

Abstract
Light‐responsive materials have been extensively studied due to the attractive possibility of manipulating their properties with high spatiotemporal control in a non‐invasive fashion. This stimulated the development of a series of photo‐deformable smart devices. However, it remained a challenge to reversibly modulate the stiffness and toughness of bulk materials. Here, we present bioengineered protein fibers and their optomechanical manipulation by employing electrostatic interactions between supercharged polypeptides (SUPs) and an azobenzene (Azo)‐based surfactant. Photo‐isomerization of the Azo moiety from the E‐ to Z‐form reversibly triggered the modulation of tensile strength, stiffness, and toughness of the bulk protein fiber. Specifically, the photo‐induced rearrangement into the Z‐form of Azo possibly strengthened cation–π interactions within the fiber material, resulting in an around twofold increase in the fiber's mechanical performance. The outstanding mechanical and responsive properties open a path towards the development of SUP‐Azo fibers as smart stimuli‐responsive mechano‐biomaterials.
Keywords: azobenzene, mechanical behavior, photo-modulating, protein fibers, supercharged polypeptides
Bioengineered protein fibers were prepared through electrostatic complexation of a positively charged polypeptide and a negatively charged azobenzene‐based surfactant. The photo‐isomerization of the azobenzene moiety from E‐ to Z‐isomer reversibly triggered a modulation of the bulk protein fiber's mechanical performance.

Introduction
Endowing (macro)molecules and structures assembled from them with the ability to respond to stimuli with reversible or irreversible transitions is widely recognized as a promising route towards the creation of smart and interactive materials.[ 1 , 2 , 3 ] For this purpose, temperature, [4] mechanical force, [5] light, [6] electrical, [7] or magnetic fields [8] have been successfully applied. Light often outperforms other stimuli due to its high spatiotemporal and energetic resolution in combination with its non‐invasive character. The use of photo‐switches for material preparation is in turn advancing the design of molecular motors, [9] drug delivery devices, [10] (bio)sensors, [11] actuators,[ 12 , 13 ] optical transistors, [14] as well as energy storage‐, [15] biomedical‐ [16] and classical polymer systems. [17]
Azobenzene and its derivatives arguably constitute the most popular class of photo‐switches as they exhibit outstanding reversibility, high conversion at the photo‐stationary state (PSS), and excellent fatigue resistance. [18] Specifically, the large geometrical change upon UV‐induced isomerization from the E‐ to the Z‐isomer and the facile modification of the azobenzene core with different moieties render them ideal for applications in optomechanics. [19] By anchoring azobenzene or its derivatives within liquid‐crystalline networks (LCNs), high‐speed actuation on the micro‐ and nanoscale was successfully realized. [19] In addition to actuation,[ 20 , 21 , 22 ] artificial muscle contraction, [23] shape memory, [24] phase transition, [25] and motion in liquids, [26] azobenzenes were employed in more constrained environments, such as molecular crystals,[ 27 , 28 ] thin films, [29] and hydrogels. [30]
As opposed to widely‐reported photo‐induced mechanical motion, the photo‐modulation of the mechanical performance, for example, stiffness and toughness, of bulk materials remains a considerable challenge.[ 30 , 31 , 32 ] Thus, it is of great interest to translate photo‐induced molecular rearrangements into altered mechanical performances of materials. Creating molecular architectures whose mechanical performance can be tuned on demand is promising for solid‐state photonic switches, optical interconnects, artificial muscles, chemical sensors, and drug delivery.
Research on stimuli‐responsive fibers has been flourishing due to their potential applications in many fields.[ 33 , 34 , 35 ] Ikeda and co‐workers achieved photo‐mobility of crosslinked liquid‐crystalline polymer (CLCP) fibers containing azobenzene units. [36] The CLCP fibers are capable of three‐dimensional movements upon UV‐light irradiation. Minko and co‐workers reported single‐walled carbon nanotube (SWCNT)‐alginate based composite fibers that reversibly deform during swelling and shrinking. [37] This behavior leads to tunable electro‐conductive properties of composite fibers. Zhao and co‐workers reported photo‐thermally responsive graphene oxide (GO)/N‐isopropylacrylamide (NIPAAm) composite fibers with an NIR‐irradiation‐triggered water collection ability. [38] However, to the best of our knowledge, stimuli‐induced modulation of fiber mechanical performance has not been achieved yet. [39]
Here, we describe the preparation, structure, and reversible bulk photo‐modulation of the mechanical performance of bioengineered protein fibers. We fabricated these fibers from photo‐switchable azobenzene units non‐covalently assembled with a supercharged polypeptide (SUP). Highly positively charged SUPs were complexed with negatively charged surfactants containing azobenzene (Azo) moieties by electrostatic interactions. It was found that the reversible photo‐induced E‐ to Z‐isomerization of azobenzene changes the local packing of the intermolecular microenvironment and alters cation‐π interactions, allowing for modulation of the non‐covalently assembled complexes and hence the mechanics of the resulting fibers.
Results and Discussion
The fabrication of photo‐switchable bioengineered protein‐surfactant fibers consists of two crucial components: a cationic supercharged polypeptide (SUP) forming the structural basis of the fiber material and an anionic sulfonated surfactant containing an azobenzene moiety (Azo) (Figure 1 A). The SUPs are derived from natural elastin and were expressed recombinantly in E. coli.[ 40 , 41 , 42 , 43 , 44 ] The high net charge of SUPs is encoded in the pentapeptide repeat unit (VPGKG)n in which the fourth position is consisting of a lysine residue (K). Regarding the SUP used for fiber fabrication, we chose K108cys that contains two cysteines at the N‐ and C‐terminus, respectively. The digit of K108cys denotes the number of positive charges along the polypeptide backbone (Figure S1–S3, Table S1). Regarding the surfactant, we synthesized a non‐symmetrically substituted azobenzene (Azo) bearing a sodium sulfonate group in the p‐position of one phenyl ring and a triethylene glycol monomethyl ether moiety or a n‐butyl group in the p‐position of the second phenyl ring (Scheme S1). The Azo surfactants were characterized by nuclear magnetic resonance spectroscopy (NMR) (Figure S4) and high‐resolution mass spectrometry (HR‐MS). In principle, Azo surfactant functionalized with the n‐butyl group should provide an additional hydrophobic interaction in the system. However, Azo with butyl substituents exhibited poor solubility in water even with such a short chain length, which hence prevented us for the preparation of the SUP‐Azo coacervate. Therefore, we functionalized the Azo surfactant with a triethylene glycol monomethyl ether moiety to increase the solubility in aqueous solutions. K108cys and Azo were mixed in aqueous solution in a 1:1 molar ratio of lysine to surfactant. As a result, the solution became turbid, a liquid‐liquid phase transition occurred, and after centrifugation a protein‐surfactant coacervate was obtained at the bottom of the tube (experimental details can be found in the Supporting Information). It should be noted that short poly‐d‐lysine (average mol wt 30 000–70 000) was also investigated. No coacervate was observed which suggests appropriate electrostatic interactions between lysine amino acids and surfactant is important for the formation of coacervation and the following fibers. From this SUP‐Azo complex, fibers were produced simply by introducing a needle into the coacervate and drawing the needle out of the material. Finally, the resulting fibers were kept at ambient conditions for 2 h before further characterization.
Figure 1.
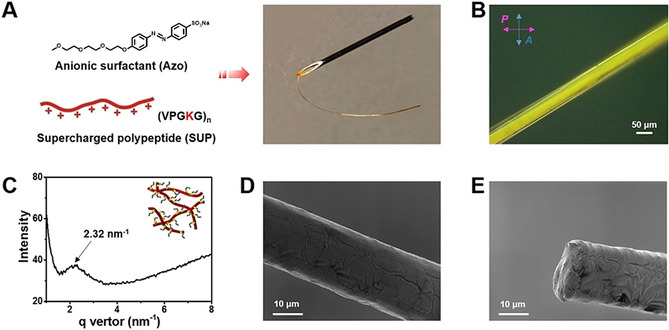
Preparation and characterization of the mechanically responsive SUP‐Azo fibers. A) SUP‐Azo fibers assembled by electrostatic interactions between genetically engineered cationic K108cys (SUP) and anionic azobenzene surfactant (Azo). B) POM analysis of SUP‐Azo fiber. Scale bar is 50 μm. The birefringent properties suggested an ordered structure. C) SAXS analysis of SUP‐Azo fiber. The average distance of the formed nematic phase of the SUP‐Azo complex is around 2.7 nm. The insert schematic graph represents the nematic model of the SUP‐Azo complex. D–E) Scanning electron microscope (SEM) images of SUP‐Azo fiber before UV‐light irradiation. Scale bars are 10 μm. D) Analysis of the surface morphology of SUP‐Azo fiber. E) Analysis at a cross‐section to examine the SUP‐Azo fiber internal core.
Initially, a quantitative component determination of the SUP‐Azo complexes was carried out by 1H‐NMR spectroscopy. Therefore, a lower molar mass SUP variant was employed. For the K18‐Azo complex, a stoichiometry of K18:Azo of 1:16 was measured, which indicates that ca. 90 % of the positive lysine residues are complexed with anionic surfactant molecules (Figure S5). This observation might indicate that besides electrostatic interactions and π‐stacking, cation‐π interactions of the non‐complexed lysine moieties in the SUP and the phenyl groups in the Azo are present within the fiber system. To gain more insight into the SUP‐Azo fibers, we performed polarized optical microscopy (POM) revealing significant birefringence of the fiber under cross‐polarized light indicating the presence of structural order in the material (Figure 1 B). Further characterization of SUP‐Azo fiber was conducted by small‐angle X‐ray scattering (SAXS). A weak broad diffraction peak at q=2.32 nm−1 corresponds to the d spacing of 2.7 nm (d=2πq −1) (Figure 1 C). Combined with POM analysis, this confirms the nematic ordering of the SUP‐Azo complex with an average diameter of 2.7 nm. The scanning electron microscopy (SEM) image of SUP‐Azo fibers in Figure 1 D revealed a uniform, cylindrical, and smooth surface morphology. In addition, the cross‐section analysis showed the solid internal core of the SUP‐Azo fiber (Figure 1 E).
To assess the photo‐switching capabilities, we initially performed irradiation experiments in combination with UV‐vis spectroscopy on Azo alone in aqueous solution. Firstly, the pristine Azo (in E‐configuration) was tested regarding its stability in solution without irradiation. No changes in the absorption spectrum were detected over the course of almost 30 h indicating the stability of Azo in an aqueous environment over this period of time (Figure S6). The E‐isomer shows the typical π‐π* and n‐π* transitions at ca. 350 nm and 430 nm, respectively. As shown in Figure S7, upon irradiation with UV‐light (λ=365 nm, 0.5 mW cm−2), the characteristic decrease of the π‐π* absorption band with a concomitant hypsochromic shift to ca. 300 nm accompanied by an increase in absorption of the n‐π* transition was observed. This E‐ to Z‐isomerization is fully reversible either thermally or by irradiation with visible light (λ=450 nm, 30 μW cm−2). Azo was found to exhibit a thermal half‐life (t 1/2) of 177 h at 25 °C in H2O, indicating that the Z isomer shows excellent thermal stability (Figure S8 and S9). Photo‐switching between both isomers resulted in good conversions at the PSS. 95 % of the Z‐isomer were obtained for the E‐ to Z‐transition while 78 % E‐isomer were obtained for the Z‐ to E‐isomerization, as determined by 1H‐NMR spectroscopy (Figure S10, Table S2).
Regarding the photo‐isomerization characterization of the Azo component in the bulk materials, solid‐state UV‐vis spectroscopy was conducted. It should be noted that it was not possible to investigate this process within the fiber due to limitations of the sensitivity of the instrument. Thus, we examined the SUP‐Azo complex in the bulk after spin‐coating films on glass substrates (Figure S11). The analysis of solid‐state UV‐vis spectra of SUP‐Azo complexes revealed that a significant decrease of the π‐π* absorption (≈360 nm) and an increase of n‐π* absorption (≈450 nm) occurred after exposure to UV‐irradiation (λ=365 nm, 0.5 mW cm−2, 2 h), indicating the E‐ to Z‐isomerization in bulk samples. After a period of 2 h in the dark, a small increase of the characteristic π‐π* absorption band was observed. This effect became more obvious after resting of the sample overnight, confirming the thermally reversible Z‐to E‐ isomerization. These results indicate that the internal structural properties of SUP‐Azo bulk materials can be modulated by light irradiation.
Afterwards, the mechanical properties of SUP‐Azo fibers were studied by uniaxial tensile testing on a universal testing machine (Figure 2 and S12). In the absence of light, that is, with all azobenzenes in the E‐state, Young's moduli of 3.5±0.4 GPa were recorded in the elastic region of 0–3 % applied strain (Figure 2 A). Most notably, after irradiating the fiber with UV‐light (λ=365 nm, 0.5 mW cm−2, 2 h) and isomerizing the azobenzenes to the Z‐state, the moduli increased significantly to 6.8±0.6 GPa. The original moduli could be recovered after keeping the same sample in the dark for 2 h, which can be attributed to the thermal back‐isomerization of the azobenzene molecules to the E‐state (Figure S12, Table S3). These results indicate that the stiffness of SUP‐Azo fibers can be modulated by reversible structural changes in the complex.
Figure 2.
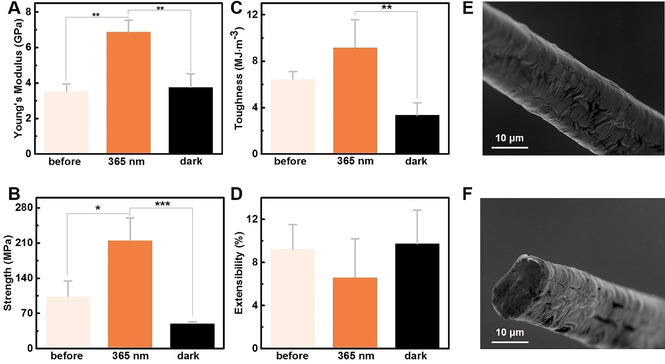
Characterization of the mechanical properties of SUP‐Azo fibers by uniaxial tensile testing before UV‐irradiation, after UV‐irradiation (λ=365 nm, 0.5 mW cm−2, 2 h), and after UV‐irradiation and 2 h in the dark. Three parallel tensile testing experiments were performed for each sample. P‐values were calculated using Student's t‐test. ns, no significant difference; *, p<0.1; **, p<0.05, ***, p<0.01. A) Young's moduli, B) breaking strength, C) toughness, and D) breaking strain. E,F) SEM images of SUP‐Azo fiber (UV‐irradiation (λ=365 nm, 0.5 mW cm−2, 2 h) after breaking (scale bars are 10 μm). E) Analysis of the surface morphology of SUP‐Azo fiber. F) Analysis at a cross‐section to examine the SUP‐Azo fiber's internal morphology.
We observed a similar trend for the tensile strengths of the fibers. In their native and non‐irradiated state (Azo in E‐form), an average breaking strength of 103.7±30.6 MPa was recorded. This value increased significantly to 215.3±44.7 MPa when the fiber was illuminated with UV‐light (λ=365 nm, 0.5 mW cm−2, 2 h) and rearrangement of the complexed Azo surfactant to the Z‐state partially or completely occurred (Figure 2 B). Leaving the UV‐illuminated, non‐extended samples in the dark for 2 h revealed a decreased breaking strengths of 50.0±3.0 MPa suggesting that the consecutive E‐ to Z‐ to E‐reconfiguration within the fiber by the photo‐induced molecular structural changes of the azobenzene moiety recovered the original state (Figure 2 B and S12, Table S3). Analogous observations were made regarding the fiber toughness revealing photo‐modulation from an average of 6.4±0.6 MJ m−3 before irradiation over 9.2±2.4 MJ m−3 after UV‐irradiation to 3.4±1.1 MJ m−3 for the thermally back‐isomerized non‐extended samples (Figure 2 C).
Interestingly, we could not observe this modulation for the breaking strain that remained unaffected within the margin of experimental error before and after irradiation (Figure 2 D). In general, the distance between the two p‐positions of the aromatic rings of azobenzene decreases from the E‐state (9.0 Å) to the Z‐state (5.5 Å) during the photo‐isomerization process. [45] This subtle difference between the E‐ and Z‐isomers can be used to explain the macroscopically negligible elongation rate under experimental conditions. To verify the reproducibility of the photomechanical behavior, a second batch of experiments was conducted confirming the results mentioned above (Figure S13 and Table S4). In addition, comparing to previous light‐responsive soft materials,[ 20 , 21 , 22 ] our SUP‐Azo bulk fibers didn't show any macroscopic motion under light irradiation. This behavior might be due to the inherent high stiffness that makes it too hard to be deformed. Moreover, we performed fractography employing SEM on the intact and fractured samples after UV‐irradiation (λ=365 nm, 0.5 mW cm−2, 2 h) (Figures 2 E‐F, and S14). On all fibers, pristine, UV‐irradiated, or thermally back‐isomerized, we identified a comparable uniform cylindrical diameter as well as surface morphology giving no further insight on changes of the material, indicating the stability of the fibers’ bulk structure under different treatment conditions.
Hereafter, we investigated the mechanical properties of the SUP‐Azo fibers at the nanoscale using atomic force microscopy (AFM) nano‐indentation experiments (Figures 3 and S16). [46] Initially, we determined the elastic limit by indenting with a high force above 1 μN (Figure S17A–C). The observed fiber deformed elastically up to ≈400 nN and plastically above this limit. Following this, we indented at lower force in the nN range and within the elastic regime (Figure S17D–F). The indentation experiments were performed on the same fiber but in pristine, UV‐irradiated (λ=365 nm, 0.5 mW cm−2), and thermally back‐isomerized states. Analogously to the SEM‐based fractography, AFM also did not reveal changes of the fiber surface topography in the different switching states of the azobenzene (Figures 3 A,D, and G). However, the alteration of the recorded stiffness from ≈92 N m−1 in the pristine state over ≈224 N m−1 after UV‐irradiation to ≈83 N m−1 after resting in the dark qualitatively aligns with the results from macroscopic fiber tensile testing (Figures 3 C,F, and I). We hypothesize that this observation of no apparent macroscopic topological changes and the evident alteration of mechanical properties after UV‐induced isomerization stems from minuscule but effective molecular rearrangements within the material.
Figure 3.
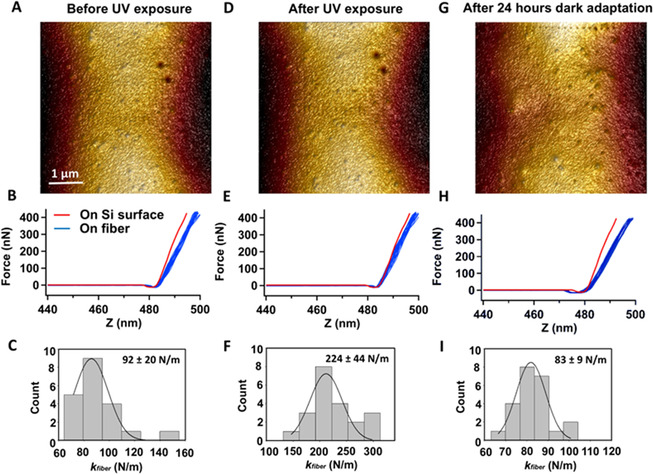
AFM‐based nano‐indentation on the SUP‐Azo fiber. A) Surface morphology of the fiber before UV irradiation. B) Superposition of force‐distance curves recorded on the image area. C) Histogram of calculated spring constant of the fiber obtained from force curves from (B) (n=20). D–F) Identical fiber portion as in (A) but after 2 h of UV‐irradiation (λ exc=365 nm, 0.5 mW cm−2). G–I) Identical fiber portion as in (A) but after 2 h of UV‐irradiation (λ exc=365 nm, 0.5 mW cm−2) and subsequent 24 h in the dark.
As discussed above, the cation‐π interactions might play an important role in the formation of the SUP‐Azo fibers. This attempt of an interpretation is strongly supported by the preparation of a SUP‐Azo complex in a 1:5 lysine:surfactant stoichiometry leaving no free cationic lysine residues on the SUP, as demonstrated by 1H‐NMR spectroscopy (Figure S18). With the resulting coacervate, no fiber manufacturing was possible at all, highlighting the importance of free cationic lysine moieties for the mechanical integrity of the material. Moreover, the experiments with excess surfactant implied that π‐π stacking might not have a significant effect in manipulating the mechanical behavior of the fiber. Thus, we speculate that the alteration of cation‐π interactions upon photo‐isomerization of the azobenzene surfactant is a major mechanism for the observed effects as non‐covalent interactions between electron‐rich species and adjacent cations have been reported (Figure 4).[ 47 , 48 ] This hypothesis bases on the geometrical rearrangement of the azobenzene units during the E‐ to Z‐isomerization, effectively altering the distance between the distant azobenzene phenyl rings and unoccupied cationic lysine residues of neighboring SUP‐Azo complexes. The isomerization process from E‐ to Z‐configuration decreases the distance between the two azobenzene phenyl rings, thereby strengthening the cation‐π interactions since in the Z‐isomer possibly both phenyl rings can interact with the protonated ϵ‐amino group of lysine. [49] As a consequence, this change might improve the fiber's breaking strength since the cohesion forces within the material are strengthened. Meanwhile, the negligible variation of strain would contribute to the increase of fiber's Young's modulus and toughness. In turn, after recovering to theE‐form, the mechanical properties of SUP‐Azo fiber are restored due to the decrease of the relevant cation‐π interactions. Therefore, altering the strength of intermolecular forces within the fiber might lead to an overall alteration of the mechanical properties of the macroscopic material.
Figure 4.
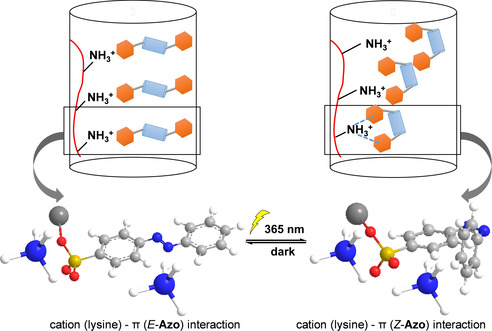
Schematic representation of a plausible mechanism for SUP‐Azo fibers with photo‐switchable mechanical properties. The geometrical rearrangement of the azobenzene units during photo‐isomerization effectively contributed to alterations of the interactions between azobenzene phenyl rings and unoccupied cationic lysine residues of neighboring SUP‐Azo complexes. Therefore, the mechanical properties of SUP‐Azo fibers might be tuned by light. (The blue/white spheres represent NH4 +, the red curve represents the SUP backbone, the red/yellow spheres represent SO3 −, the orange hexagon represents the phenyl rings of azobenzene, and the light blue cylinder represents the N=N bond). The geometrical rearrangement of the azobenzene units during the isomerization process, effectively altering the distance between the external azobenzene phenyl ring and unoccupied cationic lysine residues of neighboring SUP‐Azo complexes. The isomerization from E‐ to Z‐configuration decreases the distance between the two azobenzene phenyl rings, thereby strengthening the cation‐π interactions since in the Z‐isomer both phenyl rings can interact with the protonated ϵ‐amino group of lysine. As a result, this change might improve the fiber's mechanics (e.g. tensile strength, Young's modulus, and toughness) since the cohesion forces within the material are strengthened.
Generally, azobenzene‐induced mechanical modulation is limited to liquid crystals (LC), films and gels, which only exhibited negligible variation (hundreds of Pa) in their mechanical performance.[ 29 , 30 , 31 ] In stark contrast to these efforts, twofold increase in the fiber's mechanical properties (hundreds of MPa) was achieved by light in the bulk SUP‐Azo fibers presented here. Specifically, Young's modulus and breaking strength, as high as 6.8 GPa and 215 MPa were reached in fibers after irradiation treatment, respectively. Evenly important is the reversibility of this process.
Conclusion
We here presented the design and manufacturing of protein fibers assembled from supercharged polypeptides and azobenzene surfactants. These biosynthetic hybrid fibers exhibit excellent mechanical properties. For the first time, modulation of the fiber's mechanical performance, including Young's modulus, breaking strength, and toughness, was achieved by light as an external trigger. The mechanical properties of the SUP‐Azo fibers increased after the photo‐isomerization of azobenzene from the E‐ to Z‐state in the solid‐state. This behavior was possibly induced by increasing the strength of cation‐π interactions in the Z‐state of Azo due to the photo‐induced geometrical rearrangement of the azobenzene motif. Importantly, the fiber's mechanical properties can be restored after photo‐isomerization from the Z‐ to E‐state. In the future, the promising properties of this biosynthetic hybrid material might be translated into technological applications in which in situ mechanical manipulation is required. In addition, this work represents a milestone on the way to protein‐based and biocompatible smart and interactive mechanical materials.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the European Research Council (ERC Advanced Grant SUPRABIOTICS, Grant No. 694610), Chinese Academy of Sciences President's International Fellowship Initiative (Grant No. 2018VBA0008), the Scientific Instrument Developing Project of the Chinese Academy of Sciences (Grant No. ZDKYYQ20180001), K. C. Wong Education Foundation (GJTD‐2018‐09), the National Natural Science Foundation of China (Grant No. 21704099, 21877104, 21834007, and 21907088), the National Key R&D Program of China (2018YFA0902600). J.S. is grateful for financial support from the China Scholarship Council (grant number 201507720025). R.G. is grateful for support by a Freigeist‐Fellowship of the Volkswagen Foundation (No. 92888).
J. Sun, C. Ma, S. Maity, F. Wang, Y. Zhou, G. Portale, R. Göstl, W. H. Roos, H. Zhang, K. Liu, A. Herrmann, Angew. Chem. Int. Ed. 2021, 60, 3222.
Contributor Information
Prof. Kai Liu, Email: kailiu@tsinghua.edu.cn.
Prof. Andreas Herrmann, Email: herrmann@dwi.rwth-aachen.de.
References
- 1. Ashutosh Tiwari H. K., Responsive Materials and Methods State-of-the-Art Stimuli- Responsive Materials and Their Applications, Wiley-Scrivener, New York, 2013, pp. 3–104. [Google Scholar]
- 2. Theato P., Sumerlin B. S., O'Reilly R. K., T. H. Epps III , Chem. Soc. Rev. 2013, 42, 7055. [DOI] [PubMed] [Google Scholar]
- 3. Cychosz K. A., Thommes M., Engineering 2018, 4, 559–566. [Google Scholar]
- 4. Roy D., Brooks W. L. A., Sumerlin B. S., Chem. Soc. Rev. 2013, 42, 7214–7243. [DOI] [PubMed] [Google Scholar]
- 5. Simon Y. C., Craig S. L., Mechanochemistry in Materials, Royal Society Of Chemistry, Cambridge, 2017. [Google Scholar]
- 6. Goulet-Hanssens A., Eisenreich F., Hecht S., Adv. Mater. 2020, 32, 1905966. [DOI] [PubMed] [Google Scholar]
- 7. Yang Z., Wei J., Sobolev Y. I., Grzybowski B. A., Nature 2018, 553, 313–318. [DOI] [PubMed] [Google Scholar]
- 8. Nakayama M., Kajiyama S., Kumamoto A., Nishimura T., Ikuhara Y., Yamato M., Kato T., Nat. Commun. 2018, 9, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huck N. P. M., Jager W. F., de Lange B., Feringa B. L., Science 1996, 273, 1686–1688. [Google Scholar]
- 10. Mura S., Nicolas J., Couvreur P., Nat. Mater. 2013, 12, 991–1003. [DOI] [PubMed] [Google Scholar]
- 11. Qian Z., Kang S., Rajaram V., Cassella C., McGruer N. E., Rinaldi M., Nat. Nanotechnol. 2017, 12, 969–973. [DOI] [PubMed] [Google Scholar]
- 12. Van Oosten C. L., Bastiaansen C. W. M., Broer D. J., Nat. Mater. 2009, 8, 677–682. [DOI] [PubMed] [Google Scholar]
- 13. Li C., Iscen A., Sai H., Sato K., Sather N. A., Chin S. M., Álvarez Z., Palmer L. C., Schatz G. C., Stupp S. I., Nat. Mater. 2020, 19, 900–909. [DOI] [PubMed] [Google Scholar]
- 14. Hou L., Zhang X., Cotella G. F., Carnicella G., Herder M., Schmidt B. M., Pätzel M., Hecht S., Cacialli F., Samorì P., Nat. Nanotechnol. 2019, 14, 347–353. [DOI] [PubMed] [Google Scholar]
- 15. Gerkman M. A., Gibson R. S. L., Calbo J., Shi Y., Fuchter M. J., Han G. G. D., J. Am. Chem. Soc. 2020, 142, 8688–8695. [DOI] [PubMed] [Google Scholar]
- 16. Kadem L. F., Holz M., Suana K. G., Li Q., Lamprecht C., Herges R., Selhuber-Unkel C., Adv. Mater. 2016, 28, 1799–1802. [DOI] [PubMed] [Google Scholar]
- 17. Fuhrmann A., Göstl R., Wendt R., Kötteritzsch J., Hager M. D., Schubert U. S., Brademann-Jock K., Thünemann A. F., Nöchel U., Behl M., et al., Nat. Commun. 2016, 7, 13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dugave C., Demange L., Chem. Rev. 2003, 103, 2475–2532. [DOI] [PubMed] [Google Scholar]
- 19. White T. J., Broer D. J., Nat. Mater. 2015, 14, 1087–1098. [DOI] [PubMed] [Google Scholar]
- 20. Aßhoff S. J., Lancia F., Iamsaard S., Matt B., Kudernac T., Fletcher S. P., Katsonis N., Angew. Chem. Int. Ed. 2017, 56, 3261–3265; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3309–3313. [Google Scholar]
- 21. Molla M. R., Rangadurai P., Antony L., Swaminathan S., De Pablo J. J., Thayumanavan S., Nat. Chem. 2018, 10, 659–666. [DOI] [PubMed] [Google Scholar]
- 22. Iamsaard S., Aßhoff S. J., Matt B., Kudernac T., Cornelissen J. J. L. M., Fletcher S. P., Katsonis N., Nat. Chem. 2014, 6, 229–235. [DOI] [PubMed] [Google Scholar]
- 23. Iwaso K., Takashima Y., Harada A., Nat. Chem. 2016, 8, 625–632. [DOI] [PubMed] [Google Scholar]
- 24. Conjugated T. M., Langer R., Nature 2005, 434, 879–882. [DOI] [PubMed] [Google Scholar]
- 25. Wu Z., Ji C., Zhao X., Han Y., Müllen K., Pan K., Yin M., J. Am. Chem. Soc. 2019, 141, 7385–7390. [DOI] [PubMed] [Google Scholar]
- 26. Ichimura K., Oh S. K., Nakagawa M., Science 2000, 288, 1624–1626. [DOI] [PubMed] [Google Scholar]
- 27. Baroncini M., D'Agostino S., Bergamini G., Ceroni P., Comotti A., Sozzani P., Bassanetti I., Grepioni F., Hernandez T. M., Silvi S., et al., Nat. Chem. 2015, 7, 634–640. [DOI] [PubMed] [Google Scholar]
- 28. He J., Aggarwal K., Katyal N., He S., Chiang E., Dunning S. G., Reynolds J. E., Steiner A., Henkelman G., Que E. L., et al., J. Am. Chem. Soc. 2020, 142, 6467–6471. [DOI] [PubMed] [Google Scholar]
- 29. Verploegen E., Soulages J., Kozberg M., Zhang T., McKinley G., Hammond P., Angew. Chem. Int. Ed. 2009, 48, 3494–3498; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3546–3550. [Google Scholar]
- 30. Rosales A. M., Mabry K. M., Nehls E. M., Anseth K. S., Biomacromolecules 2015, 16, 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kulikovska O., Goldenberg L. M., Stumpe J., Chem. Mater. 2007, 19, 3343–3348. [Google Scholar]
- 32. Zhang L., Maity S., Liu K., Liu Q., Göstl R., Portale G., Roos W. H., Herrmann A., Small 2017, 13, 1701207. [DOI] [PubMed] [Google Scholar]
- 33. Huang C., Braeckmans K., De Smedt S. C., Chem. Soc. Rev. 2011, 40, 2417–2434. [DOI] [PubMed] [Google Scholar]
- 34. Bai Y., Zhang J., Chen X., ACS Appl. Mater. Interfaces 2018, 10, 14017–14025. [DOI] [PubMed] [Google Scholar]
- 35. Kartha K. K., Allampally N. K., Politi A. T., Prabhu D. D., Ouchi H., Albuquerque R. Q., Yagai S., Fernández G., Chem. Sci. 2019, 10, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshino B. T., Kondo M., Mamiya J., Kinoshita M., Yu Y., Ikeda T., Adv. Mater. 2010, 22, 1361–1363. [DOI] [PubMed] [Google Scholar]
- 37. Grigoryev A., Sa V., Gopishetty V., Tokarev I., Kornev K. G., Minko S., Adv. Funct. Mater. 2013, 23, 5903–5909. [Google Scholar]
- 38. Shang L., Wang Y., Yu Y., Wang J., Zhao Z., Xu H., Zhao Y., J. Mater. Chem. A 2017, 5, 15026–15030. [Google Scholar]
- 39. Sun J., Su J., Ma C., Göstl R., Herrmann A., Liu K., Zhang H., Adv. Mater. 2020, 32, 1906360. [DOI] [PubMed] [Google Scholar]
- 40. Veeregowda D. H., Kolbe A., Van Der Mei H. C., Busscher H. J., Herrmann A., Sharma P. K., Adv. Mater. 2013, 25, 3426–3431. [DOI] [PubMed] [Google Scholar]
- 41. Kolbe A., Mercato L., Abbasi A. Z., Gil P. R., Gorzini S. J., Huibers W. H. C., Poolman B., Parak W. J., Herrmann A., Macromol. Rapid Commun. 2011, 32, 186–190. [DOI] [PubMed] [Google Scholar]
- 42. He H., Yang C., Wang F., Wei Z., Shen J., Chen D., Fan C., Zhang H., Liu K., Angew. Chem. Int. Ed. 2020, 59, 4344–4348; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 4374–4378. [Google Scholar]
- 43. Ma C., Su J., Li B., Herrmann A., Zhang H., Liu K., Adv. Mater. 2020, 32, 1907697. [DOI] [PubMed] [Google Scholar]
- 44. Li Y., Li J., Sun J., He H., Li B., Ma C., Liu K., Zhang H., Angew. Chem. Int. Ed. 2020, 59, 8148–8152; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 8225–8229. [Google Scholar]
- 45. Merino E., Ribagorda M., Beilstein J. Org. Chem. 2012, 8, 1071–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Snijder J., Uetrecht C., Rose R. J., Sanchez-Eugenia R., Marti G. A., Agirre J., Guérin D. M. A., Wuite G. J. L., Heck A. J. R., Roos W. H., Nat. Chem. 2013, 5, 502–509. [DOI] [PubMed] [Google Scholar]
- 47. Gebbie M. A., Wei W., Schrader A. M., Cristiani T. R., Dobbs H. A., Idso M., Chmelka B. F., Waite J. H., Israelachvili J. N., Nat. Chem. 2017, 9, 473–479. [DOI] [PubMed] [Google Scholar]
- 48. Ma J. C., Dougherty D. A., Chem. Rev. 1997, 97, 1303–1324. [DOI] [PubMed] [Google Scholar]
- 49. Kumar K., Woo S. M., Siu T., Cortopassi W. A., Duarte F., Paton R. S., Chem. Sci. 2018, 9, 2655–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary