

Abstract
Background
Severe Combined Immune Deficiency (SCID) is an inherited defect in lymphocyte development and function that results in life-threatening opportunistic infections in early infancy. Data on SCID from developing countries are scarce.
Objective
To describe clinical and laboratory features of SCID diagnosed at immunology centers across India.
Methods
A detailed case proforma in an Excel format was prepared by one of the authors (PV) and was sent to centers in India that care for patients with primary immunodeficiency diseases. We collated clinical, laboratory, and molecular details of patients with clinical profile suggestive of SCID and their outcomes. Twelve (12) centers provided necessary details which were then compiled and analyzed. Diagnosis of SCID/combined immune deficiency (CID) was based on 2018 European Society for Immunodeficiencies working definition for SCID.
Results
We obtained data on 277 children; 254 were categorized as SCID and 23 as CID. Male-female ratio was 196:81. Median (inter-quartile range) age of onset of clinical symptoms and diagnosis was 2.5 months (1, 5) and 5 months (3.5, 8), respectively. Molecular diagnosis was obtained in 162 patients - IL2RG (36), RAG1 (26), ADA (19), RAG2 (17), JAK3 (15), DCLRE1C (13), IL7RA (9), PNP (3), RFXAP (3), CIITA (2), RFXANK (2), NHEJ1 (2), CD3E (2), CD3D (2), RFX5 (2), ZAP70 (2), STK4 (1), CORO1A (1), STIM1 (1), PRKDC (1), AK2 (1), DOCK2 (1), and SP100 (1). Only 23 children (8.3%) received hematopoietic stem cell transplantation (HSCT). Of these, 11 are doing well post-HSCT. Mortality was recorded in 210 children (75.8%).
Conclusion
We document an exponential rise in number of cases diagnosed to have SCID over the last 10 years, probably as a result of increasing awareness and improvement in diagnostic facilities at various centers in India. We suspect that these numbers are just the tip of the iceberg. Majority of patients with SCID in India are probably not being recognized and diagnosed at present. Newborn screening for SCID is the need of the hour. Easy access to pediatric HSCT services would ensure that these patients are offered HSCT at an early age.
Keywords: severe combined immune deficiency, India, hematopoietic stem cell transplantation, newborn screening, BCG
Introduction
Severe Combined Immune Deficiency (SCID) is an inborn error of immunity characterized by defect in T lymphocyte development and function. Children with SCID often develop life-threatening opportunistic fungal, bacterial, or viral infections in early infancy. SCID is considered a medical emergency and affected children often succumb to severe infections if diagnosis and definitive treatment are delayed. The estimated incidence of SCID is 1 in 50,000 to 100,000 live births (1). Recent data also suggest an incidence of SCID as high as 1 in 3,000 live births in countries with high consanguinity rates (2). However, due to lack of awareness and diagnostic facilities in developing countries, diagnosis is often missed. Hematopoietic stem cell transplantation (HSCT) is the definitive management for SCID. Early diagnosis and management are essential for successful outcomes. Several countries such as United States of America, Israel, Germany, Switzerland, Sweden, Norway, Iceland, New Zealand, and Taiwan have initiated newborn screening for SCID based on quantification of T-cell receptor excision circles (TRECs) to facilitate early diagnosis (3).
Opportunistic infections in SCID are recurrent, typically start in early infancy, and result in failure to thrive. Common infection patterns seen in SCID include oral thrush, disseminated BCGosis, disseminated cytomegalovirus, and life-threatening bacterial and fungal infections. Non-infective manifestations of SCID include Omenn syndrome (OS), graft versus host reaction, autoimmunity, and hemophagocytic lymphohistiocytosis (4). CD3+ T lymphocyte numbers are usually decreased in SCID (T-). However, in cases of maternal T-cell engraftment or OS, CD3+ T cell numbers can be normal or increased. The expanded T cells are autoreactive in OS, whereas, they are alloreactive in cases with transplacental-acquired maternal T-cell engraftment. T lymphocyte function and naïve T cell numbers are reduced in such cases. T- SCID can be classified based on presence or absence of B lymphocytes and natural killer cells as T-B-NK+, T-B-NK-, T-B+NK-, and T-B+NK+. Combined immunodeficiencies (CID) are also characterized by presence of opportunistic infections and immune dysregulation; however, the age of onset is little older and have a milder immunodeficiency compared to SCID (5).
Until date, 58 different monogenic defects have been identified to result in immunodeficiencies affecting both cellular and humoral immunity and 18 amongst these are known to result in SCID (5). Molecular defects in SCID can be broadly classified as abnormalities in VDJ recombination (RAG1, RAG2, DCLRE1C, NHEJ1, LIG4, PRKDC), abnormalities of cytokine signaling (IL2RG, JAK3, IL7RA), toxic metabolite accumulation (ADA, PNP), defective survival of hematopoietic precursors (AK2, RAC2), abnormalities of T-cell receptor and signaling (PTPRC, CD3D, CD3E, CD3Z, LAT), and abnormalities of actin cytoskeleton (CORO1A). While X-linked SCID due to defect in IL2RG is considered to be the commonest form of SCID in the US, Canada, and Europe, autosomal recessive form of SCID due to defects in RAG1/2 are the commonest forms of SCID in countries where consanguinity rates are high (6–8). However, after initiation of newborn screening program, defects in RAG1/RAG2 are now increasingly being identified even in countries like US and Canada where consanguinity rates are low (9).
Reports of clinical data and outcomes of SCID from developing nations are scarce. Being a tropical nation with universal coverage of BCG vaccination in newborns, microbiological pattern of infections in SCID in India is expected to be different from other cohorts. Molecular spectrum is also expected to be different considering high rates of consanguinity and endogamous marriages in India (6–8). A recent cohort of 57 patients from Mumbai, India showed a high incidence of autosomal recessive forms of SCID with RAG1/2 defects being the commonest (7). We aim to describe the clinical, immunological, and molecular features of children with SCID in this large multicentric cohort from India.
Methods
A detailed case proforma in an Excel format was prepared by one of the authors (PV) and was sent to centers that are recognized as Foundation for Primary Immunodeficiency Diseases (FPID) centers for care of primary immunodeficiencies in India. The format was also sent to tertiary-care centers that manage patients with primary immunodeficiency diseases (PIDs). Information on clinical, laboratory, and molecular details of patients with SCID and their outcomes was sought and collated. Twelve (12) centers provided details of 319 patients that were then compiled and analyzed. Fifteen (15) patients from 2 other centers with either flow-cytometry or mutation-proven SCID are not included in final analysis as data were incomplete. Twenty-three (23) children did not fulfil the criteria for clinical definition for SCID and were not included for analysis. Duplicate entries (n=4) were also noted and excluded.
Data of 277 children who had a clinical profile suggestive of SCID were taken for final analysis ( Supplementary Table 1 ). Children were categorized as SCID/OS/CID/atypical SCID as per the European Society for Immunodeficiencies (ESID) working definition (10). Three (3) patients were classified as possible SCID as they did not fulfil the complete ESID definition, however, the treating team had a high index of suspicion based on clinical and immunological features ( Table 1 ).
Table 1.
Clinical and immunological features of children with clinical features suggestive of SCID in our cohort.
| S No | Age/Sex | Clinical features | Organisms isolated | Absolute lymphocyte count | Immunoglobulin profile | Lymphocyte subsets | Molecular defect | ESID Working Definition |
|---|---|---|---|---|---|---|---|---|
| Pt. 1 | 8 months/male | Recurrent episodes of diarrhea, failure to thrive, pneumonia, meningitis | Stool: Clostridium difficile toxin assay positive | 2.260 | IgG <1.64 g/L IgA <0.36 g/L IgM- 0.25 g/L |
CD3- 0.3% (No: 6-7) CD19- 66% (No: 1492) CD56- 30% (No: 675) |
Not done | SCID |
| Pt. 2 | 5 months/male | Recurrent episodes of pneumonia, diarrhea, failure to thrive, elder male sibling expired at 6 months due to severe infections | Blood culture: Alcaligens faecalis | 0.410 | IgG <2.26 g/L IgA <0.1 g/L IgM<0.2 g/L |
CD3- 0.15% (No: 0-1) CD19- 0% (No: 0) CD56- 84% (No: 345) |
Not done | SCID |
| Pt. 3 | 6.5 months/male | Recurrent episodes of pneumonia, meningitis, hepatosplenomegaly, pancytopenia, transaminitis (HLH), 3 elder male siblings died at early infancy due to recurrent infections | Blood culture: Pseudomonas aeruginosa
Disseminated BCGosis and angioinvasive aspergillosis in lungs in autopsy |
0.940 | IgG<2.99 g/L IgA- 0.49 g/L IgM- 0.88 g/L |
CD3- 0% CD19- 86% (808) CD56- 0.3% |
IL2RG | SCID |
| Pt. 4 | 5 months/male | 2 episodes of pneumonia, recurrent diarrhea, umbilical sepsis, failure to thrive, 3 elder male siblings died at early infancy due to recurrent infections | N.A. | 2.050 | IgG- 2.64 g/L IgA <0.46 g/L IgM- 0.18 g/L |
CD3- 0% CD19- 96.1% (1968) CD56- 0% |
IL2RG | SCID |
| Pt. 5 | 3 months/male | Erythroderma, generalized adenopathy, diarrhea, lymphocytosis, eosinophilia (Omenn syndrome), failure to thrive, elder male sibling died due to eczema and pneumonia at 3rd month | N.A. | 18.540 | N.A. | CD3- 70.94% (13,124) CD19- 0.1% CD56- 7% (1,295) |
RAG2 | Omenn syndrome |
| Pt. 6 | 6 months/male | Persistent pneumonia, oral thrush, 7 maternal uncles died at early infancy due to recurrent infections | N.A. | 2.322 | IgG- 0.65 g/L IgA- 0.22 g/L IgM- 0.24 g/L |
CD3- 0% CD19- 96.75% (2,245) CD56- 3.2% (74) |
IL2RG | SCID |
| Pt. 7 | 3 months/male | Recurrent episodes of pneumonia, diarrhea, meningitis, generalized erythroderma (incomplete Omenn), elder male sibling died at early infancy due to rash and pneumonia | N.A. | 1.566 | IgG- 2.14 g/L IgM- 0.24 g/L |
CD3- 74.79% (1167) CD19- 0.27% (42) CD56- 23% (360) CD3+45RA+ 45RO-: 18.65% compared to 82% in control |
Not done | Omenn syndrome |
| Pt. 8 | 10 months/male | Recurrent episodes of diarrhea, pneumonia, otitis media, failure to thrive, BCG site ulceration, hepatosplenomegaly, generalized adenopathy, erythroderma, eosinophilia (Omenn syndrome), 5 maternal uncles died at early infancy due to recurrent infections | Disseminated BCGosis, disseminated Mycobacterium avium, disseminated CMV, and Aspergillus pneumonia in autopsy | 3.600 | IgG- 1.04 g/L IgA- 0.07 g/L IgM- 0.31 g/L IgE- 700 U/L (Normal: 0.-6 U/L) |
CD3- 95.79% (3,448) CD19- 0.2% (7) CD56- 1% (36) |
IL2RG | Omenn syndrome |
| Pt. 9 | 2 months/female | Recurrent episodes of oral thrush, failure to thrive, 1 elder male sibling expired due to sepsis in early infancy | N.A. | 0.648 | IgG- 2.72 g/L IgA- 0.09 g/L IgM- 0.41 g/L |
CD3- 1.1% (7) CD19- 0.2% (1) CD56- 93.6% (607) |
DCLRE1C | SCID |
| Pt. 10 | 3 months/male | Recurrent episodes of pneumonia, diarrhea, rickets, nephrocalcinosis, distal renal tubular acidosis, oral thrush, failure to thrive | N.A. | 0.896 | IgG- 0.88 g/L IgA <0.06 g/L IgM- 0.19 g/L |
CD3- 1.1% (10) CD19- 92.2% (8,26) CD56- 6.4% (57) |
IL7RA | SCID |
| Pt. 11 | 6 months/male | Pustulosis, hepatosplenomegaly, BCG site ulceration, transfusion-associated GVHD, elder male sibling died at 5 months due to pneumonia | Disseminated BCGosis, Blood culture: Enterobacter sp. | 1.462 | N.A. | CD3- 1.25% (183) CD19- 95% (1389) CD56- 0.45% (6-7) |
No gene variants found in IL2RG, JAK3, RAG1, RAG2 | SCID |
| Pt. 12 | 4 months/male | Recurrent episodes of pneumonia, diarrhea, failure to thrive, meningitis, oral thrush, hepatosplenomegaly, rash, eosinophilia (Omenn phenotype), one elder female sibling expired in early infancy | Pneumocystis jirovecii pneumonia, disseminated CMV in autopsy | 4.176 | IgG- 2.06 g/L IgA- 0.08 g/L IgM- 0.41 g/L |
CD3- 71.6% (2,993) CD19- 1.0% (42) CD56- 12% (504) CD3+45RO-45RA+: 24% as compared to 82% in control |
Not done | Omenn syndrome |
| Pt. 13 | 6 months/male | Persistent pneumonia, diarrhea, oral thrush, erythematous rash, hepatosplenomegaly (incomplete Omenn), nephrotic range proteinuria, two elder siblings (one male and other female) expired in early infancy | Blood culture: Acinetobacter sp.; Pneumonia and meningitis due to Aspergillus sp. and ventriculitis due to CMV in autopsy | 1.404 | IgG- 2.46 g/L IgA- 0.37 g/L IgM- 1.38 g/L |
CD3- 93.7% (1,312) CD19- 0.2% (3) CD56- 5.6% (78) |
Not done | Omenn syndrome |
| Pt. 14 | 7 months/male | Recurrent pneumonia, failure to thrive, oral thrush | Endotracheal aspirate: Klebsiella sp.; RSV pneumonia and disseminated CMV in autopsy | 0.156 | IgG- 7.86 g/L IgA- 0.61 g/L IgM <0.11 g/L |
CD3- 45.7% (72) CD19- 1.6% (2-3) CD56- 21.7% (34) |
PNP | SCID |
| Pt. 15 | 6 months/male | Recurrent episodes of pneumonia, failure to thrive, 5 elder siblings died at early infancy | N.A. | 1.391 | IgG <0.93 g/L IgA <0.16 g/L IgM <0.11 g/L |
CD3- 0% CD19- 0% CD56- 92.2% (1,291) |
RAG2 | SCID |
| Pt. 16 | 6 months/female | Recurrent pneumonia, diarrhea, failure to thrive, hepatosplenomegaly | Blood culture: Candida sp. | 0.785 | IgG- 6.33 g/L IgA- 0.07 g/L IgM <0.11 g/L |
CD3- 0.2% (1-2) CD19- 38.9% (312) CD56- 52.2% (407) |
IL7RA | SCID |
| Pt. 17 | 4 months/male | Recurrent pneumonia, pus discharging sinuses in neck, generalized rash (incomplete Omenn), 3 elder siblings (one female and 2 male) died in early infancy | CMV PCR+, Blood culture: Enterococcus cloacae | 1.800 | IgG <0.95 g/L IgA <0.17 g/L IgM- 0.12 g/L |
CD3- 83.3% (1,499) CD19- 0.2% (3-4) CD56- 14.3% (257) CD3+45RO-45RA+: 27.5% compared to 82% in control |
RAG2 | Omenn syndrome |
| Pt. 18 | 9 months/male | 2 episodes of pneumonia, failure to thrive, meningoencephalitis and hydrocephalus, MRI Brain: multiple tuberculomas noted over parietal and occipital area, 2 elder male siblings expired in early infancy | N.A. | 0.612 | IgG <0.95 g/L IgA <0.17 g/L IgM <0.15 g/L |
CD3- 4.2% CD19- 0.2% CD56- 85% |
RAG1 | SCID |
| Pt. 19 | 2 months/male | Recurrent episodes of pneumonia, failure to thrive | N.A. | 0.655 | IgG- 2.02 g/L IgA <0.16 g/L IgM <0.11 g/L |
CD3- 0% CD19- 0.13% (1) CD56- 72% (468) |
RAG1 | SCID |
| Pt. 20 | 5 months/male | Generalized rash, alopecia, loose stools (incomplete Omenn), failure to thrive, meningitis | N.A. | 1.372 | IgA <0.17 g/L IgM <0.12 g/L |
CD3- 69.6% (954) CD19- 0.15% (2) CD56- 10.7% (147) |
DCLRE1C | Omenn syndrome |
| Pt. 21 | 5 months/male | Younger sibling of Pt. 15, recurrent episodes of pneumonia, diarrhea, failure to thrive | CMV DNA PCR positive | 0.480 | IgG <0.94 g/L IgA- 0.18 g/L IgM <0.12 g/L |
CD3- 21.7% (109) CD19- 1% (5) CD56- 86% (430) |
RAG2 | SCID |
| Pt. 22 | 1.5 months/male | Persistent pneumonia, diarrhea, elder female sibling expired at early infancy | Blood culture: Candida sp. | 0.328 | IgG <2.02 g/L IgA <0.17 g/L |
CD3- 75% (248) CD19- 8.3% (27) CD56- 7.1% (23) |
ADA | SCID |
| Pt. 23 | 5 months/male | Oral thrush, pneumonia, meningitis, one elder female sibling expired due to anemia and pneumonia in early infancy | Disseminated CMV and early invasive pulmonary aspergillosis in autopsy | 0.788 | IgG- 2.49 g/L | CD3- 0.79% (6) CD19- 1.02% (8) CD56- 92.7% (744) |
RAG1 | SCID |
| Pt. 24 | 2 years/male | Recurrent pneumonia, diarrhea, otitis media, failure to thrive, esophageal candidiasis | N.A. | 8.567 | IgG- 5.19 g/L IgA <0.17 g/L IgM- 0.85 g/L |
CD3- 25.76% (2,236) CD3+CD4+- 33.5% (737) CD3+CD8+- 50.2% (1,104) CD19- 51.95% (4,451) CD56- 11.6% (994) CD3+45RA+45RO-: 31.7% compared to 74% in control |
No gene variants found | CID |
| Pt. 25 | 4 months/male | Younger sibling of pt. 8, recurrent episodes of pneumonia and diarrhea, failure to thrive | N.A. | 5.280 | IgG <2.05 g/L IgM <0.25 g/L |
CD3- 0.23% (12) CD19- 94.6% (4,995) CD56- 0.47% (25) |
IL2RG | SCID |
| Pt. 26 | 10 months/male | Recurrent episodes of pneumonia, failure to thrive | N.A. | 0.378 | N.A. | CD3- 2.7% CD19- 2.15% CD56- 85.2% |
DCLRE1C | SCID |
| Pt. 27 | 2.5 months/female | Recurrent pneumonia, otitis media, oral thrush, diarrhea, erythroderma, hepatosplenomegaly, eosinophilia (incomplete Omenn syndrome), elder female sibling expired in early infancy | N.A. | 1.650 | IgG- 1.23 g/L IgA <0.17 g/L IgM <0.25 g/L |
CD3- 7.67% (127) CD19- 0.69% (11) CD56- 82.7% (1,365) CD3+45RA+45RO-: 6.42% compared to 72% of control |
RAG2 | SCID/Omenn syndrome |
| Pt. 28 | 5 months/male | Recurrent episodes of pneumonia, failure to thrive | BAL: Pseudomonas sp.; Blood culture: Candida sp. | 0.360 | IgG <2.05 g/L IgA- 0.07 g/L IgM <0.05 g/L |
CD3- 2.3% (8) CD19- 3.8% (14) CD56- 92.2% (332) |
RAG1 | SCID |
| Pt. 29 | 8 months/female | Persistent pneumonia, recurrent episodes of diarrhea, failure to thrive, chorioretinitis, hepatosplenomegaly | Disseminated CMV; Blood culture: Acinetobacter baumanii | 1.316 | IgG- 4.17 g/L IgA- 0.22 g/L |
CD3- 11.3% (149) CD19- 69.8% (921) CD56- 1.75% (23) |
JAK3 | SCID |
| Pt. 30 | 1.5 months/male | Recurrent episodes of pneumonia, diarrhea, failure to thrive, elder male sibling died in early infancy | Blood culture: Acinetobacter baumanii | 0.204 | IgG- 1.96 g/L IgM <0.25 g/L |
CD3- 54% (108) CD19- 24% (48) CD56- 20% (40) |
ADA | SCID |
| Pt. 31 | 4 years/male | Recurrent episodes of pneumonia since early infancy, failure to thrive | N.A. | 0.116 | IgG- 4.73 g/L IgA- 1.05 g/L IgM- 1.12 g/L |
CD3- 64.8% (78) CD19- 4% (5) CD56- 7% (8-9) |
ADA | Atypical SCID |
| Pt. 32 | 9 months/male | Recurrent episodes of pneumonia, diarrhea, failure to thrive | N.A. | 0.154 | IgG- 2.27 g/L IgA- 0.27 g/L IgM <0.25 g/L |
CD3- 44.4% (67) CD19- 38.5% (58) CD56- 5.7% (9) |
Not done | SCID |
| Pt. 33 | 2 months/female | Recurrent episodes of pneumonia, diarrhea, failure to thrive | N.A. | 0.977 | IgG- 2.45 g/L IgA- 0.23 g/L IgM- 0.29 g/L |
CD3- 32% (314) CD19- 57% (559) CD56- 1.2% (12) |
Not done | SCID |
| Pt. 34 | 5 months/male | Recurrent diarrhea, failure to thrive, BCG site ulceration, pneumonia, erythroderma, eosinophilia, alopecia (Omenn syndrome) | Blood culture: Enterococcus sp.; Pneumocystis jirovecii pneumonia and disseminated BCGosis in autopsy | 2.498 | IgG <2.05 g/L IgM- 0.34 g/L IgE- 369 kU/L (up to 7.3) |
CD3- 78.01% (1,950) CD19- 4.44% (110) CD56- 13.12% (325) CD3+45RA+RO-: 2.26% compared to 83.7% in control CD3+HLA-DR+: 86.25% compared to 8.6% in control |
No gene variants found | Omenn syndrome |
| Pt. 35 | 6 months/male | Recurrent pneumonia, failure to thrive, elder male sibling expired in early infancy due to pneumonia | N.A. | 0.868 | N.A. | CD3- 3% (26) CD19- 94% (818) CD56- 0.4% (3) |
IL2RG | SCID |
| Pt. 36 | 1.5 months/female | Anasarca, nephrotic range proteinuria, pneumonia, failure to thrive, erythematous rash (incomplete Omenn), elder male sibling expired in early infancy | N.A. | 0.722 | IgG- 8.29 g/L IgA- 0.75 g/L |
CD3- 89% CD3+CD4+- 8% CD3+CD8+- 85.1% CD19- 0.3% CD56- 0.8% CD3+45RA+45RO-: 30% compared to 90% in control CD3+45RA-45RO+: 79.14% compared to 19.24% in control CD3+HLA-DR+: 90.13% compared to 12.7% in control |
ADA | Omenn syndrome |
| Pt. 37 | 5 months/female | Recurrent pneumonia, diarrhea, failure to thrive, BCG site ulceration | N.A. | 0.861 | IgG <2.0 g/L IgA <0.17 g/L |
CD3- 34% (292) CD3+CD4+- 29.7% (89) CD3+CD8+- 55.3% (165) CD19- 45% (387) CD56- 12.1% (103) |
Not done | SCID |
| Pt. 38 | 5 months/male | Recurrent pneumonia, diarrhea, failure to thrive | N.A. | 0.140 | N.A. | CD3- 9.6% (13) CD19- 8.7% (12) CD56- 80% (112) |
RAG1 | SCID |
| Pt. 39 | 2 months/male | Recurrent episodes of diarrhea, failure to thrive, sacral abscess, 2 elder siblings died in early infancy due to repeated infections | Blood culture: Staphylococcus aureus; Disseminated CMV in autopsy | 0.06 | N.A. | CD3- 50% (30) CD19- 7.7% (4-5) CD56- 34.6% (21) |
ADA | SCID |
| Pt. 40 | 2.5 months/male | Recurrent pneumonia, otitis media, failure to thrive, 6 maternal uncles and 2 elder male siblings died at early infancy due to repeated infections | N.A. | 1.406 | N.A. | CD3- 0.07% (01) CD19- 91.5% (1,598) CD56- 1.8% (33) |
Not done | SCID |
| Pt. 41 | 4 years/female | Eczematoid eruptions and chronic otitis media since early infancy, autoimmune hemolytic anemia, generalized adenopathy | N.A. | 1.922 | IgG- 21.56 g/L IgA- 4.77 g/L IgM- 0.57 g/L IgE- 933 U/L (Normal: up to 60) |
CD3- 24.79% (912) CD3+CD4+- 21.2% (193) CD3+CD8+- 55% (500) CD19- 42.3% (812) CD56- 2.7% (58) CD3+45RA+RO-: 45% compared to 76% in control CD3+CD4+45RA+RO-: 14.9% compared to 67% in control CD3+CD8+45RA+45RO-: 35.8% compared to 72% in control |
STK4 | CID |
| Pt. 42 | 4 months/male | Recurrent pneumonia, diarrhea, failure to thrive, oral thrush, 1 maternal uncle died at 2 years due to repeated infections | Blood culture: Moraxella sp. | 1.302 | N.A. | CD3- 1.3% (17) CD19- 85.16% (1,109) CD56- 2.9% (37) |
Not done | SCID |
| Pt. 43 | 2.5 months/male | Chronic diarrhea, failure to thrive, esophageal candidiasis, maternal cousin (male) expired at early infancy due to pneumonia | N.A. | 0.415 | N.A. | CD3- 3.8% (15) CD19- 84% (336) CD56- 3% (12) |
IL2RG | SCID |
| Pt. 44 | 3 months/male | Recurrent pneumonia, diarrhea, failure to thrive, erythroderma, eosinophilia, hepatosplenomegaly (maternal T cell engraftment), 1 maternal uncle died at early infancy due to pneumonia | Blood culture: Weisella confusa | 7.457 | N.A. | CD3- 15.9% (1,192) CD19- 76.4% (5,692) CD56- 1.9% (142) CD3+45RA+RO-: 5.43% compared to 59% in control CD3+45RA-45RO+: 96.9% compared to 60% in control CD3+HLA-DR+: 83.5% compared to 15.7% in control |
IL2RG | Atypical SCID |
| Pt. 45 | 4 months/male | Recurrent pneumonia, diarrhea, failure to thrive, oral thrush, BCG site ulceration | N.A. | 2.831 | IgG <0.87 g/L IgA <0.16 g/L |
CD3- 0.2% (5-6) CD19- 97.7% (2,765) CD56- 0.48% (13-14) |
No gene variants found | SCID |
| Pt. 46 | 5 months/male | Recurrent fever, BCG site ulceration, hepatosplenomegaly, oral thrush | Disseminated BCGosis | 2.086 | IgG <1.46 g/L | CD3- 0.6% (13) CD19- 97.8% (2,044) CD56- 0.2% (4) |
Not done | SCID |
| Pt. 47 | 15 days/male | Younger sibling of pt. 31, pneumonia, recurrent diarrhea, failure to thrive | Blood culture: Candida sp. | 0.094 | N.A. | CD3- 42% (38) CD19- 40% (36) CD56- 16% (14) |
ADA | SCID |
| Pt. 48 | 4 months/male | Younger sibling of pt. 27, recurrent pneumonia, diarrhea, failure to thrive, erythroderma, hepatosplenomegaly, eosinophilia (Omenn syndrome) | N.A. | 1.896 | N.A. | CD3- 74% (1,406) CD19- 0.4% (8) CD56- 22% (418) CD3+45RA+45RO-: 16% compared to 71% in control |
RAG2 | Omenn syndrome |
| Pt. 49 | 3 years/male | Recurrent sinopulmonary infections, diarrhea, failure to thrive, 1 episode of liver abscess, intra-cranial B cell lymphoma, defective T lymphocyte proliferation on stimulation with PHA. | N.A. | 3.265 | IgG- 4.02 g/L | CD3- 45.14% (1,467) CD3+CD4+- 6.9% (103) CD3+CD8+- 70.3% (1,033) CD19- 6.83% (222) CD56- 25.01% (816) CD3+45RA+45RO-: 71.06% compared to 64% in control CD3+CD4+45RA+45RO-: 3.6% compared to 72% in control CD3+CD8+45RA+45RO-: 75.3% compared to 68% in control |
CORO1A | Atypical SCID |
| Pt. 50 | 6 months/male | Recurrent episodes of pneumonia, failure to thrive | N.A. | 0.411 | IgG <0.95 g/L IgA <0.17 g/L IgM <0.25 g/L |
CD3- 20% (80) CD19- 73% (292) CD56- 1.4% (5-6) |
JAK3 | SCID |
| Pt. 51 | 10 months/male | Pneumonia, diarrhea, failure to thrive, meningoencephalitis | Endotracheal aspirate: Klebsiella pneumoniae | 0.810 | IgG <0.95 g/L IgA <0.17 g/L IgM <0.25 g/L |
CD3- 4% (32) CD19- 95% (760) CD56- 1% (8) |
IL2RG | SCID |
| Pt. 52 | 3 months/male | Recurrent pneumonia, diarrhea, failure to thrive | N.A. | 0.199 | N.A. | CD3- 0.82% (2) CD19- 1.17% (2-3) CD56- 88.9% (178) |
DCLRE1C | SCID |
| Pt. 53 | 5 months/male | Pneumonia, failure to thrive, complicated otitis media with facial nerve palsy, transfusion-associated GVHD | N.A. | 0.292 | N.A. | CD3- 0.2% (0-1) CD19- 29% (87) CD56- 60% (180) |
Not done | SCID |
| Pt. 54 | 3.5 years/male | Severe eczema since early infancy, pustules, otitis media, pneumonia, chest wall abscess, eosinophilia (incomplete Omenn) | Pus culture- Staphylococcus aureus | 1.244 | IgG- 1.64 g/L IgA- 1.56 g/L IgE- 4269 kU/L (upto 32) IgG1- 1.01 g/L IgG2- 0.95 g/L IgG3- 0.23 g/L IgG4- 0.71 g/L |
CD3- 60% (744) CD3+CD4+- 17.3% (128) CD3+CD8+- 71.5% (529) CD19- 2.3% (28) CD56- 15% (186) CD3+45RA-45RO-: 36.6% compared to 65% in control CD3+45RA-45RO+: 67% compared to 31% in control CD3+HLA-DR+: 64.2% compared to 19.3% in control |
No gene variants found | Omenn syndrome |
| Pt. 55 | 5 months/male | Recurrent pneumonia, diarrhea, failure to thrive, hyperferritinemia, hypofibrinogenemia, pancytopenia (HLH) | ET aspirate: Klebsiella pneumoniae, Acinetobacter baumanii; PCR positivity for H1N1 | 1.547 | IgG- 2.32 g/L IgA <0.2 g/L IgM- 0.22 g/L |
CD3- 1.74% (27) CD19- 91.6% (1,426) CD56- 5% (78) |
IL2RG | SCID |
| Pt. 56 | 6 months/female | Pneumonia, failure to thrive, diarrhea, BCG site ulceration | N.A. | 1.098 | IgG- 0.54 g/L IgA <0.2 g/L IgM <0.17 g/L |
CD3- 0% (0) CD19- 2% (22) CD56- 79% (869) |
DCLRE1C | SCID |
| Pt. 57 | 7 months/male | Pneumonia, diarrhea, failure to thrive, hepatosplenomegaly, BCG site ulceration | N.A. | 0.855 | N.A. | CD3- 5.1% (43) CD19- 77.5% (667) CD56- 17% (146) |
Not done | SCID |
| Pt. 58 | 11 months/male | Recurrent pneumonia, failure to thrive, hepatosplenomegaly, generalized adenopathy, BCG site ulceration, erythematous rash (incomplete Omenn), meningitis with hydrocephalus | Disseminated BCGosis, CMV DNA PCR positive, Endotracheal aspirate: Klebsiella pneumoniae | 1.832 | IgG- 7.74 g/L IgA- 0.36 g/L IgM- 2.42 g/L |
CD3- 68% (1,244) CD3+CD4+- 7.8% (97) CD3+CD8+- 45.1% (558) CD19- 5.6% (102) CD56- 23.5% (430) CD3+CD4+45RA-45RO+: 90.4% compared to 30.2% in control CD3+HLA-DR+: 67.9% compared to 5.8% in control |
RAG1 | Omenn syndrome |
| Pt. 59 | 5 months/male | BCG site ulceration, persistent diarrhea, generalized papular rash | M. bovis | 0.931 | IgA<0.10 g/L | CD3- <1% CD19- 97% (902) CD56- <1% |
IL2RG | SCID |
| Pt. 60 | 6 months/male | BCG site ulceration, oral thrush, septicemia | Candida sp. | 2.129 | N.A | CD3- 29% (617) CD19- 62% (1320) CD56- 8% (170) |
No gene variants identified | SCID |
| Pt.61 | 5 months/female | BCG site ulceration, pneumonia, erythroderma, alopecia, CMV DNA PCR- positive | CMV, M. bovis | 1.144 | IgG-9.03 g/L IgA-0.17 g/L IgM-0.41 g/L |
CD3-70.70% (806) CD19-0.14% (2) CD56-17.70% (202) CD3+45RA+-12.57% compared to 86% in control |
RAG2 | Omenn syndrome |
| Pt. 62 | 4 months/male | Severe pneumonia, CT chest: diffuse bilateral ground glass opacities with multifocal consolidation | Nil | 0.507 | IgG- <2.02 g/L IgA- <0.17 g/L IgM- <25 g/L |
CD3- 57.23% (290) CD19-0.05% (1) CD56-35.08% (179) |
RAG1 | SCID |
| Pt. 63 | 5 months/male | Severe pneumonia, CT chest: bilateral small random nodules | Nil | 1.236 | IgG- <2.03 g/L IgA- <0.17 g/L IgM- <0.25 g/L |
CD3- 0.28% (4) CD19-96.20% (1193) CD56-0.51% (6) |
IL2RG | SCID |
| Pt. 64 | 5 months/female | Persistent pneumonia- pneumothorax, oral thrush | Candida sp. | 0.180 | IgG- <2.03 g/L IgA- <0.17 g/L IgM- 0.33 g/L |
CD3-0.16% (1) CD19-0.16% (1) CD56-74.40% (134) |
DCLRE1C | SCID |
| Pt. 65 | 1.5 months/female | Left ear complicated otitis media, pneumonia, diarrhea | S. aureus | 2.443 | IgG-8.04 g/L IgA-0.75 g/L IgM-1.38 g/L |
CD3-22.87% (559) CD19-73.60% (1776) CD56-1.43% (34) |
No gene variants identified | SCID |
| Pt. 66 | 7 months/male | BCG adenitis, encephalitis | M. bovis | 0.506 | IgG- <2.05 g/L IgA- <0.17 g/L IgM- <0.26 g/L |
CD3-18.19% (93) CD19-0.08% (1) CD56-77.24% (394) |
DCLRE1C | SCID |
| Pt. 67 | 8 months/female | Recurrent diarhea, failure to thrive, pneumonia, axillary adenopathy | Nil | 6.864 | IgG-<2.03 g/L IgM->4 g/L |
CD3-69.75% (4785) CD3+CD4+ - 32% of CD3+ lymphocytes (1530) CD3+CD8+ - 62% of CD3+ lymphocytes (2967) CD3+45RA+ - 24.3% compared to 85% in healthy control CD19-8.95% (617) CD56-2.57% (178) |
No gene variants identified | SCID |
| Pt. 68 | 10 months/male | recurrent gastroenteritis, pneumonia, DCT+ autoimmune hemolytic anemia | CMV | 2.200 | IgG-3.33 g/L IgA- <0.17 g/L IgM- 1.19 g/L |
CD3-86.75% (1910) CD19-0.64% (13) CD56-6.50% (143) CD3+CD45RA+ -38.2% compared to 79% in control |
NHEJ1 | Atypical SCID |
| Pt. 69 | 5 months/male | persistent pneumonia, absent BCG scar | Nil | 0.287 | IgG-3.47 g/L IgA- 0.21 g/L IgM-0.76 g/L |
CD3-51.62% (150) CD19-31.30% (91) CD56-9.46% (28) |
No gene variants identified | SCID |
| Pt. 70 | 4 months/male | Recurrent pneumonia, diarrhoea, generalized erythematous macular rash, CMV retinitis, seizures, GVHD skin lesions | CMV | 4.921 | IgG- <1.99 g/L IgA- <0.36 g/L IgM- <0.25 g/L |
CD3-0.54% (25) CD19-0.54% (25) CD56-91.74% (4512) |
DCLRE1C | SCID |
| Pt. 71 | 6 months/male | Recurrent pneumonia, otitis media, ulceration at BCG site, hepatosplenomegaly | Enterococcus sp. | 0.816 | IgA-0. 56 g/L | CD3-1.39% (12) CD19-90.95% (746) CD56-5.4% (44) |
IL2RG | SCID |
| Pt. 72 | 11 months/male | Skin pustule and abscess, generalized erythematous macular rash, oral thrush | Nil | 1.118 | IgG-<0.90 g/L IgA- <0.21 g/L |
CD3-24.80% (278) CD19-6.20% (69) CD56-67.6% (757) |
NHEJ1 | SCID |
| Pt. 73 | 3.5 months/male | Recurrent pneumonia, diarrhoea, generalized erythematous macular rash | Nil | 2.420 | IgG- 1.62 g/L IgA- 0.09 g/L IgM- 0.48 g/L |
CD3-5% (121) CD19- 47% (1137) CD56- 42% (1016) |
No gene variants identified | SCID |
| Pt. 74 | 3 months/male | Recurrent pneumonia, diarrhoea | Nil | 1.643 | IgG- 3.09 g/L IgA- <0.07 g/L |
CD3-87.90% (1442) CD19-1.7% (28) CD56-2.2% (33) CD3+45RA+ -1.6% compared to 78% in control |
ADA | Atypical SCID |
| Pt. 75 | 3 months/male | Recurrent pneumonia, diarrhoea, failure to thrive | Nil | 2.862 | IgG- 2.14 g/L IgA- <0.20 g/L |
CD3-35.90% (1026) CD19-3.11% (89) CD56- 38% (1087) |
No gene variants identified | SCID |
| Pt. 76 | 12 months/female | Recurrent pneumonia, diarrhoea, oral thrush | Nil | 4.300 | IgG- 3.28 g/L IgA- 1.46 g/L IgM- 2.99 g/L |
CD3-3.29% (142) CD19-79.37% (3414) CD56-9.63% (413) |
No gene variants identified | SCID |
| Pt. 77 | 1.5 months/female | Recurrent pneumonia, otitis media, generalized erythematous macular rash | Pichia fermentans; E. coli | 4.720 | IgG- 4.27 g/L IgA- <0.16 g/L IgM- 0.35 g/L |
CD3-49.03% (2303) CD19-1.27% (61) CD56-37.44% (1765) CD3+45RA+ - 1.46% compared to 73% in control |
RAG1 | Omenn syndrome |
| Pt. 78 | 6 months/male | Recurrent pneumonia, diarrhoea, generalized erythematous macular rash | Nil | 1.808 | IgG- <2.02 g/L IgA- 0.20 g/L IgM-1.71 g/L |
CD3-95.65% (1732) CD19-1.78% (32) CD56-0.53% (9) CD3+45RA+ - 11% compared to 86% in control |
IL2RG | Atypical SCID |
| Pt. 79 | 6.5 months/male | Recurrent pneumonia, diarrhoea | Nil | 0.600 | IgG- 3.65 g/L IgA- 0.38g/L IgM- 0.41 g/L |
CD3-29.54% (177) CD19-41.13% (247) CD56-18.87% (114) |
No gene variants identified | SCID |
| Pt. 80 | 15 months/male | Recurrent pneumonia, diarrhoea, oral thrush | Klebsiella pneumoniae, CMV | 7.191 | IgG-2.02 g/L IgA-0.18 g/L IgM-0.46 g/L |
CD3-12.36% (892) CD19-0.74% (53) CD56-51.5% (3703) CD3+45RA+ - 14.29% (decreased) |
No gene variants identified | SCID |
| Pt. 81 | 42 months/female | Recurrent pneumonia, oral thrush | Nil | 1.615 | IgG-21.77 g/L IgA-1.23 g/L IgM-1.81 g/L |
CD3-37.40% (606) CD4- 5.7% CD8- 14.7% CD19-22.6% (366) CD56- 46% (743) |
No gene variants identified | SCID |
| Pt. 82 | 3 months/male | Recurrent pneumonia, failure to thrive, oral thrush, one elder female sibling expired due to pneumonia in early infancy | Nil | 2.492 | IgG-2.72 g/L IgA-0.09 g/L IgM-0.73 g/L |
CD3-30% (747) CD19-9.10% (227) CD56- 41% (1021) |
No gene variants identified | SCID |
| Pt. 83 | 3 months/female | Recurrent pneumonia, diarrhoea, otitis media, oral thrush | Nil | 2.608 | IgG-<2.02 g/L IgA-<0.17 g/L IgM-0.90 g/L |
CD3-32.68% (853) CD19-29.79% (783) CD56-33.41% (872) CD3+ 45RA+ -2.23% (decreased) |
No gene variants identified | SCID |
| Pt. 84 | 4 months/male | Recurrent pneumonia, diarrhoea, otitis media, ulceration at BCG site | Nil | 0.663 | IgG-<2.02 g/L IgA-<0.17 g/L IgM-<0.25 g/L |
CD3-87.66% (579) CD19-0.05% (1) CD56-10.22% (66) |
RAG1 | SCID |
| Pt. 85 | 4 months/male | Recurrent pneumonia, severe erythroderma, developmental delay | Nil | 1.441 | IgG-<2.02 g/L IgA-<0.17 g/L |
CD3-0.16% (3) CD19-94.81% (1365) CD56-0.67% (10) |
IL2RG | SCID |
| Pt. 86 | 15 months/male | Recurrent pneumonia, generalized eczematoid macular rash, developmental delay, myopathy | Nil | 14.84 | IgG-13.16 g/L IgA-1.70 g/L IgM<0.26 g/L IgE- 8423 U/L |
CD3-92.20% (13,683) CD19-2.85% (416) CD56-3.21% (475) CD4+45RA+ - 12.17% compared to 56% in control CD8+45RA+ - 18.6% compared to 72% in control |
STIM1 | CID |
| Pt. 87 | 6 months/male | Recurrent pneumonia, extensive eczematoid rash | CMV | 6.556 | IgG-13.75 g/L IgA-0.42 g/L IgM-1.88 g/L IgE- 622 U/L |
CD3-53.90% (3536) CD4- 4.9% (320) CD8- 30.2% (1968) CD19-25.9% (1706) CD56-5.6% (368) |
No gene variants identified | Omenn syndrome |
| Pt. 88 | 2.5 months/female | Recurrent pneumonia, diarrhoea, BCG site abscess | Nil | 0.055 | IgG-2.33 g/L IgA<0.17 g/L IgM<0.25 g/L |
CD3-95.58% (48) CD19-0.07% (1) CD56-0.79% (1) |
ADA | SCID |
| Pt. 89 | 4 months/female | Recurrent diarrhoea, otitis media, generalized erythematous macular rash, ulceration at BCG site | Enterococcus faecalis | 2.352 | IgG<2.02 g/L IgM<0.21 g/L |
CD3-68.95% (1621) CD19-0.05% (1) CD56-25.37% (597) CD3+ 45RA+ -6.79% compared to 70% in control HLA DR in CD3+ - 74.2% compared to 15% in control |
RAG1 | Omenn syndrome |
| Pt. 90 | 4.5 months/male | Recurrent pneumonia, | Nil | 0.724 | IgG-<0.95 g/L IgA<0.17 g/L |
CD3-2.41% (17) CD19-0.45% (4) CD56-90.91% (655) |
DCLRE1C | SCID |
| Pt. 91 | 4 months/male | Recurrent pneumonia, chorioretinitis, failure to thrive, 3 maternal uncles died at early infancy due to severe infections | Blood CMV PCR positive | 1.760 | IgG < 2.7 g/L IgA <0.4 g/L IgM- 1.07 g/L |
CD3- 1% (18) CD19- 61% (1,098) CD56- 1% (18) |
IL2RG | SCID |
| Pt. 92 | 4 months/male | Oral thrush, pneumonia, failure to thrive | N.A. | 0.650 | N.A. | CD3- 0.4% (2-3) CD19- 97% (631) CD56- 0.4% (2-3) |
IL2RG | SCID |
| Pt. 93 | 6 months/female | Recurrent pneumonia, CMV chorioretinitis | Blood CMV PCR positive | 2.200 | IgG <2.7 g/L IgA <0.4 g/L IgM <0.25 g/L |
CD3- 7.6% (167) CD19- 1% (22) CD56- 40% (880) |
RAG2 | SCID |
| Pt. 94 | 4.5 months/male | Persistent pneumonia, failure to thrive, elder sibling died at 6 months due to severe pneumonia | N.A. | 1.750 | IgG <1.37 g/L IgA <0.26 g/L IgM <0.16 g/L |
CD3- 4% (70) CD19- 47% (823) CD56- 25% (438) |
IL7RA | SCID |
| Pt. 95 | 6 months/male | Recurrent pneumonia, failure to thrive | N.A. | 2.000 | IgG <1.37 g/L IgA <0.26 g/L IgM- 0.53 g/L |
CD3- 20% (400) CD19- 80% (1,600) CD56- 0.1% (2) |
IL2RG | SCID |
| Pt. 96 | 3 months/female | Oral thrush, septicemia | N.A. | 0.720 | IgG- 0.8 g/L IgA <0.26 g/L IgM <0.18 g/L |
CD3- 5% (36) CD19- 7% (50) CD56- 53% (382) |
RAG2 | SCID |
| Pt. 97 | 5 months/male | Recurrent episodes of pneumonia, diarrhea, failure to thrive, oral thrush, BCG site ulceration, elder female sibling expired at 6 months due to recurrent infections | N.A. | 0.850 | IgG <2.7 g/L IgA <0.4 g/L IgM <0.25 g/L |
CD3- 5% (43) CD19- 2.3% (20) CD56- 46% (391) |
Not done | SCID |
| Pt. 98 | 3 months/female | Recurrent pneumonia, failure to thrive | N.A. | 0.700 | IgG <2.7 g/L IgA <0.4 g/L IgM <0.25 g/L |
CD3- 6.5% (46) CD19- 3.6% (25) CD56- 34% (238) |
Not done | SCID |
| Pt. 99 | 3 months/female | Recurrent pneumonia, failure to thrive | N.A. | 0.680 | IgG- 0.65 g/L IgA <0.5g/L IgM <0.25 g/L |
CD3- 2% (14) CD19- 4% (27) CD56- 53% (360) |
DCLRE1C | SCID |
| Pt. 100 | 3.5 months/male | Persistent pneumonia, failure to thrive | N.A. | 4.911 | IgG- 0.82 g/L IgA <0.26 g/L IgM <0.18 g/L |
CD3- 9.4% (461) CD19- 0.4% (20) CD56- 90% (4,410) |
Not done | SCID |
| Pt. 101 | 6 months/female | Persistent pneumonia, failure to thrive | N.A. | 5.756 | IgG- 6.7 g/L IgA <0.26 g/L IgM- 0.78 g/L |
CD3- 2% (116) CD19- 41% (2,362) CD56- 47% (2,707) |
Not done | SCID |
| Pt. 102 | 7.5 months/male | Recurrent pneumonia, diarrhea, failure to thrive | N.A. | 0.750 | IgG- 0.76 g/L IgA- 0.08 g/L IgM- 0.08 g/L |
CD3- 26% (195) CD19- 67% (503) CD56- 6% (45) |
JAK3 | SCID |
| Pt. 103 | 1 month/male | Erythroderma, loss of eyelashes, eosinophilia (incomplete Omenn), failure to thrive, two siblings (one male and one female) expired in early infancy due to erythroderma, generalized lymphadenopathy, and severe infections | N.A. | 3.358 | IgG- 5.86 g/L IgA <0.26 g/L IgM- 0.36 g/L IgE >2,500 U/L |
CD3- 9% (302) CD19- 48% (1,613) CD56- 11% (370) |
No variants identified | Omenn syndrome |
| Pt. 104 | 5 months/female | Recurrent pneumonia, otitis media, failure to thrive, oral thrush, pancytopenia, hepatosplenomegaly, seizures, encephalopathy (HLH) | N.A. | 1.890 | IgG <2.7 g/L IgA <0.4 g/L IgM <0.25 g/L |
CD3- 32% (605) CD3+CD4+- 87% (528) CD3+CD8+- 13% (78) CD19- 65% (1,229) CD56- 3% (57) |
SP110 | SCID |
| Pt. 105 | 11 months/male | Chronic diarrhea, failure to thrive | Stool culture: Acinetobacter sp. | 0.691 | IgG- 3.0 g/L IgA- 0.52 g/L IgM- 0.39 g/L |
CD3- 13.6% (94) CD19- 56% (386) CD56- 28% (193) |
RAG1 | SCID |
| Pt. 106 | 4.5 months/male | Oral thrush, recurrent pneumonia, diarrhea, failure to thrive, BCG site ulceration, 3 maternal uncles died at early infancy due to repeated infections | N.A. | 0.900 | IgG- 1.1 g/L IgA- 0.05g/L IgM- 0.07g/L |
CD3- 5% (45) CD19- 91% (819) CD56- 2% (18) |
IL2RG | SCID |
| Pt. 107 | 5 months/male | Recurrent pneumonia, oral thrush, failure to thrive, BCG site ulceration, encephalopathy | N.A. | 1.120 | IgG <1.4g/L IgA <0.17g/L IgM <0.19g/L |
CD3- 6% (66) CD19- 92% (1,012) CD56- 1% (11) |
JAK3 | SCID |
| Pt. 108 | 8 months/male | Recurrent pneumonia, diarrhea, failure to thrive | N.A. | 0.380 | IgG <1.4g/L IgA <0.17g/L IgM <0.19g/L |
CD3- 0% CD19- 94% (357) CD56- 4% (15) |
IL7R | SCID |
| Pt. 109 | 4.5 months/male | Persistent pneumonia, failure to thrive | N.A. | 2.092 | IgG <1.4g/L IgA <0.17g/L IgM- 0.24g/L |
CD3- 0.8% (17) CD19- 98% (2,048) CD56- 0% |
Not done | SCID |
| Pt. 110 | 4.5 months/male | Persistent pneumonia, recurrent diarrhea, skin abscess, failure to thrive, situs inversus, one elder sibling died at early infancy due to pneumonia | N.A. | 0.780 | IgG- 1.92 g/L IgA- 0.04 g/L IgM- 0.02 g/L |
CD3- 0.04% CD19- 0.29% CD56- 96% |
Not done | SCID |
| Pt. 111 | 5.5 months/female | Recurrent pneumonia, failure to thrive, 3 elder male siblings died within first year of life due to severe infections | N.A. | 1.425 | IgG- 0.42 g/L IgA <0.03 g/L IgM- 0.34 g/L |
CD3- 1.2% (17) CD19- 71% (1,012) CD56- 25% (356) |
CD3D | SCID |
| Pt. 112 | 6 months/female | Recurrent pneumonia, failure to thrive | N.A. | 0.336 | IgG <1.36 g/L IgA <0.25 g/L IgM <0.18 g/L |
CD3- 0.2% (0-1) CD19- 0% CD56- 99% (335) |
Not done | SCID |
| Pt. 113 | 3.5 months/female | Recurrent pneumonia, failure to thrive, one elder female sibling died at 4 months due to a probable infection | N.A. | 2.210 | IgG <1.36 g/L IgA <0.25 g/L IgM <0.18 g/L |
CD3- 0.8% (18) CD19- 97.4% (2,153) CD56- 1% (22) |
No variants identified | SCID |
| Pt. 114 | 8 months/male | Recurrent pneumonia, BCG site ulceration, failure to thrive, one elder male sibling died at early infancy due to pneumonia | Disseminated BCGosis | 1.650 | IgG- 0.15 g/L IgA <0.24 g/L IgM- 0.2 g/L |
CD3- 0% CD19- 61% (1,007) CD56- 38% (627) |
IL7R | SCID |
| Pt. 115 | 7 months/female | Persistent pneumonia, failure to thrive, two elder siblings (one male, one female) died in early infancy due to severe infections, one had disseminated BCGosis | N.A. | 0.870 | IgG <2.0 g/L IgA <0.3 g/L IgM <0.2 g/L |
CD3- 15% (131) CD19- 0% CD56- 40% (350) |
DCLRE1C | SCID |
| Pt. 116 | 7 months/female | Persistent pneumonia, failure to thrive, autoimmune hemolytic anemia | Disseminated CMV, pulmonary aspergillosis | 1.700 | IgG- 3.2 g/L IgA- 0.38 g/L IgM- 0.4 g/L |
CD3- 4% (68) CD19- 0% CD56- 72% (1,224) |
RAG2 | SCID |
| Pt. 117 | 8 months/male | Recurrent pneumonia, persistent diarrhoea, BCG site ulceration | Disseminated BCGosis | 2.400 | IgG- 2.9 g/L IgA- 0.32 g/L IgM- 0.24 g/L |
CD3- 0% CD19- 70% (1,680) CD56- 24% (576) |
CD3E | SCID |
| Pt. 118 | 5 months/male | Recurrent pneumonia, failure to thrive | Pneumocystis jirovecii from endotracheal aspirate | 3.200 | IgG- 3.64 g/L IgA- 0.42 g/L IgM- 0.38 g/L |
CD3- 2% (64) CD19- 64% (2,048) CD56- 1% (32) |
IL2RG | SCID |
| Pt. 119 | 3.5 months/male | Recurrent pneumonia, septicemia | None | 0.064 | IgG- 1.49 g/L IgA- <0.26 g/L IgM- <0.16 g/L |
CD3- 63% (27) CD19- 2.4% (1) CD56- 2.4% |
ADA | SCID |
| Pt. 120 | 1 month 8 days/female | Recurrent pneumonia, cupping of ribs with blunting of lower end of scapula in radiology | None | 0.160 | IgG- 3.54 g/L IgA- <0.05 g/L IgM- <0.03 g/L |
CD3- 32% (51) CD19- 8.9% (14) CD56- 58% (93) |
ADA (probable); Gene sequencing not done | SCID |
| Pt. 121 | 10 months/male | Recurrent pneumonia, persistent diarrhoea, oral candidiasis | Adenovirus | 0.582 | IgG- 3.54 g/L IgA- <0.05 g/L IgM- <0.03 g/L |
CD3-11% (47.8) CD19-68.7% (298.7) CD56-18% (79.2) |
JAK3 | SCID |
| Pt. 122 | 7 months/female | Recurrent pneumonia, persistent diarrhoea, septicemia | Rhinovirus, Blood- Candida sp. | 0.952 | IgG- 16.55 g/L IgA- 0.29 g/L IgM- 1.12 g/L |
CD3-4.7% (12) CD19-0% CD56-91% (231) |
Not done | SCID |
| Pt. 123 | 6 months/male | Recurrent pneumonia, persistent diarrhoea | Nil | 0.780 | IgG- 3.06 g/L IgA- 0.26 g/L IgM- 0.30 g/L |
CD3-85% (665) CD19-3% (26) CD56-11% (87) |
ADA | SCID |
| Pt. 124 | 6 months/male | Recurrent pneumonia, persistent diarrhoea, cellulitis, hepatosplenomegaly, panniculitis | M. bovis | 0.370 | IgG- 0.19 g/L IgA- <0.01 g/L IgM- 0.16 g/L |
CD3-4.94% (22) CD19-84% (404) CD56-0.09% (3) |
Not done | SCID |
| Pt. 125 | 36 months/male | Recurrent pneumonia, persistent diarrhoea | Nil | 0.480 | IgG- 11.80 g/L | CD3-33.3% (156.5) CD19-33.4% (156.3) CD56- 28.3% (112) |
Not done | SCID |
| Pt. 126 | 7 months/female | Recurrent pneumonia, persistent diarrhoea, septicemia | Blood- Acinetobacter baumanni, Candida sp. | 1.090 | IgG- 9.80 g/L IgA- 0.17 g/L IgM- 0.43 g/L |
CD3-0.35% (4) CD19-82.7% (1048) CD56- 4.56% (58) |
Not done | SCID |
| Pt. 127 | 11 months/male | Recurrent pneumonia, persistent diarrhoea, otitis media | Ear pus- P. aeruginosa | 0.824 | IgG- 12.40 g/L | CD3-3.0% (26) CD19-84% (682) CD56- 3.56% (48) |
Not done | SCID |
| Pt. 128 | 4 months/female | Recurrent pneumonia, persistent diarrhoea, otitis media, cellulitis | BAL- Adenovirus | 0.160 | IgG- 7.30 g/L IgA- 0.34 g/L IgM- 0.92 g/L |
CD3-0% CD19-30% (75) CD56- 42% (103) |
Not done | SCID |
| Pt. 129 | 8 months/male | Recurrent pneumonia, persistent diarrhoea, otitis media, septicemia | Blood- S. aureus, P. aeruginosa | 0.340 | IgG- 3.30 g/L IgA- 0.24 g/L IgM- 0.17 g/L |
CD3-2.0% (6) CD19-93% (310) CD56- 4% (18) |
JAK3 | SCID |
| Pt. 130 | 22 months/male | Recurrent pneumonia, persistent diarrhoea, septicemia | Blood- Streptococcus pneumoniae | 0.357 | IgG- 18.80 g/L IgA- 1.62 g/L IgM- 0.85 g/L |
CD3-5% (18) CD19-27% (97) CD56- 72% (222) |
Not done | SCID |
| Pt. 131 | 60 months/female | Recurrent pneumonia, persistent diarrhoea, septicemia, microcephaly | Nil | 0.760 | IgG- 13.80g/L IgA- 0.34 g/L IgM- 1.20 g/L |
CD3-4.0% (24) CD19-92.0% (696) CD56- 3% (18) |
Not done | SCID |
| Pt. 132 | 4 months/male | Recurrent pneumonia, persistent diarrhoea | Nil | 1.378 | IgG- 9.60 g/L IgA- 0.22 g/L IgM- 0.55 g/L |
CD3-4.0% (44) CD19-91% (986) CD56- 5.3% (58) |
Not done | SCID |
| Pt. 133 | 30 months/male | Recurrent pneumonia, persistent diarrhoea, otitis media, septicemia | CMV | 0.357 | IgG- 10.70 g/L IgA- 0.30 g/L IgM- 0.79 g/L |
CD3-5.0% (17.5) CD19-12% (93) CD56- 34.6% (124) |
Not done | SCID |
| Pt. 134 | 8 months/male | Recurrent pneumonia, otitis media, septicemia | Nil | 3.485 | IgG- 6.80 g/L IgA- 0.31 g/L IgM- 0.43 g/L |
CD3-1.0% (6) CD19-82% (2830) CD56- 18% (654) |
Not done | SCID |
| Pt. 135 | 5 months/male | Recurrent pneumonia, persistent diarrhoea, septicemia | Candida sp. | 3.240 | IgG- 2.80 g/L IgA- 0.18 g/L IgM- 0.26 g/L |
CD3-12% (388) CD19-0% CD56- 86% (2786) |
Not done | SCID |
| Pt. 136 | 6 month/male | Two elder male sibling death at early infancy | Nil | 0.300 | N.A. | CD3-0.7% (1) CD19-97.6% (290) CD56-0.4% (1) |
IL2RG | SCID |
| Pt. 137 | 6 months/male | Recurrent pneumonia, septicemia, eczematoid rash | Candida sp. | 2.436 | IgG- 2.70 g/L IgA- 0.35 g/L IgM- 0.36 g/L IgE- 24,200 U/L |
CD3-66% (1610) CD19-26% (634) CD56-8% (195) CD3+45RO+ - 97.5% (elevated) |
CD3D | Omenn syndrome |
| Pt. 138 | 2 months/female | Recurrent pneumonia, cellulitis, OS, abscess | Candida sp. | 32.600 | IgG- <0.33 g/L IgA- <0.06 g/L IgM- <0.04 g/L |
CD3-87% (28,362) CD19-0% CD56-7.6% (2478) |
Not done | Omenn syndrome |
| Pt. 139 | 1 months/female | Cellulitis, rash | N.A | 1.230 | IgG- 9.40 g/L IgA- <0.25 g/L IgM- N.A |
CD3-1% (12) CD19-N.A. CD56-N.A. |
Not done | SCID |
| Pt. 140 | 36 months/male | Recurrent pneumonia, persistent diarrhoea | Clostridium difficle, CMV | 3.024 | IgG- 12.90 g/L IgA- 1.53 g/L IgM- 0.56 g/L |
CD3-56% (1680) CD19-1.4% (42) CD56-26% (780) |
RAG1 | Atypical SCID |
| Pt. 141 | 6 months/female | Recurrent pneumonia, septicemia | Candida sp., Staphylococcus sp. | 2.405 | IgG- <0.75 g/L IgA- 0.24 g/L IgM- N.A |
CD3-0.6% (14) CD19-62.4% (1504) CD56-22.9% (552) |
Not done | SCID |
| Pt. 142 | 8 months/male | Recurrent pneumonia, persistent diarrhoea, septicemia | candida | 2.075 | IgG- 0.09 g/L IgA- <0.26 g/L IgM- <0.16 g/L |
CD3-1% (21) CD19-93% (1934) CD56-0.2% (4) |
Not done | SCID |
| Pt. 143 | 8 months/male | Recurrent pneumonia | Nil | 2.650 | IgG- 7.62 g/L IgA- 0.25 g/L IgM- 0.64 g/L |
CD3-18.36% (488) CD19-5% (133) CD56- N.A. |
Not done | SCID |
| Pt. 144 | 7 months/female | Recurrent pneumonia, persistent diarrhoea, BCG site ulceration | Nil | 1.090 | IgG- 0.10 g/L IgA- 0.02 g/L IgM- N.A |
CD3- 1.1% (12) CD19- 0% CD56- 22% (231) |
Not done | SCID |
| Pt. 145 | 5 months/male | Recurrent pneumonia, | Nil | 0.060 | IgG- 1.08 g/L IgA- 0.10 g/L IgM- 0.14 g/L |
CD3-0.01% (1) CD19-NA (151) CD56- 62.33% (206) |
Not done | SCID |
| Pt. 146 | 7 months/male | Recurrent pneumonia, | Nil | 4.200 | N.A | CD3-18.31% (838) CD19-51.69% (2682) CD56- 15.9% (826) |
Not done | SCID |
| Pt. 147 | 9 months/female | Recurrent pneumonia, Septicemia, disseminated BCGosis | E. coli, M. bovis | 0.994 | IgG- 0.06 g/L IgA- 0.26 g/L IgM- 0.30 g/L |
CD3-1.53% (10) CD19-84.69% (692) CD56- 2.76% (23) |
Not done | SCID |
| Pt. 148 | 16 months/male | Recurrent pneumonia, Septicemia | Candida sp. | 1.316 | IgG- 12.20 g/L IgA- 1.08 g/L IgM- 6.54 g/L |
CD3-0.54% (2) CD19-0.64% (3) CD56- 10.93% (46) |
Not done | SCID |
| Pt. 149 | 5 months/male | Recurrent pneumonia, persistent diarrhea, BCG site ulceration | Nil | 1.5 | IgG- 0.02 g/L IgA- 0.37 g/L IgM- 0.21 g/L |
CD3- 0.1% (2) CD19- 0% CD56- 95% (1425) |
Not done | SCID |
| Pt. 150 | 7 months/male | Recurrent pneumonia, | BAL – M. tuberculosis, Pseudomonas sp. | 3.000 | IgG- <2.0 g/L IgA- 0.10 g/L IgM- 0.90 g/L |
CD3-0.20% (6) CD19-70% (2100) CD56- 36% (1080) |
CD3E | SCID |
| Pt. 151 | 6 months/male | Recurrent pneumonia, oral thrush | Enterococcal sepsis | 0.6 | IgG- 1.24 g/L IgA- <0.01 g/L IgM- <0.01 g/L |
CD3- 47.2% (283) CD19- 0.1% (1) CD56- 46% (276) |
RAG1 | SCID |
| Pt. 152 | 5 months/male | Recurrent pneumonia, persistent diarrhoea | Nil | 1.099 | IgG- <0.75 g/L IgA- <0.10 g/L IgM- 0.35 g/L |
CD3-0% CD19-93% (1015) CD56- 2% (23) |
IL2RG | SCID |
| Pt. 153 | 2 months/male | Recurrent pneumonia, septicemia | Candida sp. (blood) | 0.080 | IgG- 1.90 g/L IgA- <0.05 g/L IgM- <0.05 g/L |
CD3-10.4% (2.4) CD19-5.6 (1.29) CD56- 64% (14.84) |
ADA | SCID |
| Pt. 154 | 3 months/male | Acute fever, cough | Nil | 0.323 | IgG- <1.46 g/L IgA- <0.24 g/L IgM- 0.97 g/L |
CD3-26% (84) CD19-65% (202) CD56-40% (129) |
ADA | SCID |
| Pt. 155 | 7 months/male | Persistent diarrhoea | Nil | 2.538 | IgG-1.59 g/L IgA- <0.24 g/L IgM- <0.17 g/L |
CD3-0% (0) CD19-86% (2183) CD56%-11% (279) |
IL7R | SCID |
| Pt. 156 | 21 months/male | Meningoencephalitis, right chorioretinitis, left vitreal hemorrhage | CMV | 0.508 | N.A. | CD3-5% (25) CD19-12% (61) CD56-62% (315) |
PNP | SCID |
| Pt. 157 | 5 months/male | Pneumonia | Citrobacter sp. | 1.520 | IgG- <1.34 g/L IgA- <0.28 g/L IgM- 0.25 g/L |
CD3-0% (0) CD19-0% (0) CD56-96% (146) |
RAG1 | SCID |
| Pt. 158 | 24 months/male | Recurrent pneumonia, diarrhoea, meningoencephalitis | E. coli | 16.624 | N.A. | CD3-85% (14130) CD4- 7% (1164) CD8- 70% (11637) CD19-10% (1662) CD56- 4% (665) HLA-DR expression on B cells- 0% |
RFXANK | CID |
| Pt. 159 | 5 months/male | Pneumonia, diarrhoea, rash | S. epidermidis | 0.320 | N.A. | CD3-1% (1) CD19-32% (102) CD56-14% (45) |
ADA | SCID |
| Pt. 160 | 5 months, female | Pneumonia | P. jirovecii, H1N1 | 1.967 | N.A. | CD3-2% (39) CD19-0% (0) CD56-95% (1869) |
Not done | SCID |
| Pt. 161 | 3 months, female | Pneumonia, diarrhoea | Nil | 0.203 | N.A. | CD3-0% (0) CD19-0% (0) CD56- 82% (166) |
Not done | SCID |
| Pt. 162 | 12 months/male | Recurrent diarrhoea, left empyema | Nil | 1.958 | IgG- 15.7 g/L IgA- 3.94 g/L IgM- 2.13 g/L |
CD3-43% (842) CD4- 2% (39) CD8- 30% (387) CD19-15% (294) CD56-38% (744) |
Not done | SCID |
| Pt. 163 | 7 months, male | Pneumonia, global developmental delay | M. tuberculosis | 0.979 | NA | CD3-7% (69) CD19-86% (842) CD56-3% (29) |
Not done | SCID |
| Pt. 164 | 2 months, male | Scaly erythrodermic rash (OS) | Nil | 3.854 | IgG-1.89 g/L IgA-0.28 g/L IgM-2.08 g/L |
CD3-55% (2120) CD4- 33% (1272) CD8- 11% (424) CD4+ 45RA+ - 3% (decreased) CD19-18% (694) CD56-25% (964) |
Not done | Omenn syndrome |
| Pt. 165 | 12 months, male | Abscesses in lung, liver, oral thrush | Nil | 0.548 | IgG-5.63 g/L IgA- <0.70 g/L IgM- <1.07 g/L |
CD3-42% (230) CD19-18% (694) CD56-20% (110) |
Not done | SCID |
| Pt. 166 | 2 months, male | Recurrent pneumonia, rash | Acinetobacter sp. | 1.620 | N.A. | CD3-68% (1102) CD4- 12% (194) CD8- 36% (583) CD4+ 45RA+ - 0% CD19-21% (340) CD56-6% (97) |
Not done | Omenn syndrome |
| Pt. 167 | 10 months, male | Chronic fever, pneumonia, hepatomegaly, pancytopenia | Nil | 0.954 | IgG- <1.34 g/L IgA- <0.28 g/L IgM- <0.17 g/L |
CD3-24% (229) CD19-63% (601) CD56-7% (67) |
Not done | SCID |
| Pt.168 | 12 months, male | N.A. | N.A. | 3.080 | IgG- <0.29 g/L IgA- 0.64 g/L IgM- 0.34 g/L |
CD3- 4.5% (138) CD19- 71.3% (2197) CD56- 1.9% (60) |
IL2RG | SCID |
| Pt.169 | 60 months, male | NA | NA | 0.870 | IgG- <0.10 g/L IgA- <0.001 g/L IgM- <0.01 g/L |
CD3- 0.5% (6) CD19- 89.7% (1076) CD56- 7.8% (94) |
IL2RG | SCID |
| Pt.170 | 7 months, male | NA | NA | 0.330 | IgG- 0.06 g/L IgA- 0.001 g/L IgM- 0.002 g/L |
CD3- 7% (23) CD19- 0.3% (1) CD56- 80.9% (267) |
RAG1 | SCID |
| Pt.171 | 48 months, male | NA | NA | 1.160 | IgG-0.59 g/L IgA-0.005 g/L IgM-0.007 g/L |
CD3- 31.6% (367) CD19- 35.3% (410) CD56- 31.4% (364) |
RAG1 | SCID |
| Pt.172 | 12 months, female | NA | NA | 7.220 | IgG-2.36 g/L IgA-0.002 g/L IgM-0.01 g/L |
CD3- 0.3% (22) CD19- 0.7% (54) CD56- 56% (4044) |
RAG2 | SCID |
| Pt.173 | 48 months, male | Otitis media, recurrent pneumonia since early infancy | NA | 1.800 | IgG-1.16 g/L IgA-0.008 g/L IgM-0.006 g/L |
CD3- 9.4% (169) CD19- 58.3% (1049) CD56- 22.5% (406) |
DOCK2 | CID |
| Pt.174 | 4 months, female | Chronic diarrhoea, pneumonia, failure to thrive, absent thymus | E. coli, Cryptosporidium | 1.63 | IgG- 5.35 g/L IgA- 0.31 g/L IgM- 1.82 g/L |
CD3- 89.7% (1462) CD19- 1.2% (19) CD56- 4% (65) |
Not done | Possible SCID*** |
| Pt.175 | 30 months, male | Recurrent pneumonia, diarrhoea, failure to thrive | Nil | 0.35 | IgG-7.02 g/L IgA-1.31 g/L IgM- 0.82 g/L |
CD3- 56.3% (197) CD19- 0.8% (3) CD56- 39.4% (138) |
Not done | SCID |
| Pt.176 | 9 months, male | Chronic diarrhoea, pneumonia, failure to thrive | P. aeruginosa, Candida sp. | 0.60 | IgG- 0.99 g/L IgA-0.7 g/L IgM-0.4 g/L |
CD3- 35.8% (215) CD19- 6.7% (40) CD56- 47.8% (287) |
Not done | SCID |
| Pt.177 | 14 months, female | Recurrent pneumonia, diarrhoea, failure to thrive | Nil | 2.63 | IgG-1.46 g/L IgA- <0.25 g/L IgM- <0.18 g/L |
CD3- 74% (1947) CD4- 14% (368) CD8- 34% (895) CD19- 2% (53) CD56- 23% (605) |
Not done | CID |
| Pt.178 | 3 months, male | Recurrent pneumonia, fungal skin infection, 2 early sibling death | P. aeruginosa, Streptococcus sp. | 2.12 | IgG-2.52 g/L IgA- <0.25 g/L IgM- 1.12 g/L |
CD3- 57.3% (1215) CD4- 0.5% (11) CD8- 56.3% (1194) CD19- 38.2% (810) CD56- 1% (21) |
Not done | SCID |
| Pt.179 | 7 months, male | Pneumonia, scaly erythrodermic rash | Nil | 0.42 | IgG- 0.18 g/L IgA-0.52 g/L IgM-0.42 g/L |
CD3- 83.3% (350) CD19- 1% (4) CD56- 1% (4) |
Not done | Omenn syndrome |
| Pt.180 | 14 months, male | Recurrent pneumonia, eczematoid rash, failure to thrive | Cryptosporidium | 2.19 | IgG- 2.16 g/L IgA-1.21 g/L IgM-1.22 g/L |
CD3- 41% (897) CD4- 4% (88) CD8- 17% (372) CD19- 1% (22) CD56- 17% (372) |
Not done | CID |
| Pt.181 | 6 months, male | Chronic diarrhoea, failure to thrive, septicemia | E. coli, Candida sp. | 1.37 | IgG-9.52 g/L IgA-1.79 g/L IgM-0.26 g/L |
CD3- 87.1% (1194) CD4- 34% (466) CD8- 53.1% (727) CD19- 0.2% (3) CD56- 12% (165) |
Not done | SCID |
| Pt.182 | 3 months, male | Meningitis, pneumonia, oral thrush, early sibling death | P. aeruginosa | 3.55 | N.A. | CD3- 35% (1244) CD4- 10% (355) CD8- 15% (533) CD19- 0.1% (4) CD56- 56% (1990) |
Not done | SCID |
| Pt.183 | 16 months, male | Pneumonia, eczematoid rash, Varicella infection, early sibling death due to pneumonia | Acinetobacter sp., Pseudomonas sp. | 2.55 | N.A. | CD3- 46% (1174) CD4- 14% (357) CD8- 33% (842) CD19- 4% (102) CD56- 50% (1276) |
Not done | CID |
| Pt.184 | 3 months, female | Pneumonia, abdominal distension, diarrhoea, failure to thrive | Nil | 1.80 | IgG-1.63 g/ IgA- <0.06 g/L IgM- <0.16 g/L |
CD3- 35% (630) CD19- 0.8% (14) CD56- 63% (1134) |
Not done | SCID |
| Pt.185 | 3 months, male | Pneumonia, failure to thrive | M. tuberculosis | 0.50 | IgG-9.03 g/L IgA- 0.39 g/L IgM-2.23 g/L |
CD3- 45% (315) CD19- 50% (350) CD56- 1.4% (10) |
Not done | SCID |
| Pt.186 | 9 months, male | Persistent diarrhoea, pneumonia, left forearm abscess | Nil | 2.43 | IgG-4.0 g/L IgA-0.74 g/L IgM- 1.1 g/L |
CD3- 43.9% (1068) CD3+CD4+- 26% (631) CD3+CD8+- 14% (340) CD19- 54.9% (1335) CD56- 1% (24) |
Not done | Possible SCID*** |
| Pt.187 | 4 months, male | Developmental delay, pneumonia, diarrhoea, failure to thrive, 1 early sibling death | E. coli | 1.50 | IgG-2.95 g/L IgA-0.07 g/L IgM- 1.04 g/L |
CD3- 44.9% (674) CD19- 44.9% (674) CD56- 10% (150) |
Not done | Possible SCID*** |
| Pt.188 | 3 months, female | Otitis media, oral thrush, failure to thrive | Nil | 1.86 | IgG-2.95 g/L IgA-0.07 g/L IgM- 1.04 g/L |
CD3- 9% (167) CD19- 0.5% (9) CD56- 87.8% (1633) |
Not done | SCID |
| Pt.189 | 96 months, female | Recurrent pneumonia, ear discharge, failure to thrive | Nil | 1.21 | IgG-2.14 g/L IgA- 7.05 g/L IgM-1.54 g/L |
CD3- 39% (473) CD19- 16% (194) CD56- 41.2% (498) |
Not done | CID |
| Pt.190 | 24 months, female | Ear discharge, diarrhoea, scaly rash (Omenn phenotype) | Nil | 8.75 | N.A. | CD3- 80% (7003) CD4- 5% (438) CD8- 30% (2626) CD19- 2% (175) CD56- 14% (1226) |
Not done | CID |
| Pt.191 | 1 month, female | Septicemia, 3 early siblings died at early infancy | Nil | 2.89 | N.A. | CD3- 63.9% (1847) CD4- 55.9% (1616) CD8- 8% (231) CD19- 18% (520) CD56- 15% (433) |
Not done | SCID |
| Pt.192 | 6 months, male | Multiple hypodense lesions in liver and spleen, necrotic retroperitoneal lymph nodes | Nil | 0.01 | N.A. | CD- 0 CD19- 0 CD56- 0 |
Not done | SCID |
| Pt.193 | 7 months, male | Recurrent pneumonia, diarrhoea, early sibling death due to disseminated BCGosis | Acid-fast bacilli, Candida sp. (BAL) | 2.15 | N.A. | CD3- 0% CD19- 98.9% (2128) CD56- 0.3% (6) |
Not done | SCID |
| Pt.194 | 2.5 months, male | Diarrhoea, ear discharge, pneumonia, dermatitis, knee joint swelling, axilla abscess, 1 elder sibling expired due to SCID | Blood, pus: S. aureus (Methicillin sensitive) | 2.85 | N.A. | CD3- 57% (1624) CD4- 17% (484) CD8- 31% (883) CD19- 0.3% (9) CD56- 40% (1140) |
Not done | SCID |
| Pt.195 | NA, male | Pneumonia, otitis media, septicemia | Pseudomonas sp. | 1.51 | IgG-4.0 g/L IgA-0.52 g/L IgM-0.32 g/L |
CD3- 0.3% (5) CD19- 0 CD56- 4% (62) |
Not done | SCID |
| Pt.196 | 3 months, male | Pneumonia, oral thrush | Nil | 0.01 | N.A. | N.A. | ADA | SCID |
| Pt.197 | 3 months, male | Recurrent pneumonia, 1 early sibling death | Nil | 2.86 | N.A. | N.A. | IL2RG | SCID |
| Pt.198 | 2 months, female | Pneumonia, colitis | Nil | 4.40 | IgG-0.89 g/L IgA- <0.24 g/L IgM- <0.17 g/L |
CD3- 0.5% (22) CD19- 87.3% (3839) CD56- 2% (88) |
JAK3 | SCID |
| Pt.199 | 1.5 months, female | Pneumonia, oral thrush, 2 elder female siblings died at early infancy | Nil | 1.50 | N.A. | CD3- 0 CD19- 63.1% (947) CD56- 34.7% (521) |
Not done | SCID |
| Pt.200 | 8 months, male | Recurrent pneumonia, BCGosis | Nil | N.A. | N.A. | CD3- 0% CD19- 64% (443) CD56- 31% (214) |
IL7RA | SCID |
| Pt.201 | 8 months, female | Recurrent pneumonia, oral thrush, BCGosis | Nil | 0.55 | IgG- <0.06 g/L IgA- <0.24 g/L IgM- <0.17 g/L |
CD3- 0 CD19- 16.7% (92) CD56- 65.3% (359) |
Not done | SCID |
| Pt.202 | 7 months, female | Recurrent pneumonia, diarrhoea | CMV viremia, Candida sp. | 2.48 | IgG-0.97 g/L IgA-1.82 g/L |
CD3- 70.8% (1755) CD4- 2.6% (65) CD8- 59% (1463) CD19- 30.2% (748) CD56- 22.3% (553) |
Not done | SCID |
| Pt.203 | 24 months, male | Recurrent pneumonia, otitis media | S. aureus (Methicillin resistant) | 1.60 | IgG-1.61 g/L IgA-0.29 g/L IgM-0.29 g/L |
CD3- 22% (352) CD19- 58% (928) CD56- 13% (208) |
Not done | SCID |
| Pt.204 | 24 months, female | Chronic diarrhoea, pneumonia | Corona virus 229E, Alpha hemolytic streptococci (blood), esophageal candidiasis | 1.16 | IgG-1.14 g/L IgA-0.15 g/L IgM-0.27 g/L |
CD3- 23.4% (272) CD19- 9.1% (105) CD56- 42.3% (491) |
RAG1 | SCID |
| Pt.205 | 192 months, male | Recurrent pneumonia, varicella infection, madarosis, Hodgkin lymphoma | Epstein Barr viremia | N.A. | N.A. | N.A. | RAG1 | Atypical SCID |
| Pt.206 | 5 months, male | Recurrent pneumonia, diarrhoea, elder male sibling died in early infancy, 4 maternal uncles expired < 6 months age | Adenovirus | N.A. | N.A. | NA; CD132 expression very low in monocytes (0.2%) compared to normal expression in controls | Not done | SCID |
| Pt.207 | 5 months, male | Pneumonia, diarrhoea, ear discharge, oral thrush, rash, early sibling death | VAPP in stool, Enterovirus, Klebsiella (BAL), CSF- Enterovirus, Mycoplasma | 0.39 | IgG- <1.46 g/L IgA- <0.28 g/L IgM- 0.17 g/L |
CD3- 28% (109) CD19- 1% (4) CD56- 68% (265) |
RAG2 | SCID |
| Pt.208 | 20 days, male | Pneumonia, diarrhoea, rash, renal abscess | Corona OC43, Rhinovirus | 0.25 | N.A. | CD3- 19% (47) CD19- 0 CD56- 24.4% (61) |
ADA | SCID |
| Pt.209 | 5 months, female | Chest wall abscess, recurrent pneumonia, oral thrush, diarrhoea | P. jirovecii, Rotavirus (stool), Mycoplasma (nasopharyngeal aspirate) | 0.97 | IgG- <1.46 g/L IgA- <0.17 g/L IgM- <0.28 g/L |
CD3- 1.3% (13) CD19- 0 CD56- 60% (581) |
RAG2 | SCID |
| Pt.210 | 6 months, male | Recurrent pneumonia, diarrhoea, scalp abscess, 1 male sibling death | CMV, Rhinovirus, Enterovirus | N.A. | IgG-0.26 g/L IgA-0.02 g/L IgM- 1.70 g/L |
N.A. | CIITA | CID |
| Pt.211 | 84 months, female | Recurrent diarrhoea, oral ulcer, pneumonia, colitis | Nil | 0.84 | IgG-4.97 g/L IgA- <0.67 g/L IgM-1.7 g/L |
CD3- 77% (649) CD19- 15.5% (130) CD56- 3% (28) HLA-DR expression in B cells- 0% |
RFX5 | CID |
| Pt.212 | 18 months, male | Recurrent pneumonia, diarrhoea, failure to thrive | VDPV, M. tuberculosis, Cryptosporidium, Enterobacter sp. (blood) | 3.75 | IgG- <1.41 g/L IgA- <0.24 g/L IgM-0.20 g/L |
CD3- 53.04% (1989) CD4- 22% (826) CD19- 4% (150) CD56- 42% (1576) |
Not done | CID |
| Pt.213 | 132 months, female | Recurrent pneumonia, diarrhoea, oral thrush, otitis media, meningitis | Hemophilus influenzae (CSF) | 2.94 | IgG-0.22 g/L IgA- <0.24 g/L IgM-0.44 g/L |
CD3- 34.7% (1022) CD4- 16.7% (490) CD8- 13.7% (405) CD19- 34% (1001) CD56- 2.2% (64) |
Not done | CID |
| Pt.214 | 4 months, male | Failure to thrive, recurrent pneumonia, diarrhoea | Nil | 1.22 | NA | NA | JAK3 | SCID |
| Pt.215 | 7 months, male | Otitis media, septicemia | Staphylococcus aureus | 6.23 | IgG- <0.3 g/L IgA- <0.05 g/L IgM- 0.11 g/L |
NA | IL2RG | SCID |
| Pt.216 | 8 months, male | Pneumonia, diarrhoea, rash | Nil | 5.02 | IgG- <0.11 g/L IgA- <0.05 g/L IgM- <0.11 g/L |
NA | IL2RG | SCID |
| Pt.217 | 1 month, male | Failure to thrive, persistent diarrhea, perianal rash | Nil | 0.97 | IgG- 0.42 g/L IgA- 0.06 g/L IgM- 0.59 g/L |
CD3- 4% (39) CD19- 39% (378) CD56- 54% (524) |
Not done | SCID |
| Pt.218 | 2 months, female | Recurrent episodes of pneumonia and diarrhoea, failure to thrive, doing well after HSCT | Nil | NA | NA | CD3- 3476 (Very low CD4 counts with CD4/CD8 reversal) CD19- 1765 CD56- 156 |
Probable MHC Class 2 defect | CID |
| Pt.219 | 1 month, female | Recurrent episodes of pneumonia and diarrhoea | Nil | NA | NA | NA | IL7R | SCID |
| Pt.220 | 1 month, male | Recurrent episodes of diarrhoea and failure to thrive | Nil | NA | NA | NA | IL2RG | SCID |
ESID, European Society for Immunodeficiencies; CMV, Cytomegalovirus; BCG, Bacillus Calmette-Guerin; BAL, Bronchoalveolar lavage; CSF, Cerebrospinal fluid; OS, Omenn syndrome; PJP, Pneumocystis jirovecii pneumonia; EBV, Epstein-Barr virus; VDPV, Vaccine-derived polio virus; VZV, Varicella zoster virus; AIHA, Autoimmune hemolytic anemia; VAPP, Vaccine-associated paralytic polio; CID, Combined Immune Deficiency.
Clinical details of patients 221-277 are previously reported (7).
***Possible SCID is categorized if patients did not fulfil the complete ESID definition, however, the treating team had a high index of suspicion based on clinical and immunological features.
Clinical profile of all patients was obtained along with family history and other demographic details. Clinical features included number of infections, type of infections, site of infections, organism involved, age of presentation, age of onset, presence of skin rash, BCG ulceration, history of administration of vaccines and complications, if any. Basic hematological, biochemistry, and immunological investigations including immunoglobulin profile and lymphocyte subsets were also recorded.
Analysis of lymphocyte subsets by flow cytometry had been carried out in most patients. Methodology for laboratory assay of lymphocyte subsets, naïve, memory T cells, HLA-DR expression, CD132 expression, CD127 expression, and lymphocyte proliferation assays at Post Graduate Institute of Medical Education and Research (PGIMER), Chandigarh and National Institute of Immunohematology (NIIH), Mumbai have been previously described (11, 12). Other centers performed conventional lymphocyte subsets (CD3, CD19, CD4, CD8, CD56) by flow cytometry in private laboratories.
Adenosine deaminase (ADA) levels and percentage of deoxyadenosine nucleotides (%dAXP) from dried blood filter paper spot were assayed at Duke University, North Carolina for patients with ADA deficiency SCID who were diagnosed at PGIMER, Chandigarh.
Molecular Assays
Before the facility for in-house next-generation sequencing was made available in 2018, centre at PGIMER, Chandigarh had established academic collaborations with centers at Hong Kong (The University of Hong Kong), Japan (Kazusa DNA Research Institute, Kisarazu, Chiba; National Defense Medical College, Saitama), and USA (Duke University, North Carolina) for molecular work-up of patients. The centre at Hong Kong provided final molecular diagnosis for 12 patients (Pt. 8-10, Pt. 14-19, Pt. 21, Pt. 50-51) ( Table 1 ). Molecular diagnosis for 4 patients was established at Kazusa DNA Research Institute, Japan (Pt. 3–6). Thirty-four (34) patients (Pt. 59–90, pt. 119, pt. 127) with SCID were worked-up for molecular diagnosis using NGS at National Defense Medical College, Saitama and Tokyo Medical and Dental University, Tokyo, Japan (Kato T et al. manuscript in submission). Final molecular diagnosis of a patient with ADA defect (pt. 22) was also established at Duke University, North Carolina.
Sanger sequencing for IL2RG and RAG1/2 genes were initiated at PGIMER, Chandigarh (North India) in 2016. Sanger sequencing for patients with SCID at NIIH, Mumbai (West India) was previously described by Aluri et al. (7). Methodology for NGS at Christian Medical College, Vellore (South India) was described previously (13).
Next-Generation Sequencing (NGS) at PGIMER, Chandigarh
Next-generation sequencing (Ion Torrent, Thermo Fisher Scientific India Pvt. Ltd.) for clinical care was started in July 2018 at the Advanced Pediatrics Centre, PGIMER, Chandigarh. A targeted PID gene panel comprising 44 genes was used that covered 6 genes for SCID—ADA, RAG1, RAG2, IL2RG, IL7RA, and LIG4. Preparation of DNA target amplification reaction using 2-primer pools, amplification of target, combination of target amplification reactions, ligating adaptors to the amplicons and their purification was carried out as per the manufacturer’s protocol using Ion AmpliSeq™ Library kit plus (Catalog numbers 4488990, A35121 A31133, A31136, A29751, 4479790). Amplified library was quantified using Qubit™ 2.0 fluorometer instrument. Dilution that results in a concentration of ~100pm was then determined. Template preparation on Ion One Touch™ Instrument, recovery, washing and enrichment of template-positive ISPs was done as per the manufacturer’s protocol using Ion 520™ and Ion 530™ Kit-OT2 (catalog number A27751). Ion S5™ sequencer instrument was then initialized. Annealing of primers to enriched ISPs and chip loading was carried out using Ion 520 and 530 Loading Reagents OT2 Kit. Sequencing run was initiated and Torrent Browser was used to review results. Raw data were analyzed on Ion Reporter software and on integrative genome viewer.
NGS using a targeted gene panel was also performed for some patients (n = 6) in private laboratories (Medgenome Labs Pvt. Ltd., India).
NGS at Other Centers
Other centers in India obtained molecular testing results from private laboratories (Medgenome Labs Pvt. Ltd., India; Strand Genomics Pvt. Ltd., India; Neuberg Anand Diagnostics Pvt. Ltd., India). Illumina platform was used for sequencing in private laboratories with coverage of >80X. Sanger sequencing was used to confirm variants obtained by NGS.
Multiplex Ligation Probe Amplification (MLPA) Assay for DCLERC1 Exon 1-3 Deletion at PGIMER, Chandigarh
SALSA MLPA probe-mix P368 DCLRE1C kit was used in this protocol. MLPA was performed according to the instructions provided by the manufacturer (MRC Holland). 50–100ng/µL of DNA was denatured in thermocycler and hybridized with 1.5 µL of probe-mix along with 1.5µL of MLPA buffer. Content was mixed and incubated for 1 min at 95°C followed by incubation at 60°C for 18 h. After hybridization, probes were ligated using a ligase mix at 54°C for 15 min. Ligase was inactivated at 98°C for 5 min. PCR was performed using PCR primers, polymerase, buffers and required amount of water. Following conditions were used for amplifications—95°C for 20 s, 65°C for 80 s, for 35 cycles, followed by a final extension for 20 min at 72°C. ABI 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) was used for capillary electrophoresis. Later, 0.7µL of PCR reaction, 8.9µL of HI-DI formamide, and 0.4µL of DNA standard LIZ 600 provided by GeneScan were mixed and then denatured for 2 min at 95°C. The sample was then loaded and MLPA data were analyzed using a Coffalyser software.
Results
Current study included data of patients diagnosed and managed at centers in Northern, Southern, and Western parts of India. Amongst the 277 patients, 254 were categorized as SCID (208 – SCID; 17 – atypical SCID; 26 – OS; 3 – possible SCID) and 23 as CID ( Table 1 ). A steady increase in number of diagnosed cases was noted over last 10 years. The unit at PGIMER, Chandigarh (North India) diagnosed its first case of SCID in year 2001. Only 14 cases of SCID were identified until 2011 and an exponential rise in number of cases was noted after 2011 ( Figure 1 ). Rise in number of cases over years paralleled the expansion of available manpower resources and laboratory facilities for pediatric immunology at Advanced Pediatrics Centre, PGIMER (North India). Ninety (90) children (Pt. 1-90) with SCID have been diagnosed at PGIMER, Chandigarh until date. Fifty-eight (58) and 27 cases of SCID were enrolled from Bai Jerbai Wadia Children’s Hospital, Mumbai (West India) and Aster CMI, Bengaluru (South India), respectively.
Figure 1.
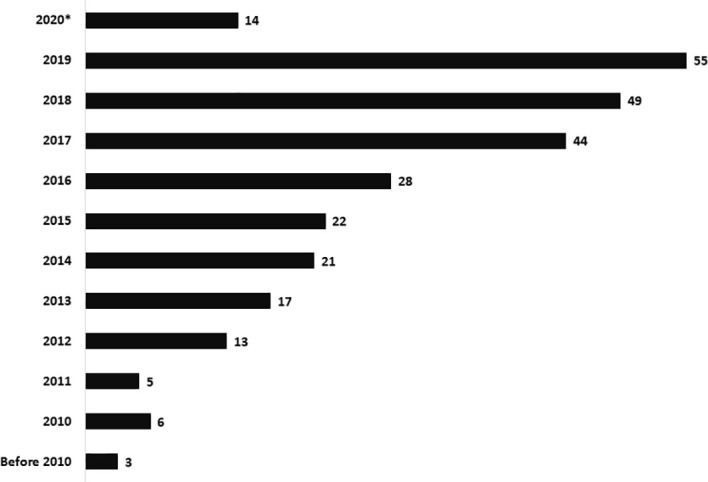
Bar graph depicting the rise in number of cases diagnosed over last 10 years.
Male-female ratio was 196:81 ( Table 1 ). Median [inter-quartile range (IQR)] age of onset of clinical symptoms and diagnosis was 2.5 months (1, 5) and 5 months (3.5, 8), respectively. Consanguinity was noted in 78 families (28.2%), and was noticeably more in Southern region (32.3%) of our country compared to Northern (22.4%). Family history of early childhood deaths was noted in 120 children (43.3%). Median (IQR) age at diagnosis in children who had a positive family history was 4.5 months (3, 6) compared to 6 months (4, 9) in children who did not have a family history, p<0.05 (Mann-Whitney U test).
Opportunistic infections were the presenting manifestation in most patients. These included pneumonia (82%), diarrhoea (43.7%), oral thrush (18.4%), BCG site ulceration (17%), otitis media (12.6%), and meningitis (4%) ( Figures 2 , 3 ). Blood-culture proven septicemia was seen in 63 children (23%)—Candida sp. (16), Staphylococcus sp. (10), Escherichia coli (5), Acinetobacter sp. (5), Pseudomonas aeruginosa (8), Klebsiella pneumoniae (5), Enterococcus sp. (3), Enterobacter sp. (2), Streptococcus sp. (1), Pichia fermentans (1), Burkholderia cepacia (1), Chryseobacterium sp. (1), Bacillus subtilis (1), Citrobacter sp. (1), Moraxella sp. (1), Alcaligens faecalis (1), and Weisella confusa (1). Bacteria isolated from respiratory tract included Mycobacterium bovis (15), Klebsiella pneumoniae (5), P. aeruginosa (4), M. tuberculosis (3), atypical mycobacterium (1), E. coli (1), Staphylococcus aureus (1), and Acinetobacter sp. (1). Microbiology proven disseminated BCG infection was noted in 27 patients (9.7%). Apart from oral thrush and candidemia, other fungal infections noted were pneumonia due to Pneumocystis jirovecii (8), invasive aspergillosis (5), esophageal candidiasis (5), and pulmonary cryptcoccosis (1). Disseminated cytomegalovirus (CMV) infection was documented in 23 (8.3%) children and 6 amongst these had evidence of CMV retinitis. Intestinal lymphangiectasia due to CMV was noted on autopsy of a child with X-linked SCID (pt.8). Prolonged excretion of vaccine-derived poliovirus was documented in a child with leaky SCID at Mumbai (14, 15). Vaccine-associated paralytic poliovirus strain was also isolated in a child with RAG1 defect at Mumbai. He had presented with persistent diarrhea, developmental delay, and hypotonia.
Figure 2.

Bar graph depicting the clinical manifestations and microbiological profile. (A) Clinical manifestations noted at first clinical presentation; (B–D) Microbiological profile of the organisms isolated—bacteria (B), fungi (C), and viruses (D).
Figure 3.

Clinical manifestations of children with SCID. (A, B) BCG site ulceration and pus discharge (Pt. 46 and 34); (C–F) Features of Omenn syndrome such as generalized erythema, scaling, loss of hair, and eyebrows (Pt. 34); (G, H) Chest radiograph of a child with ADA SCID showing radiological abnormalities—scapular spur and flattening of lower border of scapula (Pt. 39); (I) Chest radiograph of a child with CORO1A defect showing normal thymus shadow (Pt. 49).
Clinical features of OS were seen in 33 children (11.9%)—classical OS in 11 and incomplete OS in 22 ( Figure 3 ). Molecular defects associated with OS include RAG1 (7), RAG2 (5), ADA (2), NHEJ1 (1), IL2RG (1), JAK3 (1), STIM1 (1), CD3D (1), DCLRE1C (1), and RFXANK (1). Two children with IL2RG defect had features of engraftment of transplacental-acquired maternal T cells that mimicked clinical features of OS ( Figure 4 ). Warm autoimmune hemolytic anemia (AIHA) requiring immunosuppressive medications was observed in 5 children. While anemia responded to intravenous (IV) methylprednisolone pulses in 2 patients (RAG1 and NHEJ1 defect each), pt.42 with STK4 defect received IV rituximab (375 mg/m2 2 doses) for control of AIHA and she did not have further relapse of AIHA for next 1.5 years. Transfusion-associated graft-vs-host reaction was documented in 4 patients (2 X-linked SCID; 2 AR-SCID); all had development of rash and transaminitis following transfusion of non-irradiated blood products. Four (4) children had features of hemophagocytic lymphohistiocytosis (HLH). Possible triggers for HLH included disseminated BCG (2) and H1N1 (1) infections. The child with SP110 defect did not have any identifiable trigger for HLH (pt.104). Hodgkin lymphoma and intra-cranial B cell lymphoma were noted in children with RAG1 and CORO1A defects, respectively.
Figure 4.

Chimerism analysis using dual colour FISH probes targeting centromeres of X (DXZ1; green) and Y (DYZ1, orange) chromosomes in a male child suspected with transplacental-acquired maternal T cell engraftment (Pt. 44). (A) Immunomagnetically sorted CD19 positive cells (B cells) showing XY pattern in all cells while; (B) Immunomagnetically sorted CD3 positive cells showing XX pattern in two out of three cells suggesting maternal T cell engraftment. Inset shows XX pattern in a lymphocyte and XY pattern in neutrophils.
Four of 18 children with ADA defect were noted to have radiographic abnormalities—scapular spurring and flattening of lower end of scapula ( Figure 3 ). Glomerular involvement was seen in 4 children—3 children with OS and 1 with atypical/leaky SCID. Nephrotic range proteinuria was noted in 3 patients and one child (pt.13) had features of mesangial sclerosis on autopsy. Another child (pt. 12) with OS had features of focal segmental glomerulosclerosis on autopsy. One child (pt.10) with IL7RA defect had features of distal renal tubular acidosis and nephrocalcinosis. This patient had deletion of exons 2–5 of CAPSL along with exon 4–8 deletion of IL7RA in chromosome 5p13.2. A child with PNP defect (pt.14) had evidence of horse-shoe kidney at autopsy (16).
Median (IQR) absolute lymphocyte count (ALC) observed was 1.33 × 109/L (0.6, 2.5). Normal ALC (≥ 3 × 109/L) was observed in 51 children (18.4%)—of these 26 had OS, 2 had transplacental-acquired maternal T-cell engraftment, and 23 had leaky SCID/combined immunodeficiency. Eosinophilia was observed in 37 children, and 26 amongst these had features of OS. One child (Pt. 105) with RAG1 defect had unexplained monocytosis (2.7-3.0 × 109/L) that resolved after HSCT. Results of immunoglobulin profile was available for 198 children. Fifty-five (55) children had normal or elevated levels of IgM levels—30 in SCID (14.2%), 7 in atypical SCID (41.2%), 8 in OS (30.8%), and 10 in CID (43.5%). We observed elevated levels of IgE in 12 children—8 had OS, 1 had eczema and STK4 defect, and 3 had unexplained eosinophilia.
Immunophenotyping by flow cytometry showed the following distribution: T-B-NK- (32), T-B+NK- (67), T-B+NK+ (33), T-B-NK+ (84). T+ SCID is observed in 20 children with OS—T+B-NK+ (17), T+B-NK- (2), T+B+NK+ (1) and 2 children with transplacental-acquired maternal T-cell engraftment—T+B+NK- (1), T+B-NK- (1). Genetic defects observed under each category are summarized ( Supplementary Table 2 ). We observed decreased naïve (CD3+CD45RA+) and elevated memory (CD3+CD45RO+) CD3 lymphocytes in 24 children with OS. We noted elevated HLA-DR expression in CD3+ lymphocytes in 15 children with OS. CD132 expression by flow cytometry showed reduced expression in lymphocytes or monocytes in 8 children with suspected X-linked SCID ( Table 2 ) (17–19). Levels of ADA and %dAXP were measured in 7 children with ADA SCID and 2 heterozygous carriers of ADA mutation ( Table 3 ).
Table 2.
CD132 expression by flow cytometry in children with X-linked SCID.
| Patient | Molecular defect in IL2RG | Protein change | Type of mutation | Novel or previously reported | Clinical and Immunological phenotype | CD132 expression in case | CD132 expression in control | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lymphocyte | Monocyte | Neutrophils | Lymphocyte | Monocyte | Neutrophils | ||||||
| Pt. 25 | c.202G>T (hemizygous); Mother - heterozygous carrier | p.E68X | Nonsense | Previously reported (17) | X-linked family history (5 maternal uncles died at early infancy), 1 elder male sibling (pt. 8) died at early infancy. T-B+NK- SCID |
41.5% | 94.1% | 62.9% | 50.9% | 78.2% | 39% |
| Pt. 35 | c.170T>A (hemizygous); Mother -heterozygous carrier | p.L57H | Missense | Novel | Male child, T-B+NK- SCID, low CD132 expression | – | 12.2% | – | – | 87% | – |
| Pt. 40 | – | – | – | – | X-linked family history (6 maternal uncles died at early infancy), 2 elder male siblings died at early infancy. T-B+NK- SCID; low CD132 expression |
30.4% | 60.3% | 22.1% | 84.1% | 87.6% | 30.5% |
| Pt. 43 | c.455T>C (hemizygous); Mother -heterozygous carrier | p.V152A | Missense | Previously reported (18) | X-linked family history. Cousin brother of pt. 78. T-B+NK- SCID with low CD132 expression |
– | 15.8% | – | – | 88.2% | – |
| Pt. 44 | c.752C>G (hemizygous); Mother -heterozygous carrier | p.S251X | Nonsense | Novel | 1 elder male sibling died at early infancy due to opportunistic infections. T+B+NK- SCID with low naïve CD3 cells and low CD132 expression | 24.3% | 25.8% | 26.8% | 48.3% | 81.7% | 77.5% |
| Pt. 46 | c.596_598delinsTGGATTAT (hemizygous); Mother -heterozygous carrier | p.E199VfsX76 | Frameshift | Novel | Male infant with T-B+NK- SCID with low CD132 expression | 25.2% | 98.2% | 17.5% | 83.5% | 99.5% | 66.3% |
| Pt. 59 | c.8_9insA (hemizygous); Mother -heterozygous carrier | p.P4AfsX31 | Frameshift | Novel (Kato et al., Manuscript in submission) | Male infant with T-B+NK- SCID with low CD132 expression; low naïve CD3 cells | 51.1% | 67.2% | 66% | 95.2% | 98.9% | 99.8% |
| Pt. 63 | c.854G>A (hemizygous); Mother -heterozygous carrier | p.R285Q | Missense | Previously reported (19) (Kato et al., Manuscript in submission) | Male infant with T-B+NK- SCID with low CD132 expression | 48.8% | 50.4% | 21.7% | 84.1% | 90.7% | 85.1% |
| Pt. 78 | c.455T>C (hemizygous); Mother -heterozygous carrier | p.V152A | Missense | Previously reported (18) (Kato et al., Manuscript in submission) | X-linked family history. Cousin brother of pt. 43. T-B+NK- SCID | 67.8% | 59.7% | 24.8% | 88.4% | 89% | 29.3% |
| Pt. 85 | c.116-1G>T (hemizygous); Mother -heterozygous carrier | – | Splice-site | Novel (Kato et al., Manuscript in submission) | X-linked family history – 2 maternal uncles died at early infancy with severe infections. T-B+NK- SCID with low CD132 expression | 53.7% | 55.7% | 55.1% | 96.1% | 92% | 90.8% |
Table 3.
Erythrocyte ADA levels and % dAXP measured in dried blood spots.
| Patient | Molecular defect in ADA | ADA levels (nmol/h/mg) | % dAXP | PNP levels (nmol/h/mg) |
|---|---|---|---|---|
| Normal levels | 26.4 ± 10.0 | <1.0 | 1354 ± 561 | |
| Pt. 22 | c.301C>T c.461 G>T |
0.1 | 63.9 | 1025 |
| Mother of pt. 22 | c.461 G>T (heterozygous carrier) | 10.8 | 0 | 808 |
| Father of pt. 22 | c.301C>T (heterozygous carrier) |
9.6 | 0 | 834 |
| Pt. 30 | c.646G>A | 0 | 51.4 | 1264 |
| Pt. 31 | c.478+6T>A | 0.3 | 6.8 | 1151 |
| Pt. 36 | c.407G>A | 0 | 21.1 | 1532 |
| Pt. 39 | c.845G>T | 0 | 54.2 | 1316 |
| Pt. 74 | c.466C>T | 0 | NA | NA |
| Pt. 88 | c.845G>T | 0 | NA | 929 |
ADA, adenosine deaminase; AXP (dAXP), total adenosine (deoxyadenosine) nucleotides; PNP, purine nucleoside phosphorylase; NA, Not available.
% dAXP= (dAXP/AXP+dAXP) x100.
Molecular diagnosis was obtained in 162 patients—IL2RG (36), RAG1 (26), ADA (19), RAG2 (17), JAK3 (15), DCLRE1C (13), IL7RA (9), PNP (3), RFXAP (3), CIITA (2), RFXANK (2), NHEJ1 (2), CD3E (2), CD3D (2), RFX5 (2), ZAP70 (2), STK4 (1), CORO1A (1), STIM1 (1), PRKDC (1), AK2 (1), DOCK2 (1), and SP100 (1). Of the 176 molecular variants, 51 were identified to be novel in this study ( Table 4 , Supplementary Table 3 ) (7, 13, 17–49). A novel variant in RAG1 (c.1758_1760delinsGAATC) was identified in 2 unrelated North Indian families. Deletion of exons 1-3 (8947bp) in DCLRE1C was observed in 11 children (9 from North and 2 from South India). MLPA confirmed EX1_EX3del in DCLRE1C in 7 children from North India ( Figure 5 ). Targeted clinical exome sequencing by NGS did not identify pathogenic variants in 25 patients. Whole exome sequencing was performed in 5 children, and pathogenic variants were detected in 2 amongst these (pt. 50 and 51).
Table 4.
Molecular defects in genes associated with SCID/CID in our cohort.
| Pt No | Gene | Type of mutation | Exon | cDNA position | Protein change | Novel or previously reported | References |
|---|---|---|---|---|---|---|---|
| 1. SCID | |||||||
| Pt. 3 | IL2RG | Hemizygous- missense | Exon 4 | c.515T>G | p.L172R | Novel | Current study |
| Pt. 4 | IL2RG | Hemizygous- nonsense | Exon 5 | c.737G>A | p.W246X | Previously reported | (20) |
| Pt. 6 | IL2RG | Hemizygous- missense | Exon 2 | c.185G>A | p.C62Y | Novel | Current study |
| Pt. 8 and Pt. 25 | IL2RG | Hemizygous- nonsense | Exon 2 | c.202G>T | p.E68X | Previously reported | (17) |
| Pt. 35 | IL2RG | Hemizygous- missense | Exon 2 | c.170T>A | p.L57H | Novel | Current study |
| Pt. 43 and Pt. 78 | IL2RG | Hemizygous- missense | Exon 4 | c.455T>C | p.V152A | Previously reported | (18); (Kato T et al. Manuscript in submission) |
| Pt. 44 | IL2RG | Hemizygous- nonsense | Exon 5 | c.752C>G | p.S251X | Novel | Current Study |
| Pt. 46 | IL2RG | Hemizygous- frameshift | Exon 5 | c.596_598delinsTGGATTAT | p.E199VfsX76 | Novel | Current study |
| Pt. 51 | IL2RG | Hemizygous- nonsense | Exon 7 | c.865C>T | p.R289X | Previously reported | (21) |
| Pt. 55 | IL2RG | Hemizygous- nonsense | Exon 8 | c.964C>T | p.Q322X | Previously reported | (22) |
| Pt. 59 | IL2RG | Hemizygous- frameshift | Exon 1 | c.8_9insA | p.P4AfsX31 | Novel | (Kato T et al. Manuscript in submission) |
| Pt. 63 | IL2RG | Hemizygous- missense | Exon 2 | c.854G>A | p.R285Q | Previously reported | (19); (Kato T et al. Manuscript in submission) |
| Pt. 71 | IL2RG | Hemizygous- splice site | Exon 4 | c.594+5G>T | Previously reported | (22); (Kato T et al. Manuscript in submission) | |
| Pt. 85 | IL2RG | Hemizygous- splice site | Exon 2 | c.116-1G>T | Novel | (Kato T et al. Manuscript in submission) | |
| Pt. 91 | IL2RG | Hemizygous- missense | Exon 2 | c.854G>A | p.R285Q | Previously reported | (19) |
| Pt. 92 | IL2RG | Hemizygous- nonsense | Exon 4 | c.505C>T | p.Q169X | Novel | Current study |
| Pt. 95 | IL2RG | Hemizygous- missense | Exon 5 | c.677G>A | p.R226H | Previously reported | (23) |
| Pt. 106 | IL2RG | Hemizygous- nonsense | Exon 1 | c.67delC | p.L23X | Novel | Current study |
| Pt. 118 | IL2RG | Hemizygous- splice-site | Intron 2 | c.269+1G>T | Novel | Current study | |
| Pt. 152 | IL2RG | Hemizygous- missense | Exon 4 | c.520T>A | p.W174R | Novel | Current study |
| Pt. 168 | IL2RG | Hemizygous- missense | Exon 5 | c.664C>T | p.R222C | Previously reported | (19) |
| Pt. 169 | IL2RG | Hemizygous- missense | Exon 3 | c.314A>G | p.Y105C | Previously reported | (24) |
| Pt. 197 | IL2RG | Hemizygous- frameshift | Exon 3 | c.359dupA | p.E121GfsX47 | Novel | Current study |
| Pt. 216 | IL2RG | Hemizygous- missense | Exon 5 | c.670C>T | p.R224W | Previously reported | (22) |
| Pt. 220 | IL2RG | Hemizygous- missense | Exon 5 | c.664C>T | p.R222C | Previously reported | (19) |
| Pt. 18 | RAG1 | Homozygous- frameshift | Exon 2 | c.1758_1760delinsGAATC | p.D587NfsX5 | Novel | Current study |
| Pt. 19 | RAG1 | Homozygous- frameshift | Exon 2 | c.908delC | p.P303LfsX42 | Novel | Current study |
| Pt. 23 | RAG1 | Homozygous- frameshift | Exon 2 | c.1758_1760delinsGAATC | p.D587NfsX5 | Novel | Current study |
| Pt. 28 | RAG1 | Homozygous- missense | Exon 2 | c.2147G>A | p.R716Q | Previously reported | (25) |
| Pt. 38 | RAG1 | Homozygous- frameshift | Exon 2 | c.1178delG | p.G393AfsX10 | Previously reported | (17) |
| Pt. 58 | RAG1 | Compound heterozygous- frameshift, missense | Exon 2 Exon 2 |
c.2849delT c.1421G>A |
p.I950MfsX28 p.R474H |
Previously reported Previously reported |
(7) |
| Pt. 62 | RAG1 | Homozygous- missense | Exon 2 | c.2210G>A | p.R737H | Previously reported | (26); (Kato T et al. Manuscript in submission) |
| Pt. 77 | RAG1 | Homozygous- missense | Exon 2 | c.2923C>T | p.R975W | Previously reported | (27); (Kato T et al. Manuscript in submission) |
| Pt. 84 | RAG1 | Homozygous- missense | Exon 2 | c.2923C>T | p.R975W | Previously reported | (27); (Kato T et al. Manuscript in submission) |
| Pt. 89 | RAG1 | Homozygous- missense | Exon 2 | c.1211G>A | p.R404Q | Previously reported | (28); (Kato T et al. Manuscript in submission) |
| Pt. 105 | RAG1 | Homozygous- nonsense | Exon 2 | c.310C>T | p.Q104X | Novel | Current study |
| Pt. 140 | RAG1 | Homozygous- missense | Exon 2 | c.2333G>A | p.R778Q | Previously reported | (27) |
| Pt. 151 | RAG1 | Homozygous- missense | Exon 2 | c.1331C>T | p.A444V | Previously reported | (29) |
| Pt. 157 | RAG1 | Homozygous- missense | Exon 2 | c.1871G>A | p.R624H | Previously reported | (30) |
| Pt. 170 | RAG1 | Homozygous- missense | Exon 2 | c.2326C>T | p.R776W | Previously reported | (31) |
| Pt. 171 | RAG1 | Homozygous- nonsense | Exon 2 | c.424C>T | p.R142X | Previously reported | (32) |
| Pt. 204 | RAG1 | Compound heterozygous- missense, missense | Exon 2 | c.1421G>A; c.1442G>A | p.R474H; p.C481Y |
Previously reported; Novel |
(29) |
| Pt. 205 | RAG1 | Compound heterozygous- missense, missense | Exon 2 | c.323G>A; c.1228C>T | p.R108Q; p.R410W |
Previously reported; Previously reported |
(33, 34) |
| Pt. 5 | RAG2 | Homozygous- missense | Exon 2 | c.1247G>T | p.W416L | Previously reported | (35) |
| Pt. 15 and Pt. 21 | RAG2 | Homozygous- nonsense | Exon 2 | c.921G>A | p.W307X | Previously reported | (29) |
| Pt. 17 | RAG2 | Homozygous- missense | Exon 2 | c.1247G>T | p.W416L | Previously reported | (35) |
| Pt. 27 and Pt. 48 | RAG2 | Homozygous- missense | Exon 2 | c.1247G>T | p.W416L | Previously reported | (35) |
| Pt. 61 | RAG2 | Homozygous- missense | Exon 2 | c.1247G>T | p.W416L | Previously reported | (35) (Kato T et al. Manuscript in submission) |
| Pt. 93 | RAG2 | Homozygous- missense | Exon 2 | c.95G>A | p.G32E | Novel | Current study |
| Pt. 96 | RAG2 | Homozygous- missense | Exon 2 | c.608G>A | p.G203E | Novel | Current study |
| Pt. 116 | RAG2 | Homozygous- missense | Exon 2 | c.644C>T | p.T215I | Previously reported | (36) |
| Pt. 172 | RAG2 | Homozygous- frameshift | Exon 2 | c.1056delA | p.D353MfsX91 | Previously reported | (13) |
| Pt. 207 | RAG2 | Homozygous- missense | Exon 2 | c.329T>C | p.M110T | Novel | Current study |
| Pt. 209 | RAG2 | Compound heterozygous- missense, frameshift | Exon 2 | c.303T>G; c.171delG | p.N101K; p.K58SfsX73 |
Novel; Previously reported |
Current study; (7) |
| Pt. 9 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37) | |
| Pt. 20 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37) | |
| Pt. 26 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37) | |
| Pt. 52 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37) | |
| Pt. 56 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37) | |
| Pt. 64 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37); (Kato T et al. Manuscript in submission) | |
| Pt. 66 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37); (Kato T et al. Manuscript in submission) | |
| Pt.70 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37); (Kato T et al. Manuscript in submission) | |
| Pt. 90 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37); (Kato T et al. Manuscript in submission) | |
| Pt. 99 | DCLRE1C | Homozygous- frameshift | Exon 10 | c.874dupA | p.M292NfsX33 | Novel | Current study |
| Pt. 115 | DCLRE1C | Homozygous- large deletion | Exon 1-3 | EX1_EX3del | Previously reported | (37) | |
| Pt. 22 | ADA | Compound heterozygous- missense, missense | Exon 4 Exon 5 |
c.301C>T c.461 G>T |
p.R101W p.C154F |
Previously reported; Novel |
(38); Current study |
| Pt. 30 and Pt. 47 | ADA | Homozygous- missense | Exon 7 | c.646G>A | p.G216R | Previously reported | (39) |
| Pt. 31 | ADA | Homozygous- splice-site | Intron 6 | c.478+6T>A | Novel | Current study | |
| Pt. 36 | ADA | Homozygous- missense | Exon 5 | c.407G>A | p.G136D | Novel | Current study |
| Pt. 39 | ADA | Homozygous- missense | Exon 9 | c.845G>T | p.R282L | Previously reported | (40) |
| Pt. 74 | ADA | Homozygous- missense | Exon 5 | c.466C>T | p.R156C | Previously reported | (41); (Kato T et al. Manuscript in submission) |
| Pt. 88 | ADA | Homozygous- missense | Exon 9 | c.845G>T | p.R282L | Previously reported | (40); (Kato T et al. Manuscript in submission) |
| Pt. 119 | ADA | Homozygous- deletion | Exon 2 | EX2_del | Novel | (Kato T et al. Manuscript in submission) | |
| Pt. 123 | ADA | Homozygous- missense | Exon 11 | c.1028T>C | p.L343P | Previously reported | (42) |
| Pt. 153 | ADA | Homozygous- splice-site | Intron 10 | c.975+2T>G | Novel | Current study | |
| Pt. 154 | ADA | Compound heterozygous- nonsense, missense | Exon 6 Exon 8 |
c.523C>T; c.716G>A |
p.Q175X; p.G239D |
Previously reported; Previously reported | (7, 43) |
| Pt. 196 | ADA | Compound heterozygous- nonsense, missense | Exon 6 Exon 8 |
c.523C>T; c.716G>A |
p.Q175X; p.G239D |
Previously reported; Previously reported |
(7, 43) |
| Pt. 208 | ADA | Homozygous- nonsense | Exon 6 | c.523C>T | p.Q175X | Previously reported | (7) |
| Pt. 29 | JAK3 | Compound heterozygous- missense, missense | Exon 8 Exon 6 |
c.1048C>T; c.704T>C |
p.R350W; p.M235T |
Novel Novel |
Current study Current study |
| Pt. 50 | JAK3 | Compound heterozygous- frameshift, missense | Exon 2 Exon 10 |
c.115delC c.T1289C |
p.Q39SfsX108 p.I430T |
Novel Novel |
Current study Current study |
| Pt. 102 | JAK3 | Homozygous- nonsense | Exon 19 | c.2605C>T | p.Q869X | Novel | Current study |
| Pt. 107 | JAK3 | Homozygous- frameshift | Exon 22 | c.3049_3050delCT | p.L1017VfsX24 | Novel | Current study |
| Pt. 121 | JAK3 | Homozygous- missense | Exon 11 | c.1442A>G | p.E481G | Previously reported | (44) |
| Pt. 198 | JAK3 | Homozygous- missense | Exon 13 | c.1765G>T | p.G589C | Novel | Current study |
| Pt. 10 | IL7RA | Homozygous- large deletion | Exon 4-8 | EX4_EX8del | Novel | Current study | |
| Pt. 16 | IL7RA | Homozygous- nonsense | Exon 5 | c.616C>T | p.R206X | Previously reported | (45) |
| Pt. 94 | IL7RA | Homozygous- frameshift | Exon 5 | c.623delT | p.I208TfsX244 | Novel | Current study |
| Pt. 108 | IL7RA | Homozygous- splice-site | Intron 5 | c.707-1G>T | Novel | Current study | |
| Pt. 114 | IL7RA | Homozygous- missense | Exon 4 | c.509G>C | p.R170P | Novel | Current study |
| Pt. 155 | IL7RA | Homozygous- large deletion | Exon 4-8 | EX4_EX8del | Novel | Current study | |
| Pt. 200 | IL7RA | Homozygous- missense | Exon 3 | c.324T>G | p.C108W | Novel | Current study |
| Pt. 219 | IL7RA | Homozygous- nonsense | Exon 5 | c.616C>T | p.R206X | Previously reported | (45) |
| Pt. 14 | PNP | Homozygous- nonsense | Exon 3 | c.244C>T | p.Q82X | Previously reported | (46) |
| Pt. 156 | PNP | Homozygous- splice-site | Intron 3 | c.286-18G>A | Previously reported | (47) | |
| Pt. 111 | CD3D | Homozygous- nonsense | Exon 2 | c.158C>A | p.S53X | Novel | Current study |
| Pt. 137 | CD3D | Homozygous- splice-site | Intron 2 | (IVS2-2A>G) | Previously reported | (48) | |
| Pt. 117 | CD3E | Homozygous- nonsense | Exon 6 | c.288T>A | p.Y96X | Novel | Current study |
| Pt. 150 | CD3E | Homozygous- splice-site | Intron 6 | c.352+1G>A | Novel | Current study | |
| Pt. 68 | NHEJ1 | Homozygous- frameshift | Exon 5 | c.544_545delGA | p.E182TfsX3 | Novel | (Kato T et al. Manuscript in submission) |
| Pt. 72 | NHEJ1 | Homozygous- frameshift | Exon 3 | c.221_222delGT | p.C74SfsX4 | Novel | (Kato T et al. Manuscript in submission) |
| Pt. 49 | CORO1A | Homozygous- splice-site | Intron 7 | c.862-2A>G | Novel | Current study | |
| 2. CID | |||||||
| Pt. 210 | CIITA | Homozygous- nonsense | Exon 16 | c.3122C>A | p.S1041X | Novel | Current study |
| Pt. 158 | RFXANK | Homozygous- frameshift | Exon 6 | c.430dupC | p.L144PfsX37 | Novel | Current study |
| Pt. 211 | RFX5 | Homozygous- missense | Exon 7 | c.446G>A | p.R149Q | Previously reported | (49) |
| Pt. 173 | DOCK2 | Homozygous- nonsense | Exon 34 | c.3430C>T | p.R1144X | Previously reported | (13) |
| Pt. 41 | STK4 | Homozygous- nonsense | Exon 10 | c.1165C>T | p.Q389X | Novel | Current study |
| Pt. 104 | SP110 | Homozygous- nonsense | Exon 8 | c.855G>A | p.W285X | Novel | Current study |
| Pt. 86 | STIM1 | Homozygous- missense | Exon 10 | c.1285C>T | p.R429C | Novel | (Kato T et al. Manuscript in submission) |
Molecular analysis results of patients 221–277 are previously reported (7).
Figure 5.

MLPA data was analyzed using Coffalyser software. (A) Healthy control sample having a Dosage Quotient (DQ) between 0.80 and 1.20; (B) A patient with T-B-NK+ SCID (pt. 66) showing a homozygous deletion and DQ=0; (C, D) Parents of index child showing a heterozygous deletion with DQ Score in range of 0.40 to 0.65.
Majority of patients (n=198) in this cohort succumbed to overwhelming infections as HSCT could not be carried out in them ( Figure 6 ). Twenty-three patients (8.3%) underwent hematopoietic stem cell transplantation (HSCT) and 11 are doing well post-HSCT. The centre at South India (Apollo Children’s Hospitals, Chennai) has performed HSCT for 32 children with SCID until now and 17 are alive and doing well on follow-up. However, only 4 children are included in this analysis, as flow cytometry and mutation details were not available for other children. Another centre in South India (Aster CMI Hospitals, Bengaluru) has carried out HSCT for 9 children with SCID in the last 3 years ( Table 5 ).
Figure 6.
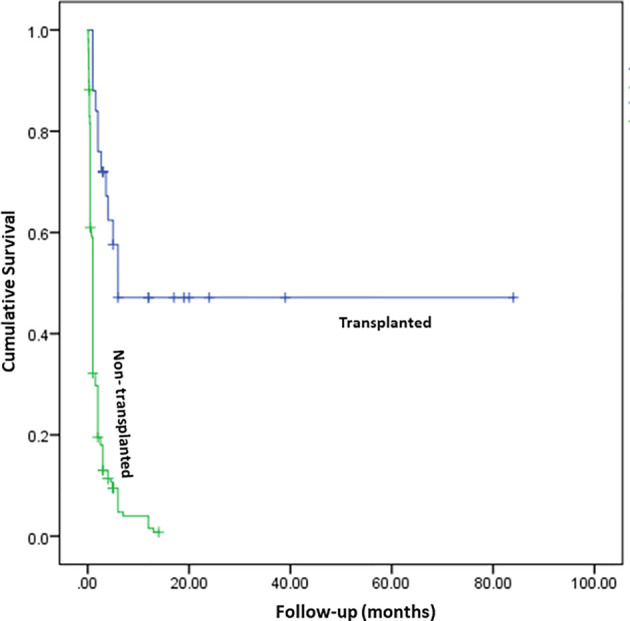
Survival curve comparing the outcome of children who underwent HSCT (n=23) and children who were not transplanted (n=213), p < 0.001 (Log Rank Mantel-Cox). Total person follow-up months—629.13 months.
Table 5.
Details of hematopoietic stem cell transplantation of 13 children with SCID.
| Patient | Type of SCID | Molecular defect | Centre | Age at transplantation | Donor characteristics | Outcome |
|---|---|---|---|---|---|---|
| Pt. 19 | T-B-NK+ | RAG1 | PGIMER, Chandigarh | 3.5 months | Father who is a complete HLA match with child | Developed BCG IRIS post-HSCT (D+90)- successfully treated with isoniazid, rifampicin, and ethambutol. He successfully engrafted and is currently doing well at 3rd year follow-up. |
| Pt. 31 | T-B-NK- | ADA | Diagnosed at PGIMER, Chandigarh; transplanted at Apollo Hospitals, Chennai | 4 years | Fully matched unrelated donor | Engrafted and doing well at 1st year follow-up. |
| Pt. 80 | T-B-NK+ | - | PGIMER, Chandigarh | 18 months | Haploidentical donor | Developed graft failure. Underwent second transplant at 3 years (details not available). |
| Pt. 94 | T-B+NK+ | IL7RA | Aster CMI Hospitals, Bengaluru | 5 months | Haploidentical (Mother) | Delayed graft failure (6 months post-HSCT). Underwent second HSCT with father being donor. Successfully engrafted and doing well at 20 months follow-up. |
| Pt. 103 | T-B+NK+ | N.A. | Aster CMI Hospitals, Bengaluru | 1.5 months | Haploidentical (Mother) | Failed engraftment. Underwent second HSCT with father being donor- successfully engrafted, however, died after 1.5 months due to fulminant sepsis. |
| Pt. 105 | T-B-NK+ | RAG1 | Aster CMI Hospitals, Bengaluru | 11 months | Haploidentical (Mother) | Successfully engrafted. Doing well 1.5 years post-HSCT. |
| Pt. 106 | T-B+NK- | IL2RG | Aster CMI Hospitals, Bengaluru | 5 months | Haploidentical (Mother) | Successfully engrafted. Doing well 1.4 years post-HSCT. |
| Pt. 108 | T-B+NK- | IL7RA | Aster CMI Hospitals, Bengaluru | 8.5 months | Fully matched sibling | Died D+20 of HSCT- MDR Klebsiella sepsis. |
| Pt. 110 | T-B-NK+ | N.A. | Aster CMI Hospitals, Bengaluru | 5 months | Haploidentical (Mother) | Successfully engrafted. Doing well 10 months post-HSCT. |
| Pt. 111 | T-B+NK+ | CD3D | Aster CMI Hospitals, Bengaluru | 6 months | Haploidentical (Mother) | Expired D+9 due to pulmonary haemorrhage. |
| Pt. 114 | T-B+NK+ | IL7R | Aster CMI Hospitals, Bengaluru | 9 months | Haploidentical (Father) | Developed severe gut GVHD and died D+60. |
| Pt. 115 | T-B-NK+ | DCLRE1C | Aster CMI Hospitals, Bengaluru | 7.5 months | Haploidentical (Father) | Successfully engrafted. Doing well 2 months post-HSCT. |
| Pt. 152 | T-B+NK- | IL2RG | Aditya Birla Memorial Hospital, Pune | 5 months | Matched family donor | Doing well at 7th year follow-up. |
Discussion
We describe the largest multi-centric cohort of patients with SCID from India. We included patients from 12 different tertiary care centers located in Northern, Southern, and Western parts of India. Patients from Eastern parts of India are usually referred to the centers located in other areas of India due to lack of availability of facilities for immunological investigations in that region. We witnessed an exponential rise in the number of cases with SCID after 2013 at multiple centers across India. We attribute this steady increase in cases to 2 factors—establishment of Indian Council of Medical Research Centers for Advanced Research in PIDs at PGIMER, Chandigarh (North India) and NIIH, Mumbai (West India) and expansion of laboratory facilities for pediatric immunology at other centers. The Pediatric Immunology and Bone Marrow Transplant Unit at Aster CMI Hospital, Bengaluru (South India) was established in 2017. Twenty-seven cases of SCID (Pt. 60–84) were diagnosed between 2017 and 2020, reflecting rise in awareness amongst referring pediatricians and better availability of diagnostic facilities at Bengaluru (South India).
Based on data from Sample Registration System of India, we estimated around 221 million live births from January 2011 to June 2020 (50). An estimated 257 patients with SCID have been diagnosed in this time period, which suggests a rough incidence of SCID at 0.12 per 100,000 live births. Though we have included data from most of the centers that care for patients with SCID in India, the estimated incidence from this study may not reflect true incidence of the country because of retrospective nature of the study and some patients diagnosed at other centers may have been missed. Nation-wide registry for SCID is needed for an accurate estimation of incidence. Nevertheless, if we extrapolate our current data on to the U.S. incidence figures of SCID (i.e. 1:58,000 live births), estimated number of children with SCID in India would be around 3,822 during the period 2011–June 2020 (1). Moreover, incidence of SCID in India is expected to be even higher than the U.S. considering high rates of consanguinity within the country. This suggests that though we have been increasingly diagnosing these children over the last few years, the diagnosis is still missed in almost 93% of these children. This is clearly unacceptable and mandates urgent intervention of health care professionals.
We observed a higher incidence of autosomal recessive forms of SCID (78.4%) compared to X-linked SCID. This is similar to reports from several other countries where consanguinity rates are high ( Table 6 ) (7, 8, 51–59). Though consanguinity rate of 28.2% observed in our study is lower than that of Saudi Arabia and Iran, practice of endogamous and intra-community marriages is, perhaps, responsible for high proportion of autosomal recessive forms of SCID in India (2, 6). Median age at diagnosis of SCID in our study is 5 months. This is similar to reports from other countries such as China, Turkey, and U.S.A ( Table 6 ). Children who had a family history of SCID had an earlier age of diagnosis (median:4.5 months) compared to children who did not have a suggestive family history (median:6 months). Our observation is similar to the report by Luk et al. that suggested the importance of family history for an early diagnosis of SCID (17).
Table 6.
Comparison of our cohort with published multicentric studies on SCID from other countries in last 10 years.
| Study (Year) | Place | No. of patients | Age of onset and diagnosis | Clinical manifestations | Molecular defects | Outcome |
|---|---|---|---|---|---|---|
| Yao et al. (51) | Shanghai, China | 44; Male: female – 40:4 |
Mean age of onset – 3.56 ± 3.91 months Mean age at diagnosis – 7.1 ± 7.96 months |
BCG-related complications noted in 14 children (31.8%). Three (3) had disseminated BCG infection. Two (2) had CMV infection | Defect in IL2RG noted in 25 children (56.8%). | Mortality seen in 37 children (84%). Six (6) children underwent HSCT and 1 of them had survived. |
| Pasic et al. (52) | Serbia and Montenegro | 21 | Median age of onset – 2 months | BCG-related complications in 41%. Pneumonia noted in 15 children (PJP- 5, CMV- 3, BCG-2, respiratory virus- 5). OS noted in 6 children. | 17 had proven molecular defect (81%). RAG1/2 commonest (12) followed by IL2RG (3), JAK3 (2), DCLRE1C (1) | Mortality seen in 16 children (76.2%). Eight (8) children underwent HSCT and 5 of them survived. |
| Lee et al. (53) | South East Asia | 42; Male: female – 30:12 |
Median age of onset and diagnosis – 2 and 4 months, respectively. | BCG-related complications in 10 children (23.8%) – 6 had localized reaction; 3 had regional adenitis; 3 had disseminated BCGosis. Oral thrush (12), CMV (2), and PJP (2) are other documented infections. OS noted in 4 children. | 26 had proven molecular defect (61.9%). IL2RG commonest (19) followed by IL7RA (2), JAK3 (2), RAG1/2 (2), DCLRE1C (1) | 12 children underwent HSCT and 8 of them survived. |
| Abolhassani et al. (54) | Iran | 169; Male: female – 96:73 |
Mean age of onset and diagnosis – 4.2 and 8.6 months, respectively. | BCG-related complications noted in 23 (13.6%). Other infections noted are PJP (13), CMV (15), EBV (8), VDPV (6), Cryptococcus (6), and VZV (6). OS noted in 11 children. | 37 had proven molecular defect (21.9%). RAG1/2 commonest (19) followed by IL2RG (3), JAK3 (3), DCLRE1C (3), ADA (2), IL7RA (2), CD3E (1), CD3D (1), PRKDC (1), NHEJ1 (1), PTPRC (1) | NA |
| Rozmus et al. (55) | Canada | 40 | Mean age at diagnosis – 4.2 months. | Oral thrush (8), CMV (6), PJP (6), RSV (1), and adenovirus (1) are the infections noted. | 20 had proven molecular defect (50%). ADA commonest (10) followed by IL2RG (4), RAG1 (2), ZAP70 (2), and MHC Class II defects (2). | Mortality observed in 12 children (30%). Fifteen (15) underwent HSCT and 10 of them survived. |
| Ikinciogullari et al. (8) | Turkey | 234 (transplanted patients); Male: female – 145:89 |
Median age at diagnosis – 5 months. | Infections noted are oral thrush (51.5%), CMV (13.5%), bacterial infections (7.4%), BCG-related complications (2.2%), and respiratory viruses (4.4%) | 42.3% had proven molecular defects – RAG1/2 (15.4%), JAK3 (6.8%), IL2RG (6%), DCLRE1C (5.6%) | Survival at 20 years is 65.7% |
| Mazzucchelli et al. (56) | Brazil | 70; Male: female – 49:21 |
Mean age of onset and diagnosis – 3.3 and 6.7 months, respectively. | BCG-related complications seen in 39 children (55.7%) – disseminated form in 29 and localized in 10. Features of OS noted in 8 children. | NA | Mortality seen in 35 patients (50%). Thirty (30) underwent HSCT and 18 of them survived. |
| de Pagter et al. (57) | Netherlands | 43 | Median delay in diagnosis in typical and atypical SCID – 2 and 27 months, respectively. | Infections noted are bacterial sepsis (11), PJP (11), CMV (8), and BCGitis (6). AIHA seen in 5 children with atypical SCID. | IL2RG (21%), RAG1 (21%), RAG2 (5%), ADA (12%), DCLRE1C (7%), PNP (7%), and IL7RA (5%) | Mortality observed in 18 children (41.9%). Thirty-two (32) underwent HSCT and 24 of them survived. Two (2) underwent gene therapy and 1 survived. |
| Haddad et al. (58) | USA and Canada | 662 (transplanted patients); Male: female – 471:191 |
Median age at diagnosis – 141.5 days (4.7 months) | NA | IL2RG (187), RAG1/2 (52), ADA (45), IL7RA (40), DCLRE1C (28), JAK3 (24), CD3 receptor defects (7), PNP (1), AK2 (1), CD45 (1) | Survival is better in children transplanted less than 3.5 months. Survival at 10 years is 71% and is higher with matched sibling donors compared to other donor types. |
| Micho et al. (59) | Greece | 30; Male: female – 19:11 |
Median age at diagnosis – 6.2 months | NA | DCLRE1C (3), IL2RG (2), JAK3 (2), RAG1 (2), ADA (2), PNP (1) | Mortality is observed in 15 children (50%). Twenty-two (22) underwent HSCT and 14 of them are doing well. |
| Aluri et al. (7) | India | 57; Male: female – 40:17 |
Median age of onset and diagnosis – 2 and 5.1 months, respectively | Infections observed include oral thrush (21%), BCG-related complications (12%), and PJP (1). OS noted in 4 children | 49 children had proven molecular defects (86%). RAG1/2 commonest (12), followed by JAK3 (9), IL2RG (9), MHC Class II defects (6), ADA (5), DCLRE1C (2), ZAP70 (2), IL7RA (1), PRKDC (1), PNP (1), and AK2 (1) | Mortality observed in 47 children (82.5%). Four (4) underwent HSCT and none survived. |
| Present study (2020) | India | 277 (23 CID, 254 SCID); Male: female – 196:81 |
Median age of onset and diagnosis – 2.5 and 5 months, respectively | BCG-related complications in 47 patients (17%) – localized form (20) and disseminated BCGosis (27). Other common infections include bacteria (72), CMV (23), Candida sp. (23), PJP (8), Aspergillus sp. (5), VAPP/VDPV (2). OS noted in 33 children. AIHA and lymphoreticular malignancy observed in 5 and 2 children, respectively. | 162 patients had proven molecular defects (58.5%) - RAG1/2 (43), IL2RG (36), ADA (19), JAK3 (15), DCLRE1C (13), IL7RA (9), PNP (3), CIITA (2), RFXAP (3), RFXANK (2), NHEJ1 (2), CD3E (2), CD3D (2), RFX5 (2), ZAP70 (2), STK4 (1), CORO1A (1), STIM1 (1), PRKDC (1), AK2 (1), DOCK2 (1), and SP100 (1) | Mortality noted in 210 children (75.8%). Twenty-three (23) underwent HSCT and 11 of them are doing well. |
BCG, Bacillus Calmette-Guerin; CMV, Cytomegalovirus; HSCT, Hematopoietic stem cell transplantation; OS, Omenn syndrome; PJP, Pneumocystis jirovecii pneumonia; EBV, Epstein-Barr virus; VDPV, Vaccine-derived polio virus; VZV, Varicella zoster virus; AIHA, Autoimmune hemolytic anemia; VAPP, Vaccine-associated paralytic polio; CID, Combined Immune Deficiency; SCID, Severe Combined Immune Deficiency.
Opportunistic infections in SCID are life-threatening and must be identified and treated adequately before HSCT. We documented a higher incidence of microbiologically-proven infections in our cohort compared to a previous report published from India (7). Amongst the bacterial infections, BCG was the commonest organism isolated. BCG-site ulceration has been noted in 47 children, however, disseminated BCGosis could be proven in 27 children only. BCG adenitis was noted in one child at D+90 post-HSCT as a part of immune reconstitution inflammatory syndrome. Lack of microbiological confirmation of BCG infection in many patients could have accounted for low rates of disseminated BCGosis in our cohort ( Table 6 ) (51, 52). Infants with SCID who had received BCG vaccination and had not developed disseminated infection, are generally started on prophylactic medications—isoniazid and rifampicin at age-appropriate doses, that is generally continued until successful engraftment following HSCT.
Septicemia due to unusual organisms such as W. confusa and A. faecalis was also noted in our cohort. These are environmental bacteria and usually do not cause invasive infections in immunocompetent hosts. We also noted a high rate of disseminated CMV infection (8.3%) in our cohort. However, several amongst these were identified only on autopsy (60). This underscores the importance of vigilant screening and preventive measures for CMV infection in children with SCID. Cytomegalovirus infection, in our cohort, was possibly transfusion-acquired as most of the children received blood transfusions that are not always leuko-depleted and screened for active CMV infection. Though many patients had clinical features suggestive of P. jirovecii pneumonia (tachypnea, hypoxemia, interstitial pneumonia) and were treated for the same, microbiological or histopathological confirmation was possible in only 8 of them.
Thirty-three children had features of OS in our cohort. One child with OS (pt.54) was being treated as severe eczema for 3 years with multiple topical and systemic immunosuppressive agents, and diagnosis of SCID was made only after he developed severe infections. This highlights the importance of early identification of clinical phenotype of OS based on clinical features (generalized erythematous rash with scaling and partial loss of scalp hairs and eyebrows) and referral for appropriate immunological workup. Twenty-eight (28) children with OS had normal or high ALC and 2 children with transplacental-acquired maternal T-cell engraftment had elevated ALC. Laboratory assay of naïve T cells, memory T cells, and HLA-DR expression in T lymphocytes necessary for the diagnosis of OS are currently being performed only in two centers (PGIMER, Chandigarh and NIIH, Mumbai).
Twenty-three children in our cohort who did not have OS had normal ALC (>3 × 109/L). However, lymphocyte subsets and naïve T cell estimation revealed diagnosis of SCID in them, thereby highlighting the importance of clinical suspicion and immunological investigations in infants with severe and life-threatening infections even if ALC is normal. Expansion of B cells or NK cells, engraftment of transplacental-acquired maternal T cells, or partial genetic defects allowing selective clone of T cell expansion could be the possible reasons for normal ALC in SCID. Aluri et al. have previously highlighted the importance of assessment of naïve T helper and cytotoxic T cells in children with severe infections and normal ALC to characterise MHC class II and ZAP70 defects, respectively (7). A child with IL7RA defect in our cohort had a T-B+NK- phenotype, similar to the report by Aluri et al. (7). Also, two children with IL2RG defect had a T+B-NK- phenotype (1- OS, 1- transplacental-acquired maternal T cell engraftment). A possible explanation for low B cells is the depletion of B cells due to high inflammatory milieu secondary to OS and severe infections (61).
CD132 expression by flow cytometry is currently carried out at only two centers—PGIMER, Chandigarh (North India) and NIIH, Mumbai (West India). At PGIMER, Chandigarh, we found low CD132 expression in lymphocytes by flow cytometry as an inexpensive and rapid method of confirmation of diagnosis of X-linked SCID in 7 children. Two (2) children with X-linked SCID and previously reported variants in IL2RG (pt. 25 and pt. 78) had a normal expression of CD132 in lymphocytes ( Table 2 ). We could not assay phosphorylated STAT5 in activated T-cells by flow cytometry to determine the functionality of IL2Rγ in many patients due to absent or very low amounts of T cells, however, naïve T cells by flow cytometry and TREC levels by RT-PCR have been assayed in some of them ( Table 2 ). Only a handful centers in India (e.g. PGIMER, Chandigarh, North India, and NIIH, Mumbai, West India) have the wherewithal to perform functional studies. Both the centers have performed flow cytometry tests for samples received from other centers, however, timely transportation of viable blood samples from far off places, especially during hot summers remains a significant problem (11, 12). Lack of state-of-the-art facilities to do functional assays in all patients with SCID is one of the limitations of our study. Establishment of more clinical immunology laboratories, training of necessary manpower, and improvement in existing laboratory services are needed to overcome these barriers (11, 12).
Genetic confirmation of diagnosis of SCID is necessary for identification of pattern of inheritance and genetic counselling of affected families. Eighty-two (82) patients did not undergo a molecular analysis for confirmation of diagnosis due to lack of easy access to molecular diagnostics and financial difficulties. With the establishment of commercial NGS laboratories and reduction in costs involved for genetic sequencing over last few years, NGS-based diagnostics have become feasible in India (7, 13). In-house NGS facility for molecular diagnosis of PID is currently available only at PGIMER, Chandigarh (North India) and Christian Medical College, Vellore (South India). Most of the patients with SCID present in a critically-ill state and convincing families for genetic studies is often challenging due to significant financial and social constraints. It must be noted that expenses for molecular diagnosis are borne by the families in India most of the times as it is not covered by state or insurance schemes. Despite these challenges, we have been able to perform genetic studies in 195 patients. Academic collaborations with institutes at Hong Kong, Japan, and USA helped the centre at PGIMER, Chandigarh (North India) to get free molecular diagnosis for the families who cannot afford for costly molecular tests. We prefer to store blood samples in terminally-ill patients and later call the family for counselling to undergo genetic tests, as confirmation of molecular diagnosis has helped the families to undergo antenatal testing in subsequent pregnancies.
Defects in RAG1/2 were found to be commonest in our cohort followed by IL2RG, DCLRE1C, and ADA. This is similar to the previous reports from Turkey, Iran, and Serbia ( Table 6 ) (8, 52, 54). MHC Class II defect and defects in STIM1, DOCK2, SP110, ZAP70, and STK4 genes are categorized as combined immunodeficiencies as per the 2019 International Union of Immunological Societies Expert Committee classification of human inborn errors of immunity (IEI) (5). However, we have included children with these defects in our cohort because they had severe infections from early infancy mimicking the clinical presentation of SCID (7).
Clinical phenotype of patients with RAG1/2 defects in our cohort was very heterogenous. This included classical SCID, OS, atypical/leaky SCID phenotype, autoimmunity in form of AIHA, and development of hematological malignancy such as Hodgkin lymphoma. Wide spectrum of clinical manifestations could be due to difference in VDJ recombination activity or influence of other genetic or environmental factors (34, 62). Other reported clinical phenotypes in RAG1/2 such as cutaneous granulomas, CVID-like phenotype or elevated γδ T cells were not seen in our cohort.
Low or undetectable ADA levels and elevated %dAXP levels were seen in 7 and 5 children with ADA defect, respectively. We noted that %dAXP levels in 2 children (pt. 31 and 36) were lower compared to other 3 children. While pt.31 had a milder clinical phenotype, pt.36 had features of OS. This suggests that low levels of accumulation of dAXP with residual ADA activity in lymphocytes may play a role in development of restricted T-cell clones that could be responsible for partial immunity and development of OS (63).
One child with ADA defect (pt. 36) had evidence of nephrotic syndrome along with OS. Renal abnormalities described with ADA defect (such as diffuse mesangial sclerosis) could result in nephrotic syndrome (64). However, renal involvement in OS manifesting as nephrotic syndrome has also been previously reported (65). We could not perform renal biopsy in this child due to severe ascites. Two other children with OS in our cohort also had renal involvement at autopsy—focal segmental glomerulosclerosis and mesangial sclerosis. Both of them also had severe infections—disseminated CMV in one and invasive aspergillosis in other. Whether the renal abnormalities are the result of genetic defect, inflammatory phenotype of OS, or severe infections is not clear and further research is needed in this regard.
Identification of radiosensitive forms of SCID is essential in B-NK+ SCID before HSCT as these children are prone to toxicity by chemotherapeutic drugs and radiation (66). Amongst the radiosensitive forms of SCID, molecular defects are predominantly noted in DCLRE1C in our cohort. Moreover, only mutation in DCLRE1C observed in North Indian children (n=9) was EX1_EX3 del. Initial MLPA screening for DCLRE1C exon 1-3 deletion before NGS in children with B-NK+ SCID was found to be more cost-effective than subjecting these children to NGS without a MLPA screen. The former approach is preferred at Chandigarh (North India) because of two reasons—NGS can miss large deletions and patients identified to have EX1_EX3 del in DCLRE1C by MLPA do not need to undergo NGS that is four to five times more expensive than MLPA in India. We also describe molecular defects in STK4, CORO1A, CD3D, CD3E, and SP110 for the first time in India. Clinical phenotype of eczema, AIHA, and CD4 lymphopenia noted in STK4 defect (pt. 41) has been previously described (67). Moshous et al. have described EBV-induced B cell lymphoma and naïve T-cell lymphopenia in patients with a hypomorphic missense variant in CORO1A (c.717G>A) (68). Our patient (pt. 49) with a novel splice-site defect in CORO1A (c.862-2A>G) had CD3 and CD4 lymphopenia, and developed an intracranial B cell lymphoma at 3.5 years of age.
A significant proportion of children (n=254) could not be subjected to HSCT due to medical and social reasons and succumbed to the illness. Presence of fulminant infections at time of diagnosis and lack of financial support dissuaded many families to undergo a costly procedure like HSCT. At present, facilities for pediatric HSCT for IEI are available at very few centers in India. Two centers in India have carried out most of the transplants for SCID – Apollo Children’s Hospitals, Chennai (South India) and Aster CMI Hospitals, Bengaluru (South India). Establishment of such dedicated pediatric HSCT units and development of manpower for HSCT services across the country are the need of the hour to ensure easy access to these services for affected patients. Provision of financial support from the government to affected families to undergo HSCT will also be required for successful outcomes. Studies from Western countries have shown that children with SCID transplanted below the age of 3.5 months of age had a significantly better outcome compared to children who underwent transplantation later (58). Though the age at diagnosis in our cohort is similar to countries where newborn screening has not been initiated, delayed referrals, presence of life-threatening infections at presentation, and lack of easy access to pediatric HSCT accounted for the unacceptable mortality rates in our cohort ( Figure 6 ) ( Table 6 ) (51, 52). We also realise that diagnosis of SCID is still being missed in most babies in India. Institution of universal newborn screening for SCID would provide more accurate estimates of incidence of SCID in our country and would also facilitate early diagnosis and treatment. However, financial implications and cost-effectiveness of implementing such a programme in a country as large, and as diverse, as India need to be worked out by health planners (69).
To conclude, we describe the largest multicentric cohort of SCID from India and document several novel mutations. Number of children with molecular diagnosis and those who have undergone HSCT has increased significantly in last decade. However, we are only too aware of our limitations. Improvement in awareness amongst physicians and pediatricians, expansion of diagnostic laboratories, institution of newborn screening, development of pediatric HSCT services, and financial support to the families to undergo HSCT are essentially needed for a better diagnosis and outcome of affected patients in the country.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material .
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
PV, AS, AGum, JN, AJ, DS, AGup, AlK, MD, PT, VG, AP, SagB, SR, RC, MeS, DM, SarB, ArR, AA, FN, BJ, AM, HL, RU, RR, SanB, and SuS—Clinical management of patients; provided necessary clinical details for compilation. AmR, RK, MaS, AnK, BS, RM, KaS, AD, NJ, PK, MM, AV, KoS, SrS, YO, TK, KI, KC, DL, OO, SN, MH, and Y-LL—Laboratory work-up of patients; provided necessary laboratory results for compilation. KG—Provided necessary histopathology details. PV, RK, AS, AGum, MaS, AnK, and JN—Compiled the data and framed the initial draft and editing of manuscript. PV, RK—Literature search. PV, AmR, and SuS—Editing of manuscript at all stages of preparation and final approval. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the support provided by the Indian Council of Medical Research and Department of Health Research, Government of India; Foundation of Primary Immunodeficiency Diseases (FPID), United States of America; Prof. Sudhir Gupta, Professor of Medicine, Pathology & Laboratory Medicine, and Microbiology & Molecular Genetics, University of California at Irvine, Irvine, CA, United States of America.
The authors thankfully acknowledge Centre d’Etude des Déficits Immunitaires, Hôpital Necker-Enfants Malades, Paris, France for carrying out the molecular analysis for one of the patients (Case 152).
The authors also thankfully acknowledge Mr. Jitendra Kumar Shandilya, Ms. Jhumki Das, and Ms. Kanika Arora, PhD students in Allergy Immunology Unit, Advanced Pediatrics Centre, Post Graduate Institute of Medical Education and Research, Chandigarh, India for assisting in flow cytometry experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.619146/full#supplementary-material
References
- 1. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA (2014) 312(7):729–38. 10.1001/jama.2014.9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Al-Mousa H, Al-Dakheel G, Jabr A, Elbadaoui F, Abouelhoda M, Baig M, et al. High Incidence of Severe Combined Immunodeficiency Disease in Saudi Arabia Detected Through Combined T Cell Receptor Excision Circle and Next Generation Sequencing of Newborn Dried Blood Spots. Front Immunol (2018) 9:782. 10.3389/fimmu.2018.00782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van der Burg M, Mahlaoui N, Gaspar HB, Pai SY. Universal Newborn Screening for Severe Combined Immunodeficiency (SCID). Front Pediatr (2019) 7:373. 10.3389/fped.2019.00373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers (2015) 1:15061. 10.1038/nrdp.2015.61 [DOI] [PubMed] [Google Scholar]
- 5. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. 10.1007/s10875-019-00737-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shahbazi Z, Yazdani R, Shahkarami S, Shahbazi S, Hamid M, Sadeghi-Shabestari M, et al. Genetic mutations and immunological features of severe combined immunodeficiency patients in Iran. Immunol Lett (2019) 216:70–8. 10.1016/j.imlet.2019.10.001 [DOI] [PubMed] [Google Scholar]
- 7. Aluri J, Desai M, Gupta M, Dalvi A, Terance A, Rosenzweig SD, et al. Clinical, Immunological, and Molecular Findings in 57 Patients With Severe Combined Immunodeficiency (SCID) From India. Front Immunol (2019) 10:23. 10.3389/fimmu.2019.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ikinciogullari A, Cagdas D, Dogu F, Tugrul T, Karasu G, Haskologlu S, et al. Clinical Features and HSCT Outcome for SCID in Turkey. J Clin Immunol (2019) 39(3):316–23. 10.1007/s10875-019-00610-x [DOI] [PubMed] [Google Scholar]
- 9. Dvorak CC, Haddad E, Buckley RH, Cowan MJ, Logan B, Griffith LM, et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010-2018). J Allergy Clin Immunol (2019) 143(1):405–7. 10.1016/j.jaci.2018.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. European Society for Immunodeficiencies Registry Working Party Diagnosis Criteria (2019). Available at: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria (Accessed 10 October 2020).
- 11. Rawat A, Arora K, Shandilya J, Vignesh P, Suri D, Kaur G, et al. Flow Cytometry for Diagnosis of Primary Immune Deficiencies-A Tertiary Center Experience From North India. Front Immunol (2019) 10:2111. 10.3389/fimmu.2019.02111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Madkaikar MR, Shabrish S, Kulkarni M, Aluri J, Dalvi A, Kelkar M, et al. Application of Flow Cytometry in Primary Immunodeficiencies: Experience From India. Front Immunol (2019) 10:1248. 10.3389/fimmu.2019.01248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arunachalam AK, Maddali M, Aboobacker FN, Korula A, George B, Mathews V, et al. Primary Immunodeficiencies in India: Molecular Diagnosis and the Role of Next-Generation Sequencing. J Clin Immunol (2020) 23. 10.1007/s10875-020-00923-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohanty MC, Madkaikar MR, Desai M, Aluri J, Varose SY, Taur P, et al. Natural Clearance of Prolonged VDPV Infection in a Child With Primary Immunodeficiency Disorder. Front Immunol (2019) 10:1567. 10.3389/fimmu.2019.01567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mohanty MC, Madkaikar MR, Desai M, Taur P, Nalavade UP, Sharma DK, et al. Poliovirus Excretion in Children with Primary Immunodeficiency Disorders, India. Emerg Infect Dis (2017) 23(10):1664–70. 10.3201/eid2310.170724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gupta K, Singh S, Sharma D, Singh MP, Singh M. An infant with repeated respiratory infections and failure to thrive. Indian Pediatr (2014) 51(10):819–26. 10.1007/s13312-014-0509-6 [DOI] [PubMed] [Google Scholar]
- 17. Luk ADW, Lee PP, Mao H, Chan KW, Chen XY, Chen TX, et al. Family History of Early Infant Death Correlates with Earlier Age at Diagnosis But Not Shorter Time to Diagnosis for Severe Combined Immunodeficiency. Front Immunol (2017) 8:808. 10.3389/fimmu.2017.00808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izuhara K, Heike T, Otsuka T, Yamaoka K, Mayumi M, Imamura T, et al. Signal transduction pathway of interleukin-4 and interleukin-13 in human B cells derived from X-linked severe combined immunodeficiency patients. J Biol Chem (1996) 271(2):619–22. 10.1074/jbc.271.2.619 [DOI] [PubMed] [Google Scholar]
- 19. Clark PA, Lester T, Genet S, Jones AM, Hendriks R, Levinsky RJ, et al. Screening for mutations causing X-linked severe combined immunodeficiency in the IL-2R gamma chain gene by single-strand conformation polymorphism analysis. Hum Genet (1995) 96(4):427–32. 10.1007/BF00191801 [DOI] [PubMed] [Google Scholar]
- 20. Vihinen M, Arredondo-Vega FX, Casanova JL, Etzioni A, Giliani S, Hammarström L, et al. Primary immunodeficiency mutation databases. Adv Genet (2001) 43:103–88. 10.1016/s0065-2660(01)43005-7 [DOI] [PubMed] [Google Scholar]
- 21. Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell (1993) 73(1):147–57. 10.1016/0092-8674(93)90167-o [DOI] [PubMed] [Google Scholar]
- 22. Puck JM, Pepper AE, Henthorn PS, Candotti F, Isakov J, Whitwam T, et al. Mutation analysis of IL2RG in human X-linked severe combined immunodeficiency. Blood (1997) 89(6):1968–77. 10.1182/blood.V89.6.1968 [DOI] [PubMed] [Google Scholar]
- 23. Pepper AE, Buckley RH, Small TN, Puck JM. Two mutational hotspots in the interleukin-2 receptor gamma chain gene causing human X-linked severe combined immunodeficiency. Am J Hum Genet (1995) 57(3):564–71. [PMC free article] [PubMed] [Google Scholar]
- 24. Puck JM. IL2RGbase: a database of gamma c-chain defects causing human X-SCID. Immunol Today (1996) 17(11):507–11. 10.1016/0167-5699(96)30062-5 [DOI] [PubMed] [Google Scholar]
- 25. National Center for Biotechnology Information ClinVar; [VCV000304495.5]. Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000304495.5 (Accessed Oct. 10, 2020).
- 26. Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell (1998) 93(5):885–96. 10.1016/s0092-8674(00)81448-8 [DOI] [PubMed] [Google Scholar]
- 27. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med (2008) 358(19):2030–8. 10.1056/NEJMoa073966 [DOI] [PubMed] [Google Scholar]
- 28. Noordzij JG, de Bruin-Versteeg S, Verkaik NS, Vossen JM, de Groot R, Bernatowska E, et al. The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG-deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood (2002) 100(6):2145–52. 10.1182/blood.V100.6.2145 [DOI] [PubMed] [Google Scholar]
- 29. Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood (2001) 97(1):81–8. 10.1182/blood.v97.1.81 [DOI] [PubMed] [Google Scholar]
- 30. Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science (1996) 274(5284):97–9. 10.1126/science.274.5284.97 [DOI] [PubMed] [Google Scholar]
- 31. Xiao Z, Yannone SM, Dunn E, Cowan MJ. A novel missense RAG-1 mutation results in T-B-NK+ SCID in Athabascan-speaking Dine Indians from the Canadian Northwest Territories. Eur J Hum Genet (2009) 17(2):205–12. 10.1038/ejhg.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kato M, Kimura H, Seki M, Shimada A, Hayashi Y, Morio T, et al. Omenn syndrome–review of several phenotypes of Omenn syndrome and RAG1/RAG2 mutations in Japan. Allergol Int (2006) 55(2):115–9. 10.2332/allergolint.55.115 [DOI] [PubMed] [Google Scholar]
- 33. National Center for Biotechnology Information ClinVar; [VCV000968838.1]. Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000968838.1 (Accessed Oct. 11, 2020).
- 34. Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol (2014) 133(4):1099–108. 10.1016/j.jaci.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sobacchi C, Marrella V, Rucci F, Vezzoni P, Villa A. RAG-dependent primary immunodeficiencies. Hum Mutat (2006) 27(12):1174–84. 10.1002/humu.20408 [DOI] [PubMed] [Google Scholar]
- 36. Meshaal S, El Hawary R, Elsharkawy M, Mousa RK, Farid RJ, Abd Elaziz D, et al. Mutations in Recombination Activating Gene 1 and 2 in patients with severe combined immunodeficiency disorders in Egypt. Clin Immunol (2015) 158(2):167–73. 10.1016/j.clim.2015.04.003 [DOI] [PubMed] [Google Scholar]
- 37. Alsmadi O, Al-Ghonaium A, Al-Muhsen S, Arnaout R, Al-Dhekri H, Al-Saud B, et al. Molecular analysis of T-B-NK+ severe combined immunodeficiency and Omenn syndrome cases in Saudi Arabia. BMC Med Genet (2009) 10:116. 10.1186/1471-2350-10-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akeson AL, Wiginton DA, Dusing MR, States JC, Hutton JJ. Mutant human adenosine deaminase alleles and their expression by transfection into fibroblasts. J Biol Chem (1988) 263(31):16291–6. [PubMed] [Google Scholar]
- 39. Hirschhorn R, Chakravarti V, Puck J, Douglas SD. Homozygosity for a newly identified missense mutation in a patient with very severe combined immunodeficiency due to adenosine deaminase deficiency (ADA-SCID). Am J Hum Genet (1991) 49(4):878–85. [PMC free article] [PubMed] [Google Scholar]
- 40. Hellani A, Almassri N, Abu-Amero KK. A novel mutation in the ADA gene causing severe combined immunodeficiency in an Arab patient: a case report. J Med Case Rep (2009) 3:6799. 10.1186/1752-1947-3-6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hirschhorn R. Identification of two new missense mutations (R156C and S291L) in two ADA- SCID patients unusual for response to therapy with partial exchange transfusions. Hum Mutat (1992) 1(2):166–8. 10.1002/humu.1380010214 [DOI] [PubMed] [Google Scholar]
- 42. GnomAD browser Single nucleotide variant: 20-43248990-A-G (GRCh37) (2020). Available at: https://gnomad.broadinstitute.org/variant/20-43248990-A-G?dataset=gnomad_r2_1 (Accessed 11 October 2020).
- 43. Adams SP, Wilson M, Harb E, Fairbanks L, Xu-Bayford J, Brown L, et al. Spectrum of mutations in a cohort of UK patients with ADA deficient SCID: Segregation of genotypes with specific ethnicities. Clin Immunol (2015) 161(2):174–9. 10.1016/j.clim.2015.08.001 [DOI] [PubMed] [Google Scholar]
- 44. Candotti F, Oakes SA, Johnston JA, Giliani S, Schumacher RF, Mella P, et al. Structural and functional basis for JAK3-deficient severe combined immunodeficiency. Blood (1997) 90(10):3996–4003. 10.1182/blood.V90.10.3996 [DOI] [PubMed] [Google Scholar]
- 45. Jo EK, Kook H, Uchiyama T, Hakozaki I, Kim YO, Song CH, et al. Characterization of a novel nonsense mutation in the interleukin-7 receptor alpha gene in a Korean patient with severe combined immunodeficiency. Int J Hematol (2004) 80(4):332–5. 10.1532/ijh97.04026 [DOI] [PubMed] [Google Scholar]
- 46. Somech R, Lev A, Simon AJ, Hanna S, Etzioni A. T- and B-cell defects in a novel purine nucleoside phosphorylase mutation. J Allergy Clin Immunol (2012) 130(2):539–42. 10.1016/j.jaci.2012.03.038 [DOI] [PubMed] [Google Scholar]
- 47. Markert ML, Finkel BD, McLaughlin TM, Watson TJ, Collard HR, McMahon CP, et al. Mutations in purine nucleoside phosphorylase deficiency. Hum Mutat (1997) 9(2):118–21. [DOI] [PubMed] [Google Scholar]
- 48. Takada H, Nomura A, Roifman CM, Hara T. Severe combined immunodeficiency caused by a splicing abnormality of the CD3delta gene. Eur J Pediatr (2005) 164(5):311–4. 10.1007/s00431-005-1639-6 [DOI] [PubMed] [Google Scholar]
- 49. Nekrep N, Jabrane-Ferrat N, Wolf HM, Eibl MM, Geyer M, Peterlin BM. Mutation in a winged-helix DNA-binding motif causes atypical bare lymphocyte syndrome. Nat Immunol (2002) 3(11):1075–81. 10.1038/ni840 [DOI] [PubMed] [Google Scholar]
- 50. Sample Registration System Sample Registration System Statistical Report (2018). Available at: https://censusindia.gov.in/Vital_Statistics/SRS_Report_2018/SRS_Statistical_Report_2018.pdf (Accessed 11 October 2020).
- 51. Yao CM, Han XH, Zhang YD, Zhang H, Jin YY, Cao RM, et al. Clinical characteristics and genetic profiles of 44 patients with severe combined immunodeficiency (SCID): report from Shanghai, China (2004-2011). J Clin Immunol (2013) 33(3):526–39. 10.1007/s10875-012-9854-1 [DOI] [PubMed] [Google Scholar]
- 52. Pasic S, Vujic D, Veljković D, Slavkovic B, Mostarica-Stojkovic M, Minic P, et al. Severe combined immunodeficiency in Serbia and Montenegro between years 1986 and 2010: a single-center experience. J Clin Immunol (2014) 34(3):304–8. 10.1007/s10875-014-9991-9 [DOI] [PubMed] [Google Scholar]
- 53. Lee PP, Chan KW, Chen TX, Jiang LP, Wang XC, Zeng HS, et al. Molecular diagnosis of severe combined immunodeficiency–identification of IL2RG, JAK3, IL7R, DCLRE1C, RAG1, and RAG2 mutations in a cohort of Chinese and Southeast Asian children. J Clin Immunol (2011) 31(2):281–96. 10.1007/s10875-010-9489-z [DOI] [PubMed] [Google Scholar]
- 54. Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol (2018) 141(4):1450–8. 10.1016/j.jaci.2017.06.049 [DOI] [PubMed] [Google Scholar]
- 55. Rozmus J, Junker A, Thibodeau ML, Grenier D, Turvey SE, Yacoub W, et al. Severe combined immunodeficiency (SCID) in Canadian children: a national surveillance study. J Clin Immunol (2013) 33(8):1310–6. 10.1007/s10875-013-9952-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mazzucchelli JT, Bonfim C, Castro GG, Condino-Neto AA, Costa NM, Cunha L, et al. Severe combined immunodeficiency in Brazil: management, prognosis, and BCG-associated complications. J Investig Allergol Clin Immunol (2014) 24(3):184–91. [PubMed] [Google Scholar]
- 57. de Pagter AP, Bredius RG, Kuijpers TW, Tramper J, van der Burg M, van Montfrans J, et al. Overview of 15-year severe combined immunodeficiency in the Netherlands: towards newborn blood spot screening. Eur J Pediatr (2015) 174(9):1183–8. 10.1007/s00431-015-2518-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood (2018) 132(17):1737–49. 10.1182/blood-2018-03-840702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Michos A, Tzanoudaki M, Villa A, Giliani S, Chrousos G, Kanariou M. Severe combined immunodeficiency in Greek children over a 20-year period: rarity of γc-chain deficiency (X-linked) type. J Clin Immunol (2011) 31(5):778–83. 10.1007/s10875-011-9564-0 [DOI] [PubMed] [Google Scholar]
- 60. Gupta K, Rawat A, Agrawal P, Jindal A, Nada R, Saikia B, et al. Infectious and non-infectious complications in primary immunodeficiency disorders: an autopsy study from North India. J Clin Pathol (2018) 71(5):425–35. 10.1136/jclinpath-2017-204708 [DOI] [PubMed] [Google Scholar]
- 61. Cain D, Kondo M, Chen H, Kelsoe G. Effects of acute and chronic inflammation on B-cell development and differentiation. J Invest Dermatol (2009) 129(2):266–77. 10.1038/jid.2008.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Delmonte OM, Schuetz C, Notarangelo LD. RAG Deficiency: Two Genes, Many Diseases. J Clin Immunol (2018) 38(6):646–55. 10.1007/s10875-018-0537-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Flinn AM, Gennery AR. Adenosine deaminase deficiency: a review. Orphanet J Rare Dis (2018) 13(1):65. 10.1186/s13023-018-0807-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Whitmore KV, Gaspar HB. Adenosine Deaminase Deficiency - More Than Just an Immunodeficiency. Front Immunol (2016) 7:314. 10.3389/fimmu.2016.00314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rybojad M, Cambiaghi S, Moraillon I, Vignon-Pennmen MD, Morel P, Baudoin V, et al. Omenn’s reticulosis associated with the nephrotic syndrome. Br J Dermatol (1996) 135(1):124–7. 10.1046/j.1365-2133.1996.d01-948.x [DOI] [PubMed] [Google Scholar]
- 66. Dvorak CC, Cowan MJ. Radiosensitive severe combined immunodeficiency disease. Immunol Allergy Clin North Am (2010) 30(1):125–42. 10.1016/j.iac.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Halacli SO, Ayvaz DC, Sun-Tan C, Erman B, Uz E, Yilmaz DY, et al. STK4 (MST1) deficiency in two siblings with autoimmune cytopenias: A novel mutation. Clin Immunol (2015) 161(2):316–23. 10.1016/j.clim.2015.06.010 [DOI] [PubMed] [Google Scholar]
- 68. Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol (2013) 131(6):1594–603. 10.1016/j.jaci.2013.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leung D, Chua GT, Mondragon AV, Zhong Y, Nguyen-Ngoc-Quynh L, Imai K, et al. Current Perspectives and Unmet Needs of Primary Immunodeficiency Care in Asia Pacific. Front Immunol (2020) 11:1605. 10.3389/fimmu.2020.01605 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material .