

Abstract
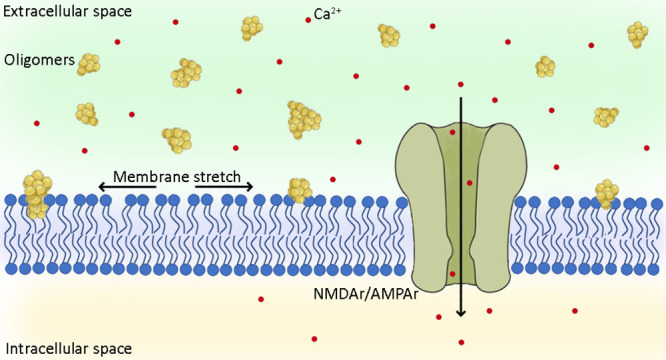
Alzheimer’s disease, which is the most common form of dementia, is characterized by the aggregation of the amyloid β peptide (Aβ) and by an impairment of calcium homeostasis caused by excessive activation of glutamatergic receptors (excitotoxicity). Here, we studied the effects on calcium homeostasis caused by the formation of Aβ oligomeric assemblies. We found that Aβ oligomers cause a rapid influx of calcium ions (Ca2+) across the cell membrane by rapidly activating extrasynaptic N-methyl-d-aspartate (NMDA) receptors and, to a lower extent, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. We also observed, however, that misfolded oligomers do not interact directly with these receptors. Further experiments with lysophosphatidylcholine and arachidonic acid, which cause membrane compression and stretch, respectively, indicated that these receptors are activated through a change in membrane tension induced by the oligomers and transmitted mechanically to the receptors via the lipid bilayer. Indeed, lysophosphatidylcholine is able to neutralize the oligomer-induced activation of the NMDA receptors, whereas arachidonic acid activates the receptors similarly to the oligomers with no additive effects. An increased rotational freedom observed for a fluorescent probe embedded within the membrane in the presence of the oligomers also indicates a membrane stretch. These results reveal a mechanism of toxicity of Aβ oligomers in Alzheimer’s disease through the perturbation of the mechanical properties of lipid membranes sensed by NMDA and AMPA receptors.
Keywords: Protein misfolding, neurodegenerative diseases, senile plaques, memantine, calcium homeostasis, membrane destabilization
Introduction
Alzheimer’s disease affects over 30 million people worldwide.1 According to the amyloid cascade hypothesis, a central role in the etiology of the disease is played by the self-assembly of the amyloid β peptide (Aβ) into senile plaques.2−4 In particular, many lines of evidence suggest that oligomeric forms of Aβ accumulating as intermediates in the aggregation process or released by mature fibrils have a variety of pathogenic effects as they are able to interact with a number of biological targets.3,5,6
An early biochemical modification in neurons is the disruption of calcium homeostasis, resulting in increased concentrations of calcium ions (Ca2+) in the intracellular space.7−11 Many mechanisms have been described through which Aβ oligomers cause an increase of Ca2+ levels in neurons. Aβ oligomers have been found to induce the release of glutamate by astrocytes, as well as to downregulate and inactivate excitatory amino acid transporters 1 and 2 (EAAT-1 and EAAT-2), which are responsible for glutamate reuptake by the same cells.12 These processes cause an excessive glutamate concentration in the perisynaptic space, which activates ionotropic glutamate receptors acting as Ca2+ channels on neuronal membranes, particularly extrasynaptic N-methyl-d-aspartate (NMDA) receptors, leading to a rise in intraneuronal Ca2+ concentration.9,10,12 This phenomenon, known as excitotoxicity, is not, however, the only mechanism through which Aβ oligomers induce a disruption of Ca2+ homeostasis in neurons. Aβ oligomers have also been found to induce directly a glutamate-independent Ca2+ influx from the extracellular space to the cytosol in cultured neuroblastoma cells and primary neurons, by interacting directly with, and destabilizing, lipid membranes and their protein components.13−25
The glutamate-independent Ca2+ influx, through which Aβ oligomers can induce directly the entry of Ca2+ ions in neurons across cell membranes, was described in terms of many mechanisms including the destabilization or perforation of the lipid bilayer,16,25−28 the activation of ionotropic glutamate receptors acting as Ca2+ channels, such as the NMDA receptors17,19−25,29,30 and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors,13,17−19 the activation of voltage-dependent Ca2+ channels,31,32 the activation of transient receptor potential melastatin 2 (TRPM2),33 and the activation of transient receptor potential A1 (TRPA1).34 It is not yet clear, however, whether Aβ oligomers activate the Ca2+ channels only by interacting with them directly causing their opening or also through other mechanisms mediated by unknown protein factors. The analysis that we report here allowed us to identify a molecular mechanism by which Aβ oligomers activate extrasynaptic NMDA and AMPA receptors, which is based on an interaction of the oligomers with lipid membranes that perturb their mechanical properties, which is sensed by the receptors through their mechanosensitivity, in the absence of any direct or protein-mediated interaction with the oligomers.
Results
Model Oligomers Are Toxic to SH-SY5Y Cells in a Ca2+-Dependent Manner
We started our investigations by using model oligomers formed by the protein HypF-N, since Aβ oligomers are difficult to isolate and study systematically. In particular, certain HypF-N oligomers were previously found to have effects similar to those of Aβ oligomers in cell and animal models.35−38 These HypF-N oligomers (known as type A) are ideally suited for analysis, as they are highly stable, versatile, and easy to isolate and have a nontoxic counterpart (known as type B), which is useful as a negative control.
In order to shed light on the importance of the Ca2+ influx event in determining the cellular toxicity mediated by these model oligomers, we first performed a cell stress readout assay in the presence or absence of extracellular Ca2+. The cellular stress induced by type A and type B oligomers and native proteins was assessed on SH-SY5Y neuroblastoma cells after 24 h treatment using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay, which is an indicator of the mitochondrial activity.39 Results of MTT reduction assays obtained with this cell and oligomer types have been found to correlate well with those of other cell stress readout assays.36 The aggregates were transferred from the solution in which they were formed into the cultured medium, a transition that is known to maintain their morphological and structural properties,35 and then added to the cells. The treatment of the cells with the native protein or with type B oligomers (12 μM monomer equivalents) did not affect the ability of the cells to reduce the MTT molecule, whereas the same dose of type A oligomers caused a 27 ± 5% reduction of mitochondrial activity (Figure 1A), confirming previous results.35,36 A 29 ± 6% decrease of MTT reduction was also obtained with the Ca2+ influx positive control ionomycin, suggesting that the Ca2+ influx causes a reduction of the metabolic activity (Figure 1A). Such a reduction was not observed when the experiment with type A oligomers was repeated in extracellular medium without Ca2+ (Figure 1B). This result was also confirmed in another cell line, the N13 murine microglial cells, which showed a higher susceptibility to lower concentration of type A oligomers compared to SH-SY5Y cells (Figure S1A). Overall, the data indicate that the cellular toxicity induced by the oligomers is mediated by extracellular Ca2+ levels.
Figure 1.

MTT reduction and intracellular free Ca2+ levels in SH-SY5Y cells treated with type A and type B oligomers. MTT reduction (A) in cells treated for 24 h with native HypF-N (12 μM), type B and type A oligomers (12 μM, monomer equivalents), and ionomycin (1 μM) and (B) in medium without Ca2+, with or without 24 h treatment with type A oligomers. (C) Representative confocal scanning microscopy images following the treatment with 1 μM ionomycin, no treatment, 12 μM native HypF-N for 15 min, 12 μM type B oligomers for 15 min, 12 μM type A oligomers for 5, 10, 15, 30, 60, 90, 120, and 180 min. The green fluorescence arises from Ca2+ binding to the Fluo-4 probe. (D) Semiquantitative analysis of intracellular free Ca2+-derived fluorescence. (E) Representative confocal scanning microscopy images showing the levels of intracellular free Ca2+ following the treatment with 12 μM type A oligomers for 10 and 60 min in a medium without Ca2+ and semiquantitative analysis of intracellular free Ca2+-derived fluorescence. Variable numbers of cells (12–22) in three different experiments were analyzed for each condition. Data are represented as the mean ± SEM. The double (∗∗) and triple (∗∗∗) asterisks refer to p values of <0.01 and <0.001 relative to the untreated cells, respectively.
Type A Oligomers Induce Ca2+ Dyshomeostasis
To investigate the mechanism of oligomer-induced and calcium-mediated toxicity, we monitored the change of intracellular Ca2+ concentration in SH-SY5Y cells following the treatment with type A oligomers (12 μM monomer equivalents) over time (Figure 1C,D). The data show a gradual increase of the intracellular Ca2+ levels, which reached maximum levels after 180 min of treatment (Figure 1C,D). By contrast, type B oligomers or the native protein did not have such effect (Figure 1C,D). When these experiments were carried out in a Ca2+-free medium, the cytoplasmic Ca2+ did not increase to any detectable extent (Figure 1E), indicating the extracellular origin of Ca2+.
The experiments were repeated on the N13 microglial cells, where we observed a more rapid increase in the intracellular Ca2+ levels within the first 5 min of treatment with type A oligomers, which then remained almost constant up to 60 min (Figure S1B,C). After 5 min of treatment with type A oligomers in the Ca2+-free medium, the intracellular Ca2+ levels were slightly lower than those observed after the treatment in the medium with Ca2+ but significantly higher than those of untreated cells (Figure S2A,B), suggesting that the cytosolic Ca2+ increment in the microglial cells could arise in a large part from intracellular compartments. To assess this possibility, we treated the N13 cells with type A oligomers after the inhibition of specific channels involved in the mechanism of release of Ca2+ from the intracellular compartments. We used, in particular, the benzothiazepine CGP-37157 to inhibit the mitochondrial Na+/Ca2+ exchange and sarcoplasmic reticulum calcium stimulated ATPase (SERCA),40,41 and 2-aminoethyl diphenylborinate (2APB) to inhibit the intracellular d-myo-inositol 1,4,5-trisphosphate (IP3)-induced Ca2+ release from the endoplasmic reticulum.42 Both the inhibitors determined a reduction of the intracellular Ca2+ levels following the treatment with type A oligomers for 5 min, compared with the treatment without inhibitors. This reduction was further exacerbated when the treatment was performed with both inhibitors at the same time and even more using both inhibitors and the medium without extracellular Ca2+, where low levels of cytosolic Ca2+ comparable to the untreated cells were observed (Figure S2A,B). When the cytosolic Ca2+ levels were measured after 60 min of treatment with type A oligomers, the contribution of Ca2+ from the extracellular space was found to be higher and that from the internal sources lower than that found after 5 min treatment (Figure S2C,D). Again, the pretreatment with both 2APB and CGP-37157 inhibitors and the use of the Ca2+ free medium did not cause any increase of cytosolic Ca2+ following the treatment with type A oligomers.
Hence, type A oligomers appear to induce an increase of cytosolic Ca2+ in both neuroblastoma SH-SY5Y and microglial N13 cells. However, in the first case the increase is due almost exclusively to the extracellular Ca2+ crossing the cell membrane, whereas in the second case the source of cytosolic Ca2+ is both the extracellular medium and the intracellular stores.
Glutamate Receptors Mediate Early Ca2+ Influx Induced by Type A Oligomers
In the search for potential Ca2+ channels mediating the influx of Ca2+ from the extracellular medium upon exposure to type A oligomers, we first considered the extensive literature reporting that ligand-gated Ca2+ channels of glutamatergic type, such as the NMDA and AMPA receptors, are involved in the influx of Ca2+ mediated by Aβ oligomers.13,17−24,29,30 In order to verify the presence of the NMDA and AMPA receptors on the SH-SY5Y cellular membrane, we probed them on the cells with their specific antibodies, observing their expression (Figure S3A). To assess whether the expressed receptors were also functional and active, we activated them using their specific agonists NMDA and AMPA, finding that both small molecules are able to induce an increase of the intracellular Ca2+ levels (Figure S3B), an influx that was inhibited if the cells were pretreated with the NMDA receptor uncompetitive inhibitor memantine and the AMPA receptor competitive antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), respectively (Figure S3B).
We therefore pretreated the SH-SY5Y neuroblastoma cells with CNQX or with memantine, and then we treated the cells with type A oligomers for different time intervals (Figure 2). CNQX determined a slight reduction of the cytoplasmic Ca2+ levels in the early stages, up to 10 min, compared to the normal time course without inhibitors (Figure 2). This reduction was more significant following treatment with memantine. Indeed, we observed Ca2+ levels similar to those of untreated cells up to 10 min of treatment and then a gradual increase which remained significantly different from that observed without inhibitors starting from 15 min, reaching comparable levels at 60 min of treatment (Figure 2). These results suggest that in the early stages of the Ca2+ influx from the extracellular space, extrasynaptic AMPA and NMDA receptors play an important role, particularly the latter, whereas in the late stages of the influx of Ca2+ these receptors do not seem to give a significant contribution to the massive entrance of Ca2+ ions into the cells. This finding legitimates the use of SH-SY5Y cells for this study, as they do not contain synaptic NMDA or AMPA receptors that might interfere with the analysis.
Figure 2.

Intracellular free Ca2+ levels in SH-SY5Y cells treated with type A oligomers, after the inhibition of AMPA and NMDA receptors. (A) Representative confocal scanning microscopy images following the treatment with no inhibitors (first row), 5 μM CNQX (second row), and 10 μM memantine (third row) and analyzed after 5, 10, 15, 30, 60, 90, 120, and 180 min of treatment with 12 μM (monomer equivalents) type A oligomers. (B) Semiquantitative analysis of intracellular free Ca2+-derived fluorescence. Variable numbers of cells (12–22) in three different experiments were analyzed for each condition. Data are represented as the mean ± SEM. The double (∗∗) and triple (∗∗∗) asterisks refer to p values of <0.01 and <0.001, respectively, relative to untreated cells. The single (§), double (§§), and triple (§§§) symbols refer to p values of <0.05, < 0.01, and <0.001, respectively, relative to type A oligomers without inhibitors. (C) Kinetic plots showing the fluorescence versus time as reported in panel B.
The involvement of extrasynaptic AMPA and NMDA receptors in the early stages of the influx of Ca2+ induced by type A oligomers was confirmed in the N13 cells (Figure S4). After 5 min of treatment with CNQX or memantine, we observed a reduction of the cytosolic free Ca2+ in the cells, compared to those without inhibitors (Figure S4). This reduction was similar to that observed in the treatment without Ca2+ described before at corresponding times (Figure S2B). After 60 min of treatment with the type A oligomers, the cytosolic Ca2+ levels were the same with or without the inhibitors (Figure S4).
Type A Oligomers Do Not Directly Interact with AMPA and NMDA Receptors
To explore the mechanism of activation of extrasynaptic AMPA and NMDA receptor channels by type A oligomers, we checked if there was a direct interaction between the oligomers and the receptors using a fluorescence resonance energy transfer (FRET) analysis. Therefore, the AMPA and NMDA receptors were labeled with the donor probe (D) primary antibody labeled with ATTO488, and the type A oligomers were labeled with the acceptor probe (A) Alexa Fluor 555 covalently linked to a cysteine residue of the HypF-N sequence.
First, we ruled out the possible cross-reaction between the oligomers and the anti-NMDA antibody testing the labeled antibody on the oligomers attached to the coverslip in the absence of cells (Figure S5). A positive control was also performed, testing the rabbit anti-HypF-N antibody on the oligomers attached to the coverslip and then adding the labelled anti-rabbit secondary antibody (Figure S5). A negative control was finally carried out using the labeled rabbit polyclonal anti-NMDA receptor antibody on a coverslip in the absence of the oligomers (Figure S5).
In the presence of only D, SH-SY5Y cells were excited at 488 nm (D ex wavelength) and the fluorescence was observed in the 499–535 nm range (D em range, Figure 3A, left). Then, we excited the sample at 543 nm (A ex wavelength) and observed the absence of emission in the 560–610 nm range (A em range, Figure 3A, center). By exciting the samples at 488 nm and monitoring the emission in the 560–610 nm range (A em range), we observed the bland contribution of D in the FRET channel (Figure 3A, right). In the presence of only A, we excited at 488 nm (D ex wavelength) and monitored the emission in the 499–535 nm range (D em range, Figure 3B, left), where we observed a low level of fluorescence. We then excited at 543 nm (A ex wavelength) and observed the emission in the 560–610 nm range (Figure 3B, center). We finally observed the bland emission in the 560–610 nm range in the FRET channel exciting at 488 nm (D ex wavelength, Figure 3B, right). The fluorescence observed in the donor channel in the presence of only the acceptor is due to a technical spillover of the acceptor fluorescence which can be excited at 488 nm to a low degree. This aspect was considered in our analysis quantifying such fluorescence and reducing the FRET fluorescence of the same amount.
Figure 3.

FRET analysis of the lack of direct interaction of type A oligomers with NMDA receptors in SH-SY5Y cells. (A) Cells treated with unlabeled oligomers in the presence of primary antibody against NMDA receptors labeled with D on the donor channel (left, excitation 488 nm; emission 499–535 nm), acceptor channel (center, excitation 543 nm; emission 560–610 nm), and FRET channel (right, excitation 488; emission 560–610 nm). (B) Cells treated with the oligomers labeled with A, in the absence of D on the donor channel (left, excitation 488 nm; emission 499–535 nm), acceptor channel (center, excitation 543 nm; emission 560–610 nm), and FRET channel (right, excitation 488; emission 560–610 nm). (C) Cells treated with the D and A: from left to right the donor channel, the acceptor channel, the FRET channel, and the colocalization image, obtained by overlapping the donor and acceptor channels. (D) Semiquantitative analysis of the oligomers-derived fluorescence in the FRET channel in the presence of only A (type A oligomers labeled A) or both D and A (primary antibody against NMDA receptors labeled with D and type A oligomers labeled with A). (E) Representative confocal scanning microscopy image of D in the presence of both D and A (left). The blue and yellow circles indicate representative areas without oligomers (FD) and with type A oligomers (FDA), respectively. Semiquantitative analysis of the NMDA receptors-derived fluorescence in the areas corresponding to the oligomers compared with the surrounding areas (right). Data are represented as the mean ± SEM.
In the presence of both D and A, we acquired images in the donor channel (ex 488 nm, em 499–535 nm), in the acceptor channel (ex 543 nm, em 560–610 nm), and in the FRET channel (ex 488 nm, em 560–610 nm) and finally overlapped the donor and acceptor channels to obtain the colocalization image (Figure 3C). The comparison between the FRET channel with only A or both D and A (Figure 3D) did not show any variation of the A fluorescence. Moreover, the comparison of the D fluorescence in the areas enriched with the A-labeled oligomers (FDA, Figure 3E, yellow circle) with the surrounding areas (FD, Figure 3E, blue circle) showed similar values of D fluorescence. By calculating the FRET efficiency (E) value as
| 1 |
we obtained a value close to 0, suggesting the lack of any direct interaction between type A oligomers and NMDA receptors. The experiments were performed also with the AMPA receptors and the type A oligomers, and similarly, we did not detect any direct interaction between the receptors and the oligomers (Figure S6).
To rule out technical issues in the experiments described above (Figures 4 and S6) that could be responsible for the lack of FRET signal, we carried out a positive control of FRET using an anti-AMPA receptors antibody labeled with ATTO488 and the Alexa Fluor 594-conjugated anti-rabbit secondary antibody as D and A, respectively (Figure S7A–C). In the presence of only A, the fluorescence in the FRET channel was lower compared to that in the presence of both D and A (Figure S7E); moreover, the D fluorescence in the donor channel was higher with only D compared to that with both D and A (Figure S7D), proving the presence of energy transfer between D and A.
Figure 4.

Intracellular free Ca2+ in SH-SY5Y cells treated with type A oligomers after the enrichment with lysophosphatidylcholine and arachidonic acid. (A) Representative confocal scanning microscopy images following the treatment for 2 h with 0, 0.05, 0.1, 1.0, and 2.0 μM lysophosphatidylcholine (LPC) with or without type A oligomers and (B) semiquantitative analysis of intracellular free Ca2+-derived fluorescence. (C) Cells treated with 0 and 10 μM arachidonic acid (AA) with or without type A and after pretreatment with 10 μM memantine and (D) semiquantitative analysis of intracellular free Ca2+-derived fluorescence. Variable numbers of cells (12–22) in three different experiments were analyzed for each condition. Data are represented as the mean ± SEM. The double (∗∗) and triple (∗∗∗) asterisks refer to p values of <0.01 and <0.001, respectively, relative to untreated cells.
Type A Oligomers Activate NMDA Receptors by Mechanical Stimuli through the Lipid Bilayer
The activation of extrasynaptic NMDA and AMPA receptors in the absence of a direct interaction with the oligomers may suggest a specific response involving other proteins or messengers. However, the rapidity of the activation, highlighted by the absence of a lag phase in the time-dependent rise of intracellular Ca2+ ions (Figure 2), rather suggests the alternative hypothesis that the membrane deformation caused by the interaction with the oligomers is transmitted mechanically to the receptors via the lipid bilayer. In fact, NMDA receptors are known to be mechanosensitive, potentiated by mechanical stimuli such as membrane depression, hypotonic solutions, and lateral membrane stretch and inhibited by the opposite stimuli.43−47 To assess this possibility, we enriched the membranes of SH-SY5Y cells with lysophosphatidylcholine and arachidonic acid, which are known to cause a membrane compression and stretch, respectively, because of their opposite shapes. Lysophosphatidylcholine has the shape of a cone (large hydrophilic head and narrow hydrophobic tail), whereas arachidonic acid has that of an inverted cone (narrow hydrophilic head and large, highly unsaturated, hydrophobic tail).44,48 They therefore cause an inhibition and potentiation of the NMDA receptors, respectively.44 It was previously observed that the use of lysophosphatidylcholine can cause an increase in intracellular Ca2+ levels through the signaling of GPR55,49 a receptor, which is, however, not expressed (Human Protein Atlas source) or weakly expressed (Harmonizome source) in SH-SY5Y cells.
SH-SY5Y cells were treated with various concentrations of lysophosphatidylcholine (0–2 μM) for 2 h, washed with PBS, and then incubated with 12 μM type A oligomers for 10 min. A concentration of 2 μM lysophosphatidylcholine was found to be enough to inhibit the oligomer-induced Ca2+ entry through the NMDA receptors (Figure 4A,B). The ability of a bilayer-embedded compound, such as lysophosphatidylcholine, to counteract the oligomer-induced NMDA receptors opening, despite the lack of any specific interaction between the lipid and the receptors, suggests that the oligomers induce a mechanical stimulus upon binding to the membrane, which is then transmitted down to the NMDA receptors via the lipid bilayer, thus causing their opening. The opposing force exerted by lysophosphatidylcholine effectively inhibits the mechanical signal generated by the action of the oligomers onto the membrane.
We repeated the experiments by incubating the cells with 10 μM arachidonic acid before the addition of the oligomers. Arachidonic acid alone was found to be enough to activate an NMDA receptors-mediated influx of Ca2+ ions, even in the absence of oligomers (Figure 4C,D), in agreement with the literature reporting its mechanical action on the membrane.43,44 The influx of Ca2+ ions was not further increased when 12 μM type A oligomers were added in the presence of arachidonic acid pretreatment (Figure 4C,D), in agreement with the idea that an occlusion exists if two mechanical stimuli of the same type are applied to the membrane to modulate NMDA receptors.44 Importantly, all three effects are inhibited by memantine indicating that they all result from an ionic influx involving the NMDA receptors (Figure 4C,D).
Ion Channels Interacting with Type A Oligomers Are Not Involved in the Ca2+ Influx
In the search for others potential Ca2+ channels responsible for the observed influx of Ca2+ from the extracellular medium, we took advantage of a recent interactome-wide study in which more than 2500 membrane proteins of N13 cells interacting with type A oligomers were identified.50 Among them, we did not find any subunits of NMDA and AMPA receptors, in agreement with our FRET results, but we identified five different Ca2+ channels (Table 1): anoctamin-6 (SCAN channel), transient receptor potential cation channel subfamily V member 2 (TrpV2), P2X purinoceptor 4 (P2X4), piezo-type mechanosensitive ion channel component 1 (FAM38A), and transient receptor potential cation channel subfamily M member 7 (LTrpC-7). Four of them were found to be expressed also in SH-SY5Y cells (Table 1) except for TrpV2 [Human Protein Atlas source].51
Table 1. Five Ca2+ Channels Interacting with Type A Oligomers.
| protein name | gene name | protein identifier | inhibitor | N13 expression | SH-SY5Y expression |
|---|---|---|---|---|---|
| anoctamin-6 (SCAN channel) | Ano6 | Q6P9J9 | anti-ANO6 antibody | + | + |
| transient receptor potential cation channel subfamily V member 2 (TrpV2) | Trpv2 | Q9WTR1 | tranilast | + | – |
| P2X purinoceptor 4 (P2X4) | P2rx4 | Q9JJX6 | anti-P2RX4 antibody | + | + |
| piezo-type mechanosensitive ion channel component 1 (FAM38A) | Piezo1 | E2JF22 | GsMTx4 | + | + |
| transient receptor potential cation channel subfamily M member 7 (LTrpC-7) | Trpm7 | Q923J1 | FTY720 | + | + |
To investigate the possible role of these four channels in the influx of Ca2+ induced by type A oligomers in SH-SY5Y cells, we inhibited them one by one, using specific inhibitors, before the treatment for 5, 10, or 60 min with the type A oligomers (Figure S8A,B). In all cases, we did not observe any variation in the influx of Ca2+, indicating that none of the four Ca2+ channels are involved directly, either in the early or in the late stages of Ca2+ influx.
We also investigated other channels, which were not found to interact directly with type A oligomers in the interactome study50 but are known to be important in some circumstances for Ca2+ homeostasis and thus potentially relevant in the present context: the voltage-gated calcium channels (VGCCs), the calcium release-activated channels (CRACs), the nicotinic acetylcholine receptor α7 (α7nAChR), and the cystine/glutamate transporter (xCT).31,32,52−54 In these cases, we pretreated the SH-SY5Y cells with Cd2+ (VGCCs inhibitor), with Synta66 (CRAC channel inhibitor), with α-bungarotoxin (α7nAChR inhibitor), or with erastin (xCT inhibitor), and then we treated the cells with type A oligomers for 5, 10, or 60 min. The pattern of Ca2+ influx was similar to that observed without inhibitors at all time points (Figure S8A,B), ruling out their involvement in the Ca2+ influx induced by type A oligomers.
The entire analysis was also performed on N13 cells, confirming the same results (Figure S9). On this cell line we also used combinations of inhibitors but failed to find significant variations (Figure S10A,B). As a positive control, we treated N13 cells with probenecid and with a combination of probenecid and tranilast, two small molecule drugs acting as specific activator and inhibitor of the TrpV2 channel, respectively, in both cases in the absence of HypF-N oligomers. In these experiments we observed an increase of intracellular Ca2+ and a restoration to the levels observed in the untreated cells, respectively (Figure S10C,D).
Zinc-Stabilized Aβ40 Oligomers and Aβ42 ADDLs Activate NMDA and AMPA Receptors
To establish whether the results obtained with the HypF-N model oligomers can be extended to Aβ oligomers, we studied zinc-stabilized Aβ40 oligomers55 at the concentration of 5 μM monomer equivalents and Aβ42-derived diffusible ligands (ADDLs)56 at the concentration of 1 μM. The treatment of SH-SY5Y cells with the zinc-stabilized Aβ40 oligomers induced an increase of intracellular Ca2+ levels, which reached the maximum value after 180 min of treatment (Figures 5A,B and S11A). The use of specific inhibitors of AMPA and NMDA receptors (CNQX and memantine, respectively) confirmed the involvement of these channels in the early stage of the influx of Ca2+, up to 10 min of treatment for the AMPA receptors and 15 min for the NMDA receptors, as the inhibitors slowed down the kinetics of the Ca2+ increase (Figures 5A,B and S11A). Moreover, we analyzed the four Ca2+ channels found to interact with the type A oligomers in the N13 cells, as well as the VGCCs, the CRAC channels, the α7nAChR, and the xCT, using their respective specific inhibitors and protocols described before. The results confirmed those observed with the type A oligomers, that is, the lack of involvement of these channels in the influx of Ca2+ (Figure S12).
Figure 5.

Intracellular free Ca2+ levels in SH-SY5Y cells treated with zinc-stabilized Aβ40 and Aβ42 ADDLs oligomers, after the inhibition of AMPA and NMDA receptors. (A, C) Semiquantitative analysis of intracellular free Ca2+-derived fluorescence following the treatment with no inhibitors, 5 μM CNQX, and 10 μM memantine and analyzed after 5, 10, 15, 30, 60, 90, 120, and 180 min of treatment with (A) 5 μM (monomer equivalents) zinc-stabilized Aβ40 oligomers and (C) 1 μM (monomer equivalents) Aβ42 ADDLs oligomers. Variable numbers of cells (12–22) in three different experiments were analyzed for each condition. Data are represented as mean ± SEM. The single (∗), double (∗∗), and triple (∗∗∗) asterisks refer to p values of <0.05, < 0.01, and <0.001, respectively, relative to untreated cells. The single (§), double (§§), and triple (§§§) symbols refer to p values of <0.05, < 0.01, and <0.001, respectively, relative to oligomers without inhibitors. (B) Kinetic plots showing the fluorescence values versus time as reported in panel A. (D) Kinetic plots showing the fluorescence values versus time as reported in panel C.
The Aβ42 ADDLs had a similar behavior, causing an increase in the intracellular Ca2+ levels up to 180 min after the treatment of the SH-SY5Y cells (Figures 5C,D and S11B). Similar to the zinc-stabilized Aβ40 oligomers described above, the inhibition of the AMPA and NMDA receptors determined a reduction of Ca2+ up to 10 and 15 min of treatment with the oligomers, respectively (Figures 5C,D and S11B), whereas inhibition of any of the other Ca2+ channels did not have any such effects (Figure S13)
The effect on Ca2+ dyshomeostasis induced by Aβ42 ADDLs after 10 and 60 min of treatment was tested also in primary rat cortical neurons (Figure 6). Along the same lines, 10 min of treatment with the oligomers was sufficient to induce an increase of the intracellular Ca2+ levels, which increased further after 60 min of treatment. The inhibition of the AMPA and NMDA receptors determined a significant reduction of the Ca2+ levels after 10 min of treatment with the Aβ42 ADDLs, confirming the involvement of the receptors in the Ca2+ influx.
Figure 6.

Intracellular free Ca2+levels in primary rat cortical neurons treated with Aβ42 ADDLs oligomers after the inhibition of AMPA and NMDA receptors. (A) Representative confocal scanning microscopy images of untreated cells and cells treated with 1 μM ionomycin (first row) and following the treatment with no inhibitors (second row), 5 μM CNQX (third row), and 10 μM memantine (fourth row) and analyzed after 10 and 60 min of treatment with 1 μM (monomer equivalents) Aβ42 ADDLs oligomers. (B) Semiquantitative analysis of intracellular free Ca2+-derived fluorescence. Variable numbers of cells (12–22) in three different experiments were analyzed for each condition. The single double (∗∗) and triple (∗∗∗) asterisks refer to p values of <0.01 and <0.001, respectively, relative to untreated cells. The single (§) and double (§§) symbols refer to p values of <0.05 and <0.01, respectively, relative to Aβ42 ADDLs oligomers without inhibitors.
To confirm these results with a different technique, we pretreated SH-SY5Y cells with the siRNA against the subunit 2B of the NMDA receptor. A significant reduction of the NMDA receptor was observed in the silenced cells (Figure S14A), and significantly lower intracellular Ca2+ levels were observed in cells pretreated with siRNA after 10 min of treatment with the Aβ42 ADDLs oligomers, compared with cells without siRNA treatment (Figure S14B), confirming the results observed with memantine. After 60 min of ADDL treatment, the Ca2+ levels increased, but they were still lower than the cells without siRNA (Figure S14B).
Aβ42 ADDLs Oligomers Activate NMDA Receptors through Mechanical Stimuli
We then repeated the experiments described on SH-SY5Y cellular membranes enriched with lysophosphatidylcholine and arachidonic acid, using Aβ42 ADDLs rather than HypF-N model oligomers. SH-SY5Y cells were treated with various concentrations of lysophosphatidylcholine (0–2 μM) for 2 h, washed with cell medium, and then incubated with 1 μM Aβ42 ADDLs oligomers for 10 min. The enrichment with 2 μM lysophosphatidylcholine caused a complete inhibition of the ADDL-induced Ca2+ entry through the NMDA receptors (Figure 7A,B), in agreement with the results obtained with the model oligomers and lysophosphatidylcholine-enriched membranes. Moreover, the application of 1 μM Aβ42 ADDLs oligomers to SH-SY5Y cells pretreated with 10 μM arachidonic acid produced a slightly higher, but not significant, increase of Ca2+ influx with respect to cells pretreated with arachidonic acid alone, again in agreement with observations obtained with the model oligomers and arachidonic acid-enriched membranes (Figure 7C,D). All these effects are inhibited by memantine, indicating that they all result from an ionic influx involving the NMDA receptors (Figure 7C,D).
Figure 7.

Intracellular free Ca2+ in SH-SY5Y cells treated with Aβ42 ADDLs oligomers after the enrichment with lysophosphatidylcholine and arachidonic acid. (A) Representative confocal scanning microscopy images following the treatment for 2 h with 0, 0.05, 0.1, 1.0, and 2.0 μM lysophosphatidylcholine (LPC) with or without Aβ42 ADDLs oligomers and (B) semiquantitative analysis of intracellular free Ca2+-derived fluorescence. (C) Cells treated with 0 or 10 μM arachidonic acid (AA) with or without Aβ42 ADDLs oligomers and after pretreatment with 10 μM memantine and (D) semiquantitative analysis of intracellular free Ca2+-derived fluorescence. Variable numbers of cells (12–22) in three different experiments were analyzed for each condition. Data are represented as the mean ± SEM. The double (∗∗) and triple (∗∗∗) asterisks refer to p values of <0.01 and <0.001, respectively, relative to untreated cells.
When repeated on primary rat cortical neurons, the results confirmed the inhibition of the ADDL-induced Ca2+ entry in lysophosphatidylcholine-enriched cells and a slight, but not significant, increase of Ca2+ influx in arachidonic acid-enriched cells with respect to cells pretreated with arachidonic acid alone (Figure 8). Memantine treatment again inhibited the Ca2+ entry in neurons treated with arachidonic acid and ADDLs, confirming the involvement of the NMDA receptors (Figure 8).
Figure 8.

Intracellular free Ca2+ levels in primary rat cortical neurons treated with Aβ42 ADDLs oligomers after the enrichment with lysophosphatidylcholine and arachidonic acid. (A) Representative confocal scanning microscopy images following the treatment for 2 h with no additives, 2.0 μM lysophosphatidylcholine (LPC), 10 μM arachidonic acid (AA), and 10 μM arachidonic acid (AA) after pretreatment with 10 μM memantine, with or without 10 min treatment with 1 μM (monomer equivalents) Aβ42 ADDLs oligomers. (B) Semiquantitative analysis of intracellular free Ca2+-derived fluorescence. Variable numbers of cells (12–22) in three different experiments were analyzed for each condition. The double (∗∗) and triple (∗∗∗) asterisks refer to p values of <0.01 and <0.001, respectively, relative to untreated cells.
Aβ42 ADDLs Oligomers Change the Cellular Membrane Fluidity
In order to gain a second independent piece of evidence that Aβ42 ADDLs activate the NMDA receptors via mechanical stimuli through the membrane assimilable to a bilayer stretching, we analyzed the effects of the oligomers on the rotational diffusion of the 1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate (TMA-DPH) probe, which is known to insert in the polar head group region of the cell membrane due to its charged group.57 The incorporation of the TMA-DPH in the plasma membrane was observed analyzing its colocalization with the Alexa Fluor 594-labeled trodusquemine, a small molecule that belongs to a family of compounds known to bind the membrane58 (Figure 9A). In the absence and presence of a 10 min treatment with ADDLs, the anisotropy of TMA-DPH was 0.22 ± 0.01 and 0.18 ± 0.01, respectively (Figure 9B), indicating a higher increase of the rotational freedom of the fluorophore, in the presence of the oligomers, resulting from a gain of free space between the membrane lipids and fluidity of the whole membrane bilayer.
Figure 9.
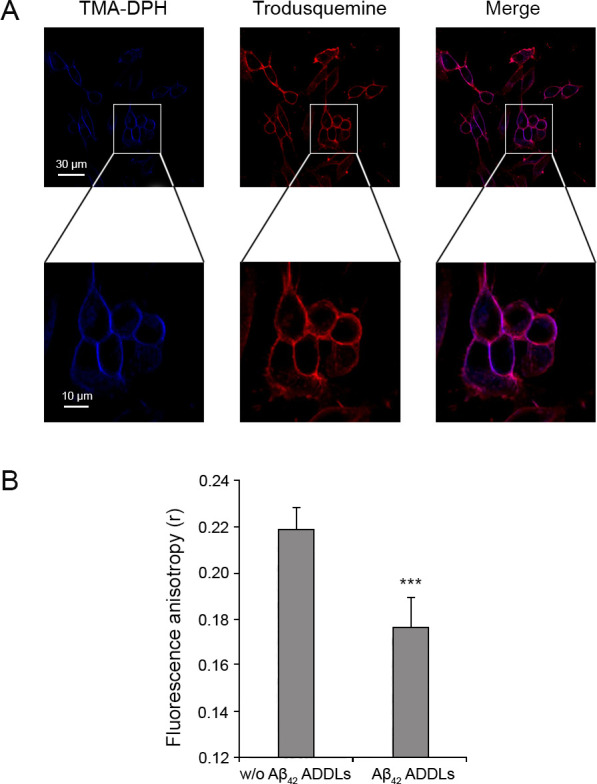
Incorporation of TMA-DPH in the SH-SY5Y cellular membrane and measurement of fluorescence anisotropy. (A) Representative confocal scanning microscopy images following the treatment for 5 min with 2.0 μM TMA-DPH and for 10 min with 5 μM trodusquemine labeled with Alexa Fluor 594. (B) Fluorescence anisotropy (r) of TMA-DPH obtained in the absence and presence of 1 μM (monomer equivalents) Aβ42 ADDLs oligomers. Data are represented as the mean ± SEM. The triple (∗∗∗) asterisks refer to p values of <0.001 relative to the results in the absence of oligomers.
Discussion
A key process in neurodegeneration, particularly in Alzheimer’s disease, is the disruption of calcium homeostasis in neurons of the neocortex and hippocampus.7−11 Indeed, one of the few drugs approved for this disease by the American Food Drug Agency (FDA), the European Medicine Agency (EMA), and other governmental agencies is memantine, an inhibitor of extrasynaptic NMDA receptors and of the Ca2+ current through the cell membrane of neurons mediated by this receptor.59 Aβ oligomers interact with, and destabilize, the cell membrane and its protein components, with a consequent Ca2+ influx from the extracellular space to the cytosol of neurons. This influx has been proposed to occur through a destabilization or perforation of the lipid bilayer16,25,26,28 or activation of ionotropic receptors acting as calcium channels.13,17−20,22−25,29−34
Our results show that zinc-stabilized Aβ40 oligomers and Aβ42 oligomeric ADDLs, as well as toxic HypF-N oligomers mimicking Aβ oligomers, which were all previously found to be toxic to neuronal cells,17,35,36,38,55,56,60,61 cause an influx of Ca2+ on neuroblastoma cells by activating rapidly extrasynaptic NMDA receptors and, to a lower extent, AMPA receptors and also by causing, on a slower time scale, a perforation or destabilization of the lipid bilayer of the membrane. This result was also confirmed on cultured rat primary neurons. Indeed, the rise of cytosolic Ca2+ ions detected within the first 10 min following the exposure of the cells to the oligomers is caused by NMDA and, to a lower extent, AMPA receptors as their specific inhibition with memantine and CNQX, or knockdown with a siRNA against the NMDA receptor GRIN2B subunit, causes the appearance of a lag phase in the time-dependent rise of intracellular Ca2+, with no significant increase within the first 10 min. Later, the intracellular Ca2+ levels start to increase despite the persistent inactivation of the two receptors and after 60–120 min they reach the same levels observed in the absence of any inhibition. In this late stage, the influx of Ca2+ from the extracellular to the intracellular space is caused by the direct passage of the ions through the cell membrane after the interaction of the oligomers with the lipid bilayer and a consequent destabilization and perforation.16,25,26,28,62
These results reinforce the idea that extrasynaptic NMDA receptors play a central role in the pathogenesis of Alzheimer’s disease, not just because they are activated aberrantly in the disease as a result of the excitotoxicity induced by Aβ but also because of the interaction of the peptide in aggregated forms with the lipid membranes. Our results also provide an explanation as to why blockage of the receptors is beneficial, but not sufficient, to re-establish calcium homeostasis, as the oligomers also cause a nonspecific entry of Ca2+ through the destabilized membrane. This concept finds confirmation in the clinical effects that are exerted by memantine as a drug, which is symptomatic and helps improve the cognitive performance of the patients but is not enough to reverse the disease or block its progression. Early diagnosis of the disease and therapeutic strategies that also interfere with the association of Aβ in oligomeric forms with lipid membranes may add considerable value in the fight against the disease.
Interestingly, none of the Ca2+ channels that have been found in a previous interactome study to interact with HypF-N oligomers, namely, SCAN, TrpV2, P2X4, FAM38A, and LTrpC-7,50 can participate in the observed Ca2+ influx induced by HypF-N, Aβ40 and Aβ42 oligomers. Similarly, other Ca2+ channels previously suggested to be involved in the Aβ-induced alteration of Ca2+ homeostasis in some circumstances, such as VGCCs, CRAC, α7nAChR, and xCT,31,32,52−54 were not found to be involved. Indeed, their specific inhibition in either microglial N13 and neuroblastoma SH-SY5Y cells does not recover the Ca2+ dyshomeostasis, at either the early or late times.
It is remarkable to observe that none of the protein subunits forming the AMPA and NMDA receptors are present in the HypF-N oligomer interactome studied in N13 cells, despite the presence of these two receptors in this cell line.50 Such a lack of interactions was confirmed here by the absence of any significant FRET between the oligomers and receptors. This finding is not in contrast with the observation that Aβ or HypF-N oligomers colocalize with NMDA or AMPA receptors as observed with confocal microscopy or coimmunoprecipitation.17,18,25,30,63 Both species are indeed found to bind to the lipid rafts of the membrane appearing closely associated in these studies. However, within such membrane domains noncovalent interactions between the two species appear to be absent or labile.
How can misfolded protein oligomers activate rapidly extrasynaptic AMPA and NMDA receptors in the absence of any interaction between the two species? Specific or generic responses involving other proteins and messengers as mediators might in principle take place. It is indeed increasingly accepted that the Fyn/PrPC signal transduction is involved in the phosphorylation and downregulation of synaptic NMDA receptors.64,65 However, the extrasynaptic localization of the two AMPA and NMDA receptors in SH-SY5Y cells, the rapidity of their activation (in particular the absence of a lag phase in the time-dependent rise of intracellular Ca2+ ions upon addition of the oligomers), and their well-established mechanosensitivity43−47 suggest that the change in membrane tension caused by the oligomers is transmitted energetically to the receptors via the lipid bilayer. We indeed found that lysophosphatidylcholine, a lipid that mimics a mechanical compression of the membrane and inhibits the NMDA receptors without acting as a specific ligand,44 is able to neutralize the oligomer-induced activation of the NMDA receptors, suggesting that this is the case. Similarly, the occlusion observed between the effects of the oligomers and that of arachidonic acid, that by contrast mimics a mechanical stretch and activates the receptors,43,44 also corroborates the hypothesis that the oligomers act with this mechanism. Finally, the decreased fluorescence anisotropy of the TMA-DPH probe embedded within the polar region of the bilayer in the presence of the oligomers is a direct indication of an enhanced increase of free space between the various lipids in the bilayer and membrane fluidity upon interaction with the oligomers.
In conclusion, Aβ oligomers were found to induce an influx of Ca2+ ions across the cell membrane via an early activation of AMPA and NMDA receptors, particularly the latter, and via a later nonspecific destabilization of lipid bilayers. The mechanism of action through which Aβ oligomers activate the glutamatergic receptors was found to be based on a change of the mechanical properties of the membrane following the insertion of the oligomers in the bilayer that is transmitted down to the receptors, which are therefore activated due to their mechanosensitivity.
Materials and Methods
Preparation of HypF-N Oligomers, Aβ42 ADDLs, and Zinc-Stabilized Aβ40 Oligomers
Wild-type and C7S/C65A HypF-N (carrying a single cysteine residue at position 40) were prepared and purified as described previously35 and stored at −80 °C in 20 mM potassium phosphate buffer, pH 7.0, with 2 mM dithiothreitol (DTT) for the wild-type form or in 20 mM potassium phosphate buffer, pH 7.0, with 2 mM tris (2-carboxyethyl)phosphine hydrochloride (TCEP) for the mutant form. HypF-N and its mutant were converted into toxic (type A) and nontoxic (type B) oligomers as previously described.35 For the FRET experiments, type A oligomers were formed by mixing mutant HypF-N labeled with Alexa Fluor 555 and unlabeled HypF-N, at a molar ratio of 1:5. Each type of oligomer was immediately diluted at the monomer equivalent concentration of 12 μM when added to the SH-SY5Y cell culture media and of 0.1 μM when added to the N13 cell culture media.
Lyophilized Aβ42 (Bachem, Bubendorf, Switzerland) was dissolved in HFIP to 1.0 mM and incubated for 1 h at room temperature to allow complete peptide monomerization. Then, Aβ42 was converted into ADDLs as previously described56 and immediately diluted in the appropriate medium and added to the SH-SY5Y cell culture media at the concentration of 1 μM. ADDLs were chosen as representative Aβ42 oligomers because they are widely used and their morphology and purity were routinely verified in our labs.66−68
Expression and purification of Aβ40 was carried out as previously described,55 lyophilized in 50 mM ammonium acetate, pH 8.5, and stored at −80 °C. 1 mg of lyophilized Aβ40 was converted in Zn2+-stabilized Aβ40 oligomers at a final concentration of 100 μM,55 diluted in the appropriate medium, and added to the SH-SY5Y cell culture media at the concentration of 5 μM. The zinc-stabilized Aβ40 oligomers morphology and size were routinely verified in our laboratories.69−71
Labeling of HypF-N Variant with Alexa Fluor 555 Maleimide
The HypF-N mutant was diluted to 120 μM in 20 mM potassium phosphate buffer, 2 mM TCEP, pH 7.0. Alexa Fluor 555 maleimide (Life Technologies/Thermo Fisher Scientific, Waltham, MA, USA) was added to the sample to a final concentration of 360 μM. The reaction mixture was left in the dark on a mechanical shaker for 2 h at room temperature and then overnight at 4 °C. It was dialyzed in the dark (using a membrane with a cutoff of 3 kDa) against 1 L of 20 mM potassium phosphate buffer, pH 7.0, for 8 h and then 1 L of the same buffer overnight. The sample was then centrifuged to remove any precipitate. The concentration of the dye was determined spectrophotometrically, using ε555 = 158 000 M–1 cm–1. The protein concentration was also determined spectrophotometrically using ε280 = 12 490 M–1 cm–1 after subtraction of the absorbance contribution of the probe at the same wavelength of 280 nm. We estimate this contribution as
| 2 |
where x represents the molar concentration of the dye and y is the absorbance of the dye at 280 nm.
Cell Cultures
Human SH-SY5Y neuroblastoma cells (A.T.C.C., Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and F-12 HAM with 25 mM N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES) and NaHCO3 (1:1) and supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 1% antibiotics. Cell cultures were maintained in a 5% CO2 humidified atmosphere at 37 °C and grown until they reached 80% confluence for a maximum of 20 passages.
Murine N13 microglial cells (RRID CVCL_5G93) were cultured in DMEM and F-12 HAM with 25 mM HEPES and NaHCO3 (1:1) and supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 1% antibiotics, and 1% nonessential amino acids. Cell cultures were maintained in a 5% CO2 humidified atmosphere at 37 °C and grown until they reached 80% confluence for a maximum of 20 passages.
Primary rat cortical neurons (Life Technologies/Thermo Fisher Scientific) were plated in 24-well plate at the density of 200 000 cells per well and maintained in neuronal basal plus medium (Life Technologies/Gibco, Thermo Fisher Scientific) supplemented with GlutaMAX (Gibco) at the concentration of 0.5 mM and 2% (v/v) B-27 serum-free complement (Gibco) at 37 °C in a 5.0% CO2 humidified atmosphere. Every 4 days the medium was partially replaced with fresh one. All the experiments were performed 12–16 days after plating.
MTT Reduction Assay
The MTT reduction assay was performed in SH-SY5Y and N13 cells seeded in 96-well plate at a density of 10 000 cells per well. SH-SY5Y and N13 cells were treated with 1 μM ionomycin, type A and type B oligomers, and the native protein at concentrations of 12 μM and 0.1 μM, respectively, in normal cultured medium or in a medium without Ca2+. 24 h after the treatment the cells were incubated at 37 °C with 0.5 mg/mL of MTT solution in RPMI for 4 h and then were incubated for 1–3 h at 37 °C with the lysis buffer (20% SDS, 50% N,N-dimethylformamide, pH 4.7). Cell viability was determined with the absorbance at 595 nm and expressed as the percentage of MTT reduction relative to the untreated cells, taken as 100%.
Expression of AMPA and NMDA Receptors
The expression of AMPA and NMDA receptors was checked in SH-SY5Y cells plated in six-well plates containing coverslips at a density of 40 000 cells per well. After 24 h they were washed with PBS, fixed with 2% (v/v) paraformaldehyde for 10 min at room temperature, and incubated for 1 h at 37 °C in PBS and 1% FBS with 1:150 diluted rabbit polyclonal anti-AMPA receptors antibody or with 1:100 diluted rabbit polyclonal anti-NMDA receptor antibody, both labeled with ATTO488 (Alomone Labs, Jerusalem, Israel).
Measurement of Cytosolic Free Ca2+ Levels
The cytosolic Ca2+ levels were measured in living SH-SY5Y cells and N13 cells loaded with 4 μM Fluo-4 AM (Life Technologies/Thermo Fisher Scientific) after excitation at 488 nm by a TCS SP5 scanning confocal microscopy system equipped with an argon laser source (Leica Microsystems, Mannheim, Germany). A series of 1 μm thick optical sections (1024 × 1024) was taken through the cell depth for each sample using a Leica Plan Apo 63× oil immersion objective and projected as a single composite image by superimposition (Leica). 10–22 cells, in three different experiments, were analyzed using ImageJ software. In one set of experiments, SH-SY5Y cells were treated with 12 μM native HypF-N for 15 min, 12 μM type B oligomers (monomer equivalents) for 15 min, 12 μM type A oligomers for 5–180 min, 1 μM Aβ42 ADDLs for 5–180 min, and 5 μM zinc-stabilized Aβ40 oligomers for 5–180 min. In a separate set of experiments, the SH-SY5Y cells were treated for 10 or 60 min with 12 μM type A oligomers in a medium with or without Ca2+.
In another set of experiments, the SH-SY5Y cells were treated for 10 min with 1 mM NMDA or with 50 μM AMPA, with or without pretreatment for 60 min with 10 μM memantine (NMDA receptor inhibitor) or 5 μM CNQX (AMPA receptor inhibitor), respectively.
In another set of experiments, before the treatment with the various oligomers, the SH-SY5Y cells were pretreated for 1 h with 5 μM CNQX (AMPA receptor antagonist), 10 μM memantine (NMDA receptor inhibitor), 1 μg/mL anti-ANO6 antibody (SCAN channel inhibitor), 0.6 μg/mL anti-P2RX4 antibody (P2X4 inhibitor), 5 μM GsMTx4 (FAM38A inhibitor), 3 μM FTY720 (LTrpC-7 inhibitor), 10 μM Cd2+ (VGCCs inhibitor), 10 μM Synta66 (CRAC inhibitor), 100 nM α-bungarotoxin (α7nAChR inhibitor), or 100 μM erastin (xCT inhibitor). Before the treatment with 12 μM type A oligomers or 1 μM Aβ42 ADDLs for 10 min, the SH-SY5Y cells were also pretreated for 2 h with 0.05, 0.1, 1.0, and 2.0 μM l-α-lysophosphatidylcholine or 10 μM arachidonic acid.
The experiments described above were performed also using N13 cells with the difference that 0.1 μM type A oligomers were used. In addition, in this case the N13 cells were also treated for 5 or 60 min with 0.1 μM type A oligomers in a medium without Ca2+ after 60 min of pretreatment with 10 μM CGP-37157 or with 100 μM 2APB or with both or with both inhibitors in a medium without Ca2+.
The experiments with 1 μM Aβ42 ADDLs for 10 and 60 min with or without 5 μM CNQX or 10 μM memantine and with or without 2.0 μM l-α-lysophosphatidylcholine or 10 μM arachidonic acid were also performed in the primary rat cortical neurons.
RNA Interference
SH-SY5Y cells were plated in six-well plates containing coverslips at a density of 40 000 cells per well. After 24 h, they were washed with PBS and transfected using 25 nM Stealth RNAi siRNA against glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B) (Life Technologies/Thermo Fisher Scientific), 7 μL of Lipofectamine, 10 μL of 5 mg/L transferrin in DMEM for 3 h in a 5% CO2 humidified atmosphere at 37 °C. The cells were also transfected with vehicle (transfection mix without siRNA) and with 25 nM Stealth RNAi siRNA negative controls (Life Technologies/Thermo Fisher Scientific). 3 h after transfection the DMEM was replaced with fresh complete medium, and the cells were incubated for 72 h. The cells were then washed and incubated for 60 min at 37 °C with 1:400 diluted mouse monoclonal anti-NMDA receptor 2B antibody (Life Technologies/Thermo Fisher Scientific) in PBS and 1% FBS and for 90 min with 1:1000 diluted Alexa Fluor 488-conjugated anti-mouse secondary antibodies (Life Technologies/Thermo Fisher Scientific) in PBS and 1% of FBS. The cytosolic Ca2+ levels after the treatment with 1 μM Aβ42 ADDLs were also measured in silenced cells after 72 h of incubation, using the protocol described above.
FRET Analysis
SH-SY5Y cells were treated with type A oligomers (12 μM monomer equivalents) labeled with Alexa Fluor 555 (Life Technologies/Thermo Fisher Scientific) as an acceptor (A) for 15 min, then fixed with 2% buffered paraformaldehyde for 10 min at room temperature and then permeabilized with PBS, 0.5% (v/v) Triton-X and 0.5% bovine serum albumin (BSA) for 7 min at room temperature. After washing, the cells were stained with 1:150 diluted rabbit polyclonal anti-AMPA receptors antibody or with 1:100 diluted rabbit polyclonal anti-NMDA receptor antibody, both labeled with ATTO488 (Alomone Labs) as a donor (D) in PBS and 1% FBS for 60 min at 37 °C. A positive control was performed in the absence of oligomers using the same experimental protocol described above and then incubating the cells for 60 min at 37 °C with 1:900 diluted Alexa Fluor 594-conjugated anti-rabbit secondary antibody (Life Technologies/Thermo Fisher Scientific). Cells were analyzed using a Leica TCS SP5 confocal scanning microscopy (Leica Microsystems), as described above. The optical sections were acquired in the donor channel (excitation at 488 nm and emission at 499–535 nm), in the acceptor channel (excitation at 543 nm and emission at 560–610 nm), and in the FRET channel (excitation at 488 nm and emission at 560–610 nm).
In another experimental set, unlabeled type A oligomers (12 μM monomer equivalents) were incubated for 1 h at 37 °C in cultured medium without cells, in wells containing glass coverslips. The coverslips were then fixed with 2% (v/v) paraformaldehyde for 10 min at room temperature and incubated with 0.5% BSA for 5 min at room temperature for 30 min at 37 °C with 1:800 diluted rabbit anti-HypF-N antibodies and then for 30 min at 37 °C with 1:1000 Alexa Fluor 488-conjugated anti-rabbit secondary antibody (Life Technologies/Thermo Fisher Scientific). Alternatively, they were incubated for 30 min at 37 °C with 1:100 diluted rabbit polyclonal anti-NMDA receptor antibody labeled with ATTO488 (Alomone Labs). The latter experiment was repeated on a glass coverslip without oligomers. The first and third experiments represent the positive and negative controls, respectively.
TMA-DPH Labeling and Anisotropy
SH-SY5Y cells, seeded on glass coverslips, were loaded for 15 min with 5 μM trodusquemine labeled with Alexa Fluor 594 succinimidyl ester in a 10:1 (trodusquemine:dye) molar ratio to label the membrane, and in the last 5 min 2 μM TMA-DPH from Thermo Fisher scientific was added. Cells were analyzed using a Leica TCS SP5 confocal scanning microscopy as described above. Cells loaded with 2 μM TMA-DPH, with or without a treatment with 1 μM Aβ42 ADDLs for 10 min, were recovered after trypsination using PBS with MgCl2 and CaCl2, and the fluorescence anisotropy (r) values were measured at 430 nm, after excitation at 355 nm, using a 10 × 4 path length quartz cuvette and an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA) equipped with a thermostated cell holder attached to a Agilent PCB 1500 water Peltier system.
Statistical Analysis
All data were expressed as the mean ± SEM (standard error of the mean). Comparisons between the different groups were performed by Student’s t test. The single, double and triple symbols refer to p values of <0.05, <0.01, and <0.001, respectively.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00811.
Additional figures showing all the results performed on N13 microglial cells (Figures S1, S2, S4, S9, S10), functional expression of AMPA and NMDA (Figure S3), immunostaining of immobilized oligomers (Figure S5), FRET analysis between type A oligomers and AMPA receptors and the positive control (Figures S6, S7), intracellular free Ca2+ levels in cells treated with type A oligomers (Figure S9) or with zinc-stabilized Aβ40 oligomers (Figure S12) or with Aβ42 ADDLs oligomers (Figure S13) after the inhibition of various Ca2+ channels, the intracellular free Ca2+ levels in cells treated with zinc-stabilized Aβ40 oligomers and Aβ42 ADDLs oligomers, after the inhibition of AMPA and NMDA receptors (Figure S11), and the NMDA receptor-derived fluorescence and intracellular free Ca2+ levels in cells treated with siRNA against NMDA (PDF)
Author Contributions
G.F. performed the cell experiments and the associated data analysis. B.M. and G.V. performed the interactome study and data analysis. G.F., F.C., C.C., R.C., M.V., and C.M.D. were principally involved in the design of the study. G.F. and F.C. wrote the paper. All authors were involved in the analysis of the data and editing of the manuscript.
This work was supported by Regione Toscana (FAS-Salute 2014), Project Supremal (F.C. and C.C.), by the Progetto Dipartimento di Eccellenza “Gender Medicine” (C.C.), and by Fondi di Ateneo (C.C. and F.C.).
The authors declare no competing financial interest.
Supplementary Material
References
- Patterson C. (2018). World Alzheimer Report 2018, The State of the Art of Dementia Research: New Frontiers, Alzheimer’s Disease International, London. [Google Scholar]
- Jack C. R. Jr; Knopman D. S.; Jagust W. J.; Petersen R. C.; Weiner M. W.; Aisen P. S.; Shaw L. M.; Vemuri P.; Wiste H. J.; Weigand S. D.; Lesnick T. G.; Pankratz V. S.; Donohue M. C.; Trojanowski J. Q. (2013) Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J.; Hardy J. (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun H.; Marriott I.; Lee C. J.; Cho H. (2018) Elucidating the interactive roles of glia in Alzheimer’s disease using established and newly developed experimental models. Front. Neurol. 9, 797. 10.3389/fneur.2018.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I.; Karran E.; De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Chakroborty S.; Stutzmann G. E. (2014) Calcium channelopathies and Alzheimer’s disease: insight into therapeutic success and failures. Eur. J. Pharmacol. 739, 83–95. 10.1016/j.ejphar.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Agostini M.; Fasolato C. (2016) When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s Disease. Cell Calcium 60, 289–298. 10.1016/j.ceca.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Li P.; Feng J.; Wu M. (2016) Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol Sci. 37, 1039–1047. 10.1007/s10072-016-2546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Reddy P. H. (2017) Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer's Dis. 57, 1041–1048. 10.3233/JAD-160763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong B. C.; Wu A. J.; Li M.; Cheung K. H. (2018) Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta, Mol. Cell Res. 1865, 1745–1760. 10.1016/j.bbamcr.2018.07.018. [DOI] [PubMed] [Google Scholar]
- Acosta C.; Anderson H. D.; Anderson C. M. (2017) Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 95, 2430–2447. 10.1002/jnr.24075. [DOI] [PubMed] [Google Scholar]
- Tozaki H.; Matsumoto A.; Kanno T.; Nagai K.; Nagata T.; Yamamoto S.; Nishizaki T. (2002) The inhibitory and facilitatory actions of amyloid-beta peptides on nicotinic ACh receptors and AMPA receptors. Biochem. Biophys. Res. Commun. 294, 42–45. 10.1016/S0006-291X(02)00429-1. [DOI] [PubMed] [Google Scholar]
- Ye C. P.; Selkoe D. J.; Hartley D. M. (2003) Protofibrils of amyloid beta-protein inhibit specific K+ currents in neocortical cultures. Neurobiol. Dis. 13, 177–190. 10.1016/S0969-9961(03)00068-8. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Sokolov Y.; Edmonds B.; McIntire T. M.; Milton S. C.; Hall J. E.; Glabe C. G. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279, 46363–46366. 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- Demuro A.; Mina E.; Kayed R.; Milton S. C.; Parker I.; Glabe C. G. (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300. 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- De Felice F. G.; Velasco P. T.; Lambert M. P.; Viola K.; Fernandez S. J.; Ferreira S. T.; Klein W. L. (2007) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Zhao W. Q.; Santini F.; Breese R.; Ross D.; Zhang X. D.; Stone D. J.; Ferrer M.; Townsend M.; Wolfe A. L.; Seager M. A.; Kinney G. G.; Shughrue P. J.; Ray W. J. (2010) Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J. Biol. Chem. 285, 7619–7632. 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberdi E.; Sánchez-Gómez M. V.; Cavaliere F.; Pérez-Samartín A.; Zugaza J. L.; Trullas R.; Domercq M.; Matute C. (2010) Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 47, 264–272. 10.1016/j.ceca.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Decker H.; Jürgensen S.; Adrover M. F.; Brito-Moreira J.; Bomfim T. R.; Klein W. L.; Epstein A. L.; De Felice F. G.; Jerusalinsky D.; Ferreira S. T. (2010) N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-β peptide oligomers. J. Neurochem. 115, 1520–1529. 10.1111/j.1471-4159.2010.07058.x. [DOI] [PubMed] [Google Scholar]
- Rönicke R.; Mikhaylova M.; Rönicke S.; Meinhardt J.; Schröder U. H.; Fändrich M.; Reiser G.; Kreutz M. R.; Reymann K. G. (2011) Early neuronal dysfunction by amyloid β oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 32, 2219–2228. 10.1016/j.neurobiolaging.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Texidó L.; Martín-Satué M.; Alberdi E.; Solsona C.; Matute C. (2011) Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium 49, 184–90. 10.1016/j.ceca.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Sinnen B. L.; Bowen A. B.; Gibson E. S.; Kennedy M. J. (2016) Local and use-dependent effects of β-Amyloid oligomers on NMDA receptor function revealed by optical quantal analysis. J. Neurosci. 36, 11532–11543. 10.1523/JNEUROSCI.1603-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbel-Ornath M.; Hudry E.; Boivin J. R.; Hashimoto T.; Takeda S.; Kuchibhotla K. V.; Hou S.; Lattarulo C. R.; Belcher A. M.; Shakerdge N.; Trujillo P. B.; Muzikansky A.; Betensky R. A.; Hyman B. T.; Bacskai B. J. (2017) Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 12, 27. 10.1186/s13024-017-0169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella R.; Evangelisti E.; Bigi A.; Becatti M.; Fiorillo C.; Stefani M.; Chiti F.; Cecchi C. (2017) Soluble oligomers require a ganglioside to trigger neuronal calcium overload. J. Alzheimer's Dis. 60, 923–938. 10.3233/JAD-170340. [DOI] [PubMed] [Google Scholar]
- Kawahara M.; Kuroda Y.; Arispe N.; Rojas E. (2000) Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 275, 14077–14083. 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- Evangelisti E.; Wright D.; Zampagni M.; Cascella R.; Fiorillo C.; Bagnoli S.; Relini A.; Nichino D.; Scartabelli T.; Nacmias B.; Sorbi S.; Cecchi C. (2013) Lipid rafts mediate amyloid-induced calcium dyshomeostasis and oxidative stress in Alzheimer’s disease. Curr. Alzheimer Res. 10, 143–53. 10.2174/1567205011310020004. [DOI] [PubMed] [Google Scholar]
- Sepúlveda F. J.; Fierro H.; Fernandez E.; Castillo C.; Peoples R. W.; Opazo C.; Aguayo L. G. (2014) Nature of the neurotoxic membrane actions of amyloid-β on hippocampal neurons in Alzheimer’s disease. Neurobiol. Aging 35, 472–481. 10.1016/j.neurobiolaging.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Snyder E. M.; Nong Y.; Almeida C. G.; Paul S.; Moran T.; Choi E. Y.; Nairn A. C.; Salter M. W.; Lombroso P. J.; Gouras G. K.; Greengard P. (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8, 1051–1058. 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Deshpande A.; Kawai H.; Metherate R.; Glabe C. G.; Busciglio J. (2009) A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 29, 4004–4015. 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K.; Shinohara S.; Yagami T.; Asakura K.; Kawasaki K. (1997) Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J. Neurochem. 68, 265–271. 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- Quan Q. K.; Li X.; Yuan H. F.; Wang Y.; Liu W. L. (2016) Ginsenoside Rg1 inhibits high-voltage-activated calcium channel currents in hippocampal neurons of beta-amyloid peptide-exposed rat brain slices. Chin. J. Integr. Med. 10.1007/s11655-015-2301-4. [DOI] [PubMed] [Google Scholar]
- Ostapchenko V. G.; Chen M.; Guzman M. S.; Xie Y. F.; Lavine N.; Fan J.; Beraldo F. H.; Martyn A. C.; Belrose J. C.; Mori Y.; MacDonald J. F.; Prado V. F.; Prado M. A.; Jackson M. F. (2015) The transient receptor potential melastatin 2 (TRPM2) channel contributes to β-Amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 35, 15157–15169. 10.1523/JNEUROSCI.4081-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson A.; Paumier A.; Boisseau S.; Jacquier-Sarlin M.; Buisson A.; Albrieux M. (2017) TRPA1 channels promote astrocytic Ca2+ hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-β peptide. Mol. Neurodegener. 12, 53. 10.1186/s13024-017-0194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S.; Mannini B.; Zampagni M.; Pensalfini A.; Parrini C.; Evangelisti E.; Relini A.; Stefani M.; Dobson C. M.; Cecchi C.; Chiti F. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147. 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- Zampagni M.; Cascella R.; Casamenti F.; Grossi C.; Evangelisti E.; Wright D.; Becatti M.; Liguri G.; Mannini B.; Campioni S.; Chiti F.; Cecchi C. (2011) A comparison of the biochemical modifications caused by toxic and non-toxic protein oligomers in cells. J. Cell Mol. Med. 15, 2106–2116. 10.1111/j.1582-4934.2010.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelisti E.; Cecchi C.; Cascella R.; Sgromo C.; Becatti M.; Dobson C. M.; Chiti F.; Stefani M. (2012) Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 125, 2416–2427. 10.1242/jcs.098434. [DOI] [PubMed] [Google Scholar]
- Tatini F.; Pugliese A. M.; Traini C.; Niccoli S.; Maraula G.; Ed Dami T.; Mannini B.; Scartabelli T.; Pedata F.; Casamenti F.; Chiti F. (2013) Amyloid-β oligomer synaptotoxicity is mimicked by oligomers of the model protein HypF-N. Neurobiol. Aging 34, 2100–2109. 10.1016/j.neurobiolaging.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Neumann J. T.; Diaz-Sylvester P. L.; Fleischer S.; Copello J. A. (2011) CGP-37157 inhibits the sarcoplasmic reticulum Ca2+ ATPase and activates ryanodine receptor channels in striated muscle. Mol. Pharmacol. 79, 141–7. 10.1124/mol.110.067165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R.; Sekler I. (2012) The mitochondrial Na(+)/Ca(2+) exchanger. Cell Calcium 52, 9–15. 10.1016/j.ceca.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Maruyama T.; Kanaji T.; Nakade S.; Kanno T.; Mikoshiba K. (1997) 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 122, 498–505. 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- Paoletti P.; Ascher P. (1994) Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron 13, 645–655. 10.1016/0896-6273(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Casado M.; Ascher P. (1998) Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity. J. Physiol. 513, 317–330. 10.1111/j.1469-7793.1998.317bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaban V. V.; Li J.; Ennes H. S.; Nie J.; Mayer E. A.; McRoberts J. A. (2004) N-methyl-D-aspartate receptors enhance mechanical responses and voltage-dependent Ca2+ channels in rat dorsal root ganglia neurons through protein kinase C. Neuroscience 128, 347–357. 10.1016/j.neuroscience.2004.06.051. [DOI] [PubMed] [Google Scholar]
- Kloda A.; Lua L.; Hall R.; Adams D. J.; Martinac B. (2007) Liposome reconstitution and modulation of recombinant N-methyl-D-aspartate receptor channels by membrane stretch. Proc. Natl. Acad. Sci. U. S. A. 104, 1540–1545. 10.1073/pnas.0609649104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. P.; Arora S.; Bhardwaj A.; Srivastava S. K.; Kadakia M. P.; Wang B.; Grizzle W. E.; Owen L. B.; Singh S. (2012) CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase- and Akt kinase-mediated activation of nuclear factor κB: implications for bidirectional tumor-stromal interactions. J. Biol. Chem. 287, 39115–39124. 10.1074/jbc.M112.409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L. V.; Vogel S. S.; Sokoloff A.; Onaran H. O.; Leikina E. A.; Zimmerberg J. (1993) Lysolipids reversibly inhibit Ca(2+)-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 318, 71–76. 10.1016/0014-5793(93)81330-3. [DOI] [PubMed] [Google Scholar]
- Drzazga A.; Sowinska A.; Krzeminska A.; Rytczak P.; Koziolkiewicz M.; Gendaszewska-Darmach E. (2017) Lysophosphatidylcholine elicits intracellular calcium signaling in a GPR55-dependent manner. Biochem. Biophys. Res. Commun. 489, 242–247. 10.1016/j.bbrc.2017.05.145. [DOI] [PubMed] [Google Scholar]
- Mannini B.; Vecchi G.; Labrador-Garrido A.; Fabre B.; Fani G.; Franco J. M.; Lilley K.; Pozo D.; Vendruscolo M.; Chiti F.; Dobson C. M.; Roodveldt C. (2019) Differential interactome and innate immune response activation of two structurally distinct misfolded protein oligomers. ACS Chem. Neurosci. 10, 3464–3478. 10.1021/acschemneuro.9b00088. [DOI] [PubMed] [Google Scholar]
- Uhlén M.; Fagerberg L.; Hallström B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson Å.; Kampf C.; Sjöstedt E.; Asplund A.; Olsson I.; Edlund K.; Lundberg E.; Navani S.; Szigyarto C. A.; Odeberg J.; Djureinovic D.; Takanen J. O.; Hober S.; Alm T.; Edqvist P. H.; Berling H.; Tegel H.; Mulder J.; Rockberg J.; Nilsson P.; Schwenk J. M.; Hamsten M.; von Feilitzen K.; Forsberg M.; Persson L.; Johansson F.; Zwahlen M.; von Heijne G.; Nielsen J.; Pontén F. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Qin S.; Colin C.; Hinners I.; Gervais A.; Cheret C.; Mallat M. (2006) System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1–40. J. Neurosci. 26, 3345–3356. 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova M.; Sanz-Blasco S.; Zhang X.; Xia P.; Akhtar M. W.; Okamoto S.; Dziewczapolski G.; Nakamura T.; Cao G.; Pratt A. E.; Kang Y. J.; Tu S.; Molokanova E.; McKercher S. R.; Hires S. A.; Sason H.; Stouffer D. G.; Buczynski M. W.; Solomon J. P.; Michael S.; Powers E. T.; Kelly J. W.; Roberts A.; Tong G.; Fang-Newmeyer T.; Parker J.; Holland E. A.; Zhang D.; Nakanishi N.; Chen H. S.; Wolosker H.; Wang Y.; Parsons L. H.; Ambasudhan R.; Masliah E.; Heinemann S. F.; Piña-Crespo J. C.; Lipton S. A. (2013) Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U. S. A. 110, E2518–2527. 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S. (2019) CRAC channels and disease - From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 80, 112–116. 10.1016/j.ceca.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannini B.; Habchi J.; Chia S.; Ruggeri F. S.; Perni M.; Knowles T. P. J.; Dobson C. M.; Vendruscolo M. (2018) Stabilization and characterization of cytotoxic Aβ40 oligomers isolated from an aggregation reaction in the presence of zinc ions. ACS Chem. Neurosci. 9, 2959–2971. 10.1021/acschemneuro.8b00141. [DOI] [PubMed] [Google Scholar]
- Lambert M. P.; Viola K. L.; Chromy B. A.; Chang L.; Morgan T. E.; Yu J.; Venton D. L.; Krafft G. A.; Finch C. E.; Klein W. L. (2001) Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 79, 595–605. 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- Illinger D.; Duportail G.; Mely Y.; Poirel-Morales N.; Gerard D.; Kuhry J. G. (1995) A comparison of the fluorescence properties of TMA-DPH as a probe for plasma membrane and for endocytic membrane. Biochim. Biophys. Acta, Biomembr. 1239, 58–66. 10.1016/0005-2736(95)00135-P. [DOI] [PubMed] [Google Scholar]
- Zasloff M.; Adams A. P.; Beckerman B.; Campbell A.; Han Z.; Luijten E.; Meza I.; Julander J.; Mishra A.; Qu W.; Taylor J. M.; Weaver S. C.; Wong G. C. (2011) Squalamine as a broad-spectrum systemic antiviral agent with therapeutic potential. Proc. Natl. Acad. Sci. U. S. A. 108, 15978–83. 10.1073/pnas.1108558108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung S. Y.; Fu W. M. (2017) Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 24, 47. 10.1186/s12929-017-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M. P.; Barlow A. K.; Chromy B. A.; Edwards C.; Freed R.; Liosatos M.; Morgan T. E.; Rozovsky I.; Trommer B.; Viola K. L.; Wals P.; Zhang C.; Finch C. E.; Krafft G. A.; Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U. S. A. 95, 6448–6453. 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelisti E.; Cascella R.; Becatti M.; Marrazza G.; Dobson C. M.; Chiti F.; Stefani M.; Cecchi C. (2016) Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 6, 32721. 10.1038/srep32721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relini A.; Torrassa S.; Rolandi R.; Gliozzi A.; Rosano C.; Canale C.; Bolognesi M.; Plakoutsi G.; Bucciantini M.; Chiti F.; Stefani M. (2004) Monitoring the process of HypF-N fibrillization and liposome permeabilization by protofibrils. J. Mol. Biol. 338, 943–957. 10.1016/j.jmb.2004.03.054. [DOI] [PubMed] [Google Scholar]
- Pellistri F.; Bucciantini M.; Relini A.; Nosi D.; Gliozzi A.; Robello M.; Stefani M. (2008) Nonspecific interaction of prefibrillar amyloid aggregates with glutamatergic receptors results in Ca2+ increase in primary neuronal cells. J. Biol. Chem. 283, 29950–29960. 10.1074/jbc.M803992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um J. W.; Nygaard H. B.; Heiss J. K.; Kostylev M. A.; Stagi I. M.; Vortmeyer A.; Wisniewski T.; Gunther E. C.; Strittmatter S. M. (2012) Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhao Y.; Zhang L.; Yu W.; Wang Y.; Chang W. (2019) Cellular prion protein as a receptor of toxic amyloid-β42 oligomers is important for Alzheimer’s disease. Front. Cell. Neurosci. 13, 339. 10.3389/fncel.2019.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbocker R.; Chia S.; Ruggeri F. S.; Perni M.; Cascella R.; Heller G. T.; Meisl G.; Mannini B.; Habchi J.; Michaels T. C. T.; Challa P. K.; Ahn M.; Casford S. T.; Fernando N.; Xu C. K.; Kloss N. D.; Cohen S. I. A.; Kumita J. R.; Cecchi C.; Zasloff M.; Linse S.; Knowles T. P. J.; Chiti F.; Vendruscolo M.; Dobson C. M. (2019) Trodusquemine enhances Aβ42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 10, 225. 10.1038/s41467-018-07699-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani Boroujeni E.; Hosseini S. M.; Fani G.; Cecchi C.; Chiti F. (2020) Soluble Prion Peptide 107–120 Protects Neuroblastoma SH-SY5Y Cells against Oligomers Associated with Alzheimer’s Disease. Int. J. Mol. Sci. 21, 7273. 10.3390/ijms21197273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchelli M.; Cascella R.; D’Andrea C.; Cabaj L.; Osticioli I.; Ciofini D.; Li M. S.; Skupień K.; de Angelis M.; Siano S.; Cecchi C.; Pini R.; La Penna G.; Chiti F.; Matteini P. (2020) Nanoscopic insights into the surface conformation of neurotoxic amyloid β oligomers. RSC Adv. 10, 21907–21913. 10.1039/D0RA03799K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbocker R.; Mannini B.; Cataldi R.; Chhangur S.; Wright A. K.; Kreiser R. P.; Albright J. A.; Chia S.; Habchi J.; Sormanni P.; Kumita J. R.; Ruggeri F. S.; Dobson C. M.; Chiti F.; Aprile F. A.; Vendruscolo M. (2020) Rationally designed antibodies as research tools to study the structure-toxicity relationship of Amyloid-β oligomers. Int. J. Mol. Sci. 21, 4542. 10.3390/ijms21124542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbocker R.; Mannini B.; Ruggeri F. S.; Cascella R.; Xu C. K.; Perni M.; Chia S.; Chen S. W.; Habchi J.; Bigi A.; Kreiser R. P.; Wright A. K.; Albright J. A.; Kartanas T.; Kumita J. R.; Cremades N.; Zasloff M.; Cecchi C.; Knowles T. P. J.; Chiti F.; Vendruscolo M.; Dobson C. M. (2020) Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 3, 435. 10.1038/s42003-020-01140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T.; Aprile F. A.; Sormanni P.; Ruggeri F. S.; Perni M.; Heller G. T.; Haas C. P.; Middel C.; Limbocker R.; Mannini B.; Michaels T. C. T.; Knowles T. P. J.; Dobson C. M.; Vendruscolo M. (2020) A rationally designed bicyclic peptide remodels Aβ42 aggregation in vitro and reduces its toxicity in a worm model of Alzheimer’s disease. Sci. Rep. 10, 15280. 10.1038/s41598-020-69626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.