]
- 15. Joerger A.C., Fersht A.R.. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008; 77:557–582. [DOI] [PubMed] [Google Scholar]
- 16. Espinosa J.M. Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene. 2008; 27:4013–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vousden K.H. p53: death star. Cell. 2000; 103:691–694. [DOI] [PubMed] [Google Scholar]
- 18. Espinosa J.M., Emerson B.M.. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell. 2001; 8:57–69. [DOI] [PubMed] [Google Scholar]
- 19. Gomes N.P., Bjerke G., Llorente B., Szostek S.A., Emerson B.M., Espinosa J.M.. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 2006; 20:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morachis J.M., Murawsky C.M., Emerson B.M.. Regulation of the p53 transcriptional response by structurally diverse core promoters. Genes Dev. 2010; 24:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zilfou J.T., Lowe S.W.. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009; 1:a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fischer M., Steiner L., Engeland K.. The transcription factor p53: not a repressor, solely an activator. Cell Cycle. 2014; 13:3037–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischer M., Quaas M., Steiner L., Engeland K.. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016; 44:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017; 36:3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018; 25:114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Menendez D., Resnick M.A., Haran T.E.. Transactivation by low and high levels of human p53 reveals new physical rules of engagement and novel super-transactivation sequences. Cell Cycle. 2012; 11:4287–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vyas P., Beno I., Xi Z., Stein Y., Golovenko D., Kessler N., Rotter V., Shakked Z., Haran T.E.. Diverse p53/DNA binding modes expand the repertoire of p53 response elements. Proc. Natl. Acad. Sci. USA. 2017; 114:10624–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weinberg R.L., Veprintsev D.B., Fersht A.R.. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004; 341:1145–1159. [DOI] [PubMed] [Google Scholar]
- 29. Rajagopalan S., Huang F., Fersht A.R.. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011; 39:2294–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gaglia G., Guan Y., Shah J.V., Lahav G.. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:15497–15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bareket-Samish A., Cohen I., Haran T.E.. Signals for TBP/TATA box recognition. J. Mol. Biol. 2000; 299:965–977. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y., Crothers D.M.. High-throughput approach for detection of DNA bending and flexibility based on cyclization. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:3161–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y., Crothers D.M.. Statistical mechanics of sequence-dependent circular DNA and its application for DNA cyclization. Biophys. J. 2003; 84:136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Senear D.F., Brenowitz M.. Determination of binding constants for cooperative site-specific protein-DNA interactions using the gel mobility-shift assay. J. Biol. Chem. 1991; 266:13661–13671. [PubMed] [Google Scholar]
- 35. Faiger H., Ivanchenko M., Cohen I., Haran T.E.. TBP flanking sequences: asymmetry of binding, long-range effects and consensus sequences. Nucleic Acids Res. 2006; 34:104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Faiger H., Ivanchenko M., Haran T.E.. Nearest-neighbor non-additivity versus long-range non-additivity in TATA-box structure and its implications for TBP-binding mechanism. Nucleic Acids Res. 2007; 35:4409–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jegga A.G., Inga A., Menendez D., Aronow B.J., Resnick M.A.. Functional evolution of the p53 regulatory network through its target response elements. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tomso D.J., Inga A., Menendez D., Pittman G.S., Campbell M.R., Storici F., Bell D.A., Resnick M.A.. Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:6431–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Storici F., Resnick M.A.. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 2006; 409:329–345. [DOI] [PubMed] [Google Scholar]
- 40. Sikorski R.S., Hieter P.. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989; 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Inga A., Storici F., Darden T.A., Resnick M.A.. Differential transactivation by the p53 transcription factor is highly dependent on p53 level and promoter target sequence. Mol. Cell. Biol. 2002; 22:8612–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andreotti V., Ciribilli Y., Monti P., Bisio A., Lion M., Jordan J., Fronza G., Menichini P., Resnick M.A., Inga A.. p53 transactivation and the impact of mutations, cofactors and small molecules using a simplified yeast-based screening system. PLoS One. 2011; 6:e20643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nguyen T.T., Grimm S.A., Bushel P.R., Li J., Li Y., Bennett B.D., Lavender C.A., Ward J.M., Fargo D.C., Anderson C.W.et al.. Revealing a human p53 universe. Nucleic Acids Res. 2018; 46:8153–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bourdon J.C., Deguin-Chambon V., Lelong J.C., Dessen P., May P., Debuire B., May E.. Further characterisation of the p53 responsive element–identification of new candidate genes for trans-activation by p53. Oncogene. 1997; 14:85–94. [DOI] [PubMed] [Google Scholar]
- 45. Menendez D., Nguyen T.A., Freudenberg J.M., Mathew V.J., Anderson C.W., Jothi R., Resnick M.A.. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013; 41:7286–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huarte M., Guttman M., Feldser D., Garber M., Koziol M.J., Kenzelmann-Broz D., Khalil A.M., Zuk O., Amit I., Rabani M.et al.. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010; 142:409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sullivan K.D., Galbraith M.D., Andrysik Z., Espinosa J.M.. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018; 25:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Berg O.G., von Hippel P.H.. Selection of DNA binding sites by regulatory proteins. Statistical- mechanical theory and application to operators and promoters. J. Mol. Biol. 1987; 193:723–750. [DOI] [PubMed] [Google Scholar]
- 49. Berg O.G., von Hippel P.H.. Selection of DNA binding sites by regulatory proteins. II. The binding specificity of cyclic AMP receptor protein to recognition sites. J. Mol. Biol. 1988; 200:709–723. [DOI] [PubMed] [Google Scholar]
- 50. Stormo G.D. DNA binding sites: representation and discovery. Bioinformatics. 2000; 16:16–23. [DOI] [PubMed] [Google Scholar]
- 51. O’Flanagan R.A., Paillard G., Lavery R., Sengupta A.M.. Non-additivity in protein-DNA binding. Bioinformatics. 2005; 21:2254–2263. [DOI] [PubMed] [Google Scholar]
- 52. Zhao Y., Ruan S., Pandey M., Stormo G.D.. Improved models for transcription factor binding site identification using nonindependent interactions. Genetics. 2012; 191:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Efron B. Nonparametric estimates of standard error: the jackknife, the bootstrap and other methods. Biomtlrika. 1981; 68:589–509. [Google Scholar]
- 54. Balasubramanian S., Xu F., Olson W.K.. DNA sequence-directed organization of chromatin: structure-based computational analysis of nucleosome-binding sequences. Biophys. J. 2009; 96:2245–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nettling M., Treutler H., Cerquides J., Grosse I.. Detecting and correcting the binding-affinity bias in ChIP-seq data using inter-species information. BMC Genomics. 2016; 17:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Menendez D., Inga A., Resnick M.A.. The expanding universe of p53 targets. Nat. Rev. Cancer. 2009; 9:724–737. [DOI] [PubMed] [Google Scholar]
- 57. Hafner A., Bulyk M.L., Jambhekar A., Lahav G.. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell. Biol. 2019; 20:199–210. [DOI] [PubMed] [Google Scholar]
- 58. Golovenko D., Brauning B., Vyas P., Haran T.E., Rozenberg H., Shakked Z.. New insights into the role of DNA shape on its recognition by p53 proteins. Structure. 2018; 26:1237–1250. [DOI] [PubMed] [Google Scholar]
- 59. Malecka K.A., Ho W.C., Marmorstein R.. Crystal structure of a p53 core tetramer bound to DNA. Oncogene. 2009; 28:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Petty T.J., Emamzadah S., Costantino L., Petkova I., Stavridi E.S., Saven J.G., Vauthey E., Halazonetis T.D.. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 2011; 30:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vainer R., Cohen S., Shahar A., Zarivach R., Arbely E.. Structural basis for p53 Lys120-acetylation-dependent DNA-binding mode. J. Mol. Biol. 2016; 428:3013–3025. [DOI] [PubMed] [Google Scholar]
- 62. Nicholls C.D., McLure K.G., Shields M.A., Lee P.W.. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J. Biol. Chem. 2002; 277:12937–12945. [DOI] [PubMed] [Google Scholar]
- 63. El-Deiry W.S., Tokino T., Velculescu V.E., Levy D.B., Parsons R., Trent J.M., Lin D., Mercer W.E., Kinzler K.W., Vogelstein B.. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993; 75:817–825. [DOI] [PubMed] [Google Scholar]
- 64. Chen H.H., Rau D.C., Charney E.. The flexibility of alternating dA-dT sequences. J. Biomol. Struct. Dyn. 1985; 2:709–719. [DOI] [PubMed] [Google Scholar]
- 65. Okonogi T.M., Alley S.C., Reese A.W., Hopkins P.B., Robinson B.H.. Sequence-dependent dynamics of duplex DNA: the applicability of a dinucleotide model. Biophys. J. 2002; 83:3446–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang Y., Xi Z., Hegde R.S., Shakked Z., Crothers D.M.. Predicting indirect readout effects in protein-DNA interactions. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:8337–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Olson W.K., Gorin A.A., Lu X.J., Hock L.M., Zhurkin V.B.. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ma L., Wagner J., Rice J.J., Hu W., Levine A.J., Stolovitzky G.A.. A plausible model for the digital response of p53 to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:14266–14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang Y.V., Wade M., Wong E., Li Y.C., Rodewald L.W., Wahl G.M.. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:12365–12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hunziker A., Jensen M.H., Krishna S.. Stress-specific response of the p53-Mdm2 feedback loop. BMC Syst. Biol. 2010; 4:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lokshin M., Tanaka T., Prives C.. Transcriptional regulation by p53 and p73. Cold Spring Harb. Symp. Quant. Biol. 2005; 70:121–128. [DOI] [PubMed] [Google Scholar]
- 72. Saenger W. Principles of Nucleic Acid Structure. 1984; NY: Springer-Verlag. [Google Scholar]
- 73. Peters J.P., Mogil L.S., McCauley M.J., Williams M.C., Maher L.J. 3rd. Mechanical properties of base-modified DNA are not strictly determined by base stacking or electrostatic interactions. Biophys. J. 2014; 107:448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jordan J.J., Menendez D., Inga A., Nourredine M., Bell D., Resnick M.A.. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008; 4:e1000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Menendez D., Krysiak O., Inga A., Krysiak B., Resnick M.A., Schonfelder G.. A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:1406–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Menendez D., Inga A., Snipe J., Krysiak O., Schonfelder G., Resnick M.A.. A single-nucleotide polymorphism in a half-binding site creates p53 and estrogen receptor control of vascular endothelial growth factor receptor 1. Mol. Cell Biol. 2007; 27:2590–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Menendez D., Inga A., Resnick M.A.. Estrogen receptor acting in cis enhances WT and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Menendez D., Shatz M., Azzam K., Garantziotis S., Fessler M.B., Resnick M.A.. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLos Genet. 2011; 7:e1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yan J., Menendez D., Yang X.P., Resnick M.A., Jetten A.M.. A regulatory loop composed of RAP80-HDM2-p53 provides RAP80-enhanced p53 degradation by HDM2 in response to DNA damage. J. Biol. Chem. 2009; 284:19280–19289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ly E., Kugel J.F., Goodrich J.A.. Single molecule studies reveal that p53 tetramers dynamically bind response elements containing one or two half sites. Sci. Rep. 2020; 10:16176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Friedman P.N., Chen X., Bargonetti J., Prives C.. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:3319–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. McLure K.G., Lee P.W.K.. How p53 binds DNA as a tetramer. EMBO J. 1998; 17:3342–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Natan E., Hirschberg D., Morgner N., Robinson C.V., Fersht A.R.. Ultraslow oligomerization equilibria of p53 and its implications. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:14327–14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Haran T.E., Crothers D.M.. Cooperativity in A-tract structure and bending properties of composite TnAn blocks. Biochemistry. 1989; 28:2763–2767. [DOI] [PubMed] [Google Scholar]
- 85. Haran T.E., Mohanty U.. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009; 42:41–81. [DOI] [PubMed] [Google Scholar]
- 86. Merling A., Sagaydakova N., Haran T.E.. A-tract polarity dominate the curvature in flanking sequences. Biochemistry. 2003; 42:4978–4984. [DOI] [PubMed] [Google Scholar]
- 87. Schone S., Jurk M., Helabad M.B., Dror I., Lebars I., Kieffer B., Imhof P., Rohs R., Vingron M., Thomas-Chollier M.et al.. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat. Commun. 2016; 7:12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chaudhari H.G., Cohen B.A.. Local sequence features that influence AP-1 cis-regulatory activity. Genome Res. 2018; 28:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Levo M., Zalckvar E., Sharon E., Dantas Machado A.C., Kalma Y., Lotam-Pompan M., Weinberger A., Yakhini Z., Rohs R., Segal E.. Unraveling determinants of transcription factor binding outside the core binding site. Genome Res. 2015; 25:1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gordan R., Shen N., Dror I., Zhou T., Horton J., Rohs R., Bulyk M.L.. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013; 3:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Le D.D., Shimko T.C., Aditham A.K., Keys A.M., Longwell S.A., Orenstein Y., Fordyce P.M.. Comprehensive, high-resolution binding energy landscapes reveal context dependencies of transcription factor binding. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E3702–E3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rudnizky S., Khamis H., Malik O., Squires A.H., Meller A., Melamed P., Kaplan A.. Single-molecule DNA unzipping reveals asymmetric modulation of a transcription factor by its binding site sequence and context. Nucleic Acids Res. 2018; 46:1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Verfaillie A., Svetlichnyy D., Imrichova H., Davie K., Fiers M., Kalender Atak Z., Hulselmans G., Christiaens V., Aerts S.. Multiplex enhancer-reporter assays uncover unsophisticated TP53 enhancer logic. Genome Res. 2016; 26:882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Younger S.T., Rinn J.L.. p53 regulates enhancer accessibility and activity in response to DNA damage. Nucleic Acids Res. 2017; 45:9889–9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Catizone A.N., Uzunbas G.K., Celadova P., Kuang S., Bose D., Sammons M.A.. Locally acting transcription factors regulate p53-dependent cis-regulatory element activity. Nucleic Acids Res. 2020; 48:4195–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schneider T.D., Stephens R.M.. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990; 18:6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.