

Abstract
1,4‐Dihydropyridines (DHP), the most commonly used antihypertensives, function by inhibiting the L‐type voltage‐gated Ca2+ (Cav) channels. DHP compounds exhibit chirality‐specific antagonistic or agonistic effects. The structure of rabbit Cav1.1 bound to an achiral drug nifedipine reveals the general binding mode for DHP drugs, but the molecular basis for chiral specificity remained elusive. Herein, we report five cryo‐EM structures of nanodisc‐embedded Cav1.1 in the presence of the bestselling drug amlodipine, a DHP antagonist (R)‐(+)‐Bay K8644, and a titration of its agonistic enantiomer (S)‐(−)‐Bay K8644 at resolutions of 2.9–3.4 Å. The amlodipine‐bound structure reveals the molecular basis for the high efficacy of the drug. All structures with the addition of the Bay K8644 enantiomers exhibit similar inactivated conformations, suggesting that (S)‐(−)‐Bay K8644, when acting as an agonist, is insufficient to lock the activated state of the channel for a prolonged duration.
Keywords: cryo-electron microscopy, inhibitors, nanodiscs, structural biology, voltage-gated calcium ion channels
High‐resolution cryo‐EM structures of nanodisc‐embedded Cav1.1 in complex with antagonists Levamlodipine, (R)‐(+)‐Bay K8644, and dual agonist/antagonist (S)‐(−)‐Bay K8644 reveal the molecular basis of the allosteric modulation of the voltage‐gated Ca2+ channel Cav1.1 by dihydropyridine (DHP) drugs. Advanced structural understanding of the stereo‐selective mode of action of DHP will facilitate drug discovery targeting Cav channels.
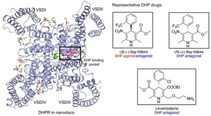
Introduction
Voltage‐gated Ca2+ (Cav) channels (VGCC) are responsible for a broad spectrum of physiological events, including muscle contraction, secretion, and synaptic signal transduction. [1] In mammals, 10 subtypes of Cav channels are classified to three subfamilies: Cav1 (Cav1.1‐Cav1.4), Cav2 (Cav2.1‐Cav2.3), and Cav3 (Cav3.1‐Cav3.3). Cav1 channels, also known as the L‐type VGCCs or dihydropyridine (DHP) receptors (DHPRs), are composed of a core α1 subunit and three auxiliary subunits, α2δ, β, and γ. [2] The α1 subunit is a single polypeptide of ≈2000 amino acids, folding into four homologous repeats I–IV. Each repeat contains six transmembrane segments (S1–S6) that form two functional entities: segments S1–S4 in each repeat constitute the peripheral voltage sensing domains (VSDs), and the S5–S6 segments from the four repeats, along with the intervening pore helices P1 and P2, enclose the central ion‐permeating pore domain (PD). The short fragments between P1 and P2 from the four repeats serve as the molecular sieve, known as the selectivity filter (SF), which discriminates Ca2+ from other ions.[ 1 , 2 ]
Dysfunctional Cav channels are associated with various pathophysiological conditions ranging from cardiovascular disorders to psychiatric and neurological syndromes, such as cardiac arrhythmias, seizures, epilepsy, autism, and Parkinson's disease.[ 1 , 2 ] Antagonists of DHPRs have demonstrated excellent efficacy in clinical practice for the treatment of specific conditions, including hypertension, cardiac ischemia, pain and tremor. [3]
Among the Cav antagonists, DHP compounds are the most widely prescribed drugs, which, exemplified by amlodipine and nifedipine, have been the world's bestsellers for decades. [4] Benzothiazepines (BTZ) and phenylalkylamines (PAA) represent the other two major classes of DHPR‐targeting drugs.[ 4a , 5 ] While BTZ and PAA compounds directly block ion conduction by traversing the central cavity of the pore domain, DHP antagonists bind to the fenestration, which is the portal on the PD side wall, on the interface of repeats III and IV for allosteric modulation. [5b]
While DHP drugs act as antagonists, some DHP compounds exhibit agonistic effects on DHPRs. Even more intriguingly, the mode of action (MOA) of some DHP compounds is stereo‐selective; enantiomers of these DHP ligands possess opposite pharmacological profiles. [6] For instance, whereas compound (S)‐(−)‐Bay K8644 is a potent agonist for DHPRs, its enantiomer, (R)‐(+)‐Bay K 8644 displays antagonistic activity (Figure 1 A). [7] It is also noted that (S)‐(−)‐Bay K8644 produces a biphasic dose‐response effect, whereby the agonist is turned to an antagonist when applied at high concentrations. [8] The biphasic property is also observed for modulators of other membrane proteins, such as dopamine D‐2 and cannabinoid receptors, for which the usage dose should be finely regulated to achieve the desired efficacy. [9]
Figure 1.

Cryo‐EM analysis of Cav1.1 bound separately to RBK, SBK, and amlodipine in nanodiscs. A) Chemical structures of (S)‐(−)‐Bay K8644 (SBK), (R)‐(+)‐Bay K8644 (RBK), and levamlodipine (amlodipine). B) Last step purification for rCav1.1 in MSP2N2‐surrounded nanodiscs. Shown here is a representative size‐exclusion chromatogram (SEC) and Coomassie blue‐stained SDS‐PAGE. C) Local resolution maps and densities for the bound ligands. Left: Resolution heatmap for the overall 3DEM reconstruction of rCav1.1‐100S. The unit for the scale bar is Å. Right: The local resolutions for the bound ligands. The density of the ligand in each reconstruction is highlighted with an orange square in the upper row. The densities in the lower row, shown as blue mesh, are contoured at 3σ and prepared in PyMol.
An in‐depth understanding of the MOA of these modulators requires high‐resolution structures. Owing to the advances of the resolution revolution of single‐particle cryogenic electron microscopy (cryo‐EM), we were able to resolve the structures of the rabbit Cav1.1 (rCav1.1) channel complex and human Cav3.1 alone and in the presence of various small molecule ligands. [10] Specifically, the atomic structures of rCav1.1 bound to representative DHP, BTZ, and PAA drugs, nifedipine, diltiazem, and verapamil, respectively, have elucidated the molecular details of the drugs’ action. [10c]
Notwithstanding these exciting structural advances, there are a number of outstanding questions. First, all the reported structures of eukaryotic Cav channels and the closely related voltage‐gated sodium ion (Nav) channels are of proteins purified in detergent micelles. It is unclear whether the detergents have altered local structures of these highly dynamic channels. As such, structural elucidation in a more physiologically relevant environment, such as in nanodiscs, is required. Second, in the structure of Cav1.1 complexed with the agonist (S)‐(−)‐Bay K8644, the overall structure conforms to what is expected to be an inactivated state. [10c] (S)‐(−)‐Bay K8644 was applied at 100 μM, a dose at which the compound may function as an antagonist. It remains to be tested whether the agonist at lower concentrations may lock the channel in an activated state. Last but not least, both DHP compounds, (S)‐(−)‐Bay K8644 and nifedipine, have relatively small chemical groups. It has yet to be seen how the bulkier groups of some DHP drugs, such as levamlodipine (the pharmacologically active enantiomer S‐amlodipine; referred to as amlodipine for short hereafter), are coordinated by DHPR.
To address these remaining questions, we sought to resolve the structures of rabbit Cav1.1 reconstituted in nanodiscs with addition of representative DHPR compounds. Here we report high‐resolution cryo‐EM structures of nanodisc‐embedded Cav1.1 bound to two antagonists, amlodipine and (R)‐(+)‐Bay K 8644, and a titration of (S)‐(−)‐Bay K8644 (Figure 1 A). These structures together provide advanced knowledge on the modulation of DHPRs by DHP compounds. For simplicity, we will refer to the enantiomers of Bay K8644 as RBK and SBK.
Results and Discussion
Structures of Cav1.1 in Lipid Nanodiscs are Nearly Identical to Those in Detergent Micelles
The endogenous rCav1.1 complex isolated from the rabbit skeletal muscle was purified following our published protocols.[ 10a , 11 ] Please refer to the Supporting Information for details of nanodisc reconstitution with the membrane scaffold protein 2N2 (MSP2N2) and 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC) (Figure 1 B and Supporting Information, Figure S1). The mono‐dispersed peak fractions of rCav1.1 nanodiscs from size‐exclusion chromatography (SEC) were pooled and incubated with the target molecules, amlodipine or RBK at a final concentration of 100 μm, and SBK at 100 μm or 10 μm, before cryo‐sample preparation. For SBK at 1 μm, the compound was added during nanodisc reconstitution and included in the SEC running buffer.
The cryo‐grids were made following a standard protocol and images were collected on a Titan Krios G3 cryo‐electron microscope equipped with the spherical aberration (Cs) image corrector and GIF quantum electron energy filter. The workflow for data processing is described in the Supporting Information (Supporting Information, Figures S2 and S3). The overall resolutions of the channel complexes, all embedded in nanodiscs, were determined at 2.9 Å with amlodipine, 3.2 Å with RBK, and 3.4 Å, 3.4 Å, and 3.0 Å with SBK at 1 μm, 10 μm, and 100 μm, respectively (PDB ID: 7JPX, 7JPW, 7JPV, 7JPL, and 7JPK, respectively). For simplicity, we will refer to these five structures as rCav1.1‐100A (with 100 μm amlodipine), 100R (with 100 μm RBK), and 1S/10S/100S (with SBK applied at three different concentrations). Our published structure of digitonin‐embedded rCav1.1 in the presence of 200 μm nifedipine, used as structural reference several times in this manuscript, will be referred to as rCav1.1‐200N (PDB code: 6JP5). [10c] The excellent map quality and high local resolutions allowed for accurate assignment of the DHP ligands (Figure 1 C and Supporting Information, Figure S4).
The overall structures of rCav1.1‐100A and rCav1.1‐100R are nearly identical to that of rCav1.1‐200N, with root‐mean‐square deviation (RMSD) values both of 0.52 Å over 1115 Cα atoms for the α1 subunit (Figure 2 A,B). The structure of rCav1.1‐100S in nanodiscs is also nearly identical to that in glyco‐diosgenin (GDN) micelles (PDB code: 6JP8), [10c] with the RMSD of 0.55 Å over 1115 Cα atoms for α1 subunit (Figure 2 C).
Figure 2.
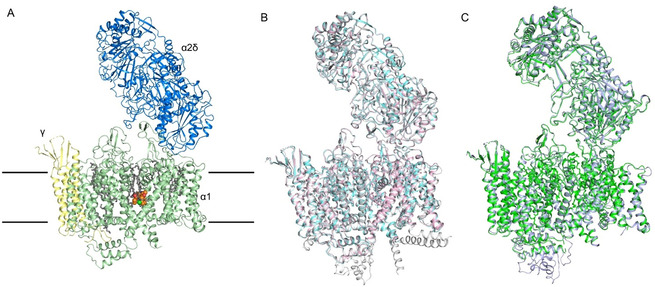
Nearly identical conformations of rCav1.1 in the presence of DHP compounds in detergents and in nanodiscs. A) Overall structure of the amlodipine‐bound rCav1.1 complex (rCav1.1‐100A) at 2.9 Å resolution. The overall structure of the channel complex is colored for different subunits. The β1 subunit is omitted throughout the manuscript because of its poor resolution. Amlodipine is shown as orange spheres and the bound lipids are shown as gray sticks. The approximate position of the membrane is indicated by black lines. B) The structures of rCav1.1‐100A (with 100 μm amlodipine, pink) and rCav1.1‐100R (with 100 μm RBK, cyan) in nanodiscs and rCav1.1‐200N (with 200 μm nifedipine, gray) in digitonin are nearly identical. C) Structures of rCav1.1‐100S (with 100 μm SBK) in nanodisc (green) and in digitonin (light purple) are nearly identical.
These observations alleviate the concerns about the potential interference with the structural integrity and conformations of Cav1.1, and probably all other single‐chain Cav and Nav channels whose structures have been resolved, by detergents.
Nearly Identical Conformations of rCav1.1 with SBK Titrations
We applied SBK to rCav1.1 with a concentration gradient in the hope to capture different channel states. However, the three structures of SBK‐bound rCav1.1 in nanodiscs are nearly identical. The intracellular gate is closed and the four VSDs exhibit depolarized conformations in all three structures, characteristic of the putative inactivated state (Figure 3 A).
Figure 3.
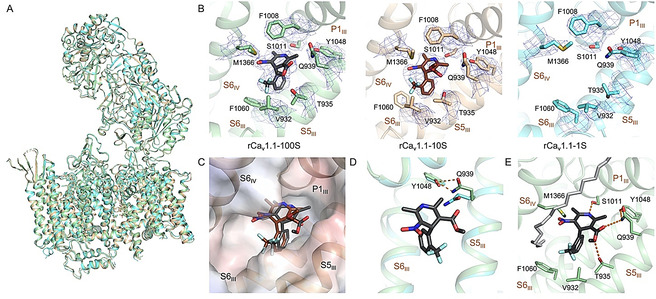
The conformation of nanodisc‐embedded rCav1.1 remains nearly unchanged with SBK applied at different conformations. A) Structural comparison of rCav1.1‐100S (green), 10S (gold), and 1S (cyan). Despite different concentrations of SBK applied, the overall structure remains nearly unchanged. B) There is no density of SBK in the EM reconstruction of rCav1.1‐1S. The densities for SBK and its surrounding residues, all contoured at 7σ in PyMol, are shown as blue mesh. C) Slight displacement of SBK in rCav1.1‐100S (black) and 10S (brown). The electrostatic surface potential of rCav1.1‐100S, in a semi‐transparent presentation, was calculated in PyMol. D) Gln939 on S5III moves toward Tyr1048 in the presence of SBK, as seen between rCav1.1‐100S (green) and rCav1.1‐1S (cyan). This local shift is important for accommodating the ligand. E) Coordination of SBK by both polar and hydrophobic residues. Potential H bonds are shown as dashed lines. A nearby lipid (gray sticks) that blocks the III–IV fenestration with one hydrophobic tail was resolved.
It is of particular note that the fenestration on the interface between repeats III and IV (the III–IV fenestration), which is the expected DHP binding site, is empty in rCav1.1‐1S, although 1 μm SBK was added during nanodisc reconstitution and the subsequent SEC purification. In contrast, SBK was resolved in rCav1.1‐10S/100S, although the density for SBK in rCav1.1‐10S is worse than that in rCav1.1‐100S, implying less stable coordination (Figure 3 B).
In both rCav1.1‐100S and 10S, SBK is nestled into the same III‐IV fenestration site, surrounded by residues on the segments P1III, S5III, S6III, and S6IV (Figure 3 B). A close examination shows a slight displacement of SBK in rCav1.1‐100S and 10S. In particular, the C3‐ester group appears to rotate by ≈90° in these two structures (Figure 3 C). This minor difference may not change SBK′s function as an antagonist to rCav1.1 in this particular conformation, but it remains to be elucidated whether the change of position and conformation of SBK in rCav1.1‐10S is reminiscent of that bound to an activated channel.
Due to the lack of ligand density, rCav1.1‐1S represents the state of an apo channel, hence providing a good control to trace the conformational changes of rCav1.1 in nanodiscs upon SBK binding. The only difference occurs in Gln939 on S5III (Figure 3 D). Upon SBK entry, Gln939 rotates toward Tyr1048 of S6III and mediates the essential hydrogen bond (H‐bond) network between the ester group of SBK and Tyr1048. It was reported that single point mutations corresponding to Y1048F and Y1048A resulted in reduced affinity with DHP by ≈10‐ and ≈1,000‐fold, respectively. [12] Our structural comparison suggests that Tyr1048 may be required for DHP association through stabilizing Gln939. Rotation of Gln939 facilitated by Tyr1048 clears the path for the entry of DHP ligands, followed by the stabilization from the hydrophobic and H‐bond interactions with the surrounding residues.
Because the coordination of SBK is largely identical to rCav1.1‐100S in GDN, [10c] it will not be elaborated here. Of note, a density corresponding to a phospholipid was well‐resolved outside the fenestration in the nanodisc‐embedded channel (Figure 3 E and Supporting Information, Figure S5 A). One aliphatic tail of the lipid blocks the fenestration. This arrangement may prevent the ligand from exiting the binding site. The role of phospholipid in ligand binding to membrane proteins has rarely been discussed. Our structural finding provides a clue to understanding the sophisticated ligand binding within membrane.
Comparison for DHP Enantiomers
SBK and RBK are a pair of DHP enantiomers with a chiral center on the C4 atom of the dihydropyridine ring (Figure 1 A). In rCav1.1‐100R, RBK is positioned in the same binding site as for SBK in rCav1.1‐10S/100S (Figure 4 A). Compared to SBK in rCav1.1‐100S, the CF3‐substituted phenyl ring of RBK projects into the hydrophobic pocket formed by Thr935, Val932, and Phe1060, while the dihydropyridine flips over by ≈180°. In this way, the nitro group of RBK is H‐bonded to Tyr1048/Gln939 and Thr935, similar to the role of C3‐ester in SBK (Figure 4 B).
Figure 4.
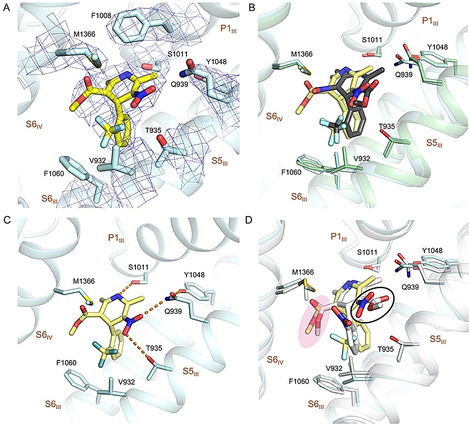
Coordination of the antagonistic RBK by rCav1.1. A) The densities for the RBK binding site in rCav1.1‐100R. RBK is shown as yellow sticks. The densities, shown as blue mesh, are contoured at 7σ and prepared in PyMol. B) Overlapping binding site for RBK and SBK. The structures of rCav1.1‐100R (blue) and −100S (green) are superimposed. RBK and SBK are colored yellow and black, respectively. C) Coordination of RBK. Potential H bonds are shown as dashed lines. D) Comparison of RBK and nifedipine coordination. The structure rCav1.1‐100R (yellow) and nifedipine (gray) are superimposed. The NO2 group in RBK is highlighted with a black circle. The C3‐ester groups of RBK and nifedipine are highlighted a with pink shadow.
On the other side, the C3‐ester of RBK, identical to that in nifedipine, is positioned in the vicinity of the hydrophobic residues, Met1366, Val932, and Phe1060 (Figure 4 C,D). Placement of the more polar nitro group in SBK to this hydrophobic surrounding is likely to underlie the unfavored binding of SBK to an inactivated conformation. [10c] The distinctive binding modes of DHP enantiomers, RBK and SBK, demonstrates the stereo‐selective activities of DHP drugs. Compared to the agonist SBK, RBK shares more conserved conformation with DHP antagonists (Figure 4 D).
Coordination of Amlodipine
Amlodipine is the most potent DHP antagonist that displays a pH‐dependent efficacy. [13] As expected, it is also accommodated in the III–IV fenestration of the PD. The bulky substituent in amlodipine, the ethanolamine group, points to the central cavity of the PD and is H‐bonded to the carbonyl group of Ser1011 (Figure 5 A,B). Except for this group, the interactions between amlodipine and the surrounding residues in rCav1.1 are nearly identical with those for nifedipine and RBK, including two H‐bonds between the C3‐ester and Tyr1048/Gln939/Thr935, and one between the N1 atom and Ser1011.
Figure 5.
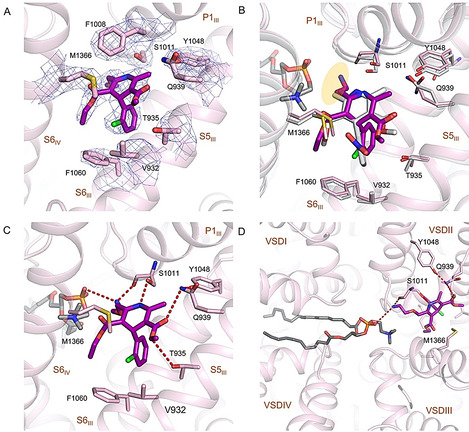
Specific interactions between rCav1.1 and amlodipine. A) The densities for amlodipine binding site in rCav1.1‐100A. Amlodipine is shown as magenta sticks. The densities, shown as blue mesh, are contoured at 7σ and prepared in PyMol. B) Comparison of amlodipine (magenta) and nifedipine (gray) coordination. The ethanolamine group in amlodipine is highlighted with a yellow shadow. C) Coordination of amlodipine by rCav1.1. The coordinating residues are shown as pink sticks. Potential H bonds are indicated by dashed lines. D) A transverse lipid in the central cavity interacts with amlodipine. The phosphate group of the lipid, assigned as a phosphatidylcholine, and Ser1011 coordinate the ethanolamine group of amlodipine from two opposite sides. Please refer to Figure S5 B in the Supporting Information for the density of the lipid.
In addition to the compound and surrounding residues, EM densities corresponding to lipids were resolved in the central cavity of DHP‐bound rCav1.1. It is noteworthy that such densities were also observed in the digitonin ‐solubilized conditions for both ligand‐free and antagonists‐bound rCav1.1 reconstructions. Although the resolution is insufficient to identify specific lipids, the contour allowed for putative docking of the lipid 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphoethanolamine (16:0–18:1 PE), a major component of eukaryotic plasma membrane. [10b]
The lipid appears to directly coordinate amlodipine. Specially, the phosphate group of the lipid points to the fenestration site and interacts with the ethanolamine substituent of amlodipine (Figure 5 C,D, Figure S5B). Although the exact identity of this specific lipid cannot be identified, the phosphate group is common in all phospholipids, suggesting that most phospholipids, if traversing the central cavity of PD under physiological condition, can interact with the bound amlodipine (Figure 5 D).
Amlodipine displays longer duration of action than nifedipine, allowing a once‐a‐day dosage regimen in human. [14] Although amlodipine forms one more H‐bond between its ethanolamine group and Ser1011 (Figure 5 C), its binding affinity remains at the same level compared to other C2‐methyl substituted DHPs. [15] The change of the pharmacokinetic properties and binding kinetics may be attributed to the positive charge carried by the ethanolamine group under physiological pH of approximately 7.4. The DHP modulators get access to the binding site, which is in the mid‐height of the membrane, from the lateral side. To reach the fenestration, they have to diffuse through the lipid bilayer. Increased polarity or extra charge may impede the diffusion of amlodipine in the hydrophobic bilayer, resulting in decreased on/off rate with regard to association with the polar site in Cav1 channels. [16] Supporting this analysis, the on/off rate of amlodipine became similar to that of nisoldipine at pH 10, under which condition the ethanolamine is neutral. [13]
Conclusion
rCav1.1 was the first single‐chain VGIC (voltage‐gated ion channels) whose structure was determined.[ 10a , 10b ] We have been employing it as a prototype for cryo‐EM analysis of VGIC members. Before this study, all the structures of rCav1.1, as well as the closely related human Cav3.1 and multiple eukaryotic Nav channels, were solved for purified proteins embedded in detergent micelles. [17] We had some concerns with potential structural perturbation of these highly dynamic molecular machines by detergents. The structural similarity of rCav1.1 in nanodiscs and in detergent micelles, which was also observed in our recently published NaChBac, [11] alleviates this concern and consolidates the structural findings obtained from the detergent‐embedded channels. More satisfyingly, the coordination of the backbones of all resolved DHP antagonists, nifedipine, [10c] amlodipine, and RBK, is conserved regardless of the surrounding milieu of detergents or nanodiscs.
The high resolutions of rCav1.1 in complex with different DHP compounds allowed for detailed analyses of ligand binding, providing the basis to understand the distinct efficacy of the drugs and functional groups of the channel. The direct participation of a phospholipid in the coordination of amlodipine is intriguing in that it is unclear whether a phospholipid can enter the central cavity in the intact membrane. Although our structure was obtained in nanodiscs, the native membrane was disrupted during protein extraction and purification. Therefore, this remains to be an enigma that awaits further investigation. Multiple biophysical and computational approaches are needed to dissect the mechanism and the function of the transverse lipids in the physiology and pharmacology of VGIC channels.
We had aimed to utilize the well‐characterized agonist SBK to capture the channel in an activated state. Despite our attempt to apply SBK at different doses and in different environments, the channel remains in the same inactivated state. Notably, when applied at 1 μm, there was no SBK density observed in the reconstruction. It was shown that SBK, applied at 3 μm, [18] could prolong rat Cav1.1 opening. The lack of SBK density at 1 μm supports our previous analysis that the inactivated conformation may not favor SBK binding. Therefore, a high‐concentration of SBK is required to compensate for the penalty and stabilize the ligand binding to the inactivated conformation. [10c] It is also noted that the rat Cav1.1 channels closed within 1 s even in the presence of 3 μm SBK, [18] suggesting that SBK is insufficient to keep the channel in an activated conformation, which may represent a transient intermediate upon depolarization. Therefore, other strategies have to be developed for structural determination of Cav1.1 in the activated state. Because it is impractical to conveniently introduce point mutations to the endogenous rCav1.1 channels, new methods may be required to lock voltage‐gated ion channels in distinct functional states.
Despite the remaining questions, our structural analyses reported here and previously [10c] provide a more comprehensive understanding of the MOA of DHP drugs. The updated knowledge will facilitate drug discovery targeting other Cav channels and additional VGICs in general.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the cryo‐EM facility at Princeton Imaging and Analysis Center, which is partially supported by the Princeton Center for Complex Materials, a National Science Foundation (NSF)‐MRSEC program (DMR‐1420541). The work was supported by grant from NIH (5R01GM130762).
S. Gao, N. Yan, Angew. Chem. Int. Ed. 2021, 60, 3131.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.1101/2020.08.13.250340).
Contributor Information
Dr. Shuai Gao, Email: shuaig@princeton.edu.
Prof. Nieng Yan, Email: nyan@princeton.edu.
References
- 1.
- 1a. Catterall W. A., Cold Spring Harbor Perspect. Biol. 2011, 3, a003947; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Clapham D. E., Cell 2007, 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Ertel E. A., Campbell K. P., Harpold M. M., Hofmann F., Mori Y., Perez-Reyes E., Schwartz A., Snutch T. P., Tanabe T., Birnbaumer L., Tsien R. W., Catterall W. A., Neuron 2000, 25, 533–535; [DOI] [PubMed] [Google Scholar]
- 2b. Nowycky M. C., Fox A. P., Tsien R. W., Nature 1985, 316, 440–443. [DOI] [PubMed] [Google Scholar]
- 3. Zamponi G. W., Striessnig J., Koschak A., Dolphin A. C., Pharmacol. Rev. 2015, 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Zamponi G. W., Nat. Rev. Drug Discovery 2016, 15, 19–34; [DOI] [PubMed] [Google Scholar]
- 4b. Sorkin E. M., Clissold S. P., Brogden R. N., Drugs 1985, 30, 182–274. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Dolphin A. C., Br. J. Pharmacol. 2006, 147 S56–62; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Hockerman G. H., Peterson B. Z., Johnson B. D., Catterall W. A., Annu. Rev. Pharmacol. Toxicol. 1997, 37, 361–396. [DOI] [PubMed] [Google Scholar]
- 6. Goldmann S., Stoltefuss J., Angew. Chem. Int. Ed. Engl. 1991, 30, 1559–1578; [Google Scholar]; Angew. Chem. 1991, 103, 1587–1605. [Google Scholar]
- 7.
- 7a. Franckowiak G., Bechem M., Schramm M., Thomas G., Eur. J. Pharmacol. 1985, 114, 223–226; [DOI] [PubMed] [Google Scholar]
- 7b. Schramm M., Thomas G., Towart R., Franckowiak G., Nature 1983, 303, 535–537. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Dubé G. P., Baik Y. H., Schwartz A., J. Cardiovasc. Pharmacol. 1985, 7, 377–389; [DOI] [PubMed] [Google Scholar]
- 8b. Bechem M., Hoffmann H., Pfluegers Arch. 1993, 424, 343–353. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Monti J. M., Hawkins M., Jantos H., D′Angelo L., Fernández M., Psychopharmacology 1988, 95, 395–400; [DOI] [PubMed] [Google Scholar]
- 9b. Rey A. A., Purrio M., Viveros M.-P., Lutz B., Neuropsychopharmacology 2012, 37, 2624–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Wu J., Yan Z., Li Z., Yan C., Lu S., Dong M., Yan N., Science 2015, 350, aad2395; [DOI] [PubMed] [Google Scholar]
- 10b. Wu J., Yan Z., Li Z., Qian X., Lu S., Dong M., Zhou Q., Yan N., Nature 2016, 537, 191–196; [DOI] [PubMed] [Google Scholar]
- 10c. Zhao Y., Huang G., Wu J., Wu Q., Gao S., Yan Z., Lei J., Yan N., Cell 2019, 177, 1495–1506; [DOI] [PubMed] [Google Scholar]
- 10d. Zhao Y., Huang G., Wu Q., Wu K., Li R., Lei J., Pan X., Yan N., Nature 2019, 576, 492–497. [DOI] [PubMed] [Google Scholar]
- 11. Gao S., Valinsky W. C., On N. C., Houlihan P. R., Qu Q., Liu L., Pan X., Clapham D. E., Yan N., Proc. Natl. Acad. Sci. USA 2020, 117, 14187–14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peterson B. Z., Tanada T. N., Catterall W. A., J. Biol. Chem. 1996, 271, 5293–5296. [DOI] [PubMed] [Google Scholar]
- 13. Kass R. S., Arena J. P., J. Gen. Physiol. 1989, 93, 1109–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arrowsmith J. E., Campbell S. F., Cross P. E., Stubbs J. K., Burges R. A., Gardiner D. G., Blackburn K. J., J. Med. Chem. 1986, 29, 1696–1702. [DOI] [PubMed] [Google Scholar]
- 15. Nayler W. G., Gu X. H., J. Cardiovasc. Pharmacol. 1991, 17, 587–592. [DOI] [PubMed] [Google Scholar]
- 16. Mason R. P., Campbell S. F., Wang S. D., Herbette L. G., Mol. Pharmacol. 1989, 36, 634–640. [PubMed] [Google Scholar]
- 17.
- 17a. Shen H., Zhou Q., Pan X., Li Z., Wu J., Yan N., Science 2017, 355, eaal4326; [DOI] [PubMed] [Google Scholar]
- 17b. Yan Z., Zhou Q., Wang L., Wu J., Zhao Y., Huang G., Peng W., Shen H., Lei J., Yan N., Cell 2017, 170, 470–482; [DOI] [PubMed] [Google Scholar]
- 17c. Shen H., Li Z., Jiang Y., Pan X., Wu J., Cristofori-Armstrong B., Smith J. J., Chin Y. K. Y., Lei J., Zhou Q., King G. F., Yan N., Science 2018, 362, eaau2596; [DOI] [PubMed] [Google Scholar]
- 17d. Pan X., Li Z., Zhou Q., Shen H., Wu K., Huang X., Chen J., Zhang J., Zhu X., Lei J., Xiong W., Gong H., Xiao B., Yan N., Science 2018, 362, eaau2486; [DOI] [PubMed] [Google Scholar]
- 17e. Pan X., Li Z., Huang X., Huang G., Gao S., Shen H., Liu L., Lei J., Yan N., Science 2019, 363, 1309–1313; [DOI] [PubMed] [Google Scholar]
- 17f. Shen H., Liu D., Wu K., Lei J., Yan N., Science 2019, 363, 1303–1308. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Affolter H., Coronado R., Biophys. J. 1985, 48, 341–347; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Bechem M., Schramm M., J. Mol. Cell. Cardiol. 1987, 19, 63–75; [DOI] [PubMed] [Google Scholar]
- 18c. Brown A. M., Kunze D. L., Yatani A., Nature 1984, 311, 570–572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary