

Abstract
The outbreak of Coronavirus infection (COVID-19) has prompted the World Health Organisation (WHO) to declare the outbreak, a Public Health Emergency of International concern. As part of the efforts to discover lead compounds for clinical use, 53 molecules were screened using molecular docking and dynamic simulations (MDS) techniques to identify potential inhibitors of SARS-CoV-2 spike protein (COVID-19 Sgp) and main protease (COVID-19 Mpro) or both. Lopinavir (LPV), nelfinavir (NEF), hydroxychloroquine (HCQ), remdesivir (RDV) and an irreversible inhibitor of SARS-CoV (N3) were used as standard drugs for COVID-19 Mpro, while zafirlukast (ZFK) and cefoperazone (CSP)) as standard drugs for COVID-19 Sgp. After 100 ns of MDS, with reference to standard drugs (N3, −52.463 Kcal/mol, NEF, −51.618 Kcal/mol, RDV, −48.780 Kcal/mol, LPV, −46.788 Kcal/mol, DRV, −33.655 Kcal/mol and HCQ, −21.065 Kcal/mol), five molecules, HCR, GRN, C3G, EGCG, and K7G were predicted to be promising inhibitors of COVID-19 Mpro with binding energies of −53.877 kcal/mol, −50.653 Kcal/mol, −48.600 kcal/mol, −47.798 kcal/mol and −46.902 kcal/mol, respectively. These lead molecules were then docked at receptor-binding domain (RBD) of COVID-19 Sgp to examine their inhibitory effects. C3G, GRN and K7G exhibited higher binding energies of −42.310 kcal/mol, −32.210 kcal/mol, −26.922 kcal/mol than the recorded values for the reference drugs (CSP, −35.509 kcal/mol, ZFK, −24.242 kcal/mol), respectively. The results of the binding energy and structural analyses from this study revealed that C3G, GRN and K7G could serve as potential dual inhibitors of COVID-19 Sgp and COVID-19 Mpro, while HCR and EGCG would be inhibitors of COVID-19 Mpro.
Communicated by Ramaswamy H. Sarma
Keywords: Coronavirus infection, molecular dynamic simulations, spike (S) glycoprotein, main protease enzyme, inhibitors
Introduction
Coronavirus disease (COVID-19) remains one of the most severe and current outbreak culminating into global pandemic. COVID-19 is caused by the infection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (WHO, 2020). SARS-CoV-2 belongs to the same class of other coronaviruses (Family-Coronaviridae) such as Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) that have been implicated in respiratory and neurological diseases (de Wit et al., 2016; Shen et al., 2020). The first case of COVID-19 was reported in Wuhan, Hubei province of China on December 31, 2019 and has since spread across all continents of the world (Zhu et al., 2020; Li et al., 2020). As of September 19, 2020, over 30.5 million confirmed cases had been reported, with more than 950,000 deaths confirmed world-wide (https://coronavirus.jhu.edu/map.html).
SARS-CoV-2 infected patients may be asymptomatic, while those with confirmed cases showed common symptoms such as fever, cough, fatigue, and shortness of breath (CDC, 2020; Chen et al., 2020). The report of the WHO-China joint mission on COVID-19 has shown that 88%, 68%, 38% and 33% of infected patients showed fever, dry cough, fatigue, and sputum production symptoms, respectively (WHO, 2020). The outbreak of the disease is currently the world’s main health challenge, which has prompted WHO to declare the outbreak a Public Health Emergency of International Concern (WHO, 2020).
As the world is facing the scourge of COVID-19 outbreak, it is, therefore, a matter of urgency and necessity to develop or repurpose drugs or therapeutics to cure the disease. Although, there is currently no specific and permanent treatment for COVID-19, however, preventive, and supportive therapies are being implemented for its management to avert further complications and pathological damage (Rodríguez-Morales et al., 2020). Researchers are working towards the development of effective treatments, and there have been reports that existing drugs, such as anti-malarials (chloroquine and hydroxychloroquine), antiretrovirals (Lopinavir–ritonavir) and Ebola drugs (remdesivir) could be effective against COVID-19 (Cao et al., 2020; CDC, 2020; Wang et al., 2020). An in silico study on four drugs (praziquantel, perampanel, pitavastatin and nelfinavir) against COVID-19 main protease enzyme (COVID-19 Mpro) showed that nelfinavir (NEF) exhibited the most promising and best inhibitory activity against the enzyme (Xu et al., 2020). Studies have also reported drugs/compounds such as zafirlukast, cefoperazone, Hesperidin, Curcumin and andrographolide as potential inhibitors of COVID-19 Sgp (Basu et al., 2020; Maurya et al., 2020; Senathilake et al., 2020). Several other studies, including clinical trials on vaccines and drugs, are ongoing.
The COVID-19 Sgp is vital in the entry mechanism of SARS-CoV-2 into the host cell. SARS-CoV-2 binds to the angiotensin-converting enzyme 2 (ACE-2) receptor of the host cell using the COVID-19 Sgp (Wrapp et al., 2020). The receptor-binding domain (RBD) of COVID-19 Sgp is used in binding with the ACE-2 receptor on the host cell to form the needed complex of RBD-ACE-2 that facilitate the viral entry (Wrapp et al., 2020). High-affinity binding of the RBD-ACE-2 has been suggested to be one of the factors enhancing the quick and widespread of the disease (Wrapp et al., 2020). Likewise, COVID-19 Mpro is essential in viral replication and transcription (Yang et al., 2003; Pillaiyar et al., 2016). Jin et al. (2020) further reported that the COVID-19 Mpro through extensive proteolytic processing is responsible for the release of functional polypeptides from the virus polyproteins. The COVID-19 Mpro and COVID-19 Sgp are, therefore, attractive, and invaluable therapeutic targets for the development of drug candidates for COVID-19 treatment.
Inhibition of the COVID-19 Sgp and COVID-19 Mpro will lead to a reduction in the spread of the infection to vulnerable hosts or cells and production of non-infectious virions. As part of the efforts to discover novel lead compounds for clinical use, this study employed combined structure-based drug design and molecular dynamic simulations to identify new leads that could target either the spike (S) glycoprotein (COVID-19 Sgp) and COVID-19 Mpro or both. Fifty-three molecules with reported antiviral activities were screened as potential inhibitors of COVID-19 Sgp and COVID-19 Mpro or dual inhibitors. Antiretroviral drugs (lopinavir, nelfinavir and darunavir), anti-malaria drug (hydroxychloroquine), Ebola drug (remdesivir) and an irreversible inhibitor, N3 (peptide-like inhibitor) of SARS-CoV and MERS-CoV, (Liu et al., 2020; Ren et al., 2013; Wang et al., 2016) were used as standard drugs for COVID-19 Mpro, while for COVID-19 Sgp, the anti-asthmatic drug (zafirlukast) and cefoperazone were used as standard drugs (Senathilake et al., 2020).
Methods
Proteins (COVID-19 sgp and COVID-19 mpro) acquisition and preparation
The X-ray crystal structures of the COVID-19 Mpro (PDB codes: 6LU7) and COVID-19 Sgp (PDB code: 6LZG) were obtained from the RSCB Protein Data Bank (Burley et al. 2018; Jun et al., 2020). The structures of the two proteins were then prepared on the UCSF Chimera software package (Yang et al., 2012). The structure of the proteins were prepared removing water molecules, nonstandard naming, protein residue connectivity. The missing atoms of sidechains and protein backbone were added in the protein structure before the molecular docking.
Ligand acquisition and preparation
The standard drugs nelfinavir (NEF), lopinavir (LPV), remdesivir (RDV), N3, hydroxychloroquine (HCQ), zafirlukast (ZFK) and cefoperazone (CSP) as well as the test molecules, were accessed from PubChem (Kim et al., 2016) and the 3-D structures prepared on the Avogadro software package (Hanwell et al., 2012).
Molecular docking
The molecular docking software utilised in this study was the Autodock available on Chimera (Grosdidier et al., 2011), with default docking parameters. Before docking, Gasteiger charges were added to the molecules, and the non-polar hydrogen atoms were merged to carbon atoms. The molecules were then docked into the binding pocket of the proteins; COVID-19 Mpro and COVID-19 Sgp by defining the grid box with a spacing of 1 Å each and size (27 × 20 × 30) and (17 × 43 × 27) pointing in x, y and z directions, respectively. The standard drug systems, as well as the 11 molecules with the best docking scores, were then subjected to molecular dynamics simulations. The best five molecules with good binding free energy were subsequently docked into the RBD of COVID-19 Sgp (Figures 1 and 2).
Figure 1.

Amino acid residues at the catalytic site of the COVID-19 Mpro (A) and Receptor binding domain of COVID-19 Sgp (B).
Figure 2.

Superpositions of the crystalized structures of (A) inhibitor (N3) of COVID-19 Mpro (in red) with other ligands, and (B) ligands at the Receptor binding domain of COVID-19 Sgp with their respective RMSD values.
Molecular dynamic (MD) simulations
The MD simulation was performed as described by Idowu et al. (2019). The simulations were performed using the GPU version provided with the AMBER package (AMBER 18), in which the FF18SB variant of the AMBER force field (Nair & Miners, 2014) was used to describe the systems.
ANTECHAMBER was used to generate atomic partial charges for the ligand by utilising the Restrained Electrostatic Potential (RESP) and the General Amber Force Field (GAFF) procedures. The Leap module of AMBER 18 allowed for the addition of hydrogen atoms, as well as Na + and Cl- counter ions to COVID-19 Mpro and COVID-19 Sgp, respectively to neutralise all systems. The amino acids were numbered, numbering residues 1-303 for COVID-19 Mpro and 1-223 for COVID-19 Sgp. The systems were then suspended implicitly within an orthorhombic box of TIP3P water molecules such that all atoms were within 8 Å of any box edge (Jorgensen et al., 1983).
An initial minimization of 2000 steps were carried out with an applied restraint potential of 500 kcal/mol for both solutes, were performed for 1000 steps using the steepest descent method followed by 1000 steps of conjugate gradients. An additional full minimization of 1000 steps were further carried out using the conjugate gradient algorithm without restraint. A gradual heating MD simulation from 0 K to 300 K was executed for 50 ps, such that the systems maintained a fixed number of atoms and fixed volume. The solutes within the systems were imposed with a potential harmonic restraint of 10 kcal/mol and collision frequency of 1.0 ps. Following heating, an equilibration estimating 500 ps of each system was conducted; the operating temperature was kept constant at 300 K. Additional features such as several atoms and pressure were also kept constant mimicking an isobaric-isothermal ensemble. The system's pressure was maintained at 1 bar using the Berendsen barostat (Basconi & Shirts, 2013; Gonnet, 2007).
The total time for the MD simulations conducted were 100 ns. In each simulation, the SHAKE algorithm was employed to constrict the bonds of hydrogen atoms (Ryckaert et al., 1977). The step size of each simulation was 2fs, and an SPFP precision model was used. The simulations coincided with the isobaric-isothermal ensemble (NPT), with randomized seeding, the constant pressure of 1 bar maintained by the Berendsen barostat (Basconi & Shirts, 2013), a pressure-coupling constant of 2 ps, a temperature of 300 K and Langevin thermostat (Izaguirre et al., 2001) with a collision frequency of 1.0 ps.
Post-dynamic analysis
Analysis of root mean square deviation (RMSD), root means square fluctuation (RMSF), solvent accessible surface area (SASA) and radius of gyration (RoG) was done using the CPPTRAJ module employed in the AMBER 18 suit. All raw data plots were generated using the Origin data analysis software (Seifert, 2014).
Binding free energy calculations
To estimate and compare the binding affinity of the systems, the free binding energy was calculated using the Molecular Mechanics/GB Surface Area method (MM/GBSA) (Ylilauri & Pentikäinen, 2013). Binding free energy was averaged over 100000 snapshots extracted from the 100 ns trajectory. The free binding energy (ΔG) computed by this method for each molecular species (complex, ligand, and receptor) can be represented as:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
The term Egas denotes the gas-phase energy, which consists of the internal energy Eint; Coulomb energy Eele and the van der Waals energies Evdw. The Egas was directly estimated from the FF14SB force field terms. Solvation free energy, Gsol, was estimated from the energy contribution from the polar states, GGB, and non-polar states, G. The non-polar solvation energy, SA. GSA, was determined from the solvent-accessible surface area (SASA), using a water probe radius of 1.4 Å. In contrast, the polar solvation, GGB, the contribution was estimated by solving the GB equation. S and T denote the total entropy of the solute and temperature, respectively.
Pharmacokinetic properties analysis
For the calculation/prediction of the pharmacokinetic properties of the lead compounds and the standard drugs, SwissADME server was employed (Daina et al., 2017). The server predicts the target of small molecules.
Results and discussion
Molecular docking scores for COVID-19 mpro
Fifty-three molecules and five FDA-approved drugs were docked into the active site of COVID-19 Mpro to calculate the affinity of the molecules for the enzyme. The results of the molecular docking analysis for all the molecules are presented in Table S1 (Supplementary materials). Docking score is a measure of the fitness of the molecule into the catalytic active site pocket of an enzyme, and the more negative the value, the better the fitness of the molecule (Idowu et al., 2020).
Table 1.
2D structure and docking scores of the standard drugs against COVID-19 Mpro.
| Molecules | 2D Structure | Docking Score |
|---|---|---|
| Nelfinavir (NEF) |  |
 |
| Remdesivir (RDV) |  |
 |
| Lopinavir (LPV) | 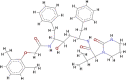 |
 |
| N3 | 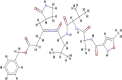 |
 |
| Hydroxychloroquine (HCQ) | 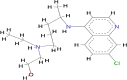 |
 |
The scoring functions permitted the calculation and prediction of the binding affinities of individual molecules, from which the best binding molecules were identified and selected (Abdullahi et al., 2018). Eleven molecules out of the 53 compounds showed better docking scores for COVID-19 Mpro that fall within or higher than the docking scores of the standard drugs except for hydroxychloroquine (HCQ) with the lowest docking score of −5.8 kcal/mol. Kumi et al. (2020), suggested that selected molecules might generally display favourable binding mode within the active catalytic site of the enzyme relative to standard drugs and as well might enhance their interactions and stability within the active site. Tables 1 and 2 showed the results of the respective molecular docking analysis of the standard drugs and the five lead inhibitory molecules against COVID-19 Mpro. The antiretroviral drugs, LPV (-7.1 kcal/mol) and NEF (-7.6 kcal/mol) (HIV protease inhibitors (PIs)) had higher docking scores than the recorded score for HCQ. Remdesivir, an anti-Ebola drug and N3, which had previously been reported to be an inhibitor of COVID-19 Mpro, (Jin et al., 2020) showed docking score values of −7.4 kcal/mol and −6.7 kcal/mol, respectively (Table 1). Nine molecules (BA, CA, EGCG, GRN, HCR, K7G, OAA, P3G, UAA) showed better docking scores ranging from −7.7 kcal/mol to −8.8 kcal/mol, which were higher than the highest binding score for the standard drugs (-7.6 kcal/mol) (Table S1 supplementary materials). Additional two compounds (C3G and MA) exhibited docking scores that fall within the range of the docking scores for the standard drugs. As molecular docking only measures the geometric fitness of molecules at the active site of a protein, molecular dynamics simulations study was carried out on the standard drugs and the ten molecules (molecules with better docking scores) to investigate their binding affinities/energy and structural evaluation.
Table 2.
2D structure and docking scores for the top five hit molecules towards COVID-19 Mpro.
| Molecules | 2D Structure | Docking Score |
|---|---|---|
| Geraniin (GRN) |  |
 |
| 6-Hydroxylcyanidin-3- rutinoside (HCR) |  |
 |
| Epigallocatechin gallate (EGCG) |  |
 |
| Kaempferol-7-glucoside (K7G) |  |
 |
| Cyanindin-3-glucoside (C3G) |  |
 |
Binding energies of the hit molecules and standard drugs for COVID-19 mpro and COVID-19 sgp
In this study, molecular mechanics/generalised born surface area (MMGBSA) computational technique was employed to estimate the binding free energies (ΔGbind) of the standard drugs and the eleven hit molecules towards COVID-19 Mpro, and five lead molecules (molecules with the best binding free energy from COVID-19 Mpro) toward COVID-19 Sgp. Table S2 (Supplementary materials) showed the binding free energies of all the ten molecules. Table 3 showed the results of the binding free energy of the best five molecules toward COVID-19 Mpro, with N3 and NEF molecules exhibiting the highest binding energies of −52.463 kcal/mol and −51.618 kcal/mol among the standard drugs. This agrees with previous studies that reported the inhibitory activities of the two drugs against COVID-19 Mpro. Xu et al., in an in silico study reported NEF to the most promising and potent inhibitor of COVID-19 Mpro among the four tested drugs (praziquantel, perampanel, pitavastatin, and nelfinavir) after it exhibited the best binding energy against the enzyme (Xu et al., 2020). Both in silico and in vitro studies have reported the inhibitory activities of N3 not only against COVID-19 Mpro but also against SARS-CoV and MERS-CoV (Yang et al., 2005; Wang et al., 2016). RDV and LPV showed a relatively similar ΔGbind, but lower than ΔGbind recorded for NEF and N3. HCQ had the lowest ΔGbind of −21.065 kcal/mol, which might suggest that HCQ could not be a protease inhibitor as it exhibits distinct mechanism of action from the mechanism of actions of known FDA-approved protease inhibitors. Although HCQ was listed by Centre for Disease Control (CDC) among drugs use in treating COVID-19, and drugs under clinical trial (CDC, 2020), its mechanism of actions has been reported to include alteration of protein degradation by acidic hydrolases in the lysosome, assembly of macromolecules in the endosomes, and increase intracellular pH (Robert, 1993). This study assumes HCQ is not an inhibitor of COVID-19 Mpro but suggests it inhibits SARS-CoV-2 through any of the mechanisms reported by Robert, 1993. As shown in Table 3, HCR exhibited the highest binding free energies relative to the standard drugs, while GRN had the second-highest value of −50.653 kcal/mol among the tested molecules but lesser than the values recorded for N3 and NEF. EGCG, C3G and K7G exhibited relatively similar binding free energy values comparable to those for RDV and LPV, while P3G exhibited the lowest value of −32.049 kcal/mol.
Table 3.
Thermodynamic Binding Free Energy Profiles for the hit molecules and standard drugs towards COVID-19 Mpro.
| Energy Components (kcal/mol) | |||||
|---|---|---|---|---|---|
| Complex | Δ EvdW | ΔEelec | ΔGgas | ΔGsolv | ΔGbind |
| N3 | −61.685 ± 5.383 | −139.335 ± 17.103 | −201.020 ± 19.795 | 148.556 ± 12.436 | −52.463 ± 4.937 |
| NEF | −55.881 ± 4.046 | −151.517 ± 11.509 | −207.399 ± 11.671 | 155.780 ± 9.653 | −51.618 ± 4.906 |
| RDV | −57.869 ± 3.670 | −21.668 ± 5.969 | −79.537 ± 6.245 | 30.757 ± 4.669 | −48.780 ± 3.804 |
| LPV | −62.806 ± 8.979 | −19.616 ± 3.098 | −82.422 ± 13.158 | 5.634 ± 6.298 | −46.788 ± 7.840 |
| DRV | −46.018 ± 3.040 | −16.176 ± 4.849 | −62.195 ± 5.640 | 28.539 ± 4.232 | −33.655 ± 3.374 |
|
HCQ |
−18.548 ± 0.381 |
−48.905 ± 1.240 |
−53.453 ± 1.596 |
45.388 ± 1.328 |
−21.065 ± 0.293 |
|
Lead Molecules | |||||
| HCR | −56.0056 ± 5.126 | −146.740 ± 16.786 | −202.748 ± 15.220 | 152.870 ± 11.894 | −53.877 ± 4.886 |
| GRN | −54.3287 ± 4.186 | −30.887 ± 6.205 | −85.2167 ± 7.681 | 38.563 ± 4.326 | −50.653 ± 3.140 |
| EGCG | −36.4632 ± 4.615 | −72.369 ± 10.499 | −108.832 ± 9.460 | 62.232 ± 5.336 | −48.600 ± 5.217 |
| C3G | −48.8334 ± 5.099 | −140.163 ± 20.932 | −189.001 ± 20.053 | 143.202 ± 15.368 | −47.798 ± 4.231 |
| K7G | −45.9164 ± 4.053 | −35.158 ± 12.099 | −81.075 ± 10.496 | 37.172 ± 8.693 | −46.902 ± 5.288 |
| P3G | −37.5272 ± 3.485 | −128.148 ± 14.775 | −165.678 ± 15.444 | 133.629 ± 12.379 | −32.049 ± 4.760 |
ΔEele electrostatic energy, ΔEvdW van der Waals energy, ΔGbind total binding free energy, ΔGsol solvation free energy, ΔEgas gas-phase free energy.
From the binding free energy results, HCR, GRN, EGCG, C3G and K7G seemed to be promising and potential inhibitors of COVID-19 Mpro. These five lead molecules were then docked into the RBD of COVID-19 Sgp. Table S3 (supplementary materials) showed the result of the molecular docking analysis against COVID-19 Sgp. C3G had the highest binding free energy of −42.310 kcal/mol higher than the binding energy values of the reference inhibitors (CSP, −35.509 kcal/mol and ZFK, −24.242 kcal/mol) of COVID-19 Sgp. In addition to C3G molecule, K7G and GRN (-32.210 kcal/mol and −26.922 kcal/mol, respectively) had binding energies higher than ZFK but lower than CSP. Consequent upon these data, it could be logically inferred that, C3G, K7G and GRN might possess dual inhibitory activities against COVID-19 Mpro and COVID-19 Sgp. A further probe to examine and establish the mechanistic inhibitory characteristics of the molecules through the interaction between the molecules and the active site amino residues (enzyme-ligand interaction plots) of the proteins (COVID-19 Mpro and COVID-19 Sgp) was performed (Table 4).
Table 4.
Thermodynamic Binding Free Energy Profiles for the molecules and standard drugs towards COVID-19 Sgp.
| Energy Components (kcal/mol) | |||||
|---|---|---|---|---|---|
| Complex | Δ EvdW | ΔEelec | ΔGgas | ΔGsolv | ΔGbind |
| CSP | −46.272 ± 3.763 | −27.793 ± 0.3550 | −74.065 ± 8.3195 | 38.556 ± 6.734 | −35.509 ± 3.368 |
|
ZFK |
−34.010 ± 4.284 |
−13.058 ± 3.432 |
−47.0716 ± 8.102 |
22.829 ± 4.564 |
−24.242 ± 4.372 |
|
Lead Molecules | |||||
| C3G | −45.927 ± 5.534 | −16.494 ± 2.686 | −62.423 ± 4.644 | 20.112 ± 1.459 | −42.310 ± 6.436 |
| K7G | −35.013 ± 3.533 | −33.445 ± 4.295 | −68.459 ± 5.168 | 36.249 ± 3.855 | −32.210 ± 3.218 |
| GRN | −36.302 ± 2.984 | −17.337 ± 4.67 | −53.642 ± 6.4552 | 26.719 ± 4.500 | −26.922 ± 2.926 |
| EGCG | −25.606 ± 2.581 | −16.849 ± 2.878 | −42.455 ± 5.547 | 22.623 ± 3.860 | −19.832 ± 3.385 |
| HCR | −31.520 ± 3.943 | 54.832 ± 6.743 | 23.309 ± 0.648 | −40.648 ± 3.463 | −17.338 ± 3.831 |
ΔEele electrostatic energy, ΔEvdW van der Waals energy, ΔGbind total binding free energy, ΔGsol solvation free energy, ΔEgas gas-phase free energy.
Comparative ligand-receptor interaction profiles of hit molecules and standard drugs
Receptor-ligand interaction examined the molecular interactions between the bound molecules and the amino acid residues at the active sites of the enzyme (Chetty et al., 2016; Idowu et al., 2019). Figures 3–5 showed the 2 D visualisation of the interactions between the molecules and active site residues of COVID-19 Mpro, COVID-19 Sgp and the types of interactions (such as hydrogen bond, π-sigma, π-cation, π-Sulfur, π-alkyl, π-π stacked interaction, donor-donor interactions and Van der Waals (vdW) overlaps) observed in the interaction plots. For COVID-19 Mpro, although, N3 and RDV had the same and the highest number of interactions (20 bonds) (Figure 1), but the binding free energy of N3 is higher than that of RDV. This might be attributed to higher number of der Waals force (12), Hydrogen bond (7) and π-Sulfur bond (1) in N3 than RDV with 10 van der Waals force, 1 Hydrogen bond and no π-Sulphur bond (Figure 3). LPV exhibited the second highest number of interactions among the standard drugs but lower binding free energy than NEF with a total number of 16 bonds. This might be associated with the presence of attractive charge and donor-donor interactions observed between NEF and the active site amino acid residues, which are absent in LPV interaction plot.
Figure 3.

Interaction types and receptor (COVID-19 Mpro)-ligand interactions plots of a) LPV, b) RDV, c) N3, and d) NEF with COVID-19 Mpro, respectively.
Figure 4.

Interaction types and receptor (COVID-19 Mpro)-ligand interactions plots of a) C3G, b) GRN c) EGCG d) K7G e) HCR.
Figure 5.

Interaction types and receptor(COVID-19 Sgp)-ligand interactions plots of a) C3G, b) CSP, c) GRN, d) ZFK, e) K7G.
From the interaction plots for the lead molecules (Figure 4), K7G, EGCG, GRN, and HCR had total number of interactions of 23, 22, 18 and 17 bonds, respectively. Although, HCR with the lowest number of bonds, had the highest binding energy due to the presence of noncovalent and π-π stacked interactions between the aromatic ring of HCR and amino acid residue ARG188. This interaction (π-π stacked) had been previously reported to be important in drug development (Babine & Bender, 1997) and has been used in the development of acetylcholinesterase (AChE) inhibitor for treatment against Alzheimer's disease (da Silva et al., 2006). This noncovalent and π-π stacked interaction was also observed in two of the standard drugs; between the aromatic ring of NEF and amino acid residue HIS 41, and aromatic ring of RDV and amino acid residue HIS 163.
Novel coronavirus' main protease enzyme (COVID-19 Mpro), like other coronaviruses' main protease enzyme, has a Cys–His catalytic dyad (Jin et al., 2020; St. John et al., 2015). Residues HIS 41 and CYS 148 at the active site of MERS protease enzyme have been implicated to be essential in inhibiting the specific activity of the enzyme (St. John et al., 2015). Although, the features of the catalytic binding pocket of main protease enzymes of coronaviruses are conserved (Yang et al., 2005), sequence alignment has shown that the CYS 148 has been replaced with CYS 145 in COVID-19 Mpro (Jin et al., 2020). Furthermore, a study has reported that the HIS 41 and CYS 145 play a vital role and are essential with regards to the inhibitory activity of any potential and promising inhibitors of COVID-19 Mpro (Jin et al., 2020). In this study, the interaction of all the five lead molecules and the standard drugs (except LPV, that interacted with only HIS 41) with both HIS 41 and CYS 145 residues of the enzyme, is a further indication that they might be potent inhibitors of COVID-19 Mpro.
For the COVID-19 Sgp bound systems, C3G has the highest number of interactions (21), which justified its high binding free energy in this study. The two standard drugs had the same number of interactions (12 interactions each), but CSP had a higher binding energy which could be attributed to five carbon-hydrogen bonds in its interaction plot, which was absent in ZFK. GRN and K7G had a total of 9 and 11 interactions, respectively. Three carbon-hydrogen bonds interactions were also observed in C3G. A study on the analysis of the interaction surface of RBD-ACE2 revealed that strong H bonding was involved in the binding of RBD of COVID-19 Sgp to ACE-2 (Xu et al., 2020). This might suggest that H bonding between RBD and a potential inhibitor is crucial for the development of COVID-19 Sgp inhibitor. This could also justify the high binding free energy for C3G, (with a total of 18 H bonds), K7G (10) and CSP (9).
Structural dynamics of bound COVID-19 mpro and COVID-19 sgp systems
Binding of an inhibitor to a specific biological target is usually associated with structural and conformational changes, which could influence the biological activity of the target (Sindhu & Srinivasan, 2015). To establish the stability and proper equilibration of the investigated systems, RMSD, RoG and RMSF of alpha carbon (Cα) atoms were monitored and analysed along with the entire duration of 100 ns of the MD simulation for the apo-enzyme (unbound) and the bound systems. RMSD is a measurement of the systems convergence and stability (Hess, 2002). The deviation produced by a protein during simulation is a factor determining the protein's stability. For the COVID-19 Mpro, all the systems (including the apo-enzyme) achieved convergence between 10-12 ns MD simulation, signifying all the systems reached structural stability and maintained stable conformations after the convergence till the end of the simulation (Figure 6). C3G, EGCG, HCR, K7G and GRN had average RMSD values of 1.249 Å, 1.387 Å, 1.501 Å, 1.511 Å, 1.511 Å and 2.211 Å, while the standard drugs, LPV, NEF, N3 and RDV had average values of 1.861 Å, 1.901 Å, 3.367 Å, and 4.031 Å, respectively (Figure 6). The confirmation of the apo-enzyme (COVID-19 Mpro) that served as control exhibited an average RMSD value of 2.471 Å. The result, therefore, showed that the binding of the five lead molecules, NEF and LPV induced more structural stability on COVID-19 Mpro and this is indicative of promising prospect for the identified lead compounds as COVID-19 Mpro inhibitors. The RMSD for the COVID-19 Sgp systems revealed that all the systems, including the standard drugs, achieved convergence at 20 ns and maintained structural stability and stable conformation (Figure 7a). C3G, K7G and GRN had average RMSD values of 4.217 Å, 4.131 Å, and 4.89 Å, while the standard drugs, CSP and ZFK had average values of 4.414 Å and 3.949, respectively. The binding of the ligands brought more stability to the enzyme, as the average RMSD values of the ligand are lower than the average RMSD of the apo-enzyme (4.821 Å)
Figure 6.

Comparative RMSD plots of alpha C atoms of the COVID-19 Mpro, N3, NFE, RDV and LPV with molecules, a) HCR, b) C3G, c) EGCG, d) K7G and e) GRN systems calculated throughout 100 ns MD simulations.
Figure 7.

Comparative plots of alpha C atoms of the COVID-19 Sgp, N3, NFE, RDV and LPV with molecules, a) RMSD plots b) RoG plots systems calculated throughout 100 ns MD simulations.
To evaluate the overall structural compactness of the enzymes upon binding of ligands, the RoG was also calculated. The alteration of structural compactness of a protein induced by the binding of the ligand could influence the biological activity of the protein (Sindhu & Srinivasan, 2015), and the lower the RoG value, the more stable the system. In the COVID-19 Mpro systems, the apo-enzyme exhibited an average RoG value of 21.841 Å. The lead molecules had marginally lower average RoG values (C3G 21.412 Å, K7G 21.616 Å, HCR 21.873 Å, GRN 21.904 Å and EGCG 22.207 Å) than the standard drugs (NEF; 22.223 Å, LPV; 22.354 Å, RDV; 22.267 Å, and N3; 22.531 Å), which correlated with the RMSD results where all the five lead molecules induced more structural stability and compactness on the enzyme than the standard drugs (Figure 8). The highest RoG value in the COVID-19 Sgp systems was observed with ZFK (22.427 Å), with the lowest RoG value exhibited by K7G (20.523 Å). The three lead molecules (C3G, K7G and GRN) exhibited more structural stability and compactness than the two standard drugs (Figure 7b).
Figure 8.

Comparative RoG plots of alpha C atoms of the COVID-19 Mpro, N3, NEF, RDV and LPV with molecules, a) HCR, b) C3G, c) EGCG, d) K7G and e) GRN systems calculated throughout 100 ns MD simulations.
RMSF is a measure of the effect of the binding of the ligand on the behaviour of the active site residues of a protein. Higher and lower fluctuation values indicate more and less flexible movements, respectively. For the COVID-19 Mpro, the highest average RMSF values were observed in N3 (2.215 Å) and NEF (2.096 Å), followed by LPV (2.002 Å), HCR (1.932 Å), RDV (1.865), GRN (1.821 Å), C3G (1.785 Å) and EGCG (1.801 Å). The lowest value was recorded in K7G systems with an average value of (1.791 Å) (Figure 9). As shown in Figure 10, similar pattern of fluctuations was observed in residues: 41-55, 155-165, 185-195 and 275-290 in all the systems, indicating more flexible movement and unrestricted fluctuations at the residues. The unbound system of COVID-19 Sgp (Apo-enzyme) and K7G exhibited the highest and the lowest residual flexibility with average RMSF values of 4.301 Å, and 2.411 Å, respectively. ZFK, CSP, GRN and C3G had average values of 4.033 Å, 3.874 Å, 3.272 Å and 3.121 Å, respectively. The binding of the ligands (both in the lead molecules and the standard drugs) decreased the enzyme residues flexibility. At residues 165-171 and 217-220 (except for K7G at 165-171), the similarity in the pattern of fluctuations was observed for the unbound system and the molecules (Figure 10).
Figure 9.

RMSF plots of Residue-based average alpha Carbon fluctuations of COVID-19 Mpro, N3, NFE, RDV and LPV with molecules, a) HCR, b) C3G, c) EGCG, d) K7G and e) GRN systems calculated throughout 100 ns MD simulations.
Figure 10.

RMSF plots of Residue-based average alpha Carbon fluctuations of COVID-19 Sgp bound with ZFK, CSP, GRN, K7G and C3G systems calculated throughout 100 ns MD simulation.
Assessing the drug-likeness of the hit molecules and the standard drugs
By employing the SwissADME server, the pharmacokinetic properties of the standard drugs and the lead molecules were assessed (Daina et al., 2017). Yamashita and Hashida (2004), reported that the in-silico absorption, distribution, metabolism, and excretion (ADME) evaluation gives an in vivo overview of the drug interaction, and eventually decrease the risk of disapproval of drug development. The Lipinski's rule of five (Ro5) is a set of rules used to evaluate the drug-likeness of compounds with pharmacological activity. The Ro5 examines both the physical and chemical properties of a compound to ascertain its safety as an orally active drug (Lipinski et al., 2012). The rule stated that the molecular weight of a potential compound should be less than 500 daltons and the lipophilicity of the compound (partition coefficient (logP)) should not be greater than 5. The higher value of logP usually means the probability of a compound to permeate the lipid membrane is low (Remko et al., 2011). As shown in Table 5, the hit molecules and the standard drugs had logP values lesser than 5, indicating they could permeate the lipid bilayers. However, the results of this study further showed that all the standard drugs (except RDV) passed the Ro5, and none of the hit molecules passed the Ro5.
Table 5.
Pharmacokinetic and physicochemical properties of the standard drugs and the lead molecules.
| Molecule | M. Formula | M. Weight (g/mol) |
Lipophilicity (iLOGP) |
Water Solubility |
GIT Absorption |
BBB Permeability |
Bioavailability Score |
Drug likeness (Lipinski) |
|---|---|---|---|---|---|---|---|---|
| NEF | C32H45N3O4S | 567.78 | 4.33 | Poor | Low | No | 0.55 | Yes |
| RDV | C27H35N6O8P | 602.58 | 1.50 | Moderate | Low | No | 0.17 | No (2) |
| LPV | C37H48N4O10 | 628.8 | 3.44 | Poor | High | No | 0.55 | Yes |
| HCQ | C18H26ClN3O | 335.87 | 3.37 | Moderate | High | Yes | 0.55 | Yes |
| ZFK | C31H33N3O6S | 575.68 | 4.66 | Poor | Low | No | 0.55 | Yes |
| CSP | C15H21N3O7S | 387.41 | −1.17 | High | Low | No | 0.11 | Yes |
| EGCG | C22H18O11 | 458.37 | 1.83 | High | Low | No | 0.17 | No (2) |
| K7G | C21H20O11 | 448.38 | 1.55 | High | Low | No | 0.17 | No (2) |
| GER | C30H24O10 | 544.51 | 2.14 | Moderate | Low | No | 0.17 | No (2) |
| C3G | C21H21O11 | 449.38 | −1.16 | High | Low | No | 0.17 | No (2) |
| HCR | C27H31O16 | 611.53 | −2.72 | High | Low | No | 0.17 | No (3) |
The maintenance of optimal concentrations of a drug in the systemic circulation is a dependant factor of the absorption of such drug in the Gastrointestinal tract (GIT). This is therefore important for such a drug to get to its site of action with enough concentration to exert its maximum therapeutic effects (Kremers, 2002). LPV and HCQ are highly absorbed in the GIT, while the other molecules had low absorption rate. The blood-brain barrier (BBB) is a protective gate that prevents the passage of toxic compounds into the brain or the central nervous system (Begley & Brightman, 2011). Compounds with larger molecular weight (higher than 400 Dalton) or not lipid-soluble cannot pass through the BBB, but smaller and lipid-soluble molecules can pass through the BBB (Begley & Brightman, 2011).
Of all the lead molecules and the standard drugs, only HCQ (lower molecular weight of 335.87 g/mol) was predicted to permeate through the BBB. The bioavailability score of a drug is the measurement of the degree or rate of absorption and quantity of a given amount of unchanged drug that goes to the systemic circulation (Heaney, 2001). Intravenously administered drugs have a higher bioavailability score than an orally administered drug. In the calculation of drug dosage, it is an essential pharmacokinetic property that needs to be carefully considered. Higher bioavailability score correlates with a higher optimal concentration of drug in the systemic circulation and enhanced maximum therapeutic effects. NEF, LPV, HCQ and ZFK had the highest bioavailability scores of 0.55, and CSP exhibited the lowest score of 0.11.
Conclusion
Considering the vital roles of COVID-19 Sgp and COVID-19 Mpro in the entry mechanism and replication of SARS-CoV-2, inhibition, and the discovery of potent inhibitors of these proteins are important in the treatment of COVID-19. Therefore, this study employed molecular docking and molecular dynamic simulation techniques to identify potential inhibitors of these proteins. The results of the binding free energy revealed that five molecules (HCR, GRN, K7G, EGCG and C3G) and three molecules (GRN, K7G and C3G) are high affinity binders of COVID-19 Mpro and COVID-19 Sgp, respectively. Structural dynamics analyses of the two proteins upon inhibitor binding, evaluated by the calculation of RMSD, RoG and RMSF of C-α atoms, revealed that binding of five molecules (HCR, GRN, K7G, EGCG and C3G) and three molecules (GRN, K7G and C3G) conferred stability and compactness on the structural architecture of COVID-19 Mpro and COVID-19 Sgp, respectively. GRN, K7G and C3G might possess dual inhibitory activities against COVID-19 Sgp and COVID-19 Mpro. The study further revealed that the lead molecules possess similar pharmacokinetic and physicochemical properties with the FDA-approved standard drugs. These lead molecules will serve as prerequisite towards the development of inhibitors of COVID-19 Sgp and COVID-19 Mpro upon additional in vitro and in vivo evaluations.
Supplementary Material
Acknowledgements
The authors are grateful to our colleagues for their useful contributions to the technical discussions on SARS-CoV-2.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Abdullahi, M., Olotu, F. A., & Soliman, M. E. (2018). Allosteric inhibition abrogates dysregulated LFA-1 activation: Structural insight into mechanisms of diminished immunologic disease. Computational Biology and Chemistry, 73, 49–56. 10.1016/j.compbiolchem.2018.02.002 [DOI] [PubMed] [Google Scholar]
- Babine, R. E., & Bender, S. L. (1997). Molecular recognition of proteinminus signLigand complexes: Applications to drug design. Chemical Reviews, 97(5), 1359–1472. 10.1021/cr960370z [DOI] [PubMed] [Google Scholar]
- Basconi, J. E., & Shirts, M. R. (2013). Effects of temperature control algorithms on transport properties and kinetics in molecular dynamics simulations. Journal of Chemical Theory and Computation, 9(7), 2887–2899. [DOI] [PubMed] [Google Scholar]
- Basu, A., Sarkar, A., & Maulik, U. (2020). Computational approach for the design of potential spike protein binding natural compounds in SARS- CoV2. Pharmacodynamics, 4, 1–22. 10.21203/rs.3.rs-33181/v1[Epub ahead of print]. [DOI] [Google Scholar]
- Begley, D. J., & Brightman, M. W. (2011). Structural and functional aspects of the blood-brain barrier. Peptide Transport and Delivery into the Central Nervous System’, 61(5), 39–78. [DOI] [PubMed] [Google Scholar]
- Burley, S. K., Berman, H. M., Christie, C., Duarte, J. M., Feng, Z., Westbrook, J., Young, J., & Zardecki, C. (2018). RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Science : A Publication of the Protein Society, 27(1), 316–330. 10.1002/pro.3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, B., Wang, Y., Wen, D., Liu, W., Wang, J., Fan, G., Ruan, L., Song, B., Cai, Y., Wei, M., Li, X., Xia, J., Chen, N., Xiang, J., Yu, T., Bai, T., Xie, X., Zhang, L., Li, C., … Wang, C. (2020). A trial of lopinavir–ritonavir in adults hospitalized with severe COVID. New England Journal of Medicine, 382(19), 1787–1799. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centres for Disease Control and Prevention . (2020). Information for clinicians on therapeutic options for COVID-19 patients. Retrieved September 21, 2020, from https://www.cdc.gov/coronavirus/2019-ncov/hcp/therapeutic-options.html
- Chen, N., Zhou, M., Dong, X., Qu, J., Gong, F., Han, Y., Qiu, Y., Wang, J., Liu, Y., Wei, Y., Xia, J., Yu, T., Zhang, X., & Zhang, L. (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. The Lancet), 395(10223), 507–513. 10.1016/S0140-6736(20)30211-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetty, S., Bhakat, S., Martin, A. J. M., & Soliman, M. E. S. (2016). Multi-drug resistance profile of PR20 HIV-1 protease is attributed to distorted conformational and drug binding landscape: Molecular dynamics insights. Journal of Biomolecular Structure & Dynamics, 34(1), 135–151. 10.1080/07391102.2015.1018326 [DOI] [PubMed] [Google Scholar]
- da Silva, C. H. T. P., Campo, V. L., Carvalho, I., & Taft, C. A. (2006). Molecular modeling, docking and ADMET studies applied to the design of a novel hybrid for treatment of Alzheimer's disease. Journal of Molecular Graphics & Modelling, 25(2), 169–175. 10.1016/j.jmgm.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717. 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit, E., van Doremalen, N., Falzarano, D., & Munster, V. J. (2016). SARS and MERS: Recent insights into emerging coronaviruses. Nature Reviews Microbiology, 14(8), 523–534. 10.1038/nrmicro.2016.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnet, P. (2007). P-SHAKE: A quadratically convergent SHAKE. Journal of Computational Physics, 220(2), 740–747. 10.1016/j.jcp.2006.05.032 [DOI] [Google Scholar]
- Grosdidier, A., Zoete, V., & Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res, 39(suppl_2), W270–7. 10.1093/nar/gkr366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeerschd, T., Zurek, E., & Hutchison, G. R. (2012). Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. Journal of Cheminformatics, 4(8), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaney, R. P. (2001). Factors influencing the measurement of bioavailability, taking calcium as a model. The Journal of Nutrition, 131(4 Suppl), 1344S–1348S. 10.1093/jn/131.4.1344S [DOI] [PubMed] [Google Scholar]
- Hess, B. (2002). Convergence of sampling in protein simulations. Physical Review, 65(3), 031910. https://coronavirus.jhu.edu/map.html [DOI] [PubMed] [Google Scholar]
- Idowu, K., Ramharack, P., Nlooto, M., & Gordon, M. (2019). The pharmacokinetic properties of HIV-1 protease inhibitors: A computational perspective on herbal phytochemicals. Heliyon, 5(10), e02565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idowu, K., Ramharack, P., Nlooto, M., & Gordon, M. (2020). Molecular dynamic mechanism(s) of inhibition of bioactive antiviral phytochemical compounds targeting cytochrome P450 3A4 and P-glycoprotein. Journal of Biomolecular Structure and Dynamics, 3, 1221–1233. [DOI] [PubMed] [Google Scholar]
- Izaguirre, J. A., Catarello, D. P., Wozniak, J. M., & Skeel, R. D. (2001). Langevin stabilization of molecular dynamics. The Journal of Chemical Physics, 114(5), 2090–2098. 10.1063/1.1332996 [DOI] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79(2), 926–935. 10.1063/1.445869 [DOI] [Google Scholar]
- Jun, L., Jiwan, G., Jinfang, Y., Sisi, S., Huan, Z., Shilong, F., Qi, Z., Xuanling, S., Qisheng, W., Linqi, Z., & Xinquan, W. (2020). Crystal structure of the 2019-1 nCoV spike receptor-binding domain bound with the ACE2 receptor. 10.1101/2020.02.19.956235 [DOI]
- Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., Han, L., He, J., He, S., Shoemaker, B. A., Wang, J., Yu, B., Zhang, J., & Bryant, S. H. (2016). PubChem substance and compound databases. Nucleic Acids Research, 44(D1), D1202–1213. 10.1093/nar/gkv951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers, P. (2002). In vitro tests for predicting drug-drug interactions: The need for validated procedures. Pharmacology & Toxicology, 91(5), 209–217. 10.1034/j.1600-0773.2002.910501.x [DOI] [PubMed] [Google Scholar]
- Kumi, R. O., Soremekun, O. S., Issahaku, A. R., Fisayo, C. A., Olotu, A., & Soliman, M. E. S. (2020). Exploring the ring potential of 2,4-diaminopyrimidine derivatives towards the identification of novel caspase-1 inhibitors in Alzheimer’s disease therapy. Journal of molecular modeling, 26(68), 1–17. [DOI] [PubMed] [Google Scholar]
- Li, Q., Guan, X., Wu, P., Wang, X., Zhou, L., Tong, Y., Ren, R., Leung, K. S. M., Lau, E. H. Y., Wong, J. Y., Xing, X., Xiang, N., Wu, Y., Li, C., Chen, Q., Li, D., Liu, T., Zhao, J., Liu, M., … Feng, Z. (2020). Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. New England Journal of Medicine, 382(13), 1199–1207. 10.1056/NEJMoa2001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (2012). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 64, 4–17. 10.1016/j.addr.2012.09.019 [DOI] [PubMed] [Google Scholar]
- Liu, C., Zhou, Q., Li, Y., Garner, L. V., Watkins, S. P., Carter, L. J., Smoot, J., Gregg, A. C., Daniels, A. D., Jervey, S., & Albaiu, D. (2020). Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Central Science, 6(3), 315–331. 10.1021/acscentsci.0c00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurya, V. K., Kumar, S., Bhatt, M. L. B., & Saxena, S. K. (2020). Therapeutic development and drugs for the treatment of COVID-19. Nature Public Health Emergency Collection, 109–126. 10.1007/978-981-15-4814-7_10 [DOI] [Google Scholar]
- Nair, P. C., & Miners, J. O. (2014). Molecular dynamics simulations: From structure function relationships to drug discovery. In Silico Pharmacology, 2(4), 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remko, M., Boháč, A., & Kováčiková, L. (2011). Molecular structure, pKa, lipophilicity, solubility, absorption, polar surface area, and blood brain barrier penetration of some antiangiogenic agents. Structural Chemistry, 22(3), 635–648. 10.1007/s11224-011-9741-z [DOI] [Google Scholar]
- Ren, Z., Yan, L., Zhang, N., Guo, Y., Yang, C., Lou, Z., & Rao, Z. (2013). The newly emerged SARS-Like coronavirus HCoV-EMC also has an "Achilles' heel": Current effective inhibitor targeting a 3C-like protease. Protein & Cell, 4(4), 248–250. 249. 10.1007/s13238-013-2841-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19). (2020, February 16–24). World Health Organisation. [Google Scholar]
- Robert, I. F. (1993). Mechanism of action of hydroxychloroquine as antirheumatic drug. Seminar in Arthritis and Rheumatism, 23(2), 82–91. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Morales, P., Alfonso, J., MacGregor, K., Kanagarajah, S., Patel, D. & Schlagenhauf, (2020). Going global – Travel and the 2019 novel coronavirus. Travel Medicine and Infectious Disease, 33, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert, J. P., Ciccotti, G., & Berendsen, H. J. (1977). Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. Journal of Computational Physics, 23(3), 327–341. 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- Seifert, E. (2014). OriginPro 9.1: Scientific data analysis and graphing software-software review. Journal of Chemical Information and Modeling, 54(5), 1552–1552. 10.1021/ci500161d [DOI] [PubMed] [Google Scholar]
- Senathilake, K., Samarakoon, S., & Tennekoon, K. (2020). Virtual screening of inhibitors against spike glycoprotein of 2019 novel corona virus: A drug repurposing approach. 10.20944/preprints202003.0042.v1) [DOI]
- Shen, M., Zhou, Y., Ye, J., Al-Maskri, A. A. A., Kang, Y., Zeng, S., & Cai, S. (2020). Recent advances and perspectives of nucleic acid detection for coronavirus. Journal of Pharmaceutical Analysis, 10(2), 97–101. 10.1016/j.jpha.2020.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindhu, T., & Srinivasan, P. (2015). Exploring the binding properties of agonists interacting with human TGR5 using structural modelling, molecular docking and dynamics simulations. RSC Advances, 5(19), 14202–14213. 10.1039/C4RA16617E [DOI] [Google Scholar]
- St. John, S. E., Tomar, S., Stauffer, S. R., & Mesecar, A. D. (2015). Targeting zoonotic viruses: Structure-based inhibition of the 3C-like protease from bat coronavirus HKU4-The likely reservoir host to the human coronavirus that causes Middle East Respiratory Syndrome (MERS)). Bioorganic & Medicinal Chemistry, 23(17), 6036–6048. 10.1016/j.bmc.2015.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F., Chen, C., Tan, W., Yang, K., & Yang, H. (2016). Structure of main protease from human coronavirus NL63: Insights for wide spectrum anti-coronavirus drug design. Scientific Reports, 6, 22677–22677. 10.1038/srep22677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M., Cao, R., Zhang, L., Yang, X., Liu, J., Xu, M., Shi, Z., Hu, Z., Zhong, W., & Xiao, G. (2020). Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research, 30(3), 269–271. 10.1038/s41422-020-0282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. (2020). Coronavirus disease 2019 (COVID-19): Situation report, 38. https://apps.who.int/iris/handle/10665/331226
- Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C., Abiona, O., Graham, B. S., & McLellan, J. S. (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 367(6483), 1260–1263. 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J., Zhao, S., Teng, T., Abdalla, A. E., Zhu, W., Xie, L., Wang, Y., & Guo, X. (2020). Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses, 12(2), 244. 10.3390/v12020244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita, F., & Hashida, M. (2004). In silico approaches for predicting ADME properties of drugs. Drug Metabolism and Pharmacokinetics, 19(5), 327–338. 10.2133/dmpk.19.327 [DOI] [PubMed] [Google Scholar]
- Yang, H. T., Xie, W., Xue, X., Yang, K., Ma, J., Liang, W., Zhao, Q., Zhou, Z., Pei, D., Ziebuhr, J., & Hilgenfeld, R. (2005). Design of wide-spectrum inhibitors targeting coronavirus main proteases. Plos Biology, 3, 2044–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Yang, M., Ding, Y., Liu, Y., Lou, Z., Zhou, Z., Sun, L., Mo, L., Ye, S., Pang, H., Gao, G. F., Anand, K., Bartlam, M., Hilgenfeld, R., & Rao, Z. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences of the United States of America, 100(23), 13190–13195. 10.1073/pnas.1835675100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., Lasker, K., Schneidman-Duhovny, D., Webb, B., Huang, C. C., Pettersen, E. F., Goddard, T. D., Meng, E. C., Sali, A., & Ferrin, T. E. (2012). UCSF Chimera, MODELLER, and IMP: An integrated modeling system. Journal of Structural Biology, 179(3), 269–278. 10.1016/j.jsb.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylilauri, M., & Pentikäinen, O. T. (2013). MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. Journal of Chemical Information and Modeling, 53(10), 2626–2633. 10.1021/ci4002475 [DOI] [PubMed] [Google Scholar]
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., Tan, W; China Novel Coronavirus Investigating and Research Team. (2020). A novel coronavirus from patients with pneumonia in China, 2019. New England Journal of Medicine, 382(8), 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.