

Abstract
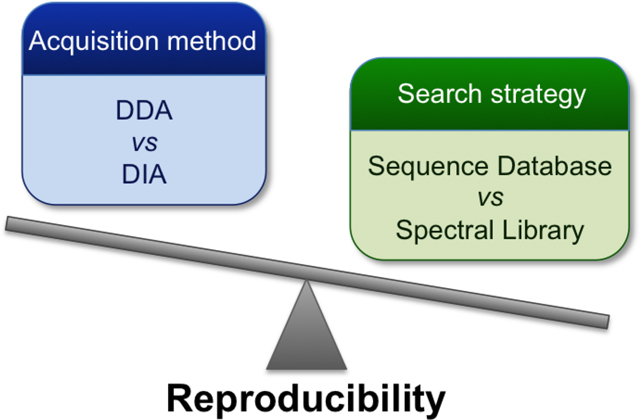
Data-independent acquisition (DIA) is a promising technique for the proteomic analysis of complex protein samples. A number of studies have claimed that DIA experiments are more reproducible than data-dependent acquisition (DDA), but these claims are unsubstantiated since different data analysis methods are used in the two methods. Data analysis in most DIA workflows depends on spectral library searches whereas DDA typically employs sequence database searches. In this study, we examined the reproducibility of DIA and DDA results using both sequence database and spectral library search. The comparison was first performed using a cell lysate and then extended to an interactome study. Protein overlap among the technical replicates in both DDA and DIA experiments was 30% higher with library-based identifications than with sequence database identifications. The reproducibility of quantification was also improved with library search compared to database search, with the mean of the coefficient of variation decreasing more than 30% and a reduction in the number of missing values of more than 35%. Our results show that regardless of the acquisition method, higher identification and quantification reproducibility is observed when library search was used.
Keywords: Reproducibility, data-independent acquisition (DIA), database search, library search, mass spectrometry, proteomics
Graphical Abstract
Introduction
Proteomic studies provide essential and comprehensive information that helps to decipher the molecular portraits of cells, but reproducibility is one of the central hurdles that has limited the potential of proteomics. Proteomic analysis is challenging 1, 2 due to the considerable complexity of the proteome, which includes a diverse array of post-translational modifications and other higher order features such as protein interactions, protein localization and protein folding. With continued improvements in mass spectrometry technology and data analysis methods, the identification and quantification of a complete proteome 3–5 is within reach, but technical reproducibility remains a challenge in large-scale studies 6–9.
Proteomic analysis relies principally on mass spectrometry technology. The first strategy developed for the automation of MS data acquisition was data-dependent acquisition (DDA), which selects precursor ions for fragmentation based on their abundances in MS1 scans. Since the peptides selected for fragmentation are usually the most abundant peaks in the survey scan, there can be a stochastic nature to data acquisition as peptide mixtures become more complex 10. Data-independent acquisition 11 (DIA) is an alternative strategy in which all precursor ions within a m/z window are fragmented regardless of their intensity and the m/z window is systematically scanned across a mass range. Venable et al 11 and Dong et al. 12 showed that DIA produced quantitative results with better signal to noise than DDA, and that fragment ion signals in DIA could be used for peptide quantification and protein identification with better reproducibility. Although Venable et al. used a database search to analyze DIA data, later strategies employed spectral library search 13. As the use of DIA in proteomics has grown, tools specific for DIA analysis 14 such as Skyline 15, Spectronaut 16, or DIA-Umpire 17 have emerged. Recent studies comparing the two scan methods have determined DIA to be more reproducible than DDA 18–20, but these studies employed different data analysis methods for each acquisition method, and thus the influence of the type of search on the results is not clear.
In a previous study we explored the use of libraries to analyze DDA data in a fashion similar to DIA data analysis strategies 21. That study showed a clear improvement in reproducibility between replicates. In this study, we assess how much a library-centric data analysis strategy improves DDA analysis relative to data collected by DIA using similar software tools. Aliquots of a cell lysate were independently measured by DIA and DDA on a Q-Exactive mass spectrometer, followed by protein and peptide identification and quantification using a sequence database and spectral library search in parallel. Subsequently, we extended our comparison to biological replicates by applying the same analysis methods to an interactome dataset.
Experimental procedures
Sample preparation.
HEK293 cells (which have not been recently checked for mycoplasma contamination) were cultured in DMEM (Gibco) supplemented with 10% v/v FBS, 1% v/v penicillin, 1% v/v streptomycin (Gibco). Cell pellets were lysed on ice using a lysis buffer composed of 4 mM HEPES, pH 7, 150 mM NaCl, 0.5 % v/v Triton X-100, 0.5% v/v NP-40, 0.01% w/v deoxycholate, protease and phosphatase inhibitors (Roche). After 24h of incubation at 4 °C, the cell extract was centrifuged at 18,000g for 30 min at 4 °C. The cleared lysate was transferred to a new tube, and the protein was quantified using a Pierce™ BCA Protein Assay Kit (Thermo Scientific). Protein mixtures were precipitated by the addition of four volumes of methanol, 1 volume of chloroform, and three volumes of water, followed by vortexing and centrifugation at 18,000xg for 2 min at room temperature. Most of the upper layer was removed, and the samples were pelleted by the addition of three volumes of methanol with centrifugation at 18,000g for 2 min. Protein pellets were air-dried for 10 min, and resuspended in 8 M urea, 100 mM Tris pH 8.5. Cysteines were reduced with 5 mM TCEP (Sigma-Aldrich) for 20 min at room temperature and alkylated with 14 mM chloroacetamide (Fluka) for 15 min in the dark at room temperature. The solution was diluted with four volumes of 100 mM Tris-HCl, pH 8.5, and digested with trypsin (sequencing grade, Promega) at an enzyme/substrate ratio of 1:50 overnight at 37 °C.
Mass spectrometry data acquisition.
The HEK293 protein digest was analyzed on a Q-Exactive mass spectrometer (Thermo) interfaced with an UHPLC EASY-nLC 1000 system (Thermo). Peptides (3 μg) were separated by reverse phase chromatography on a self-packed emitter column (ACQUITY UPLC BEH C18 1.7 μm resin, 130-Å x 100 μm x 50 cm) at 50 °C. The system was operated with the following buffers: buffer A (5% acetonitrile, 0.1% formic acid), and buffer B (80% acetonitrile, 0.1% formic acid). The UPLC delivered the following gradient at 300 nL/min: linear 1 – 45 % B in 170 min, up to 100% B in 30 min, isocratic at 100% B for 30 min, return to 1% B in 5 min, and isocratic at 1% B for 5 min. The peptide digest was independently analyzed by data-dependent acquisition (DDA) and data-independent acquisition (DIA) with the same gradient conditions and the same amounts of sample. A total of eleven DDA technical replicates were acquired; three of them were used for identification purpose only, while the other eight measurements were used for the generation of spectral libraries (Figure 1). A total of three technical replicates were acquired with the DIA method.
Figure 1. Experimental workflow.

HEK293 lysate was analyzed with DDA and DIA methods. Three technical replicates were acquired for protein and peptide identification using sequence database (ProLuCID) and spectral library search (SpectraST for DDA, and Skyline for DIA) in parallel. For DIA data, Raw Converter was applied to predict the precursors before performing the sequence database with ProLuCID. Eight additional replicates were acquired with DDA to generate the spectral libraries using SpectraST for DDA library search, and Skyline for DIA library search.
For DDA acquisition, MS1 spectra were collected in the range of 400–1200 m/z at 70,000 (FWHM) resolution. The 10 most intense precursors were selected for fragmentation with a 3 m/z isolation window at 17,500 (FWHM) resolution, for a maximum fill time of 120 ms. Precursor ions were fragmented with a normalized collision energy of 25%, and the precursors were dynamically excluded for reselection for 5s. A complete description of the method is included in Supplementary Table 1.
For DIA acquisition, a 4 m/z isolation window was used, and a total set of 200 windows was used to cover the range of 400–1200 m/z. MS2 were collected at 17,500 (FWHM) resolution for a maximum fill time of 60 ms. After 20 MS2 scans an additional MS1 scan was recorded at 70,000 (FWHM) resolution for 60 ms of maximum fill time until the mass range of 400–1200 m/z was covered, resulting in a duty cycle of ~ 2.1 s. A normalized collision energy of 25% was applied for fragmentation of the ions (see Supplementary Table 1 for further details). The raw DDA data used for identification only (three technical replicates) was previously used as one of the datasets in a recent publication 21.
Peptide and protein identification using sequence database search.
Raw files were converted to ms2 files in a centroid format using RawConverter 22 version 1.1.0.19 (available at http://fields.scripps.edu/rawconv/) with the option “select monoisotopic m/z” in DDA for the DDA data, and the option “predict precursors in DIA” for the DIA data. MS2 spectra were searched using ProLuCID 23 against the UniProt_Human_reviewed_05–05-2016 concatenated to a reverse decoy database 24. Carbamidomethyl was set as a static cysteine modification. Fully-tryptic peptides and peptides with up to three missed cleavages were allowed. The precursor-ion mass tolerance was set to 50 ppm, and the fragment-ion mass tolerance was set to 600 ppm. Database results were assembled and filtered using the DTASelect 25, 26 program (version 2.0). The peptides were filtered at 1% FDR. A complete description of the parameters used in the search is provided in Supplementary Table 2.
Spectral library generation for the DIA data search.
Raw files from the eight DDA replicates for the spectral library building were converted to ms2 files using RawConverter version 1.1.0.19 with the option “select monoisotopic m/z” in DDA. Mass spectra were searched using ProLuCID with the same parameters described in the previous paragraph. FDR at the protein level was calculated using DTASelect program (version 2.0). Results were exported to MZID files through IP2-Integrated Proteomics Pipeline version 5.0.1 (http://www.integratedproteomics.com/). A total of four spectral libraries were built using Skyline version 3.7.0.10940 with one, three, six, and eight DDA MZID files. The cut-off score was set at 0.99, and the option “keep redundant library” unchecked (see Supplementary Table 2). The total number of spectra and distinct peptide ions included in each library are provided in Supplementary Table 3.
Spectral library generation for the DDA data search.
Raw files from the eight DDA replicates used for library building were converted to mzXML files in centroid format using RawConverter version 1.1.0.19. Comet 27 [via the Trans-Proteomic Pipeline (TPP) 28 version 5.0.0] was used to query mass spectra against the database UniProt_Human_reviewed_05–05-2016 concatenated to reverse sequence decoys. The search parameters were set as follows: carbamidomethyl as a static cysteine modification, fully-tryptic digest, up to three missed cleavages, precursor peptide mass tolerance 50 ppm, and the fragment bin tolerance 0.05 m/z. Identification results were processed by PeptideProphet 29 and iProphet 30 (TPP version 5.0.0) with the following options: minimum peptide length of six, minimum PeptideProphet probability of 0.05, accurate mass binning using PPM, use decoy hits (Reverse) to pin down the negative distribution with a non-parametric model. The four spectral libraries were built using SpectraST version 5.0 with the iProphet results from one, three, six, and eight DDA files, the same combination of files used for the library built using Skyline. Results were filtered at 1% protein FDR and a non-redundant consensus library was built including shuffled decoys using the following options in SpectraST: -cP0.9; -cJU -cAC; -cAQ; -cAD -cc -cy1. All the parameters used in Comet and SpectraST are included in Supplementary Table 4. A summary of the total number of spectra and distinct peptide ions included in each library is Supplementary Table 5.
Spectral library search for DIA data.
The three DIA raw files were converted to mzXML files in centroid format using RawConverter version 1.1.0.19. The targeted data analysis was carried out using Skyline version 3.7.0.10940. The parameters used are described in Supplementary Table 6. Briefly, for the peptide settings tryptic digestion with up to three missed cleavages were allowed, and a background proteome was used with UniProt_Human_reviewed_05–05-2016 (described above). Carbamidomethyl was selected as a static modification for cysteine. A minimum peptide length of six was considered, and no retention time predictor was used. For the transition settings, the ion match tolerance was set to 0.05 m/z, 5 product ions and 3 peaks from ms1 were selected. For the retention time filtering, the option “use only scans within 5 min of ms/ms IDs” was selected. Targets lists were created independently for each spectral library by adding the spectral library with the proteins associated from the background proteome, and adding decoys using the shuffle method. The three DIA files were searched against the four spectral libraries in parallel. All detected peaks were reintegrated independently for each library using the mProphet peak-scoring model adding a q-value to each peak. Results were exported with the q-value, and filtered at 1% peptide FDR.
Spectral library search for DDA data.
The three DDA raw files selected for identification purpose only were converted to mzXML files in centroid format using RawConverter version 1.1.0.19. The DDA search against the different libraries was performed individually using SpectraST through TPP version 5.0.0. UniProt_Human_reviewed_05–05-2016 was used to associate the proteins with the results. Precursor tolerance was set to 0.05 m/z, and the default parameters were applied (See Supplementary Table 7). Results were filtered using PeptideProphet with the following options: minimum peptide length of six, minimum PeptideProphet probability of 0.05, accurate mass binning using PPM, use decoy hits (DECOY) to pin down the negative distribution with a non-parametric model. Results with a 1% peptide FDR were exported to a txt file.
Comparison of the results.
All results were compared using PACOM 31 (https://github.com/smdb21/PACOM), an in-house developed software,. Results with an FDR at 1% or below at the peptide level were individually saved in tab delimited format and imported in the software. PACOM reported (individually and globally) the number of proteins and peptides identified along with the peptide spectrum matches in each data set. The number of proteins reported by PACOM corresponds to the number of protein groups identified with the PAnalyzer 32 protein grouping algorithm after aggregation of the individual peptides of each dataset. Reproducibility was measured in two different ways: coefficients of variation and overlap within the technical replicates at protein and peptide level. The coefficients of variation of the number of proteins or peptides identified among the replicates were computed manually as the ratio of the standard deviation to the mean of the number of proteins or peptides identified with each method, and expressed in %. The protein and peptide overlap were automatically calculated with PACOM.
Quantification methods.
All the peptides that passed the identification filter of 1% FDR at peptide level were considered for quantification if detected in at least one replicate. Protein abundances were inferred by summing all the peptide intensities. The quantification of DIA data searched with library was performed by summing MS1 and MS2 peak areas per each peptide with Skyline. DIA data searched with sequence database and DDA data searched with sequence database and library were quantified by MS1 intensities using Census 33 and Progenesis QI (v4.1, Nonlinear Dynamics) respectively. All intensities were normalized by dividing them by the total intensity per sample and multiplying by the average of the total intensities for the three replicates.
Interactome data set.
HEK 293 cells were transfected over 48 h with the plasmid GFP-Flag-xCT using the X-tremeGENE HP DNA Transfection Reagent (Roche) in Opti-MEM Reduced Serum Medium (Thermo Fisher Scientific). This protocol was also used to transfect the cells with GFP-Flag as a control. After 48h the cells were washed with PBS and pelleted by centrifugation for 3 min at 500g at 4 °C. Pellets were resuspended on ice in the same lysis buffer described for the HEK293 lysate experiment. After 1 h of incubation at 4 °C, cell extracts were centrifuged at 18,000g for 30 min at 4 °C. The cleared lysates were transferred to new tubes, and the protein was quantified using the PierceTM BCA Protein Assay Kit (Thermo Scientific). Enrichment of the Flag-tagged proteins was performed by immunoprecipitation (IP) with GFP-Trap_A beads (Chromotek). The beads were washed with PBS, and 5 mg of protein from each of the cleared lysates was incubated with 25 μl of beads overnight at 4 °C on an end over end tube rotator. Beads were washed 3x with 1 ml of lysis buffer (4 mM HEPES, 200 mM NaCl, at 4 °C), and the bait complexes were released from the beads by incubation twice with 50 μl of 5% SDS at 100 °C for 10 min. The tubes were centrifuged 2 min at 500 g, and the two supernatants were collected and combined in a new tube. Protein was precipitated using MeOH/CHCl3 precipitation. Briefly, 400 μl of methanol was added to the tubes followed by 100 μl of chloroform. After vortexing, 300 μl of H2O was added, the sample was vortexed again and centrifuged. Most of the upper layer was removed, and 300 μl of MeOH was added. Sample was centrifuged and the supernatant was carefully removed allowing the pellet air dry. Protein pellets were resuspended in 8 M urea, 0.2% ProteasMAX™ surfactant trypsin enhancer (Promega). Cysteines were reduced with 5 mM TCEP for 20 min at 55 °C on a shaking incubator, and alkylated with 10 mM chloroacetamide for 20 min at dark room temperature. The sample was diluted with 25 μl of 50 mM ammonium bicarbonate and 1% ProteasMAX™. Protein was digested by addition of 1 μg of trypsin (Promega, sequencing grade modified) during 3 h at 37 °C on a shaking incubator. The samples were stored at −80 °C until analysis.
Protein digest from the bait (XCT) and control (GFP) samples were analyzed on the same instrument described for the HEK293 cell lysate, a Q-Exactive mass spectrometer interfaced with an UHPLC (Thermo). Approximately 2.3 μg of peptides were separated by reverse phase chromatography on a self-packed emitter column (ACQUITY UPLC BEH C18 1.7 μm resin, 130-Å x 100 μm x 50 cm) at 50 °C. The system was operated with the same buffers described for the HEK293 lysate. The UPLC delivered the following gradient at 300 nL/min: linear 1 – 45 % B in 140 min, up to 100% B in 30 min, isocratic at 100% B for 30 min. Samples were independently analyzed by DDA and DIA with same gradient conditions and amount of sample. Three biological replicates were acquired with DIA and DDA, for the bait and control samples. For DDA, two technical replicates were acquired per each sample, half of the technical replicates were used for identification purpose only, while the other half were used for the spectral library generation. Acquisition conditions on the mass spectrometer for DDA and DIA were the same as described for the HEK293 cell lysate analysis (see Supplementary Table 1). The raw DDA data used for identification only was previously used as one of the datasets in a recent publication 21.
Interactome data analysis.
DDA and DIA raw data were analyzed using the same pipeline used for the HEK293 lysate for sequence database and spectral library search (Figure 1) except only one spectral library was generated per condition with three DDA runs. The sequence database used for this experiment was UniProt_HumanXctGFP_03–25-2015 with the bait and tag added and reverse sequences appended. All other search settings were the same as described for the HEK293 lysate experiment. For the targeted search, two spectral libraries were built using Skyline and SpectraST following the same steps described for the HEK293 cell lysate; one for the bait (xCT) and the other for the control (GFP) samples. The description of the libraries is in Supplementary Table 11. Searches against the spectral libraries were performed with same parameters used for the HEK293 experiment. Peptides were filtered at 1% FDR. SAINTexpress 34 version 3.6.1 was used to compare biological triplicates of the xCT results with GFP controls to identify true protein interactions, using spectral counts and default settings. The SAINT analysis was performed separately for each combination of acquisition and identification methods. Interactors with 1% FDR were further considered for comparison between methods. A heatmap was created of all the interactors detected in at least one method with a 1% FDR or less using Heatmapper 35 (http://www1.heatmapper.ca/) (Fig. 3c).
Figure 3. Quantitative reproducibility.

(a) The coefficients of variation (CV) were computed from the quantitative data for the three replicates of each method at peptide and at protein levels. The line represents the median. A T-test was performed at protein and peptide level between the database and library results for DDA and DIA data independently. The three asterisks indicate that the p-value obtained from the T-test was lower than 2.2e-16. (b) The CVs were calculated for the lowest-intensity tertile of the proteins quantified with all the methods. The percentage of proteins quantified with CVs under defined thresholds are shown. (c) The missing intensity values were calculated for each method at peptide (red) and protein (blue) level considering a missing value when the intensity was not found in at least one of the three replicates. DB: sequence database search; SL: spectral library search.
Code availability.
The PACOM software code is available in Github at https://github.com/smdb21/PACOM.
Results and discussion
A trypsin digested HEK293 cell lysate was analyzed by DDA on a Q-Exactive mass spectrometer to collect data for an MS/MS spectral library. Triplicate datasets were collected using DDA and DIA methods and were searched with parallel sequence database and spectral library searches so the results could be compared (Fig. 1, and Supplementary Table 1). Spectral library generation and searches were performed using Skyline for the DIA data and SpectraST 36 for the DDA data (see Experimental procedures and Supplementary Tables 2-7). The need for two different spectral library formats for DIA and DDA data was due to incompatibilities between the library formats and the two software tools (Supplementary Note).
Reproducibility of identification
To study the identification reproducibility of the data analysis strategies, we calculated the coefficients of variation (CV) of the number of proteins and peptides identified for the three technical replicates, and then we calculated the protein and peptide overlap. Coefficients of variation were calculated as the ratio of the standard deviation to the mean of the number of proteins or peptides identified with each method, and expressed in %. The CVs for all the conditions tested were under 10% (Fig. 2a and Supplementary Table 8). Library searching decreased the variability (CVs) of protein and peptide identifications for both DDA and DIA by 30 and 80% (respectively) compared to sequence database identifications.
Figure 2. Reproducibility of DDA and DIA identification results obtained with spectral library and database search from the HEK293 cell lysate.

The reproducibility of the proteins and peptides identified between the three technical replicates was calculated for each DDA and DIA results obtained using spectral library (SpecLib) and database (Seq_DB) search in parallel. SpecLib_1, SpecLib_3, SpecLib_6, SpecLib_8, were the libraries built with one, three, six, and eight DDA files, respectively. (a) Coefficients of variation of the number of proteins or peptides identified within the technical replicates. The coefficients of variation were calculated as the ratio of the standard deviation to the mean of the number of proteins or peptides identified with each method, and expressed in percentage (%). (b) Protein and peptide overlap among the technical replicates expressed in percentage for DDA and DIA with the different identification methods. (c) The protein overlap between the methods was calculated with the total number of proteins identified combining the three technical replicates of each method. For this comparison, only the results from the library built with three DDA files were considered. The number of proteins overlapping and the number of proteins identified in only one condition are shown. The percentage of protein overlap is also included in each Venn diagram. SL: spectral library search; DB: sequence database search.
Protein overlap between the technical replicates was higher for library-based identifications for both DDA and DIA data (Fig. 2b). The overlap between the proteins identified with database search was 59% for DDA, and 57% for DIA, which is lower for both acquisition methods than when a library search was used (78–85% for DDA, and 93–98% for DIA). Therefore, protein overlap increased by more than 30% with library search compared to database search independent of the acquisition method used. Similar results were observed for the peptide overlap (Fig. 2b). These results showed that for both DIA and DDA, higher reproducibility is observed when a library search is used.
Influence of Search space size
The influence of search space size on identification results was also studied to explain the differences shown in the identification reproducibility. Four libraries were created with 1, 3, 6 or 8 DDA files increasing the search space (from 10,361 to 19,497 unique peptides for the libraries built with Skyline; 12,104 to 21,182 for the libraries built with SpectraST) (Supplementary Tables 3 and 5). However, the database search space was notably bigger (60,651,325 unique peptides). Higher protein and peptide overlap, together with lower CVs were observed when the search space decreased (Fig. 2a, b). These results suggest that the smaller search space of the library strategy helps to improve the reproducibility of the identification. Higher reproducibility of library-based over database identifications has been related to differences in the search space for DDA data 37, 38. Accordingly, in a recent reproducibility study about DIA quantification, higher CVs were observed when library size was increased 20, but the protein or peptide overlap was not computed among the replicates. However, Zhang et al. noted that library searching was more sensitive than database searching even when the size of the library and database are controlled 39.
Overlap between methods
We analyzed the protein overlap between the different acquisition and searching methods, combining the identification results from the three technical replicates and using the results from the library built with three files. Both datasets had the same overlap between data analysis methods 67% (Fig. 2c). The highest overlap (86%) was observed between DDA and DIA when spectral library was used for identification. The overlap between the proteins identified with all methods was 51% (2,085 proteins) (Supplementary Figure 1a). Similar results were observed at the peptide level (Supplementary Figure 1b).
Reproducibility of quantification
The quantification reproducibility was also evaluated using the results from the library built with three files and the database results. Proteins and peptides quantified from DIA data using library searching were obtained from MS1 and MS2 metrics, while the rest of the quantified data was based on MS1 measurements. The average of the CVs at the protein level was decreased by more than 30% when library search was used compared to database search for both acquisition methods (Fig. 3a, Supplementary tables 9 and 10). Similar results were observed for the peptide CVs. The improvement in quantification reproducibility when a library was used compared to a database search was statistically significant with a p-value < 2.2e-16 for both proteins and peptides. The difference in the reproducibility was even bigger for low-abundance proteins. In the lowest-intensity tertile, the number of irreproducibly quantified proteins with a CV > 100 was decreased by more than 50% when comparing library to database searches (63 to 31% for DDA, and 85 to 6% for DIA) (Fig. 3b). To better understand the differences in the reproducibility between library and database searches, the number of missing values was also computed. There were 39% fewer missing values detected for DDA when the library search was used compared to database search, and a 93% decrease was observed for the DIA data (Fig. 3c, Supplementary Table 10). The decrease in the number of missing values when a spectral library search was used explains the higher reproducibility of the quantification results obtained with this method compared to a database strategy.
While the reproducibility of DIA was comparable to DDA for identification and quantification when database search was used, it was markedly better with DIA-library search compared to DDA-library search (Fig. 2 and Fig. 3). This can be partially explained by the differences in the software tools used. Skyline uses retention time alignment whereas SpectraST does not implement this option unless internal retention time standards are used. In addition, Skyline retrieved the quantitative values summing the MS2 and MS1 areas while the DDA results were obtained at MS1 level only. It was previously shown that MS2 level quantification improves the reproducibility of the results compared to MS1-based 13, 40. The other reason the quantification reproducibility of DIA-library search was better than DDA is that the DIA search acquired a higher number of scans than DDA (119,598 ± 345 for DIA and 88,712 ± 2,400 for DDA, average ± standard deviation), thus providing more information for quantification.
xCT interactome
Finally, this comparison was applied to an interactome experiment of the xCT cystine/glutamate antiporter protein (SLC7A11), which plays an important role in the anti-oxidative defense mechanism 41. This protein has not been studied in BioPlex 2.0 42. We expressed xCT-GFP in HEK293 cells and compared the interactions to GFP as a negative control (Supplementary Figure 2, Supplementary Tables 11 and 12). Results obtained with spectral library search showed a higher overlap for proteins and peptides between replicates (Fig. 4a and Supplementary Figure 3), as they did in the HEK293 experiment. As we observed in the HEK293 data, the highest overlap (81%) among methods was observed between DDA and DIA using spectral library for identification (Supplementary Figure 4). To sort specific from non-specific interactors, the data was analyzed with SAINTexpress 34. A higher number of significant interactors (SAINT FDR < 0.01) was detected with library compared to database search (523 to 377 for DDA, 626 to 215 for DIA) (Fig. 4b and Supplementary Tables 13 and 14). The 4F2 heavy chain (SLC3A2), the only known binding partner of the xCT protein, was detected with all the methods with FDR < 0.01. Most of the significant interactors obtained with a database search (86% for DDA, and 71% for DIA) were also detected with a spectral library (Fig. 4c and Supplementary Table 13). Therefore, we observed not only similar biological information with a library search, but also the number of significant interactors was increased by more than 35% compared to the conventional DDA-database search workflow (Fig. 4b, c).
Figure 4. Reproducibility of the results from the xCT interactome analysis.

Reproducibility of the proteins and peptides identified for the bait (xCT) and the control (GFP) samples among the three biological replicates was addressed first. In addition, the high confident proteins identified in the bait and the control were analyzed by SAINTexpress to obtain the significant xCT interactors for all the methods in parallel. Finally, protein interactors with a SAINT probability greater than 0.99 were compared between all the methods. (a) Protein and peptide overlap among the biological replicates per each condition and method. Database is the sequence database search, and Spec_Library is the spectral library search. (b) Number of significant proteins interacting with xCT per each combination of acquisition and identification methods. (c) Heatmap with the 853 protein interactors obtained when the SAINT results are combined. The color code represents the SAINT probability for each protein. SL: spectral library search; DB: sequence database search.
Conclusions
Quantitative proteomics is an invaluable tool for addressing important biological questions. Despite the increasing number of proteomics studies in molecular biology 9, 43, poor reproducibility has limited some applications of the method. DIA has emerged as a data acquisition strategy reported to have better reproducibility and more accurate quantitation than the more widely used DDA. However, direct comparisons between DIA and DDA results 16, 20, 44, 45 have not been performed using the same identification approach. Whereas DDA data is commonly searched with a sequence database, DIA is typically searched using spectral libraries. We examined the impact of the identification method on the reproducibility of DIA and DDA results by comparing DDA and DIA data obtained from the same sample using both identification strategies in parallel. In studies with technical and biological replicates we showed higher reproducibility of library-based identification and quantification despite the acquisition method selected. In terms of reproducibility, DIA outperformed DDA, but these differences could be offset by better “fit for purpose” library software tools for DDA data. Additionally, it has been shown that TMT has reproducibility rates about equal to that of DIA and has similar “missing value” numbers 46. These results show that reproducibility of data relies on data analysis strategies. DDA can be improved relative to DIA by employing library searching strategies which strongly suggests that better “fit for purpose” tools should be developed to employ library methods for DDA data.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health grants 5R01 MH067880 and P41 GM103533. We thank Dr. Claire Delahunty for critical reading of the manuscript.
Footnotes
Supporting information
The supporting information is available free of charge.
Data availability.
All raw data files, libraries, and results for all the datasets presented in this manuscript have been deposited to ProteomeXchange consortium with the identifier PXD008235 via the MassIVE repository with the identifier MSV000081719.
References
- 1.Mallick P; Kuster B, Proteomics: a pragmatic perspective. Nat Biotechnol 2010, 28 (7), 695–709. [DOI] [PubMed] [Google Scholar]
- 2.Wilhelm M; Schlegl J; Hahne H; Gholami AM; Lieberenz M; Savitski MM; Ziegler E; Butzmann L; Gessulat S; Marx H; Mathieson T; Lemeer S; Schnatbaum K; Reimer U; Wenschuh H; Mollenhauer M; Slotta-Huspenina J; Boese JH; Bantscheff M; Gerstmair A; Faerber F; Kuster B, Mass-spectrometry-based draft of the human proteome. Nature 2014, 509 (7502), 582–7. [DOI] [PubMed] [Google Scholar]
- 3.Picotti P; Clement-Ziza M; Lam H; Campbell DS; Schmidt A; Deutsch EW; Rost H; Sun Z; Rinner O; Reiter L; Shen Q; Michaelson JJ; Frei A; Alberti S; Kusebauch U; Wollscheid B; Moritz RL; Beyer A; Aebersold R, A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 2013, 494 (7436), 266–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagaraj N; Wisniewski JR; Geiger T; Cox J; Kircher M; Kelso J; Paabo S; Mann M, Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 2011, 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsumoto M; Matsuzaki F; Oshikawa K; Goshima N; Mori M; Kawamura Y; Ogawa K; Fukuda E; Nakatsumi H; Natsume T; Fukui K; Horimoto K; Nagashima T; Funayama R; Nakayama K; Nakayama KI, A large-scale targeted proteomics assay resource based on an in vitro human proteome. Nat Methods 2017, 14 (3), 251–258. [DOI] [PubMed] [Google Scholar]
- 6.Collins BC; Hunter CL; Liu Y; Schilling B; Rosenberger G; Bader SL; Chan DW; Gibson BW; Gingras A-C; Held JM, Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nature communications 2017, 8 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabb DL; Vega-Montoto L; Rudnick PA; Variyath AM; Ham A-JL; Bunk DM; Kilpatrick LE; Billheimer DD; Blackman RK; Cardasis HL, Repeatability and reproducibility in proteomic identifications by liquid chromatography− tandem mass spectrometry. Journal of proteome research 2010, 9 (2), 761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudnick PA; Clauser KR; Kilpatrick LE; Tchekhovskoi DV; Neta P; Blonder N; Billheimer DD; Blackman RK; Bunk DM; Cardasis HL, Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Molecular & Cellular Proteomics 2010, 9 (2), 225–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H; Han J; Pan J; Liu T; Parker CE; Borchers CH, Current trends in quantitative proteomics - an update. J Mass Spectrom 2017, 52 (5), 319–341. [DOI] [PubMed] [Google Scholar]
- 10.Liu H; Sadygov RG; Yates JR, A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Analytical chemistry 2004, 76 (14), 4193–4201. [DOI] [PubMed] [Google Scholar]
- 11.Venable JD; Dong M-Q; Wohlschlegel J; Dillin A; Yates JR, Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nature methods 2004, 1 (1), 39–45. [DOI] [PubMed] [Google Scholar]
- 12.Dong M-Q; Venable JD; Au N; Xu T; Park SK; Cociorva D; Johnson JR; Dillin A; Yates JR, Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 2007, 317 (5838), 660–663. [DOI] [PubMed] [Google Scholar]
- 13.Gillet LC; Navarro P; Tate S; Rost H; Selevsek N; Reiter L; Bonner R; Aebersold R, Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 2012, 11 (6), O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navarro P; Kuharev J; Gillet LC; Bernhardt OM; MacLean B; Röst HL; Tate SA; Tsou C-C; Reiter L; Distler U, A multicenter study benchmarks software tools for label-free proteome quantification. Nature biotechnology 2016, 34 (11), 1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ, Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruderer R; Bernhardt OM; Gandhi T; Miladinović SM; Cheng L-Y; Messner S; Ehrenberger T; Zanotelli V; Butscheid Y; Escher C, Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Molecular & Cellular Proteomics 2015, 14 (5), 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsou C-C; Avtonomov D; Larsen B; Tucholska M; Choi H; Gingras A-C; Nesvizhskii AI, DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nature methods 2015, 12 (3), 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muntel J; Xuan Y; Berger ST; Reiter L; Bachur R; Kentsis A; Steen H, Advancing urinary protein biomarker discovery by data-independent acquisition on a quadrupole-orbitrap mass spectrometer. Journal of proteome research 2015, 14 (11), 4752–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsou CC; Tsai CF; Teo GC; Chen YJ; Nesvizhskii AI, Untargeted, spectral library‐free analysis of data‐independent acquisition proteomics data generated using Orbitrap mass spectrometers. Proteomics 2016, 16 (15–16), 2257–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barkovits K; Pacharra S; Pfeiffer K; Steinbach S; Eisenacher M; Marcus K; Uszkoreit J, Reproducibility, Specificity and Accuracy of Relative Quantification Using Spectral Library-based Data-independent Acquisition. Mol Cell Proteomics 2020, 19 (1), 181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez-Costa C; Martinez-Bartolome S; McClatchy D; Yates JR 3rd, Improving Proteomics Data Reproducibility with a Dual-Search Strategy. Anal Chem 2020, 92 (2), 1697–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He L; Diedrich J; Chu Y-Y; Yates JR III, Extracting accurate precursor information for tandem mass spectra by RawConverter. Analytical chemistry 2015, 87 (22), 11361–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu T; Park S; Venable J; Wohlschlegel J; Diedrich J; Cociorva D; Lu B; Liao L; Hewel J; Han X, ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. Journal of proteomics 2015, 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elias JE; Gygi SP, Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods 2007, 4 (3), 207–214. [DOI] [PubMed] [Google Scholar]
- 25.Cociorva D; L. Tabb D,; Yates JR, Validation of tandem mass spectrometry database search results using DTASelect. Current Protocols in Bioinformatics 2006, 16 (1), 13.4. 1–13.4. 14. [DOI] [PubMed] [Google Scholar]
- 26.Tabb DL; McDonald WH; Yates JR, DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. Journal of proteome research 2002, 1 (1), 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eng JK; Jahan TA; Hoopmann MR, Comet: an open‐source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–24. [DOI] [PubMed] [Google Scholar]
- 28.Deutsch EW; Shteynberg D; Lam H; Sun Z; Eng JK; Carapito C; von Haller PD; Tasman N; Mendoza L; Farrah T, Trans‐Proteomic Pipeline supports and improves analysis of electron transfer dissociation data sets. Proteomics 2010, 10 (6), 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keller A; Nesvizhskii AI; Kolker E; Aebersold R, Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry 2002, 74 (20), 5383–5392. [DOI] [PubMed] [Google Scholar]
- 30.Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI, iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Molecular & cellular proteomics 2011, 10 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martínez-Bartolomé S; Medina-Aunon JA; López-García M. A. n.; González-Tejedo C; Prieto G; Navajas R; Salazar-Donate E; Fernández-Costa C; Yates JR III; Albar JP, PACOM: A Versatile Tool for Integrating, Filtering, Visualizing, and Comparing Multiple Large Mass Spectrometry Proteomics Data Sets. Journal of proteome research 2018, 17 (4), 1547–1558. [DOI] [PubMed] [Google Scholar]
- 32.Prieto G; Aloria K; Osinalde N; Fullaondo A; Arizmendi JM; Matthiesen R, PAnalyzer: a software tool for protein inference in shotgun proteomics. BMC bioinformatics 2012, 13 (1), 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park SK; Venable JD; Xu T; Yates JR III, A quantitative analysis software tool for mass spectrometry–based proteomics. Nature methods 2008, 5 (4), 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teo G; Liu G; Zhang J; Nesvizhskii AI; Gingras A-C; Choi H, SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. Journal of proteomics 2014, 100, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babicki S; Arndt D; Marcu A; Liang Y; Grant JR; Maciejewski A; Wishart DS, Heatmapper: web-enabled heat mapping for all. Nucleic acids research 2016, 44 (W1), W147–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam H; Deutsch EW; Eddes JS; Eng JK; King N; Stein SE; Aebersold R, Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 2007, 7 (5), 655–667. [DOI] [PubMed] [Google Scholar]
- 37.Wang J; Perez-Santiago J; Katz JE; Mallick P; Bandeira N, Peptide identification from mixture tandem mass spectra. Mol Cell Proteomics 2010, 9 (7), 1476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deutsch EW, Tandem mass spectrometry spectral libraries and library searching. Methods Mol Biol 2011, 696, 225–32. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X; Li Y; Shao W; Lam H, Understanding the improved sensitivity of spectral library searching over sequence database searching in proteomics data analysis. Proteomics 2011, 11 (6), 1075–85. [DOI] [PubMed] [Google Scholar]
- 40.Weisbrod CR; Eng JK; Hoopmann MR; Baker T; Bruce JE, Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification. J Proteome Res 2012, 11 (3), 1621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewerenz J; Hewett SJ; Huang Y; Lambros M; Gout PW; Kalivas PW; Massie A; Smolders I; Methner A; Pergande M, The cystine/glutamate antiporter system xc− in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants & redox signaling 2013, 18 (5), 522–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huttlin EL; Bruckner RJ; Paulo JA; Cannon JR; Ting L; Baltier K; Colby G; Gebreab F; Gygi MP; Parzen H, Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545 (7655), 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W; Sue AC; Goh WWB, Feature selection in clinical proteomics: with great power comes great reproducibility. Drug Discov Today 2017, 22 (6), 912–918. [DOI] [PubMed] [Google Scholar]
- 44.Lambert JP; Ivosev G; Couzens AL; Larsen B; Taipale M; Lin ZY; Zhong Q; Lindquist S; Vidal M; Aebersold R; Pawson T; Bonner R; Tate S; Gingras AC, Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat Methods 2013, 10 (12), 1239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ting YS; Egertson JD; Bollinger JG; Searle BC; Payne SH; Noble WS; MacCoss MJ, PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat Methods 2017, 14 (9), 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muntel J; Kirkpatrick J; Bruderer R; Huang T; Vitek O; Ori A; Reiter L, Comparison of Protein Quantification in a Complex Background by DIA and TMT Workflows with Fixed Instrument Time. J Proteome Res 2019, 18 (3), 1340–1351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.