

Abstract
Objective
To investigate the association between obesity‐related traits and risk of rheumatoid arthritis (RA).
Methods
We conducted genetic correlation analysis and a 2‐sample Mendelian randomization (MR) study, using genome‐wide genetic data based on >850,000 individuals of European ancestry. Summary statistics were collected from the largest genome‐wide association study conducted to date for body mass index (BMI; n = 806,810), waist‐to‐hip ratio (WHR; n = 697,734), WHR adjusted for BMI (WHRadjBMI; n = 694,649), and RA (ncase = 14,361, ncontrol = 43,923). We conducted cross‐trait linkage disequilibrium score regression and ρ‐HESS analyses to quantify genetic correlation between pairs of traits (causal overlap). For each obesity‐related exposure, we utilized independent, genome‐wide significant single‐nucleotide polymorphisms (P < 5 × 10−9) as instruments to perform MR analysis (causal relationship). We interrogated the causal relationship both in the general population and in a sex‐specific manner and calculated odds ratios (ORs) and 95% confidence intervals (95% CIs). Sensitivity analyses were performed to validate MR model assumptions.
Results
Despite a negligible overall genetic correlation between the 3 obesity‐related traits and RA, we found significant local genetic correlations at several regions on chromosome 6 (positions 28–29M, 30–35M, and 50–52M), highlighting a shared genetic basis. We further observed an increased risk of RA per SD increment (4.8 kg/m2) in genetically predicted BMI (OR 1.22 [95% CI 1.09–1.37]). The effect was consistent across sensitivity analyses and comparable between sexes (OR 1.22 [95% CI 1.04–1.44] in male subjects and 1.19 [95% CI 1.04–1.36] in female subjects). However, we did not find evidence supporting a causal role of either WHR (OR 0.98 [95% CI 0.84–1.14]) or WHRadjBMI (OR 0.90 [95% CI 0.79–1.04]) in RA.
Conclusion
Genetically predicted BMI significantly increases RA risk. Future studies are needed to understand the biologic mechanisms underlying this link.
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory joint disease that is more prevalent in Nordic countries and among women (1). Although genome‐wide association study meta‐analysis (meta‐GWAS) has revealed >100 RA‐associated genetic loci and large‐scale epidemiologic investigations have identified several environmental risk factors, the mechanistic developmental processes of RA remain incompletely understood (2).
Obesity represents a state of low‐grade inflammation and has been considered as a potential risk factor for RA (3). Three meta‐analyses of longitudinal studies based on >400,000 subjects have jointly demonstrated higher body mass index (BMI) to be significantly associated with a 3–12% increased risk of RA (4, 5, 6). A sex‐specific effect has also been observed, in which BMI heightens RA risk to a greater extent in women, with an odds ratio (OR) of 1.12 per 5 kg/m2 increment (95% confidence interval [95% CI] 1.07–1.18) than in men (OR 0.90 per 5 kg/m2 increment [95% CI 0.81–1.01]) (4). In addition to BMI, researchers have proposed that abdominal obesity, commonly measured by waist circumference (WC) and/or waist‐to‐hip ratio (WHR), can serve as more appropriate indicators of RA risk (7). A significant yet modest elevation of RA risk (2–5%) among individuals with higher WC has been reported (7, 8).
Unlike genetic components, environmental triggers are usually difficult to pinpoint. Results from epidemiologic studies can be affected by measurement error, confounding, and reverse causality. For example, chronic inflammation during the long preclinical course of RA may lead to lifestyle changes that result in altered body composition (9), complicating efforts to elucidate the observed obesity–RA relationship. Mendelian randomization (MR) is a novel statistical tool that uses genetic variants as instrumental variables (IVs) to make causal inferences between exposure(s) and outcome(s), based on the fact that allocation of genetic variants at meiosis is independent of confounders and always prior to disease onset; results are therefore less susceptible to confounding and reverse causation (10). Three important model assumptions need to be satisfied for MR to yield unbiased estimates, i.e., that IVs are robustly associated with exposure (relevance), affect outcome only through exposure (exclusion restriction), and are not associated with confounders in the exposure–outcome relationship (exchangeability) (10).
To date, only 1 MR study of BMI and RA has been reported (11). This study used a small number of index single‐nucleotide polymorphisms (SNPs) (nIV = 65), had limited samples for outcome (nRA = 7,480), and lacked sensitivity analyses to verify model assumptions. Recent genetic discoveries related to several obesity‐related traits including BMI, WHR, and WHR adjusted for BMI (WHRadjBMI), involving hundreds of thousands of participants (12), have provided an unprecedented opportunity to explore a causal relationship between these factors and RA. In the current study, we substantially extended previous findings by incorporating summary statistics from the largest meta‐GWAS conducted on exposures (n > 699,000; nIV > 600) and outcome (nRA = 14,361; ncontrol = 43,923) to date. We performed the analysis both in the general population and in male and female populations separately. In addition, to explore a causal relationship, we quantified genetic correlation, a measure of shared genetic overlap, between obesity‐related traits and RA.
MATERIALS AND METHODS
We carried out the current study using a 2‐sample MR design, which extracts IV–exposure and IV–outcome associations from 2 independent non‐overlapping populations (13) (for a conceptual framework, see Supplementary Figure 1, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract).
IV–exposure
We selected index SNPs from the largest meta‐GWAS conducted to date on human anthropometric traits, meta‐analyzing UK Biobank (UKB) and Genetic Investigation of Anthropometric Traits Consortium data totaling ~700,000 individuals (806,810 participants for BMI, 697,734 for WHR, and 694,649 for WHRadjBMI), all of European ancestry (12). In the original meta‐GWAS, to identify independent genetic association signals, a clumping strategy of P < 5 × 10−9 and a linkage disequilibrium window of ±5 Mb (r2 > 0.05) were first applied to obtain clumping‐based loci, followed by a proximal joint and conditional analysis to determine primary and secondary hits (additional independent signals conditioning on the primary signals) within each clumping‐based locus. The GWAS was conducted both in the general population and by sex.
We performed MR analyses using primary signals only (670 BMI‐associated index SNPs, 316 WHR‐associated index SNPs, and 346 WHRadjBMI‐associated index SNPs) as well as primary and secondary signals combined (806, 382, and 463 index SNPs for BMI, WHR, and WHRadjBMI, respectively). We further extracted IV–exposure associations in men and women separately.
IV–outcome
IV–outcome associations were obtained from a meta‐GWAS of 18 cohorts totaling 14,361 RA cases and 43,923 controls of European ancestry (14) (Supplementary Table 1, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract). To our knowledge, none of the participants in these 18 studies overlapped with participants in the exposure meta‐GWAS.
We matched our IVs (primary and secondary SNPs for the 3 obesity‐related exposures) with summary data from outcome GWAS (RA meta‐GWAS). Finally, 793 SNPs for BMI, 372 SNPs for WHR, and 445 SNPs for WHRadjBMI were available (Supplementary Tables 2–4, http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract). Overall, we were able to capture >96% of IVs, a virtually complete coverage.
Genome‐wide genetic data
MR leverages information on a small number of index SNPs. To quantify genetic correlation between exposures and outcome, we further obtained full sets of summary‐level genome‐wide genetic data (~10 million SNPs) for each of the 3 obesity‐related exposures and for RA.
Statistical analysis
Genetic correlation analyses
To investigate causal overlap, we first assessed genome‐wide pairwise genetic correlation for each exposure and outcome, using linkage disequilibrium score regression (LDSC), an approach that leverages GWAS summary association statistics and LD to estimate genetic correlation (15). Genome‐wide genetic correlations estimated by LDSC measure correlation of SNP effect sizes across all SNPs in the genome. It is possible that even though 2 traits show negligible overall genetic correlation, specific genomic regions contribute to both traits. We therefore examined local genetic correlation using ρ‐HESS, an algorithm that partitions the whole genome into 1,703 regions based on LD patterns in European populations and quantifies correlation between pairs of traits due to genetic variation restricted to specific regions (16). Taking into account multiple tests, P values of 1.1 × 10−3 (0.05/45) and 2.9 × 10−5 (0.05/1,703) were applied as significant thresholds for LDSC and ρ‐HESS, respectively.
Mendelian randomization analyses
To investigate a causal relationship between obesity and RA, we conducted a 2‐sample MR applying several approaches. A random‐effects inverse variance–weighted method (IVW), an MR‐Egger regression, and an MR pleiotropy residual sum and outlier (MR‐PRESSO) test were used. The random‐effects IVW pools the estimate from each genetic variant (IV) and calculates a precise causal estimation assuming all genetic variants are valid, or are invalid in such a way that the overall pleiotropy is balanced to be zero (17). MR‐Egger regression is robust even if all variants are invalid, and its intercept can be adopted as a test of unbalanced pleiotropy (17). MR‐PRESSO detects pleiotropic outlier variants and provides outlier‐corrected estimations (18). Given the major role of the HLA region in immunity, inflammation, and RA etiology (19) and to control for potential pleiotropy, we performed all analysis excluding SNPs from this region (chromosome 6: 2.9–3.3M).
Sensitivity analyses
To verify MR model assumptions, we performed additional sensitivity analyses. For each index SNP, we searched the National Human Genome Research Institute–European Bioinformatics Institute Catalog of Human Genome‐Wide Association Studies for potential associations with confounding traits and conducted analyses with these SNPs excluded. To ascertain whether our estimation was driven by any individual SNP with a large effect, we carried out leave‐one‐out analysis in which we removed one SNP at a time and performed IVW on the remaining SNPs.
The statistical power of our study was calculated using an algorithm described by Brion et al (20). After correction for tests on 3 exposures, associations with P values of <0.017 (0.05/3) were considered statistically significant; P values between 0.017 and <0.05 were considered to be suggestive of significance. All analyses were performed using LDSC software (15), ρ‐HESS software (16), and TwoSampleMR in R 3.6.0 software (21).
RESULTS
Our results indicated that obesity‐related traits were strongly genetically correlated with one another. Within each trait, there was a high degree of shared genetic similarity between men and women ( 0.92 [95% CI 0.90–0.94] for BMI, 0.73 [95% CI 0.69–0.77] for WHR, and 0.66 [95% CI 0.62–0.70] for WHRadjBMI). Across traits, BMI and WHR shared a common genetic basis, with an overall correlation of 0.59 (95% CI 0.55–0.63) ( 0.46 [95% CI 0.41–0.51] in women and 0.69 [95% CI 0.66–0.72] in men). However, for WHRadjBMI, in which the effect of BMI was removed from WHR, we observed minimal correlation with BMI ( ranging from −0.09 to 0.01), as expected. These results illustrate the challenge of studying obesity‐related traits alone and without a genetic context, as their effects are difficult to dissect. We did not find evidence of a shared genetic basis of BMI, WHR, or WHRadjBMI with RA either overall or in the male or the female population. All estimates were close to 0 ( ranging from −0.01 to 0.08, all P > 0.05), suggesting negligible genetic correlations (Figure 1).
Figure 1.
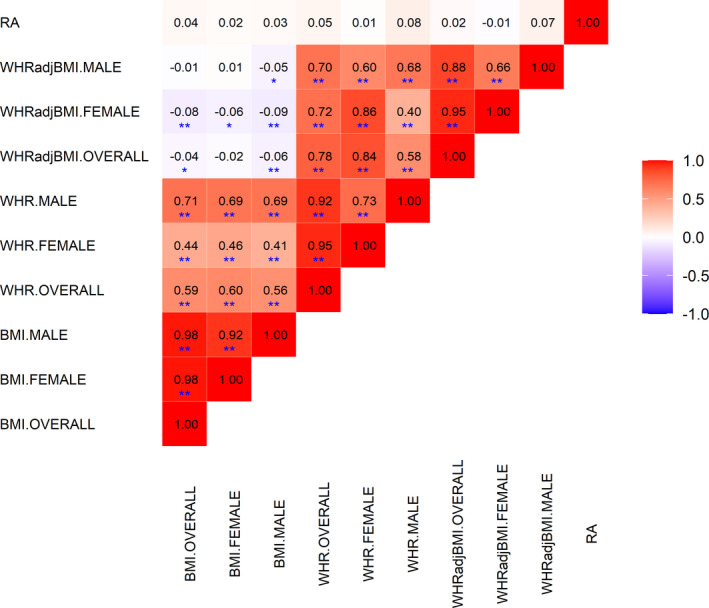
Cross‐trait genetic correlation between obesity‐related traits and rheumatoid arthritis (RA). The point estimate of genetic correlation is shown within each box, with the color of the box representing the magnitude of correlation. * = P < 0.05 (suggestive for statistical significance); ** = P (Bonferroni‐corrected) < 1.1 × 10−3. WHRadjBMI = waist‐to‐hip ratio adjusted for body mass index.
Despite an absence of genome‐wide genetic correlation, obesity‐related exposures and RA might still share pleiotropic risk loci. As shown in Figure 2, when partitioning the genome into 1,703 regions, we observed significant local genetic correlation in several regions of chromosome 6 (positions 28.0–29.0M, 30.7–35.5M, and 50.3–52.3M) and chromosome 19 (positions 9.2–11.3M). When performing separate analyses by sex, we found additional shared regions on chromosomes 8 (positions 75.4–76.5M) and 16 (positions 27.4–29.1M) in women.
Figure 2.
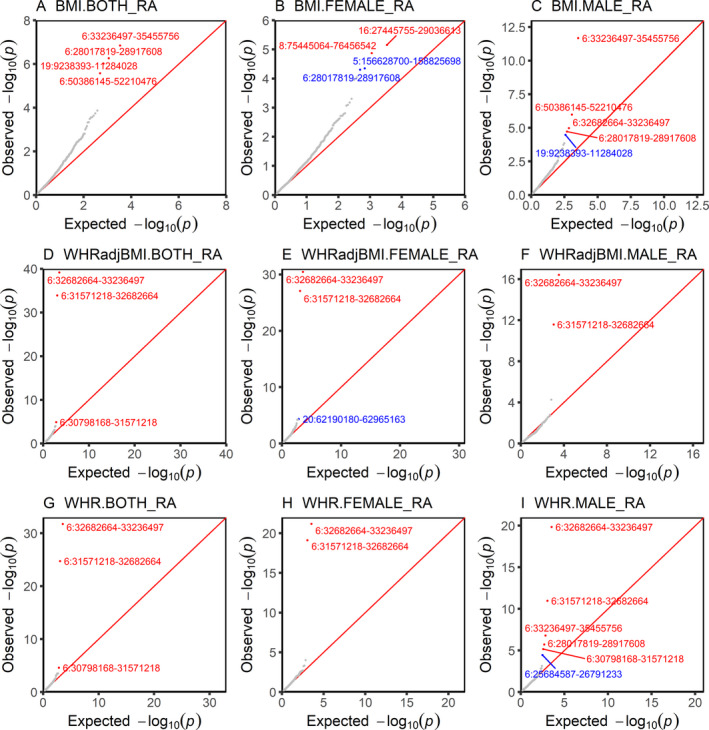
Local genetic correlation between obesity‐related traits and rheumatoid arthritis (RA). Plots show region‐specific P values for the local genetic covariance between RA and body mass index (BMI), waist‐to‐hip ratio (WHR), and WHR adjusted for BMI (WHRadjBMI) overall (both) and in female and male subjects analyzed separately. Each dot presents a specific genomic region. Red indicates significance after multiple corrections (P < 3 × 10−5 [0.05/1,703 regions compared]); blue indicates nominal significance under an arbitrary threshold of P < 5 × 10−5.
Motivated by the findings of local genetic correlations, we explored the causal relationships between different measures of obesity and RA through MR analysis. With our current sample size for outcome (n = 58,284; 24% cases) and assuming phenotypic variance of the exposures explained by IVs to be 4–8% (Supplementary Table 5, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract), our study had sufficient power to detect associations of 15% increase in risk of RA with BMI (98% power), WHR (86% power), and WHRadjBMI (96% power). The power remained acceptable in an analysis restricted to women (which had 80% power to detect a 15% increase in risk of RA associated with BMI or WHR or a 10% increase in risk of RA associated with WHRadjBMI) and decreased only slightly in an analysis restricted to men (80% power to detect a 15% increase in risk of RA associated with BMI or a 25% increase in risk of RA associated with WHR or WHRadjBMI). Since results from our ρ‐HESS analysis revealed significant correlations with the HLA region and given the complexity of that region (19), we performed all MR analysis excluding SNPs from the HLA region.
We found strong evidence supporting the notion of a causal relationship between BMI and RA. As shown in Table 1, we observed a 20% increased risk of RA per SD increment (4.8 kg/m2) in BMI (OR using random‐effects IVW 1.22 [95% CI 1.09–1.37). The effect was not altered in analyses using MR‐PRESSO (OR 1.20 [95% CI 1.07–1.34 excluding 1 outlier) and was only slightly attenuated in analyses using MR‐Egger (OR 1.13 [95% CI 0.83‐1.54), but without any apparent sign of pleiotropy (P for MR‐Egger intercept = 0.43). Sensitivity analysis with additional removal of SNPs associated with potential confounding traits, including smoking, education level, type 2 diabetes, and others (Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract) revealed similar results of even more pronounced magnitude (Table 2) (OR [95% CI] 1.27 [1.12–1.45], 1.44 [0.94–2.22], and 1.24 [1.10–1.41] in analyses using IVW, MR‐Egger, and MR‐PRESSO, respectively). Leave‐one‐out analysis demonstrated that the observed risk effect was not driven by outlying variants (Supplementary Table 6, http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract).
Table 1.
Association between genetically predicted obesity‐related traits and risk of rheumatoid arthritis in a primary analysis excluding SNPs located in the HLA region*
| Parameter, MR approach | SNPs at P < 5 × 10−9 (primary + secondary GWAS hits) | SNPs at P < 5 × 10−9 (primary GWAS hits) | ||||
|---|---|---|---|---|---|---|
|
OR (95% CI) |
P | P for heterogeneity or pleiotropy |
OR (95% CI) |
P | P for heterogeneity or pleiotropy | |
| BMI (n = 790/660) | ||||||
| Random‐effects IVW | 1.22 (1.09–1.37) | 6.1 × 10−4 | <0.001 | 1.21 (1.08–1.37) | 1.6 × 10−3 | <0.001 |
| MR‐Egger | 1.13 (0.83–1.54) | 0.43 | 0.60 | 1.11 (0.81–1.54) | 0.51 | 0.57 |
| MR‐PRESSO (detected outliers 1/NA) | 1.20 (1.07–1.34) | 1.5 × 10−3 | – | 1.21 (1.08–1.37) | 1.7 × 10−3 | – |
| WHR (n = 369/310) | ||||||
| Random‐effects IVW | 0.98 (0.84–1.14) | 0.78 | <0.001 | 0.96 (0.82–1.13) | 0.62 | <0.001 |
| MR‐Egger | 0.91 (0.60–1.37) | 0.66 | 0.71 | 0.96 (0.62–1.49) | 0.85 | 0.99 |
| MR‐PRESSO (detected outliers 2/2) | 0.93 (0.81–1.07) | 0.31 | – | 0.92 (0.79–1.08) | 0.32 | – |
| WHRadjBMI (n = 441/335) | ||||||
| Random‐effects IVW | 0.90 (0.79–1.04) | 0.15 | <0.001 | 0.85 (0.73–0.98) | 0.02 | <0.001 |
| MR‐Egger | 0.92 (0.68–1.25) | 0.60 | 0.90 | 0.99 (0.71–1.38) | 0.95 | 0.31 |
| MR‐PRESSO (detected outliers 5/3) | 0.86 (0.77–0.97) | 0.01 | – | 0.83 (0.72–0.94) | 5.6 × 10−3 | – |
N values (number of single‐nucleotide‐polymorphisms [SNPs]) for body mass index (BMI), waist‐to‐hip ratio (WHR), and WHR adjusted for BMI (WHRadjBMI) and numbers of detected outliers in the Mendelian randomization pleiotropy residual sum and outlier (MR‐PRESSO) analyses are for the analysis of primary + secondary genome‐wide association study (GWAS) hits/analysis of primary SNPs only. OR = odds ratio; 95% CI = 95% confidence interval; IVW = inverse variance–weighted method; NA = not applicable.
Table 2.
Association between genetically predicted obesity‐related traits and risk of rheumatoid arthritis in a sensitivity analysis excluding genetic instruments in the HLA region or associated with confounding traits*
| Parameter, MR approach | SNPs at P < 5 × 10−9 (primary + secondary GWAS hits) | Primary SNPs at P < 5 × 10−9 | ||||
|---|---|---|---|---|---|---|
|
OR (95% CI) |
P | P for heterogeneity or pleiotropy |
OR (95% CI) |
P | P for heterogeneity or pleiotropy | |
| BMI (n = 696/582) | ||||||
| Random‐effects IVW | 1.27 (1.12–1.45) | 2.4 × 10−4 | <0.001 | 1.28 (1.11–1.46) | 4.5 × 10−4 | <0.001 |
| MR‐Egger | 1.44 (0.94–2.22) | 0.10 | 0.55 | 1.44 (0.88–2.33) | 0.14 | 0.62 |
| MR‐PRESSO (detected outliers 1/2) | 1.24 (1.10–1.41) | 6.9 × 10−4 | – | 1.30 (1.14–1.49) | 1.2 × 10−4 | – |
| WHR (n = 307/255) | ||||||
| Random‐effects IVW | 1.05 (0.87–1.26) | 0.61 | <0.001 | 1.03 (0.86–1.24) | 0.75 | 2.0 × 10−3 |
| MR‐Egger | 0.94 (0.54–1.63) | 0.82 | 0.68 | 1.13 (0.61–2.11) | 0.70 | 0.76 |
| MR‐PRESSO (detected outliers 2/1) | 1.02 (0.87–1.21) | 0.77 | – | 1.05 (0.88–1.26) | 0.58 | – |
| WHRadjBMI (n = 348/258) | ||||||
| Random‐effects IVW | 0.92 (0.78–1.09) | 0.34 | <0.001 | 0.85 (0.71–1.01) | 0.06 | 0.01 |
| MR‐Egger | 0.98 (0.66–1.46) | 0.93 | 0.74 | 1.19 (0.72–1.97) | 0.49 | 0.16 |
| MR‐PRESSO (detected outliers 4/2) | 0.85 (0.74–0.97) | 0.02 | – | 0.80 (0.68–0.94) | 0.01 | – |
N values (number of SNPs) for BMI, WHR, and WHRadjBMI and numbers of detected outliers in the MR‐PRESSO analyses are for the analysis of primary + secondary GWAS hits/analysis of primary SNPs only. See Table 1 for definitions.
In contrast, we did not find any association between genetically predicted WHR and RA, with overall effects that were close to 1.00 and nonsignificant (OR [95% CI] 0.98 [0.84–1.14], 0.91 [0.60–1.37], and 0.93 [0.81–1.07] by IVW, MR‐Egger, and MR‐PRESSO, respectively) (Table 1). Results from sensitivity analyses removing IVs associated with potential confounders remained consistent (OR [95% CI] 1.05 [0.87–1.26], 0.94 [0.54–1.63], and 1.02 [0.87–1.21] by IVW, MR‐Egger, and MR‐PRESSO, respectively) (Table 2). Leave‐one‐out analysis did not identify any outlying variant (Supplementary Table 7, http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract).
Our findings imply a causal role of BMI, but not WHR, in the development of RA. Given the complex interrelationships between obesity‐related measurements and to validate our results, we investigated WHRadjBMI, a residual component of WHR in which the effect of BMI is removed. We anticipated observing a null or even reduced effect of WHRadjBMI on RA after eliminating the positive association of BMI from the null association of WHR. As expected, we did not observe any increased effect of WHRadjBMI on RA, with effect sizes that were either close to or slightly lower than 1.00 (OR [95% CI] 0.90 [0.79–1.04] by IVW, 0.92 [0.68–1.25] by MR‐Egger, and 0.86 [0.77–0.97] by MR‐PRESSO) (Table 1 and Supplementary Table 8, on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41517/abstract).
RA occurs in women 2–3 times more frequently than in men. To better understand this sex disparity, we performed sex‐specific analyses that were restricted to BMI and WHR. As shown in Table 3, we found comparable effects of BMI between men and women, by IVW (OR 1.22 [P = 1.5 × 10−2] and 1.19 [P = 1.2 × 10−2], respectively) and MR‐PRESSO (OR 1.19 [P = 1.4 × 10−2] and 1.19 [P = 1.2 × 10−2] respectively), with no indications of pleiotropy (P for MR‐Egger intercept = 0.62 in men and 0.40 in women). The results were consistent in sensitivity analyses in which IVs associated with potential confounders were removed (OR 1.29 [P = 9.2 × 10−3] and 1.28 [P = 3.0 × 10−3], respectively) (Table 4). For WHR, we found an increased but nonsignificant effect of WHR among men and a slightly reduced effect among women. Our sex‐specific results for WHR should be interpreted with caution given the large variations in point estimates of results from the main and sensitivity analyses (only 79 IVs for WHR in men), and substantial pleiotropy indicated in women (P for MR‐Egger intercept = 0.08 in main analysis, 0.04 in sensitivity analysis).
Table 3.
Association between genetically predicted obesity‐related traits and risk of rheumatoid arthritis by sex, excluding genetic instruments in the HLA region*
| Parameter, MR approach | SNPs at P < 5 × 10−9 (primary + secondary GWAS hits) in men | SNPs at P < 5 × 10−9 (primary + secondary GWAS hits) in women | ||||
|---|---|---|---|---|---|---|
|
OR (95% CI) |
P | P for heterogeneity or pleiotropy |
OR (95% CI) |
P | P for heterogeneity or pleiotropy | |
| BMI (n = 237/299) | ||||||
| Random‐effects IVW | 1.22 (1.04–1.44) | 1.5 × 10−2 | <0.001 | 1.19 (1.04–1.36) | 1.2 × 10−2 | <0.001 |
| MR‐Egger | 1.11 (0.71–1.72) | 0.65 | 0.62 | 1.03 (0.71–1.48) | 0.89 | 0.40 |
| MR‐PRESSO (detected outliers 4/NA) | 1.19 (1.04–1.37) | 1.4 × 10−2 | – | 1.19 (1.04–1.36) | 1.2 × 10−2 | – |
| WHR (n = 7/249) | ||||||
| Random‐effects IVW | 1.22 (0.94–1.59) | 0.13 | 0.001 | 0.86 (0.76–0.97) | 1.4 × 10−2 | 0.01 |
| MR‐Egger | 1.11 (0.41–2.97) | 0.84 | 0.84 | 1.08 (0.81–1.44) | 0.59 | 0.08 |
| MR‐PRESSO (detected outliers 1/2) | 1.16 (0.90–1.49) | 0.25 | – | 0.83 (0.74–0.93) | 2.0 × 10−3 | – |
N values (number of SNPs) for BMI, WHR, and WHRadjBMI and numbers of detected outliers in the MR‐PRESSO analyses are for the analysis of primary + secondary GWAS hits/analysis of primary SNPs only. See Table 1 for definitions.
Table 4.
Association between genetically predicted obesity‐related traits and risk of rheumatoid arthritis by sex, in a sensitivity analysis excluding genetic instruments in the HLA region or associated with confounding traits*
| Parameter, MR approach | SNPs at P < 5 × 10−9 (primary + secondary GWAS hits) in men | SNPs at P < 5 × 10−9 (primary + secondary GWAS hits) in women | ||||
|---|---|---|---|---|---|---|
|
OR (95% CI) |
P | P for heterogeneity or pleiotropy |
OR (95% CI) |
P | P for heterogeneity or pleiotropy | |
| BMI (n = 198/247) | ||||||
| Random‐effects IVW | 1.29 (1.06–1.55) | 9.2 × 10−3 | <0.001 | 1.28 (1.09–1.50) | 3.0 × 10−3 | <0.001 |
| MR‐Egger | 1.25 (0.73–2.12) | 0.42 | 0.90 | 1.83 (1.01–3.32) | 0.05 | 0.22 |
| MR‐PRESSO (detected outliers 4/NA) | 1.25 (1.06–1.46) | 6.9 × 10−3 | – | 1.28 (1.09–1.50) | 3.0 × 10−3 | – |
| WHR (n = 59/203) | ||||||
| Random‐effects IVW | 1.23 (0.92–1.63) | 0.16 | 0.02 | 0.87 (0.75–1.01) | 0.06 | 0.01 |
| MR‐Egger | 0.94 (0.34–2.64) | 0.91 | 0.61 | 1.28 (0.86–1.93) | 0.23 | 0.04 |
| MR‐PRESSO (detected outliers 1/2) | 1.14 (0.88–1.49) | 0.32 | – | 0.83 (0.72–0.95) | 8.1 × 10−3 | – |
N values (number of SNPs) for BMI, WHR, and WHRadjBMI and numbers of detected outliers in the MR‐PRESSO analyses are for the analysis of primary + secondary GWAS hits/analysis of primary SNPs only. See Table 1 for definitions.
We performed additional sensitivity analyses with restriction to only primary association signals (which are believed to be stronger IVs) (Tables 1 and 2). The results were not substantially different when different sets of IVs were used.
DISCUSSION
In the present study, we leveraged the largest genetic data set for 3 obesity‐related traits (BMI, WHR, and WHRadjBMI) published to date, together with the largest GWAS addressing the outcome of interest, to understand causal overlaps and causal relationships between obesity and risk of RA. We found a modest but significant shared genetic basis between obesity‐related exposures and RA on several genomic regions. We further identified a pronounced causal effect of BMI in the development of RA and observed that the effect of obesity on risk of RA is similar between women and men.
The present results are in accordance with findings from earlier epidemiologic investigations. Reports from 3 meta‐analyses have described a dose‐response relationship between BMI and RA (4, 5, 6). Specifically, a linear relationship with risk ratio for RA of 1.13 per 5‐kg/m2 increment in BMI has been described (4), a result that is strongly supported by our finding of an increased risk of RA with genetically predicted BMI fitting a linear model. However, studies exploring the association of WC/WHR with RA are lacking. To our knowledge, only 2 population‐based studies have examined WC and RA risk. One prospective study based on the Danish Diet, Cancer and Health cohort of 55,037 subjects has shown a modestly increased RA risk per 5‐cm WC increment in both men (OR 1.04 [95% CI 0.69–1.57]) and women (OR 1.05 [95% CI 1.01–1.09]) (8). Similar results were found in a case–control study of 557 RA cases and 1,671 controls (OR 1.02 per 1‐cm increase [95% CI 1.01–1.04]) (7). However, these findings are not supported by our current MR study, which showed no evidence of a causal relationship between WHR and RA. When we assessed the association of RA risk with WHRadjBMI‐RA, thus removing the effect of BMI from WHR, our finding of a negative WHRadjBMI–RA association further confirmed the validity of our results. It is thus reasonable to assume that conventional epidemiologic settings could not eliminate the confounding of BMI from WC/WHR, and the findings of studies exploring a relationship of WC/WHR to RA are likely to be confounded by the effect of BMI.
Our results extend findings from a previous MR study that also demonstrated a positive association between genetically predicted BMI and RA (11). However, compared to that study, which utilized 68 SNPs as instruments and 7,480 RA cases as outcomes, our current study has several advantages. We significantly improved statistical power by incorporating >600 index SNPs as IVs based on a sample of >600,000 individuals for the exposure, as well as the largest meta‐GWAS on the outcome RA. We carefully checked and controlled for bias arising from population stratification by restricting participants to individuals of European ancestry. We interrogated not only BMI but also an indicator of abdominal fat distribution (WHR) and did not find compelling evidence of a causal role of WHR. Finally, we conducted additional sensitivity analyses to verify MR model assumptions. We selected the most significant independent SNPs identified by the largest GWAS on obesity (primary signals at a stringent GWAS P threshold of 5 × 10−9), so all were robustly associated with the exposure of interest, guaranteeing the “relevance” assumption. We excluded SNPs located in the HLA region or associated with potential confounders of the exposure–outcome relationship, to satisfy “exclusion restriction” assumptions. The consistent results observed across different approaches lend further support for a putative causal relationship between BMI and RA.
Biologic mechanisms linking BMI to RA remain largely unknown, though with several well‐documented assumptions. Obesity, especially the accumulation of visceral fat, contributes to a chronic low‐grade proinflammatory state (22). Adipocytes act as an active endocrine/paracrine organ that secretes a number of adipokines involved in the regulation of inflammation and autoimmunity (9, 23). For example, leptin, an adipokine produced by adipocytes, up‐regulates phagocytic function and secretion of proinflammatory cytokines, such as tumor necrosis factor, interleukin‐6 (IL‐6), and IL‐12, in monocyte/macrophages and modifies T cell differentiation toward a proinflammatory state (24). Thus, leptin has been suggested to play a role in the pathogenesis of RA (25). Although we hypothesized the inflammatory nature of adiposity as an underlying etiology for RA risk, we did not identify an association with WHR. BMI has been found to be highly correlated with excess fat mass (r = 0.94) and abdominal visceral fat (r = 0.71) (26), while WHR poorly predicted the accumulation of visceral fat (27, 28), which may account for our significant findings with BMI but not with WHR. Moreover, studies have suggested that local adipose, such as articular adipose or infrapatellar fat pad, plays a pivotal role in the pathologic process of RA. Articular adipose tissue secretes a large amount of inflammation‐related factors (e.g., IL‐6, IL‐8, adipokines) and intensifies pathogenic activities of rheumatoid fibroblast‐like synoviocytes (29). Finally, higher BMI is associated with an increased level of estradiol (30, 31), and estrogens are suggested to have proinflammatory effects (32). Although the exact role of sex hormones in the development of RA remains to be elucidated, their involvement has been confirmed (33).
Overall, our study demonstrates a putative causal relationship between BMI and risk of RA, which provides novel insights into the disease mechanism of RA and suggests an actionable prevention strategy. However, the study had some limitations that should be taken into account. First, due to limited data, we were not able to examine RA subtypes characterized by anti–citrullinated antibody status. The current RA GWAS summary statistics were based on a mix of 88.1% seropositive, 9.3% seronegative, and 2.6% unknown‐status cases. Studies have demonstrated differing effects of BMI in the 2 distinct RA subtypes (34, 35), and future MR analyses should be designed to explore these.
Moreover, explaining and understanding the sex disparity underlying RA remains challenging, as confounding by sex and by interaction of sex with the underlying genetics in both RA and obesity is likely. Obesity‐related traits are highly correlated across men and women, with most IVs shared by both sexes. It is thus difficult to obtain sex‐specific IVs. For RA, unfortunately, the meta‐GWAS was performed without adjustment for sex to maximize statistical power; in that large‐scale genotyping collaboration across multiple countries and institutions (14), additional covariates besides blood samples were not collected (Okada Y: personal communication). Our sex‐specific analysis should be interpreted with caution.
Additionally, because the genetic code is fixed at conception, MR typically compares groups of the population having different trajectories in their distribution of exposure over time. Our analyses can therefore be interpreted as assessing the impact of long‐term elevated BMI. However, we acknowledge that incomplete information about how genetic variant changes the distribution of exposure across the life course may bias the results: if genetic associations with exposure vary over time, then MR estimates based on genetic associations with the exposure measured at a single time point can be unreliable. This was, however, a minor issue in our study; although an earlier age‐stratified GWAS identified 15 loci with age‐specific effects, these SNPs, which exert time‐varying effects on BMI, were small in amount and with modest variations across age groups (36).
Of note, despite the many solutions proposed to satisfy MR model assumptions, these falsification strategies can only detect that an assumption is violated but cannot ever confirm that it holds. For example, current identification of IV–confounder associations relies heavily on conventional techniques (genome‐wide scan) and established knowledge (through literature review and the National Human Genome Research Institute–European Bioinformatics Institute GWAS catalog). We can never be certain that all IVs associated with confounders of the exposure–outcome relationship were excluded; the association might remain to be identified or the SNP may be associated with an underlying risk factor that is unrecognized.
Future studies may include individual‐level data to investigate genetic risk score–based approaches and to test, for example, the imbalance in measured covariates across levels of the proposed instruments. Finally, there should be additional efforts to increase generalizability as almost all current MR studies in RA have been conducted in European adult populations (age >18 years). Future investigations should also use MR analysis to explore treatment strategy and prognosis of RA, in addition to disease onset.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Jiang had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Tang, Jiang.
Acquisition of data
Tang, Jiang.
Analysis and interpretation of data
Tang, Shi, Alfredsson, Klareskog, Padyukov, Jiang.
Supporting information
Supplementary Material
Acknowledgments
We are grateful to all investigators who shared genome‐wide summary statistics and to Dr. Stephen Burgess for insightful suggestions in interpreting MR results with time‐varying exposure.
Dr. Jiang’s work was supported by a starting grant from the Swedish Research Council.
No potential conflicts of interest relevant to this article were reported.
References
- 1. Kiadaliri AA, Kristensen LE, Englund M. Burden of rheumatoid arthritis in the Nordic region, 1990–2015: a comparative analysis using the Global Burden of Disease Study 2015. Scand J Rheumatol 2018;47:1–101. [DOI] [PubMed] [Google Scholar]
- 2. Malmstrom V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting [review]. Nat Rev Immunol 2017;17:60–75. [DOI] [PubMed] [Google Scholar]
- 3. Hedstrom AK, Klareskog L, Alfredsson L. Interplay between obesity and smoking with regard to RA risk. RMD Open 2019;5:e000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng J, Chen Q, Yu F, Wang Z, Chen S, Jin Z, et al. Body mass index and risk of rheumatoid arthritis: a meta‐analysis of observational studies. Medicine (Baltimore) 2016;95:e2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feng X, Xu X, Shi Y, Liu X, Liu H, Hou H, et al. Body mass index and the risk of rheumatoid arthritis: an updated dose‐response meta‐analysis. Biomed Res Int 2019;2019:3579081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin B, Yang M, Fu H, Ma N, Wei T, Tang Q, et al. Body mass index and the risk of rheumatoid arthritis: a systematic review and dose‐response meta‐analysis. Arthritis Res Ther 2015;17:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ljung L, Rantapaa‐Dahlqvist S. Abdominal obesity, gender and the risk of rheumatoid arthritis: a nested case‐control study. Arthritis Res Ther 2016;18:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Linauskas A, Overvad K, Symmons D, Johansen MB, Stengaard‐Pedersen K, de Thurah A. Body fat percentage, waist circumference, and obesity as risk factors for rheumatoid arthritis: a Danish cohort study. Arthritis Care Res (Hoboken) 2019;71:777–86. [DOI] [PubMed] [Google Scholar]
- 9. Stavropoulos‐Kalinoglou A, Metsios GS, Koutedakis Y, Kitas GD. Obesity in rheumatoid arthritis. Rheumatology (Oxford) 2010;50: 450–62. [DOI] [PubMed] [Google Scholar]
- 10. Davies NM, Holmes MV, Smith GD. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bae SC, Lee YH. Causal association between body mass index and risk of rheumatoid arthritis: a Mendelian randomization study. Eur J Clin Invest 2019;49:e13076. [DOI] [PubMed] [Google Scholar]
- 12. Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta‐analysis of genome‐wide association studies for body fat distribution in 694,649 individuals of European ancestry. Hum Mol Genet 2019;28:166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hartwig FP, Davies NM, Hemani G, Smith GD. Two‐sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol 2016;45:1717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bulik‐Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi H, Mancuso N, Spendlove S, Pasaniuc B. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am J Hum Genet 2017;101:737–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR‐Egger method. Eur J Epidemiol 2017;32:377–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weyand CM, Goronzy JJ. Association of MHC and rheumatoid arthritis: HLA polymorphisms in phenotypic variants of rheumatoid arthritis. Arthritis Res Ther 2000;2:212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol 2012;42:1497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR‐Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev 2010;11:11–8. [DOI] [PubMed] [Google Scholar]
- 23. Lago F, Dieguez C, Gómez‐Reino J, Gualillo O. Adipokines as emerging mediators of immune response and inflammation. Nat Clin Pract Rheumatol 2007;3:716–24. [DOI] [PubMed] [Google Scholar]
- 24. Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. J Immunol 2005;174:3137–42. [DOI] [PubMed] [Google Scholar]
- 25. Tian G, Liang JN, Wang ZY, Zhou D. Emerging role of leptin in rheumatoid arthritis. Clin Exp Immunol 2014;177:557–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bouchard C. BMI, fat mass, abdominal adiposity and visceral fat: where is the ‘beef’? Int J Obes (Lond) 2007;31:1552–3. [DOI] [PubMed] [Google Scholar]
- 27. Van der Kooy K, Leenen R, Seidell JC, Deurenberg P, Droop A, Bakker CJ. Waist‐hip ratio is a poor predictor of changes in visceral fat. Am J Clin Nutr 1993;57:327–33. [DOI] [PubMed] [Google Scholar]
- 28. Busetto L, Baggio MB, Zurlo F, Carraro R, Digito M, Enzi G. Assessment of abdominal fat distribution in obese patients: anthropometry versus computerized tomography. Int J Obes Relat Metab Disord 1992;16:731–6. [PubMed] [Google Scholar]
- 29. Kontny E, Plebanczyk M, Lisowska B, Olszewska M, Maldyk P, Maslinski W. Comparison of rheumatoid articular adipose and synovial tissue reactivity to proinflammatory stimuli: contribution to adipocytokine network. Ann Rheum Dis 2012;71:262–7. [DOI] [PubMed] [Google Scholar]
- 30. Rohrmann S, Shiels MS, Lopez DS, Rifai N, Nelson WG, Kanarek N, et al. Body fatness and sex steroid hormone concentrations in US men: results from NHANES III. Cancer Causes Control 2011;22:1141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McTiernan A, Wu L, Chen C, Chlebowski R, Mossavar‐Rahmani Y, Modugno F, et al. Relation of BMI and physical activity to sex hormones in postmenopausal women. Obesity 2006;14:1662–77. [DOI] [PubMed] [Google Scholar]
- 32. Cutolo M, Sulli A, Capellino S, Villaggio B, Montagna P, Pizzorni C, et al. Anti‐TNF and sex hormones. Ann N Y Acad Sci 2006;1069:391–400. [DOI] [PubMed] [Google Scholar]
- 33. Alpízar‐Rodríguez D, Pluchino N, Canny G, Gabay C, Finckh A. The role of female hormonal factors in the development of rheumatoid arthritis. Rheumatology (Oxford) 2016;56:1254–63. [DOI] [PubMed] [Google Scholar]
- 34. Wesley A, Bengtsson C, Elkan AC, Klareskog L, Alfredsson L, Wedrén S, et al. Association between body mass index and anti–citrullinated protein antibody–positive and anti–citrullinated protein antibody–negative rheumatoid arthritis: results from a population‐based case–control study. Arthritis Care Res (Hoboken) 2013;65:107–12. [DOI] [PubMed] [Google Scholar]
- 35. Pedersen M, Jacobsen S, Klarlund M, Pedersen BV, Wiik A, Wohlfahrt J, et al. Environmental risk factors differ between rheumatoid arthritis with and without auto‐antibodies against cyclic citrullinated peptides. Arthritis Res Ther 2006;8:R133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Winkler TW, Justice AE, Graff M, Barata L, Feitosa MF, Chu S, et al. The influence of age and sex on genetic associations with adult body size and shape: a large‐scale genome‐wide interaction study [corrected and republished in PLoS Genet 2016 Jun;12:e1006166]. PLoS Genet 2015;11:e1005378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material