

Abstract
Diaphyseal long bone cortical tissue from 30 patients with lethal perinatal Sillence II and progressively deforming Sillence III osteogenesis imperfecta (OI) has been studied at multiple levels of structural resolution. Interpretation in the context of woven to lamellar bone formation by mesenchymal osteoblasts (MOBLs) and surface osteoblasts (SOBLs) respectively demonstrates lamellar on woven bone synthesis as an obligate self-assembly mechanism and bone synthesis following the normal developmental pattern but showing variable delay in maturation caused by structurally abnormal or insufficient amounts of collagen matrix. The more severe the variant of OI is, the greater the persistence of woven bone and the more immature the structural pattern; the pattern shifts to a structurally stronger lamellar arrangement once a threshold accumulation for an adequate scaffold of woven bone has been reached. Woven bone alone characterizes lethal perinatal variants; variable amounts of woven and lamellar bone occur in progressively deforming variants; and lamellar bone increasingly forms rudimentary and then partially compacted osteons not reaching full compaction. At differing levels of microscopic resolution: lamellar bone is characterized by short, obliquely oriented lamellae with a mosaic appearance in progressively deforming forms; polarization defines tissue conformations and localizes initiation of lamellar formation; ultrastructure of bone forming cells shows markedly dilated rough endoplasmic reticulum (RER) and prominent Golgi bodies with disorganized cisternae and swollen dispersed tubules and vesicles, structural indications of storage disorder/stress responses and mitochondrial swelling in cells with massively dilated RER indicating apoptosis; ultrastructural matrix assessments in woven bone show randomly oriented individual fibrils but also short pericellular bundles of parallel oriented fibrils positioned obliquely and oriented randomly to one another and in lamellar bone show unidirectional fibrils that deviate at slight angles to adjacent bundles and obliquely oriented fibril groups consistent with twisted plywood fibril organization. Histomorphometric indices, designed specifically to document woven and lamellar conformations in normal and OI bone, establish ratios for: i) cell area/total area X 100 indicating the percentage of an area occupied by cells (cellularity index) and ii) total area/number of cells (pericellular matrix domains). Woven bone is more cellular than lamellar bone and OI bone is more cellular than normal bone, but these findings occur in a highly specific fashion with values (high to low) encompassing OI woven, normal woven, OI lamellar and normal lamellar conformations. Conversely, for the total area/number of cells ratio, pericellular matrix accumulations in OI woven are smallest and normal lamellar largest. Since genotype-phenotype correlation is not definitive, interposing histologic/structural analysis allowing for a genotype-histopathologic-phenotype correlation will greatly enhance understanding and clinical management of OI.
Keywords: Osteogenesis imperfecta, Histopathology, ER Golgi stress, Woven bone, Lamellar bone
Highlights
-
•
Mosaic appearance of lamellar bone composed of short lamellae at oblique angles
-
•
Histomorphometry establishes new cellularity indices for woven and lamellar bone.
-
•
Golgi bodies by TEM show irregular dispersed cisternae and swollen tubulo-vesicles.
-
•
Changes in matrix conformation along woven to lamellar continuum reflect OI severity.
1. Introduction
Osteogenesis imperfecta (OI) is an hereditary connective tissue disorder characterized by variable degrees of bone fragility leading to fractures and deformation (Nijhuis et al., 2019; Bullough et al., 1981). Phenotypic variability ranges from severe under-formation of bone incompatible with life, the lethal perinatal variant, to a benign autosomal dominant condition with full-life expectancy where there is no bone deformity and fracture rarely occurs. Molecular changes in OI are increasingly well outlined but genotype-phenotype correlations are not definitive. Structural studies have become infrequent other than formalized histomorphometry on iliac crest bone biopsies. We previously defined a positional and functional difference in osteoblasts referred to as mesenchymal osteoblasts (MOBLs) that synthesize woven bone and surface osteoblasts (SOBLs) that synthesize lamellar bone (Shapiro, 1988; Shapiro, 2008; Shapiro and Wu, 2019). The terms static and dynamic osteogenesis refer to the same phenomenon (Ferretti et al., 2002). We recognize a universal sequence of woven to lamellar bone formation by MOBLs and SOBLs respectively in the synthesis of normal, repair and pathologic bone (Shapiro and Wu, 2019). OI bone is assessed i) in the context of MOBLs and SOBLs forming woven and lamellar bone conformations respectively and ii) as a molecular disorder based on abnormalities affecting collagen synthesis. The histologic spectrum of cell and tissue changes from lethal perinatal and progressively deforming OI long bones are outlined at multiple levels of resolution by light microscopy (LM), polarizing light microscopy (PLM), transmission electron microscopy (TEM) and histomorphometric (HM) measurements designed for specific parameters of woven and lamellar bone. We also interpret observations from previous histopathologic studies in terms of the woven bone – lamellar bone continuum and current knowledge of mutations affecting collagen synthesis. Clinical severity of OI, while based on molecular abnormalities, is accurately reflected in the structure of bone along the woven to lamellar continuum. We propose that increased use of structural bone studies would translate into a meaningful bridge between molecular studies and clinical management.
2. Materials and methods
2.1. Bone specimens
2.1.1. Osteogenesis imperfecta bone
Bone specimens from 30 patients with osteogenesis imperfecta were obtained at autopsy (4 patients, Sillence II lethal peri-natal) or from intact long bones at time of osteotomy to correct deformity (26 patients, Sillence III progressively deforming). Osteotomies were done electively on intact but deformed bones, none of the tissue was obtained at time of or shortly after acute fractures and tissue removed as part of correction otherwise would have been discarded. Patients were categorized using Sillence et al. terminology (Sillence et al., 1979a) and temporal/radiographic categorization [OI Congenita A, OI Congenita B, OI Tarda A, OI Tarda B] based on congenita/tarda definitions (Shapiro, 1985; Kocher and Shapiro, 1998). Reference to the two classification systems allows histological findings from earlier studies to be incorporated into current clinical terminology; lethal perinatal Sillence II = OIC A and progressively deforming Sillence III = OIC B, OIT A and earlier more severe OIT B.
2.1.2. Normal bone
Normal bone specimens were obtained from 11 individuals. Neonatal, childhood and young adult cortical diaphyseal bone was obtained from 9 patients who died from non-skeletal disorders or had non-affected bone, that would otherwise have been discarded, removed at amputation or tumor resection for osteosarcoma. Human fetal long bone histology slides (2 fetuses) were reviewed from longstanding Orthopedic Research Laboratory collections. Cortical long bone tissue for our studies was obtained over a several year period by one author (FS) at Boston Children's Hospital, Boston, MA, from which the study had continuous IRB approval.
2.2. Tissue preparation
2.2.1. Light microscopy
Bone was prepared for light microscopy examination by one of three methods: paraffin embedding, JB4 plastic embedding, and epon embedding (tissue being prepared for transmission electron microscopy). For paraffin and plastic embedding techniques, tissues were fixed initially in 10% neutral buffered formalin and then decalcified in 25% formic acid. After embedding, they were sectioned at 5 μ thickness and stained either with hematoxylin and eosin (paraffin) or 1% toluidine blue (plastic). The sections embedded in epon were sectioned at 1 μ thickness and stained with 1% toluidine blue. Assessments included: osteocyte cellularity in relation to matrix structure (woven or lamellar), proportions and appearances of woven and lamellar bone when both were present on the same tissue section, and structure of lamellar tissue deposition.
2.2.2. Transmission electron microscopy
OI bone tissue was obtained from intact femurs or tibias undergoing osteotomy for deformity correction. Bone samples were fixed in the operating room for maximum preservation of cell detail. Bone spicules approximately 1 mm × 1 mm × 1 mm were fixed in a mixture of one part 2.5% glutaraldehyde to two parts 1.0% osmium tetroxide and 0.1 M cacodylate buffer. Decalcification was performed in 7.5% EDTA and 2.5% glutaraldehyde. Tissues were embedded in Epon, sectioned at 1 μ thickness, stained with 1% toluidine blue and examined by light microscopy to choose areas for ultrastructural assessment. Blocks were trimmed, sectioned at 60 nm and stained with lead citrate and uranyl acetate.
2.3. Tissue examination
2.3.1. Light microscopy
Tissue was examined from all 30 OI and 11 normal individuals. Multiple tissue samples were examined since several bones per individual were often obtained (autopsies) and several OI patients had more than one operative procedure. LM assessment initially used a Zeiss photomicroscope with polarization capability and subsequently an Olympus BX50 photomicroscope equipped for polarization studies. Zeiss-examined tissue was photographed using film while tissue examined on the Olympus microscope was recorded by a digital camera system.
2.3.2. Polarizing light microscopy
All tissue sections examined by LM were also examined under polarization. Tissue sections were examined by PLM on the Zeiss microscope using polarization sheets and on an Olympus B50 microscope with polarization adaptation using a γ 137 nm U-TP137 quarter wave test (phase) plate. For demonstration and documentation of the polarization of any specific matrix, a light microscopy section photomicrograph was taken followed immediately by a polarization view of the same section. Both plastic and paraffin embedded sections were examined.
2.3.3. Transmission electron microscopy
TEM assessments of OI bone were performed on 16 patients, encompassing 20 groups of specimens due to repeat procedures on some. Examination was performed on a JEOL 100C or JEOL 1200 electron microscope at 60 kV. Specific attention was directed to: i) mesenchymal osteoblasts (MOBLs) of woven bone, surface osteoblasts (SOBLs) synthesizing lamellar bone and osteocytes within the specific matrices and ii) collagenous matrix in terms of its conformation (woven or lamellar), fibril appearance and fibril diameters.
2.4. Correlations between histology and radiographic structure
2.4.1. Samples assessed
Femoral and tibial cortical bone samples (41) were assessed in a subset of 17 OI patients in relation to their corresponding long bone radiographs. Diaphyseal bone tissue was assessed from 4 type II patients (lethal perinatal) undergoing autopsy and 13 type III patients (progressively deforming) undergoing osteotomies. Some of the type III patients had more than one long bone operated or repeated procedures to account for the number of samples assessed.
2.4.2. Histologic grading
Following light and polarizing microscopic examination bone sections were graded semi-quantitatively. Criteria were developed to reflect specific features of OI cortical bone: cortical compaction, determined as <50%, 50% to 75%, and > 75%; lamellar pattern, discontinuous (mosaic) pattern, referring to short segments of lamellar bone positioned obliquely and or at right angles to adjacent segments or continuous (normal) pattern; and percentage of woven and lamellar bone, grade 1: all woven, grade 2: woven and lamellar (2a woven > lamellar, 2b woven = lamellar, 2c woven < lamellar), and grade 3: all lamellar (3a partially compacted, 3b fully compacted).
2.4.3. Radiographic grading
Radiographs of the bone from which specimens were obtained were assessed. Grading criteria reflected major determinants of structure: cortical thickness, thin or thick and long bone diaphyseal shape, mildly or markedly bowed.
2.5. Histomorphometry
2.5.1. Light microscopy
Histomorphometry studies involved assessment of multiple histologic sections from OI femoral and tibial cortical diaphyseal bone in 12 patients with type II OI (OIC A) and type III OI (OIC B or OIT A) and from normal cortical diaphyseal bone in 11 individuals (2 approximately 4-5 month fetuses [3 femurs, 2 humeri], 2 newborn femurs from autopsies for non-skeletal disorders and 7 children and adolescents undergoing amputation or wide tumor resection for osteosarcoma).
Quantitation was performed in specimens with areas clearly definable as woven or lamellar bone. Cell and matrix outlines were performed either on standardized photomicrographs taken at 16× magnification, printed at 8″ X 10″ size, and traced on the Zeiss Interactive Digital Analysis System (Zidas) or directly from microscopic images using the digitized histomorphometry application on the Olympus B50 microscope. LM and PLM examination defined woven bone in lethal perinatal Sillence II (OIC A) and fetal specimens, mixtures of woven and lamellar bone in progressively deforming Sillence III (OIC B and OIT A) and newborn normal bone and lamellar bone alone in normal childhood and adolescent specimens. Two or three histologic sections were made from each of several regions in a bone tissue specimen. Values from the 2 or 3 slides were quantitated and a mean value was determined to serve as the value from that localized area. A mean value was then determined from several regions of a specimen to serve as the value for the bone/specimen.
2.5.2. Quantitation of woven and lamellar bone areas
Quantitation of woven and lamellar bone areas involved: i) total area of the specific tissue being assessed (woven or lamellar); ii) area of the cells in the specific tissue (woven or lamellar measured as the area of the lacunae) and iii) number of cells in the specific area (woven or lamellar). Two types of ratio were then documented: a) Cell area/total area ratios of woven and lamellar bone were determined and converted to more clearly understood percent tissue involvement as cell area/total area X 100. These valuations serve as an index of cellularity of tissue types, referred to as a cellularity index. b) Total area/number (#) of cells ratios of woven and lamellar bone were determined. These valuations serve as an indication of the relative amount (area) of a tissue type associated with/supported by an osteocyte within it, referred to as the spatial pericellular domain.
2.5.3. Statistical analyses
Statistical analyses were performed using the GraphPad Prism 5 software (GraphPad Software, San Diego, CA). Data were evaluated using either student's t-test or two-way ANOVA with multiple comparisons and Bonferroni's test as the post-hoc analysis. All data are shown either as mean ± SD unless indicated.
2.5.4. Ultrastructural quantitation of collagen fibril diameters from OI and normal bone
Collagen fibril diameters in bone tissue were measured on numerous cross-sections from multiple bone spicules from 9 patients with OI and 2 normal controls. A mean number of 16 (range 10–26) fibers from 2 or 3 different regions of the same bone were measured. Fiber diameters were measured from newly synthesized osteoid as these were sufficiently discrete to allow for accurate quantitation. Electron micrographs of 35,400 magnification or greater were used to enhance accuracy. Fibril diameters were measured using a millimeter scale examined through an 8× magnification loupe (Agfa – Lupe 8×, Agfa-Gevaert AG, Munich, Germany) and millimeter values were converted to nanometers using the Slide Guide (Dunne and Riedmann, Pacific Palisades, CA).
3. Results
3.1. Light microscopy findings related to clinical classification
Light microscopic images of osteogenesis imperfecta bone are presented in Fig. 1a–g.
Fig. 1.



Osteogenesis imperfecta bone tissues assessed by light microscopy photomicrographs are shown.
a. i) Anteroposterior and lateral views of a femoral specimen X-ray from an OI Sillence II (OIC A) patient who died a few weeks after birth are shown; ii) Cross-section of the femur diaphysis (same specimen) shows periosteum (P) at top, a narrow collection of discontinuous cortical woven bone (W) and an extensive marrow cavity (M) without trabeculae below. Plastic embedded 1% toluidine blue stained section.
b. i) Woven bone from tibial cortex of a type II (OIC A) patient is surrounded by cellular tissue with vessels. Paraffin embedded hematoxylin and eosin stained (black and white) section. ii) A separate section of tibial cortex in a type II (OIC A) patient shows thin woven bone spicules separated by wide minimally cellular spaces. Plastic embedded 1% toluidine blue stained section.
c. i) Woven bone alone is present in femoral cortex from type II (OIC A) patient. The tissue is well vascularized (arrows). Woven bone cells are round to oval and closely packed. Plastic embedded 1% toluidine blue stained section. ii) Woven bone (W) accumulations, even in type II bone, when sufficiently extensive can lead to coverage by surface osteoblasts (SOBLs). MOBLs = mesenchymal osteoblasts. Plastic embedded 1% toluidine blue stained section.
d. i) Lamellar on woven bone pattern of tissue deposition is seen with close observation in specimen from type III patient. Cell shape is distinctive in each tissue conformation; woven bone cells are round to oval and lamellar bone osteocytes are elliptical and elongated along the long axis of the lamellae. W = woven bone, L = lamellar bone and SOBLs = surface osteoblasts. Plastic embedded 1% toluidine blue stained section. ii) Higher power LM shows lamellar (L) on woven (W) bone deposition in specimen from type III patient. Plastic embedded 1% toluidine blue stained section.
e. Four photomicrographs (i-iv) from tibial cortical bone in different type III patients demonstrate the mosaic pattern of lamellar OI bone formation in severe to moderate cases. The lamellae are short and obliquely positioned to one another. Figures i, ii and iv are also examples of partial compaction of the forming lamellar bone. Plastic embedded 1% toluidine blue stained sections.
f. i) Rudimentary (Rud) osteon formation begins as lamellar (L) bone on woven (W) bone formation increases. Tissue shown is from a severe type III (OIC B) femur. Plastic embedded 1% toluidine blue stained section. ii) The amount of lamellar bone is greatly increased in this type III patient and compaction has increased from rudimentary to partial. Plastic embedded 1% toluidine blue stained section. iii) Section illustrates the mechanism of osteonal formation from SOBLs synthesizing lamellar tissue and closing in on longitudinal blood vessels (BV). Lamellar (L) tissue has been deposited on woven (W) bone scaffolds in type III patient. Plastic embedded 1% toluidine blue stained section.
g. i) Osteoid (Os) tissue layer is prominent in this type II tissue section. The osteoid has been synthesized by the SOBLs on the woven (W) bone scaffold. Plastic embedded 1% toluidine blue stained section. ii) Lamellar bone from a type III patient shows a dense concentration of canaliculi linking adjacent osteocytes. Plastic embedded 1% toluidine blue stained section.
3.1.1. Lethal perinatal OI (Sillence II, OIC A)
Bone from 4 patients with lethal perinatal OI was assessed with similar findings in each. Spicules of highly cellular woven bone only without lamellar tissue are present in all femoral and tibial sections (Fig. 1a–c). Osteocytes are large, oval to irregularly shaped and separated by small amounts of matrix. Femoral and tibial cortices are composed of isolated segments of woven bone often with no continuity. Bone is well vascularized and marrow is cellular with few to absent trabeculae (Fig. 1a ii, c ii). Surface osteoblasts are seen occasionally on the more prominent collections of woven bone tissue but linear series of surface osteoblasts completely covering all woven bone accumulations are not seen (Fig. 1c ii). Where present, the SOBLs synthesize surface collections of parallel fibered matrix (osteoid) but these contain no osteocytes (Fig. 1g i). Within metaphyseal regions many cartilage cores remain uncovered by any bone or are covered only by discontinuous collections of bone. The epiphyseal, articular and physeal cartilage regions are structurally normal.
3.1.2. Progressively deforming OI (Sillence III, OIC B, OIT A)
In each patient, the cortex consists of both woven and lamellar bone with the lamellar bone having been synthesized on woven scaffolds (Fig. 1d). Sub-periosteal regions, where the most recently synthesized bone is present, are highly woven and regions deeper within the cortex show increasingly more lamellar bone since they had been synthesized earlier. The intermixture of these two conformational patterns is noted in all age groups and in both ambulatory and non-ambulatory patients. The lamellar bone is either parallel fibered and non-osteonal or, if osteonal, is either rudimentary or partially compacted only (Fig. 1e). Rudimentary osteons surround longitudinal vessels but are initially small and embedded in large accumulations of woven bone (Fig. 1f i). As development towards normal occurs, there are more and larger osteons and diminished woven bone tissue but only partial compaction as relatively large intracortical spaces, filled with cells and central vessels, persist (Fig. 1f ii). Compact fully mature osteons (Haversian bone systems) are not seen. Bone tissue is either woven or lamellar with intermediate mixtures not seen. Osteocyte size and number are diminished in lamellar as compared with woven bone. Cells communicate in both types of bone via canaliculi. In heavily stained sections, there is an abundance of canaliculi per osteocyte/osteoblast with those in woven bone randomly oriented in relation to cells and matrices while those in lamellar bone are oriented transversely or longitudinally as cells assume a shape parallel to the long axis of the lamellae (Fig. 1g ii).
3.2. Relative amounts of woven and lamellar bone deposition
A grading system was outlined based on differing matrix orientations. Grade I refers to presence of discontinuous spicules of woven bone only with a subgrade, Grade Ia, applied when microscopy shows thin collections of parallel-fibered osteoid on isolated woven bone surfaces. Grade II bone tissue specimens demonstrate both woven and lamellar components with three relative proportional groupings: Grade IIa woven bone greater in amount than lamellar bone; Grade IIb woven and lamellar bone in equal amounts; and Grade IIc lamellar bone greater in amount than woven bone and forming rudimentary osteons. Grade III refers to presence of lamellar bone exclusively with two relative proportional groupings: Grade IIIa forming partially compacted osteonal systems with considerable intracortical space between them and Grade IIIb where lamellar bone is composed of fully compacted mature osteons (Haversian systems), interosteonal lamellar bone and little to no intracortical space devoid of bone tissue. Grade I or Ia specimens are characteristic of Type II (OIC A) OI as discontinuous spicules of woven bone with or without thin surface collections of parallel fibers. Grade II specimens are characteristic of Type III (OIC B, OIT A, OIT B) OI and comprise the large majority of specimens. Grade IIIa specimens (all lamellar bone) are seen in this patient group but there are no fully compacted collections. The grade II subgroups allow for demonstration of evolving osteonal synthesis; grade IIa shows only traces of lamellae on woven bone surfaces insufficient for osteonal synthesis, grade IIb has sufficient lamellar bone to allow for beginning osteonal synthesis, and grade IIc has enough lamellar bone to enclose complete lamellation and enhance rudimentary to partial compaction (synthesizing bone progressively inwards towards the vessel).
3.3. Specific patterns of lamellar bone deposition
3.3.1. Mosaic pattern
All grade II bone tissue shows a unique pattern of disorganized lamellar deposition. It is composed of short segments of lamellae positioned either obliquely at varying angles and sometimes even at right angles to adjacent short segments. These various segments positioned at oblique multiplanar angles to one another in random array present a mosaic appearance markedly distinct from the lamellar patterns of lengthy parallel fibril collections characteristic of normal bone (Fig. 1e i-iv).
3.3.2. Tissue compaction
Tissue compaction refers to the amount of bone tissue per unit area. Lethal perinatal bone cortices show poor compaction where there are only small woven bone fragments (spicules) separated by regions of fibrous tissue and vascularity. In grade II OI bone there is partial compaction that increases progressively from areas of woven bone predominance to areas of equal woven and lamellar bone to areas where lamellar bone predominates. The lamellar bone then increasingly fills in space surrounding the vessels to further compact the osteonal systems. Although not included in this report, autosomal dominant OI bone (or normal bone which it closely approximates in mild cases) shows lamellae, osteons, and interosteonal lamellae to be densely packed filling the unit area with bone tissue and defining full or dense compaction.
3.4. Polarizing microscopy
PLM demonstrates random orientation of fibers with non-passage of light in woven bone (isotropic) and the parallel orientation of fibers with passage of light in lamellar bone (anisotropic) (Fig. 2a–c). While woven and lamellar bone conformations can be distinguished without polarization by careful examination of well-prepared histologic sections, PLM is particularly valuable in distinguishing the two conformations and defining the early stages of conformational change. Polarization demonstrates the orientation of parallel fiber deposition as the woven bone scaffold becomes sufficiently extensive to allow for SOBLs to begin early deposition of surface bone. It also better outlines the mosaic pattern of lamellar tissue deposition in grade II variants as long as the same region is assessed by microscope stage rotation since the axes of the short oblique lamellae differ.
Fig. 2.


Images show the value of using polarizing light microscopy (PLM) to highlight woven and lamellar conformations in osteogenesis imperfecta bone.
a. i) and ii) LM photomicrographs shows both lamellar and woven bone from a type III patient. The box in i outlines the region magnified in ii. Woven (W) bone is relatively hypercellular with round to oval cells compared with lamellar (L) bone where cells are elliptical and oriented along the axis of the lamellae. iii) and iv) PLM views show the same tissue seen by LM in i and ii. The box in iii outlines the region magnified in iv. Woven (W) bone regions are dark due to the random orientation of the collagen fibrils producing an isotropic effect with polarization. Lamellar (L) bone regions are light due to the parallel orientation of the fibrils producing an anisotropic effect allowing passage of light with polarization. The clear-cut differentiation of woven and lamellar tissue is highlighted by the PLM technique. Plastic embedded 1% toluidine blue stained section.
b. i) LM section of cortical bone from patient with type II OI shows mixture of woven (W) and lamellar (L) bone. At this magnification one can distinguish the 2 conformations, especially by looking at the cellularity regarding number and shape. ii) PLM view of the exact same section highlights lamellar (L) bone as light blue (anisotropic) and woven (W) bone as dark (isotropic). Thin short streaks of light blue within woven areas represent the earliest phases of lamellar bone deposition on a sufficient woven scaffold. Plastic embedded 1% toluidine blue stained section.
c. i) A section of femoral bone assessed by LM is shown; ii) the same section is assessed by PLM. MOBLs = mesenchymal osteoblasts synthesizing woven (W) bone and SOBLs = surface osteoblasts synthesizing lamellar (L) bone. An * (asterisk) = central vascular canal of forming osteon. Plastic embedded 1% toluidine blue stained section.
d. Section of mosaic pattern in OI diaphyseal bone using partial polarization allows for cellular visualization as well as lamellar matrix orientation. The bone tissue is all lamellar and partially compacted but shows multiple short lamellae in variable orientations (cross-section, longitudinal and oblique). With rotation of the slide, regions appearing dark show the parallel lamellar orientation. Plastic embedded 1% toluidine blue stained section under partial polarization.
3.5. Subgroup of patients undergoing LM histology and radiology comparisons
This assessment is based on 41 specimens from 17 OI patients. The range of radiographic findings in lower extremity long bones is seen in Fig. 3 i-v.
Fig. 3.

Five radiographs show the range of deformity and cortical thickness in lower extremity long bones with decreasing clinical severity. i) Anteroposterior view of femur in severe type III (OIC B) patient showing extreme diaphyseal and cortical thinness with marked deformity and ii – v) type III (OIT A) patients with ii) lateral tibia with marked cortical thinness; iii) and iv) lateral tibia radiographs with improved cortical thickness; and v) lateral femur with marked deformity but reasonably good cortical thickness.
3.5.1. Histology
3.5.1.1. Percentages of woven and lamellar bone
Grade I specimens (all woven bone) comprise 18%; Grade 2 (woven and lamellar components) comprise the large majority of the specimens at 82% with the sub-grading breakdown 2a (woven > lamellar) 10%, 2b (woven = lamellar) 18%, and 2c (lamellar > woven) 54%; and Grade 3 (all lamellar bone) 0%, not seen.
3.5.1.2. Compaction
<50% is present in 35% of the specimens; from 50% to 75% in 43%; and > 75% in 22%.
3.5.2. Radiology
Radiology assessments grade the cortices as thin in 42% and thick in 58% and the diaphysis as markedly bowed in 84% and only minimally bowed in 16%.
3.5.3. Radiology-histology correlations
When the percentage compositions of woven and lamellar bone are considered, Grades 1, 2a and 2b have thin cortices and Grade 2c thick cortices. While Grades 1 and 2a are markedly bowed, Grade 2b is minimally bowed and Grade 2c mildly bowed. When cortical compaction is related to radiologic findings, <50% compaction is associated with thin cortices and markedly bowed segments; 50% to 75% compaction with thin more often than thick cortices with segments usually bowed; and with >75% compaction, cortices are thick and minimally bent.
3.6. Light microscopic histomorphometry of woven and lamellar bone
Assessments involve 37,884 cells from 449 histologic sections (Table 1).
Table 1.
Histomorphology assessments in OI and normal bone.
| Number of regions | Number of histology sections | Number of cells assessed | |
|---|---|---|---|
| OI Bone | 108 | 232 | 19,362 |
| 12 Patients | [W 48, L 60] | [W 103, L 129] | [W 10871, L 18491] |
| Normal Bone | 104 | 217 | 18,522 |
| 11 Individuals | [W 25, L 79] | [W 53, L164] | [W 6525, L 11997] |
| Total | 212 | 449 | 37,884 |
| [W 73, L 139] | [W 156, L 293] | [W 17396, L 20488] |
3.6.1. General assessment of osteogenesis imperfecta bone versus normal bone and woven bone versus lamellar bone (PRISM t-test)
-
i).
cell area/total area × 100. All OI bone cells (woven and lamellar tissue combined) show greater cellularity than all normal bone cells (woven and lamellar tissue combined). The value in OI bone is 4.24 ± 2.43 (23) and in normal bone 2.13 ± 1.51 (13) (p < 0.01). All woven bone cells (OI and normal tissue combined) show greater cellularity than all lamellar bone cells (OI and normal tissue combined). The value in woven bone is 5.42 ± 2.16 (16) and in lamellar bone 1.93 ± 0.95 (20) (p < 0.0001).
-
ii).
total area/cell number. All OI bone cells (woven and lamellar combined) are surrounded by less matrix than all normal bone cells (woven and lamellar combined); OI bone 228 ± 97 (23) and normal bone 388 ± 199 (13) (p < 0.01). Similarly, all woven bone cells (OI and normal combined) are surrounded by lesser amounts of matrix than all lamellar bone cells (OI and normal combined); woven bone 166 ± 58 (16) and lamellar bone 382 ± 150 (20) (p < 0.0001) (Fig. 4a and b).
Fig. 4.

Histomorphometric studies compare woven and lamellar bone in normal and osteogenesis imperfecta bone.
a. Histomorphometric comparison between all osteogenesis imperfecta bone specimens and all normal human bone specimens is shown.
b. Histomorphometric comparison between all woven bone tissue and all lamellar bone tissue is shown. Student t-test values: for panels a and b * p < 0.05; ** p < 0.01; *** p < 0.001; and **** p < 0.0001.
c. Histomorphometric values for cell area / total area X100 assess OI and normal woven bone and OI and normal lamellar bone.
d. Histomorphometric values for total area / number of cells assess OI and normal woven bone and OI and normal lamellar bone. Key: * compared to OI woven bone, + compared to normal woven bone, ^ compared to OI lamellar bone. Data were evaluated using two-way ANOVA with multiple comparisons and Bonferroni's test as the post-hoc analysis. Significance level is the same as panels a and b for all symbols.
3.6.2. Specific assessment of four groups of tissue conformations (PRISM 2 way ANOVA)
-
i).
cell area/total area × 100. Normal woven bone (4, two fetal and two newborn individuals) registers a cell area/total area index of 4.08 ± 0.98 SD (53 histologic sections, 6525 cells) and normal lamellar bone (9, newborn to young adult) registers an index of 1.26 ± 0.57 (164 sections, 11,997 cells) (p < 0.05). Osteogenesis imperfecta woven bone (11, newborn – 18 years of age) registers an index of 5.86 ± 2.29 (103 sections, 10,871 cells) and lamellar bone (10, 2–18 years of age) registers an index of 2.47 ± 0.86 (129 sections, 8491 cells) (p < 0.0001). Also, OI woven bone is significantly more cellular than normal lamellar bone (p < 0.0001) (Fig. 4c).
-
ii).
total area/cell number (providing a relative index of pericellular matrix volume) from the same histologic sections assessed in section i above: in normal lamellar bone 482 ± 165 and normal woven bone 178 ± 19 (p < 0.001); in OI lamellar bone 300 ± 69 and in OI woven bone 162 ± 67 (p < 0.05). Also, normal lamellar bone ratio is much higher than OI lamellar bone (p < 0.01) and OI woven bone (p < 0.0001). These latter values indicate that both normal and OI lamellar bone osteocytes are surrounded by greater accumulations of collagen matrix than woven bone osteocytes (Fig. 4d).
The highest cellularity index seen is in OI woven bone (5.86); it is always higher (p < 0.0001) than the corresponding lamellar bone index in both OI and normal bone and is higher than the index in normal bone (4.08), although not at a statistically significant level. The lamellar bone index is also higher in OI versus normal bone, 2.47 and 1.26 respectively, although not at a statistically significant level indicating that the cellularity of OI bone begins to approach that of normal bone. The less severe the variant of OI is, the closer to normal values it would register. The total area/number of cells ratio, an index of the spatial pericelllular domain, is even more sensitive to group differences in the two-way ANOVA application. Normal lamellar bone has the highest pericellular domain with statistically significant differences compared to OI lamellar bone (p < 0.01), normal woven bone (p < 0.001), and OI woven bone (p < 0.0001) (Fig. 4d).
The cellularity index for woven bone varies greatly, compared to that for lamellar bone, since the stage of woven bone formation determines the concentration of mesenchymal osteoblasts in relation to the amount of surrounding collagen. The index is thus highest in areas of developing cellular differentiation where woven bone formation is either extremely limited by disease or just beginning in the normal. This is seen in bones: i) affected with perinatal lethal Sillence II OI (individual A, 9 regions, 11 histology sections) where the mean cellular index is 8.87 (range 13.25–5.85) compared to 5.86 in all 12 OI patient assessments and 5.59 in the other 11 progressively deforming Sillence III OI patients and ii) in normal groove of Ranvier region where periosteum begins to form with a value of 10.80 compared with entire woven bone mean of 4.37 and a value of 3.08 excluding the Ranvier region.
3.7. Transmission electron microscopy cell observations; mesenchymal osteoblasts (MOBLs) and surface osteoblasts (SOBLs)
The primary ultrastructural finding, in both MOBLs synthesizing woven bone and SOBLs synthesizing lamellar bone on the woven bone scaffold, is cytoplasm containing abundant amounts of rough endoplasmic reticulum (RER) that is invariably moderately to markedly dilated and filled with a mildly electron-dense homogeneous material (Fig. 5a and b). Also seen in both types of osteoblasts are enlarged circumscribed Golgi bodies with disorganized cisternae, rather than linear curvilinear collections seen in quiescent cells, and swollen dispersed tubules and vesicles (Fig. 5a–c). Mitochondrial swelling is seen only in osteoblasts with massively dilated RER (Fig. 5d).
Fig. 5.
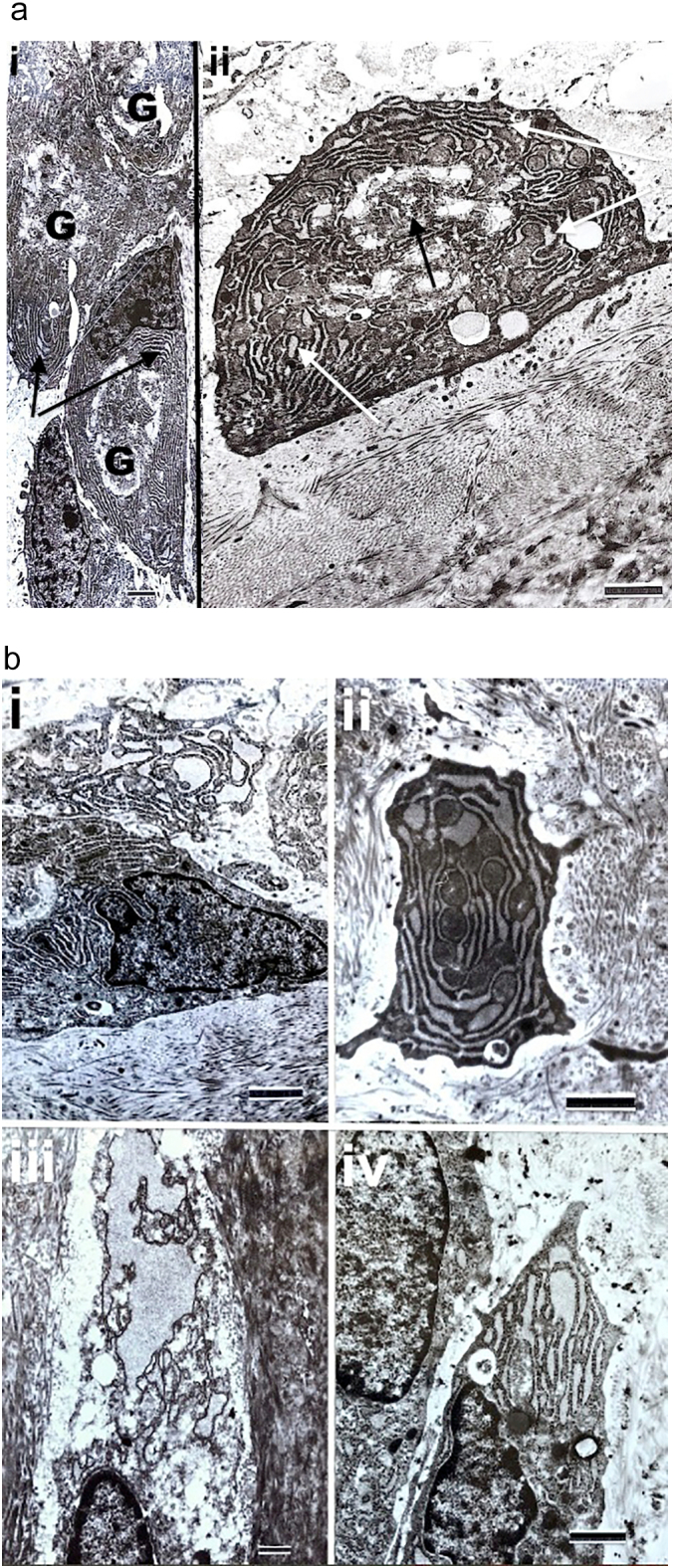

Transmission electron micrographs of osteogenesis imperfecta osteoblasts and osteocytes show specific cell features.
a. Cells are from a patient with type III OI. i) Surface osteoblasts (SOBLs) show prominent Golgi bodies (G) and abundant rough endoplasmic reticulum (arrows). Marker = 1.33 μm; ii) Single surface osteoblast (SOBL) with prominent Golgi body (dark arrow) and dilated rough endoplasmic reticulum (white arrows). The osteoblast is flat against the surface of the underlying matrix (lower 1/3rd of image) that consists of unidirectional collagen fibrils in orthogonal array (layers at right angles to one another). Marker = 1.33 μm.
b. Markedly dilated RER is seen in cells from 4 different type III patients. i) SOBL, marker = 1.33 μm; ii) MOBL, marker = 1.33 μm; iii) osteocyte, marker = 0.67 μm, iv) SOBL, marker = 1.18 μm.
c. Abnormal Golgi bodies seen in SOBLs in patients with type III OI are shown. G = Golgi body; RER = rough endoplasmic reticulum. i) Prominent Golgi body (G) is seen in SOBL in the peri-nuclear region. Dilated RER is present in adjacent SOBL at upper left. Marker = 1.18 μm; ii) Magnified image from an adjacent cell showing irregular stacked cisternae (wide white arrow) and scattered swollen tubules and vesicles (TVs) (narrower white arrows). Marker 0.77 μm; iii) Osteoblast from another type III patient showing markedly dilated RER (black arrows) and a prominent Golgi with markedly swollen tubulovesicular (TVs) components (white arrows). Marker = 1.08 μm.
d. i) MOBL from patient with type III OI shows massively dilated confluent lakes of rough endoplasmic reticulum (dark arrow) and swollen mitochondria with loss of definition of cristae (Mito, white arrows). Marker = 1.0 μm; ii) magnified image shows dilated RER (dark arrow) and highly specific bulbous mitochondrial swelling (white arrow). Marker = 1.0 μm.
3.8. Transmission electron microscopy collagen matrix observations
3.8.1. Fibrillar orientation
While fibrillar orientation is random to parallel in conformity with the light and polarizing microscopic appearances of woven and lamellar bone respectively, additional ultrastructural observations are made within both conformations (Fig. 6a).
Fig. 6.


Transmission electron micrographs of the collagenous matrix in OI woven and lamellar bone are shown.
a. i) Fibrils from woven bone in a severe type III (OIC B) patient show characteristic random fibril orientation. Marker = 0.22 μm; ii) Fibrils from lamellar bone in a different patient with severe type III OI (OIC B) are unidirectional. Marker = 0.38 μm.
b. MOBL (center left) in woven bone tissue in a patient with type III OI shows markedly dilated RER. The cell is surrounded by 6 prominent bundles of unidirectional collagen fibrils but the bundles are obliquely positioned (randomly oriented) to one another. Marker = 1.08 μm. ii) Tissue from the box region in i) is magnified in ii) to highlight the fibrillar bundle random orientation. Marker = 1.08 μm.
c. Fibrils from lamellar bone in two patients both with severe type III OI are shown. i) Collagen fibrils show unidirectional orientation at right and a clear tendency to gradual deviation in direction upwards and downwards at left. Marker = 0.45 μm; ii) Collagen fibrils with unidirectional pattern also tend to a gradual angular deviation. The fibrils show the characteristic collagen crossbanding at this TEM magnification. Marker = 0.19 μm.
d. i) Orthogonal array of collagen fibril deposition in adjacent layers in lamellar bone is readily seen at these magnifications. Patient had a severe type III OI. Marker = 0.76 μm; ii) Collagen fibrils in lamellar bone in a severe type III (OIC B) patient show unidirection at top and angular deviation at bottom. Marker = 0.55 μm.
e. TEM images are shown from lamellar bone in two patients with type III OI. i) Unidirectional collagen fibrils are present but a gradual angular deviation as great as 45° is seen. Marker = 0.45 μm; ii) A well-organized fibrillar weave (herringbone pattern) characterizes some sections of lamellar bone. Marker = 0.93 μm.
3.8.1.1. Woven bone
In woven bone, while accumulations of individual fibrils were randomly arrayed, considerable parts were composed of numerous small bundles of parallel fibrils that were obliquely oriented in relation to adjacent bundles (Fig. 6b). Much of what appears as woven bone at the LM or PLM level is actually collections of small parallel-fibered bundles of collagen that are randomly oriented to one another.
3.8.1.2. Lamellar bone
In lamellar bone fibrils tend to align in uni-directional array rather than being absolutely parallel. Several groups of fibrils deviate from adjacent fibrils to be positioned slightly oblique to one another along the long axis of a particular lamella Fig. 6c, d ii, 6e i). Orthogonal arrays of adjacent lamellae formed, as in classic descriptions, as parallel longitudinal fibers in one plane are juxtaposed at right angles with parallel transverse fibers in the immediately adjacent plane Fig. 6d i). On occasion a regular oblique interdigitation pattern of fibril bundles in a herringbone pattern is seen in lamellar bone (Fig. 6e ii). For the most part, fibrillar appearances are examples of the angular or twisted rotation of fibrils in lamellar bone rather than specific abnormal patterns due to OI. The regularity of formation makes it difficult to attribute the findings to pathologic malformation.
3.8.2. Individual collagen fibrils
Individual collagen fibrils are generally normal appearing in both woven and lamellar bone. The fibrillar periodicity is normal along the long axis of the fibrils and the fibrils are round without electron dense accumulations on their outer surfaces in cross-sectional planes. The transition from woven bone to lamellar bone is abrupt, even at the TEM level, and intermediate regions mixing the conformations are not seen.
3.8.3. Measurement of collagen fibril diameters
On measurement, collagen fibril diameters range from 45 to 90 nm with occasional fibrils as wide as 120 nm. The fibril diameters are within the low normal range. Ranges of values from 9 OI patients and 2 normal controls are shown in Table 2.
Table 2.
Collagen fiber diameters in OI and normal osteoid.
| OI type* | Number of fibers measured** | Diameter of fibers, nm (Mean ± SD) |
|---|---|---|
| S II, OIC A | 17 | 48.0 ± 1.8 |
| 14 | 70.0 ± 4.6 | |
| S II, OIC A | 20 | 50.2 ± 1.3 |
| 18 | 51.3 ± 1.3 | |
| S III, OIC B | 12 | 66.8 ± 4.2 |
| S III, OIC B | 17 | 81.9 ± 3.2 |
| 11 | 82.3 ± 3.5 | |
| S III, OIC B | 10 | 63.1 ± 2.1 |
| 12 | 72.5 ± 3.0 | |
| S III, OIC B | 20 | 66.9 ± 1.7 |
| 15 | 67.9 ± 2.2 | |
| 17 | 71.6 ± 2.3 | |
| S III, OIC B | 24 | 58.6 ± 1.7 |
| 17 | 66.5 ± 2.1 | |
| 26 | 67.5 ± 2.2 | |
| S III, OIT A | 10 | 70.8 ± 2.2 |
| 12 | 63.8 ± 2.5 | |
| N (4 years old) | 20 | 52.4 ± 1.7 |
| 14 | 63.3 ± 2.5 | |
| N (6 years old) | 20 | 89.0 ± 2.4 |
| 10 | 92.9 ± 4.2 | |
| 14 | 100.0 ± 3.0 | |
Code: * OI TYPE: S = Sillence, OIC A and B = osteogenesis imperfecta congenita A and B, and OIT A = osteogenesis imperfecta tarda A (Shapiro); ** Number of fibers measured = number of discrete fibers measured in cross-sections from newly synthesized peri-cellular (osteoblast) osteoid tissue with crossbanding in longitudinal sections from tissue section(s) from one piece of bone. (Where 2 or 3 separate values per patient are listed, each is from a separate piece of bone.); nm = nanometers thick in cross-section; SD = standard deviation; N = normal.
4. Discussion
4.1. Extent of the woven to lamellar bone formation continuum in osteogenesis imperfecta underlies the eventual maturation level of the tissue
4.1.1. Overview
Study of OI bone, in the context of the continuum of woven to lamellar bone formation, demonstrates two basic mechanisms in OI bone histogenesis: i) that lamellar on woven bone deposition is an obligate self-assembly mechanism at a hierarchical level beyond the known self-assembly of the collagen molecule itself and ii) that bone synthesis in OI follows the normal developmental pattern but shows variable delays in maturation due to synthesis of structurally abnormal or insufficient amounts of collagen. The histogenesis and structural characteristics of OI bone tissue are summarized in Table 3.
Table 3.
Histogenesis and structural characteristics of osteogenesis imperfecta bone tissue.
| General observations referable to OI bone varying in extent from severe to moderate to mild depending on the specific gene mutation |
|---|
| 1. Synthesis of OI bone follows the developmental pattern of normal bone formation but is held up at progressively earlier stages dependent on the severity of molecular changes impacting collagen synthesis. This is a “shift to the left” with the degree of shift dependent on the severity of the molecular effects. |
| 2. Invariable initial synthesis of woven bone by MOBLs is followed by increasing amounts of lamellar bone synthesis by SOBLs on a woven scaffold. Lamellar bone increasingly predominates as disease severity lessens. The deposition of lamellar bone is an obligate self-assembly phenomenon at the supramolecular level. |
| 3. Woven bone alone synthesized by MOBLs is seen in lethal perinatal cases. Occasionally, even in these variants, SOBLs begin synthesis of parallel fibered lamellar tissue (osteoid). |
| 4. Lamellar bone in moderate variants generally is deposited as shortened, irregularly oriented collections leading to a mosaic appearance in long bone cortices. |
| 5. OI bone tissue is hypercellular due to relatively decreased matrix synthesis; however, the woven and lamellar segments in OI bone are hypercellular to differing extents both in relation to each other and to normal bone. Hypercellularity of bone in OI is due primarily to its woven component although lamellar bone is still more cellular than normal lamellar bone. |
| 6. Polarizing microscopy should be used to distinguish woven from lamellar bone especially in fetal, newborn and osteogenesis imperfecta specimens. This prevents hypercellular appearing bone being interpreted as invariably woven in conformation. |
| 7. Lamellar bone shows a developmental progression. [Terminology varies, however, depending on the level of structural resolution. What are described as parallel fibers at the light microscopic level often appear to be, and are more accurately described as, not strictly parallel but uni-directional at the ultrastructural level.] As woven bone is transformed into lamellar bone, the initial orientation of lamellar fibril deposition is dependent on the position of the woven bone scaffold and the adjacent blood vessels. The initial deposits are thus often short and oblique rather than strictly parallel to the longitudinal bone axis. |
| 8. As lamellar synthesis predominates, fibril orientation at the ultrastructural level is in the twisted plywood / nested arc / herringbone pattern seen in normal bone but further complicated by mosaic patterns defined here in OI bone. |
| 9. Lamellar bone formation moves progressively from rudimentary osteons (with woven bone still predominating) to partially compacted osteons (where lamellar tissue predominates but woven bone persists with considerable spacing between osteons) to fully compacted cortical bone formed completely of densely packed osteons and interposed lamellae. |
| 10. In milder cases, lamellar bone alone is seen but often in decreased amounts with decreased compaction and irregular orientation. Fully compacted cortical bone is seen only in the mildest OI cases. |
| 11. In association with the bone tissue underdevelopment described above, both cortices and trabeculae show decreased thickness. |
Cortical bone development occurs in hierarchical fashion (Reznikov et al., 2014). Development begins with synthesis of collagen by newly differentiated mesenchymal osteoblasts. This study has assessed the supramolecular changes that occur as the genetically flawed collagen molecules proceed with bone formation. Structural findings in OI demonstrate imperfect progress in the continuum of woven to lamellar bone formation. By specifically defining bone tissue conformation as woven, woven and lamellar or fully lamellar; the lamellar component as non-osteonal or osteonal; and the osteonal component as rudimentary, partially compacted or fully compacted and relating it to the gene/molecular abnormality it becomes possible to better understand the ultimate effects of specific mutations on bone structure. Bone synthesis in OI follows the normal developmental pattern but rarely achieves fully compacted osteonal lamellar bone. The morphologic appearance of OI bone shows a continuum of change from severe lethal perinatal to progressively deforming forms. The developmental pattern in OI can be considered (borrowing a term from the hematology discipline) as showing a “shift to the left” indicating that a particular population of cells and tissue conformation is shifted towards more immature precursor patterns (Table 4).
Table 4.
Bone development sequence “left to right”.
| Bone development can be represented (left to right) as proceeding from undifferentiated mesenchymal cells ⇒⇒ mesenchymal osteoblasts (MOBLs) synthesizing woven bone ⇒⇒ surface osteoblasts (SOBLs) synthesizing lamellar bone in non-osteonal parallel array on a woven bone scaffold ⇒⇒ formation of rudimentary osteons surrounding vessels within woven bone accumulations ⇒⇒ a preponderance of lamellar bone forming arrays of osteons leading to partial compaction ⇒⇒ lamellar bone alone composed of more densely compacted (Haversian) osteons progressing to full compaction. |
The developmental pattern in OI can be considered (borrowing a term from the hematology discipline) as showing a “shift to the left” indicating that a particular population of cells and tissue conformation is shifted towards more immature precursor patterns.
The deviations in structure in OI from normal are due to variable delays in maturation. The more severe the variant of OI is, the greater the persistence of woven bone and the more immature the structural pattern. The pattern shifts to a structurally stronger lamellar arrangement once a threshold accumulation for an adequate scaffold of woven bone has been reached. As development proceeds towards normal, there is proportionately less woven bone persisting, greater amounts of lamellar bone and progressively more mature tissue deposition. OI bone does not reach the fully compacted stage in progressively deforming variants but develops variable maturity within the state of partial compaction. Although this study did not assess the mildest OI group (Sillence type I non-deforming autosomal dominant) lamellar bone from that variant has a well-developed osteonal structure approaching full compaction. Bone from children with type I OI often appeared normal in microstructure and amount (Jones et al., 1999). The histopathology relates well with the clinical and radiologic spectrum of the disorder.
4.1.2. Collagen self-assembly
Collagen self-assembly occurs on differing hierarchical levels. Collagen molecules reach their fibrillar structure by a process referred to as self-assembly where positional relationships align based on physico-chemical factors rather than biologic cellular direction (Kar et al., 2006; Gautieri et al., 2012). This process is maintained in OI but the collagen molecular mutations eventually affect fibril structure. In continuation of the hierarchy of bone formation at the supramolecular level, lamellar bone deposition on woven bone also represents an obligate self-assembly phenomenon once a sufficient amount of woven bone has been synthesized to serve as a scaffold. Lamellar on woven self-assembly is also of a physicochemical nature, not directly related to genomic information but clearly responding to the tissue aggregation presented. It is universal in bone formation occurring in normal developmental, repair and pathologic bone formation (Shapiro and Wu, 2019).
4.2. Histologic structure of OI bone from previous reports interpreted in the woven bone to lamellar bone continuum
4.2.1. Background: clinical and molecular variability in OI
We categorize lethal perinatal (type II) and progressively deforming patients (type III) using the Sillence classification (Sillence et al., 1979a) but also refer to congenita-tarda terminology incorporated into the temporal-radiographic categorization (Shapiro, 1985) to relate to earlier reports on bone histopathology. OI was defined as a single entity with variable severity referred to as osteogenesis imperfecta congenita and tarda; fractures in utero being OI congenita and fractures after birth OI tarda (Looser, 1906). Regarding OI tarda, those fracturing between birth and one year of age were tarda gravis and those initially after one year of age were tarda levis (Seedorff, 1949). The updated congenita (C)/tarda (T) approach includes radiographic-functional aspects: OIC A (lethal perinatal)---short, broad crumpled femurs and ribs; OIC B (less severe congenita variants)---bone shape and structure closer to normal, intrauterine fractures but patients generally survive; OIT A---initial fracture after birth but before walking; and OIT B---initial fracture after walking begins (Shapiro, 1985). Not all patients with intrauterine/birth fractures (congenita forms) have lethal disorders. Congenita forms have been differentiated based on skeletal radiologic differences (Spranger et al., 1982). Classification of OI beyond the congenital/tarda outline evolved to encompass gene and molecular findings. Sillence et al. developed a four-part classification, types I to IV (Sillence et al., 1979a); with recognition of different degrees of severity type II lethal perinatal group was subcategorized into types A, B and C (Sillence et al., 1984). Additional subgroups were added to the Sillence system enlarging it from 4 to 7 (Glorieux et al., 2000a; Glorieux et al., 2002; Ward et al., 2002); more groups were added as variants due to mutations in molecules other than collagen were described leading to as many as 14 types (Van Dijk et al., 2010; Forlino et al., 2011; Van Dijk and Sillence, 2014). Van Dijk and Sillence re-defined Sillence groups I to V as osteogenesis imperfecta syndromes, with group V representing the variant defined by interosseous forearm bone calcification and newer gene defect variants were placed into the most appropriate group recognizing that these phenotypic groups would sometimes be genetically heterogenous (Van Dijk and Sillence, 2014). In association with the newer findings, OI is increasingly categorized and assessed by molecular mutations.
4.2.2. Previous illustrations of abnormal structure in OI bone formation
Histologic studies of OI bone, generally defining affected bones as congenita or tarda, commented on paucity of bone, persistence of woven bone, hypercellular bone and limited development of lamellar bone. Careful examination of the histopathology reports is consistent with the woven to lamellar bone developmental pathway even though the findings were not interpreted that way at the time. Several studies outlining histopathology of lethal perinatal Sillence II (OIC A) variants illustrate accumulations of woven bone alone without progression to lamellar bone formation (Michel, 1903; Klotz, 1909; Heinrich Bauer, 1920a; Heinrich Bauer, 1920b; Weber, 1930; Jeckeln, 1931; Follis, 1953; Freda et al., 1961; Roujeau et al., 1967; Adler and Bollmann, 1973). Van der Harten et al. studied pre-natally diagnosed cases by radiology and pathology showing the spectrum of Sillence II A-C variants (van der Harten et al., 1988). Less severe but still congenita variants can be classed as severe Sillence III (progressively deforming) or OIC B patients. Histopathology of congenita cases that represent both Sillence II/OIC A and severe Sillence III/OIC B disorders have been described (Buday, 1895). Histopathology report of a child that had fractures at birth and died at 10 months shows a severe Sillence III (OIC B) variant with lamellar bone (Nichols and Lovett, 1906). Reports on OI tarda clearly highlighted lamellar bone (with woven bone persisting) in more severe forms and lamellar bone only (not fully compacted) in less severe variants. Histopathology illustrations of tarda variants show tissue sections with both woven and lamellar bone (Wilton, 1932; Jaffe, 1972; Milgram et al., 1973); others illustrate tarda variants where lamellar bone either predominates or is the only type seen (Hagenbach, 1911; Key, 1926; Engfeldt et al., 1954; Ramser and Frost, 1966): thin lamellar bone collections with poor compaction (Hagenbach, 1911); shortened multidirectional lamellae (with what we now describe as a mosaic-type appearance) (Key, 1926); and several cases characterized by hypercellular bone tissue with a mixture of woven and lamellar bone or lamellar bone only (Robichon and Germain, 1968).
4.2.3. Subgroups with histopathological findings
Three additional types of OI (V, VI and VII) were defined based on clinical and radiographic findings, histomorphometry and absence of type I collagen mutations (Glorieux et al., 2000a; Glorieux et al., 2002; Ward et al., 2002). Type V (calcification of the forearm interosseous membrane, decreased rotation, hyperplastic callus formation following fractures) is included in the expanded Sillence-VanDijk classification types I-V (Van Dijk and Sillence, 2014; Lee et al., 2006; Shapiro et al., 2013). Histologic sections show “fish-scale” or “mesh-like patterns” of irregularly arranged lamellar deposition (Glorieux et al., 2000a; Lee et al., 2006; Shapiro et al., 2013; Roughley et al., 2003); in these histologic sections what we describe as mosaic patterns of lamellation and lamellar deposition on woven cores can be seen. Type VI is distinguished histologically by prominent osteoid seams and delayed mineralization (Glorieux et al., 2002; Roughley et al., 2003; Homan et al., 2011; Venturi et al., 2012; Fratzl-Zelman et al., 2015; Hoyer-Kuhn et al., 2014). Type VII OI (with rhizomelia) has normal lamellation by histology (Ward et al., 2002). Our cases and those reported over several decades demonstrate that the woven bone to increasingly more lamellar continuum characterizes OI bone from lethal to progressively deforming forms as they diminish in severity.
4.3. Specific changes in OI bone observed at each of the structural levels of resolution
Study at each level of structural resolution has revealed new findings.
4.3.1. Synthesis of short obliquely oriented (mosaic) lamellae in OI bone
Although lamellar bone forms in OI, in progressively deforming variants it is often deposited in a mosaic pattern of short obliquely arrayed lamellae at irregular angles to one another (Fig. 1e). This tends to be seen where woven bone has been almost completely resorbed and partially compacted osteonal formation is underway (Fig. 1f). Recognition of mosaic lamellae has not been made previously in descriptions of OI histopathology and would be expected to further weaken structural support. The mosaic pattern has been described, however, in the histopathology of Paget's disease of bone (Freund, 1929; Schmorl, 1932; Jaffe, 1933).
4.3.2. Prominent osteoid seams in type II (OIC A) patient
Prominent osteoid seams are seen in this study in a type II (OIC A) patient (Fig. 1 g i). The tissue layer covering woven bone accumulations is deposited by SOBLs aligned in linear array on its surface. Ultrastructure shows osteoid deposition with parallel collagen fibrils. The finding demonstrates SOBLs aligning on a surface of woven bone once it reaches a sufficient (currently unknown) extent. There is not true lamellar bone formation since osteocytes are not embedded in the matrix. Prominent osteoid seams are seen in Type VI OI where the phenotype is milder than type II OI (Homan et al., 2011; Venturi et al., 2012; Fratzl-Zelman et al., 2015; Hoyer-Kuhn et al., 2014). They also occur in types I, III and IV although they have not been previously mentioned in type II (Glorieux et al., 2002). Osteoid seams are described by HM in OI bone as normal or decreased (Wilson et al., 1966; Ste-Marie et al., 1984; McCarthy et al., 1997; Rauch, 2006).
4.3.3. Histogenesis shown by polarizing microscopy
PLM clearly defines woven and lamellar conformations (Fig. 2a-d) and should be considered as essential in assessing OI bone. Lamellar OI bone has often been described inaccurately as woven using plain LM alone based on its increased cellularity. Also, PLM shows that lamellar bone is deposited initially as a thin collection on a woven bone scaffold when the latter reaches a certain volume. It is synthesized at randomly oriented positions within a woven mass that bears no relationship to the outer cortex or to the long axis of the bone (Fig. 2b,c). Recognition of the position of deposition (Fig. 2b,c) and the presence of mosaic patterns in lamellar bone (Fig. 2d) are important to interpret the histopathology of the disorder.
4.3.4. Cellularity of OI and normal woven and lamellar bone specifically quantified by histomorphometry
OI bone is invariably described as hypercellular in histopathologic descriptions. We observe woven bone qualitatively to be more cellular than lamellar bone in normal and OI groups. Histomorphometry has been used in this study to quantify the observation. Two specific indices were developed to assess the cellularity of woven and lamellar bone. OI bone is hypercellular but we demonstrate that it is the woven bone component that is predominantly hypercellular while lamellar bone is much less cellular and can approach the levels of normal bone. Unless specific efforts are made to distinguish woven from lamellar bone in histologic studies, this fact will be overlooked. The index of cellularity (cell area/total area × 100) shows all woven bone (normal and OI) to be more cellular than all lamellar bone and all OI bone to be more cellular than all normal bone (both p < 0.0001) (Fig. 4a,b). With more specific assessments, OI woven bone is more cellular than OI lamellar bone (p < 0.0001) and normal woven bone is more cellular than normal lamellar bone (p < 0.05) (Fig. 4c). When the four latter groups are compared, cellularity from high to low is OI woven, normal woven, OI lamellar and normal lamellar. The total area/cell number index indicates the individual pericellular matrix volume or domain characteristic of each type of bone conformation. The reverse of the previous ratio is found with each corresponding total area/cell number assessment; the lesser the cellularity per unit area, the greater the pericellular matrix domain supported by the central cell (Fig. 4d).
Standardized HM of iliac crest bone is valuable in assessing normal and pathologic bone (Dempster et al., 2013). Normal data for iliac bone in growing children have been established (Glorieux et al., 2000b) along with the value of the technique in pediatric bone disorders (Rauch, 2006). Histology studies from human OI patients have almost exclusively used iliac crest bone biopsies since these are readily obtainable and unaltered by fracture or deformity (Jones et al., 1999). Iliac bone HM in OI has established comparative values in children and adults (Ste-Marie et al., 1984), in the four clinical Sillence types (Rauch et al., 2000), and in Sillence type I adults (McCarthy et al., 1997) as well as quantifying the effects of intravenous pamidronate (Rauch et al., 2002). Techniques to optimize the evaluation of bone micro-architecture continue to be developed (Malhan et al., 2018). The most commonly used HM measurements in standardized iliac crest studies assess bone thickness and surface events and involve structural, static formation, dynamic formation, and static resorption parameters (Rauch, 2006). In an early iliac crest HM study of OI bone, hypercellularity and diminished matrix synthesis were documented (Falvo and Bullough, 1973). There are some limitations to the use of iliac crest bone in OI since it is a non-weight bearing bone not significantly affected clinically by fracture or deformation. Since long bones in OI are the sites of major clinical problems of deformation and frequent fracture, there is considerable value in assessing the structure and some HM parameters in diaphyseal cortical bone. In this study we establish two parameters to assess cellularity and pericellular matrix domains of woven and lamellar bone in OI and normal bone. These are important in understanding the pathogenesis of OI bone formation since persistence of cortical woven bone and failure of normal lamellar bone formation to reach a level of complete osteonal Haversian system compaction represent not only the histologic hallmark of OI bone but also one of the underlying reasons for its weakness.
The relatively decreased matrix associated with increased cellularity in OI bone is also associated with an increased concentration of canaliculi as seen in plastic embedded sections heavily stained with toluidine blue (Fig. 1g ii). Initial woven bone deposition in bone formation provides an endogenous scaffold along which well-oriented lamellar tissue is deposited (Shapiro, 1988; Shapiro, 2008; Shapiro and Wu, 2019; Kerschnitzki et al., 2011a). Lamellar bone formation soon replaces the woven bone scaffold. The lacunar-canalicular network mirrors the extracellular matrix conformation (Shapiro, 1988; Kerschnitzki et al., 2011b). Greater lacunar/canalicular volume contributes to increased fragility. Carriero et al. investigated cortical bone tissue porosity in a mouse model of OI, oim (Carriero et al., 2014). The oim bone (compared with a wild type) had more osteocyte lacunae per unit volume and more numerous and branched vascular canals, findings associated with increased porosity that lead to greater fragility.
4.4. Ultrastructural findings regarding OI bone cells
4.4.1. Bone cell ultrastructure
Assessment of MOBLs, SOBLs and osteocytes of OI bone at the ultrastructural level confirms cytoplasmic collections of moderate to massive amounts of dilated rough endoplasmic reticulum and prominent Golgi bodies with disorganized cisternae (saccules) and swollen, dispersed tubules and vesicles (Fig. 5a–c). RER and the Golgi body are clearly evident in normal osteoblasts (Makhoul et al., 2019; Klumperman, 2011). The swollen distorted appearance of the RER and Golgi apparatus in OI cells show that the bone forming cells in OI cannot release sufficient amounts of normal collagen into the matrix due to production of mutant forms that lead to misfolding and retention of the abnormal protein within the organelles where processing occurs. While dilated RER is seen in normal active and repair osteoblasts, the extent of the markedly dilated RER in all OI bone forming cells indicates accumulation of protein that must subsequently be degraded within the organelle or returned to the cytosol for degradation. This finding was observed in all 16 patient bone samples assessed by TEM. While isolated references have been made to Golgi body abnormalities and mitochondrial swelling in OI, this study is the first to demonstrate the nature and extent of the structural abnormalities in these organelles at a high level of ultrastructural definition (Fig. 5c and d). Mitochondrial swelling is seen in OI osteoblasts with massively dilated RER but this is interpreted as apoptosis (cell death) rather than as specific for OI. Mitochondrial swelling is defined by a specific sequence of ultrastructural appearances; the outer membrane ruptures while the inner membrane remains intact, bulges through the defect along with the mitochondrial matrix, including the cristae, and leads to malformation of the organelle (Sesso et al., 2012).
4.4.2. Relationship of TEM changes to known molecular localization
The extent and invariable finding of dilated RER defines OI at the structural level as a rough endoplasmic reticulum storage disorder (endoplasmic reticulum stress). Inclusion of OI in these stress disorders has been recognized (Kim and Arvan, 1998; Rutishauser and Spiess, 2002). Similarly, prominent Golgi bodies with disorganized cisternae and swollen tubules and vesicles also defines a Golgi stress disorder due to molecular mutations (Kulkarni-Gosavi et al., 2019; Machamer, 2015; Di Martino et al., 2019). Since normal intracellular collagen synthesis occurs with assembly, folding and passage from the endoplasmic reticulum to the Golgi apparatus (Leblond, 1989; Canty and Kadler, 2005), collagen mutations lead to intracellular molecular misfolding and delays in processing at both the RER and Golgi stages of synthesis; abnormalities reflected in clear changes of structure at the TEM level. In moderate to severe OI, such as in type II and III patients studied here, limited bone matrix formation is due to structural abnormalities of the collagen with misfolding of the nascent molecules trapping them either in the RER or Golgi apparatus and leading to intracellular destruction or slowed extrusion of abnormal fibrils into the extracellular matrix negatively impacting the amount of fibrillar material, positional orientation of adjacent fibrils (self assembly) and mineralization of the fibrils (Gautieri et al., 2012; Byers, 2001; Bryan et al., 2011; Gautieri et al., 2009; Bodian et al., 2008; Bodian et al., 2009).
The molecular components mediating the RER stress reaction are increasingly known and several mutated molecules causing severe recessive forms of OI specifically act in trafficking in the RER and the RER membrane (Ishikawa and Bächinger, 2013; Burman et al., 2018; Oakes and Papa, 2015). Study of both RER and Golgi apparatus protein processing is revealing the molecular aspects of protein transfer (Canty and Kadler, 2005; Zhao, 2012; Nabavi et al., 2012). Post-translational collagen modifications concentrated in the RER and Golgi apparatus associated with molecular mutations causing OI have been summarized (Marini et al., 2017; Boudko et al., 2013; Forlino and Marini, 2016; Morello, 2018). Several genes have been specifically localized to the ER (FKBP10, PLOD2, PPIB, P3H1, CRTAP), the ER Membrane (TMEM38B, CREB3L1/OASIS) and the ER-Golgi (MBTPS2, SERPINH1) (Forlino and Marini, 2016; Morello, 2018). Examples of mutations in RER/Golgi trafficking genes causing OI by interfering with normal collagen synthesis are becoming common along with TEM studies showing associated dilated RER. Dilated RER is seen in cultured cells from a patient with autosomal recessive OI due to mutations in RER protein FKBP65 (Alanay et al., 2010). Mutations in SEC24D (a transcriptional target of OASIS, an endoplasmic reticulum stress transducer and a component of the COPII machinery) cause a severe form of syndromic recessive OI (Cole-Carpenter syndrome) (Moosa et al., 2015; Zhang et al., 2017). COPII proteins are involved with intracellular vesicle initiation, budding and transportation cargo proteins forming RER and Golgi apparatus sites (Garbes et al., 2015; Zanetti et al., 2012). Electron microscopy of fibroblasts from an affected individual with an SEC24D mutation shows dilated RER and insufficient ER export of procollagen (Zanetti et al., 2012). OASIS is encoded by cyclic AMP responsive element binding protein 3-like 1 (CREB3L1); monoallelic and biallelic CREB3L1 variants cause mild and severe OI respectively (Keller et al., 2018; Symoens et al., 2013). OASIS mice (−/−) exhibit osteopenia and abnormally dilated RER containing a large amount of bone matrix proteins (Murakami et al., 2009).
4.4.3. Cellular changes in previous TEM OI studies in relation to our findings
Dilated RER in OI bone has been reported in several TEM studies (Albright et al., 1975; Doty and Mathews, 1971; Stöss, 1988; Cassella et al., 1996; Sarathchandra et al., 2000) (Table 5). It was initially interpreted as indicative of active osteoblasts as distinct from a pathologic finding. This study shows it to be an invariable finding in OI bone cells (Fig. 5a, b). Golgi body abnormalities have been referred to occasionally but not clearly illustrated; we show Golgi bodies to be prominent with internal disorganization characterized by irregular cisternae and dispersed dilated tubulovesicular (TV) components (Fig. 5c). Mitochondrial swelling in OI osteoblasts was first observed by Stöss (Stöss, 1988) and subsequently reported to be present in all OI osteoblasts (Sarathchandra et al., 2000); we demonstrate classic mitochondrial swelling (Fig. 5d) to be limited to osteoblasts with massively dilated RER and interpret the finding as associated with apoptosis. Electron dense accumulations are not seen in dilated RER in OI; the material is a uniform mildly electron dense homogenous accumulation. In many skeletal dysplasias electron dense material is seen as retained abnormal collagen attempts assembly while trapped in the RER (Table 6).
Table 5.
TEM studies of OI bone cells.
| ⇒ Rib biopsies, OI tarda (Sillence III/IV): markedly dilated endoplasmic reticulum in osteoblasts consistent with metabolically active cells (Albright et al., 1975) |
| ⇒ Cortical bone biopsies assessing lamellar bone, OI Tarda (Sillence III): extensive well-developed Golgi complexes containing dense striated material, abnormal RER not noted, osteocytes considered normal (Doty and Mathews, 1971) |
| ⇒ OI bone tissue showing massively dilated rough endoplasmic reticulum in osteoblasts, enlarged prominent Golgi collections, mitochondrial granules (Stöss, 1988) |
| ⇒ Range of OI types (13 patients): dilated rough endoplasmic reticulum and mitochondrial inclusions in bone cells, “persistence of dilated RER in every OI patient examined under TEM…(filled with)....amorphous material” (Cassella et al., 1996) |
| ⇒ Review of 36 OI bone samples: invariable RER distention and swollen mitochondria in all OI osteoblasts (Sarathchandra et al., 2000) |
Table 6.
Material in dilated RER—OI and skeletal dysplasias.
| ⇒ Uniform mildly electron dense homogenous accumulation: osteogenesis imperfecta; spondylo-epiphyseal dysplasia congenita (Sillence et al., 1979b; Shapiro, 1998). |
| ⇒ Circular or lamellar electron-dense accumulations characterize such disorders as: pseudoachondroplasia (Sillence et al., 1979b; Cooper et al., 1973; Unger and Hecht, 2001), multiple epiphyseal dysplasia (Fairbank) (Stanescu et al., 1993), multiple epiphyseal dysplasia due to collagen type IX abnormality (Bönnemann et al., 2000), and spondyloepiphyseal-metaphyseal dysplasia (Shapiro et al., 2006). |
4.5. Ultrastructural matrix findings in OI
4.5.1. Individual collagen fibrils; collagen fibril histomorphometry
Measurement of fibrils by ultrastructure shows values in the low normal range (Table 2). Fibril diameters were smaller in lethal perinatal Sillence II patients than in progressively deforming type III patients. These findings are consistent with previous descriptions (Table 7).
Table 7.
Collagen fibril diameters (TEM) reported in OI bone.
| ⇒ Significantly smaller collagen fibril diameters measured in OI with clear differences in the four Sillence groups; smallest fibrils in lethal perinatal group and largest in milder cases (Stöss and Freisinger, 1993) |
| ⇒ Collagen fibrils in OI type I adults consistently smaller in diameter than normal age matched subjects; OI fibrils 40–60 nm and normal controls 60–80 nm (Jones et al., 1984) |
| ⇒ Smaller fibrils in 42 OI patients (all groups): severe Sillence II lethal perinatal with smallest mean diameter, 45 nm; mildest Sillence I autosomal dominant, 57 nm; and normal control mean diameter, 73 nm (Sarathchandra et al., 1999) |
| ⇒ Mean fibril diameters in 12 OI patients, 60 nm, slightly larger than those in age and site matched controls, 53 nm (Cassella and Ali, 1992) |
| ⇒ Collagen fibers, woven bone, Sillence II patient much smaller (diameters not given) than controls (Haebara et al., 1969) |
4.5.2. Collagen fibril accumulations
The large majority of extracellular matrix fibrils appear normal to qualitative assessment in OI bone. Abnormal appearances of collagen fibers on TEM that characterize some skeletal dysplasias such as fibrillar splitting and unraveling, markedly irregular cross-sectional outlines, electron-dense deposits on the surface, or abnormal fusion of adjacent fibers to form massive accumulations are not seen in OI. On occasion, cross-sections show collagen fibrils with markedly differing diameters interspersed with one another and there can be slight irregularity from circular shapes.
In woven bone single well-formed fibrils with typical crossbanding are positioned in random array and are often densely packed. In lamellar regions multiple fibrils are generally aligned in a uni-directional array; at the ultrastructural level there is rarely strict parallelism to fibril accumulations. Other collagen fibrillar orientations in woven and lamellar bone are noted, however. In woven bone, there are short bundles of closely packed uni-directional fibrils in the extracellular matrix adjacent to the MOBL cell membranes that are positioned in oblique directions to one another such that in these areas it is not individual fibrils that are in random array but rather bundles of unidirectional fibrils that are randomly oriented. These bundles would not be seen at light microscopic magnification but would contribute to the isotropic appearance of woven bone with polarization. As for fibrils in unidirectional or parallel array in lamellar bone, we note examples of orthogonal array with fibrils at right angles to one another in adjacent layers although this is not an invariable finding. Many fibrillar groups in longitudinal array show some of the fibrils to angle away from the main group. Other sections, slightly oblique to the longitudinal plane, demonstrate a well-organized repetitive interdigitating “herringbone-like” pattern. It has been recognized for several years that, at the ultrastructural level, lamellar bone fibrils are not positioned in strict parallel and orthogonal array but, rather have a complex twist or rotation along the long axis. Giraud-Guille (Giraud-Guille, 1988) demonstrated fibrillar orientation patterns having a “twisted plywood” structure that in oblique sections appeared as a series of “nested arcs”. Weiner and colleagues (Weiner et al., 1997; Weiner et al., 1999) described the complex fibrillar rotation by defining an individual lamellar unit (thick and thin lamellae together) to be composed of five sublayers of parallel fibrils each offset by 30° in one direction (thus rotating from 0° to 30° to 60° to 90° to 120°). Marotti and colleagues also noted fibrillar changes of direction in adjacent layers (frequently offset at 30° angulation) but described alternating collagen-rich dense lamellae and collagen-poor loose lamellae all with interwoven fibrils (to the point that lamellar bone could be considered a variety of woven bone) (Marotti et al., 2013). Wagermaier et al. also note three-dimensional spiraling of fibrils in a helicoidal plywood structure in a regular sequence of 5°-25° (Wagermaier et al., 2006). Similar examples of spiral twisting patterns of fiber orientation in normal lamellar bone have been demonstrated (Reznikov et al., 2014; Yamamoto et al., 2012). Our TEM findings in OI woven and lamellar bone, therefore, appear similar to normal structure rather than pathologic variation but more clearly define and expand on bone matrix orientation (Fig. 6 c–e).
There is a close hierarchical relationship between the three-dimensional organization of bone cells in lacunae, their communication via osteocyte processes in canaliculi, the collagen matrix of bone and its mineralization (Shapiro, 1988; Shapiro and Wu, 2019; Reznikov et al., 2014; Kerschnitzki et al., 2011b; Carriero et al., 2014; Vanleene et al., 2012; Bolger et al., 2019). Histomorphometry, ultrastructure and other modalities have been used to assess mineralization of human and mouse (oim/oim) OI bone since matrix abnormality has a high likelihood of altering mineralization and further contributing to bone fragility (Fratzl-Zelman et al., 2015; Fratzl et al., 1996; Boyde et al., 1999; Roschger et al., 2008). There is a higher than normal mineralization density in iliac crest bone in OI patients in all types and independent of specific collagen mutations (Boyde et al., 1999; Roschger et al., 2008). Vanleene et al. assessed several parameters in oim mouse bone and showed (consistent with study of these variables independently): i) increased mineralization, ii) significantly smaller, highly packed and disoriented apatite crystals, and iii) lower stiffness due to the poorly organized mineral matrix (Vanleene et al., 2012). Scanning electron microscopy is valuable for assessing matrix conformation, lamellar bone and mineralization (Shah et al., 2019). For studies of matrix conformation, PLM has a spatial resolution in the range of 250 nm, SEM 1 nm and TEM 0.1 nm (Georgiadis et al., 2016).
4.6. Relationship of structural findings to gene/molecular abnormalities
4.6.1. Recognition of OI as a molecular disorder of collagen
The molecular structure of collagen (the main component of bone matrix) has been defined and the collagen superfamily now includes 28 types (Canty and Kadler, 2005; Kadler et al., 2007; Ricard-Blum, 2011). Shortly after the recognition of type I collagen as the main component of bone, ligament, tendon and skin matrices, studies of OI tissue detected molecular abnormalities of type I collagen in either COL 1A1 or COL 1A2 genes. More than 1500 mutations in type I collagen have been found and these are considered as the causative factor in 85–90% of OI patients (Flier et al., 1992; Kuivaniemi et al., 1991; Marini et al., 2007; Maioli et al., 2019). Over the past few years, mutations in other genes related to collagen formation, usually to post-translational processing of the collagen molecule, have been detected in OI patients; mutations in 18 genes have been implicated in the disorder (Marini et al., 2017; Boudko et al., 2013; Forlino and Marini, 2016; Morello, 2018). Marini et al. retained the Sillence I – IV groups but limited them to the COL1A1 and COL1A2 mutations with new genes given additional type numbers based on the mutation without clinical correlation (Marini et al., 2017). Classification of OI is now defined by the molecular mutation and its functional defect (impairment of collagen synthesis and structure, compromised bone mineralization, abnormal collagen post-translational modification, compromised collagen processing and crosslinking, and altered osteoblast differentiation and function) (Marini et al., 2017; Forlino and Marini, 2016) or based on the cellular location of the defective protein (Morello, 2018). While biologically specific regarding mutations and their effect on the various phases of collagen molecular synthesis, the genetic classifications completely bypass histologic/structural assessments.
4.6.2. Genotype-phenotype correlations
Genotype-phenotype correlations have been attempted for several decades (Maioli et al., 2019; Byers et al., 1991; Rauch et al., 2009; Ben Amor et al., 2013; Li et al., 2019). Byers pointed out the complexity of relating molecular findings to clinical severity (Byers, 2001). While several generalized findings regarding the effects of types and positions of mutations have been made, the relation of molecular defects to clinical status is far from definitive. The mildest form of OI, nondeforming Sillence type I dominant, is generally caused by quantitative defects resulting from nonsense mutations leading to decreased production of normal type I collagen molecules by the haploinsufficiency effect; less protein produced but that produced is structurally normal (Ben Amor et al., 2013; Lin et al., 2015; Zhytnik et al., 2019). Qualitative defects lead to an abnormal collagen molecule and are caused by mutations in each of the COL1A1 and COL1A2, post-translational and bone synthesis-modifying groups (Marini et al., 2017; Forlino and Marini, 2016; Morello, 2018). Most of these are autosomal recessive and tend to cause moderate to severe to lethal cases of OI. For each current syndromal OI grouping II-V and for essentially all 18 gene mutation groups variability occurs within each specific group leading to a range of clinical severity described as mild to moderate or moderate to severe and sometimes as widely as mild to lethal. This variability leads to difficulty in assessing prognosis, especially when the child is young, and can be problematic for families and clinicians planning long-term treatment programs.
4.6.3. Value of structural assessment of bone tissue in OI patients
Renewed emphasis on structural assessment of bone tissue in OI at multiple levels of resolution using LM, PLM, HM, TEM and SEM appears warranted especially in view of the variability of the clinical prognosis as classification is increasingly based solely on molecular categorization. Iliac crest bone tissue is readily obtainable and can be studied with or without an associated standardized HM assessment. Diaphyseal tissue is available in many moderately to severely involved patients at time of surgical osteotomy to correct deformation.
5. Conclusion
Assessment and interpretation of bone structure in the context of the woven to lamellar bone and the MOBL to SOBL continuum allows for recognition of any shift to the left in bone maturation; the effects of gene mutations on collagen synthesis are accurately reflected by the stage of bone development reached. In addition, much additional information regarding OI bone can be gained by further supramolecular study at differing levels of structural resolution and in relation to specific molecular subtypes such as: the effects of the mosaic lamellar structure on bone strength; assessment of woven and lamellar conformations using detailed polarization and histomorphometric assessments; more specific delineation of organelle abnormalities at RER, Golgi body and mitochondrial levels; and ultrastructural assessments of matrix conformations in comparison to the normal. Since genotype-phenotype correlation is not definitive, interposing histologic/structural analysis to allow for a genotype-histopathologic-phenotype correlation would greatly enhance biologic understanding of OI and its clinical management.
Funding
This work was partially supported from several sources including the National Institutes of Health grants [AR39965, AR37318 and AM15671], Stanford Nano Shared Facilities (SNSF), Orthopedic Research Society, Pediatric Orthopedic Society of North America, Peabody Fund (Boston MA) and Milton Fund (Harvard Medical School, Boston, MA).
Authors' Contributions
FS conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, writing – original, writing – editing;
KM conceptualization, data curation, investigation, methodology, writing – editing;
SS data curation, formal analysis, methodology, writing – original, writing – editing;
HZ conceptualization, writing – original, writing – editing;
EF conceptualization, data curation, investigation, methodology, writing – editing;
JW data curation, investigation, writing – original, writing – editing;
JYW conceptualization, formal analysis, methodology, writing – original, writing – editing;
Transparency document
Transparency document.
Declaration of competing interest
The authors declare that there are no conflicts of interest for authorship or publication of this article.
Acknowledgements
The authors acknowledge the excellent technical work of Lena Ellezian and George Malatantis in the early phases of this study.
Footnotes
The Transparency document associated with this article can be found, in online version.
References
- Adler C.P., Bollmann R. Osteogenesis imperfecta congenita (Vrolik) Med. Welt. 1973;24:2007–2012. [PubMed] [Google Scholar]
- Alanay Y., Avaygan H., Camacho N., Utine G.E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F.T., Zabel B., Superti-Furga A., Bruckner-Tuderman L., Curry C.J.R., Pyott S., Byers P.H., Eyre D.R., Baldridge D., Lee B., Merrill A.E., Davis E.C., Cohn D.H., Akarsu N., Krakow D. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright J.P., Albright J.A., Crelin E.S. Osteogenesis Imperfecta Tarda. Clin. Orthop. Relat. Res. 1975;108:204–213. doi: 10.1097/00003086-197505000-00034. [DOI] [PubMed] [Google Scholar]
- Ben Amor I.M., Roughley P., Glorieux F.H., Rauch F. Skeletal clinical characteristics of osteogenesis imperfecta caused by haploinsufficiency mutations inCOL1A1. J. Bone Miner. Res. 2013;28:2001–2007. doi: 10.1002/jbmr.1942. [DOI] [PubMed] [Google Scholar]
- Bodian D.L., Madhan B., Brodsky B., Klein T.E. Predicting the clinical lethality of osteogenesis imperfecta from collagen glycine mutations. Biochemistry. 2008;47:5424–5432. doi: 10.1021/bi800026k. [DOI] [PubMed] [Google Scholar]
- Bodian D.L., Chan T.-F., Poon A., Schwarze U., Yang K., Byers P.H., Kwok P.-Y., Klein T.E. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum. Mol. Genet. 2009;18:463–471. doi: 10.1093/hmg/ddn374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger M.W., Romanowicz G.E., Kohn D.H. Advancements in composition and structural characterization of bone to inform mechanical outcomes and modeling. Curr. Opin. Biomed. Eng. 2019;11:76–84. doi: 10.1016/j.cobme.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönnemann C.G., Cox G.F., Shapiro F., Wu J.J., Feener C.A., Thompson T.G., Anthony D.C., Eyre D.R., Darras B.T., Kunkel L.M. A mutation in the alpha 3 chain of type IX collagen causes autosomal dominant multiple epiphyseal dysplasia with mild myopathy. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1212–1217. doi: 10.1073/pnas.97.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudko S.P., Pokidysheva E.N., Bächinger H.P. Autosomal recessive osteogenesis imperfecta: a puzzle for bone formation, structure and function. Curr. Genet. Med. Rep. 2013;1:239–246. doi: 10.1007/s40142-013-0026-2. [DOI] [Google Scholar]
- Boyde A., Travers R., Glorieux F.H., Jones S.J. The mineralization density of iliac crest bone from children with Osteogenesis Imperfecta. Calcif. Tissue Int. 1999;64:185–190. doi: 10.1007/s002239900600. [DOI] [PubMed] [Google Scholar]
- Bryan M.A., Cheng H., Brodsky B. Sequence environment of mutation affects stability and folding in collagen model peptides of osteogenesis imperfecta. Biopolymers. 2011;96:4–13. doi: 10.1002/bip.21432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buday K. Beiträge zur Lehre von der Osteogenesis imperfecta. Sitzungsberichte Akad Wiss. Wien. 1895;104:61–102. [Google Scholar]
- Bullough P.G., Davidson D.D., Lorenzo J.C. The morbid anatomy of the skeleton in Osteogenesis Imperfecta. Clin. Orthop. Relat. Res. 1981;159:42–57. doi: 10.1097/00003086-198109000-00007. [DOI] [PubMed] [Google Scholar]
- Burman A., Tanjore H., Blackwell T.S. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018;68–69:355–365. doi: 10.1016/j.matbio.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers P.H. Folding defects in fibrillar collagens. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2001;356:151–158. doi: 10.1098/rstb.2000.0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers P.H., Wallis G.A., Willing M.C. Osteogenesis imperfecta: translation of mutation to phenotype. J. Med. Genet. 1991;28:433–442. doi: 10.1136/jmg.28.7.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty E.G., Kadler K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- Carriero A., Doube M., Vogt M., Busse B., Zustin J., Levchuk A., Schneider P., Müller R., Shefelbine S.J. Altered lacunar and vascular porosity in osteogenesis imperfecta mouse bone as revealed by synchrotron tomography contributes to bone fragility. Bone. 2014;61:116–124. doi: 10.1016/j.bone.2013.12.020. [DOI] [PubMed] [Google Scholar]
- Cassella J.P., Ali S.Y. Abnormal collagen and mineral formation in osteogenesis imperfecta. Bone Miner. 1992;17:123–128. doi: 10.1016/0169-6009(92)90722-p. [DOI] [PubMed] [Google Scholar]
- Cassella J.P., Stamp T.C.B., Ali S.Y. A morphological and ultrastructural study of bone in osteogenesis imperfecta. Calcif. Tissue Int. 1996;58:155–165. doi: 10.1007/bf02526881. [DOI] [PubMed] [Google Scholar]
- Cooper R.R., Ponseti I.V., Maynard J.A. Pseudoachondroplastic dwarfism. J. Bone Jt. Surg. 1973;55:475–484. doi: 10.2106/00004623-197355030-00003. [DOI] [PubMed] [Google Scholar]
- Dempster D.W., Compston J.E., Drezner M.K., Glorieux F.H., Kanis J.A., Malluche H., Meunier P.J., Ott S.M., Recker R.R., Parfitt A.M. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry nomenclature committee. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2013;28:2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Martino R., Sticco L., Luini A. Regulation of cargo export and sorting at the trans-Golgi network. FEBS Lett. 2019;593:2306–2318. doi: 10.1002/1873-3468.13572. [DOI] [PubMed] [Google Scholar]
- Doty S.B., Mathews R.S. Electron microscopic and Histochemical investigation of Osteogenesis Imperfecta Tarda. Clin. Orthop. Relat. Res. 1971;80:191–201. doi: 10.1097/00003086-197110000-00027. [DOI] [PubMed] [Google Scholar]
- Engfeldt B., Engström R., Zetterström R. Biophysical studies of the bone tissue in osteogenesis imperfecta. J. Bone Joint Surg. (Br.) 1954;36-B:654–661. doi: 10.1302/0301-620x.36b4.654. [DOI] [PubMed] [Google Scholar]
- Falvo K.A., Bullough P.G. Osteogenesis imperfecta. J. Bone Jt. Surg. 1973;55:275–286. doi: 10.2106/00004623-197355020-00003. [DOI] [PubMed] [Google Scholar]
- Ferretti M., Palumbo C., Contri M., Marotti G. Static and dynamic osteogenesis: two different types of bone formation. Anat. Embryol. (Berl). 2002;206:21–29. doi: 10.1007/s00429-002-0265-6. [DOI] [PubMed] [Google Scholar]
- Flier J.S., Underhill L.H., Prockop D.J. Mutations in collagen genes as a cause of connective-tissue diseases. N. Engl. J. Med. 1992;326:540–546. doi: 10.1056/nejm199202203260807. [DOI] [PubMed] [Google Scholar]
- Follis R.H.J. Histochemical studies on cartilage and bone. III. Osteogenesis imperfecta. Bull. Johns Hopkins Hosp. 1953;93:386–399. [PubMed] [Google Scholar]
- Forlino A., Marini J.C. Osteogenesis imperfecta. Lancet. 2016;387:1657–1671. doi: 10.1016/s0140-6736(15)00728-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlino A., Cabral W.A., Barnes A.M., Marini J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratzl P., Paris O., Klaushofer K., Landis W.J. Bone mineralization in an osteogenesis imperfecta mouse model studied by small-angle x-ray scattering. J. Clin. Invest. 1996;97:396–402. doi: 10.1172/JCI118428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratzl-Zelman N., Schmidt I., Roschger P., Roschger A., Glorieux F.H., Klaushofer K., Wagermaier W., Rauch F., Fratzl P. Unique micro- and nano-scale mineralization pattern of human osteogenesis imperfecta type VI bone. Bone. 2015;73:233–241. doi: 10.1016/j.bone.2014.12.023. [DOI] [PubMed] [Google Scholar]
- Freda V.J., Vosburgh G.J., Di Liberti C. Osteogenesis imperfecta congenita. A presentation of 16 cases and review of the literature. Obstet. Gynecol. 1961;18:535–547. [PubMed] [Google Scholar]
- Freund E. Zur Frage der Ostitis deformans Paget. Herrn Professor Tandler zum 60. Geburtstag. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1929;274:1–36. doi: 10.1007/bf02032764. [DOI] [Google Scholar]
- Garbes L., Kim K., Rieß A., Hoyer-Kuhn H., Beleggia F., Bevot A., Kim M.J., Huh Y.H., Kweon H.-S., Savarirayan R., Amor D., Kakadia P.M., Lindig T., Kagan K.O., Becker J., Boyadjiev S.A., Wollnik B., Semler O., Bohlander S.K., Kim J., Netzer C. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am. J. Hum. Genet. 2015;96:432–439. doi: 10.1016/j.ajhg.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautieri A., Uzel S., Vesentini S., Redaelli A., Buehler M.J. Molecular and mesoscale mechanisms of osteogenesis imperfecta disease in collagen fibrils. Biophys. J. 2009;97:857–865. doi: 10.1016/j.bpj.2009.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautieri A., Vesentini S., Redaelli A., Buehler M.J. Osteogenesis imperfecta mutations lead to local tropocollagen unfolding and disruption of H-bond network. RSC Adv. 2012;2:3890. doi: 10.1039/c2ra01047j. [DOI] [Google Scholar]
- Georgiadis M., Müller R., Schneider P. Techniques to assess bone ultrastructure organization: orientation and arrangement of mineralized collagen fibrils. J. R. Soc. Interface. 2016;13:20160088. doi: 10.1098/rsif.2016.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud-Guille M.M. Twisted plywood architecture of collagen fibrils in human compact bone osteons. Calcif. Tissue Int. 1988;42:167–180. doi: 10.1007/bf02556330. [DOI] [PubMed] [Google Scholar]
- Glorieux F.H., Rauch F., Plotkin H., Ward L., Travers R., Roughley P., Lalic L., Glorieux D.F., Fassier F., Bishop N.J. Type V Osteogenesis Imperfecta: a new form of brittle bone disease. J. Bone Miner. Res. 2000;15:1650–1658. doi: 10.1359/jbmr.2000.15.9.1650. [DOI] [PubMed] [Google Scholar]
- Glorieux F.H., Travers R., Taylor A., Bowen J.R., Rauch F., Norman M., Parfitt A.M. Normative data for iliac bone histomorphometry in growing children. Bone. 2000;26:103–109. doi: 10.1016/s8756-3282(99)00257-4. [DOI] [PubMed] [Google Scholar]
- Glorieux F.H., Ward L.M., Rauch F., Lalic L., Roughley P.J., Travers R. Osteogenesis Imperfecta type VI: a form of brittle bone disease with a mineralization defect. J. Bone Miner. Res. 2002;17:30–38. doi: 10.1359/jbmr.2002.17.1.30. [DOI] [PubMed] [Google Scholar]
- Haebara H., Yamasaki Y., Kyogoku M. An autopsy case of osteogenesis imperfecta congenita-histochemical and electron microscopical studies. Pathol. Int. 1969;19:377–394. doi: 10.1111/j.1440-1827.1969.tb00713.x. [DOI] [PubMed] [Google Scholar]
- Hagenbach E. Osteogenesis imperfecta tarda. Frankfurt Zeit f Path. 1911;6:398–460. [Google Scholar]
- Heinrich Bauer K. Über Osteogenesis imperfecta. Dtsch. Zeitschrift Für Chir. 1920;154:166–213. doi: 10.1007/BF02799130. [DOI] [Google Scholar]
- Heinrich Bauer K. Über Identität und Wesen der sogenannten Osteopsathyrosis idiopathica und Osteogenesis imperfecta. Dtsch. Zeitschrift Für Chir. 1920;160:289–351. doi: 10.1007/bf02801188. [DOI] [Google Scholar]
- Homan E.P., Rauch F., Grafe I., Lietman C., Doll J.A., Dawson B., Bertin T., Napierala D., Morello R., Gibbs R., White L., Miki R., Cohn D.H., Crawford S., Travers R., Glorieux F.H., Lee B. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J. Bone Miner. Res. 2011;26:2798–2803. doi: 10.1002/jbmr.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer-Kuhn H., Netzer C., Koerber F., Schoenau E., Semler O. Two years' experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J. Rare Dis. 2014;9:145. doi: 10.1186/s13023-014-0145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y., Bächinger H.P. A molecular ensemble in the rER for procollagen maturation. Biochim. Biophys. Acta, Mol. Cell Res. 2013;1833:2479–2491. doi: 10.1016/j.bbamcr.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Jaffe H.L. Osteitis deformans, Paget's disease of bone. Arch. Pathol. 1933;15:83–131. [Google Scholar]
- Jaffe H.J. Metabolic, degenerative, and inflammatory diseases of bones and joints. Ann. Intern. Med. 1972;77:1016. doi: 10.7326/0003-4819-77-6-1016_1. [DOI] [Google Scholar]
- Jeckeln E. Systemgebundene mesenchymale Erschöpfung. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1931;280:351–373. doi: 10.1007/bf02076384. [DOI] [Google Scholar]
- Jones C.J.P., Cummings C., Ball J., Beighton P. Collagen defect of bone in Osteogenesis Imperfecta (type I) Clin. Orthop. Relat. Res. 1984:208–214. doi: 10.1097/00003086-198403000-00032. [DOI] [PubMed] [Google Scholar]
- Jones S.J., Glorieux F.H., Travers R., Boyde A. The microscopic structure of bone in Normal children and patients with Osteogenesis Imperfecta: a survey using backscattered Electron imaging. Calcif. Tissue Int. 1999;64:8–17. doi: 10.1007/s002239900571. [DOI] [PubMed] [Google Scholar]
- Kadler K.E., Baldock C., Bella J., Boot-Handford R.P. Collagens at a glance. J. Cell Sci. 2007;120:1955–1958. doi: 10.1242/jcs.03453. [DOI] [PubMed] [Google Scholar]
- Kar K., Amin P., Bryan M.A., Persikov A.V., Mohs A., Wang Y.-H., Brodsky B. Self-association of collagen triple helic peptides into higher order structures. J. Biol. Chem. 2006;281:33283–33290. doi: 10.1074/jbc.m605747200. [DOI] [PubMed] [Google Scholar]
- Keller R.B., Tran T.T., Pyott S.M., Pepin M.G., Savarirayan R., McGillivray G., Nickerson D.A., Bamshad M.J., Byers P.H. Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively. Genet. Med. 2018;20:411–419. doi: 10.1038/gim.2017.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschnitzki M., Wagermaier W., Liu Y., Roschger P., Duda G.N., Fratzl P. Poorly ordered bone as an endogenous scaffold for the deposition of highly oriented lamellar tissue in rapidly growing ovine bone. Cells Tissues Organs. 2011;194:119–123. doi: 10.1159/000324467. [DOI] [PubMed] [Google Scholar]
- Kerschnitzki M., Wagermaier W., Roschger P., Seto J., Shahar R., Duda G.N., Mundlos S., Fratzl P. The organization of the osteocyte network mirrors the extracellular matrix orientation in bone. J. Struct. Biol. 2011;173:303–311. doi: 10.1016/j.jsb.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Key J.A. Brittle bones and blue sclera. Arch. Surg. 1926;13:523–567. doi: 10.1001/archsurg.1926.01130100067006. [DOI] [Google Scholar]
- Kim P.S., Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr. Rev. 1998;19:173–202. doi: 10.1210/edrv.19.2.0327. [DOI] [PubMed] [Google Scholar]
- Klotz O. Osteogenesis imperfecta. J. Pathol. Bacteriol. 1909;13:467–481. doi: 10.1002/path.1700130148. [DOI] [Google Scholar]
- Klumperman J. Architecture of the mammalian Golgi. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocher M.S., Shapiro F. Osteogenesis Imperfecta. J. Am. Acad. Orthop. Surg. 1998;6:225–236. doi: 10.5435/00124635-199807000-00004. [DOI] [PubMed] [Google Scholar]
- Kuivaniemi H., Tromp G., Prockop D.J. Mutations in collagen genes: causes of rare and some common diseases in humans. FASEB J. 1991;5:2052–2060. doi: 10.1096/fasebj.5.7.2010058. [DOI] [PubMed] [Google Scholar]
- Kulkarni-Gosavi P., Makhoul C., Gleeson P.A. Form and function of the Golgi apparatus: scaffolds, cytoskeleton and signalling. FEBS Lett. 2019;593:2289–2305. doi: 10.1002/1873-3468.13567. [DOI] [PubMed] [Google Scholar]
- Leblond C.P. Synthesis and secretion of collagen by cells of connective tissue, bone, and dentin. Anat. Rec. 1989;224:123–138. doi: 10.1002/ar.1092240204. [DOI] [PubMed] [Google Scholar]
- Lee D.Y., Cho T.-J., Choi I.H., Chung C.Y., Yoo W.J., Kim J.H., Park Y.K. Clinical and radiological manifestations of osteogenesis imperfecta type V. J. Korean Med. Sci. 2006;21:709–714. doi: 10.3346/jkms.2006.21.4.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.-J., Lyu F., Song Y.-W., Wang O., Jiang Y., Xia W.-B., Xing X.-P., Li M. Genotype-phenotype relationship in a large cohort of osteogenesis imperfecta patients with COL1A1 mutations revealed by a new scoring system. Chin. Med. J. 2019;132:145–153. doi: 10.1097/CM9.0000000000000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.-Y., Chuang C.-K., Su Y.-N., Chen M.-R., Chiu H.-C., Niu D.-M., Lin S.-P. Genotype and phenotype analysis of Taiwanese patients with osteogenesis imperfecta. Orphanet J. Rare Dis. 2015;10:152. doi: 10.1186/s13023-015-0370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looser E. Zur Kenntnis der Osteogenesis Imperfecta congenita und tarda (sogenannte idiopathische osteopsathyrosis) Mitt. Grenzgeb. Med. Chir. 1906;15:160–207. [Google Scholar]
- Machamer C.E. The Golgi complex in stress and death. Front. Neurosci. 2015;9:421. doi: 10.3389/fnins.2015.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maioli M., Gnoli M., Boarini M., Tremosini M., Zambrano A., Pedrini E., Mordenti M., Corsini S., D'Eufemia P., Versacci P., Celli M., Sangiorgi L. Genotype–phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur. J. Hum. Genet. 2019;27:1090–1100. doi: 10.1038/s41431-019-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhoul C., Gosavi P., Gleeson P.A. Golgi dynamics: the morphology of the mammalian Golgi apparatus in health and disease. Front. Cell Dev. Biol. 2019;7:112. doi: 10.3389/fcell.2019.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhan D., Muelke M., Rosch S., Schaefer A.B., Merboth F., Weisweiler D., Heiss C., Arganda-Carreras I., El Khassawna T. An optimized approach to perform bone histomorphometry. Front. Endocrinol. (Lausanne) 2018;9:666. doi: 10.3389/fendo.2018.00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini J.C., Forlino A., Cabral W.A., Barnes A.M., Antonio J.D. San, Milgrom S., Hyland J.C., Körkkö J., Prockop D.J., De Paepe A., Coucke P., Symoens S., Glorieux F.H., Roughley P.J., Lund A.M., Kuurila-Svahn K., Hartikka H., Cohn D.H., Krakow D., Mottes M., Schwarze U., Chen D., Yang K., Kuslich C., Troendle J., Dalgleish R., Byers P.H. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini J.C., Forlino A., Bächinger H.P., Bishop N.J., Byers P.H., De Paepe A., Fassier F., Fratzl-Zelman N., Kozloff K.M., Krakow D., Montpetit K., Semler O. Osteogenesis imperfecta. Nat. Rev. Dis. Prim. 2017;3 doi: 10.1038/nrdp.2017.52. [DOI] [PubMed] [Google Scholar]
- Marotti G., Ferretti M., Palumbo C. The problem of bone lamellation: an attempt to explain different proposed models. J. Morphol. 2013;274:543–550. doi: 10.1002/jmor.20114. [DOI] [PubMed] [Google Scholar]
- McCarthy E.F., Earnest K., Rossiter K., Shapiro J. Bone Histomorphometry in adults with type IA Osteogenesis Imperfecta. Clin. Orthop. Relat. Res. 1997;336:254–262. doi: 10.1097/00003086-199703000-00034. [DOI] [PubMed] [Google Scholar]
- Michel F. Osteogenesis imperfecta. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1903;173:1–35. doi: 10.1007/bf01947875. [DOI] [Google Scholar]
- Milgram J.W., Flick M.R., Engh C.A. Osteogenesis Imperfecta. J. Bone Jt. Surg. 1973;55:506–515. doi: 10.2106/00004623-197355030-00006. [DOI] [PubMed] [Google Scholar]
- Moosa S., Chung B.H.-Y., Tung J.Y.-L., Altmüller J., Thiele H., Nürnberg P., Netzer C., Nishimura G., Wollnik B. Mutations inSEC24Dcause autosomal recessive osteogenesis imperfecta. Clin. Genet. 2015;89:517–519. doi: 10.1111/cge.12678. [DOI] [PubMed] [Google Scholar]
- Morello R. Osteogenesis imperfecta and therapeutics. Matrix Biol. 2018;71–72:294–312. doi: 10.1016/j.matbio.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Saito A., Hino S., Kondo S., Kanemoto S., Chihara K., Sekiya H., Tsumagari K., Ochiai K., Yoshinaga K., Saitoh M., Nishimura R., Yoneda T., Kou I., Furuichi T., Ikegawa S., Ikawa M., Okabe M., Wanaka A., Imaizumi K. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 2009;11:1205–1211. doi: 10.1038/ncb1963. [DOI] [PubMed] [Google Scholar]
- Nabavi N., Pustylnik S., Harrison R.E. Rab GTPase mediated procollagen trafficking in ascorbic acid stimulated osteoblasts. PLoS One. 2012;7(9):e46265. doi: 10.1371/journal.pone.0046265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols R.W., Lovett E.H. Osteogenesis imperfecta. With the report of a case, with autopsy and histological examination. Br. Med. J. 1906;2:915–920. https://www.jstor.org/stable/20291485 [Google Scholar]
- Nijhuis W.H., Eastwood D.M., Allgrove J., Hvid I., Weinans H.H., R.A. Bank, Sakkers R.J. Current concepts in osteogenesis imperfecta: bone structure, biomechanics and medical management. J. Child. Orthop. 2019;13:1–11. doi: 10.1302/1863-2548.13.180190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes S.A., Papa F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramser J.R., Frost H.M. The study of a rib biopsy from a patient with Osteogenesis Imperfecta: a method using in vivoTetracycline labeling. Acta Orthop. Scand. 1966;37:229–240. doi: 10.3109/17453676608989411. [DOI] [PubMed] [Google Scholar]
- Rauch F. Watching bone cells at work: what we can see from bone biopsies. Pediatr. Nephrol. 2006;21:457–462. doi: 10.1007/s00467-006-0025-6. [DOI] [PubMed] [Google Scholar]
- Rauch F., Travers R., Parfitt A.M., Glorieux F.H. Static and dynamic bone histomorphometry in children with osteogenesis imperfecta. Bone. 2000;26:581–589. doi: 10.1016/s8756-3282(00)00269-6. [DOI] [PubMed] [Google Scholar]
- Rauch F., Travers R., Plotkin H., Glorieux F.H. The effects of intravenous pamidronate on the bone tissue of children and adolescents with osteogenesis imperfecta. J. Clin. Invest. 2002;110:1293–1299. doi: 10.1172/jci0215952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch F., Lalic L., Roughley P., Glorieux F.H. Relationship between genotype and skeletal phenotype in children and adolescents with Osteogenesis Imperfecta. J. Bone Miner. Res. 2009 doi: 10.1359/jbmr.091109. 091123190932048–30. [DOI] [PubMed] [Google Scholar]
- Reznikov N., Shahar R., Weiner S. Bone hierarchical structure in three dimensions. Acta Biomater. 2014;10:3815–3826. doi: 10.1016/j.actbio.2014.05.024. [DOI] [PubMed] [Google Scholar]
- Ricard-Blum S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011;3(1):a004978. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichon J., Germain J.P. Pathogenesis of osteogenesis imperfecta. Can. Med. Assoc. J. 1968;99:975–979. [PMC free article] [PubMed] [Google Scholar]
- Roschger P., Fratzl-Zelman N., Misof B.M., Glorieux F.H., Klaushofer K., Rauch F. Evidence that abnormal high bone mineralization in growing children with Osteogenesis Imperfecta is not associated with specific collagen mutations. Calcif. Tissue Int. 2008;82:263–270. doi: 10.1007/s00223-008-9113-x. [DOI] [PubMed] [Google Scholar]
- Roughley P.J., Rauch F., Glorieux F.H. Osteogenesis imperfecta - clinical and molecular diversity. Eur. Cells Mater. 2003;5:41–47. doi: 10.22203/ecm.v005a04. [DOI] [PubMed] [Google Scholar]
- Roujeau J., Sée G., Galian A., Guillaumin J.P., Le Gland P.F. Histological study of osteo-psathyrosis (a propos of two cases) Ann. Anat. Pathol. (Paris). 1967;12:79–89. [PubMed] [Google Scholar]
- Rutishauser J., Spiess M. Endoplasmic reticulum storage diseases. Swiss Med. Wkly. 2002;132:211–222. doi: 10.4414/smw.2002.09861. [DOI] [PubMed] [Google Scholar]
- Sarathchandra P., Pope F.M., Ali S.Y. Morphometric analysis of type I collagen fibrils in the osteoid of Osteogenesis Imperfecta. Calcif. Tissue Int. 1999;65:390–395. doi: 10.1007/s002239900719. [DOI] [PubMed] [Google Scholar]
- Sarathchandra P., Pope F.M., Kayser M.V., Ali S.Y. A light and electron microscopic study of osteogenesis imperfecta bone samples, with reference to collagen chemistry and clinical phenotype. J. Pathol. 2000;192:385–395. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH704>3.0.CO;2-U. doi:10.1002/1096-9896(2000)9999:9999<::aid-path704>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Schmorl G. Über Ostitis deformans Paget. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1932;283:694–751. doi: 10.1007/bf01887990. [DOI] [Google Scholar]
- Seedorff K.S. 1949. Osteogenesis Imperfecta. A study of clinical features and heredity based on 55 Danish families comprising 180 affected members. University of Aarhus, Denmark. [Google Scholar]
- Sesso A., Belizário J.E., Marques M.M., Higuchi M.L., Schumacher R.I., Colquhoun A., Ito E., Kawakami J. Mitochondrial swelling and incipient outer membrane rupture in preapoptotic and apoptotic cells. Anat. Rec. (Hoboken). 2012;295:1647–1659. doi: 10.1002/ar.22553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah F.A., Ruscsák K., Palmquist A. 50 years of scanning electron microscopy of bone-a comprehensive overview of the important discoveries made and insights gained into bone material properties in health, disease, and taphonomy. Bone Res. 2019;7:15. doi: 10.1038/s41413-019-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro F. Consequences of an Osteogenesis Imperfecta diagnosis for survival and ambulation. J. Pediatr. Orthop. 1985;5:456–462. doi: 10.1097/01241398-198507000-00014. [DOI] [PubMed] [Google Scholar]
- Shapiro F. Cortical bone repair. The relationship of the lacunar-canalicular system and intercellular gap junctions to the repair process. J. Bone Jt. Surg. 1988;70:1067–1081. doi: 10.2106/00004623-198870070-00016. [DOI] [PubMed] [Google Scholar]
- Shapiro F. Structural abnormalities of epiphyses in skeletal dysplasias in: Skeletal growth and development: Clinical issues and basic science advances. In: Buckwalter J.A., Ehrlich M.G., Sandell L., Trippel S.B., editors. Skelet. Growth Dev. Clin. Issues Basic Sci. Adv; American Academy of Orthopaedic Surgeons: 1998. pp. 471–489. [Google Scholar]
- Shapiro F. Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur. Cells Mater. 2008;15:53–76. doi: 10.22203/ecm.v015a05. [DOI] [PubMed] [Google Scholar]
- Shapiro F., Wu J.Y. Woven bone overview: structural classification based on its integral role in developmental, repair and pathological bone formation throughout vertebrate groups. Eur. Cells Mater. 2019;38:137–167. doi: 10.22203/ecm.v038a11. [DOI] [PubMed] [Google Scholar]
- Shapiro F., Mulhern H., Weis M.A., Eyre D. Rough endoplasmic reticulum abnormalities in a patient with Spondyloepimetaphyseal dysplasia with scoliosis, joint laxity, and finger deformities. Ultrastruct. Pathol. 2006;30:393–400. doi: 10.1080/01913120600967004. [DOI] [PubMed] [Google Scholar]
- Shapiro J.R., Lietman C., Grover M., Lu J.T., Nagamani S.C., Dawson B.C., Baldridge D.M., Bainbridge M.N., Cohn D.H., Blazo M., Roberts T.T., Brennen F.-S., Wu Y., Gibbs R.A., Melvin P., Campeau P.M., Lee B.H. Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation. J. Bone Miner. Res. 2013;28:1523–1530. doi: 10.1002/jbmr.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillence D.O., Senn A., Danks D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillence D.O., Horton W.A., Rimoin D.L. Morphologic studies in the skeletal dysplasias. Am. J. Pathol. 1979;96:813–870. [PMC free article] [PubMed] [Google Scholar]
- Sillence D.O., Barlow K.K., Garber A.P., Hall J.G., Rimoin D.L. Osteogenesis imperfecta type II delineation of the phenotype with reference to genetic heterogeneity. Am. J. Med. Genet. 1984;17:407–423. doi: 10.1002/ajmg.1320170204. [DOI] [PubMed] [Google Scholar]
- Spranger J., Cremin B., Beighton P. Osteogenesis imperfecta congenita. Pediatr. Radiol. 1982;12:21–27. doi: 10.1007/bf01221706. [DOI] [PubMed] [Google Scholar]
- Stanescu R., Stanescu V., Muriel M.-P., Maroteaux P. Multiple epiphyseal dysplasia, Fairbank type: morphologic and biochemical study of cartilage. Am. J. Med. Genet. 1993;45:501–507. doi: 10.1002/ajmg.1320450420. [DOI] [PubMed] [Google Scholar]
- Ste-Marie L.G., Charhon S.A., Edouard C., Chapuy M.C., Meunier P.J. Iliac bone histomorphometry in adults and children with osteogenesis imperfecta. J. Clin. Pathol. 1984;37:1081–1089. doi: 10.1136/jcp.37.10.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöss H. Pathologic anatomy of osteogenesis imperfecta. Light and electron microscopy of the skeletal system. Med. Klin. (Munich) 1988;83:358–362. [PubMed] [Google Scholar]
- Stöss H., Freisinger P. Collagen fibrils of osteoid in osteogenesis imperfecta: Morphometrical analysis of the fibril diameter. Am. J. Med. Genet. 1993;45:257. doi: 10.1002/ajmg.1320450220. [DOI] [PubMed] [Google Scholar]
- Symoens S., Malfait F., D'hondt S., Callewaert B., Dheedene A., Steyaert W., Bächinger H.P., De Paepe A., Kayserili H., Coucke P.J. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J. Rare Dis. 2013;8:154. doi: 10.1186/1750-1172-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger S., Hecht J.T. Pseudoachondroplasia and multiple epiphyseal dysplasia: new etiologic developments. Am. J. Med. Genet. 2001;106:244–250. doi: 10.1002/ajmg.10234. [DOI] [PubMed] [Google Scholar]
- van der Harten H.J., Brons J.T.J., Dijkstra P.F., Meijer C.J.L.M., van Geijn H.P., Arts N.F.T., Niermeijer M.F. Perinatal lethal Osteogenesis Imperfecta: radiologic and pathologic evaluation of seven prenatally diagnosed cases. Pediatr. Pathol. 1988;8:233–252. doi: 10.3109/15513818809042968. [DOI] [PubMed] [Google Scholar]
- Van Dijk F.S., Sillence D.O. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A. 2014;164A:1470–1481. doi: 10.1002/ajmg.a.36545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk F.S., Pals G., Van Rijn R.R., Nikkels P.G.J., Cobben J.M. Classification of Osteogenesis Imperfecta revisited. Eur. J. Med. Genet. 2010;53:1–5. doi: 10.1016/j.ejmg.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Vanleene M., Porter A., Guillot P.-V., Boyde A., Oyen M., Shefelbine S. Ultra-structural defects cause low bone matrix stiffness despite high mineralization in osteogenesis imperfecta mice. Bone. 2012;50:1317–1323. doi: 10.1016/j.bone.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi G., Gandini A., Monti E., Carbonare L.D., Corradi M., Vincenzi M., Valenti M.T., Valli M., Pelilli E., Boner A., Mottes M., Antoniazzi F. Lack of expression ofSERPINF1, the gene coding for pigment epithelium-derived factor, causes progressively deforming osteogenesis imperfecta with normal type I collagen. J. Bone Miner. Res. 2012;27:723–728. doi: 10.1002/jbmr.1480. [DOI] [PubMed] [Google Scholar]
- Wagermaier W., Gupta H.S., Gourrier A., Burghammer M., Roschger P., Fratzl P. Spiral twisting of fiber orientation inside bone lamellae. Biointerphases. 2006;1:1–5. doi: 10.1116/1.2178386. [DOI] [PubMed] [Google Scholar]
- Ward L.M., Rauch F., Travers R., Chabot G., Azouz E.M., Lalic L., Roughley P.J., Glorieux F.H. Osteogenesis imperfecta type VII: an autosomal recessive form of brittle bone disease. Bone. 2002;31:12–18. doi: 10.1016/s8756-3282(02)00790-1. [DOI] [PubMed] [Google Scholar]
- Weber M. Osteogenesis imperfecta congenita: a study of its histopathogenesis. Arch. Pathol. 1930;9:984–1006. [Google Scholar]
- Weiner S., Arad T., Sabanay I., Traub W. Rotated plywood structure of primary lamellar bone in the rat: orientations of the collagen fibril arrays. Bone. 1997;20:509–514. doi: 10.1016/s8756-3282(97)00053-7. [DOI] [PubMed] [Google Scholar]
- Weiner S., Traub W., Wagner H.D. Lamellar bone: structure–function relations. J. Struct. Biol. 1999;126:241–255. doi: 10.1006/jsbi.1999.4107. [DOI] [PubMed] [Google Scholar]
- Wilson R.V., Ramser J.R., Frost H.M. Thickness of osteoid seams in some diseases of man. Clin. Orthop. Relat. Res. 1966;49:119–124. doi: 10.1097/00003086-196611000-00009. [DOI] [PubMed] [Google Scholar]
- Wilton Å. Die Skeletveränderungen bei einem Spätfalle von Osteogenesis imperfecta nebst Erörterung der Entstehungsweise unter Berücksichtigung anderer Skeletkrankheiten. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1932;283:778–800. doi: 10.1007/bf01887992. [DOI] [Google Scholar]
- Yamamoto T., Hasegawa T., Sasaki M., Hongo H., Tabata C., Liu Z., Li M., Amizuka N. Structure and formation of the twisted plywood pattern of collagen fibrils in rat lamellar bone. Microscopy. 2012;61:113–121. doi: 10.1093/jmicro/dfs033. [DOI] [PubMed] [Google Scholar]
- Zanetti G., Pahuja K.B., Studer S., Shim S., Schekman R. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 2012;14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
- Zhang H., Yue H., Wang C., Gu J., He J., Fu W., Hu W., Zhang Z. Novel mutations in the SEC24D gene in Chinese families with autosomal recessive osteogenesis imperfecta. Osteoporos. Int. 2017;28:1473–1480. doi: 10.1007/s00198-016-3866-2. [DOI] [PubMed] [Google Scholar]
- Zhao H. Membrane trafficking in osteoblasts and osteoclasts: new avenues for understanding and treating skeletal diseases. Traffic. 2012;13:1307–1314. doi: 10.1111/j.1600-0854.2012.01395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhytnik L., Maasalu K., Pashenko A., Khmyzov S., Reimann E., Prans E., Kõks S., Märtson A. COL1A1/2 pathogenic variants and phenotype characteristics in Ukrainian Osteogenesis Imperfecta patients. Front. Genet. 2019;10:722. doi: 10.3389/fgene.2019.00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.