

Abstract
SARS-CoV-2, the viral particle, is responsible for triggering the 2019 Coronavirus disease outbreak (COVID-19). To tackle this situation, a number of strategies are being devised to either create an antidote, a vaccine, or agents capable of preventing its infection. To enable research on these strategies, numerous target proteins are identified where Spike (S) protein is presumed to be of immense potential. S-protein interacts with human angiotensin-converting-enzyme-2 (ACE2) for cell entry. The key region of S-protein that interacts with ACE2 is a portion of it designated as a receptor-binding domain (RBD), following whereby the viral membrane fuses with the alveolar membrane to enter the human cell. The proposition is to recognize molecules from the bundle of phytochemicals of medicinal plants known to possess antiviral potentials as a lead that could interact and mask RBD, rendering them unavailable to form ACE2 interactions. Such a molecule is called the ‘S-protein blocker’. A total of 110 phytochemicals from Withania somnifera, Asparagus racemosus, Zinziber officinalis, Allium sativum, Curcuma longa and Adhatoda vasica were used in the study, of which Racemoside A, Ashwagandhanolide, Withanoside VI, Withanoside IV and Racemoside C were identified as top five hits using molecular docking. Further, essential Pharmacophore features and their ADMET profiles of these compounds were studied following to which the best three hits were analyzed for their interaction with RBD using Molecular Dynamics (MD) simulation. Binding free energy calculations were performed using MM/GBSA, proving these phytochemicals can serve as S-protein blocker.
Keywords: SARS-CoV-2 novel coronavirus, S-Protein blocker, Antiviral phytochemicals, Molecular dynamics (MD) simulation
Graphical abstract

1. Introduction
The reparations of novel Severe Acute Respiratory Syndrome Virus-Coronavirus-2 (SARS-CoV-2) is severe, and it is hard to come over with the situation of pandemic even after a year of its onset. There has been no focused drug to stop the spread and replication of this viral particle in human host [1,2]. Regardless of being flabbergasted from the circumstance of pandemic rose by SARS-CoV-2, people of scientific and clinical society are aggregately attempting to discover an immunization or to build up a technique to battle the mind-boggling virus, SARS-CoV-2 [[3], [4], [5]]. According to the details of the World health organization (WHO), worldwide ∼25 million people have been affected by the infection of SARS-CoV-2 of which ∼0.85 million passings have occurred. (https://covid19.who.int/). According to the description of the International Committee on Taxonomy of Viruses (ICTV), this pandemic causing infection is hereditarily alike the SARS-CoV infection causing the flare-up of 2003; thus, the identified virus was assigned as SARS-CoV-2 on eleventh of February 2020 [6]. Albeit hereditarily related, the two infection strains are unique in their strength of causing disease, where the latest identified novel coronavirus is known to be quickly contracting and shows moderately lower mortality, notwithstanding, its high infectivity has made this viral infection spread all around the world in an exceptionally brief timeframe. It fundamentally causes a respiratory or gastrointestinal disease with the effects like fever, pneumonia, windedness in certain populaces [7]. It is unavoidable to recognize a vaccine or a helpful drug that can function as a counteractant or that can debilitate the virus causing a decrease in its spread.
Apart from discovering the vaccine, there exists two main strategies to control CoVs, for the researchers on working on the alternative strategy of developing the anti-viral drug, that is by identifying or developing inhibitors (i) to impede viral passage into the host cells, and (ii) to forestall viral replication [5]. For these reasons, an aggregate of twelve proteins distinguished as targets which are comprehensively delegated are (i) Structural Proteins and (ii) Non-structural protein. Structural proteins, namely Spike protein (S-protein), Envelop small membrane protein (E-protein), Membrane protein (M-protein), Nucleocapsid protein (N-protein), are significant for accomplishing the virulent viral physical conformation. Causing bending or misfolding of these proteins is focused on essentially lessening the harmfulness and infectivity of the virus. S-protein is considered to have more noteworthy structural-functional significance and it turns into a conspicuous decision to be a significant target protein. Non-structural proteins, namely Main protease (Mpro), Papain-like protease, Non-structural protein (nsp)-10, nsp-11 nsp-13, nsp-14, nsp-15 and nsp-16 are needed for replication and bundling of the virus particle into the capsid protein coat [2,6,8] thus these proteins are being viewed as targets on the grounds that hindering any of these proteins can stop viral load increase in host cell [6,8,9].
Of the clear large number of structural proteins, S-protein is foreseen to be the main protein for being a potential target. This is on the grounds that S-protein is the principal protein to communicate with the human host by interacting with human angiotensin-converting enzyme 2 (ACE2) for entering the human cell. The receptor-binding domain (RBD), of S-protein, comprises of S1 and S2 subunits. S1 subunits use human ACE2 as the receptor to infect human cells and the S2 subunit intervenes the membrane fusion process, for example fusion of viral membrane with human cell membrane [[10], [11], [12], [13]].
RBD serves as a potential target, if masked with a small molecule, its interaction with ACE2 can be halted. Interaction of RBD with ACE2 is a complex mechanism that occurs on the cell membrane of alveolar cells. The theory is to recognize molecules as a lead having capacity to explicitly interact and mask critical amino acids of RBD, making them inaccessible to interact with ACE2 at the alveolar membrane [14]. Small molecules or bioactive compounds that restrain or forbid the merging of viral membrane with the host’s cell membrane by any mechanism can be designated as ‘S-protein inhibitors’. Moreover, these small bioactive molecules are also to be accessed for their ADMET (Absorption–Distribution–Metabolism–Excretion–Toxicity) and pharmacophore properties to predict their availability on the alveolar membrane for S-protein to interact in priority to that of ACE2. One of the most well-perceived research in developing S-protein inhibitors was by developing a peptide known designated as ‘pan-coronavirus fusion inhibitor targeting peptide EK1’ works by disallowing the fusion of viral membrane with human cell’s membrane [15]. Notwithstanding, EK1 being peptide can’t be consumed orally however was proposed to be managed by nasal course making it hard to utilize [15,16]. Moreover, EK1 was also prone to protease degradation by membrane proteases reducing its shelf life and therefore, molecules with a similar function possessing capacity administered orally is being searched for. In this context, recent research is performed and Glycyrrhizic acid, a phytochemical from Glycyrrhiza glabra has shown properties of being a S-protein inhibitors predicted using Docking and Molecular Dynamics (MD) simulation [17]. Despite a modest bunch of endeavours been sought after, there exists huge degree for distinguishing more up to date S-protein inhibitors against SARS-CoV-2.
To deliver such lead compounds to hit the commercial market, it is essential for them to not show toxicity, and so selecting a library of compounds for such assessment becomes very important. One hundred ten phytochemicals were selected from various medicinal plants like Withania somnifera, Asparagus racemosus, Zinziber officinalis, Allium sativum, Curcuma longa and Adhatoda vasica based on their previous reports of showing antiviral activity against other viruses. Here we hypothesized to identify S-protein inhibitors from the library of antiviral phytochemicals that are already known for their beneficial implications on human health using Docking and Molecular Dynamics (MD) simulation.
2. Materials and methods
2.1. Spike protein assessment for identifying correct docking site
S-protein in general is an enormous protein containing with a molecular weight of 459.49 kDa (PDB ID: 6XR8). S-protein is a trimeric structure containing three indistinguishable chains where each chain is containing seven subunits. For such an enormous protein, it is fundamental to distinguish the right site for docking the ligands [18]. The protein was carefully analyzed and the correct subunit i.e., RBD of a single S-protein chain was chosen and the respective protein structure of RBD in the conformation interacting with ACE2 was retrieved from PDB (PDB ID 6M0J). The unique feature of this protein is that the conformation of RBD is open, where the amino acids interacting with ACE2 can be traced. Once the amino acids of the RBD chain of 6M0J were identified, their coordinates were chosen for performing the molecular docking of against 110 phytochemicals.
2.2. Ligands and protein preparation
A library of 110 compounds showing antiviral property from the plants W. somnifera [19], A. racemosus [20], Z. officinalis [21], A. sativum [22], C. longa [23], A. vasica [20] were constructed. All the ligands were prepared for docking by adding charges and hydrogen. These structures were energy minimized and aligned using the Amber03 force field algorithm [24].
Protein from PDB with ID 6M0J was chosen for the current study. This protein possessed two chains A and E. The resolution of the entire structure was 2.45 Å. Chain A corresponded to human ACE2 and consisted of 603 amino acids, while chain E was of RBD of monomeric chain spike protein and consisted of 229 amino acids. For further use, chain A was removed, and all the molecular docking was performed with chain E and the coordinates for the molecular docking corresponding to the interacting residues with chain ACE2 were chosen.
2.3. Molecular docking
The interactions of all the 110 ligands with the chain E (RBD of S-protein) for the protein 6M0J was performed using YASARA (Yet Another Scientific Artificial Reality Application) commercial package [25] software which employs the use of AutoDock Vina molecular docking algorithm [26]. Before performing docking, water molecules were removed from the.pdb file and then the energy minimization was performed with the help of the YASARA NOVA steepest descent conjugate method in the YASARA structure. For docking, the following parameters were set: (i) number of binding modes- 10; (ii) exhaustiveness of search- 8 and (iii) maximum energy difference- 3 kcal/mol. All the Interaction profile after performing docking so generated were also studied. Best pose based on binding energies for each ligand-protein interaction were further analyzed in ACCELRYS Discovery Studio (DS) visualizer for assessment hydrogen bond formation and for hydrophobic interactions by the functional groups of ligands with amino acids. From the interaction profile the ligands (hits) having high binding energy were further considered for the molecular dynamics approach. The binding free energy ΔG bind was estimated according to Equation (1):
| (1) |
where ΔGvdW = van der Waals term for docking energy.
ΔGHbond = H bonding term for docking energy.
ΔGelec = electrostatic term for docking energy.
ΔGtor = torsional free energy term for ligand when the ligand transits from unbounded to bounded state.
ΔGdesolv = desolvation term for docking energy.
2.4. ADMET analysis
ADMET properties of the top 5 docked compounds was performed using pkCSM [27] and TOPKAT module of ACCELRYS Discovery Studio v 20.1. It computed in-vivo Absorption parameters like; Water solubility in buffer system (SK atomic types, mg/L), in-vivo Caco2 cell permeability (Human colorectal carcinoma), Human intestinal absorption (HIA, %), in-vivo P-glycoprotein inhibition and in-vivo skin permeability (logKp, cm/hour). Metabolic parameters were determined using in-vivo Cytochrome P450 2C19 inhibition, in-vivo Cytochrome P450 2C9 inhibition, in-vivo Cytochrome P450 2D6 inhibition, in-vivo Cytochrome P450 2D6 substrate, in-vivo Cytochrome P450 3A4 inhibition and in-vivo Cytochrome P450 3A4 substrate. Distribution property included tests like Blood-Brain Barrier (BBB) penetration, Lipinski’s Rule (Rule of Five), Central Nervous System (CNS) permeability. To access the toxicity of compounds under study a range of important endpoints including, Acute algae toxicity, Ames test, 2 years carcinogenicity bioassay in mouse, 2 years carcinogenicity bioassay in rat, in-vivo Ames test results in TA100 strain (Metabolic activation by rat liver homogenate) were computed. Excretion again is a very important parameter and as many drugs are often withdrawn at clinical trial stages due to their poorer renal clearance. In this study, we included Total Renal clearance and Renal OCT2 Substrate to identify the Excretion efficacy of the proposed metabolite.
2.5. Molecular dynamics (MD) simulations study
The physical movements of atoms and molecules of protein-ligand docked complex was identified through molecular dynamics simulation. Top scoring 5 compounds were chosen for the MD simulation at 100 ns time interval each. Molecular dynamics simulations study was carried out in Desmond (Schrödinger Release 2019–3). These complexes were prepared using a protein preparation wizard to allow complex relaxation. The addition of hydrogens, water removal, bond orders assignment, fill in missing side chains and loops with optimization of hydrogen-bond assignment (sampling of water orientations and use of pH 7.0) was done. The ligands were prepared using LigPrep of Maestro which adds hydrogen atoms, generates tautomers, ionization states, ring conformations, and produces minimized 3D structures. Whereas the protein was prepared by performing restrained minimization using OPLS-2005 force field. The system for simulation was built keeping solvent model as TIP3P, the boundaries were defined with the box shape of orthorhombic with the dimension of 10 Å × 10 Å × 10 Å. This was then followed by neutralization by Cl− or Na+ counter ions. Steepest descent energy minimization was performed, and the simulation was proceeded for 100 ns with NPT (constant Number of particles, Pressure, and Temperature) with 300 K and 1.01 bar, constant volume, Smooth Particle Mesh Ewald (PME) method. On completion of simulation the trajectories were analyzed in simulation interaction diagram wizard which analyzes trajectories for root mean square deviation (RMSD), root means square fluctuation (RMSF), Ligand-protein contact profiles, for Ligand and Protein modifications [24,25,28].
2.6. Binding free energy calculations MM/GBSA
The single trajectory approach was used for the binding free energy calculation using molecular mechanics generalized Born surface area (MM/GBSA) [[29], [30], [31], [32]]. From entire Simulation, a recording interval was adjusted to 100 ps for entire 100 ns and of which 1000 number frames were gathered which were used to calculate MMGBSA in PRIME module of Maestro 11.4. MM/GBSA calculations of entire trajectory was performed with the OPLS_2005 force field that use the VSGB 2.0 solvation model.
The free energy values were calculated using the following equations (2), (3):
| (2) |
and
| (3) |
Where ΔTDS is the conformation entropic contribution, and ΔG MM is the molecular mechanics’ interaction energy (electrostatic + van der Waals interaction) between protein and ligand. ΔG PB and ΔG SA depict the polar solvation energy and the nonpolar solvation energy, respectively.
3. Results
3.1. Spike protein assessment for identifying correct docking site
S-protein as a whole consists of three identical chains. Each of this chain possess two subunits, S1 and S2. Within the S1 subunit there exist two regions, N-terminal domain that is followed by the region of RBD. Based on the structure of SARS-CoV-2, the S1 and S2 subunits of S-protein monomers form the bulbous head and stalk region (Fig. 1 a). In the pre-infection inactive state of SARS-CoV-2, S-protein exists as an inactive precursor. During its infection, target cell proteases activate the S-protein by cleaving it into S1 and S2 subunits, which is necessary for activating the membrane fusion domain after viral entry into target cells (Fig. 1b). Therefore, it is necessary to use the cleaved part of S1 to perform the docking to use the correct open conformation of RBD which ideally binds to ACE2 (Fig. 1c).
Fig. 1.

(a) S-protein of SARS-CoV-2 (PDB ID: 6XR8) consisting of homotrimer where the conformation of each chain is closed (b) S-protein of SARS-CoV-2 upon interaction with human protease, cleave occur between S1 and S2 which caused the RBD of S1 to attain open conformation (PDB ID: 6VYB) (c) Interaction of RBD with of S1 with ACE2, where the monomeric chain in is interacting with ACE2 is shown, the zoomed image is of the protein with PDB ID: 6M0J consisting of only RBD portion of monomeric chain of Spike protein interacting with ACE2.
The rationale here is to mask the RBD in open conformation with phytochemical (lead compound) to inhibit its binding with ACE2. Therefore, protein PDB with ID 6M0J was chosen as this PDB entry possesses the open RBD region interacting with ACE2. For selecting 6M0J, all the assemblies of RBD interacting with ACE2 were retrieved from PDB in the priority of resolution “Best to Worse” and the PDB IDs of the assemblies were 6M0J, 6LZG, 6VW1, 3SCI, 3D0I, 2AJF, 7C8D, 3SCK, 3SCL, 3SCJ and 3D0H. The RBD chains of all these structures were superimposed to find the variation in the structural conformation. The RMSD was found to be 0.974 with respect to the best structure 6MJ0, and this protein structure showing the best resolution, it was chosen for the further experimental analysis. The ACE2 chain (Chain A, in this case, is removed) and the open conformation of RBD (Chain E) is used to perform molecular docking. The coordinates of residues (LYS417, GLU484, THR500, GLY446, TYR449, ASN487, GLY496, TYR449, ASN487, GLN493, GLY502, TYR453, PHE486, LEU455, PHE486, TYR505, PHE486, TYR505) of RBD found to be interacting with ACE2 were chosen to perform the docking of test molecules from antiviral compounds from the plants W. somnifera, A. racemosus, Z. officinalis, A. sativum, C. longa, and A. vasica the list of molecules that were screened is provided in supplementary material file.
3.2. Molecular docking study
All the known anti-viral phytochemicals from our constructed library were docked at the specific site of the RBD. The coordinates that were chosen for performing docking involved all the amino acids that interact with ACE2 under natural conditions. The chosen site for docking involved the following amino acids: LYS417, GLU484, THR500, GLY446, TYR449, ASN487, GLY496, TYR449, ASN487, GLN493, GLY502, TYR453, PHE486, LEU455, PHE486, TYR505, PHE486, TYR505 and the set of these 18 amino acids are designated as Target Binding Locus (TBL). From all the docking analyses performed the top five compounds were screened based on two major criteria (i) Binding energy of ligand with target site on RBD and (ii) ability of the ligand to recruit the maximum number of amino acids from TBL. The top five phytochemicals possessing the ability to interact with RBD were found to be Racemoside A, Ashwagandhanolide, Withanoside VI, Withanoside IV and Racemoside C (Fig. 2 ).
Fig. 2.

(a) The interaction of RBD (Chain E of protein with PDB ID: 6M0J) with ACE2 (b) Shows the interaction of specific important amino acids of RBD with the best five docked compounds. Interaction of (c) Racemoside A (d) Ashwagandhanolide (e) Withanoside VI (f) Withanoside IV and (g) Racemoside C with RBD in 2D view.
From the docking analysis, Racemoside A showed the maximum TBL binding energy of 8.622 kcal/mol (Table 1 ) and could recruit and interact with ARG403, GLU406, GLN409, LYS417, TYR449, TYR453, LEU455, ILE472, GLY482, VAL483, GLU484, PHE490, LEU492, GLN493, SER494, TYR 495, GLY496, PHE497, GLN498, ASN501 and TYR505 from the dataset of docking obtained from YASARA. When the same best pose, pose with minimum Root Mean Square Deviation (RMSD) was viewed in DS, five hydrogen bonds, two carbon-hydrogen bonds, one pi-alkyl and one pi-pi T shaped bond formation were observed to be formed. Similarly, the second-best compound based on the binding energy was found to be Ashwagandhanolide, where the binding energy was found to be 8.346 kcal/mol and could interact with ARG403, TYR453, LEU455, PHE456, GLU484, TYR489, PHE490, GLN493, SER494, TYR495, GLY496, GLN498, THR500, ASN501 and TYR505 as per the docking dataset of YASARA (Table 1). DS showed Ashwagandhanolide to form five hydrogen bonds, three pi-pi T-shaped bonds and one unfavorable donor-donor bond formation. The third best compound based on the binding energy was Withanoside VI with the value of 8.083 kcal/mol and showed to interact with ARG403, TYR449, TYR453, LEU455, PHE456, GLU484, GLY485, PHE486, CYS488, TYR489, PHE490, GLN493, SER494, TYR495, GLY496, GLN498, ASN501 and TYR505 (Table 1). The view in DS showed this compound to make one six hydrogen bonds, one carbon hydrogen bond and two pi-alkyl interactions. Interestingly, out of all the screened compounds, this compound (Withanoside VI) is shown to be making maximum number of hydrogen bonds. The fourth screened compound is Withanoside IV which showed binding energy of 7.915 kcal/mol and is shown to make interactions with ARG403, TYR449, TYR453, LEU455, PHE456, GLU484, GLY485, PHE486, ASN487, CYS488, TYR489, PHE490, GLN493, SER494, TYR495, GLY496, GLN498, ASN501 and TYR505. When analyzed in DS, only four hydrogen bonds were being formed along with two carbon-hydrogen bonds, two pi-alkyl interactions and formed one unfavorable donor-donor bonding. Racemoside C is the fifth and the last of the screened compound, it showed 7.906 kcal/mol of binding energy and showed to interact with ARG403, TYR449, TYR453, LEU455, PHE456, TYR489, GLN493, SER494, TYR495, GLY496, GLN498, THR500, ASN501, GLY502 and TYR505. It was found to make four hydrogen bonds, three carbon-hydrogen bonds and two pi-alkyl interactions. Based on the binding energy and capabilities to form hydrogen bonds, the top three compounds Racemoside A, Ashwagandhanolide, and Withanoside VI were further analyzed for their protein interactions by performing MD simulations. However, pharmacophore modelling and ADMET analysis of all the five screened compounds were performed.
Table 1.
Binding energies and amino acid interaction profile of the top five hits obtained on performing molecular docking.
| Name | Binding energy [kcal/mol] | The rank of the compound based on binding energy | Amino acid interaction profile |
|---|---|---|---|
| Racemoside A | 8.622 | 1 | ARG403, GLU406, GLN409, LYS417, TYR449, TYR453, LEU455, ILE472, GLY482, VAL483, GLU484, PHE490, LEU492, GLN493, SER494, TYR 495, GLY496, PHE497, GLN498, ASN501, TYR505 |
| Ashwagandhanolide | 8.346 | 2 | ARG403, TYR453, LEU455, PHE456, GLU484, TYR489, PHE490, GLN493, SER494, TYR495, GLY496, GLN498, THR500, ASN501, TYR505 |
| Withanoside VI | 8.083 | 3 | ARG403, TYR449, TYR453, LEU455, PHE456, GLU484, GLY485, PHE486, CYS488, TYR489, PHE490, GLN493, SER494, TYR495, GLY496, GLN498, ASN501, TYR505 |
| Withanoside IV | 7.915 | 4 | ARG403, TYR449, TYR453, LEU455, PHE456, GLU484, GLY485, PHE486, ASN487, CYS488, TYR489, PHE490, GLN493, SER494, TYR495, GLY496, GLN498, ASN501, TYR505 |
| Racemoside C | 7.906 | 5 | ARG403, TYR449, TYR453, LEU455, PHE456, TYR489, GLN493, SER494, TYR495, GLY496, GLN498, THR500, ASN501, GLY502, TYR505 |
The structural properties of all the top five ligands are represented in Table 2 while the pharmacophore analysis of these phytochemical ligands is depicted in Fig. 3 . All the top five screened compounds (Racemoside A, Ashwagandhanolide, Withanoside VI, Withanoside IV and Racemoside C) have several excellent structural features to interact with amino acids as these compounds have maximum proton donors and acceptors which is crucial to interact with amino acids by making hydrogen bonds. LogP values of Withanoside VI and Withanoside IV are identical and much lower than other screened compounds. On the contrary Ashwagandhanolide has the maximum LogP value suggesting its hydrophobic nature (Table 2).
Table 2.
Structures and chemical properties of screened anti-viral phytochemicals.
| Descriptor | Phytochemicals |
||||
|---|---|---|---|---|---|
| Racemoside A/C |
Ashwagandhanolide |
Withanoside VI |
Withanoside IV |
Racemoside C |
|
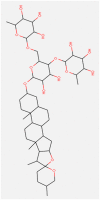 |
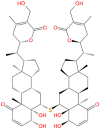 |
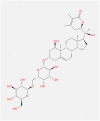 |
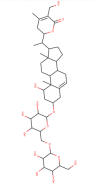 |
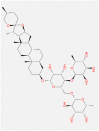 |
|
| Value | Value | Value | Value | Value | |
| Molecular Weight | 871.071 | 975.295 | 782.921 | 782.921 | 871.071 |
| LogP | 1.3216 | 6.376 | −0.0516 | −0.1941 | 1.3216 |
| #Rotatable Bonds | 7 | 8 | 8 | 9 | 7 |
| #Acceptors | 16 | 13 | 15 | 15 | 16 |
| #Donors | 8 | 6 | 9 | 9 | 8 |
| Surface Area | 359.006 | 411.756 | 321.42 | 321.42 | 359.006 |
Fig. 3.

MD simulation Protein-ligand interaction root-mean-square deviation (RMSD) profile of (a) RBD-Racemoside A (b) RBD-Ashwagandhanolide and (c) RBD-Withanoside VI complexes.
3.3. ADMET analysis
All the ADMET properties of the top five screened compounds is depicted in Table 3 . The key feature for any compound to serve as an oral drug is its movement across the intestinal epithelial barrier that determines its rate of movement and degree of human absorption which directly affects its bioavailability. For the computational model, high Caco-2 permeability would translate in predicted values greater than 0.90. Of all the compounds Ashwagandhanolide have Caco-2 value in positive range suggesting its moderate permeability, while all the other compounds (Racemoside A, Withanoside VI, Withanoside IV and Racemoside C) has a predicted value in negative range suggesting poor permeability. However, such compounds can be transported along with fat and through binding with albumin. Intestinal absorption (human) value predicts the absorption of the drug from an orally administered solution. Racemoside A, Ashwagandhanolide and Racemoside C have this value above 60% suggesting good absorption. While Withanoside VI and Withanoside IV have this value 39% and 31% respectively, suggesting their moderate to low absorption. All the compounds have highly similar skin permeability predicted values which are smaller than −2.5 log Kp suggesting poor permeability. P-glycoprotein is a component of ATP-binding cassette (ABC) transporter, the value ‘yes’ for P-glycoprotein substrate suggests the compound can get transported across the cell membrane by ABC transporter. Here, all the five compounds are predicted to transport across ABC transporter. The volume of Distribution (VDss) suggests the total volume of drugs needed to be uniformly distributed in blood. The value below −0.15 logVDss stands for a low VDss value and above 0.45 LogVDss, which stands for considerably high VDss value. In this context, all the compounds in the current study has low VDss values. Blood-Brain barrier (BBB) permeability gives an account of the compound’s ability to reach the brain or not. It is presumed that compounds with logBB values greater than 0.3 can pass BBB therefore, all the compounds are not predicted to cross BBB, moreover, BBB permeability is not needed for targeting S-protein and so is not a desirable trait for the current objective. Moreover, all the compounds is expected to have Central Nervous System (CNS) permeability as for them, their logPS values is smaller than −2.0 which is good as only the drugs for the nervous system disease. Metabolism prediction suggested that all these screened compounds do not affect cytochrome functioning in a major way, where none of the compounds out of five is predicted to show CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 inhibition. Renal excretion of all the compounds differ. None of the compounds show AMES toxicity suggesting these compounds does not possess mutagenicity. All the compounds are not expected to show any Hepatotoxicity and Skin Sensitisation. The hERG I and II are potassium channels encoded by hERG, and their inhibition can cause QT syndrome (QT refers to the peaks of heart electrocardiogram) which affects the repolarization of the heart after a heartbeat. Their inhibition properties by screened phytochemicals are depicted in Table 3 along with other essential ADMET properties.
Table 3.
ADMET properties of screened phytochemicals.
| Property | Model Name | Phytochemicals |
||||
|---|---|---|---|---|---|---|
| Racemoside A/C | Ashwagandhanolide | Withanoside VI | Withanoside IV | Unit | ||
| Absorption | Water solubility | −3.259 | −3.159 | −2.974 | −2.887 | Numeric (log mol/L) |
| Absorption | Caco2 permeability | −0.722 | 0.38 | −0.684 | −0.631 | Numeric (log Papp in 10−6 cm/s) |
| Absorption | Intestinal absorption (human) | 63.519 | 63.819 | 39.364 | 31.451 | Numeric (% Absorbed) |
| Absorption | Skin Permeability | −2.735 | −2.735 | −2.735 | −2.735 | Numeric (log Kp) |
| Absorption | P-glycoprotein substrate | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Absorption | P-glycoprotein I inhibitor | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Absorption | P-glycoprotein II inhibitor | No | Yes | No | No | Categorical (Yes/No) |
| Distribution | VDss (human) | −0.348 | −1.652 | −0.563 | −0.605 | Numeric (log L/kg) |
| Distribution | Fraction unbound (human) | 0.349 | 0.249 | 0.408 | 0.398 | Numeric (Fu) |
| Distribution | BBB permeability | −1.733 | −1.006 | −1.595 | −1.51 | Numeric (log BB) |
| Distribution | CNS permeability | −4.281 | −2.859 | −4.412 | −4.436 | Numeric (log PS) |
| Metabolism | CYP2D6 substrate | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP3A4 substrate | Yes | Yes | No | No | Categorical (Yes/No) |
| Metabolism | CYP1A2 inhibitor | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP2C19 inhibitor | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP2C9 inhibitor | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP2D6 inhibitor | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP3A4 inhibitor | No | No | No | No | Categorical (Yes/No) |
| Excretion | Total Clearance | 0.302 | −0.679 | 0.544 | 0.636 | Numeric (log ml/min/kg) |
| Excretion | Renal OCT2 substrate | No | No | No | No | Categorical (Yes/No) |
| Toxicity | AMES toxicity | No | No | No | No | Categorical (Yes/No) |
| Toxicity | Max. tolerated dose (human) | −2.635 | −0.058 | −1.925 | −1.94 | Numeric (log mg/kg/day) |
| Toxicity | hERG I inhibitor | No | No | No | No | Categorical (Yes/No) |
| Toxicity | hERG II inhibitor | Yes | No | Yes | Yes | Categorical (Yes/No) |
| Toxicity | Oral Rat Acute Toxicity (LD50) | 3.025 | 3.704 | 2.875 | 2.886 | Numeric (mol/kg) |
| Toxicity | Oral Rat Chronic Toxicity (LOAEL) | 2.511 | 1.49 | 3.401 | 3.501 | Numeric (log mg/kg_bw/day) |
| Toxicity | Hepatotoxicity | No | No | No | No | Categorical (Yes/No) |
| Toxicity | Skin Sensitisation | No | No | No | No | Categorical (Yes/No) |
| Toxicity | T. pyriformis toxicity | 0.285 | 0.285 | 0.285 | 0.285 | Numeric (log ug/L) |
| Toxicity | Minnow toxicity | 9.971 | 2.486 | 7.489 | 6.847 | Numeric (log mM) |
3.4. MD simulations
Racemoside A, Ashwagandhanolide and Withanoside VI were selected after secondary screening by assessing the properties of their interaction with RBD based on molecular docking, structural analysis based on pharmacophore profile and ADMET prediction. The complexes (i) RBD-Racemoside A, (ii) RBD-Ashwagandhanolide and (iiI) RBD- Withanoside VI were then subjected to 100 ns of MD simulations.
After performing MD simulations, the Root Mean Square Deviation (RMSD) appraisal was performed which is utilized to quantify the normal change in dislodging of an of particles for a specific frame as for a reference constant frame. It is determined for all frames of trajectory. The plots in Fig. 3 depict RMSD movements in the portions of the protein (left Y-axis). The docked pose of ligand and protein in the complex is considered as the reference frame and then the movement for this original alignment during MD simulation is gauged by aligning all the protein frames concerning time. Checking the RMSD of the protein can give knowledge into its auxiliary 3D structural movement on a graph during the simulation. RMSD examination can demonstrate if the simulation has equilibrated — its changes towards the finish of the recreation are around some thermal energetically stable conformation. Changes in the range of 1–4 Å are completely satisfactory for little, globular proteins. However, this range of value widens as the size of the protein increases. For the complex of RBD-Racemoside A (Fig. 3a) the protein backbone hovers the value of RMSD not exceeding 3.0 Å; for the RBD-Ashwagandhanolide complex (Fig. 3b), the value stays well under 4.0 Å and for the RBD-Withanoside VI complex (Fig. 3c), the value stays well under 3.5 Å for most of the simulation. Ligand RMSD (right Y-axis, plots of Fig. 3) suggests the stability of ligand posture concerning the docked position of the ligand in the binding cleft of the protein. ‘Lig fit Prot’ suggests the RMSD of a ligand for protein backbone. For this, the values slightly larger than the protein’s RMSD are considered satisfactory but if the values observed are significantly larger than the RMSD of the protein, then it is likely that the ligand acquires a different stable position than the original posture. For RBD-Racemoside A (Fig. 3a), the Lig fit Prot stays significantly lower than protein’s RMSD up to 60 ns, but thereafter at exponentially increases and then again lowering and stabilizing suggesting the reorientation of Racemoside A. For RBD-Ashwagandhanolide (Fig. 3b), the Lig fit Prot value stayshigh and then stabilizes after 40 ns, suggesting the Ashwagandhanolide changing poses up to ∼40ns and then stabilizing. For RBD- Withanoside VI (Fig. 3c), the Lig fit Prot stays significantly higher than protein’s RMSD, however stays below 4.0 Å, suggesting its constant interaction with protein. Moreover, the protein (RBD) does not have a cavity into which the ligands can go inside and bind, rather here the ligands stick on the surface of protein therefore the Lig fit Prot values does not provide significant information. Lig fit Prot values is more decipherable for lock and key sort of phenomenon for ligand binding in protein cavity.
The Root Mean Square Fluctuation (RMSF) is useful for portraying confined changes along the protein chain (Fig. 4 ). In the graph, the peaks demonstrate regions of the protein that vary the most throughout the simulation. Ordinarily, the tails (N-and C-terminal) change the maximum than other internal regions of the protein. Secondary regions of proteins like alpha helices and beta strands are generally more inflexible and rigid than the unstructured regions and hence vacillate not exactly like loop forming portions of protein. Alpha-helical and beta-strand areas are featured in red and blue foundations, separately. These districts are characterized by helices or strands that endure over 70% of the whole re-enactment. Protein deposits that contact ligand is set apart with green-hued vertical bars. It is seen that all the three buildings RBD-Racemoside A (Fig. 4a) and RBD-Ashwagandhanolide (Fig. 4b), and RBD-Withanoside VI (Fig. 4c) protein interacts with the respective phytochemicals.
Fig. 4.

MD simulation Protein-ligand interaction root-mean-square fluctuation (RMSF) profile of (a) RBD-Racemoside A (b) RBD-Ashwagandhanolide and (c) RBD-Withanoside VI.
The conduct of all the three ligands during the whole course of simulation has appeared in Fig. 5 . Root mean square deviation (RMSD) of a ligand concerning the reference compliance (ideally the first frame of trajectory is utilized as the reference and it is viewed as time t = 0). The radius of Gyration (rGyr) evaluates the ’extendedness’ of a ligand and is equal to its essential snapshot of idleness. Intramolecular Hydrogen Bonds (intraHB) shows the number of inner hydrogen bonds inside a ligand atom. Molecular Surface Area (MolSA) portrays the sub-atomic surface figuring with a 1.4 Å test sweep. This worth is proportionate to a van der Waals surface zone. Solvent Accessible Surface Area (SASA) is the surface zone of a molecule open for access to water molecules and finally, Polar Surface Area (PSA) is the dissolvable available surface territory in a particle contributed uniquely by oxygen and nitrogen iotas. Every one of these highlights is qualities of individual compound (ligand) subsequently these estimations of two unique ligands cannot be compared directly.
Fig. 5.

Ligand properties for best hits such as RMSD, the radius of gyration (rGyr), intramolecular hydrogen bonds (intraHB), Molecular Surface Area (MolSA), Solvent Accessible Surface Area (SASA), Polar Surface Area (PSA) on interacting with protein during MD simulation.
Protein interactions with the ligand can be monitored throughout the simulation. These interactions can be categorized by type and summarized, as shown in Fig. 6 a for the RBD-Racemoside A complex; Fig. 7 a for the RBD-Ashwagandhanolide complex and Fig. 8 a for RBD-Withanoside VI complex. Protein-ligand interactions are categorized into four types: Hydrogen Bonds, Hydrophobic, Ionic and Water Bridges. Every connection type contains more explicit subtypes, which can be investigated through the ‘Simulation Interactions Diagram’ board. The stacked bar outlines are standardized throughout the direction: for instance, an estimation of 0.8 recommends that 80% of the simulation time the particular collaboration is kept up. Qualities over 1.0 are conceivable as some protein build-up may make numerous contacts of the same subtype with the ligand. A timetable portrayal of the associations and contacts (Hydrogen bonds, Hydrophobic, Ionic, Water spans) is appeared in Fig. 6b for the RBD-Racemoside A complex; Fig. 7b for the RBD-Ashwagandhanolide complex; and Fig. 8a for RBD-Withanoside VI complex. These figures portray which deposits communicate with the ligand in every direction outline. A few residues make more than one explicit contact with the ligand, which is shown to by a hazier shade of orange, as indicated by the scale to one side of the plot. These plots are very crucial suggesting these three compounds viz. Racemoside A, Ashwagandhanolide, and Withanoside VI interact with the amino acids throughout the simulation and are not dissociating away from their interacting site, however, the variations in the RMSD and RMSF values of the ligand as showed in Figs. 3 and 4 respectively, suggest that these ligands may be reorienting themselves during the simulation.
Fig. 6.

Protein-Ligand interaction profile of RBD-Racemoside A complex (a) interaction profile of crucial interacting amino acids (b) timeline representation of the interactions of amino acids.
Fig. 7.

Protein-Ligand interaction profile of RBD-Ashwagandhanolide complex (a) interaction profile of crucial interacting amino acids (b) timeline representation of the interactions of amino acids.
Fig. 8.

Protein-Ligand interaction profile of RBD-Withanoside VI complex (a) interaction profile of crucial interacting amino acids (b) timeline representation of the interactions of amino acids.
3.5. MM/GBSA binding free energy calculations
Post simulation analysis of all the three the complexes RBD-Racemoside A, RBD-Ashwagandhanolide and RBD-Withanoside VI were performed by taking snapshots of the trajectory profiles developed on performing 100 ns MD simulation and is depicted in Table 4 . Binding free energy change calculation provides an insight into the ligand potential to strongly interact with the amino acids of the protein. The energy released (ΔGbind) due to bond formation, or rather interaction of the ligand with protein is in the form of binding energy and it determines the stability of any given protein–ligand complex. The free energy of a favourable reaction is negative. It is observed that all the top three ligands have ΔGBind in the negative range. All the three compounds, tend to have similar energies corresponding to van der Waals interactions represented as ΔGvdW, suggesting these compounds tends to stay in the vicinity of the interacting amino amides of RBD. All the three compounds show negative values for Coulomb energy which describe the potential energy in systems. The negative value suggests that these ligands while interacting with RBD has poor potential energy, suggesting better stability as the ligands do not have enough potential energy to get destabilized. In addition to the total energy, the contributions to the total energy from different components such as Hydrogen-bonding correction, Lipophilic energy, Pi-pi packing correction and van der Waals energy is provided in Table 4.
Table 4.
MM/GBSA profiles of Racemoside A, Ashwagandhanolide and Withanoside VI, while interacting with RBD.
| Ligand | ΔGBind (Kcal/mol) | ΔGCoulomb (Kcal/mol) | ΔGHbond (Kcal/mol) | ΔGLipo (Kcal/mol) | ΔGPacking (Kcal/mol) | ΔGvdW (Kcal/mol) |
|---|---|---|---|---|---|---|
| Ligands interacting with RBD | ||||||
| Racemoside A | −89.06 | −34.83 | −4.45 | −55.81 | −5.25 | −54.51 |
| Ashwagandhanolide | −93.22 | −37.52 | −3.32 | −70.52 | −4.29 | −57.34 |
| Withanoside VI | −78.64 | −40.12 | −2.56 | −66.23 | −3.36 | −53.49 |
Note, meaning of abbreviations used in the table are as follows: Coulomb—Coulomb energy. Hbond—Hydrogen-bonding correction. Lipo—Lipophilic energy. Packing—Pi-Pi packing correction. vdW—Van der Waals energy.
4. Discussion
The S-protein has been the hype owing to its high degree of similarity existing amongst SARS-CoV and SARS-CoV-2, where 89.8% sequence identity is found amongst their S2 subunits, which mediate the membrane fusion process [33,34]. While both of their S1 subunits utilize ACE2 as the receptor to infect human cells but the S1 subunit of SARS-CoV-2 shows 10-fold more ACE2 binding affinity. Such striking similarity is not found for other proteins of SARS-CoV and SARS-CoV-2, this has raised the speculation in the minds of researchers, that SARS-CoV-2 may possess the reengineered version of S-protein from SARS-CoV’s. After binding of RBD in the S1 subunit of S-protein with ACE2 receptor of human cell, the heptad repeat 1 (HR1) and 2 (HR2) domains in its S2 subunit of S-protein interact with each other to form a six-helix bundle (6-HB) fusion core, bringing viral and host cell membranes into proximity for fusion and infection [33]. Blocking this phenomenon has huge potentials to stop the infection of virus and finding RBD inhibitors is of huge potentials, where researchers have even tried to block RBD with Human recombinant soluble ACE2 and other similar peptides [[35], [36], [37], [38], [39], [40]]. A natural food preservative peptide nisin can interact with the SARS-CoV-2 spike protein receptor human ACE2 is reported [41]. Fusion inhibitor, and the S-protein inhibitors in current study specifically inhibit this process of membrane fusion stopping the entry of viral components entering into the host cell [15]. As an effort to develop such inhibitors, EK1 peptide was created which focused explicitly HR1 subunit and have indicated promising reactions for in vivo mice model as well [33]. With restricted extension to work with SARS-CoV-2, as it requires Biosafety Level 4 (BSL4) arrangement, increasingly more exploration is been distributed with in silico approach with docking and MD simulations being the foundation of the computational examination, regardless of such examination ailing in vitro investigations, gigantic volumes of information demonstrating the collaborations of a functioning drug compound or phytochemical with the viral protein is been brought to the analysts area, which can come very handy for those having facility of performing in vitro examinations for approving computational estimates at last sparing a ton of time.
Under current study we have first constructed a library of phytochemicals of the compounds reported to have any kind of antiviral potentials. Here we meticulously constructed a collection of 110 compounds from the medicinal plants W. somnifera [19], A. racemosus [20], Z. officinalis [21], A. sativum [22], C. longa [23], A. vasica [20]. There are reports where the RBS of S-protein is focused to veil this site communicating with ACE2, where phytochemicals from Glycyrrhiza glabra are imagined to collaborate and obstruct the ACE2 restricting amino corrosive buildups of S-protein [17]. Efforts are also made for repurposing approved drugs as inhibitors of S-protein of SARS-CoV-2 where the authors have proposed 10 FDA approved drugs showing potentials to interact and mask the RBD [42]. Of lately, strategy of repurposing of the approved small molecule drugs in order to inhibit SARS-CoV-2 S protein and human ACE2 interaction through virtual screening approaches is used where drugs, Diammonium Glycyrrhizinate, Digitoxin, Ivermectin, Rapamycin, Rifaximin, and Amphotericin B represented the most desirable features to serve as RBD blockers [43]. Similar other study showed laxative drug, Bisoxatin to have potentials act as RBD blocker [44]. S-protein is additionally focused with the most ordinarily discovered phytochemicals, where Kaempferol, Quercetin, and Fisetin had been appeared to bind and block the RBD which is validated utilizing molecular docking and MD reenactments [45]. Terpenes from plant sources like Trevesia Palmata, Aralia Dasyphylla and Glycyrrhiza glabra have shown the potentials to block and mask the RBD using computational methods is reported [46]. In another study, Epigallocatechingallate, Curcumin, Apigenin and Chrysophanol were identified to be potential hits possessing ability to serve as RBD blockers [47]. There are efforts made to mask RBD using antibodies as well [18]. Our research presented in the current studies works in same fashion where, we have identified Racemoside A, Ashwagandhanolide and Withanoside VI from medicinal plants showing antiviral properties as discussed below.
As of late, our research group proposed Pyranonigrin A and Flaviolin to have the possibilities to interact with Mpro and hinder its viral replication capacity [9]. Another gathering of specialists has comparably recognized mixes from Tea plants that have the possibilities to associate with Mpro and restrain it’s capacity [48,49]. The subsequent methodology is to check the accessible commercial drugs to market against the SARS-CoV-2. There may be an exceptionally high possibility that current medications may interfere with the biochemical cycles of the virus and restrain it. Such a methodology is called drug repurposing. Utilizing drug repurposing, Procainamide, Tetrahydrozoline, Levamisole was distinguished as medications that can meddle with the papain-like protease of SARS-CoV-2 [50]. Such a methodology utilizes molecular docking and molecular dynamics as the center strategies for the in-silico examination, and by utilizing these techniques several lead compounds are recognized to have potential to meddle with the biochemistry and life cycle of SARS-CoV-2 as of late. There are a few spaces of life sciences where the approaches of docking and MD reproductions have been of an incredible use [[51], [52], [53]]. Comparable examinations utilizing docking and MD reproductions are likewise performed for accessing the potency of hydroxychloroquine with various targets of SARS-CoV-2 [54].
Looking through compounds that can restrain SARS-CoV-2 is of the most elevated need for the scientific fraternity. To commercialize such compounds, molecules from natural plant and microbe are been looked for, because of their low harmfulness, simple extraction and being effectively acknowledged by individuals. Such medications likewise have more limited periods of preliminaries. W. somnifera is known as Ashwagandha and is an important Indian medicinal plant containing plethora of pharmacologically important triterpenoid steroidal lactones, which are collectively termed as withanolides. The importance of phytochemicals from Ashwagandha for their antiviral properties are widely described [55]. Ashwagandhanolide, a bioactive dimeric thiowithanolide isolated from the roots of Ashwagandha. Previously Ashwagandhanolide was evaluated for its ability to inhibit cell proliferation against human lung (NCI H-460), breast (MCF7), colon (HCT-116), CNS (SF-268), and gastric (AGS) tumor cell lines using the MTT cell viability assay, as described earlier [56]. More recently, several compounds of Ashwagandha has been reported to inhibit Mpro of SARS-CoV-2 [57]. Further, compounds from Ashwagandha that were able to inhibit Mpro were Withanoside II, Withanoside IV, Withanoside V and Sitoindoside IX. Under current study the other compounds that can block the RBD of S-protein are Racemoside A, Withanoside VI, Withanoside IV and Racemoside C. Withanosides are also found to be in significant amounts from Ashwagandha and they are known to improved Abeta-induced memory impairment, neurite atrophy, and synaptic loss in the cerebral cortex and hippocampus in mice. Withanoside VI is also known to facilitates the regeneration of axons and dendrites by reconstructing pre- and post-synapses in neurodegenerative diseases and preventing pathogenesis and neuronal death. Withanoside VI is important candidate for the therapeutic treatment of neurodegenerative diseases. There are at least eleven different types of Withanosides found in Ashwagandha [58]. Off lately Withanoside X, is also reported to inhibit SARS-CoV-2 viral entry in to the cell [59]. Moreover, this corroborates with our results too where we have three phytochemicals namely Ashwagandhanolide, Withanoside VI and Withanoside IV of W. somnifera to have shown exceptional potency to interact with RBD of S-protein and inhibit its interaction with ACE2 ultimately blocking the entry of viral particle in to the human host. The other two important phytochemicals to have shown similar potency belongs to the plant A. racemosus and these phytochemicals are Racemoside A and Racemoside C. Racemosides are natural steroidal saponins and are well-known to have anti-leishmanial properties [60]. Moreover, Racemosides are also reported to interact with S-protein and other structural proteins of SARS-CoV-2 [61].
To summarize the entire study, to start with we have used the standard workflow used for “receptor-based drug designing” which deals screening and selecting top hits of phytochemicals based on docking, then verifying the interactions with simulation. The interaction screened phytochemicals with human body is predicted with ADMET and as an add on to see which structural features of screened phytochemicals can interact with amino acids, we have performed pharmacophore mapping. For the study we created the library of 110 compounds from the medicinal plants W. somnifera, A. racemosus, Z. officinalis, A. sativum, C. longa and A. vasica, of which we found Racemoside A, Ashwagandhanolide and Withanoside VI to be the most potent molecules to bind RBD thereby blocking its interaction with RBD.
Compliance with ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Authors contributions
CP, DG and DJ conceptualized and designed the project. CP and DG developed methodology. CP and DG acquired data and wrote the manuscript. CP and DG analyzed and interpreted data. HP, RP and HS provided technical support. HP supervised the study. The whole manuscript was approved by all authors.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The Financial Assistance Programme – Department of Science and Technology [Grant number GSBTM/MD/JDR/1409/2017–18] and Gujarat Council on Science and Technology [GUJCOST/Supercomputer/2019–20/1359] had provided grants for the purchase of computer hardware only and is been used up. The research work is not been granted from any fund. The authors gratefully acknowledge the Department of Botany, Bioinformatics and Climate Change Impacts Management, Gujarat University for providing an opportunity to access the bioinformatics research facilities.
References
- 1.Schwartz D.A., Graham A.L. Potential maternal and infant outcomes from coronavirus 2019-NCOV (SARS-CoV-2) infecting pregnant women: lessons from SARS, MERS, and other human coronavirus infections. Viruses. 2020;12 doi: 10.3390/v12020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joshi R.S., Jagdale S.S., Bansode S.B., Shiva Shankar S., Tellis M.B., Pandya K., Chugh A., Giri A.P., Kulkarni M.J., Joshi S. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1760137. Taylor Fr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y., Guo Y., Pan Y., Zhao Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020 doi: 10.1016/j.bbrc.2020.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song Z., Xu Y., Bao L., Zhang L., Yu P., Qu Y., Zhu H., Zhao W., Han Y., Qin C. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. 2019;11 doi: 10.3390/v11010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., Yuan M.L., Zhang Y.Z.Y.Z.L., Dai F.H., Liu Y., Wang Q.M., Zheng J.J., Xu L., Holmes E.C., Zhang Y.Z.Y.Z.L. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao P., Shukla A., Parmar P., Rawal R.M., Patel B., Saraf M., Goswami D. Reckoning a fungal metabolite, Pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophys. Chem. 2020 doi: 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kong R., Yang G., Xue R., Liu M., Wang F., Hu J., Guo X., Chang S. J ArXiv Prepr.; 2020. COVID-19 Docking Server: an Interactive Server for Docking Small Molecules, Peptides and Antibodies against Potential Targets of COVID-19.https://arxiv.org/abs/2003.00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao P., Shukla A., Parmar P., Rawal R., Patel B., Saraf M., Goswami D. Proposing a fungal metabolite-Flaviolin as a potential inhibitor of 3CLpro of novel coronavirus SARS-CoV-2 identified using Docking and Molecular Dynamics. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1813202. [DOI] [PubMed] [Google Scholar]
- 10.Chen X., Li R., Pan Z., Qian C., Yang Y., You R., Zhao J., Liu P., Gao L., Li Z., Huang Q., Xu L., Tang J., Tian Q., Yao W., Hu L., Yan X., Zhou X., Wu Y., Deng K., Zhang Z., Qian Z., Chen Y., Ye L. Human monoclonal antibodies block the binding of SARS-CoV-2 spike protein to angiotensin converting enzyme 2 receptor. Cell. Mol. Immunol. 2020;17:647–649. doi: 10.1038/s41423-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020;117 doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trezza A., Iovinelli D., Santucci A., Prischi F., Spiga O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020;10:1–8. doi: 10.1038/s41598-020-70863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia S., Yan L., Xu W., Agrawal A.S., Algaissi A., Tseng C.T.K., Wang Q., Du L., Tan W., Wilson I.A., Jiang S., Yang B., Lu L. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019;5:eaav4580. doi: 10.1126/sciadv.aav4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia S., Zhu Y., Liu M., Lan Q., Xu W., Wu Y., Ying T., Liu S., Shi Z., Jiang S., Lu L. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein, Cell. Mol. Immunol. 2020;17:765–767. doi: 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinha S.K., Prasad S.K., Islam M.A., Gurav S.S., Patil R.B., AlFaris N.A., Aldayel T.S., AlKehayez N.M., Wabaidur S.M., Shakya A. Identification of bioactive compounds from Glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: a pharmacoinformatics study. J. Biomol. Struct. Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1779132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y., Yang C., feng Xu X., Xu W., wen Liu S. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020:1–9. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mwitari P.G., Ayeka P.A., Ondicho J., Matu E.N., Bii C.C. Antimicrobial activity and probable mechanisms of action of medicinal plants of Kenya: Withania somnifera, Warbugia ugandensis, Prunus africana and Plectrunthus barbatus. PloS One. 2013;8 doi: 10.1371/journal.pone.0065619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sabde S., Bodiwala H.S., Karmase A., Deshpande P.J., Kaur A., Ahmed N., Chauthe S.K., Brahmbhatt K.G., Phadke R.U., Mitra D., Bhutani K.K., Singh I.P. Anti-HIV activity of Indian medicinal plants. J. Nat. Med. 2011;65:662–669. doi: 10.1007/s11418-011-0513-2. [DOI] [PubMed] [Google Scholar]
- 21.Lamari Z., Larbi R., Negache H. Trace element content of Zingiber officinalis and Salvia officinalis medicinal plants from Algeria. J. Radioanal. Nucl. Chem. 2016;309:17–22. doi: 10.1007/s10967-016-4858-6. [DOI] [Google Scholar]
- 22.Balasubramanian G., Sarathi M., Kumar S.R., Hameed A.S.S. Screening the antiviral activity of Indian medicinal plants against white spot syndrome virus in shrimp. Aquaculture. 2007;263:15–19. doi: 10.1016/j.aquaculture.2006.09.037. [DOI] [Google Scholar]
- 23.Dao T.T., Nguyen P.H., Won H.K., Kim E.H., Park J., Won B.Y., Oh W.K. Curcuminoids from Curcuma longa and their inhibitory activities on influenza A neuraminidases. Food Chem. 2012;134:21–28. doi: 10.1016/j.foodchem.2012.02.015. [DOI] [Google Scholar]
- 24.Krieger E., Darden T., Nabuurs S.B., Finkelstein A., Vriend G. vol. 57. Wiley Online Libr; 2004. pp. 678–683. (Making Optimal Use of Empirical Energy Functions: Force-Field Parameterization in Crystal Space). [DOI] [PubMed] [Google Scholar]
- 25.Krieger E., Vriend G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel C.N., Kumar S.P., Modi K.M., Soni M.N., Modi N.R., Pandya H.A. Cardiotonic steroids as potential Na+/K+-ATPase inhibitors–a computational study. J. Recept. Signal Transduct. 2019;39:226–234. doi: 10.1080/10799893.2019.1660893. [DOI] [PubMed] [Google Scholar]
- 29.Wang W., Donini O., Reyes C.M., Kollman P.A. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct. 2001;30:211–243. doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- 30.Wang J., Hou T., Xu X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput. Aided Drug Des. 2006;2:287–306. doi: 10.2174/157340906778226454. [DOI] [Google Scholar]
- 31.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A., Cheatham T.E. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 32.Massova I., Kollman P.A. Combined molecular mechanical and continuum solvent approach (MM- PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000;18:113–135. doi: 10.1023/A:1008763014207. [DOI] [Google Scholar]
- 33.Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu R., Chen L., Lan R., Shen R., Li P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents. 2020;56 doi: 10.1016/j.ijantimicag.2020.106012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J., Petitjean S., Derclaye S., Koehler M., Zhang Q., Dumitru A., Soumillion P., Alsteens D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Res. Sq. 2020 doi: 10.21203/rs.3.rs-30468/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baig M.S., Alagumuthu M., Rajpoot S., Saqib U. Identification of a potential peptide inhibitor of SARS-CoV-2 targeting its entry into the host cells. Drugs R. 2020;20:161–169. doi: 10.1007/s40268-020-00312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong F.-C., Ong J.-H., Chai T.-T. EFood; 2020. Identification of Putative Cell-Entry-Inhibitory Peptides against SARS-CoV-2 from Edible Insects: an in Silico Study. [DOI] [Google Scholar]
- 38.Alagumuthu M., Rajpoot S., Baig M.S. Structure-based design of novel peptidomimetics targeting the SARS-CoV-2 spike protein. Cell. Mol. Bioeng. 2020 doi: 10.1007/s12195-020-00658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schütz D., Ruiz-Blanco Y.B., Münch J., Kirchhoff F., Sanchez-Garcia E., Müller J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 2020;167:47–65. doi: 10.1016/j.addr.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathod S.B., Prajapati P.B., Punjabi L.B., Prajapati K.N., Chauhan N., Mansuri M.F. Peptide modelling and screening against human ACE2 and spike glycoprotein RBD of SARS-CoV-2. Silico Pharmacol. 2020;8 doi: 10.1007/s40203-020-00055-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhattacharya R., Gupta A.M., Mitra S., Mandal S., Biswas S.R. A natural food preservative peptide nisin can interact with the SARS-CoV-2 spike protein receptor human ACE2. Virology. 2021;552:107–111. doi: 10.1016/j.virol.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Oliveira O.V., Rocha G.B., Paluch A.S., Costa L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalhor H., Sadeghi S., Abolhasani H., Kalhor R., Rahimi H. Repurposing of the approved small molecule drugs in order to inhibit SARS-CoV-2 S protein and human ACE2 interaction through virtual screening approaches. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1824816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Unni S., Aouti S., Thiyagarajan S., Padmanabhan B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020;45 doi: 10.1007/s12038-020-00102-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandey P., Rane J.S., Chatterjee A., Kumar A., Khan R., Prakash A., Ray S. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. J. Biomol. Struct. Dyn. 2020:1–11. doi: 10.1080/07391102.2020.1796811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muhseen Z.T., Hameed A.R., Al-Hasani H.M.H., Tahir ul Qamar M., Li G. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (RBD) attachment inhibitors to the human ACE2 receptor: integrated computational approach. J. Mol. Liq. 2020;320 doi: 10.1016/j.molliq.2020.114493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ravichandran K., Sankar M. In silico molecular docking analysis of targeting SARS-CoV-2 spike protein and selected herbal constituents. Artic. J. Pure Appl. Microbiol. 2020 doi: 10.22207/JPAM.14.SPL1.37. [DOI] [Google Scholar]
- 48.Bhardwaj V.K., Singh R., Sharma J., Rajendran V., Purohit R., Kumar S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1766572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhardwaj V.K., Purohit R. Targeting the protein-protein interface pocket of Aurora-A-TPX2 complex: rational drug design and validation. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1772109. [DOI] [PubMed] [Google Scholar]
- 50.Arya R., Das A., Prashar V., Kumar M. 2020. Potential Inhibitors against Papain-like Protease of Novel Coronavirus (SARS-CoV-2) from FDA Approved Drugs. [Google Scholar]
- 51.Parmar P., Shukla A., Rao P., Saraf M., Patel B., Goswami D.%J.B.C. 2020. The Rise of Gingerol as Anti-QS Molecule: Darkest Episode in the LuxR-Mediated Bioluminescence Saga. [DOI] [PubMed] [Google Scholar]
- 52.Shukla A., Parmar P., Rao P., Goswami D., Saraf M. Twin peaks: presenting the antagonistic molecular interplay of Curcumin with LasR and LuxR quorum sensing pathways. Curr. Microbiol. 2020 doi: 10.1007/s00284-020-01997-2. [DOI] [PubMed] [Google Scholar]
- 53.Kumar S.P., Patel C.N., Rawal R.M., Pandya H.A. Energetic contributions of amino acid residues and its cross-talk to delineate ligand-binding mechanism. Proteins Struct. Funct. Bioinf. 2020;88:1207–1225. doi: 10.1002/prot.25894. [DOI] [PubMed] [Google Scholar]
- 54.Mukherjee S., Dasgupta S., Adhikary T., Adhikari U., Panja S.S. Structural insight to hydroxychloroquine-3C-like proteinase complexation from SARS-CoV-2: inhibitor modelling study through molecular docking and MD-simulation study. J. Biomol. Struct. Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1804458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bomzan D.P., Shilpashree H.B., Anjali P., Kumar S.R., Nagegowda D.A. Methods Mol. Biol. Humana Press Inc.; 2020. Virus-induced gene silencing for functional genomics in withania somnifera, an important indian medicinal plant; pp. 139–154. [DOI] [PubMed] [Google Scholar]
- 56.Subbaraju G.V., Vanisree M., Rao C.V., Sivaramakrishna C., Sridhar P., Jayaprakasam B., Nair M.G. Ashwagandhanolide, a bioactive dimeric thiowithanolide isolated from the roots of Withania somnifera. J. Nat. Prod. 2006;69:1790–1792. doi: 10.1021/np060147p. [DOI] [PubMed] [Google Scholar]
- 57.Tripathi M.K., Singh P., Sharma S., Singh T.P., Ethayathulla A.S., Kaur P. Identification of bioactive molecule from Withania somnifera (Ashwagandha) as SARS-CoV-2 main protease inhibitor. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1790425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Girme A., Saste G., Pawar S., Balasubramaniam A.K., Musande K., Darji B., Satti N.K., Verma M.K., Anand R., Singh R., Vishwakarma R.A., Hingorani L. Investigating 11 Withanosides and Withanolides by UHPLC–PDA and mass fragmentation studies from Ashwagandha (Withania somnifera) ACS Omega. 2020;5:27933–27943. doi: 10.1021/acsomega.0c03266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chikhale R.V., Gurav S.S., Patil R.B., Sinha S.K., Prasad S.K., Shakya A., Shrivastava S.K., Gurav N.S., Prasad R.S. Sars-cov-2 host entry and replication inhibitors from Indian ginseng: an in-silico approach. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1778539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dutta A., Ghoshal A., Mandal D., Mondal N.B., Banerjee S., Sahu N.P., Mandal C. Racemoside A, an anti-leishmanial, water-soluble, natural steroidal saponin, induces programmed cell death in Leishmania donovani. J. Med. Microbiol. 2007;56:1196–1204. doi: 10.1099/jmm.0.47114-0. [DOI] [PubMed] [Google Scholar]
- 61.Chikhale R.V., Sinha S.K., Patil R.B., Prasad S.K., Shakya A., Gurav N., Prasad R., Dhaswadikar S.R., Wanjari M., Gurav S.S. In-silico investigation of phytochemicals from Asparagus racemosus as plausible antiviral agent in COVID-19. J. Biomol. Struct. Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1784289. [DOI] [PubMed] [Google Scholar]